 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion receptor chemistry: highlights from 2011 and 2012
Philip A.
Gale
*,
Nathalie
Busschaert
,
Cally J. E.
Haynes
,
Louise E.
Karagiannidis
and
Isabelle L.
Kirby
Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: philip.gale@soton.ac.uk; Fax: +44 (0)23 8059 6805; Tel: +44 (0)23 8059 3332
First published on 9th October 2013
Abstract
This review covers advances in anion complexation in the years 2011 and 2012. The review covers both organic and inorganic systems and also highlights the applications to which anion receptors can be applied such as self-assembly and molecular architecture, sensing, catalysis and anion transport.
 Nathalie Busschaert, Cally Haynes, Phil Gale, Louise Karagiannidis and Isabelle Kirby | Phil Gale is Professor of Supramolecular Chemistry and Head of Chemistry at the University of Southampton. His research has recently focused on understanding transmembrane lipid bilayer anion transport processes mediated by small molecules with the goal of applying anion transporters to the future treatment of diseases such as cancer and cystic fibrosis. He has won a number of research awards including the Corday Morgan medal and prize from the RSC (2005) and a Royal Society Wolfson Research Merit Award (2013–2018). He is currently the chair of the editorial board of Chemical Society Reviews. |
Nathalie Busschaert completed a BSc in chemistry at the K. U. Leuven, Belgium, and continued studying for an MSc in chemistry at the same university. She then moved to the University to Southampton in 2010 to undertake a PhD under the supervision of Professor Philip A. Gale, with research stays at A*STAR, Singapore, and the University of Ljubljana, Slovenia. She is currently working as a post doctoral researcher in the group of Phil Gale. Her research interests are anion coordination chemistry, transmembrane anion transport and medicinal applications of supramolecular systems. |
Cally J. E. Haynes completed a masters degree in chemistry at Wadham College, Oxford in 2008 and went on to study for a PhD under the supervision of Professor Philip Gale at the University of Southampton from 2009–2011. She continued post doctoral work in the group of Phil Gale until 2013, and is currently working as a publishing editor at the Royal Society of Chemistry. Her research interests are in supramolecular chemistry, focusing on the coordination and membrane transport of anions. |
Louise E. Karagiannidis completed her masters degree in chemistry at the University of Southampton in 2011. She is currently in the third year of her PhD studies, under the supervision of Professor Philip A. Gale at the University of Southampton. Her research focuses on supramolecular chemistry, in particular the influence of receptor structure on the transmembrane transport of small anions. |
Isabelle L. Kirby graduated with a masters degree in chemistry from the University of Southampton in 2010. She is currently completing her PhD studies under the joint supervision of Professor Philip Gale and Dr Simon Coles in Southampton focusing on the molecular recognition of anions and the characterisation of non-covalent interactions in the solid-state. |
Introduction
This review is the latest in a series covering recent advances in anion complexation.1–4 It highlights the developments during 2011 and 2012 in the field of new receptors, modes of binding, transport and extraction of anions, and sensing mechanisms. It is not intended to be a comprehensive overview of the area. The structure of the review is primarily according to the dominant functional group used to complex the anion and by the potential applications of these systems. Many receptors use more than one functional group to bind anions or have various applications and the organisation of this review should be regarded as a flexible guide to the classification of these systems.Ammonium-based receptors
As anions are inherently negatively charged, an intuitive way of achieving anion binding is by the employment of positively charged species and ammonium groups have been widely used for this purpose. Bencini, Lippolis and co-workers have studied the effect of the aromatic linker between two [9]aneN3 ammonium units in receptors 1–3.5 In aqueous solutions the two [9]aneN3 units are protonated and have the potential to interact with anions via hydrogen bonding and electrostatic interactions. Using potentiometric, 1H NMR and 31P NMR measurements it was found that 1 selectively binds triphosphate over di- and monophosphate, while 2 selectively binds diphosphate and 3 monophosphate in aqueous solutions over a wide pH range, an order that correlates well with the distance between the two [9]aneN3 units in 1–3. Furthermore, 1 and 2 also preferentially interact with the nucleotides ATP (adenosine triphosphate) and ADP (adenosine diphosphate) over inorganic phosphate species, with the highest associations observed for receptor 1 with ATP and ADP. This can be attributed to the presence of π-stacking and hydrophobic interactions between the adenine units of the guest and the aromatic linker in 1 and 2, an effect that is more pronounced for the larger phenanthroline spacer in receptor 1. In a subsequent paper, the groups of Bencini and Lippolis reported the anion binding properties of terpyridine analog 4.6 Potentiometric titrations and 1H NMR and fluorescence spectra in water showed that receptor 4 is only able to selectively bind and sense diphosphate over mono- and triphosphate after the addition of Zn2+ leads to a conformational change from trans–trans to cis–cis to create a convergent binding site that has optimal dimensional fitting for diphosphate (Scheme 1).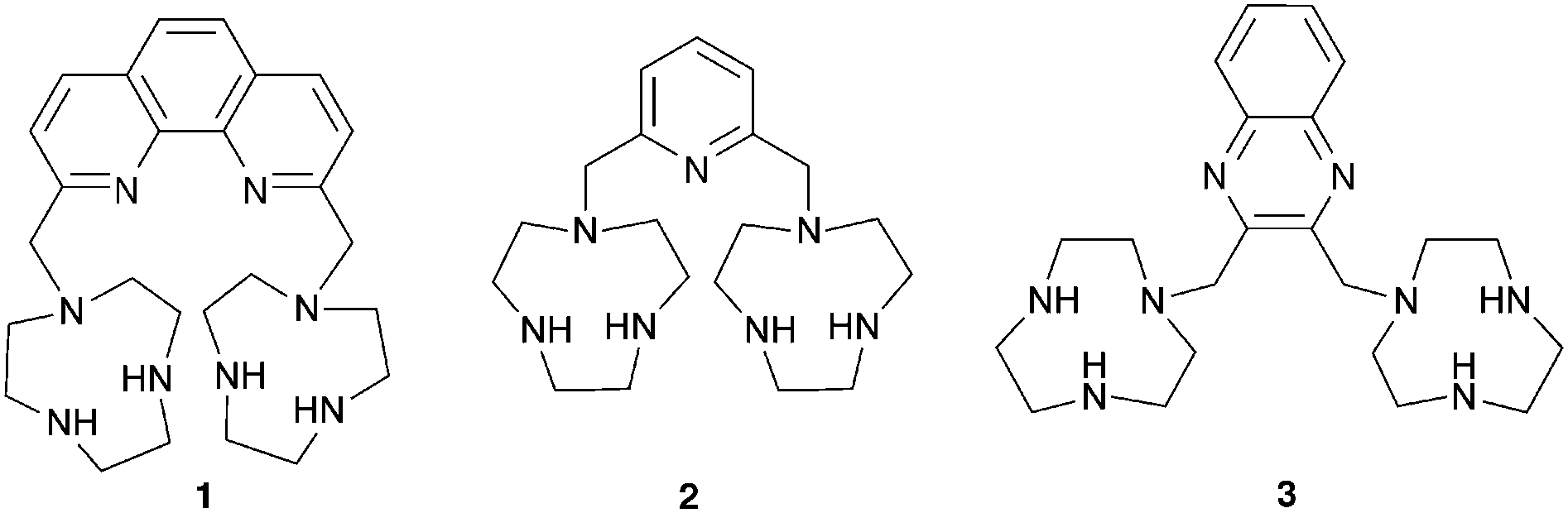
 | ||
| Scheme 1 Zn2+ induced conformational change of the terpyridine unit in receptor 4. | ||
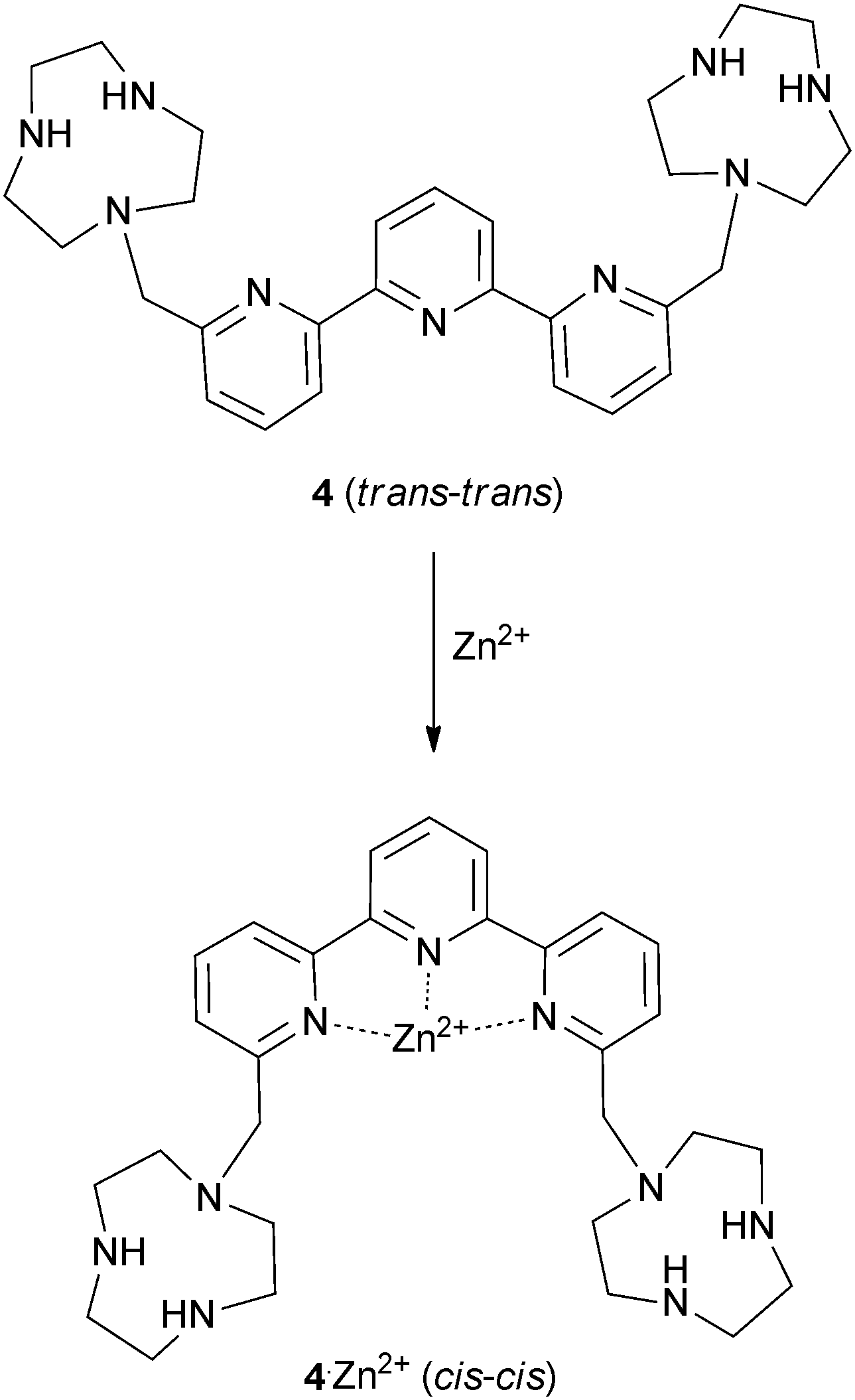
Continuing their work on [9]aneN3 based receptors, Bencini, Lippolis, Pasini and co-workers have reported the synthesis of chiral ditopic polyammonium receptor 5 based on a (S)-BINOL scaffold capable of selectively binding (S,S)-tartaric acid in water over its (R,R) or meso-forms and over other carboxylates such as (S)/(R)-malate, (S)/(R)-lactate, maleate, fumarate and succinate (Fig. 1a).7 Potentiometric, 1H NMR and fluorescence titrations were used to probe the interaction of 5 with carboxylates at a range of pH values. The compound was shown to interact selectively with (S,S)-tartrate over a range of pH values. The authors attribute the selectivity to the configuration of the stereocentres and the possibility of hydrogen bonding between the –OH groups of (S,S)-tartrate and both tertiary amine groups of receptor 5 (Fig. 1b).
 | ||
| Fig. 1 (a) Structure of receptor 5. (b) The proposed interaction mode between (S,S)-tartrate and (H25)2+. | ||
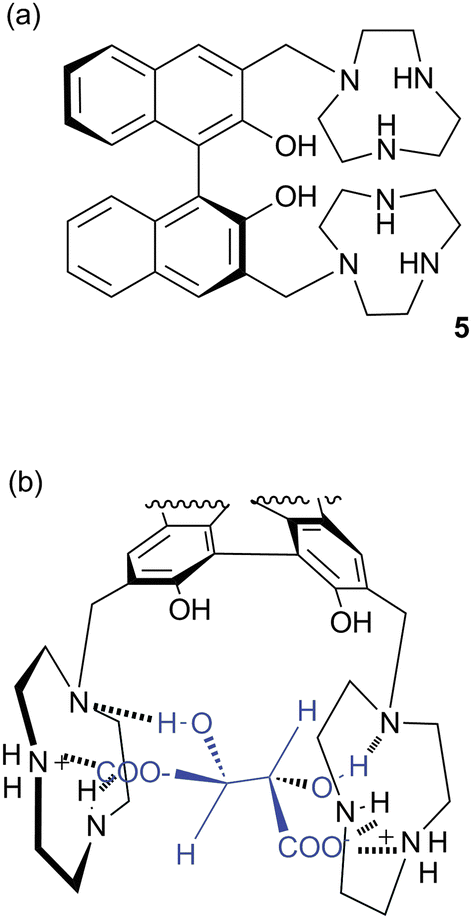
Bowman-James and co-workers have studied the role of charge on anion affinity in mixed amide–amine based macrocyclic anion receptor systems using NMR and X-ray crystallography.8 Proton NMR titrations in DMSO-d6 showed that the quaternised receptors 9, 10 and 11 showed significantly higher affinity for anions than the neutral precursors 6, 7 and 8 (Table 1). The NMR titration curves obtained predominantly fitted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry apart from those for dihydrogen phosphate and hydrogen sulfate. In the former case slow exchange was observed upon addition of this anion to 10 and the stability constant was calculated by integration of the proton resonances of the free and bound receptor. Addition of HSO4− to 9 caused significant broadening in the host's NMR resonances precluding the determination of a stability constant by this method. The results show that the pyridine containing receptors have a higher affinity in general than the compounds containing m-xylyl or thiophene spacers. This may, in part, be due to the pre-organising influence of the pyridine group on the amide NH protons forming a cavity suitable for anion complexation. This was confirmed by X-ray crystallography whereby the crystal structures of 9·Cl and 9·HSO4 showed the anions located outside the macrocycle, with one amide NH coordinating to one anion, while the crystal structure of 10·I showed that iodide was bound via two convergent amide NH groups around the pyridine spacer (Fig. 2).
:1 binding stoichiometry apart from those for dihydrogen phosphate and hydrogen sulfate. In the former case slow exchange was observed upon addition of this anion to 10 and the stability constant was calculated by integration of the proton resonances of the free and bound receptor. Addition of HSO4− to 9 caused significant broadening in the host's NMR resonances precluding the determination of a stability constant by this method. The results show that the pyridine containing receptors have a higher affinity in general than the compounds containing m-xylyl or thiophene spacers. This may, in part, be due to the pre-organising influence of the pyridine group on the amide NH protons forming a cavity suitable for anion complexation. This was confirmed by X-ray crystallography whereby the crystal structures of 9·Cl and 9·HSO4 showed the anions located outside the macrocycle, with one amide NH coordinating to one anion, while the crystal structure of 10·I showed that iodide was bound via two convergent amide NH groups around the pyridine spacer (Fig. 2).
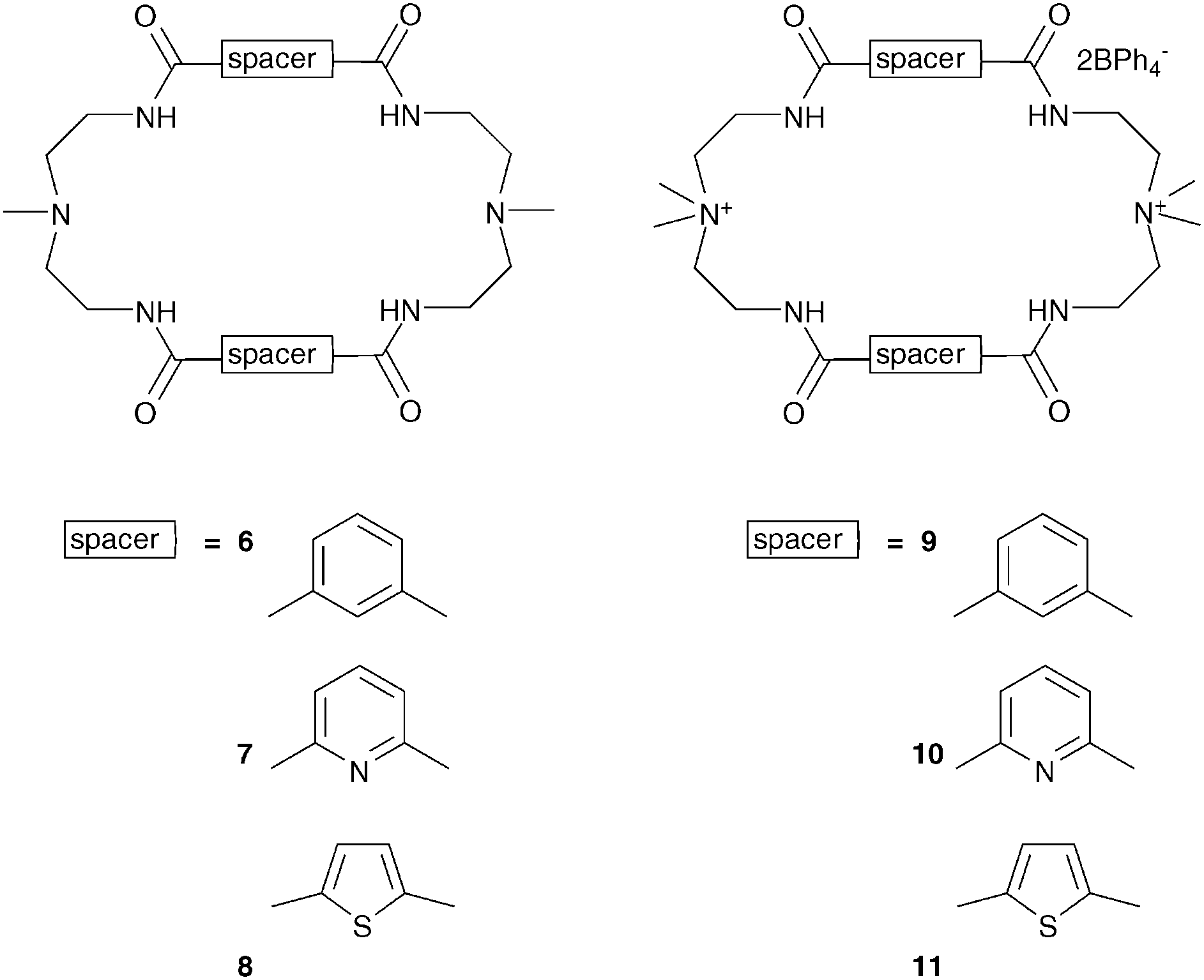
| Anions | 6 | 7 | 8 | 9 2+ | 10 2+ | 11 2+ |
|---|---|---|---|---|---|---|
| a Calculation complicated due to peak broadening after the addition of anion. b At the upper limits for NMR determination. | ||||||
| Cl− | 25 | 490 | 20 | 1700 | 56230 |
130 |
| Br− | 20 | 515 | <10 | 140 | 23990 |
>10 |
| I− | <10 | <10 | <10 | 100 | 160 | >10 |
| H2PO4− | 830 | 10960 |
100 | 10960 |
208900b |
900 |
| HSO4− | 795 | 107 | <10 | 7945 | 1500 | |
| NO3− | <10 | <10 | <10 | 45 | 210 | <10 |
| ClO4− | <10 | <10 | <10 | 40 | 250 | <10 |
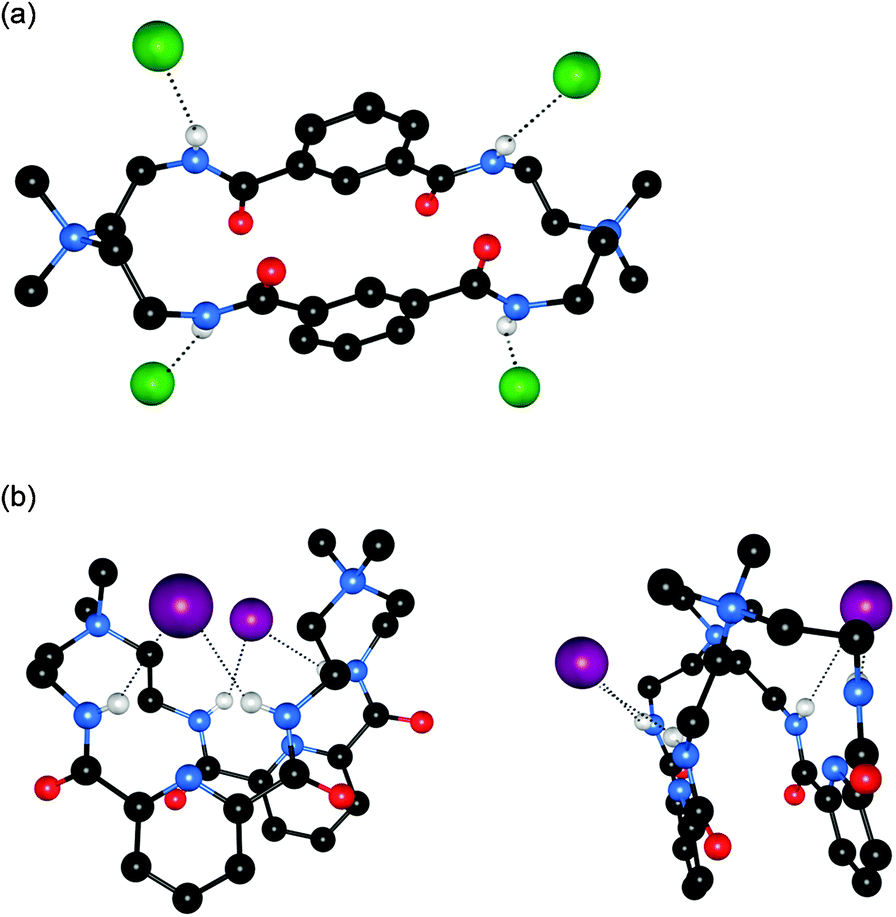 | ||
| Fig. 2 (a) Crystal structure of 92+·2Cl−·2.66H2O (b) two views on the crystal structure of 102+·2I−·2H2O. Hydrogen bonds are shown as dashed lines; solvent molecules and non-coordinating hydrogens are omitted for clarity; chloride is represented in green spacefill; iodide in purple spacefill and the receptors are shown in ball-and-stick. | ||
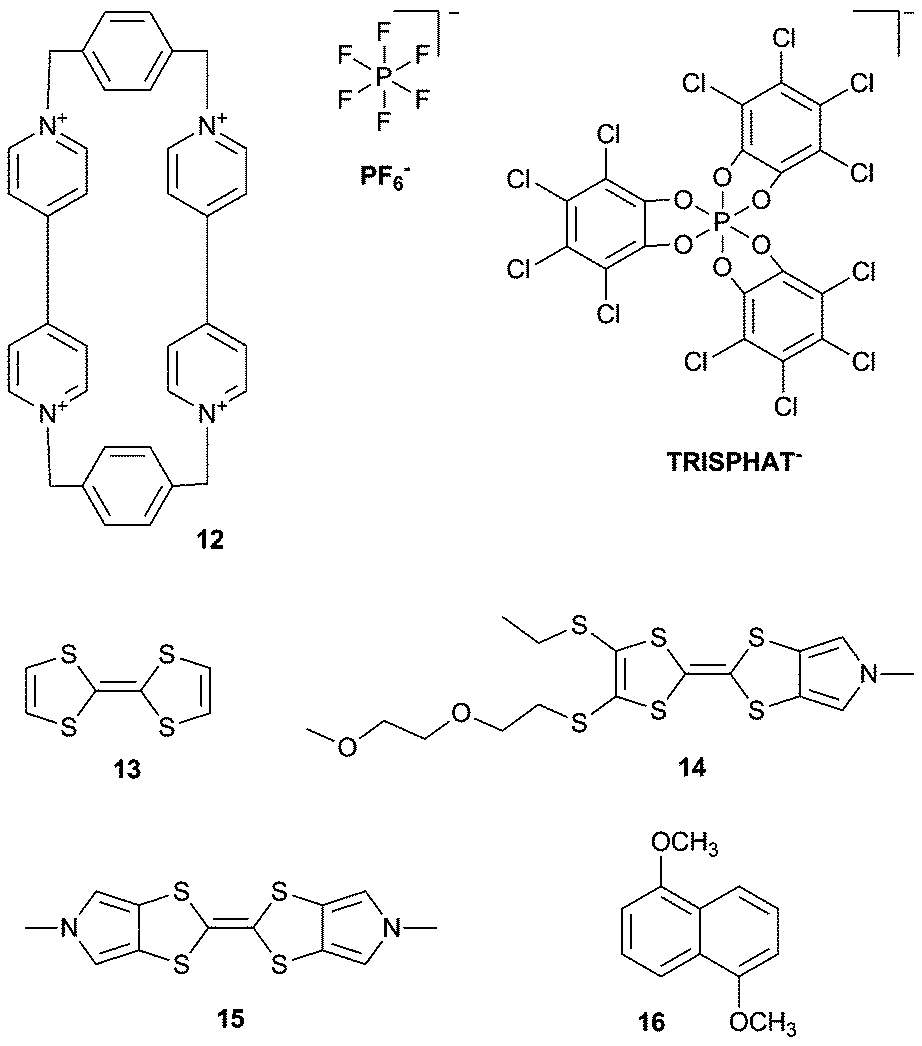
Another ammonium based macrocycle is cyclobis(paraquat-p-phenylene) (CBQT4+) 12, which is often studied as the ring component of [2]catenanes and [2]rotaxanes with π-electron donating guests,9 but the role of the counteranions on the host–guest properties of this ammonium macrocycle is less well explored. Flood, Jeppesen and co-workers have investigated the effect of different sized counterions PF6− and TRISPHAT− on the binding of 12 with various tetrathiafulvalene guests 13–15 and naphthalene guest 16.10 UV/Vis dilution experiments in MeCN at 298 K revealed that the association between 12·4TRISPHAT and 13–15 is nearly twice as strong as between 12·4PF6 and 13–15. The authors explain this observation by the larger size of the TRISPHAT− anion, leading to an increased distance between 12 and the anion, and hence stronger π-electron acceptor ability of 12. It was also shown that the opposite was true for naphthalene guest 16, where 12·4PF6 binds 16 more strongly (Ka = 2000 M−1) than 12·4TRISPHAT (Ka < 10 M−1), presumably because the interaction between 12 and 16 is governed by CH⋯π interactions and not by donor–acceptor interactions.
Amide-, urea-, thiourea- and squaramide-based anion receptors
Due to their facile synthesis and easily tunable NH acidity, amides, ureas and thioureas are probably the most widely employed hydrogen bond donor groups in anion receptor systems, and this has not changed during 2011 and 2012. Recently Beer and co-workers have used anion coordination by amide groups in the fabrication of core@shell bimetallic metal nanoparticles (Scheme 2).11 Beer's method involves coating a nanoparticle in hydrogen bond donor groups that can form complexes with chlorometallate anions. Upon reduction the anions coat the nanoparticle with a second metal. Au@Pd, Pd@Au, Pt@Pd and Pd@Pt nanoparticles were synthesised using this methodology (Scheme 2) employing imidazole (to bind to the core nanoparticle) functionalised with an alkylamide group as the hydrogen bond donor. Beer demonstrated the reduction of 2-chloronitrobenzene using Au@Pd nanoparticles produced using this methodology and showed that small nanoparticles (<5 nm) can be produced using this method.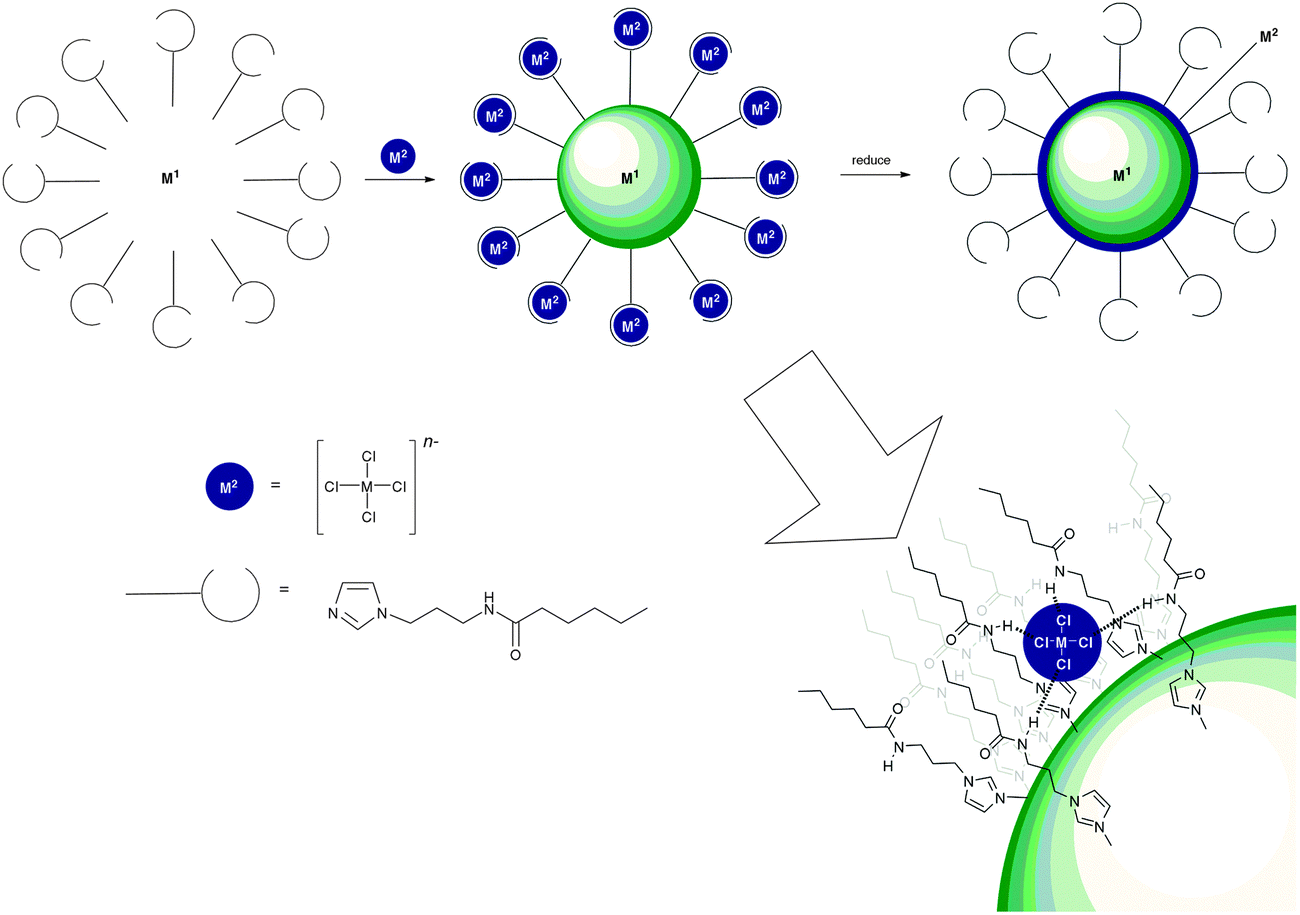 | ||
| Scheme 2 A diagrammatic representation of the synthesis of core@shell bimetallic nanoparticles using anion coordination. | ||
Acylhydrazone groups are similar to amide groups in terms of NH hydrogen bonding, but the reversible nature of this group allows it to be used in dynamic combinatorial chemistry. Beeren and Sanders have employed this strategy to develop a powerful receptor for dihydrogen phosphate.12 A dynamic combinatorial library of hydrazone linked ferrocene-based macrocycles and linear oligomers was generated and it was found that the addition of tetrabutylammonium dihydrogen phosphate (TBAH2PO4) led to the amplification of linear receptor 17, at the expense of the macrocyclic compounds. UV/Vis and NMR titrations in 94:6 chloroform:methanol with TBAH2PO4 showed that 17 binds H2PO4− in a 2:1 binding mode with positive cooperativity (K1K2 = 800000 M−2). Using NOESY NMR and CD (circular dichroism) experiments the authors were able to show that the complex of 17 with H2PO4− adopts a helical conformation with two H2PO4− anions bound by various combinations of imine CH, cyclopentyl CH and hydrazone NH hydrogen bonds (Fig. 3).
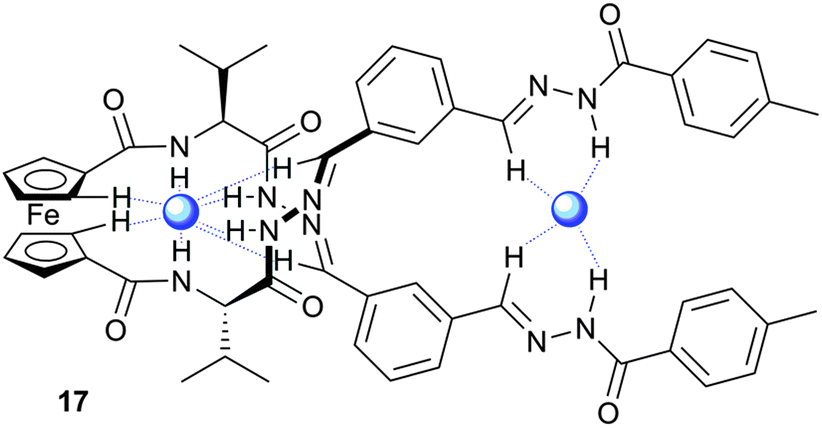 | ||
| Fig. 3 Representation of helical linear receptor 17 binding two H2PO4− anions (blue spheres) using hydrogen bonding (blue dashed lines). | ||
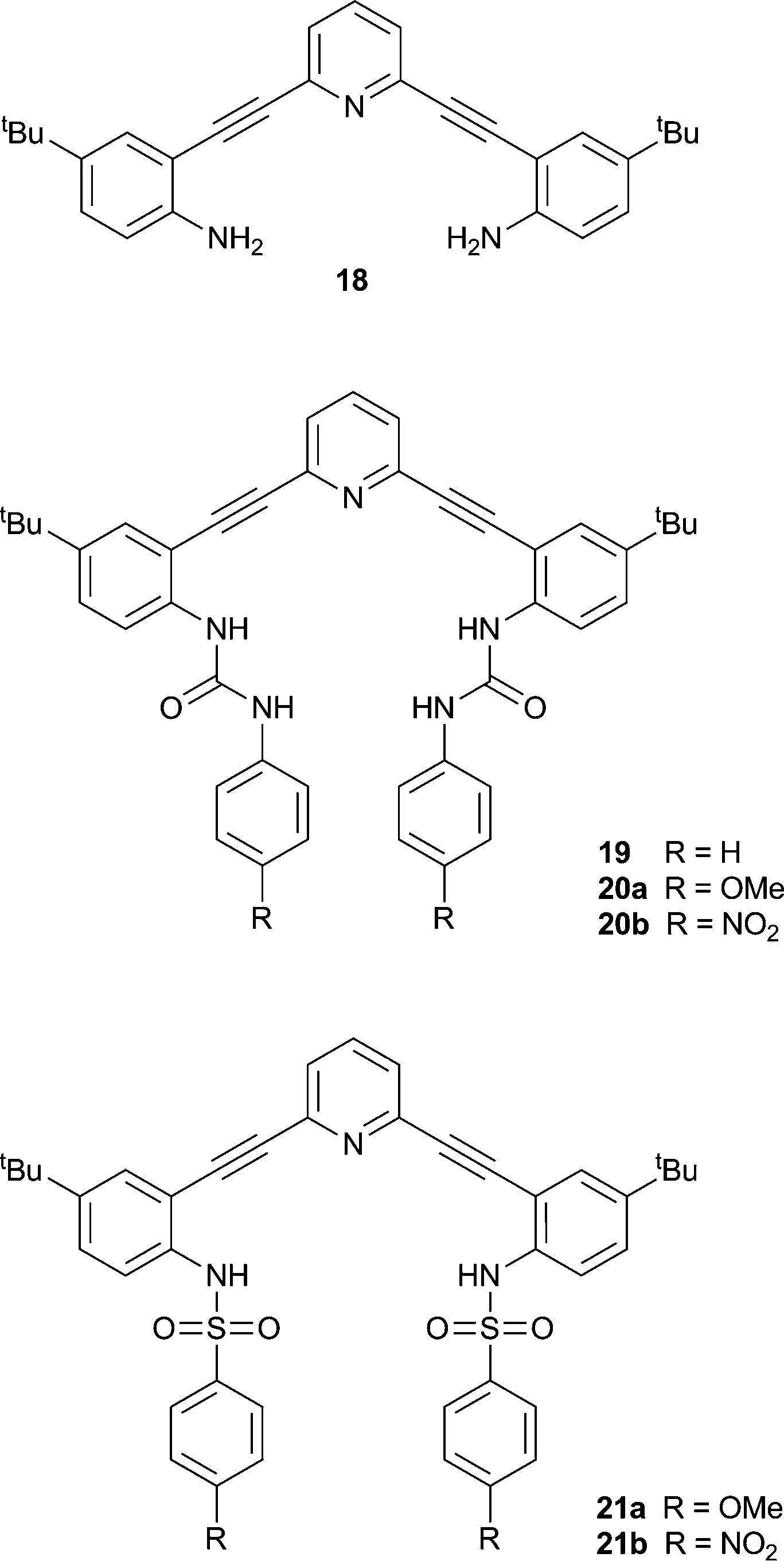
Ureas and thioureas possess two NH hydrogen bond donors and are therefore often used to coordinate anions via multiple hydrogen bonds. Johnson, Haley and co-workers have continued their work on bis(anilinoethynyl)pyridine derivatives containing urea groups and showed that these compounds can show switchable ON–OFF and OFF–ON fluorescence responses to anions.13 During previous studies it was noted that the fluorescence of compounds 18 and 19 was quenched upon protonation in chloroform, concurrently with a change in the colour of the solution from colourless to yellow.14 Derivatives of compound 19 containing either electron donating methoxy- (20a) or electron withdrawing nitro- (20b) groups were synthesised in addition to methoxy- (21a) and nitro- (21b) functionalised sulfonamide derivatives. The colour and fluorescence changes of the compounds upon exposure to acid are illustrated in Fig. 4. The solutions of the compounds go from colourless to yellow in the presence of HCl. For the compounds with electron donating methoxy substituents fluorescence is switched off in the presence of HCl, while for those with electron withdrawing nitro groups fluorescence is switched on. Using Frontier molecular orbital calculations, the authors show that in compounds 20a and 21a the electron density remains near the alkynyl groups resulting in radiative de-excitation and hence fluorescence. In the compounds containing nitro substituents (20b and 21b) a charge-transfer fluorescent state is formed resulting in quenched fluorescence. This gives rise to an ON–OFF fluorescence response for 20a and 21a, and an OFF–ON response for 20b and 21b upon the addition of acid, and the magnitude of the response depends on the nature of the acid (anion) with a larger response seen for HCl than for CF3COOH. Subsequent studies on derivatives of 19 containing various functionalities, revealed that the presence of electron withdrawing groups in 2,6-bis-(anilinoethynyl)pyridine bisureas in general transforms this class of compounds into “switch on” fluorescent chloride sensors.15
 | ||
| Fig. 4 Colourless solutions of neutral compounds in CHCl3 turn yellow upon protonation (top); fluorescence (excitation 365 nm) is quenched in electron-rich compounds 20a, 21a and is ‘turned on’ in electron poor compounds 20b, 21b (bottom). Reproduced with permission from Chem. Commun. 2011, 47, 5539–5541 © Royal Society of Chemistry. | ||
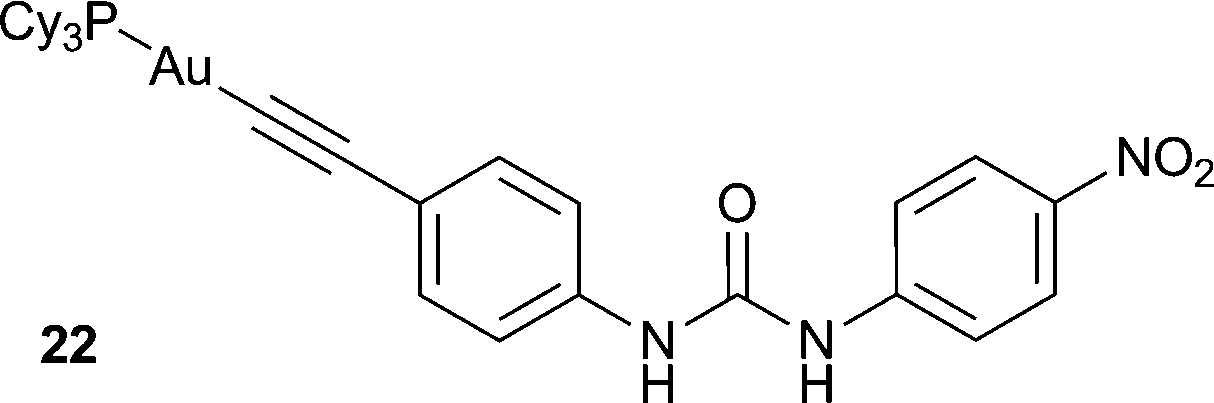
Chao and co-workers have reported the anion sensing ability of a series of mononuclear gold(I) acetylide complexes containing a urea group (e.g.22).16 These complexes exhibit intense luminescence in the solid state and in THF solutions, which was assigned to the 3(ππ*) excited state of the acetylide ligands. The authors found that changing the urea substituent could modify the anion binding strength in THF, with electron withdrawing groups (such as –NO2 in 22) resulting in the strongest anion binding due to the increased acidity of the urea NH groups. UV/Vis titrations showed that receptor 22 forms a strong 1:1 complex with F− (logKa = 6.44) in THF. The anion complexation was signalled by a decrease in the intensity of bands at 263, 276, 288, 301 and 345 nm, with the gradual appearance of bands at 292, 308 and 375 nm. In DMSO, receptor 22 was found to undergo a colour change from colourless to yellow upon the addition of F−. Analysis of the UV/Vis titration data revealed that this colour change is due to the deprotonation of the receptor by the anion, allowing for the straightforward naked eye detection of F− anions.
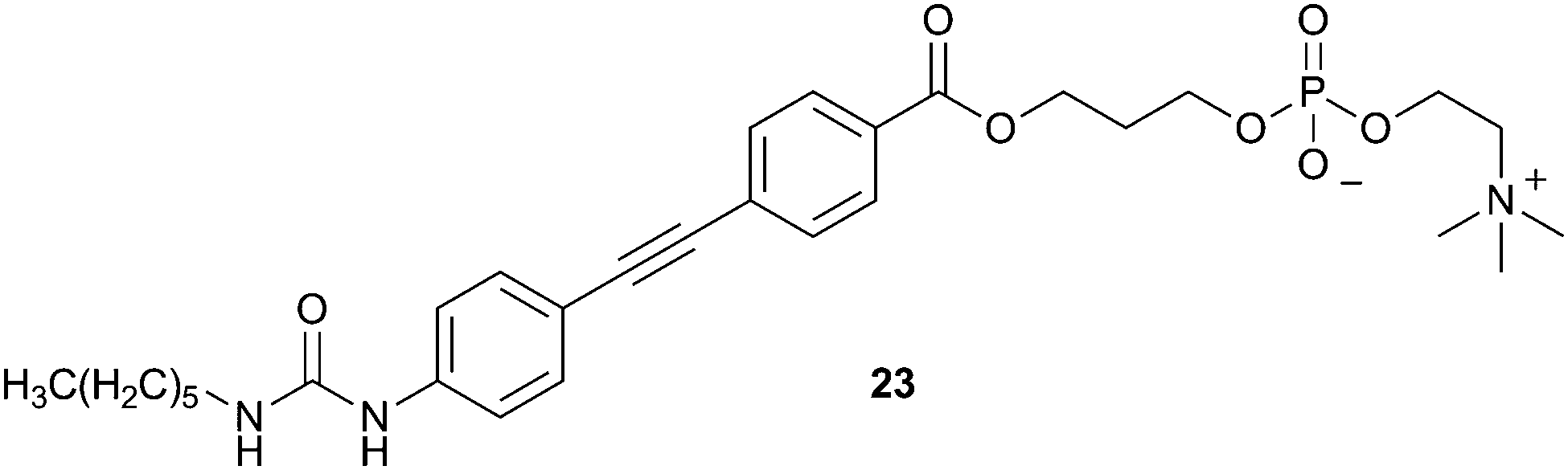
Allen, Sargent and colleagues have reported a fluorescent phospholipid analogue containing a urea group (23).17 The fluorescence of this system derives from the diarylacetylene spacer, which separates the urea group from the phosphocholine (PC) lipid head group. Consequently, the receptor forms a “head-to-tail” dimer in dichloromethane at concentrations above 2 × 10−5 M, in which the urea group of one receptor forms hydrogen bonds to the PC group of a second receptor and vice versa. The dimeric complex was identified by electrospray ionization MS in acetonitrile–water, and by 1H NMR in CD3CN, and was predicted by DFT analysis. Fluorescence experiments in dichloromethane indicated that, at high concentrations of 23 (2.6 × 10−5 M) the addition of a large excess of Cl− or NO3− did not produce a significant fluorescence response, while the addition of HCO3− or H2PO4− anions led to significant fluorescence quenching. However, at lower receptor concentrations (≤5 × 10−6 M) the addition of all of these anions was found to induce suppression of the emission intensity. This indicates that at lower receptor concentrations the dimeric species is not formed and anion binding does not compete with self-association.
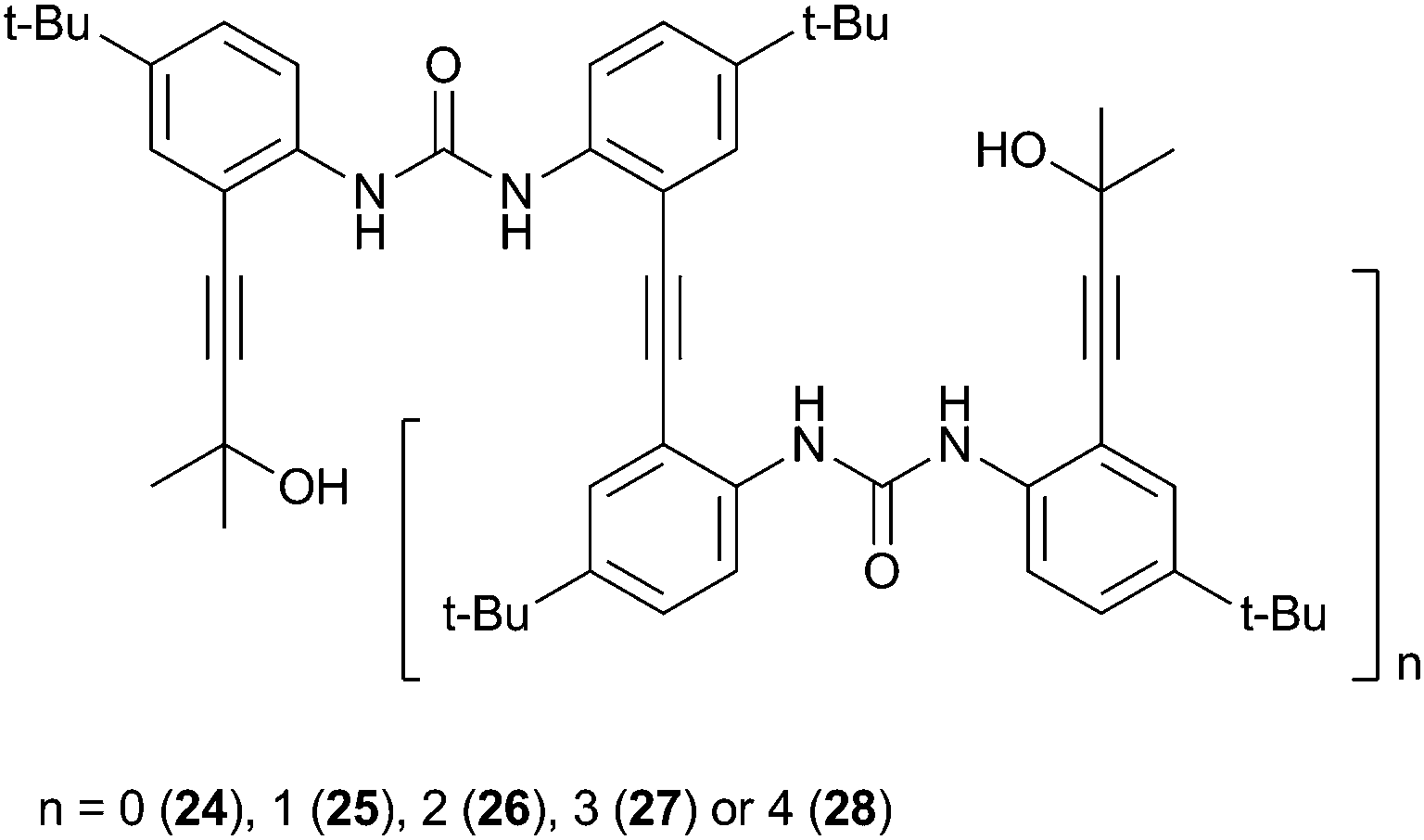
Jeong and co-workers have reported a series of diphenylureas 24–28 that form foldamer complexes with chloride and sulfate.18 The solution phase sulfate binding properties were investigated by 1H NMR titration in 20% and 40% CD3OH–DMSO-d6. In both cases the sulfate binding strength was found to increase with increasing chain length from 24–28 by approximately 4 orders of magnitude. In contrast, chloride binding studies in 15% CD3OH–acetone-d6 indicated that the binding strength reached a plateau after receptor 26 (Ka = 1340 M−1). The authors attributed this difference to the greater hydrogen bond acceptor ability of sulfate, with the tetrahedral array of oxygen atoms allowing the formation of up to 12 hydrogen bonds, whereas chloride can be coordinatively saturated by fewer urea groups. The crystal structure of 25·(Bu4N)2SO4 confirmed the encapsulation of a single SO42− anion in a 1:1 foldamer complex with each anion coordinated by four hydrogen bonds from two urea groups, and a further two hydrogen bonds from peripheral OH groups (Fig. 5).
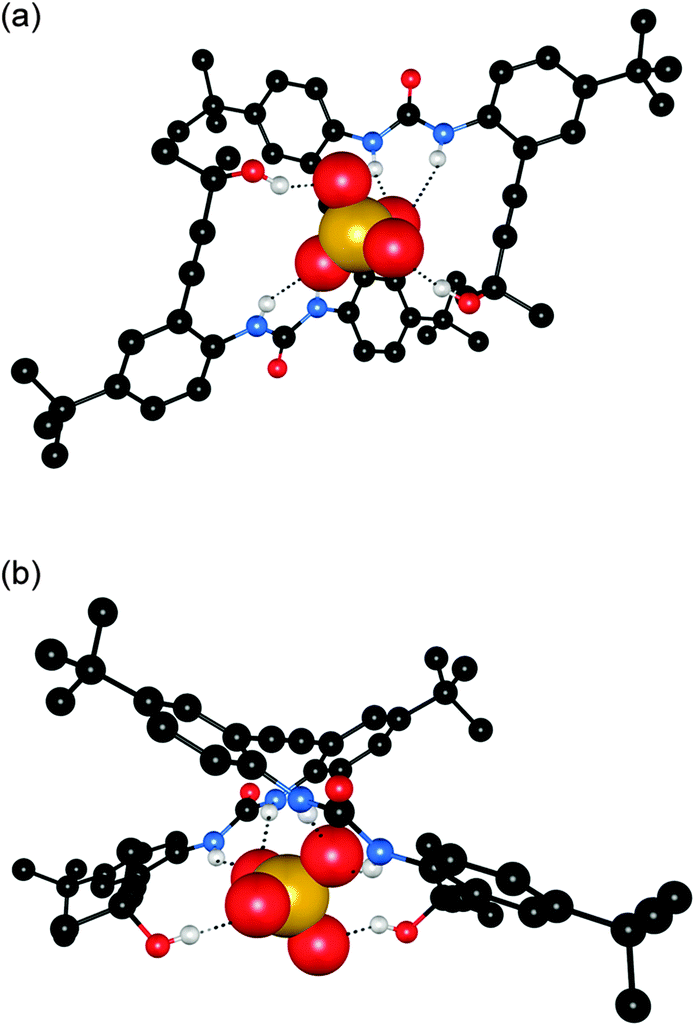 | ||
| Fig. 5 Two views of the crystal structure for complex 25·(Bu4N)2SO4. Hydrogen bonds are marked by dashed lines, non-coordinating hydrogen atoms and TBA counterions are omitted for clarity. The receptor is shown as ball-and-stick and the anion as spacefill. | ||
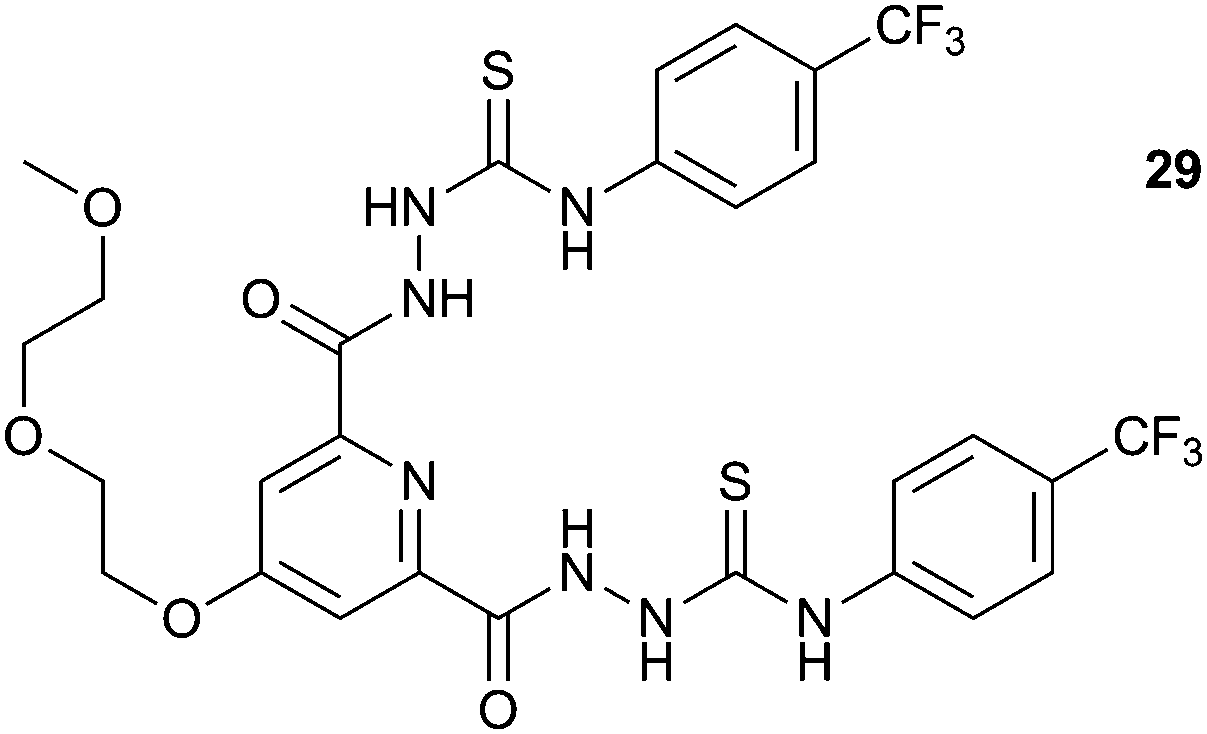
Gunnlaugsson and co-workers have reported the synthesis of a variety of pyridine-2,6-dicarboxamide-based receptors such as compound 29 (including systems containing two of these units linked via a glycol chain).19 The glycol chain confers solubility in polar solvents allowing the response of these systems to a variety of inorganic and biologically relevant organic anions to be measured in water:ethanol or methanol mixtures, or in DMSO-d6. Using UV/Vis titrations they found that receptors based on structure 29 were found to interact with AcO−, F− and H2PO4−, resulting in an increase in the ICT transition of the receptor in the presence of the anions. The data obtained in the competitive 1:1 EtOH:H2O solvent system fitted best to a 1:1 receptor:anion binding model, whereas higher order complexes were observed in titrations in DMSO:H2O and pure EtOH or MeOH solution. The authors suggested that this could be due to the formation of a hydrophobic cleft in the more strongly coordinating environment, which favours the formation of the 1:1 complex. The authors also investigated the binding of some biologically relevant phosphate anions under the same conditions. They found that AMP and ADP formed a 1:1 hydrogen bonded complex with these receptors, with binding of AMP slightly favoured over ADP. In contrast, the titration of these receptors with HP2O73− and ATP appeared to result in the deprotonation of the receptor by the anion, as indicated by a large change in the pH of the system determined by spectroscopic pH titrations.
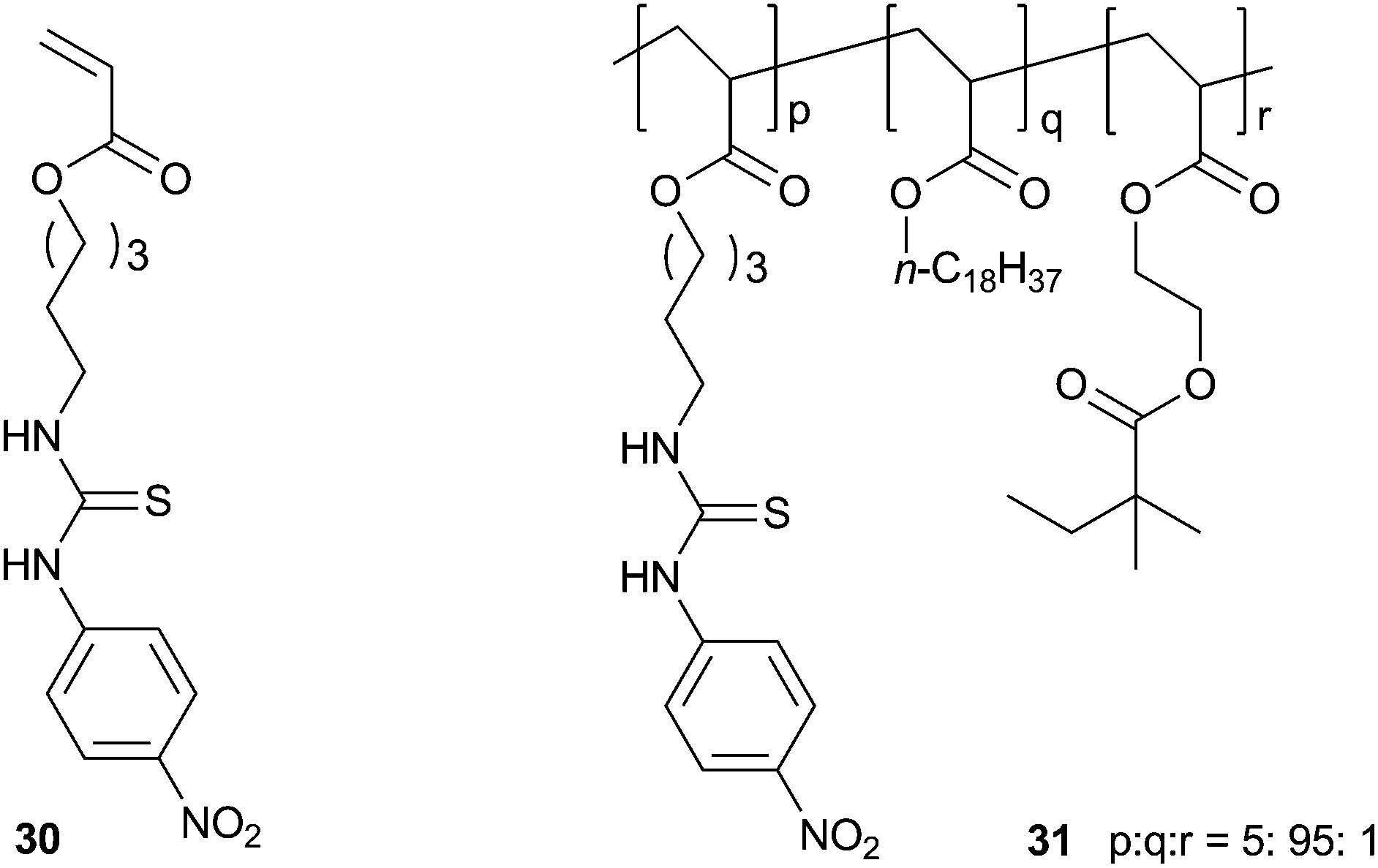
Sada and co-workers have incorporated a p-nitro-phenylthiourea group into a polymer gel network that shows selective colour change and swelling behaviour in the presence of certain anions.20 Cross-linked poly(octadecyl acrylate) containing a small number of thiourea groups (31) was prepared by radical co-polymerization gelation from the relevant monomers in a benzene:methanol mixture using azobis-isobutyronitrile (AIBN) as an initiator. The solvent was removed from the resulting gel, which was then transferred into a THF solution of the tetrabutylammonium (TBA) salts of a number of anions for 48 hours. The authors found that incubation in the presence of F− or AcO− anions leads to a significant swelling of the gel as a result of anion complexation causing the countercations to become entrapped in the gel network. This was accompanied by a colour change of the gel from pale yellow to red (F−) or yellow-green (AcO−), while other anions did not induce a colour change. Less strongly coordinating anions such as Cl− and PF6− induced a much smaller swelling effect. The F− induced swelling was found to be reversible, as immersion in THF:methanol (8:2) for 48 hours resulted in the collapse of the swelled gel. UV/Vis titration experiments in MeCN with the thiourea monomer 30 confirmed that F− and AcO− were complexed more strongly than the other anions tested, with binding constants of 3.7 × 103 M−1 and 9.2 × 103 M−1 respectively.
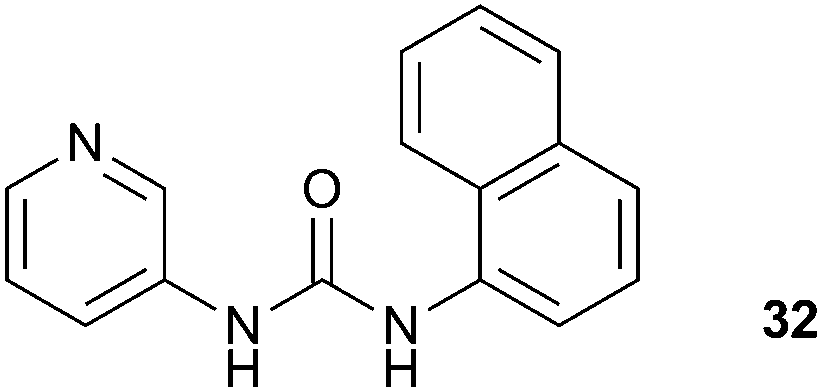
Wu and co-workers have reported a simple pyridylurea ligand 32 that assembles into a hexameric capsule, directed by the coordination of six copper(II) ions, which can encapsulate sulfate anions with six urea groups.21 The sulfate inclusion complex was generated by the reaction of CuSO4·5H2O with 3 equivalents of the free ligand 32 in DMF. The resulting complex provided single crystals suitable for X-ray analysis. The authors found that the structure contains two kinds of copper(II) environment, designated [Cu1] and [Cu2]. The [Cu1] moieties are octahedrally coordinated by six ligand molecules forming two opposing, inversion related C3-symmetric clefts. The sulfate anion is positioned within a cavity formed by two such complex units and coordinated by six urea groups, and the capsules further extend into a 1D chain (Fig. 6). Meanwhile, the [Cu2] moieties are chelated by two ligands and two DMF molecules, and adopt a Jahn–Teller distorted octahedral geometry.
 | ||
| Fig. 6 Crystal structure of the [Cu1] fragment in CuSO4 complex of 32. Sulfate and copper ions are shown as spacefill, while the receptor is shown as ball-and-stick. Hydrogen bonds are represented by dashed lines. (a) Sulfate encapsulation by multiple hydrogen bonds from six urea groups in the [SO4(32)6]2− capsule (copper ions are omitted for clarity). (b) Infinite 1D hydrogen bonded chain linked by sulfate encapsulation and copper coordination. | ||
Similarly, Custelcean et al. have synthesised symmetrical bipyridine substituted urea ligand 33 to construct a M4L6 cage using zinc(II) coordination.22 The authors hoped that the M4L6 cage would favour the coordination of tetrahedral anions within the cavity. The formation of complexes in solution was monitored using NMR techniques. The addition of zinc nitrate to a solution of 33 in 2:1 CD3OD:D2O revealed that a low-symmetry coordination complex or mixture of complexes was formed between 6 ligands and 4 zinc(II) cations within minutes.
Subsequently, the addition of a number of EO42− anions (E = S, Se, Cr, Mo, W, P) resulted in the formation of a new set of sharp NMR resonances, consistent with the formation of the desired Zn4L6 cage. The tetrahedral geometry of the templating anion proved to be crucial, as the complex was not formed in the presence of F−, Cl−, Br−, I−, NO3−, BF4−, PF6−, AcO− or CF3SO3−. Additionally, the divalent charge of the tetrahedral anions was important, as no complex formation was observed using ClO4− or ReO4−, whilst the non-tetrahedral divalent anions CO32−, SeO32− and SO32− also did not result in complex formation. Although the cages were found to not persist in the absence of the encapsulated anions, the authors were able to perform competitive binding experiments in 2:1 CD3OD:D2O using 77Se NMR to identify encapsulated and free 77SeO42−. The cages were prepared encapsulating 77SeO42−, and one or two equivalents of Na2EO4 (E = S, Cr, Mo and W) were added. These experiments were used to calculate EO42−/SeO42− exchange constants (Kex), with CrO42−complexed the strongest (Kex = 40.10) and selectivity in the order CrO42− > SO42− > SeO42− > MoO42− > WO42−. With the exception of CrO42−, this represents anti-Hofmeister selectivity. The authors also noted that CrO42−, which is the anion that does not fit the anti-Hofmeister bias, is the most basic anion, which could explain why it is bound the strongest. The crystal structures of the [Zn4(33)6(EO4)](NO3)6 complexes (E = S (Fig. 7), Cr and Mo) were found to be isomorphous, with each EO42− anion encapsulated by twelve hydrogen bonds from six urea groups. The hydrogen bonding parameters are very similar across these three structures despite the varying anion size, indicating that the cage is flexible and can accommodate anions of different sizes.
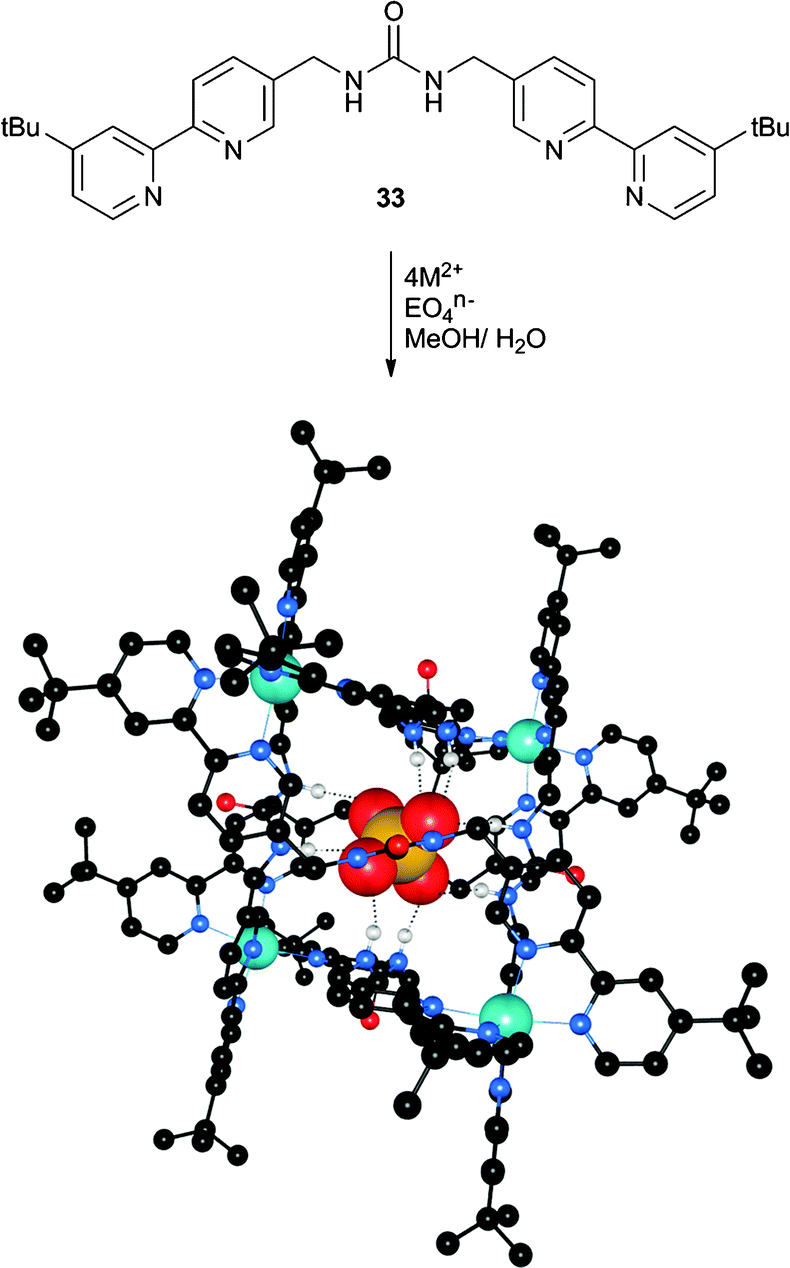 | ||
| Fig. 7 The assembly of an M4L6 cage templated by tetrahedral EO4n− anions. The figure represents the crystal structure where M2+ = Zn2+ (turquoise spheres) and EO4n− = SO42−. Hydrogen bonds are represented by dashed lines and non-coordinating hydrogen atoms and solvent molecules are omitted for clarity. Sulfate is bound by six urea groups and the four Zn2+ cations form a distorted tetrahedron. | ||
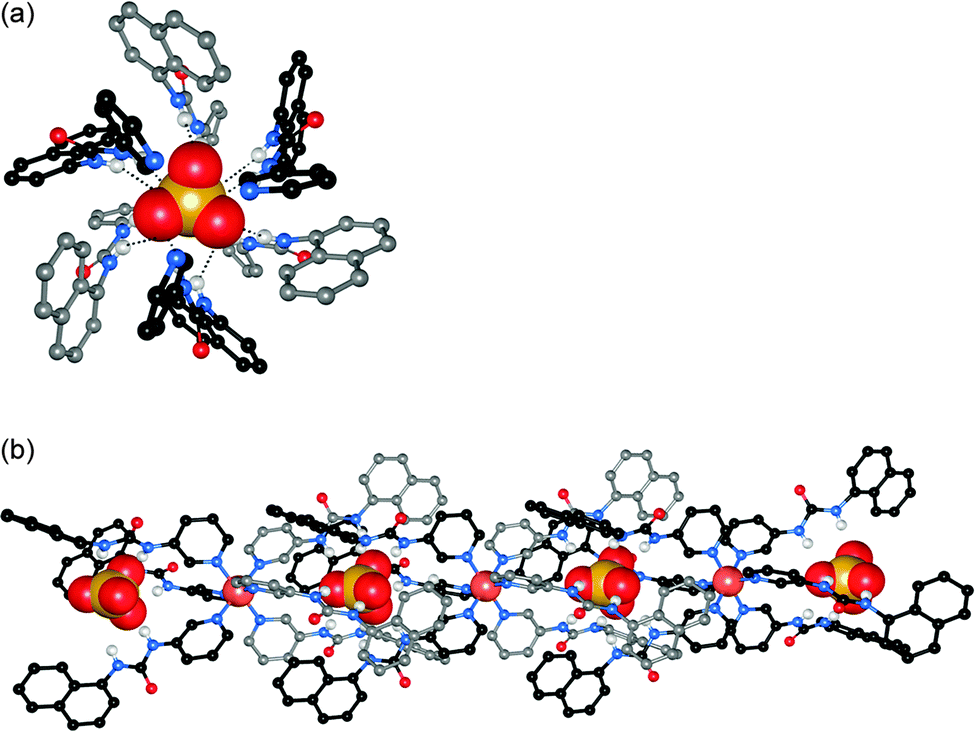
Jolliffe and co-workers have reported the synthesis of thiourea cryptands 34 and 35, based on a rigid cyclic peptide scaffold.23 They found that the yield in the final cyclisation of 34 and 35 was improved if the cyclopeptide precursor was added as the hydrogen bromide salt, implying that this procedure may be anion templated. The interaction of the cryptands with a range of monovalent anions was investigated by 1H NMR titrations in DMSO-d6–H2O 0.5%. Receptor 34 was found to interact strongly with a range of anions including F−, Cl− and AcO− (all with Ka > 104 M−1), while receptor 35 only showed a strong interaction with AcO− (Ka > 104 M−1), which was at least three times higher than any other anion studied. The authors suggested that the higher anion selectivity of 35 was due to the increased rigidity inferred by the 1,3,5-triethylbenzene cap, whereas the tren-capped cryptand 34 is more flexible, allowing it to interact strongly with a wider range of anions.
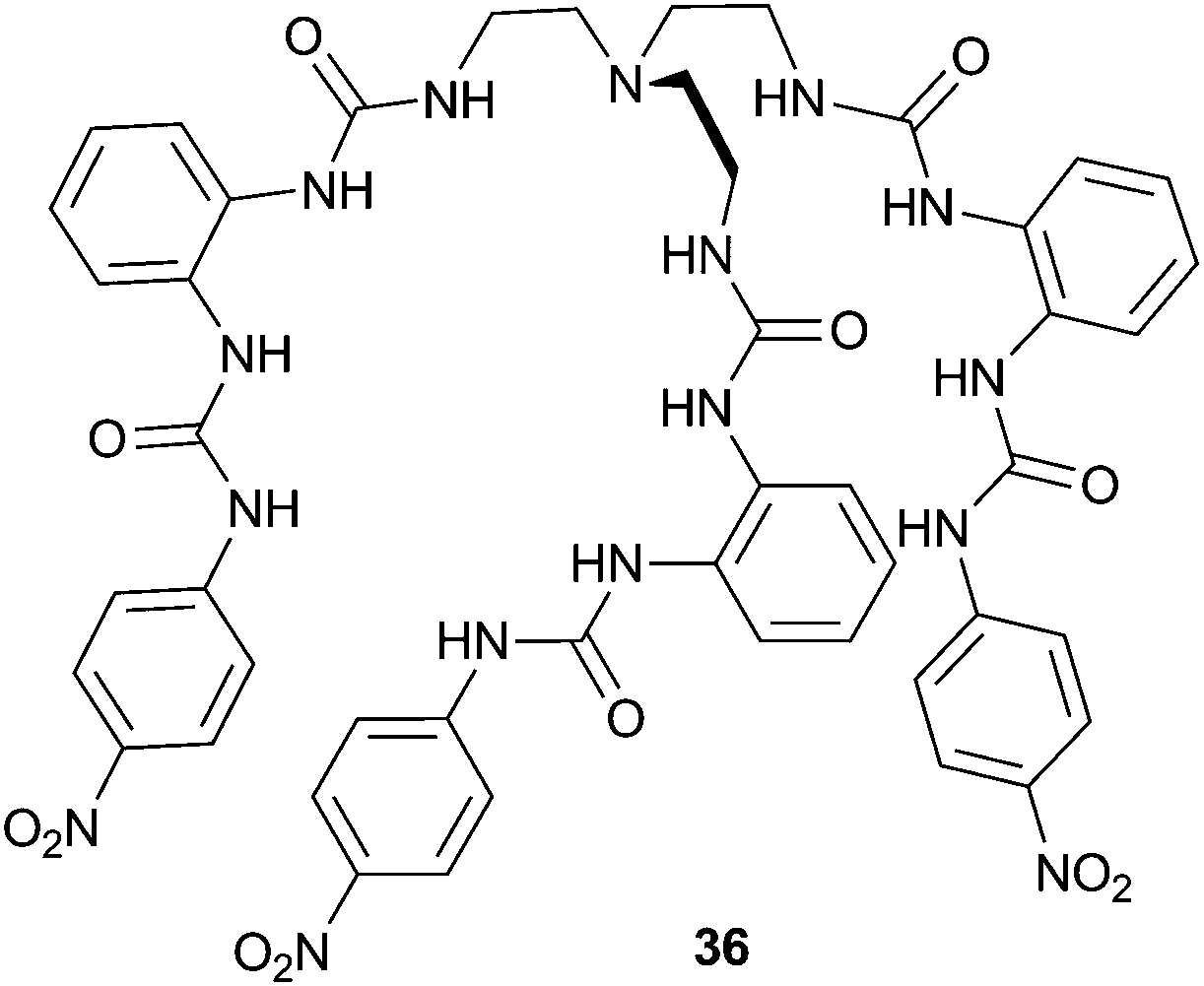
Wu and co-workers have published the strong sulfate binding and extraction properties of tripodal hexaurea 36.24 In the solid state, the authors found that a single sulfate anion could be encapsulated by 12 hydrogen bonds to the six urea groups, making 36 the first organic receptor reported to show saturated coordination of a sulfate anion by a single host. In this structure the complex forms a tetrahedral cage, with each of the three terminal arms folded along an edge of the bottom triangular face and each urea group chelating an edge of the tetrahedron (Fig. 8). The authors also investigated the solution phase anion binding properties by a number of techniques, and using 1H NMR titration estimated a sulfate binding strength of Ka > 104 M−1 in the highly competitive DMSO-d6–H2O 25%. Using NMR techniques receptor 36 was also found to be capable of efficiently extracting sulfate anions from aqueous NaNO3–Na2SO4 solutions into CDCl3 (containing tetrabutylammonium chloride for solubility purposes).
 | ||
| Fig. 8 Crystal structure of the sulfate complex of receptor 36 (top view). The anion is shown as spacefill and the receptor as ball-and-stick; hydrogen bonds are represented by dashed lines; non-coordinating hydrogen atoms, solvent and countercations (TBA) are omitted for clarity. | ||
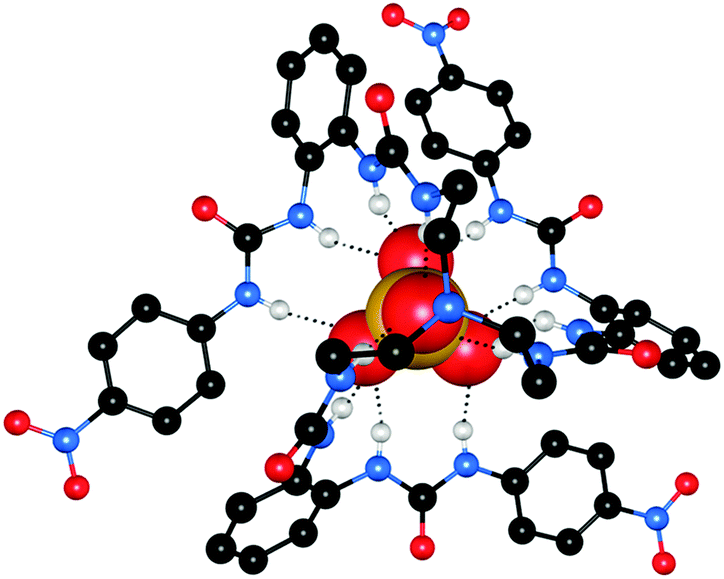
Continuing their work on receptors containing multiple urea groups, Wu and co-workers have also reported tetra- and hexa-urea ligands 3725 and 3826 incorporating repeating bis-urea units. In the solid state, receptor 37 was found to form a fascinating dinuclear triple helix structure on coordination of phosphate anions, in which three ligands are wrapped around two PO43− centres (Fig. 9a). Each phosphate anion is bound by six urea groups from three different receptors through twelve hydrogen bonds. The interaction of 37 with PO43− anions was also investigated in solution by 1H NMR titration in DMSO-d6–H2O 5%. This analysis revealed that the major species in solution was also a 3:2 receptor:anion aggregate. Meanwhile, hexaurea 38 was found to form a range of interesting structures in the solid state on complexation with a number of different chloride and bromide salts. The treatment of 38 with tetraethylammonium chloride (TEACl) resulted in a ‘molecular barrel’ complex (Fig. 9b). Two receptors dimerise by weak CH⋯O2N– hydrogen bonding interactions, and two of these associated dimers encapsulate three TEA+ cations. The urea NH groups face out of the cavity, and consequently a number of Cl− anions are associated with the outside of the barrel. When tetrapropylammonium chloride (TPACl) was used a similar barrel-like structure resulted, which encapsulates two TPA+ cations. As a result of encapsulating two TPA+vs. three TEA+ cations, the diameter of the barrel is wider and the length is shorter. However, moving to tetrabutylammonium chloride (TBACl) a helical foldamer-type structure was formed, in which one receptor molecule is threaded around two chloride anions. One Cl− accepts six hydrogen bonds from urea NH groups while the other receives four NH⋯Cl and two CH⋯Cl contacts with the cation and solvent molecules. A variety of interesting arrays were also formed using tetraalkylammonium bromide salts.
 | ||
| Fig. 9 Crystal structure of (a) triple helix formed by receptor 37 and PO43− shown in ball-and stick (left) and spacefill (right). [K(18-crown-6)]+ counterion has been omitted for clarity; (b) molecular barrel formed by receptor 38 and TEACl. Solvent molecules and non-coordinating hydrogen atoms have been omitted for clarity. Hydrogen bonds are represented by dashed lines. | ||
Wu et al. have also reported a series of oligourea receptors including 39 and 40 that coordinate chloride anions in either a mononuclear assembly or a dinuclear foldamer depending on the number of urea groups present.27 Single crystal X-ray crystallography revealed that short chain receptor 39 was found to wrap around a single chloride anion, forming mononuclear crescents. The chloride anion was complexed via four hydrogen bonds to the two peripheral urea groups, while the middle urea formed an intermolecular hydrogen bond, which connects adjacent complexes into an infinite ribbon. Longer chain analogues such as 40 form dinuclear foldamer complexes with chloride, in which two chloride anions are encapsulated within a helix. The crystal structure of [40·Cl−] is shown in Fig. 10. Each chloride anion is bound by four hydrogen bonds from two alternating urea groups. The Cl⋯Cl distance measures 3.613(9) Å, which is unusually short given that the sum of the ionic radii is 3.62 Å, indicating that a large amount of electrostatic repulsion has been overcome in the formation of this complex.
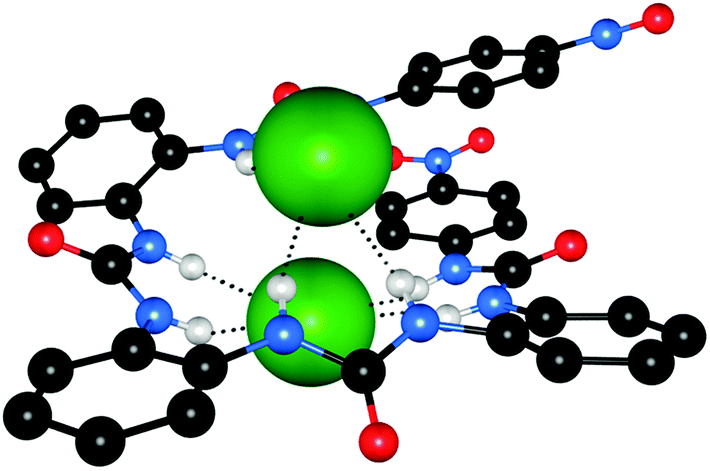 | ||
| Fig. 10 Crystal structure of the complex of receptor 40 with TBACl. The chloride anions are shown in spacefill, with the receptor in ball-and-stick, and hydrogen bonds are represented by dashed lines. Non-coordinating hydrogen atoms and countercations are omitted for clarity. | ||
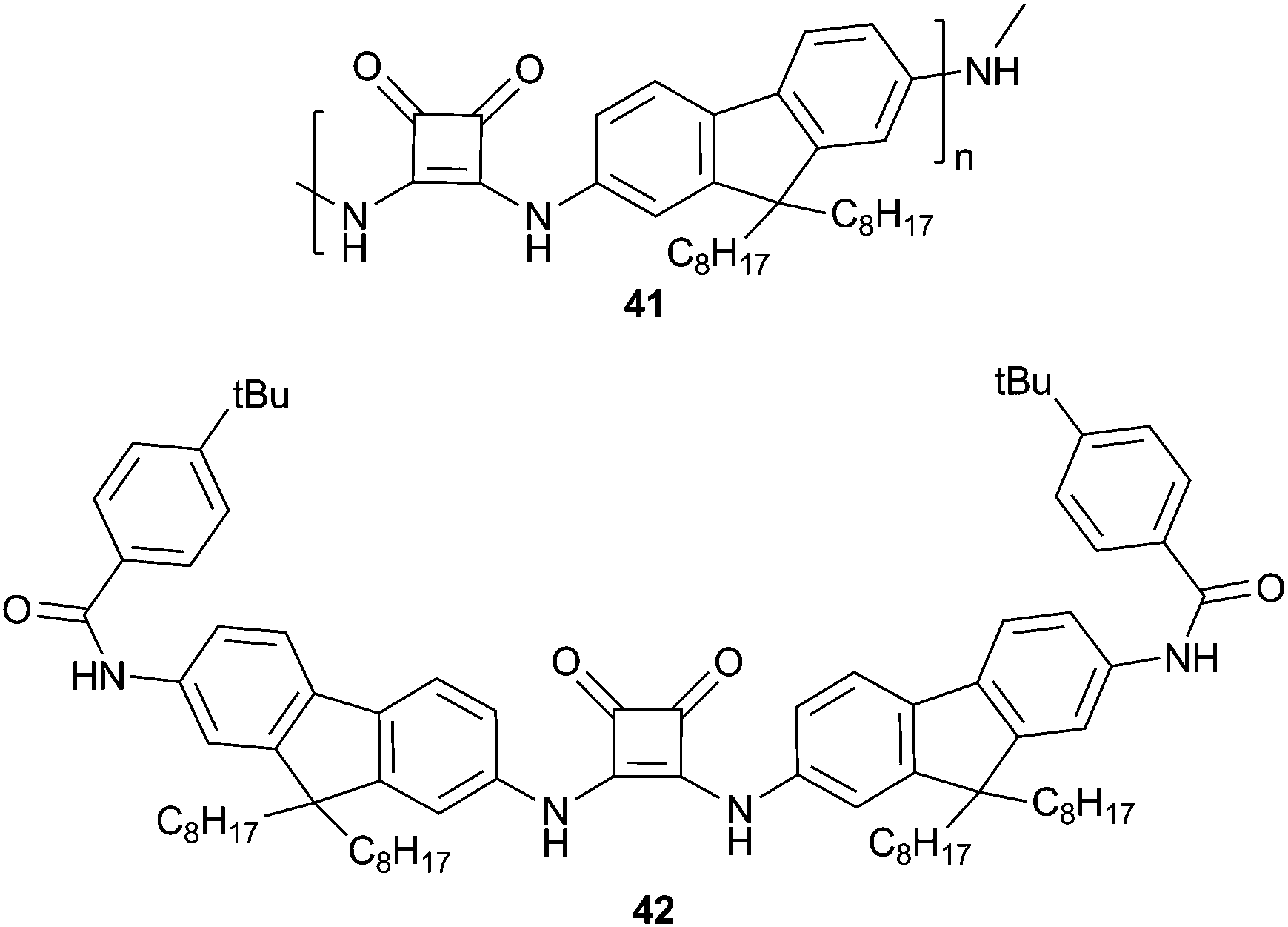
Squaramides are becoming a popular isostere for urea or thiourea anion binding sites. Taylor and co-workers have reported the synthesis of poly(squaramide) 41 by Lewis acid catalysed condensation.28 The anion complexation properties of this polymer were investigated by fluorescence spectroscopy in the competitive 10% water–NMP medium by comparison to mono-squaramide 42. The authors found that both systems showed a “turn-on” increase in fluorescence intensity on the addition of H2PO4−. However, monosquaramide 42 also displayed a significant response on the addition of F− anions, while polymer 41 was found to be completely selective for H2PO4−. Hill plot analysis also indicated that H2PO4− binding by polymer 41 was cooperative. The authors suggested that the high affinity and selectivity of the polymer system was the result of the interplay between anion binding and polymer aggregation, which was observed by GPC analysis (gel permeation chromatography), temperature dependent UV/Vis spectroscopy and transmission electron microscopy (TEM). Dynamic light scattering (DLS) indicated that the addition of phosphate anions to a heterogeneously distributed solution of 41 in DMF causes the formation of well-defined aggregates. This work highlights that the aggregation of this class of materials can be beneficial towards achieving highly selective anion sensing.
Pyrrole and indole based anion receptors
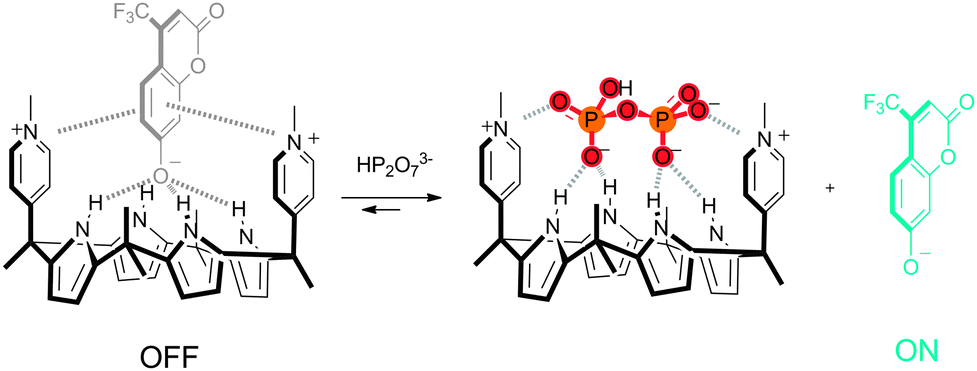
Unlike amides, (thio)ureas and squaramides, pyrrole- and indole-based anion receptors have the advantage that they do not possess any hydrogen bond acceptor moieties, avoiding aggregation and competition for hydrogen bond donor sites, rendering pyrroles and indoles popular anion binding motives. Sokkalingam et al. have employed the anion binding ability of modified calix[4]-pyrrole 43 to develop a selective sensor for fluoride using a fluorescent dye displacement assay (FDDA).29 When the fluorescent dye chromenolate 44 is bound to 43, the fluorescence of the dye is completely quenched. Subsequent addition of F− results in the recovery of the fluorescence. As no change in fluorescence was observed upon the addition of Cl−, Br−, I−, CN−, SCN−, AcO−, NO3−, PhCO2−, PF6−, HP2O73−, HSO4− and H2PO4−, the assembly consisting of 43 and 44 is a powerful and selective fluoride sensor. ITC (isothermal titration calorimetry) and UV/Vis titrations confirmed the fluoride sensing ability of the 43–44 assembly, with the affinity constant for [43·F]− (Ka = 5.69 × 106 M−1) being slightly higher than for [43·44]− (Ka = 4.69 × 106 M−1). Furthermore, the addition of Li+ to the fluorescent mixture of [43·F]− and 44, led to a “switch off” of the fluorescence, presumably due to the removal of F− from 43 by Li+ and the formation of the original non-fluorescent [43·44]− assembly.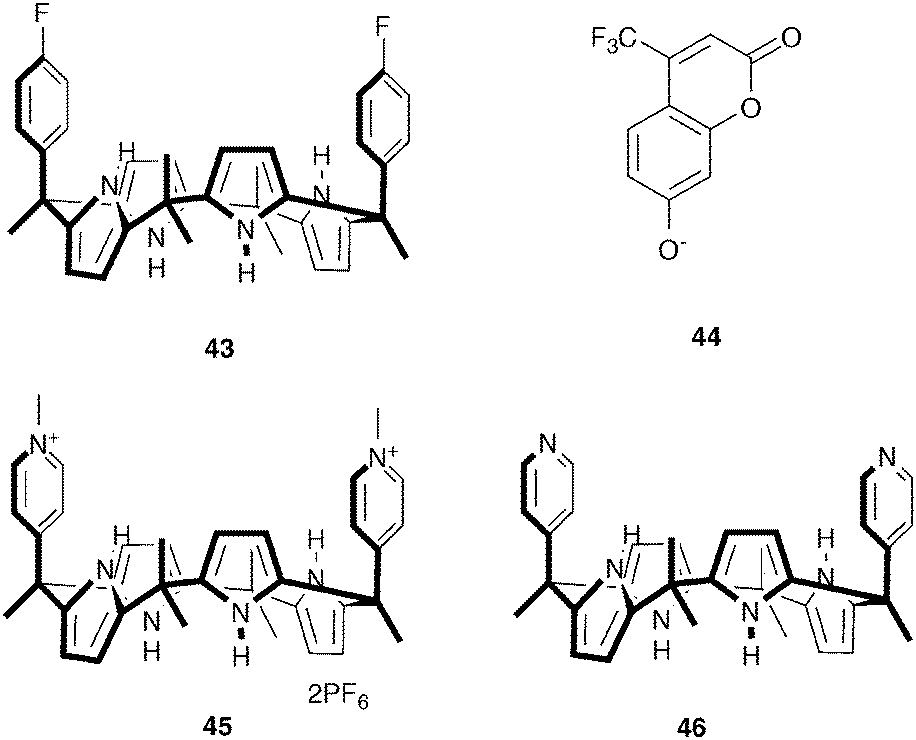
In a subsequent paper, Sokkalingam et al. have reported bis-pyridinium substituted calix[4]pyrrole 45 as a selective pyro-phosphate sensor using a similar fluorescent dye displacement assay.30 The receptor was shown to form a non-fluorescent 1:1 complex with the fluorescent chromenolate anion 44 in acetonitrile (Scheme 3), with an affinity constant of 7.25 × 106 M−1. The original fluorescence was shown to fully recover upon the addition of HP2O73−, while negligible or small enhancements of fluorescence were observed upon the addition of other anions, such as HSO4−, Br−, F−, H2PO4−, CH3COO− and Cl−. This selectivity towards HP2O73− over other anions such as F− and H2PO4− was even more pronounced when 30% aqueous acetonitrile was used as the solvent. The formation of a 1:1 receptor:HP2O73− complex was further supported by 1H NMR and MALDI-TOF MS experiments. The authors attribute the exceptionally high affinity for HP2O73− (Ka = 2.55 × 107 M−1) to the cationic charge of receptor 45, as the affinity for neutral bipyridine receptor 46 was found to be much lower (Ka = 5.25 × 105 M−1), although hydrogen bonding and anion⋯π interactions were also found to contribute to HP2O73− binding.
 | ||
| Scheme 3 Proposed Fluorescent Dye Displacement Assay (FDDA) detection of pyrophosphate anion by 45. | ||
Sessler and co-workers have reported the ion-controlled thermal electron transfer from tetrathiafulvene calix[4]pyrroles 47 and 48 to Li+@C60 in benzonitrile, consisting of an electron transfer switched on by addition of tetra-n-hexylammonium (THA) chloride and off by the addition of tetraethylammonium (TEA) chloride.31 THA chloride was shown to bind to 47 and 48 in benzonitrile with affinity constants of 1.9 × 104 M−1 and 1.7 × 104 M−1 respectively. The binding of Cl− presumably causes a conformational change of the calix[4]pyrrole to the “cone” conformation, which stabilises the close association of the Li+@C60˙− radical ion pair inside the cavity of the cone. This close association of the radical ion pair was suggested by NIR absorption and EPR spectra, DFT calculations and X-ray crystallography, which showed an overall neutral 1:1:1 binding stoichiometry of chloride and the Li+@C60˙− radical ion pair to 47. The authors also showed that the addition of TEACl to the electron transfer product obtained from 47/48, THACl and Li+@C60 in benzonitrile results in a back-electron-transfer to form the original species. It is thought that TEA+ is more effectively bound in the calix[4]pyrrole cavity, thus repelling the Li+@C60˙− radical and reversing the electron transfer process.
Sessler and co-workers have also reported the controlled recognition and cation metathesis properties of ion pair receptor 49 based on the anion binding calix[4]pyrrole unit appended with multiple cation binding sites.32 Proton NMR studies in 10% CD3OD:CDCl3, as well as gas phase energy-minimisation studies and X-ray crystallography suggest that the receptor forms stable 1:1 complexes with CsCl and CsNO3. However, the stronger association between the receptor and potassium means that the addition of KClO4 results in cation metathesis from bound caesium to bound potassium (Scheme 4). Proton NMR and radiotracer measurements showed that the receptor was able to extract CsCl and CsNO3 from D2O into nitrobenzene with the caesium cation released by contacting an aqueous KClO4 solution with the nitrobenzene phase. Recovery of the free receptor was promoted by exposure to CHCl3, suggesting the reusability of the receptor.
 | ||
| Scheme 4 Design concept underlying ion pair receptor 49 and its cation metathesis abilities. | ||
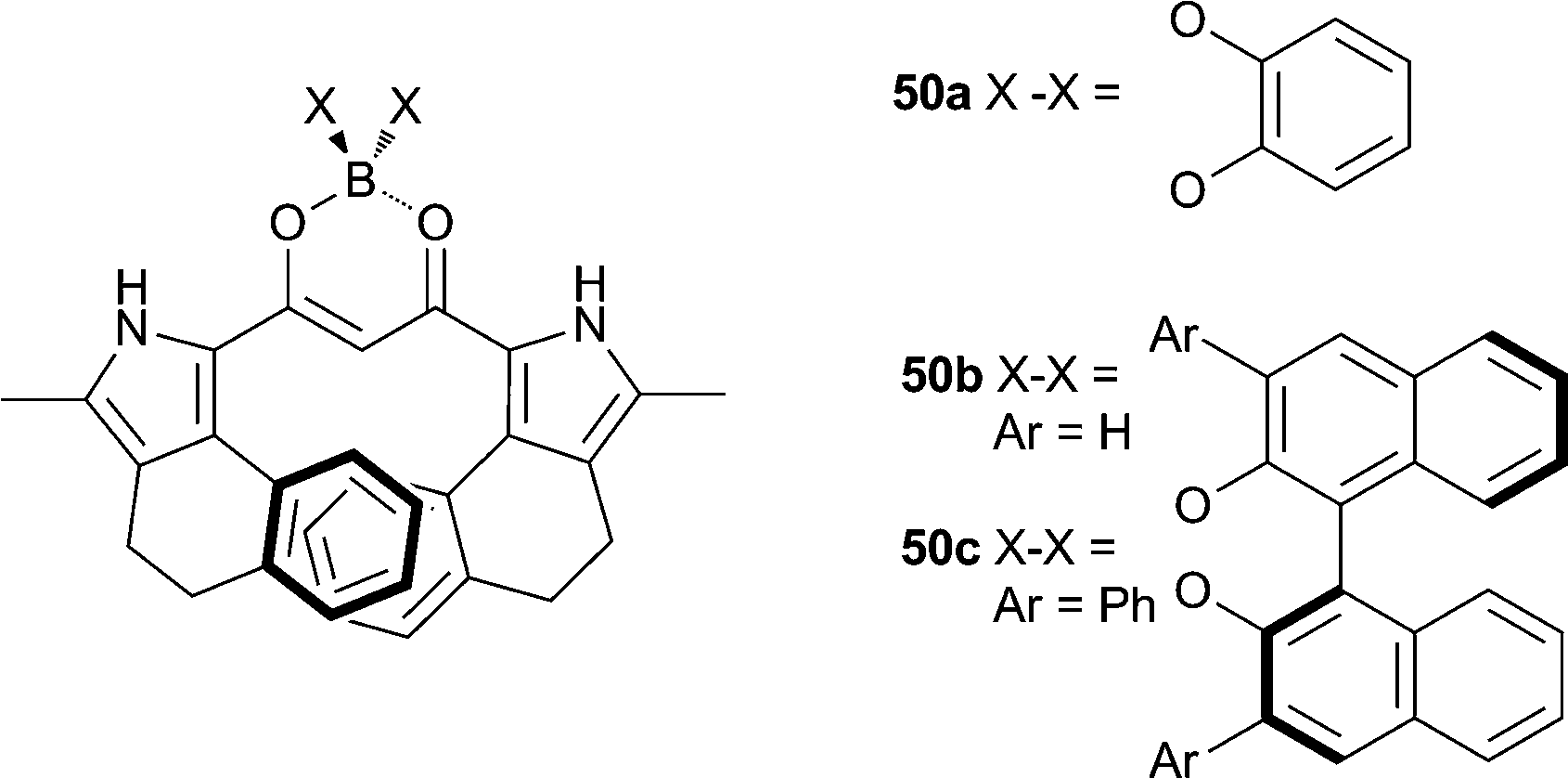
Maeda and co-workers have studied the anion recognition properties of π-conjugated dipyrrolyldiketone based anion receptors, including the anion-induced changes in the chiroptical properties of a series of chiral BINOL-boron substituted π-conjugated receptors such as receptors 50a–c.33 CD experiments suggest that the chiral boron substitution patterns (50b–c) lead to enantiomerically distorted M-like conformations. X-ray crystal structures, 1H NMR and nuclear Overhauser effect spectroscopy (NOESY) indicated that a conformational change occurs upon anion binding with the inversion of the two pyrrole rings (Fig. 11). UV/Vis titration studies in CH2Cl2 with TBA salts of acetate and chloride give binding affinities for 50c of 51000 and 8200 M−1 respectively. Anion complexation of receptors 50a–c was also shown to mediate changes to the CD, UV/Vis absorption and fluorescence spectra and result in an increased circularly polarized luminescence (CPL), with β-dihydronapthopyrrole 50c found to be most efficient at anion-driven CPL enhancement.
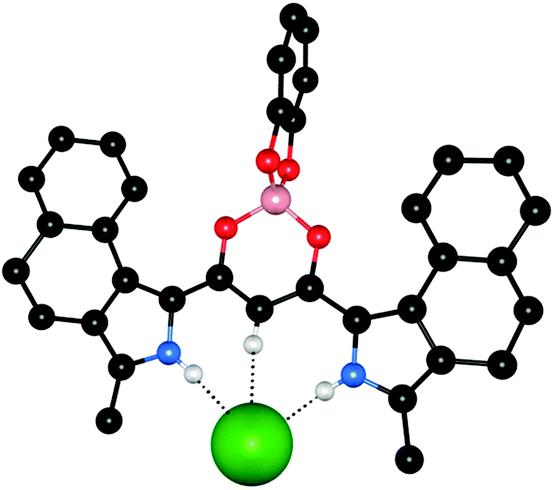 | ||
| Fig. 11 X-ray crystal structure of 50a·TBACl, non-coordinating hydrogen atoms and TBA counterions are omitted for clarity and hydrogen bonds are shown as dashed lines, the receptor is shown in ball-and-stick with the chloride anion in spacefill. | ||
Maeda et al. have also shown that the anion recognition properties of these boron-appended dipyrrolyldiketones can lead to the formation of soft materials through the introduction of different anion modules.34 Replacing spherical halide anions with aliphatic gallic carboxylate derivatives 52-n (as TBA salts) in complexes with the acyclic anion receptors 51a,b promoted the formation of ordered lamellar assemblies through the electrostatic interactions between the charged components. The binding mode between the receptor and anions was elucidated by 1H NMR, UV/Vis and DFT studies, and X-ray diffraction measurements highlighted the lamellar nature of the materials. Differential scanning calorimetry (DSC) and polarised optical microscopy (POM) suggested the presence of mesophases with properties modulated by the length of the anion's alkyl chains. Flash-photolysis time-resolved microwave conductivity (FP-TRMC) measurements were used characterise the electrical conductivity of these materials.

Maeda and Terashima have also reported that the incorporation of amide groups into the dipyrrolyldiketone BF2 based anion receptors (53a–c) can lead to the formation of various solvent-dependent supramolecular assemblies based on the hydrogen bonding properties of the amides.35 This solvent dependence is due to the balance of hydrogen bonding, π⋯π interactions and van der Waals interactions in these systems. In octane, receptors 53a–c were shown to form soluble H-aggregates, while supra-molecular gels were formed in CH2Cl2, CHCl3 and 1,4-dioxane. UV/Vis and IR spectroscopic studies suggested this was due to the different preferences for π⋯π stacking in the assemblies. Octane prefers to interact with the aliphatic side chains of the receptors and leads to tight π⋯π stacking between the receptors forming H-aggregates, while CH2Cl2, CHCl3 and 1,4-dioxane prefer the π-planes over the alkyl chains and hence these assemblies lack strong π⋯π interactions and form gels. The resulting gels were studied by scanning electron microscopy (SEM) and X-ray diffraction and were shown to be responsive to anions, with addition of both TBA and TATA (4,8,12-tripropyl-4,8,12-triazatriangulenium) salts of chloride promoting a gel–solution transition. The authors suggest that pyrrole inversion upon anion binding prevents the effective hydrogen bonding between the amide groups and hence causes a gel–solution transition upon the addition of the appropriate anion (Scheme 5).
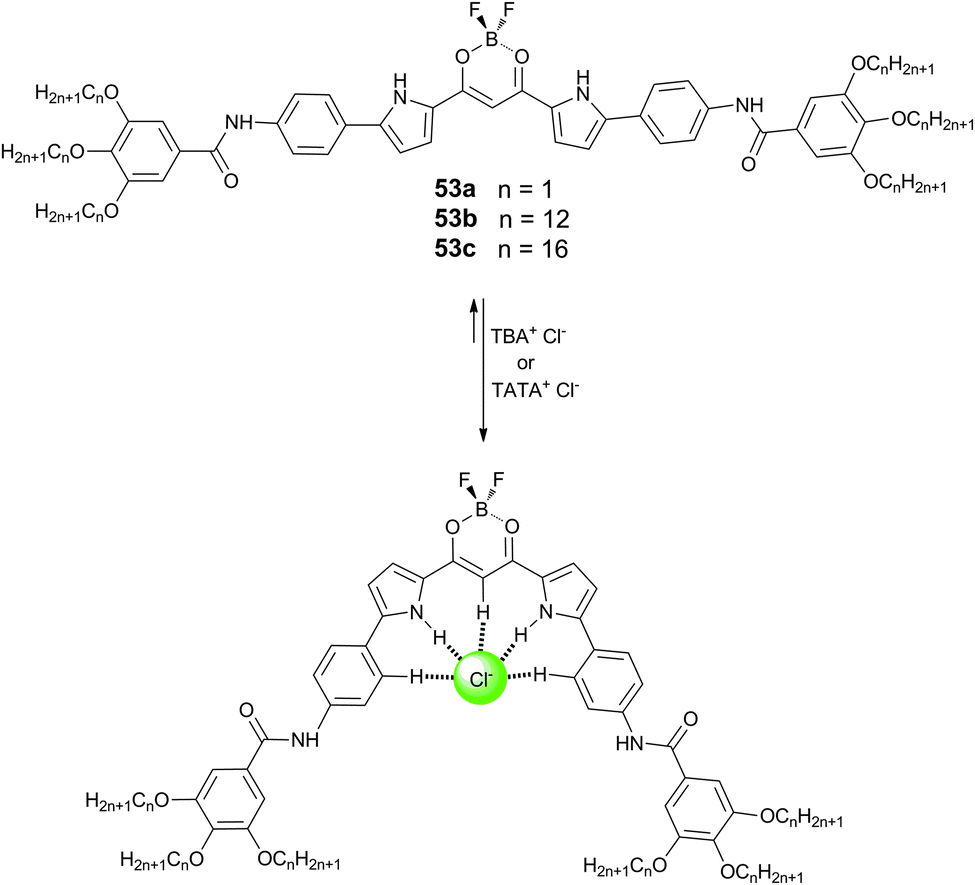 | ||
| Scheme 5 Structures of dipyrrolyldiketone BF2 complexes 53a–c and the chloride-induced pyrrole inversion. | ||
Incorporation of an anionic moiety to the π-conjugated pyrrole based scaffold has been shown by Maeda and co-workers to lead to the formation of anion binding self-assemblies.36 The neutral receptor 54 displays high affinity for carboxylates, with UV/Vis studies giving a Ka of 47000 M−1 in CH2Cl2 for the 1:1 binding stoichiometry of the receptor with acetate. Substitution of the receptor with a carboxylate at the para (55a) or meta (55b) position led to the formation of self-complementary dimers (Fig. 12), as evidenced by ESI-TOF-MS in MeCN and 1H NMR titration studies in CDCl3. DFT studies highlighted the difference in geometries of the dimers depending on the position of the carboxylate group and this was shown to lead to narcissistic self-sorting behaviour where mixing of the two dimers 55a and 55b in CDCl3 did not result in the formation of any mixed dimers, due to the higher stability of one self-complementary dimer over the other. Protic solvents were shown to induce dissociation of the self-assembled dimers due to their ability to solvate the hydrogen bond donor and acceptor groups of the receptor.
 | ||
| Fig. 12 (a) Structure of receptors 54 and 55a,b; (b) proposed dimeric structure of 55b·55b. | ||
Indoles, carbazoles and indolocarbazoles are the π-extended analogues of pyrrole and have therefore been widely used in anion-responsive foldamers. Jeong and co-workers have reported indolocarbazole dimers substituted with chiral (S)-arylethylamido groups (56 and 57), which lead to the preferential formation of one helical isomer over the other.37 The incorporation of the butadiynyl spacer promotes the formation of a helix via hydrogen bonding interactions between the indole and amide groups (Scheme 6). This folding was shown to be solvent dependent with 1H NMR confirming that the folded conformation exists in a 5% CD3CN:CD2Cl2 solvent mix, while the unfolded form is present in DMSO-d6. CD spectroscopy and polarimetry implied the formation of a left-handed (M) helix with optical rotation dependent on the solvent. The reversed chirality of the helix was observed when the amides were altered from the (S)- to the (R)-isomers, confirming that the terminal chiral units determine the helical sense. Strong association between 56 and sulfate was observed by UV/Vis titration, with an affinity constant for TBA2SO4 of 2.2 × 105 M−1 in 5% MeOH:CH3CN and binding of sulfate was shown to switch the helical sense. The sulfate dimer complex was assigned as a P-helix by CD spectroscopy, polarimetry and crystallography. The crystal structures of 56·SO42− and 57·SO42− show that the sulfate anion is situated within the cavity, stabilised by strong indole-sulfate NH⋯O hydrogen bonds complemented by weaker aromatic CH⋯O or amide NH⋯O hydrogen bonds. The switching from one helical sense to the other was shown to be reversible and cyclic, as demonstrated by the CD spectral changes upon the sequential addition of a sulfate sequestering agent and sulfate.
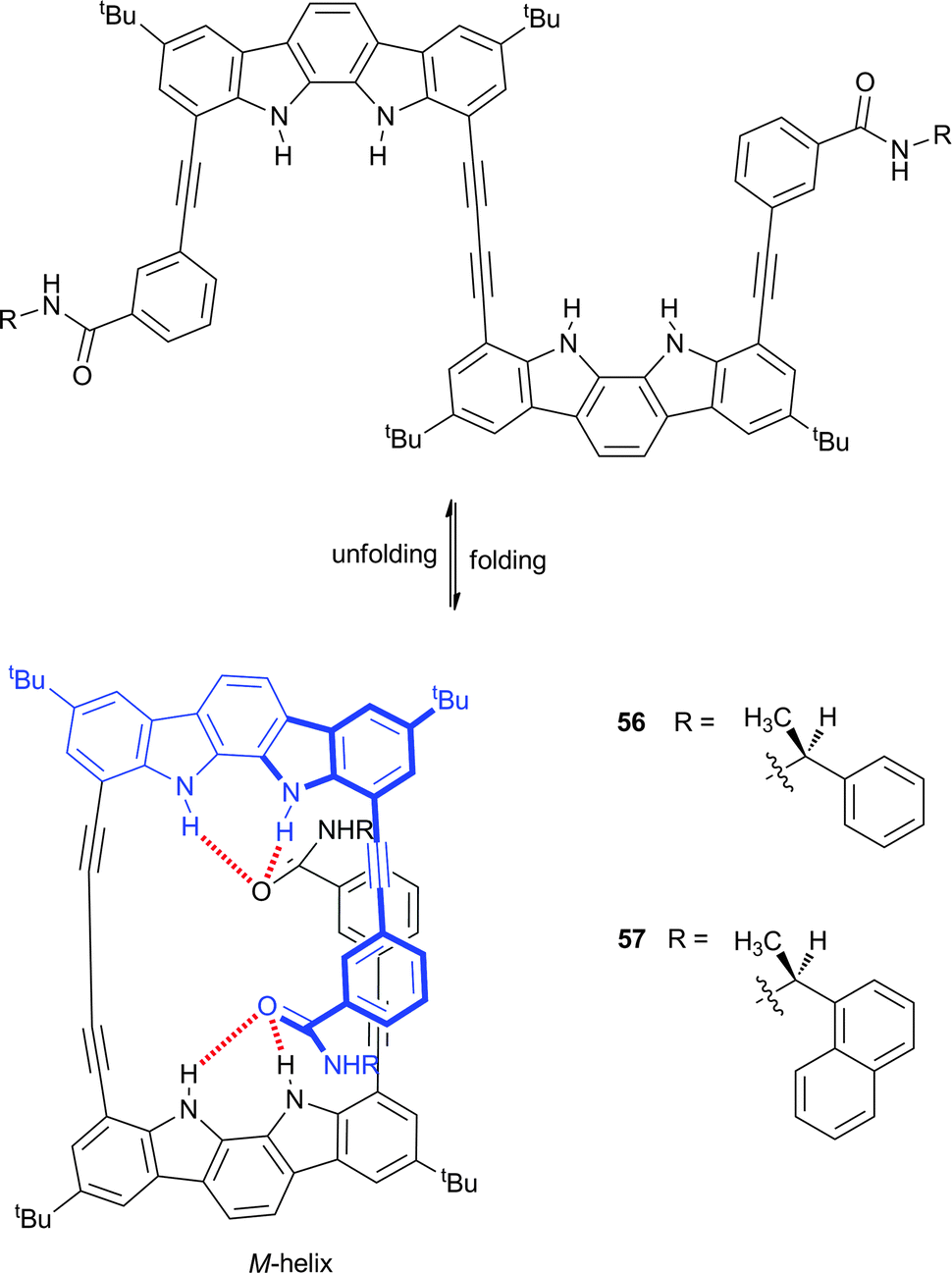 | ||
| Scheme 6 Chiral indolocarbazole dimers 56 and 57 displaying unfolding and folding behaviour. | ||
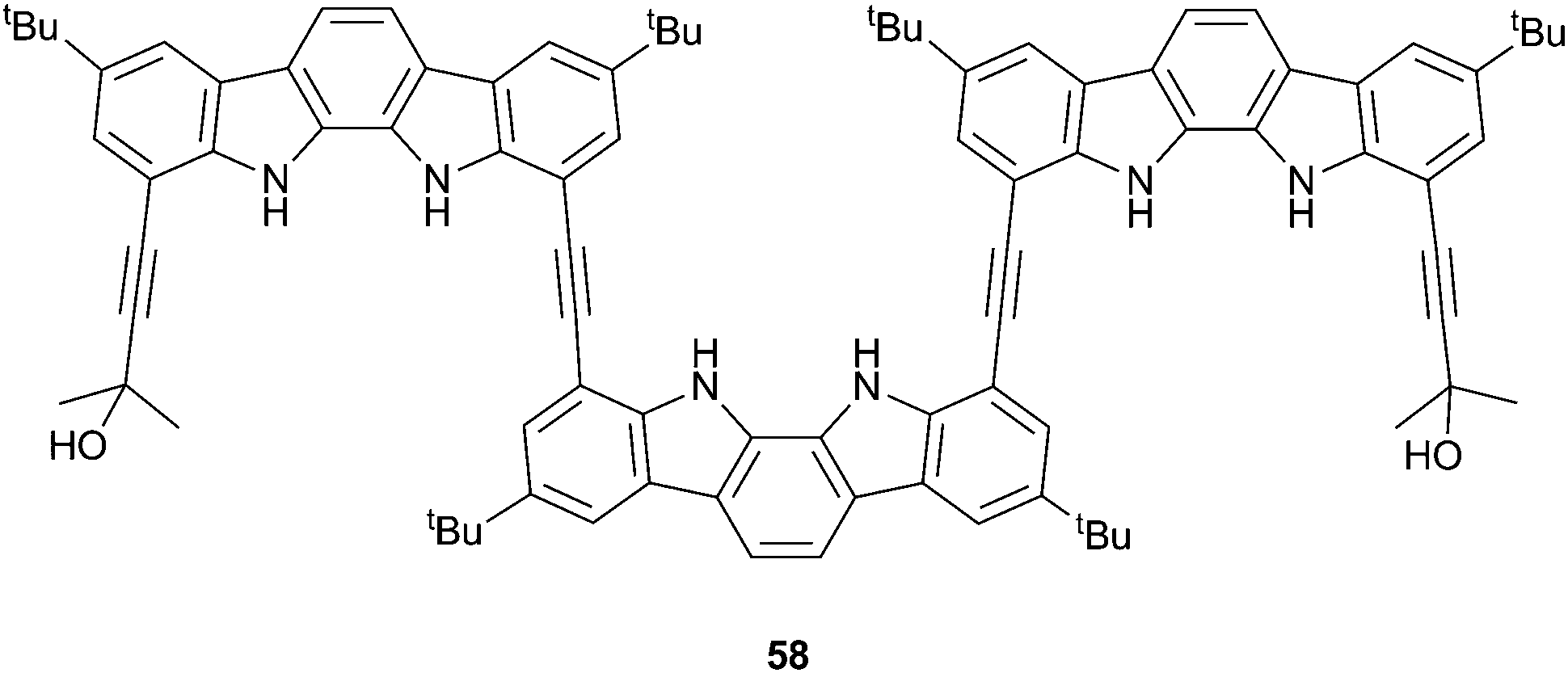
Suk, Kim and Jeong have also demonstrated the use of chiral anions to induce chirally biased helices.38 Indolocarbazole trimer 58 had previously been shown to exhibit enantiomeric helix formation upon the binding of sulfate within the cavity.39 On the other hand, CD and NMR spectroscopy in dichloromethane show that chiral camphorsulfonates promote preferential M- or P-helix formation depending upon the enantiomer added. Strong affinity between the chiral adenosine 3′,5′-cyclic monophosphate (cAMP) and 58 (Ka = 1.1 × 104 M−1), was also observed by 1H NMR titration studies in a 1:9:90 H2O:CD2Cl2:CD3CN solvent mix. CD spectroscopy in MeCN (containing 1% water) showed that the helix formed between 58 and cAMP was dissociated by the addition of CF3COOH due to protonation of the phosphate group and that this dissociation could be reversed by the addition of the base DABCO, which regenerated cAMP phosphate. This cyclic and CD observable acid–base control of helix formation offers a possibility for the development of responsive chiroptical molecular switches.
Anion receptors containing CH hydrogen bond donors
As shown in the previous sections, the majority of hydrogen bonding anion receptors employ NH functionalities as hydrogen bond donors. However, in recent years OH and CH hydrogen bond donors have also become popular. Cao et al. have reported the anion-binding and electrochemical recognition properties of ferrocene appended aryl 1,2,3-triazole 59.401H NMR titration studies in CD2Cl2 showed that the binding affinities for 1:1 complexes follow the trend of Cl− > HP2O73− > H2PO4−. However, these titration studies and DFT calculations suggest that in the case of Cl− only the aryl CH bonds of the triazole and phenyl groups are involved in hydrogen bonding to the anion, while in H2PO4− and HP2O73− the cyclopentadienyl α-protons of the ferrocene groups also contribute to the binding between the anion and the receptor. Cyclic (CV) and differential pulse voltammetry showed that this conferred electrochemical recognition selectivity of 59 for phosphate ions. Electrochemical recognition studies showed that 2.0 equivalents of H2PO4− and HP2O73− resulted in full saturation to the 1:1 receptor:phosphate anion complex with an associated cathodic shift in the CV spectrum, but no change was observed when testing the TBA salts of F−, Cl−, Br−, I−, PF6−, ClO4−, HSO4− and CH3COO−.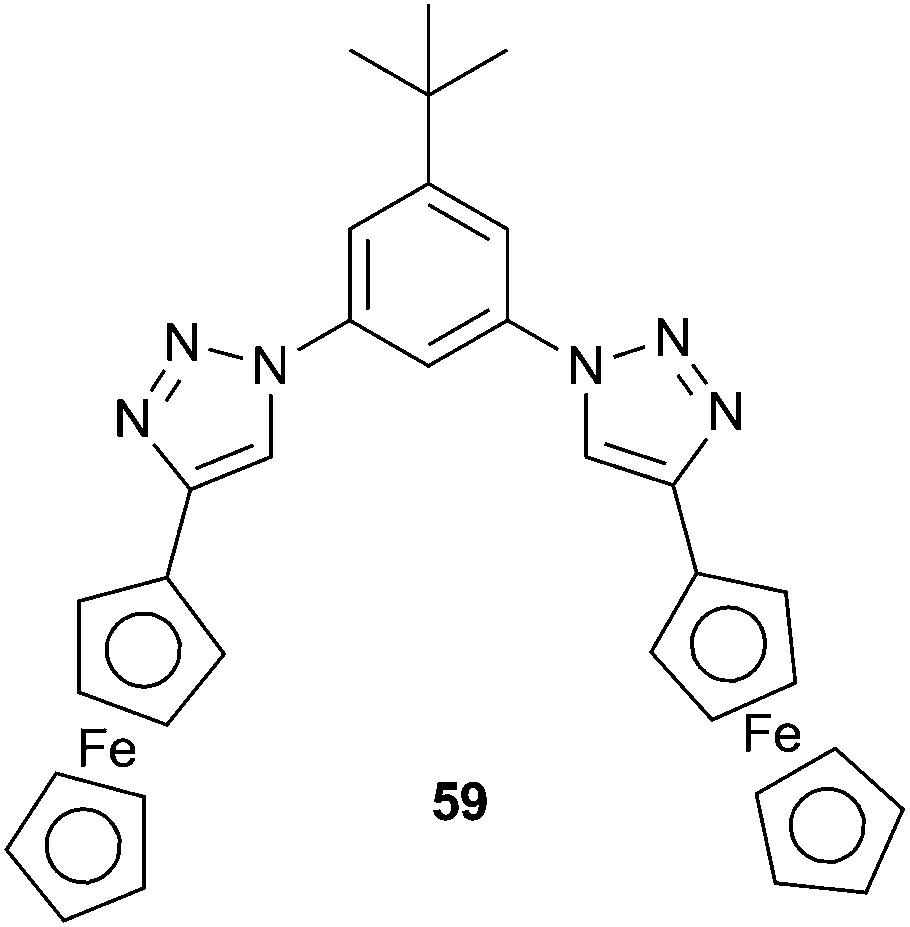
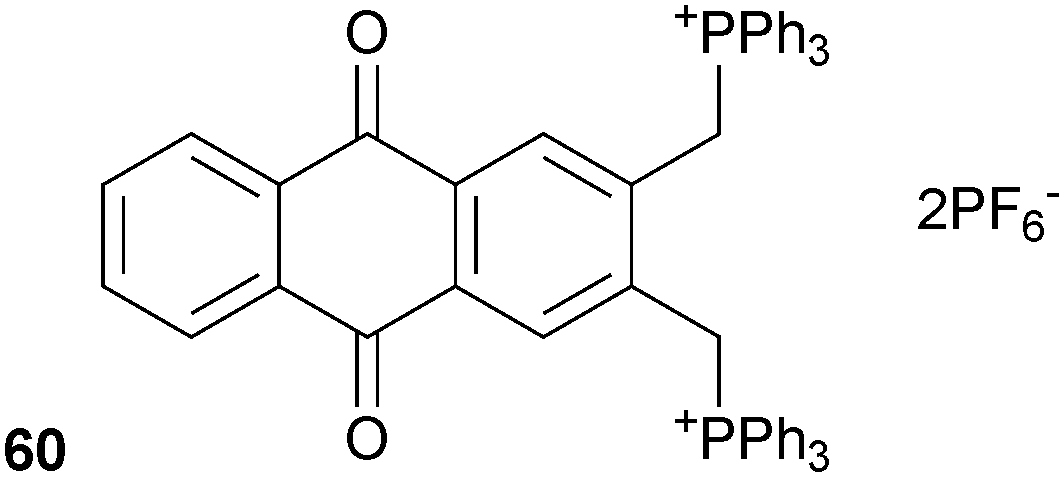
Ganguly, Das and co-workers have incorporated +PPh3 groups into an anthraquinone based receptor (60) in order to obtain an optical anion sensor through the activation of the methylene protons as C–H hydrogen bond donors that can bind anions.41 The anion binding properties of the receptor with a range of TBA salts in acetonitrile were studied by 1H and 31P NMR, electronic spectroscopy and supported by DFT calculations. Weak interactions were observed for chloride, bromide and hydrogen sulphate, with much stronger interactions detected for dihydrogen phosphate and fluoride, where the binding stoichiometry was shown to be 1:2 receptor:anion and the formation constants Kf = 2.24 × 106 M−1 for F− and Kf = 8.98 × 104 M−1 for H2PO4−. The association of these anions and the receptor was accompanied by significant changes in the electronic spectra of the receptor and a distinct colour change, most noticeable in the case of fluoride. The receptor was shown to be highly effective at extracting fluoride from aqueous solutions, with an extraction efficiency of 99.3% and effective at concentrations of F− as low as 0.06 ppm.
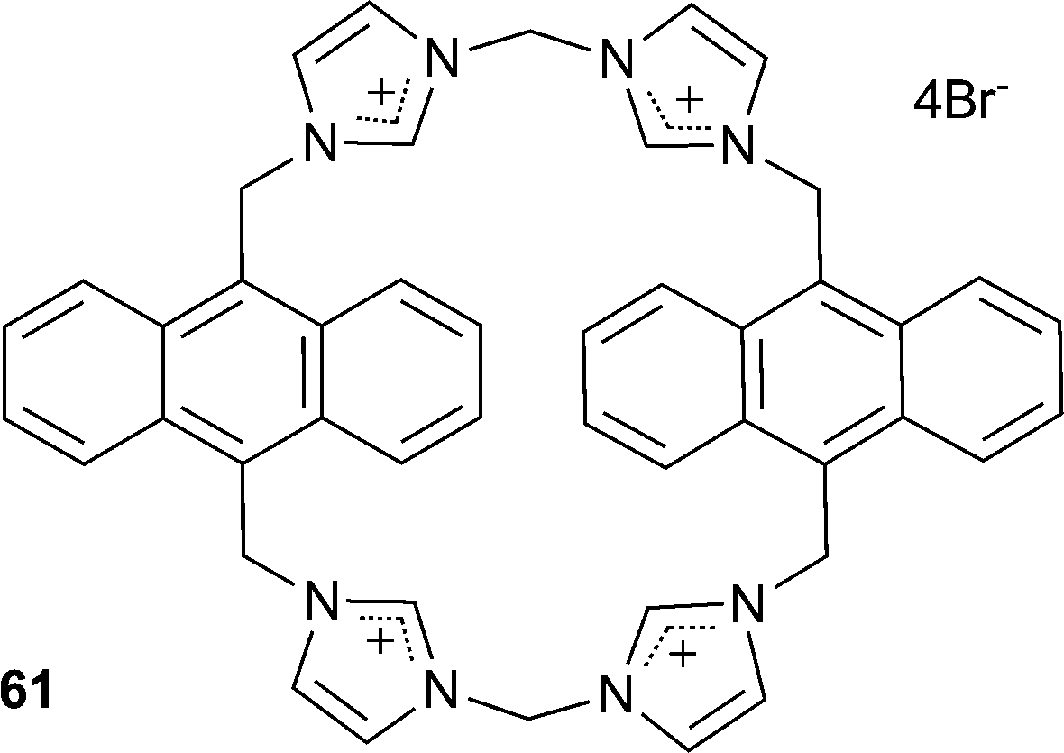
Kim and co-authors have described the use of imidazolium-anthracene based cyclophane 61 for the detection of guanosine-5′-triphosphate (GTP) and iodide in aqueous solutions at physiological pH with detection limits of 4.8 × 10−7 M and 8 × 10−5 M respectively.42 Fluorescence spectroscopy studies in 100% aqueous solution at pH 7.4 with 100 equivalents of the TBA anion salts revealed the receptor's high selectivity for iodide and a range of phosphates, most significantly GTP, with the observation of chelation-enhanced fluorescence quenching (CHEQ) of excimer emission. Different binding modes were observed between the two anions with a 2:1 and 1:1 receptor:anion stoichiometry and affinity constants of 1.1 × 103/2.1 × 106 M−1 and 1.3 × 104 M−1 for GTP and I− respectively. 1H NMR titration experiments and DFT calculations suggest that this altered binding mode is due to GTP being bound by two receptors through imidazolium CH+⋯O interactions while I− is bound within the cavity through both imidazolium CH+⋯anion and anthracene CH⋯anion interactions.
Flood and co-workers have been interested in the anion binding properties of aryl-triazole containing receptors (triazolophanes, e.g.62). These triazolophanes display high binding affinity for Cl− (> 106 M−1 in CH2Cl2), due to pre-organisation (macrocyclic effect), the presence of CH hydrogen bond donor groups and size complementarity of the receptor and chloride. The properties of these triazolophanes have been recently review by Flood and co-workers.43 In 2011, this research group also studied a variety of chloride:triazolophane complexes with varying degrees of conformational freedom (62–64) to verify the importance of structural rigidity as a pre-organisation factor paying the enthalpic cost of bringing together the electropositive CH hydrogen bond donor groups in the triazolophanes.44 Computational studies were supplemented by 1H NMR titrations to calculate the binding affinities and preparation free energies (the energy difference between the observed and ideal binding energies) for three triazolophanes (Scheme 7): a ‘‘strongly’’ preorganised macrocycle (62), a “partially” preorganised macrocycle (63) and a “poorly’’ preorganised macrocycle (64). These studies show that high rigidity (in combination with the macrocyclic effect) prevents the relaxation of close H⋯H contacts and retains the preorganised nature of the CH triazole hydrogen bond donors in 62 to maximise the strength of the receptor⋯anion interactions.
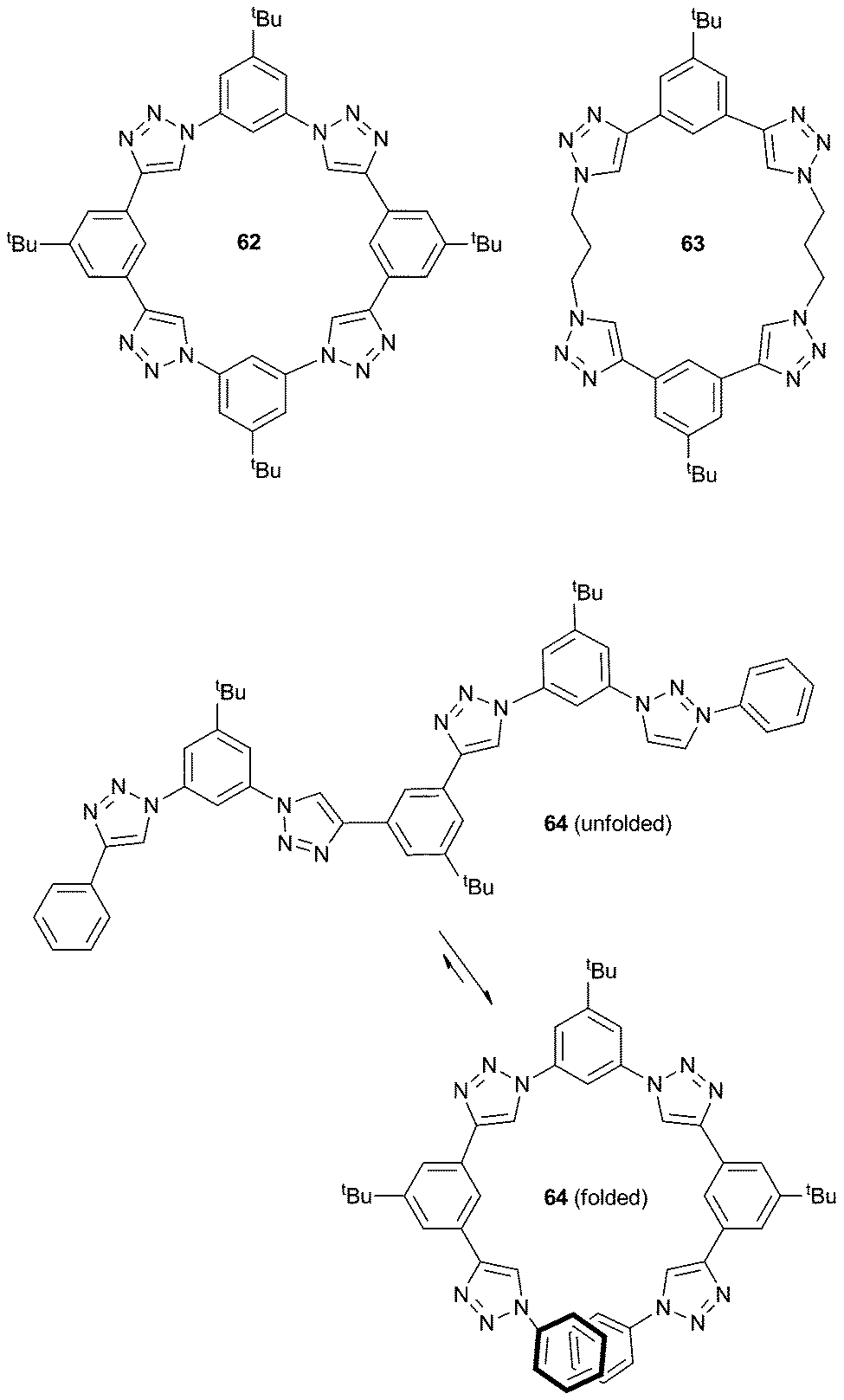 | ||
| Scheme 7 Structure of triazolophanes 62–64 with various degrees of rigidity, and the folded and unfolded conformation of 64. | ||
In a separate manuscript, Flood and co-workers have used the triazolophane scaffold to compare the strength of aromatic and aliphatic C–H hydrogen bond donors by replacing one of the phenylene groups in receptor 62 with a propylene linker in receptor 65.45 Gas phase computational studies showed that the length of the CH⋯Cl distance in the propylene based triazophane 65 was greater for the aliphatic (3.02 Å) than the aromatic (2.96 Å) CH donor, supporting its classification as a weaker interaction. The gas phase studies also suggested that the free energy of binding decreased from −194 to −182 kJ mol−1 upon substitution of the phenylene to a propylene unit and that the propylene unit interacts with chloride via a single CH⋯Cl hydrogen bond. ESI-MS and diffusion NMR spectroscopy were used to quantify all of the possible complexes present in solution, namely a 1:1 receptor:anion complex, a 2:1 sandwich complex and ion pairing (TBA+Cl− and TBA+[65·Cl]−). This allowed the deconvolution of the ion pairing events from chloride:receptor binding and hence the calculation of accurate 1:1 equilibrium binding constants for each receptor by 1H NMR and UV/Vis titrations in CH2Cl2. The free energies of binding for each receptor were shown to be within experimental error (−38 ± 2 kJ mol−1 for 62 and −39 ± 2 kJ mol−1 for 65), suggesting that the difference in CH⋯Cl hydrogen bond strength between aromatic and aliphatic donors can only be distinguished when they are competing within the same receptor.

McDonald et al. have shown that the 1,8-napthalimide unit is an effective CH hydrogen bond donor in studies on receptor 66, and quantified the strength of the anion binding from this interaction by comparison to an analogous receptor where the naphthalimide is substituted by 4-tert-butyl benzene (67).46 UV/Vis and 1H NMR titration studies in CD2Cl2 with a series of TBA salts demonstrate the high affinity of naphthalimide substituted receptor 66 for halide anions. Shifts in the 1H NMR signals for the two CH naphthalene protons (Hd and Hg, Scheme 8) support their involvement in CH⋯anion hydrogen bonding interactions. As the size of the halide anion increases, the contribution of the CH⋯anion interaction of the napthalimide unit (Hd and Hg) to the binding increases, while the contribution of the triazole unit (He) decreases (Table 2). DFT calculations show that the CH⋯Cl− distances increase from triazole (He) < napthalamide (Hd) < phenylene (Hi) and support a bifurcated CH⋯anion interaction of the napthalimide unit, analogous to that of urea, which gives an electronic binding energy greater than that of the triazole (ΔE of −80 kJ mol−1vs. −64 kJ mol−1). All these results suggest that naphthalimides possess strongly polarised CH donors suitable for anion binding.
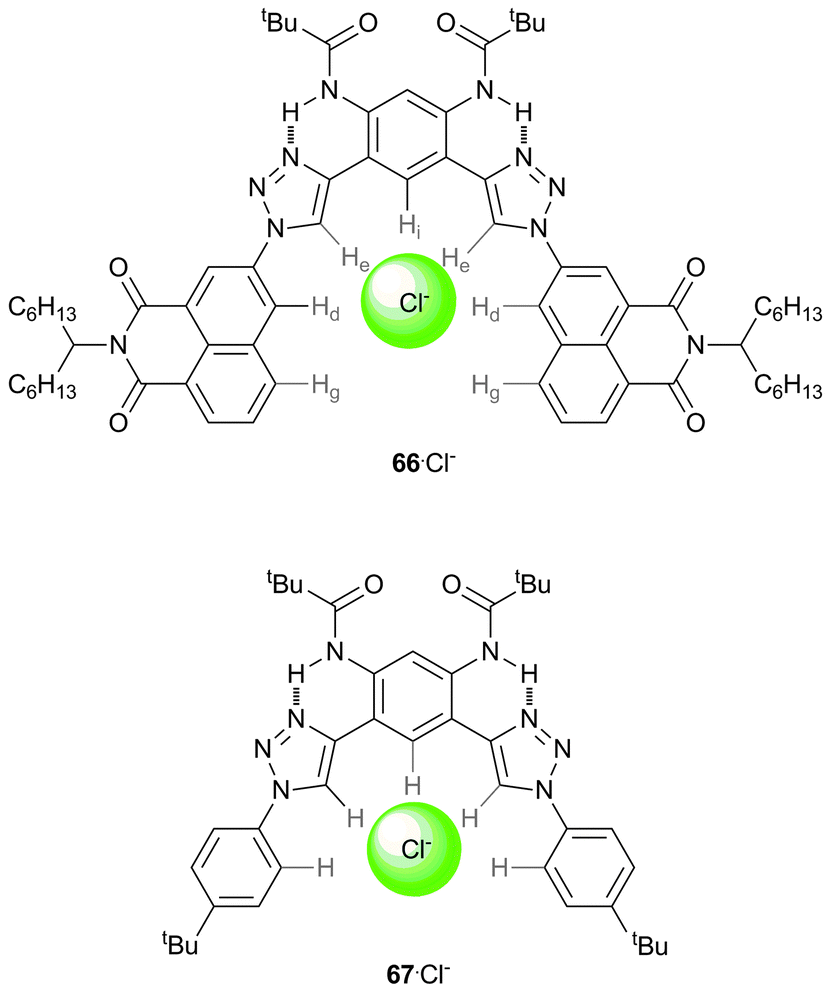 | ||
| Scheme 8 Structures of receptors 66 and 67, with the coordinating CH hydrogen atoms labelled in 66, and showing the chloride binding site. | ||
| Anion | K a (66) | K a (67) | Δδ (He) | Δδ (Hd) |
|---|---|---|---|---|
| Cl− | 1.4 ± 0.2 × 105 | 4.2 ± 0.2 × 104 | 2.2 | 0.7 |
| Br− | 3.9 ± 0.5 × 104 | 1.1 ± 0.1 × 104 | 1.9 | 0.9 |
| I− | 7.8 ± 2 × 103 | <103 | 1.6 | 1.0 |
Anion receptors employing halogen bonding, anion–π interactions and hydrophobic effects
The majority of anion receptors use electrostatic interactions or hydrogen bonding to bind anions, but in recent years more unconventional interactions such as halogen bonding, anion–π interactions and solvophobic effects have become popular. Out of these binding modes, halogen bonding, i.e. the non-covalent interaction between an electron-deficient halogen compound (Lewis acid) and a neutral or anionic Lewis base, is being increasingly exploited as an alternative to the commonly applied hydrogen bonding. Beer and co-authors have reported the anion binding properties of preorganised bidentate bromoimidazoliophane receptor 68.47 Two conformers of the receptor exist, the syn (68a) and anti (68b) forms, and they were found to exhibit different anion recognition behaviour in both solution and solid state. X-ray crystal structures of the bromide salts of the receptors show that the bromoimidazolium groups in the anti conformer 68b each bind a bromide, while in the syn conformer 68a the bromoimidazolium groups chelate a bromide to form a calix-like structure with the second bromide involved in hydrogen bonding interactions to a MeOH solvent molecule (Fig. 13). The sterics of the syn-conformer lead to a less linear halogen bonding interaction, 166.8° compared to 174° in the anti-conformer, and the increase in C–Br bond distance of the bromoimidazolium group from the anti- to syn-conformer suggests an increase in halogen bonding strength following the same trend. Proton NMR titration studies in 9:1 CD3OD:D2O found no significant binding of TBA halides in solution for the anti conformer, however, the syn conformer was shown to form 1:1 complexes with affinity constants of 889 M−1, 184 M−1, 184 M−1 and <10 M−1 for bromide, iodide, chloride and fluoride respectively. This high selectivity for bromide was not observed in the protic version of the receptor, 69, with moderate binding of ∼100 M−1 observed for chloride, bromide and iodide. This suggests that substitution of halogen atoms into hydrogen bond receptors can alter both the electronic and geometric features of the receptor and hence the exhibited anion recognition properties.
 | ||
| Fig. 13 Crystal structures of (a) 68a·2Br (non-coordinating bromide ion is omitted for clarity) and (b) 68b·2Br. Hydrogen atoms and solvent molecules are omitted for clarity; halogen bonds are represented by dashed lines; the coordinated bromide anion is shown in spacefill, with the receptor in ball-and-stick. | ||
Beer and co-workers have also incorporated the bromoimidazolium motif into catenane 70, which was synthesised via bromide anion templation.48 The receptor was designed to give a linear arrangement of the two halogen bond donor groups, resulting in cooperative halogen bonding interactions with anions. The incorporation of the naphthalene spacer allowed for the optical sensing of anions by fluorescence spectroscopy, with studies showing that while no change was observed for corresponding macrocycle 71 with the TBA salts of fluoride, chloride, bromide, iodide, acetate, dihydrogen phosphate, nitrate and the TEA salt of bicarbonate, catenane 70 displayed selective recognition of chloride and bromide, with no optical changes observed for the remaining anions. This suggests that the 1:1 binding of chloride and bromide by the catenane, with affinity constants of 3.71 × 106 M−1 and 1.48 × 105 M−1 respectively, was achieved through size complementary recognition and the preorganised cooperative nature of the halogen bond donor groups.
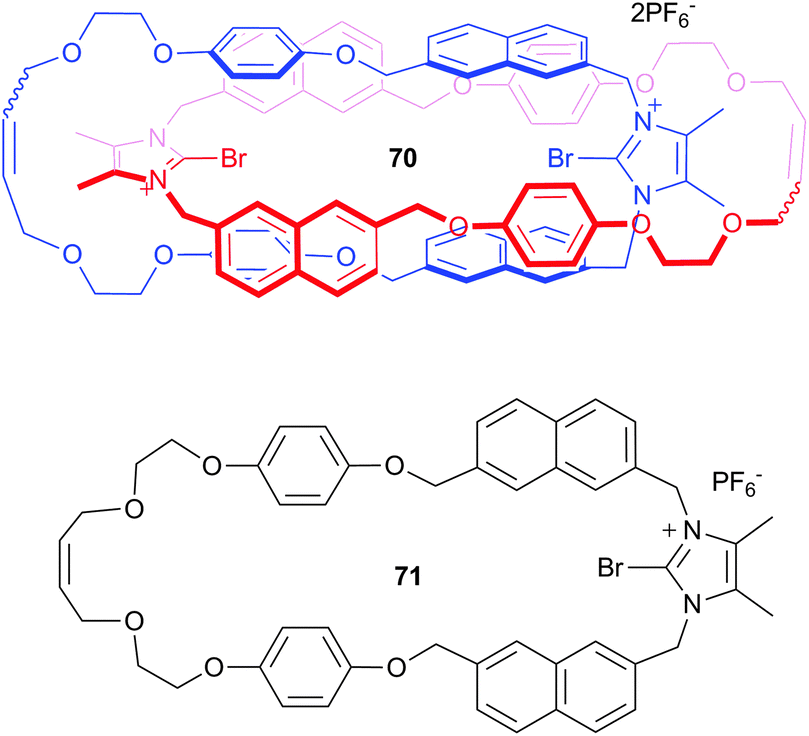
Taylor and co-workers have studied a series of urea based receptors incorporating iodoperfluorobenzoate groups (72a,b, 73a,b, 74a,b, 75a,b and 76a,b) in an attempt to use hydrogen bonding and halogen bonding in concert to bind anions.49 Solution state UV/Vis, 1H NMR and 19F NMR titration studies characterised the binding strength between the urea receptors and TBA salts of halides and oxoanions in CD3CN and suggested a 1:1 binding stoichiometry for all receptor:anion complexes. The results demonstrate that hydrogen bonding and halogen bonding can occur simultaneously and show that incorporating halogen bond donor groups into urea based systems confers halide selectivity to receptors that have high affinity for Y-shaped anions, without negating the strength of the interactions with oxoanions. Incorporation of perfluorinated benzoates into the scaffolds as controls that do not participate in halogen bonding (72b, 73b, 74b, 75b and 76b) allowed the quantification of the halogen bond contribution to the overall anion binding. For example in the symmetrical urea receptor 75a the affinity constant for the binding of halides was 30 times greater than observed with 75b, where no halogen bond donor group is present. Computational studies supported the observed dependence of halogen bond contribution to anion binding on the length of the linker, with receptors where the linker results in a more linear XB⋯anion interaction shown to give a greater halogen bonding contribution to the overall anion binding, illustrating the strong preference for linear geometry in halogen bonding. The contribution of the halogen bond to the overall thermodynamics of anion binding was found to be negligible for the majority of oxoanions, NO3−, HSO4−, TsO− (<0.2 kcal mol−1), small for BzO− and H2PO4− (0.3 and 0.4 kcal mol−1) and significant for the halides (approaching 1 kcal mol−1).
Anion⋯π interactions, the non-covalent interactions between an anion and an electron deficient π-system, are far less common than their cation⋯π counterparts but have received recent attention. Giese et al. have reported the anion binding properties of phenylbenzamide derivatives 77–80 in both the solid and solution state.50 In the crystal structure of tetraethylammonium (TEA) bromide with pentafluorobenzamide 77 an interaction between the aromatic pentafluorophenyl ring and the bromide anion was observed, with a Br⋯centroid distance of 3.67 Å. The bromide anion is offset from the centre of the ring due to the additional NH⋯Br interaction (3.431 Å) (Fig. 14). The effect of the electron withdrawing C6F5 group to promote cooperative NH⋯anion and anion⋯π binding interactions by acidifying the NH bond and providing an electron poor arene ring for increased strength anion⋯π interactions was further investigated in solution by 1H and 19F NMR titration studies of N-phenylbenzamides 78–80 with the TBA salts of Cl−, Br−, I−, BF4− and PF6−. Titrations of 78 and 79 were performed in CDCl3 while solubility issues meant the titrations of 80 were conducted in CD3CN. A 1:1 stoichiometry was confirmed for the receptors with all anions except PF6− where ambiguity between 1:1 and 2:1 receptor:anion complexation was observed. Affinity constants suggest that the strength of anion binding increases from I− < Br− < Cl−, with the binding constants for each receptor calculated from the shifts in the amide NH signals. The increase in anion binding strength from 79 < 78 < 80 was attributed to the electron-withdrawing effect of the C6F5 unit and the presence of two amide NH donor groups in 80. Low temperature NMR and computational studies suggest that anion receptor complex 80 exists as the symmetric conformer.

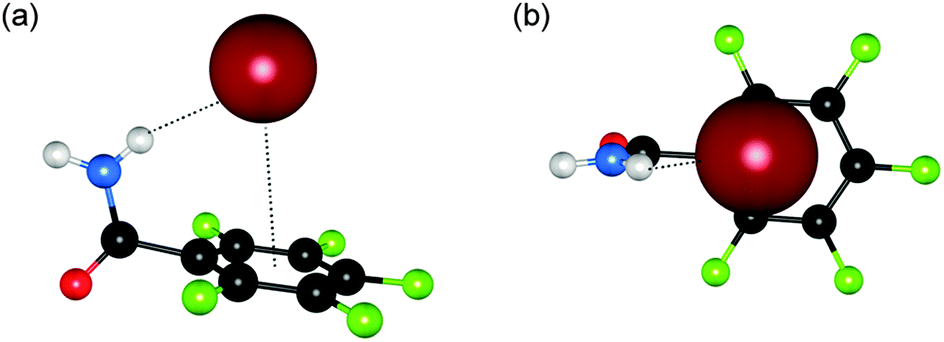 | ||
| Fig. 14 Two views (a and b) on the crystal structure of pentafluorobenzamide 77 and TEA bromide. The bromide anion is shown in spacefill with the receptor in ball-and-stick; hydrogen bonds and anion⋯π interactions are shown as dashed lines. | ||
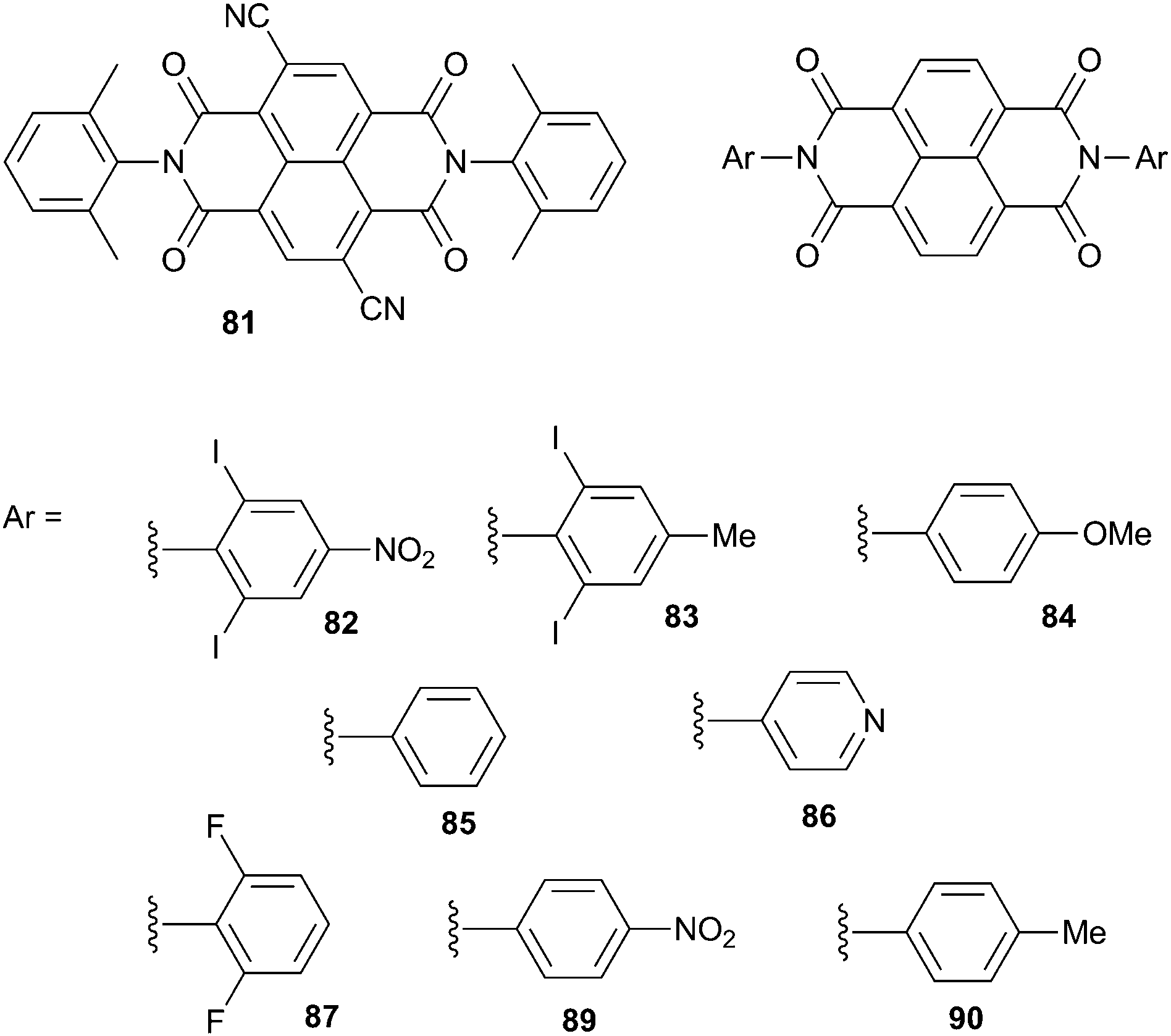
Saha and co-workers have characterised the range of interactions that can occur between electron deficient naphthalenediimides (NDIs) and a variety of anions.51 By altering a NDI scaffold by substituting either the core with electron withdrawing substituents (81) or the nitrogen centres with various electron donating or electron withdrawing groups (82–90) they were able to control the π-acidity of the NDIs. Electrochemical studies showed that core substitution enhances π-acidity more dramatically than substitution at the nitrogen centres. The interactions of the NDIs with the TBA and TEA salts of decreasing Lewis bases OH− > F− > CN− > AcO− > H2PO4− > Cl− > Br− > I− > PF6− were probed using ESI-MS and ITC (in 1,2-dichlorobenzene) and 1:1 anion:receptor complexes were observed with affinity constants shown to decrease as the π-acidity of the NDI decreased and as the Lewis basicity of the anion decreased. The interaction was shown to be dependent on the electron donor–acceptor strengths and not on CH⋯anion interactions, as NMR titrations showed that iodo- and fluoro-substituted NDIs 82, 83 and 87, with no possible CH donors, have higher binding affinities. This was complemented by computational studies where electrostatic potentials suggest that the imide rings of the NDIs are the most electron-deficient. The strength of the interaction was shown to be controlled by the strength of both the anion and NDI basicity. With the strongly π-acidic NDI 81 thermal electron transfer (ET) to form both NDI˙− and NDI2− was observed with OH−, F− and CN− in CD3CN in the dark. The method of transfer was confirmed by UV/Vis, EPR spectroscopy, 19F NMR and XPS, which provided no evidence of C–F bond formation but the sequential formation of the F˙, NDI˙− and NDI2− radical species. Switching on thermal anion-to-NDI ET was shown to be possible by maximising the π-acidity of the receptor and basicity of the anion. Moderately basic anions were shown to result in light induced PET (photo-induced electron transfer), while for the weakly basic anions such as I− and less electron deficient NDIs no ET was suggested and only anion⋯π interactions were observed. The basicity of the solvent was also shown to affect the interactions between the anion and NDI with higher solvation of anions in polar solvents hindering their electron donating ability.
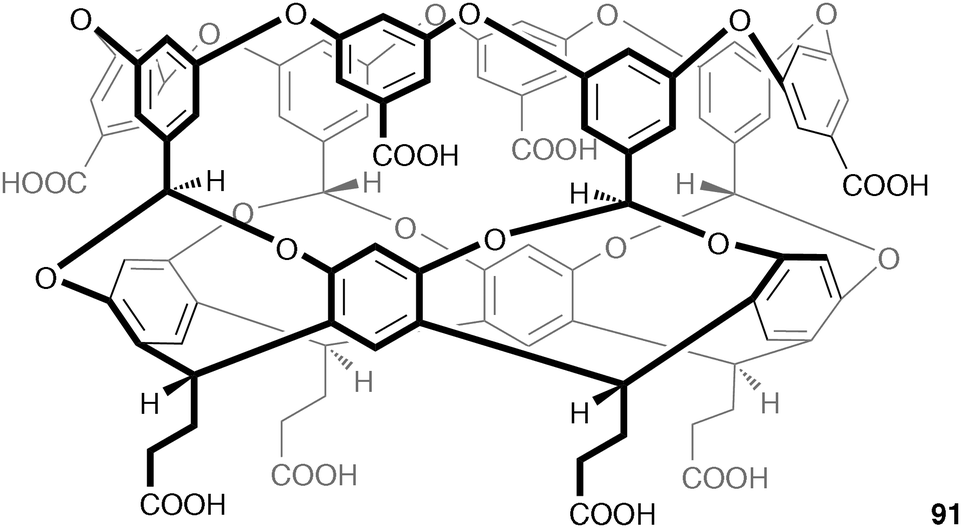
Solvophobic effects (usually hydrophobic) play an important role in supramolecular chemistry, but are often overlooked. Gibb and Gibb have studied the curved amphiphile 91 (based on the calixarene structure) with a water soluble outer coat comprised of 8 carboxylic acids, and a 8 Å × 8 Å hydrophobic pocket capable of binding a range of hydrophobic guests including adamantane carboxylic acid.521H NMR titration studies in 10 mM phosphate buffer at pH 11.3 with the sodium salts of a range of anions, showed that the amphiphile displays Hofmeister selectivity, with no association observed for strongly and moderately solvated anions (F−, SO42−, AcO−, Cl−, and Br−) while for the weaker solvated anions the signals of the benzyl protons at the base of the hydrophobic cavity were observed to shift downfield upon increasing concentration of anion. Fitting of the isotherms suggested a 1:1 ratio of anion:91, with association constants increasing across the Hofmeister series from NaNO3, <1 M−1; NaClO3, 3 M−1; NaI, 11 M−1; NaSCN, 33 M−1; to NaClO4, 95 M−1. In silico studies suggest that the perchlorate anion moves from solvation by eight waters to three or four waters upon binding within the hydrophobic cavity, supporting the hypothesis that the observed anion selectivities are related to the adaptability of the anion's solvation shell, with strongly solvated anions unable to reorganise or lose part of their solvation shell, meaning they cannot be accommodated within the cavity of the amphiphilic host. ITC experiments in 10 mM phosphate buffer at pH 11.3 in the presence of adamantane carboxylic acid show that chlorate, iodide, thiocyanate and perchlorate are able to compete with the hydrophobic guest and weaken the hydrophobic effect, with the binding of adamantane carboxylic acid 1.5 kcal mol−1 lower in the presence of perchlorate than in the presence of fluoride (the free energy of binding for adamantane carboxylic acid and 91 in the absence of salts is 9.1 kcal mol−1).
Metal and Lewis acid containing anion receptors
Metal and Lewis acid containing anion receptors have also remained popular during 2011 and 2012. Yam and co-workers have reported highly luminescent bifunctional receptor 92 containing a 1,3-alternate calix[4]-crown-5 moiety for potassium binding and Lewis acidic borane centres for the coordination of fluoride anions.53 The free receptor was found to display an intense electronic absorption band at 342 nm and bright blue fluorescence in dichloromethane. The emission was attributed to an excited state caused by intramolecular charge transfer from the phenolic oxygen atoms to the boron centres via the conjugation of the alkynyl spacers. UV/Vis and fluorescence titrations were performed with a range of anions in dichloromethane. Receptor 92 was found to interact strongly and selectively with F− by complexation through the Lewis acidic boron centres. This interaction caused significant fluorescence quenching as a result of the destruction of the intramolecular charge transfer. The binding constant for this process was determined as logKa = 4.46 ± 0.1. Additionally, the complexation of K+ cations was observed by similar means in 1:1 dichloromethane:methanol, and induced a blue shift with enhancement in the absorbance and extensive fluorescence quenching.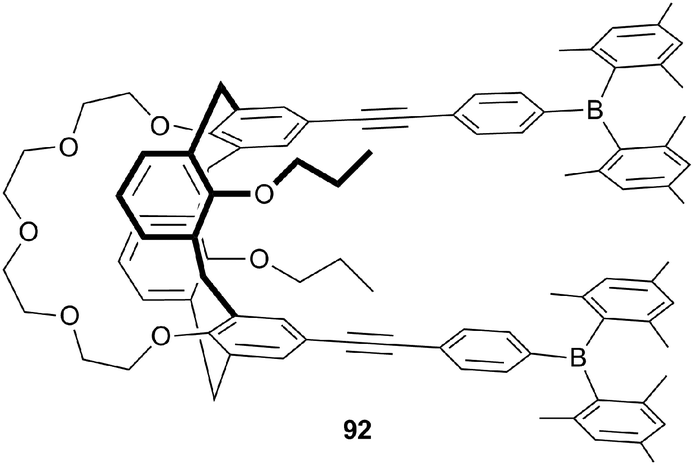
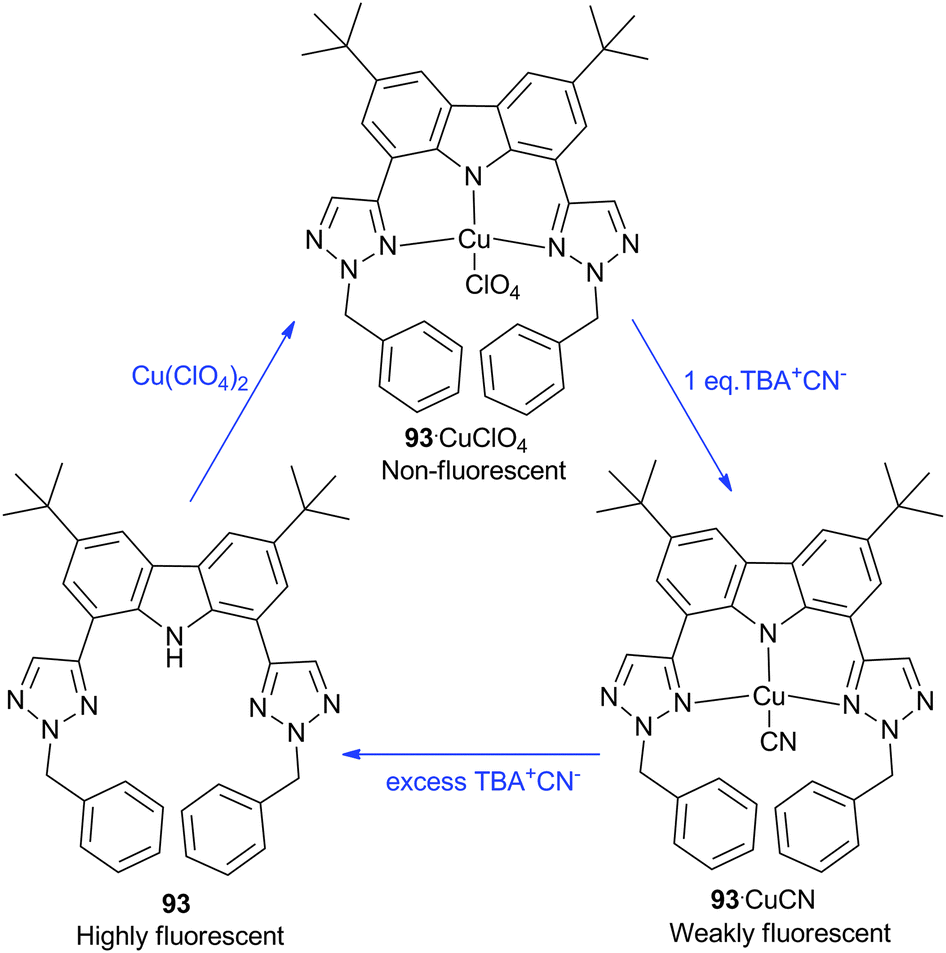
Jang and co-workers have reported the CN− sensing properties of the copper(II) complex of ligand 93.54 The free ligand was found to exhibit strong absorptions around 300 and 370 nm and fluorescence emission around 385 nm in MeCN. On the addition of copper(II)perchlorate, the fluorescence emission at 385 nm almost completely disappeared after the addition of 1 equivalent of copper(II). The authors then investigated the response to the addition of tetrabutylammonium cyanide (TBA+CN−). UV/Vis and fluorescence titrations indicated the presence of two areas of spectral change; on the addition of up to 1 equivalent of CN− the absorptions around 360 and 408 nm were found to gradually increase, with an accompanying linear increase in fluorescence intensity. When further amounts of CN− were added the fluorescence intensity increased much faster, and by the addition of 10 equivalents of anion the fluorescence of the free ligand had been largely recovered. This was attributed to the de-metallation of the 93-Cu complex in the presence of excess CN− (Scheme 9). However, the authors noted that up to the addition of 1 equivalent of cyanide this system could also be used to accurately determine micromolar concentrations of CN−.
 | ||
| Scheme 9 The proposed mechanism of CN− sensing by the 93–Cu(II) complex. | ||
Caltagirone and Lippolis have reported the anion complexation properties of the copper(II) complex of ligand 94.55 This ligand had been previously reported as a fluorescent sensor for zinc(II) ions,56 and the authors reasoned that the analogous copper(II) complex would be coordinatively unsaturated and therefore suitable for anion complexation. The solution phase response of [Cu(94)](BF4)2·MeCN to a range of anions was investigated in MeCN and H2O by UV/Vis titration. The authors found that only the addition of CN− and I− anions produced a significant response. The interaction with I− was found to follow a 1:1 binding stoichiometry and gave a binding constant of logKa = 4.8 ± 0.1 in MeCN, with weaker binding observed in H2O (logKa = 2.4 ± 0.1). The interaction with CN− was found to be more complex in MeCN, with the formation of stable 1:1 and 2:1 anion:receptor complexes, while in H2O the formation of the 1:1 CN− complex was significantly more favourable than the I− complex (logKa > 7). Remarkably, the spectral changes upon the addition of these anions allowed for the naked eye detection of I− in MeCN only and CN− in both MeCN and H2O (Fig. 15). In MeCN the formation of two different 94·CN− complexes is evident to the naked eye with a colour change from cyan (no anion) to dark blue (1 equivalent of CN−) to pink (2 equivalents of CN−). Crucially, this report represents the first example of CN− sensing by a Cu(II) complex without removal of the Cu(II) centre from the receptor complex.
 | ||
| Fig. 15 Colour change of [Cu(94)](BF4)2·MeCN (10−3 M) upon the addition of different anions in MeCN. From left to right: free receptor; +1 eq. F−; +1 eq. Cl−; +1 eq. Br−; +1 eq. CN−; +2 eq. CN−; +1 eq. I−. Reproduced with permission from Chem. Commun. 2011, 47, 3805–3807. © Royal Society of Chemistry. | ||
Lindoy and co-workers have previously reported the formation of a [Fe4L6]8+ cage (L = 95) which has been used for the encapsulation of anionic guests such as BF4− and PF6−, with selectivity for the latter guest.57 The BF4− inclusion complex could be generated by forming the cage from a mixture of ligand 95 and Fe(BF4)2·6H2O in MeCN. In a recent study the same authors have reported that attempts to form the cage using FeCl2·5H2O as a source of iron(II) in MeCN followed by anion exchange with KPF6 yielded an unprecedented complex in which the encapsulated anionic species was [FeIIICl4]−.58 Microanalysis confirmed that the stoichiometry of the deep red product was Fe4L6·(FeCl4)·(PF6)7·CH3OH (with L = 95) and the structure of this complex was elucidated by single crystal X-ray diffraction (Fig. 16). The encapsulated [FeCl4]− anion was found to have perfect tetrahedral symmetry with Fe–Cl bond lengths in the expected range for this species (2.1994(17) Å). Given that this anion is not displaced by PF6− during the purification process (when this host had previously shown good selectivity for PF6− over BF4−) the authors reasoned that the size, shape and symmetry of the [FeCl4]− guest must be highly complementary to the cavity of this cage.
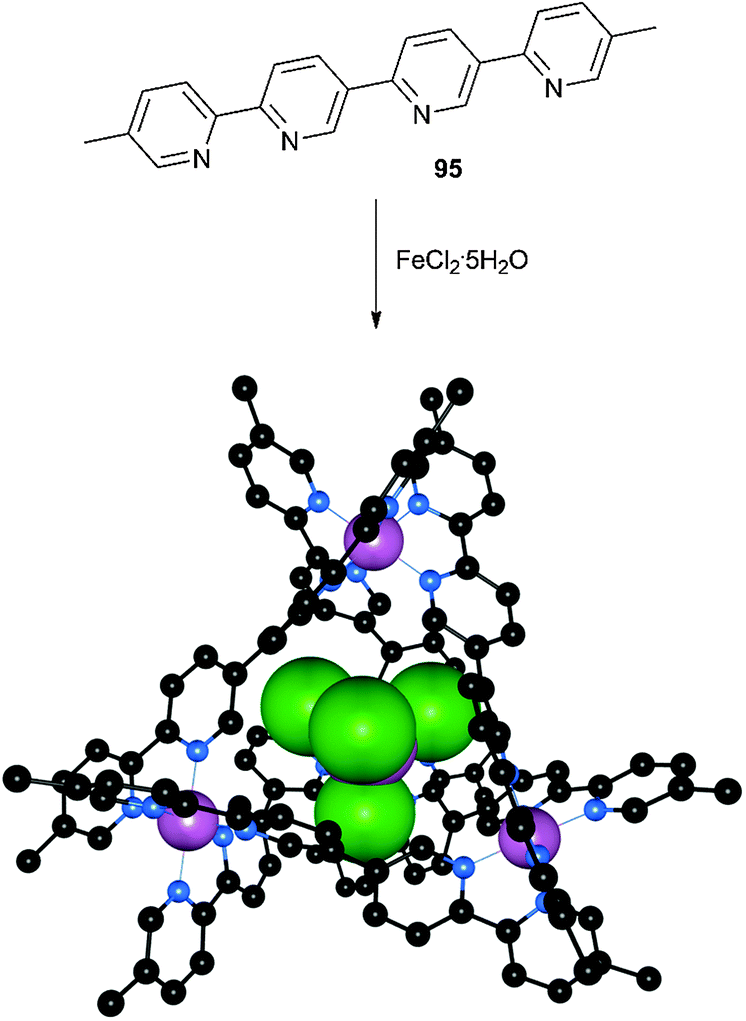 | ||
| Fig. 16 The encapsulation of [FeCl4]− within a Fe4(95)6 cage, along with the crystal structure of Fe4(95)6·(FeCl4)·(PF6)7·CH3OH. Iron and [FeCl4]− ions are shown in spacefill with ligand 95 in ball-and-stick (iron: purple, chloride: green, nitrogen: blue, carbon: black). Hydrogen atoms, solvent molecules and counterions are omitted for clarity. | ||
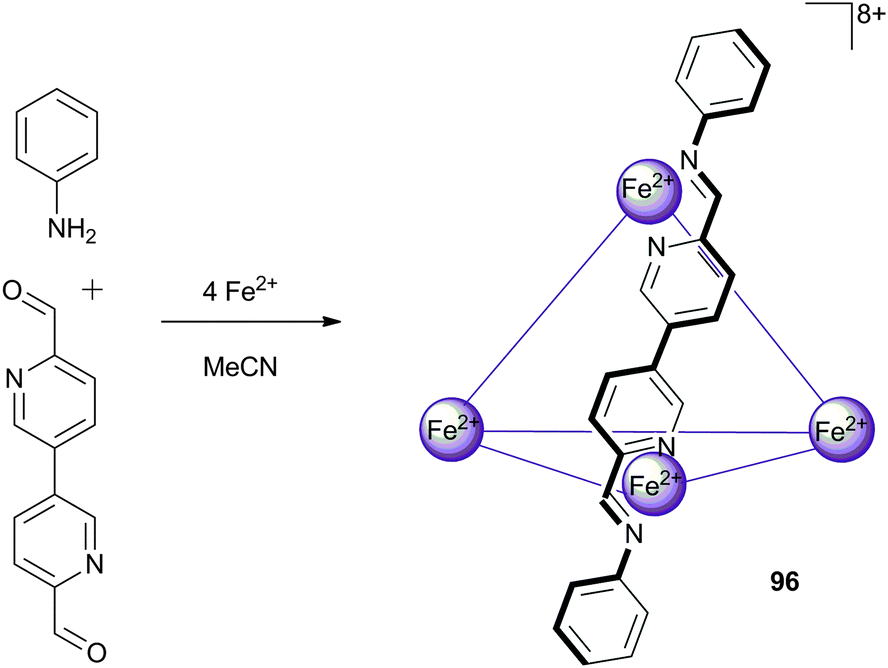
Nitschke and co-workers have continued their work on self-assembled tetrahedral cages, including a report on face-capped tetrahedral Fe4L4 cages of different sizes that can encapsulate a variety of neutral guests (depending on the size of the cage),59 and a report on a series of M4L6 cages of the form 96 in which the exterior framework of the cage is exchangeable.60 In the latter, the cage is constructed by the generation of the ligands in situ as shown in Scheme 10. As the ligand synthesis is reversible, the addition of alternative anilines into the system can lead to displacement of the original aniline and the generation of a new cage structure in a dynamic process. In general, anilines substituted with electron donating substituents appeared to be favoured; thus, the addition of p-methoxyaniline into a cage formed from p-chloroaniline resulted in the quantitative displacement of the p-chloroaniline residues, as observed by 1H NMR. If the electronic properties of the anilines used had greater similarity, a mixture of products was observed by ESI-MS analysis with varying degrees of substitution. The cages were all found to encapsulate BF4− and PF6−counteranions from the reaction media, which were difficult to remove from the cavity causing difficulties in investigating the anion binding properties of the free host. Consequently, cage 96 was synthesised with a trifluoromethanesulfonamide counteranion, which is too large to be contained within the cage, thus enabling anion complexation studies. Using NMR titration techniques in CD3CN, the authors found that BF4− and OTf− were suitable guests, with binding constants determined as Ka = 2.3 × 104 M−1 and 5.2 × 104 M−1 respectively. The binding of PF6− was found to be exceptionally strong, with a binding constant of Ka = 1.3 × 106 M−1 estimated by competitive binding experiments with BF4− and OTf−, representing the strongest example of host–guest binding for a metal–organic host that can be isolated in the absence of a guest.
 | ||
| Scheme 10 The construction of Nitschke's M4L6 cages with reconfigurable exterior. | ||
Self-assembled and anion templated molecular architectures
In the previous sections there have already been some examples of self-assembled structures, but in most of those cases it was the metal ion that drove the self-assembly. Cases where the anion functions as a template for the self-assembled structure or where the anion influences the architecture via allosteric effects are less common. Nitschke and co-workers have described a dynamic system where the addition of one anion (perchlorate) triggers a structural reorganisation to obtain a new structure with a high affinity for another anion (chloride).61 A dynamic library 97 containing a mix of coordinated complexes was created from the reaction between p-toluidine, 6,6′-diformyl-3,3′-bipyridine and cobalt(II)triflimide hydrate in MeCN. The addition of KPF6 or NaOTf to this library leads to the formation of tetrahedral Co4L68+ cage 98 as the only observed species in 1H NMR and ESI-MS spectroscopy, with the anions (PF6− or OTf−) encapsulated within the tetrahedral cage. Interestingly, the addition of LiClO4 to these tetrahedral cages or to the original dynamic library leads to the transformation into a pentagonal prism Co10L1520+ (99) as the unique species observed by 1H NMR and ESI-MS spectroscopy (Scheme 11). Single crystal X-ray crystallography of 99 revealed that it forms a “barrel-like” structure formed by 60 chemical species and that the ligands form five anion binding pockets on the outside of the barrel, ideally suited for the ClO4− anions that occupy these sites, and one binding pocket on the inside of the barrel, ideally suited for the Cl− anion that occupies this site (Fig. 17). As no chloride was added to the mixture, the authors postulate that the chloride anions were scavenged from the glassware and thus that the affinity of the Co10L1520+ pentagonal prism 99 for chloride must be extremely high. When specifically added during formation, other anions such as F−, Br−, N3−, OCN− and SCN− could also be bound inside the cavity of the Co10L1520+ pentagonal prism (but not HF2−, CN−, SeCN−, acetylene or CO2). A pentagonal prism with an empty central pocket could be obtained by replacing p-toluidine with 4-hydroxyaniline during formation, indicating that only the peripheral perchlorate anions are necessary to template the formation of the pentagonal prism, while the central anion is not required as a template but is merely bound with high affinity. The authors also note that a similar pentagonal prism could be obtained from the PF6− adduct of tetrahedral cage 98 after heating the sample for a prolonged period of time.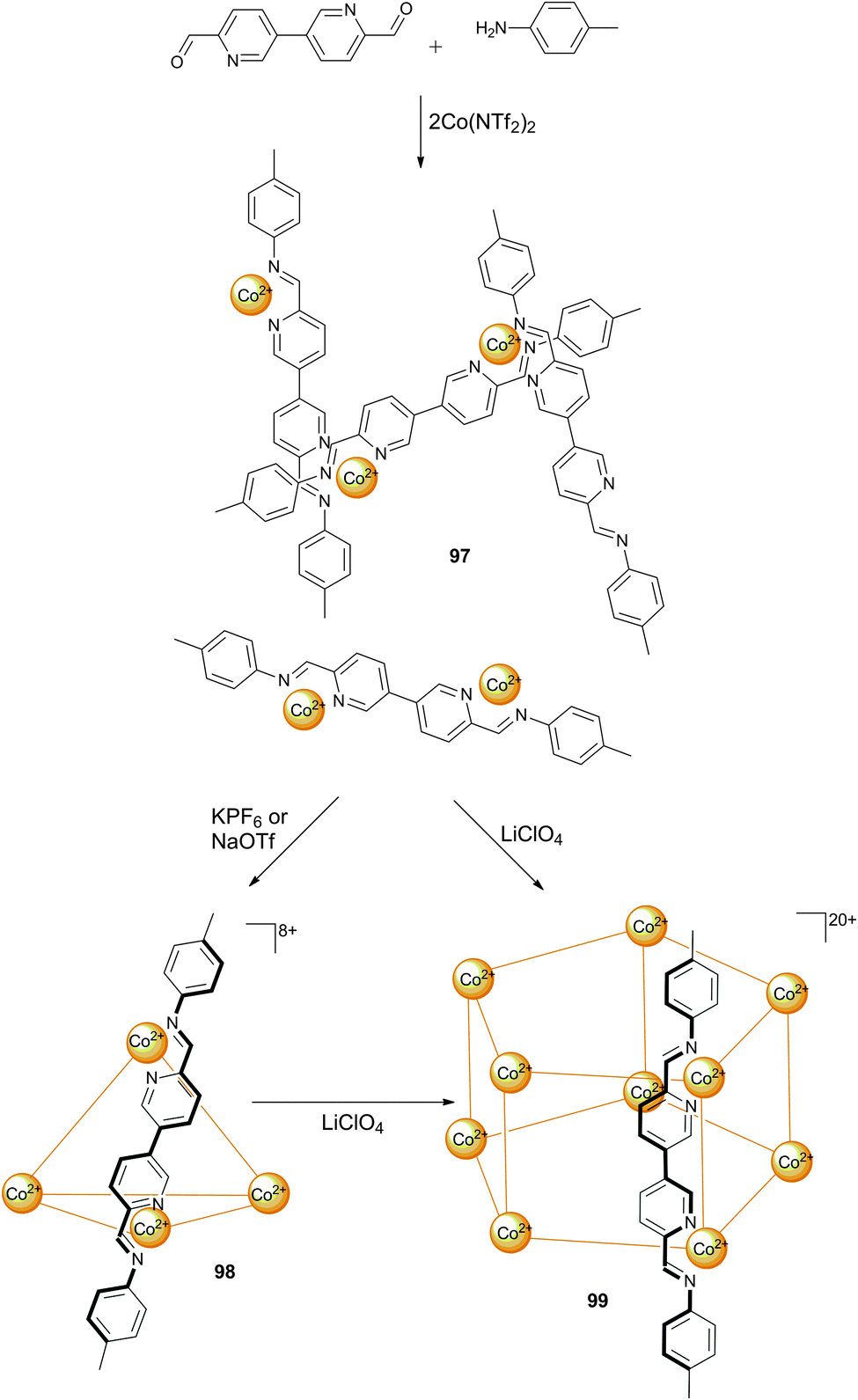 | ||
| Scheme 11 Formation of dynamic library 97, tetrahedral cage 98 or pentagonal prism 99. | ||
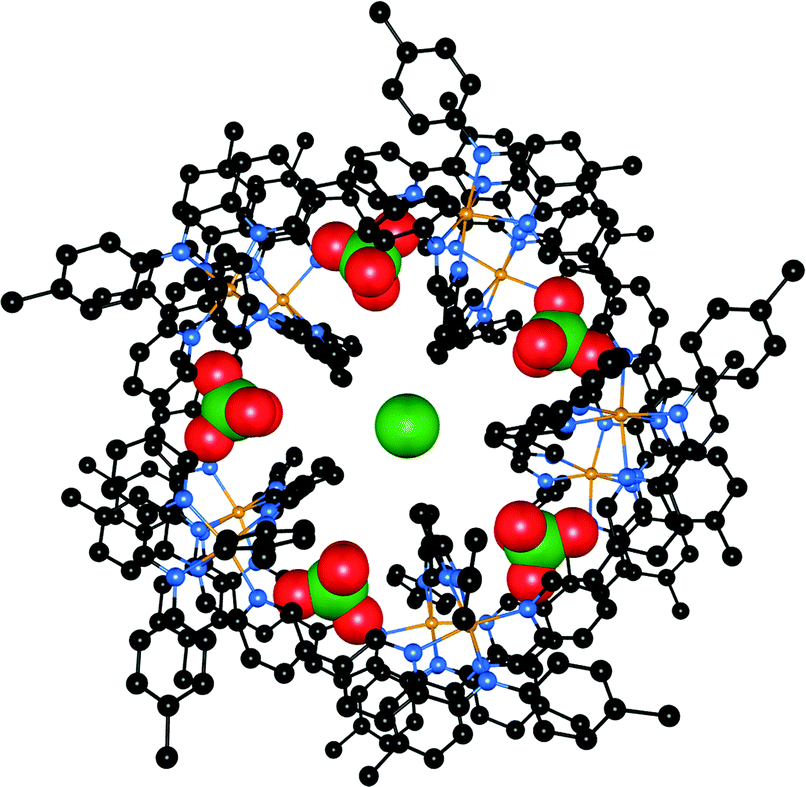 | ||
| Fig. 17 Crystal structure of 99·ClO4·Cl (top view). Perchlorate and chloride anions are in spacefill, with the ligands and cobalt ions (gold) in ball-and-stick. Hydrogen atoms and non-coordinated counterions are omitted for clarity. | ||
Sessler and co-workers have reported tetracationic macrocycle 100 that is able to form “molecular boxes”, environmentally responsive pseudorotaxanes, or metal-linked supramolecular polyrotaxanes, depending on the anion used and the external stimuli provided.62 The authors have previously reported that macrocycle 100 can bind terephthalate dianion 101 on the outside of the macrocycle.63 Alternatively, when receptor 100 is exposed to the larger dianion 102 a pseudorotaxane is formed with a 2:3 host:guest stoichiometry (pseudodimeric species of the pseudorotaxane), as confirmed by 1H NMR titrations, NOESY NMR and Job plot analysis in DMSO-d6 and ESI-MS. Furthermore, the addition of related dianion 103 to 100 results in the formation of a pure 1:1 pseudorotaxane, as confirmed by 1H NMR titrations and NOESY NMR experiments. The different supramolecular architectures that were obtained from macrocycle 100 with the varying anions 101–103 are represented by their crystal structures shown in Fig. 18a–c. The authors also showed that not only the nature of the anionic guest influences the obtained supramolecular architecture and stoichiometry, but also temperature, pH and Ag(I)+ cations. Variable temperature 1H NMR showed that increased temperature leads to de-threading of the pseudorotaxanes, while pH-dependent 1H NMR studies showed that lower pH (upon addition of CF3COOD) can also lead to de-threading and the regeneration of free 100 due to the formation of the neutral dicarboxylic acid forms of the anions (pseudorotaxane formation was still observed for the mono-protonated forms of the dianions 101–103). Subsequent addition of base (triethyl-amine) converts the acids back to their dianionic form and hence results in the formation of pseudorotaxanes and this cycle of threading and de-threading can be repeated several times by the addition of acid and base. Interestingly, the addition of Ag(I) (as a PF6− or NO3− salt) to a mixture of 100 and 103 leads to the formation of a polyrotaxane whereby the monomers are the individual 1:1 pseudorotaxanes 100·103 held together in a 1D supramolecular polymer via Ag(I) cation bridges, as observed by single crystal X-ray diffraction (Fig. 18d).

 | ||
| Fig. 18 Crystal structures of Sessler's tetracationic macrocycle with various dianions. Hydrogen atoms, solvent molecules and counterions have been omitted for clarity; the dianions are shown in spacefill with the macrocycle in ball-and-stick. (a) [100·101]2+ showing that the dianion is bound outside the macrocycle (no threading); (b) [100·102]2+ showing rotaxane formation with 2:3 stoichiometry; (c) [100·103]2+ showing rotaxane formation with 1:1 stoichiometry; (d) 100·(103)3·Ag2·16H2O showing polyrotaxane formation upon the addition of Ag(I). | ||
Singh and Sun have reported the solvent-dependent reversible nitrate binding and self-assembly properties of tripodal receptor 104.64 The HNO3 complex of 104 was observed as the sole species when nitric acid was added to a methanol suspension of 104 and the structure of 104·HNO3 was confirmed using single crystal X-ray diffraction. However, it was found that when nitric acid in aqueous methanol was added to a chloroform solution of 104, two species were observed in 1H NMR and ESI-MS experiments, namely the HNO3 adducts of 104 and of its nitro substituted analogue 105. On the other hand, when nitric acid was added to a MeCN solution of 104, the HNO3 complex of 105 was observed as the sole species, as confirmed by 1H, COSY, HSQC, HMBC and ROESY NMR and ESI-MS. Furthermore, when CHCl3 was added to a DMSO solution of the HNO3 complex of 105, the formation of self-assembled dimer (105)2 was observed using 1H NMR and ESI-MS techniques. Thus, chloroform caused the release of the bound nitric acid and the subsequent formation of homodimer (105)2via hydrogen bonding between the amide NHs and the ortho nitro substituents, and this was found to be reversible as the evaporation of chloroform from this solution resulted back in the formation of 105·HNO3. The various solvent-dependent conversions between 104, the nitrated analogue 105, HNO3 adducts of the receptors and homodimer (105)2 are shown in Scheme 12.
 | ||
| Scheme 12 Structure of receptor 104 and the nitro analogue 105 produced upon addition of HNO3, along with the various solvent-dependent conversions between receptor and complexes and dimers. | ||
Beer and co-workers have continued their work on anion templated catenanes. In a 2011 communication they reported a new synthetic pathway for preparing [2]catenanes (e.g.108a,b), based upon the association between an acyclic flexible bis-amine with a positively charged macrocycle, followed by a ring-closure reaction with an appropriate bis-acid chloride mediated by chloride anion templation (Scheme 13).65 The new catenanes thus obtained (108a and 108b) were characterized using 1H NMR, 13C NMR, 2D 1H ROESY NMR and ESI-MS and the interlocked structure of 108b was also confirmed via single crystal X-ray diffraction (Fig. 19). These experiments also showed that the chloride:catenane complexes are stabilized through hydrogen bonding interactions between chloride and the amides, π–π donor–acceptor interactions between the pyridinium surface and the hydroquinone rings and hydrogen bonding between the pyridinium methyl protons and the polyether oxygens of the neutral macrocycle. In order to determine anion binding affinities, the templating chloride anion in 108a and 108b was exchanged for the non-coordinating PF6− anion. 1H NMR titrations in 1:1 CDCl3:CD3OD revealed that 1:1 anion:receptor complexes were formed in the case of Cl−, Br−, H2PO4− and OAc− (as TBA+ salts), with the strongest interactions seen for Cl−, presumably due to the complementary size and geometry of chloride and the catenane binding cavity.
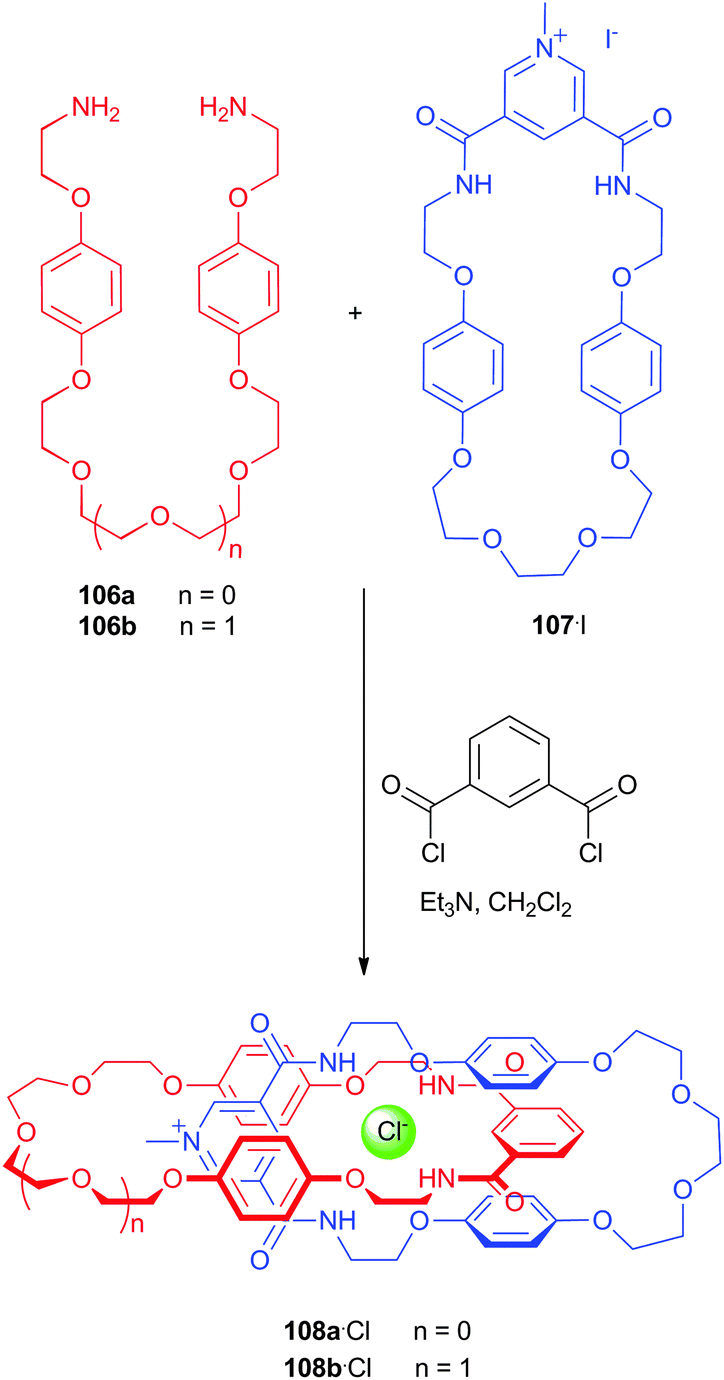 | ||
| Scheme 13 Chloride templated synthesis of [2]catenanes 108a and 108b by Beer and co-workers. | ||
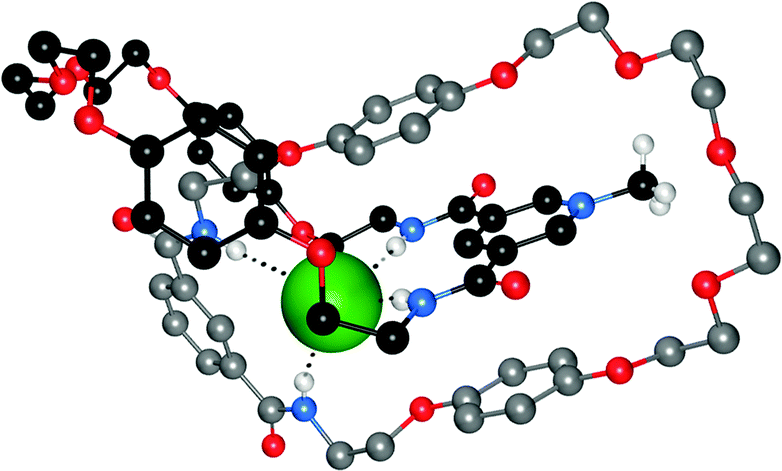 | ||
| Fig. 19 Crystal structure of 108a·Cl. The chloride anion is shown in spacefill with the macrocycles in ball-and-stick (one macrocycle is shown in a lighter shade of grey for clarity); non-coordinating hydrogen atoms, disorder and solvent molecules are omitted for clarity; hydrogen bonds with the chloride anion are represented by dashed lines. | ||
Employing a similar chloride templated synthetic strategy, Beer and colleagues have also reported the synthesis and properties of redox-active, ferrocene-containing [2]catenane 109 and the self-assembled monolayer of analogous surface-confined catenane 110.661H NMR, HRMS, ROESY NMR and X-ray crystallography were used to characterise the [2]catenanes and show that the chloride anion is bound inside the cavity via hydrogen bonding and that the structure is further stabilised through π–π interactions and hydrogen bonding between the macrocycles, in a similar fashion to the previously reported catenanes 108a and 108b. The anion binding properties of the PF6− salt of catenane 109 were determined using 1H NMR titrations in 1:1 CDCl3:CD3OD and via cyclic and square wave voltammetry in 0.1 M TBAPF6 1:1 CH2Cl2:CH3CN. The NMR results revealed that chloride was bound more strongly than oxo-anions OBz−, H2PO4− and HSO4−, while the voltammetry experiments showed only measurable cathodic shifts of the Fc/Fc+ redox couple for Cl− and H2PO4−, with chloride bound more strongly. The surface-confined catenane 110 was prepared from a pseudo-rotaxane containing a bis-thiol functionalised thread to allow absorption onto a gold electrode. Unfortunately, the bound chloride anion of this interesting surface-confined catenane could not be exchanged for PF6− and anion binding properties could therefore not be determined.
Beer and co-workers have also used chloride templated synthesis to prepare heteroditopic [2]catenanes 111 and 112.67 These catenanes contain a calix[4]diquinone unit able to bind cations, as well as an anion binding site used for templation. The chloride complexed catenanes obtained during synthesis were characterised by 1H NMR, COSY, HSQC and ESI-MS spectroscopy and X-ray diffraction and showed that both catenanes adopt the same conformation, indicating that the benzyl group does not cause rotation of the pyridinium in the calix[4]diquinone cavity and does not affect the anion binding site. After exchanging Cl− with PF6−, 1:1 cation complexation was observed for Na+, K+, NH4+ and Ba2+ using 1H NMR and UV/Vis titration in 4:1 dichloromethane:acetonitrile. It was observed that the addition of Ba(ClO4)2 leads to desymmetrisation of the catenane structure and the authors suggest that the electrostatic repulsion between Ba2+ and the positively charge pyridinium group causes an intra-ring co-conformational rotation between the two macrocycles (Fig. 20). The original symmetric catenane could subsequently be re-obtained via the addition of TBA2SO4 through decomplexation of Ba2+ and precipitation of BaSO4.
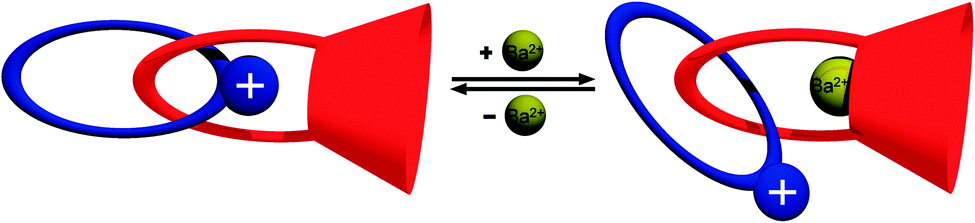 | ||
| Fig. 20 Molecular rotation in [2]catenane 111 driven by cation–cation repulsion proposed by Beer and co-workers. | ||
Ballester and co-workers have reported an exclusive self-sorting system that involves the formation of a heterodimeric assembly from tolyl calix[4]arene 113 and benzyl calix[4]pyrrole 114 in the presence of a neutral guest (trimethylamine-N-oxide) in CH2Cl2.68 This capsule is held together via hydrogen bonding between the urea functionalities of both macrocycles and the interactions with both the encapsulated guest (via calix[4]pyrrole 114) and the co-encapsulated solvent molecule (via calix[4]arene 113). Based on this information, the same group has subsequently reported the synthesis and properties of [2]catenane 117 consisting of a tetraurea calix[4]pyrrole and a tetraurea calix[4] arene.69 Bis-loop calix[4]arene 116 was synthesized according to known procedures and was mixed with calix[4]pyrrole 115 and trimethylamine-N-oxide to obtain the heterodimeric pseudo-rotaxane 115·116, according to their previous heterodimer experiments. Subsequent ring-closure metathesis and hydrogenation yielded [2]catenane 117 as a racemic mixture that was separated by HPLC chromatography with a chiral stationary phase. Using 1H NMR and GOESY NMR experiments, the authors were able to show that 117 can encapsulate N-oxides 118 and 119 along with a CHCl3 molecule to fill the void, while N-oxide 120 was large enough to be encapsulated without the additional CHCl3. Furthermore, 1H NMR experiments in CDCl3 revealed that 117 is able to accommodate ion pairs tetramethylammonium chloride, tetramethyl phosphonium chloride and tetraethyl phosphonium chloride, whereby the chloride anion is bound to the calix[4]pyrrole through hydrogen bonding and the cation sits in the calix[4]arene unit stabilised by cation⋯π and CH⋯π interactions (further confirmed through 31P NMR and 1H DOSY NMR for tetramethyl phosphonium chloride).
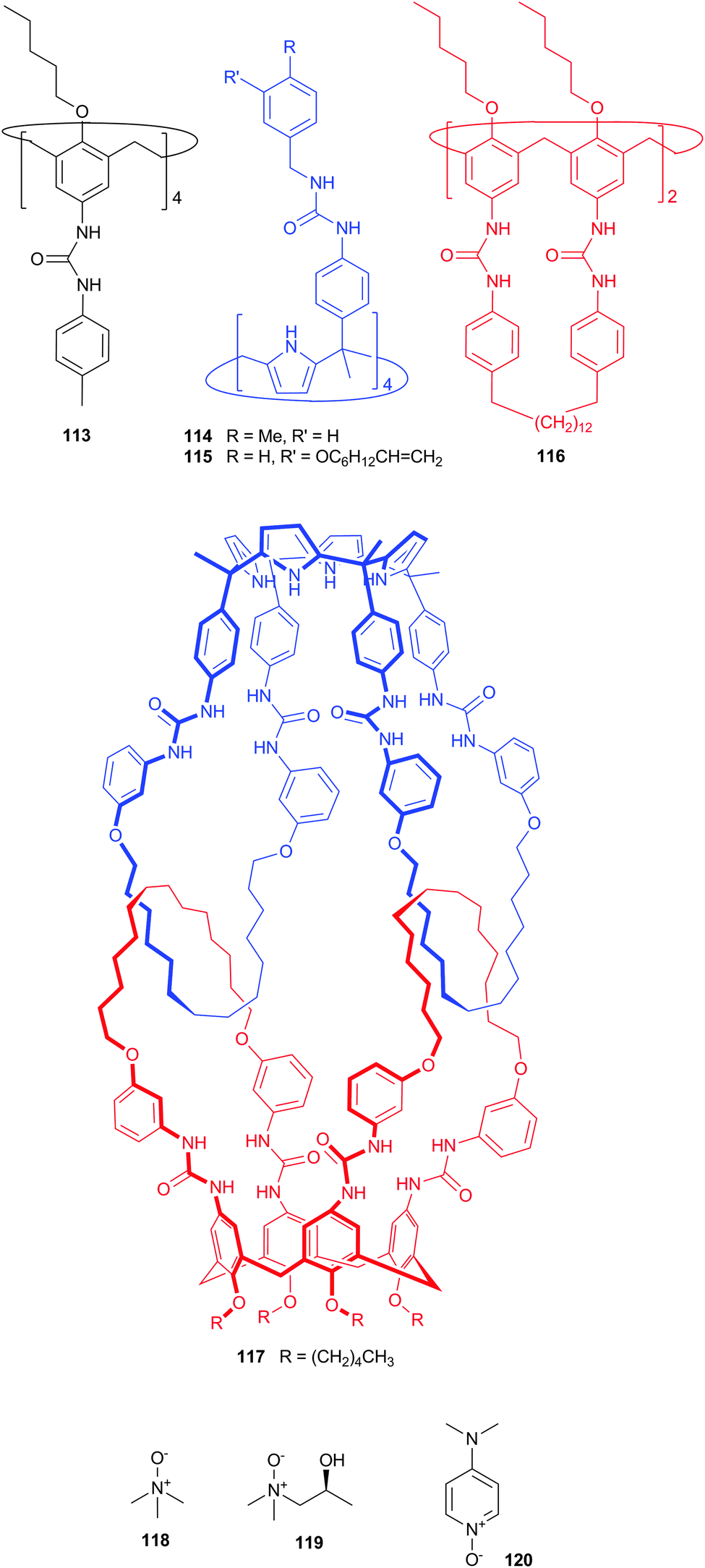
Ballester and co-workers have also described the anion binding properties of a pseudo-rotaxane consisting of bis(calix[4]pyrrole) 121 and bis(amidepyridyl-N-oxide) 122.70 This assembly is held together by hydrogen bonds between the pyrroles and the oxygen atom of the N-oxide functionality and forms a binding site containing six convergent hydrogen bond donors for anion complexation. 1H NMR experiments with TBA+OCN−, TBA+N3−, TBA+SCN− and TBA+NO3− in CDCl3 revealed that the anion is bound inside the cavity via hydrogen bonding, while the TBA+ counterion was bound in the shallow external cavity defined by calix[4]pyrrole. This ion pair complexation forming a 4-component pseudo-rotaxane was further confirmed using ROESY and DOSY NMR, ITC and X-ray crystallography (Fig. 21).
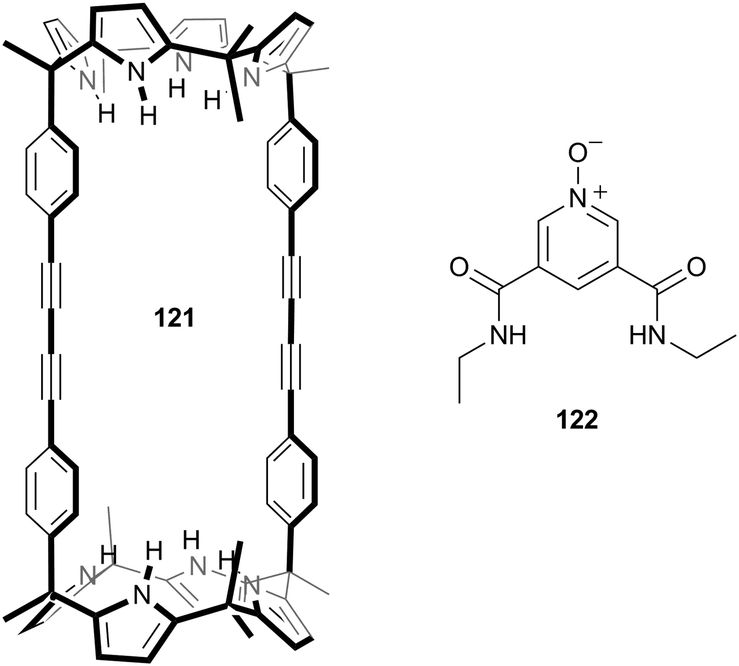
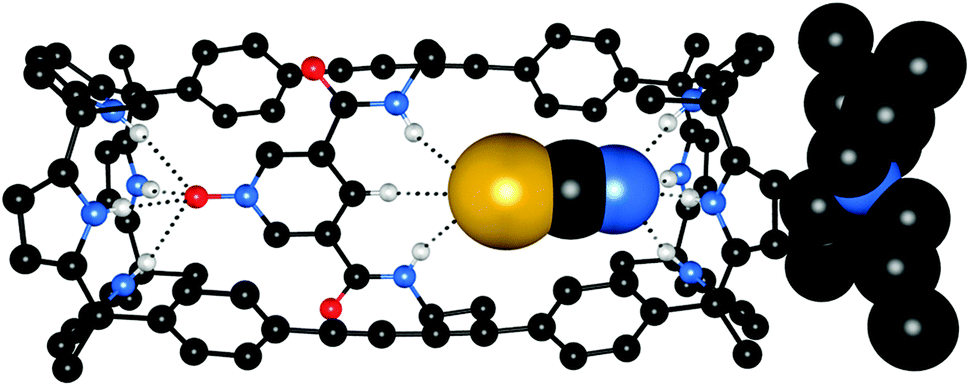 | ||
| Fig. 21 Crystal structure of the 4-component pseudo-rotaxane formed of 121, 122 and TBA+SCN−; the ion pair is shown in spacefill with the receptor assembly in ball-and-stick; non-coordinating hydrogen atoms are omitted for clarity and hydrogen bonds are shown as dashed lines. | ||
Leigh and co-workers have reported the self-assembly of five bis-aldehydes, five bis-amines, five metal cations and one chloride anion to form a 160-atom covalent pentafoil knot.71 The authors first described that the reaction of bis-aldehyde 123 and a series of amines (124a–j) in the presence of FeCl2 in DMSO at 60 °C forms a symmetric and cyclic pentafoil knot, as confirmed by 1H NMR and ESI-MS spectroscopy. It was found that anilines (124j) and amines with two substituents on the amine-bearing carbon atom (124h,i) could not form pentafoil knots with 123 and FeCl2, but all other amines tested did. Interestingly, CD spectra showed that the use of chiral amines (S)-124g or (R)-124g led to the formation of the cyclic pentafoil helicate with complete diastereoselectivity, with the handedness of the cyclic helicate determined by the enantiomer used. The authors then tried to obtain a similar, fully covalent pentafoil knot using various diamines (125a–c and 126), but only diamine 126 led to the formation of the desired cyclic pentafoil helicate. This was attributed to the preference for antiperiplanar arrangement of carbon atoms, preventing a full carbon chain (e.g.125a–c) folding back to form a loop, whereas heteroatoms are known to induce loops (e.g.126). The structure of the pentafoil knot formed by 123, 126 and FeCl2 was further characterised by 1H NMR, ESI-MS and X-ray crystallography, showing that a chloride anion is bound in the centre of the knot by ten CH⋯Cl− hydrogen bonds (Fig. 22). An empty cavity analogue of the pentafoil knot could be generated by the addition of AgPF6 in MeCN and the subsequent addition of TBACl, TBABr and TBAI showed that only the chloride anion can be bound inside the cavity and hence probably templates the formation of the pentafoil knot.
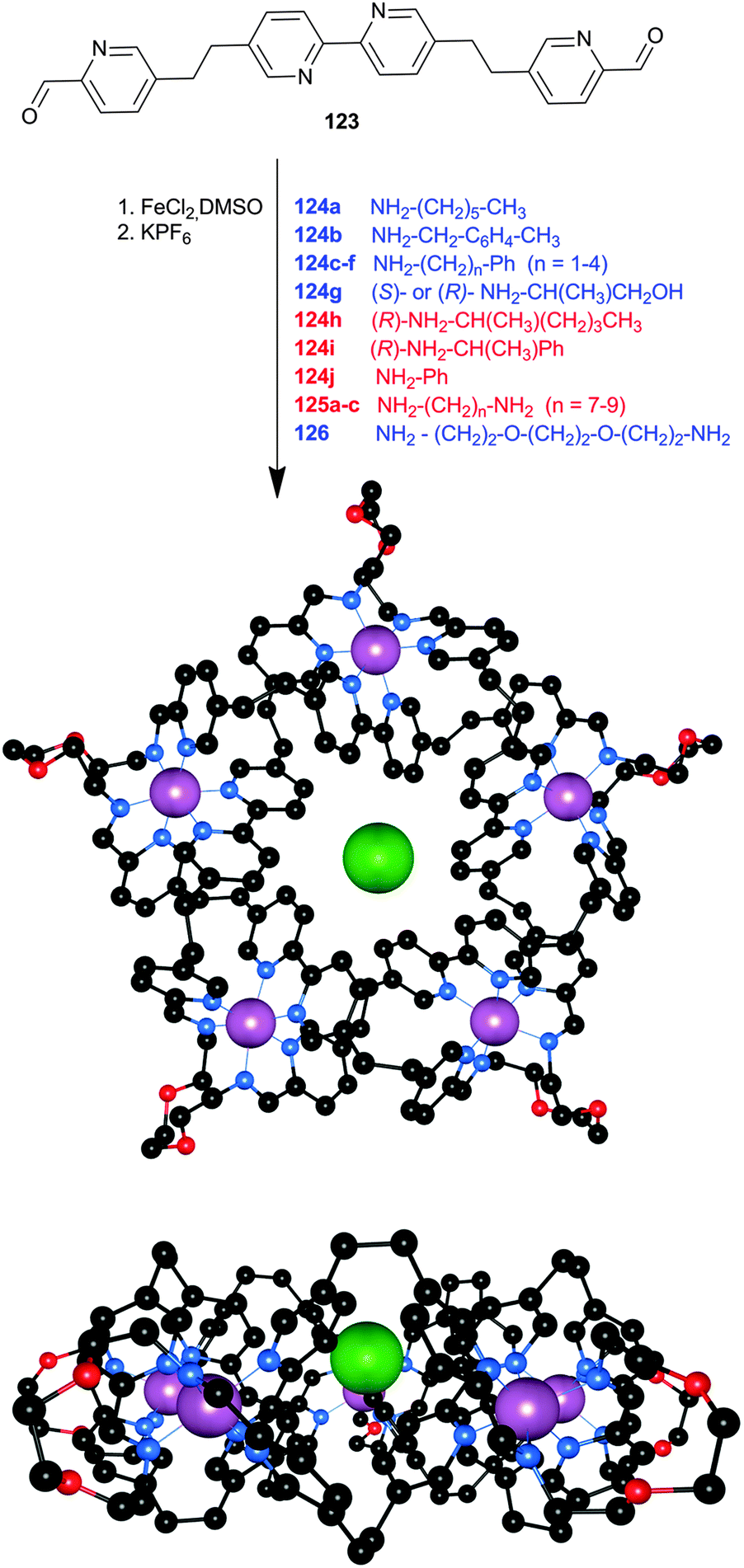 | ||
| Fig. 22 (top) Self-assembly of a pentafoil knot from aldehyde 123, FeCl2 and amines 124–126. The amines capable of forming a pentafoil knot are shown in blue, with the other amines in red. (below) Two views of the crystal structure of the knot formed between 123 and 126; hydrogen atoms, counterions and solvent molecules are omitted for clarity; chloride and iron(II) ions are shown in spacefill with the rest of the knot shown in ball-and-stick. | ||
Yashima and co-workers have studied the chiral amplification properties of positively charged amidine oligomers during the formation of double stranded helices with achiral anionic carboxylate oligomers.72 A series of chiral amidine strands were synthesised composed of (R)- or (S)-1-phenylethyl and/or achiral isopropyl amidine units, having either an all-chiral (127), edge-chiral (128) or centre-chiral (129) composition, and their duplex formation with achiral carboxylate strands (130) were investigated (Scheme 14). Using 1H NMR, UV/Vis, cold spray ionisation (CSI) MS and CD spectroscopy, it was shown that duplexes are formed between the all-chiral amidine strands 127 and the complementary carboxylic acid strands 130, whereby the handedness of the helix is determined by the enantiomer of the amidine used and the length of the oligomer, with opposite handedness seen between the 2-mers (n = 1) and the 4-/5-mers (n = 3,4). The authors also showed by CD spectroscopy that helices formed between the edge-chiral (128) or centre-chiral (129) amidine strands and the achiral carboxylic acid strands had a preferred handedness, suggesting that the single chiral monomer in the strand acts as a helical sense director and that the chirality is thus amplified through the helix. It was found that these duplexes are dynamic and that their chiroptical properties depend on solvent, temperature and oligomer length and sequence, with a larger chiral amplification effect for centre-chiral than for edge-chiral amidine strands, and the effect decreased with increasing length of the strands, as supported by 1H NMR and CD spectra and molecular dynamics (MD) simulations.
 | ||
| Scheme 14 Overview of the various amidine and carboxylic acid strands by Yashima and co-workers and their double helix formation. | ||
Anion sensors and chemodosimeters
Many examples have already been given for the optical detection of anions (colorimetric or fluorescent), but few are highly selective for a specific anion. Bai and co-workers developed a colorimetric and ratiometric fluorescent fluoride sensor based on the desilylation reaction of organosilicon compound 131.73 The addition of F− to a THF solution of 131 leads to a colour change from colourless to yellow-green and also the emission changes from modest blue to bright yellow-green (Fig. 23). UV/Vis titrations show that this colour change is due to a decrease of the 313 nm peak and the increase of the 363 nm and 410 nm peaks of the absorption spectra upon the addition of fluoride, while fluorescence studies show a decrease of the 403 nm peak and the emergence of a new peak at 520 nm, with the ratio of fluorescence intensity F520nm/F403nm exhibiting a linear change upon the addition of fluoride in the range 1–10 μM (ratiometric sensor). Furthermore, similar experiments with other anions, such as Cl−, Br−, OAc−, NO3−, H2PO4−, HSO4− and ClO4− show that F− is the only anion that can induce these optical changes. The high selectivity and sensitivity for fluoride is attributed to the selective deprotection reaction by fluoride (forming 132), followed by the formation of a hydrogen bond between F− and the newly formed hydroxy group, resulting in a charge transfer interaction between F− and the electron deficient sensor and altered emission properties. This mechanism of fluoride sensing was confirmed by 1H NMR experiments, and UV/Vis and fluorescence studies on purified deprotected product 132. | ||
| Fig. 23 Proposed mechanism of F− sensing by 131, with naked eye colour changes and under UV lamp fluorescent colour changes observed for 131 upon the addition of one mole equivalent of F− (200 μM in THF). Reproduced with permission from Chem. Commun. 2011, 47, 3957–3959. © Royal Society of Chemistry. | ||
Duke and Gunnlaugsson have reported the synthesis of a highly selective dual colorimetric and fluorescent ICT-based anion sensor for fluoride (Scheme 15).74 Compound 133 is based on a 1,8-naphthalamide skeleton in which the 3-position is functionalised with a 4-trifluoromethylphenylurea group. This system functions as a selective sensor for fluoride via a deprotonation mechanism at high fluoride concentration resulting in a dramatic enhancement in the absorption band at 327 nm and large changes within the ICT transition in the fluorescence emission spectrum of the compound in DMSO.
 | ||
| Scheme 15 Synthesis of anion sensor 133. | ||
Singh and Kaur reported receptor 134 that can remove cyanide from water and from human serum through the formation of 135.75 Upon the addition of various anions (CN−, F−, OAc−, H2PO4−, Cl−, Br−, CO32− and HCO3−) to a 1:1 water:ethanol solution of 134, a colour change from light yellow to orange/red was only observed upon the addition of NaCN. Job plot and UV/Vis titrations show that cyanide interacts with receptor 134 in a 1:1 stoichiometry with an association constant of Ka = 106 M−1. Compound 135 could be isolated from the mixture of cyanide and 134 and the authors propose, based on UV/Vis and 1H NMR studies, that the reaction shown in Scheme 16 is responsible for this observation. This reaction mechanism requires the substitution of a phenolic OH by CN and this was confirmed as no response was seen when cyanide was added to the analogous compound 136 lacking the phenolic OH group. To show the generality of the cyanide sensing abilities of 134, the UV/Vis studies were repeated in dry THF, dry MeCN, dry CHCl3 and in 1:1 water:ethanol mixtures at various pHs (pH 2–7) and similar changes in colour and absorption spectra were observed upon the addition of CN−. It was also shown that the presence of other anions or Fe3+ did not change the sensing ability towards CN− in competitive binding studies. Furthermore, the authors additionally showed that 134 is able to sense CN− in proteinaceous (and non-proteinaceous) human blood serum and that it is able to remove all CN− anions from these media. The authors believe that this property, along with the presumed low toxicity of 134 (due to the similarity to the known drug trimethoprim) and ease of synthesis, makes 134 a potential commercial antidote for cyanide.
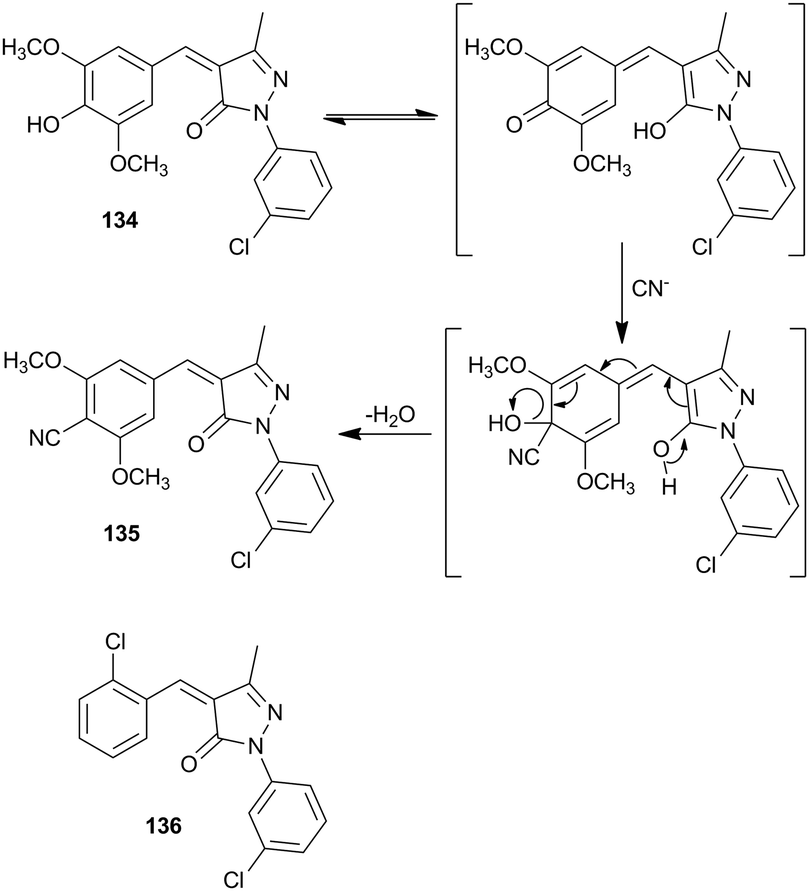 | ||
| Scheme 16 Structure of compounds 134–136 and the proposed mechanism for the conversion of 134 into 135 by the addition of cyanide. | ||
Lee, Kim and co-workers have reported coumarin derived receptor 137 that functions as a selective sensor for KCN.76 UV/Vis and fluorescence measurements with potassium salts of F−, Cl−, Br−, I−, OAc−, HSO4−, HPO42−, HCO3−, NO3−, ClO4−, CN− and SCN− in 5:95 H2O:MeCN showed that only the cyanide anion is able to induce a colour change from dark blue to yellow (naked-eye colorimetric sensor) and an increase in fluorescence intensity (turn-on fluorogenic sensor). 1H NMR and FAB-MS experiments showed that the addition of CN− to 137 results in the formation of 138. Based on DFT and time-dependent DFT calculations, the authors suggest that this reaction of 137 with CN− to produce 138 breaks the conjugation between the coumarin and the indole moiety, thereby disrupting the ICT in 137 and causing spectral changes in both absorbance and emission.
Calero et al. described the development of a quencher displacement assay (QDA) for anion sensing.77 The conventional indicator displacement assay (IDA) was modified by attaching a fluorophore (139) and a separate metal cation receptor (140) to silica nanoparticles, generating a terpyridine–sulforhodamine-functionalised nanoparticle hybrid system, in order to achieve facile regeneration of the sensing material. The system works by first loading the terpyridine units with a metal cation, which quenches the fluorescence from the nearby sulforhodamine units. The anion to be sensed is then added and competes with the terpyridine units to bind the cation, and once the cations are displaced from the terpyridine units, the fluorescence signal is regenerated (Scheme 17). The authors believe that the possibility of utilising different coordination units and metal anion quenchers can make QDA's attractive for developing new chemosensors for anions.
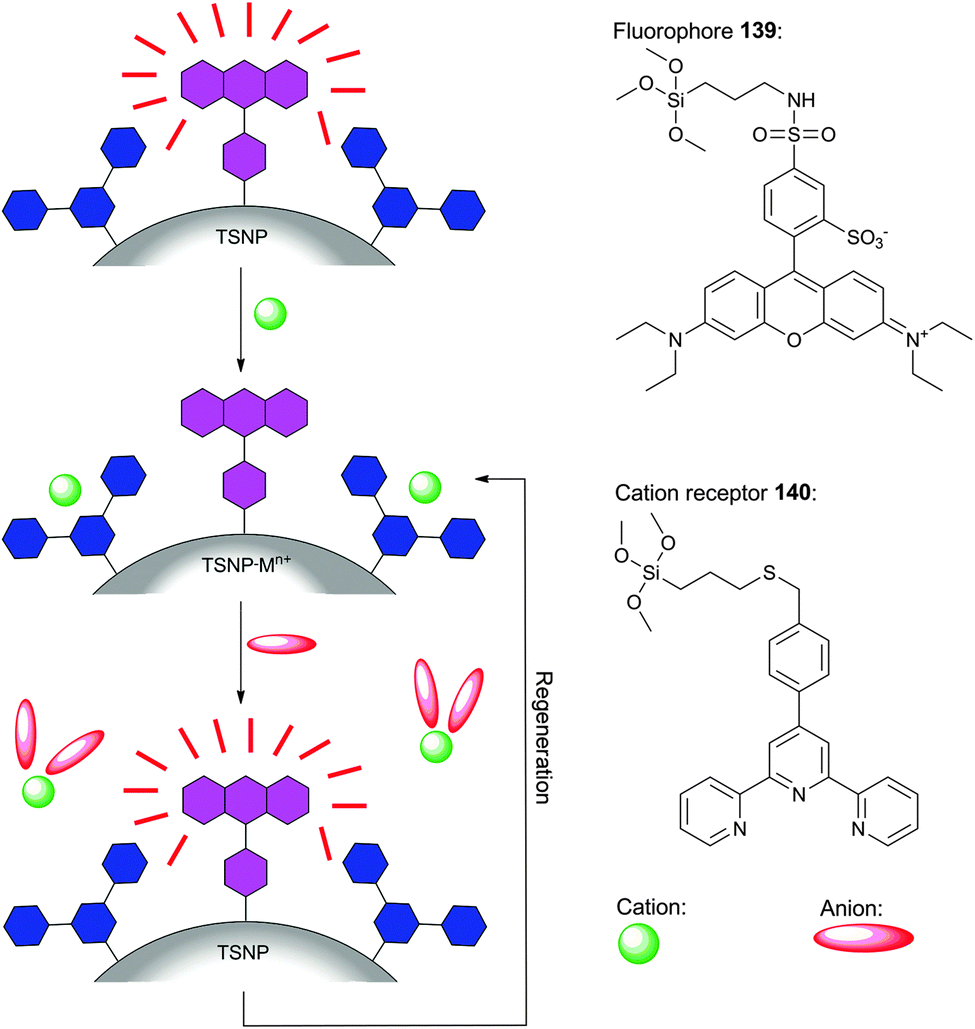 | ||
| Scheme 17 Design concept of the anion quencher displacement assay (QDA) involving terpyridine–sulforhodamine functionalised nanoparticle (TSNP) and a metal ion quencher, along with the structure of fluorescent dye 139 (magenta) and cation receptor 140 (blue). | ||
Bull, Fossey and colleagues have developed a pyridine based sensor for alkyl halides.78 Previous reports had shown that the conversion of 141 into 142 leads to a large fluorescence enhancement due to cation⋯π interactions in 142.79 Fluorescence titrations with MeI in CH2Cl2 showed that this property can be used to detect MeI with a detection limit of 1 nM. Additionally, the authors tested the sensing ability towards other alkyl halides by reacting them with 141 in CH2Cl2 under reflux for 3 hours and then measuring the fluorescence intensity (Table 3). A larger fluorescence enhancement was observed when triflate based alkylating agents were used rather than iodide based agents, presumably due to the heavy atom quenching effect of I−. Furthermore, 141 also acts as a sensor for bromide based alkylating agents, but they required a longer reaction time (hours) compared to triflate/iodide based agents (seconds) before they can be accurately detected.
| R | X | F/F0 enhancement |
|---|---|---|
| Me | I | 49 |
| Me | OTf | 114 |
| Et | I | 29 |
| Et | Br | 89 |
| Bn | Br | 89 |
| p-OMe Bn | Br | 81 |
| p-NO2 Bn | Br | 36 |
| CH2CH2Ph | Br | 99 |
| CH2CH2CH2Ph | Br | 46 |
| Propargyl | Br | 61 |
| (CH2)2–O–(CH2)2–O–CH3 | Br | 45 |
Catalysis
The link between anion binding and certain catalytic reactions has previously been suggested and reviewed, especially for thiourea based organocatalysts.80 Jacobsen and co-workers have reported the first instance of an enantioselective thiourea anion binding catalyst with fluoride.81 Pyridine derivatives have long been used to catalyse acyl transfer reactions and stabilisation of the acylpyridinium ion intermediate, via hydrogen bonding of the counterion to a specific hydrogen bond donor, could lead to increased reaction rates. Therefore, the authors investigated the enantioselective acylation of silyl ketene 144 with acyl fluorides using thiourea 143 and 4-pyrrolidinopyridine (PPY) catalysts and found that the reaction proceeds with a yield of 80% and an enantiomeric excess (ee) of 92% (Scheme 18). To test the substrate scope, a series of acylation reactions were performed with an assortment of benzoyl fluorides substituted with electron donating and electron withdrawing groups. Meta- and para substitutions were generally tolerated well, yet ortho substitutions resulted in inactivity. This observation, along with the fact that the reaction does not proceed in the absence of PPY or the thiourea catalyst, supports the concept of an acylpyridinium intermediate. Additionally, activation of 144 by fluoride is necessary for acylation to proceed, as it is observed that the use of benzoyl chloride does not generate the required product. This was attributed to the impressive hydrogen bond acceptor ability and silaphilicity of fluoride, allowing the enantioselective acylation process to move forward efficiently and afford the useful α,α-disubstituted butyrolactones, while only requiring low catalyst loadings.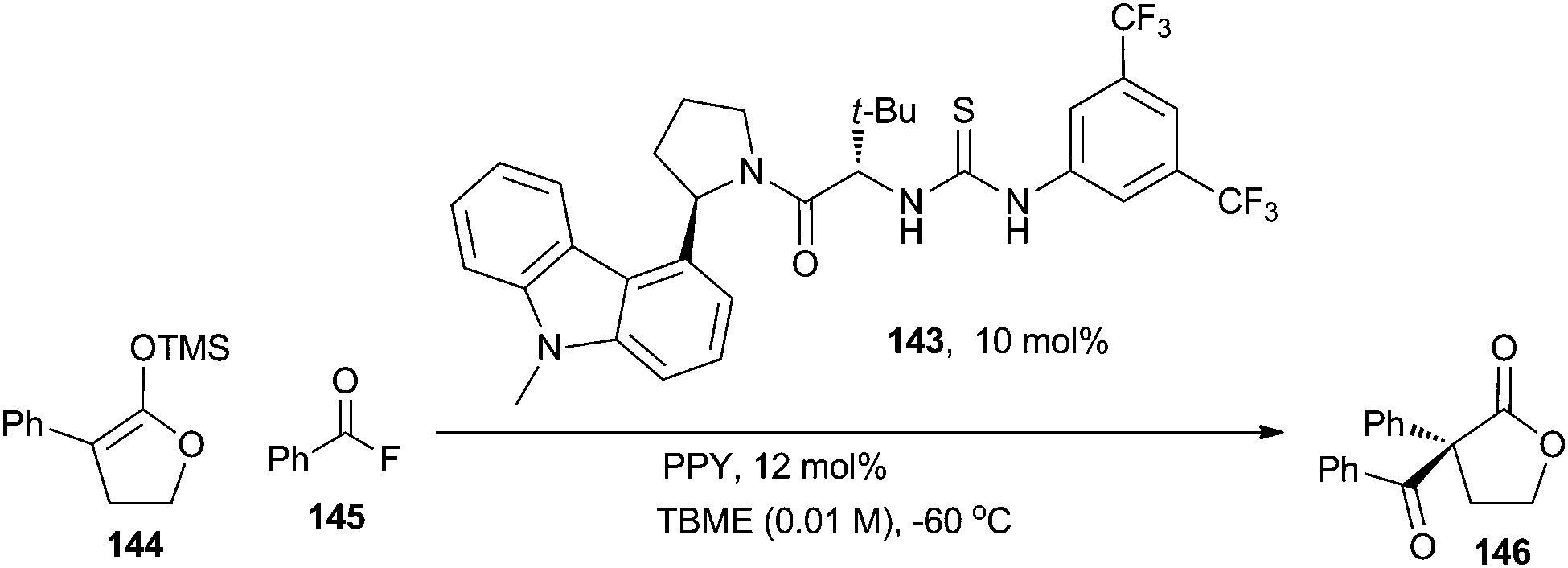 | ||
| Scheme 18 Conversion of 144 into 146 catalysed by thiourea 143 and PPY. | ||
Lin and Jacobsen have reported the use of arylpyrrolidino amido thioureas (147a–f) for the catalysis of enantioselective nucleophilic ring opening of episulfonium ions by indoles.82 Initial optimisation of the catalysis conditions showed that arylpyrrolidino amido thioureas such as 147a–f work best, that the episulfonium precursor should have a relatively non-nucleophilic leaving group and that sulfonic acids are required to co-catalyse the reaction rather than mineral acids (HCl). Furthermore, it was found that catalysts with a more extended aromatic substituent (147b–f) result in improved enantioselectivity. Based on this information and detailed kinetic analysis involving in situ infrared spectroscopy, structure–reactivity relationships and kinetic isotope effects, the authors propose a reaction mechanism as shown in Scheme 19, where the addition of indole is the rate- and enantioselectivity-determining step. The authors also propose that the transition state in this rate-determining step is stabilised by the thiourea catalyst through a number of interactions: (1) hydrogen bonding between the thiourea catalyst and the sulfonate anion – as confirmed by 1H NMR titration of 147d and dibenzylmethylsulfonium triflate in toluene-d8; (2) hydrogen bonding between the indole NH and the amide oxygen of the catalyst – as suggested by the fact that N-methylindole results in poor enantioselectivity and by the linear relationship between reaction rate and indole NH acidity; (3) cation⋯π interactions between the arene in the catalyst and the episulfonium cation – as suggested by 1H NMR experiments on the catalyst with a model sulfonium ion (dibenzylmethylsulfonium triflate) and the influence of the identity of the arene on the enantioselectivity.
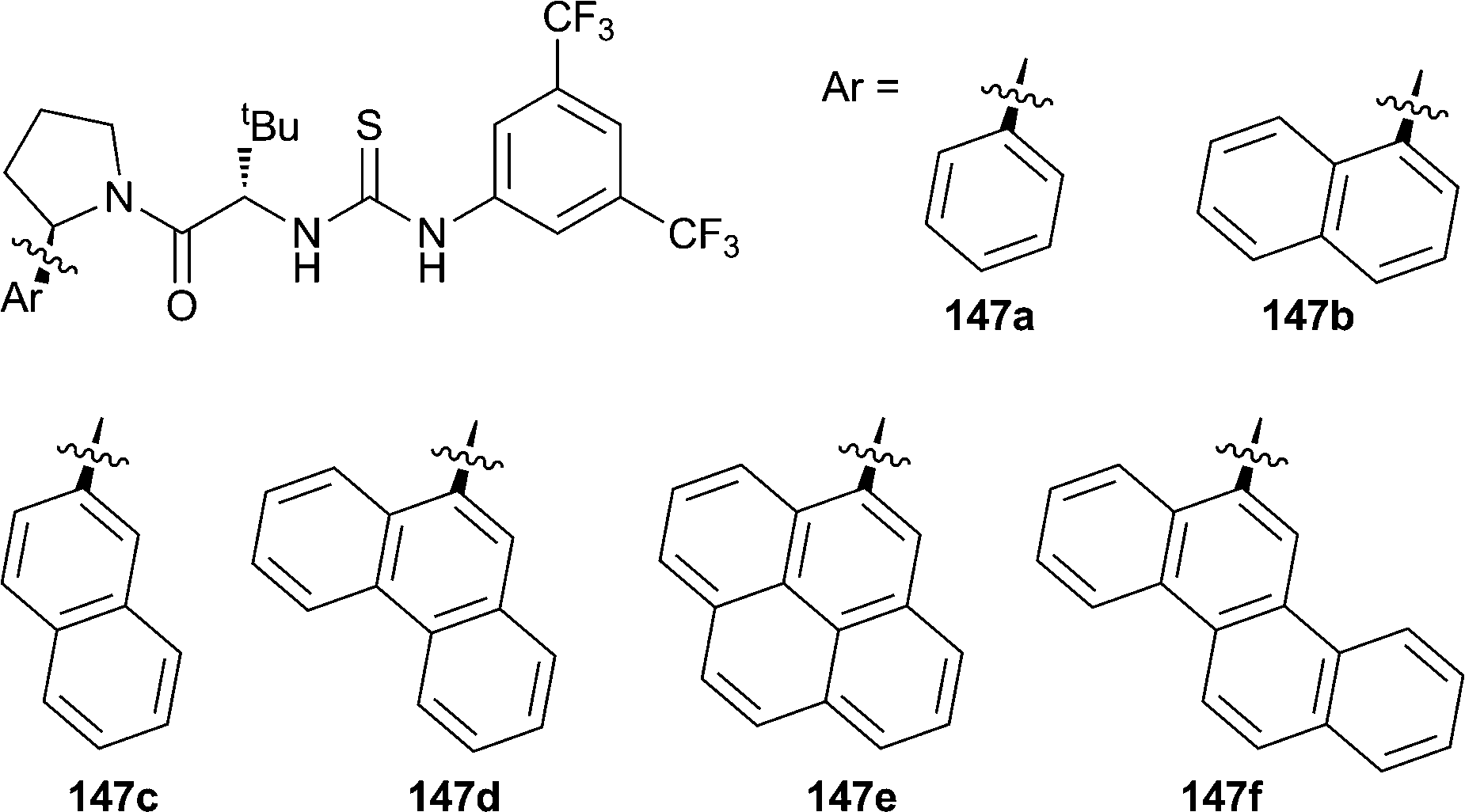
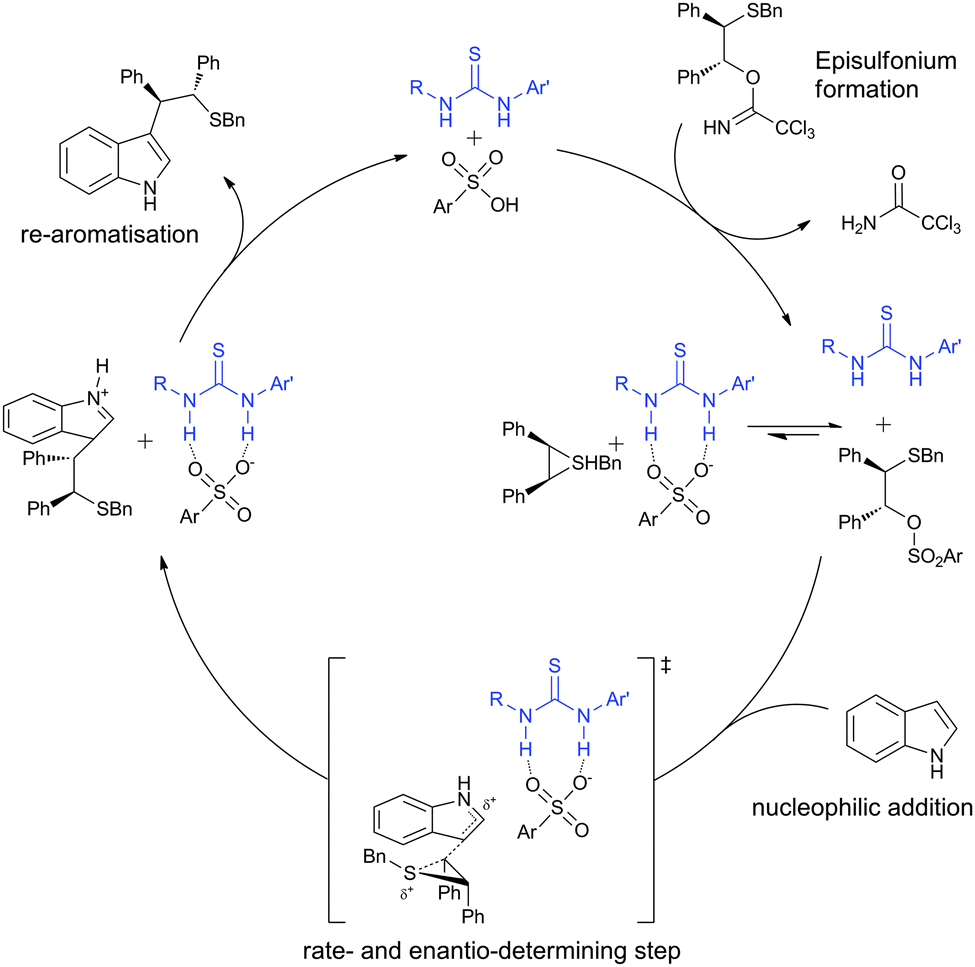 | ||
| Scheme 19 Proposed catalytic cycle for thiourea catalysed ring-opening of episulfonium ions with indole; the catalyst (147a–f) is shown in blue. | ||
Jacobsen and co-workers have also shown that a combination of an achiral thiourea (148) and a chiral primary amine (149 or a chiral aminothiourea) can be used to catalyse the intramolecular enantioselective [5+2] dipolar cycloaddition of 150 to 151 (Scheme 20).83 The reaction proceeds with 58% yield and −85% ee. They hypothesised that the thiourea can encourage ionisation to the pyrylium ion via stabilisation of the counter anion prior to the cyclisation step, which was supported by the fact that 149 is virtually inactive in the absence of thiourea 148 (7% yield). Furthermore, the primary amine in 149 is also necessary for the catalytic reaction, as a tertiary amine analogue proved to be unreactive (in both the presence and absence of thiourea 148). The authors propose a catalytic mechanism based on the initial condensation between the primary amine of 149 and the ketone of the substrate, leading to a dienamine after tautomerisation, followed by hydrogen bond donor (148) mediated abstraction of p-MeSBzO to generate the pyrylium ion that can undergo an intramolecular cycloaddition.
 | ||
| Scheme 20 Oxidopyrylium cycloaddition catalysed by 148 and 149. | ||
Ooi and co-workers have reported the development of chiral 1,2,3-triazolium salts (such as 152·Br) that function as cationic organocatalysts with anion recognition abilities.84 The increased CH-acidity of the triazolium cation versus the simple 1,4-disubstituted 1,2,3-triazole unit means an increased anion binding capacity. X-ray crystallography of 152·Cl showed that the combination of electrostatic interactions, CH⋯anion hydrogen bonding and a suitably positioned amide NH⋯anion hydrogen bond forms a structured and tightly bound ion pair. These hydrogen bonding interactions were further confirmed by comparison of the 1H NMR spectra of 152 paired with various counter anions in CD2Cl2 at 298 K. The synthesised 1,2,3-triazolium salts were investigated for their potential to catalyse the enantioselective alkylation of 3-substituted oxindoles (Scheme 21). The combination of an ortho-biphenyl substituent and a 3,5-dichlorophenyl group into the amide moiety gave the most effective catalyst (153), giving a reaction yield of 99% with 97% ee. It was also established that this alkylation was best performed in ethyl acetate.
 | ||
| Scheme 21 Structure of catalysts 152·Br and 153·Br along with the alkylation of oxindole reaction that they catalyse. | ||
Transmembrane anion transport
The control of anion concentrations is crucial to cell survival and hence the development of small molecules that are capable of transporting anions across cellular membranes has recently received a great degree of attention, mainly due to their potential therapeutic advantage.85–89 Prodiginines are small natural products that represent the most widely studied transmembrane chloride transporters due to their alleged anticancer activity.90–92 The anion binding and transport properties of obatoclax (154a, a synthetic prodiginin that is currently in clinical trials as an anticancer drug) have been investigated by Quesada and co-workers, with the aim of discovering the mechanism of its anticancer activity.93 Variations of the pyrrole substituents resulted in a series of compounds (154a,b,c–157) capable of transporting chloride and bicarbonate anions across synthetic POPC bilayers, via an antiport mechanism of transport. Compound 154b showed the highest transport ability for the chloride–bicarbonate antiport process (EC50,290s = 0.18 μM). However, modification of the number of hydrogen bond donors present in the receptors leads to receptors with reduced transport activity (155–157). The active receptors were shown to possess in vitro cytotoxic activity against the small-cell lung cancer cell line GLC4. This anticancer activity was further investigated by in vitro acridine orange staining experiments performed on the same cell line and the active receptors were shown to de-acidify the lysosomes of the cell, thereby signalling an orange to green fluorescence change. Hoechst staining revealed apoptosis to be the mechanism of cell-death. As the in vitro anticancer activity of the compounds correlates well with their anion transport ability in liposomes, it is presumed that the change in intracellular pH induced by 154a–c and 155–156 is due to bicarbonate transport facilitated by these receptors.
Quesada and co-workers have also focused on the study of transmembrane anion transport by a series of tambjamines (158–163), which are compounds structurally related to prodigiosin with proven anticancer and antimicrobial properties.94 X-ray crystal structures of the HCl salts of 159 and 163 show a flat bicyclic core and hydrogen bond interactions between chloride and the tambjamine core. 1H NMR titrations of the hydroperchlorate salts with TBACl in DMSO-d6 suggest that these hydrogen-bonding interactions also persist in solution. Chloride ion selective electrode vesicle tests were used to determine the anion transport properties of the series and it was shown that novel receptor 163 is the most efficient transporter for chloride, promoting >90% chloride efflux after 5 minutes with a carrier loading of 0.2 mol% with respect to lipid. This receptor was also shown to promote chloride efflux at carrier loadings as low as 0.025 mol% with respect to lipid. On the other hand, tambjamine 159 was the worst transporter of the series, presumably due to its low lipophilicity. Variations of the ion selective electrode tests were able to support an anion exchange transport mechanism in synthetic vesicles. In vitro activity of the tambjamines was tested on small-cell lung cancer (GLC4) cells. Using acridine orange the cell nuclei and cytoplasm fluoresced green (high pH), while some granular orange fluorescence was observed in the cytoplasm – a result of acidified lysosomes. In cells treated with tambjamine 163 the orange emission disappeared; a possible result of bicarbonate flux into the lysosome. The biological results correlate well with the synthetic transport tests and indicate the tambjamine family to be potential targets for drug development.
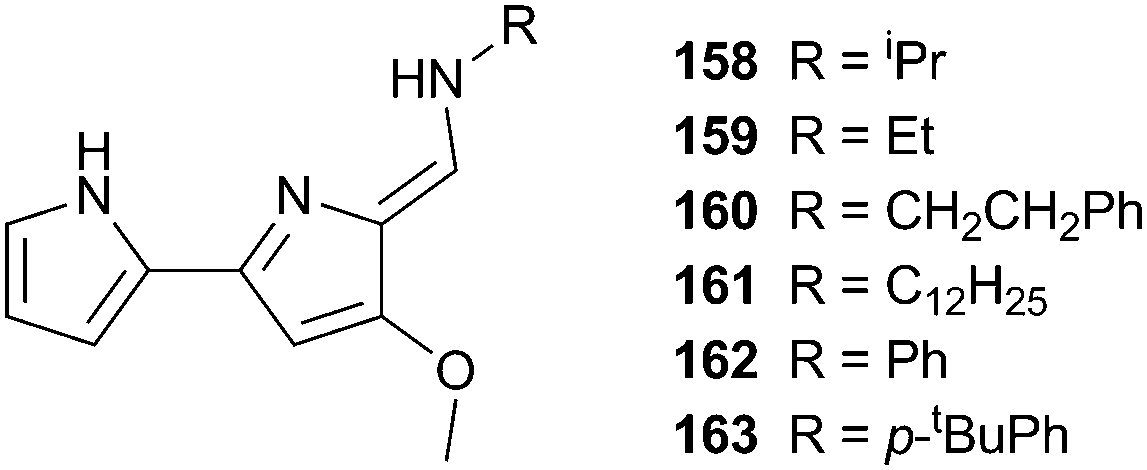
Gale and co-workers have shown that very simple compounds with urea and thiourea functionalities (164–169) can function as powerful chloride–bicarbonate antiporters at low concentrations without the need for pre-incorporation into phospholipid bilayers.951H NMR titrations with various TBA salts in DMSO-d6–0.5% water revealed that none of the six receptors interacts notably with nitrate but they do all bind chloride, the most noticeable being the interaction of indolyl urea 168 (Ka = 96 M−1). Vesicle based transport assays showed that the urea based receptors do not promote the release of chloride from POPC vesicles, in both chloride–nitrate and chloride–bicarbonate antiport assays. On the other hand, phenylthiourea 165 showed moderate chloride release, whilst 167 and 169 effectively release chloride from within the synthetic vesicles (EC50,270s = 0.109 mol% and 0.029 mol% respectively for the chloride–nitrate assay). Experiments with 30:70 cholesterol:POPC liposomes demonstrated that the receptors function as anion carriers, as opposed to forming channels, and 13C NMR experiments were used to directly confirm that the thiourea receptors are able to promote chloride–bicarbonate antiport. Additional experiments confirmed the remarkable transport activity of simple indolylthiourea 169, which is able to antiport chloride/nitrate down to ratios of 1:25000 transporter to lipid.

Gale and co-workers have also shown that simple bis-ureas of the form 170, although more flexible than their mono-urea counterparts, are able to transport anions more effectively across lipid bilayers via a mobile carrier, antiport mechanism.96 The basic modification of alkyl chain length of this type of receptor allows for tuneable anion transport activity as required. The increase in receptor flexibility with increasing alkyl chain length has no detrimental effect on transport activity; in fact the opposite is true with transport efficiency appearing to increase across the series until the optimum alkyl chain length is reached (n = 9). The receptor where n = 9 seems to strike the correct balance between enhanced membrane partitioning as a result of increased lipophilicity, and sufficient delivery capabilities through the aqueous media to the membrane, with supporting evidence being provided in the form of molecular dynamics simulations.
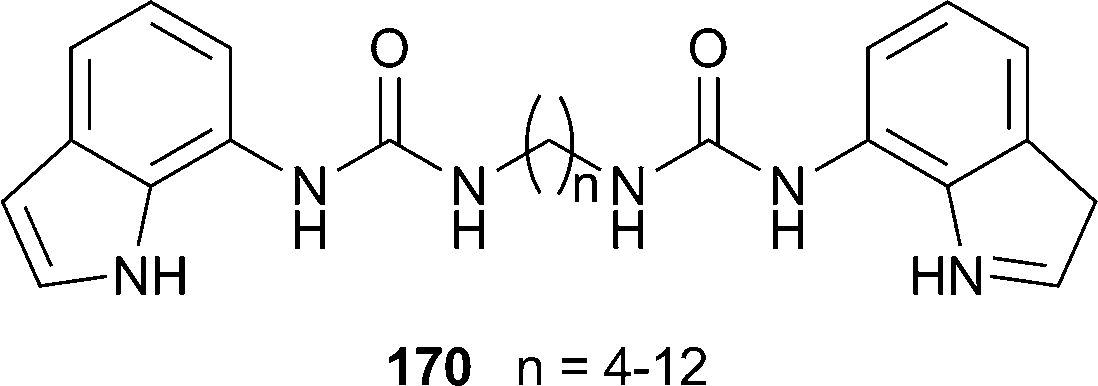
A. P. Davis and co-workers have continued their work on cholapods, which have previously been shown to be very powerful anion receptors and transporters.97 In 2011 they reported the development of smaller cholapod-like receptors based on a central trans-decalin system 171a–i, which have the advantage of smaller molecular weights and decreased lipophilicity and are therefore more “drug-like” molecules.981H NMR titrations showed a strong 1:1 interaction between the receptors and tetraethylammonium chloride, and extraction techniques were used to estimate binding affinities of >106 M−1, with the introduction of electron withdrawing fluorinated aromatic groups leading to further increased affinities. The receptors were also assessed for anion transport abilities via fluorescent lucigenin assays after pre-incorporation of 171a–i into POPC vesicles. The results demonstrated that the trans-decalin based receptors are effective chloride, and presumably nitrate, transporters with more electron deficient aromatic rings leading to enhanced transport ability – in accordance with the binding results. Furthermore, the introduction of a long octyl substituent (171g–i) results in an increased transport ability of the receptor. The most active of which, 171i, was effective at concentrations as low as 1:250000 receptor to lipid. This may be a consequence of positioning the decalin favourably within the membrane or promoting the movement of the decalin from one face of the membrane to the other during the transport processes.
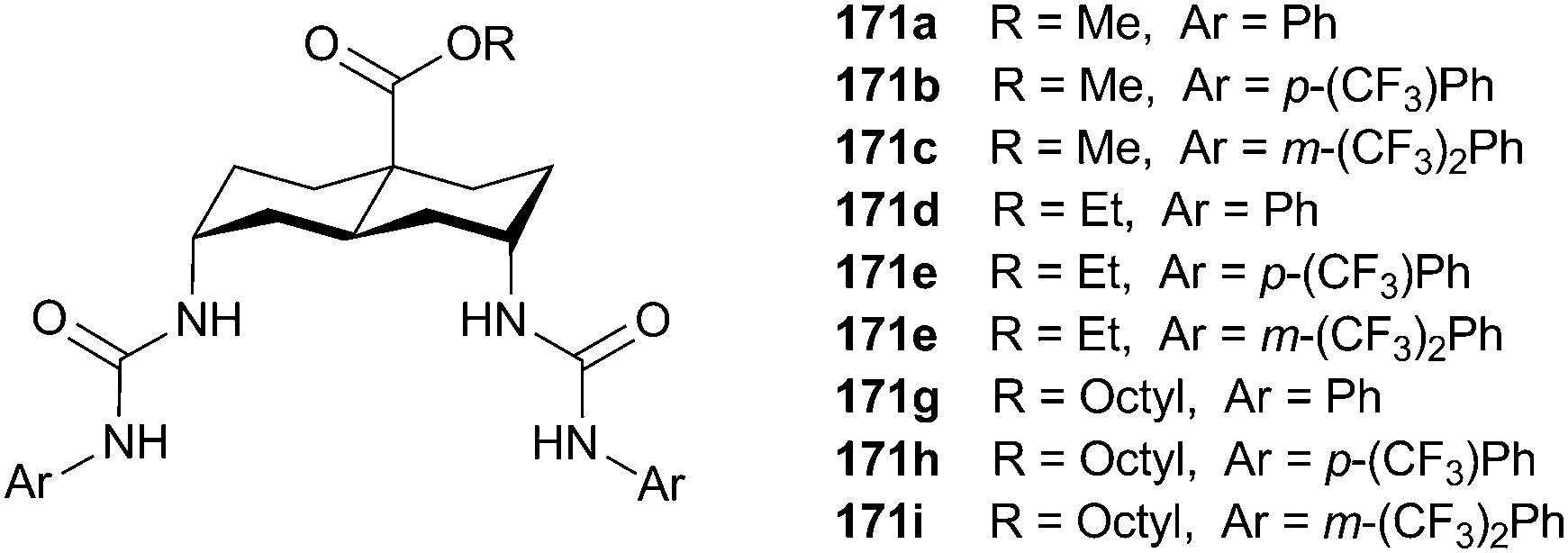
Jeong and colleagues have prepared a series anion receptors (such as 172–176) with differing side chains that possess cavities with convergent urea and hydroxyl hydrogen bond donor groups, mimicking the hydrogen bonding mode of StClC (serovarty-phimurium chloride channel).99 The binding mode of these receptors was confirmed by X-ray analysis, with the bound chloride anion shown to form four hydrogen bonds with the receptor. 1H NMR anion binding analysis in 1% H2O:CD3CN showed that receptors with chloride substituents on the phenyl rings have increased association constants, with the strongest binding observed for receptor 176 (Ka = 17000 M−1 ± 12). Receptor 176 was also shown to be the most effective transporter of the series by lucigenin fluorescence assays. This receptor has strong electron withdrawing substituents to encourage hydrogen bonding interactions with the anion and also possesses lipophilic side chains to allow for easy passage through the lipophilic POPC membrane. Mechanistic investigations of the anion transport process were also conducted and revealed that such receptors function via an antiport mechanism.

Gale and-coworkers have previously shown that the addition of fluorinated substituents on tripodal tris-ureas and tris-thioureas can greatly enhance the transmembrane anion transport ability in vesicles and in vitro cell studies.100 Building on these results, the same group has investigated a series of fluorinated indole, alkyl, and phenyl-substituted mono-ureas and mono-thioureas (e.g.177–184) for their ability to promote anion transport across synthetic phospholipid bilayers.101 Both chloride–nitrate and chloride–bicarbonate antiport processes were studied using standard POPC phospholipid vesicle model systems. The overall results indicate that fluorinated receptors exhibit greater transport activity than their unfluorinated counterparts – in accordance with increased clogP. Receptor 180 proved to be the most active receptor, facilitating chloride–nitrate antiport at receptor loadings as low as 1:20000 receptor to lipid, which corresponds to an EC50,270s value of just 0.016 mol%. The presence of a CF3 group increased anion affinity in all cases, and also increased lipophilicity sufficiently to enhance transport rates across the board, presumably by favouring partitioning of the receptor into the lipophilic phase. In vitro ionophoric activity of the active compounds was tested in A375 melanoma cells, employing the technique of acridine orange staining. After exposure to compound 180 all orange fluorescence disappeared and only green fluorescence was observed, indicating an increase in pH of previously acidic organelles by acidification of the cell cytoplasm. Subsequent studies using Hoechst 33342 staining of the same cell line showed that this reduction in cellular pH can damage cells and is able to encourage apoptosis of cancer cells.
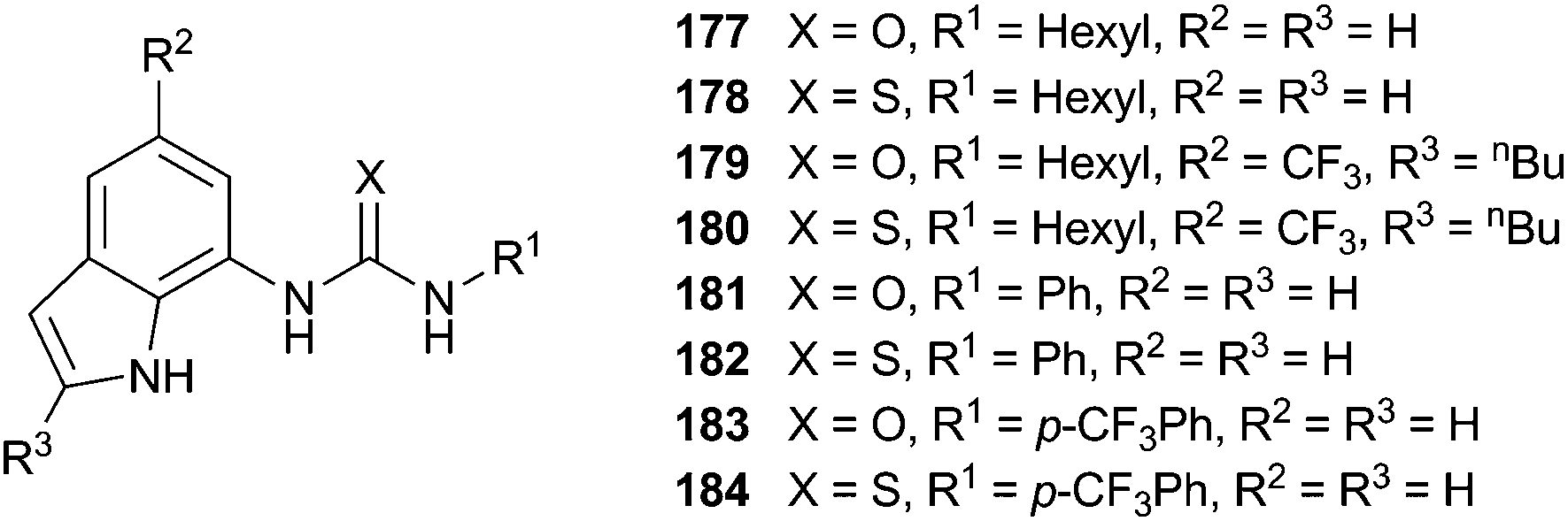
The majority of small-molecule anion transporters employ urea or thiourea based anion binding motifs, but concerns have been raised about the potential toxicity of thioureas. For this reason, Davis, Gale and co-workers compared the lipid bilayer anion transport capability of thiourea isosteres, such as receptors 185–187 based on the guanidine motif, to simple thiourea 188.102 Single crystal X-ray diffraction analysis of 185 and 186 showed that the guanidine subunit hydrogen atoms adopt the trans arrangement while the thiourea hydrogen atoms in thiourea 188 adopt the cis arrangement. 1H NMR Job plot analysis (DMSO-d6–0.5% water) indicated a 1:1 binding model for all receptors with a variety of anions. In all cases the receptors bind most strongly with sulfate anions. Chloride ion selective electrode tests with unilamellar POPC vesicles were used to test for anion transport abilities. At 2 mol% carrier loading (with respect to lipid) receptors 185 and 187 showed minimal chloride efflux (5% at 270 s), whilst receptor 186 gave a chloride efflux of 29%, comparable to thiourea 188 which shows 30% chloride efflux, and it was found that a chloride–nitrate antiport mechanism is the most likely reason for the observed chloride efflux. It was also reported that the addition of the cationophore valinomycin results in significantly enhanced anion transport for all receptors (e.g. receptor 186 increases from 29% to 58% chloride efflux). This enhancement was attributed to valinomycin bound potassium cations in the membrane either stabilising the anion–receptor complex in the membrane, or enhancing the extraction rate of anions from the aqueous phase into the membrane phase. These initial results suggest that hydrogen bonding thiourea isosteres may be optimised for anion binding and transport.
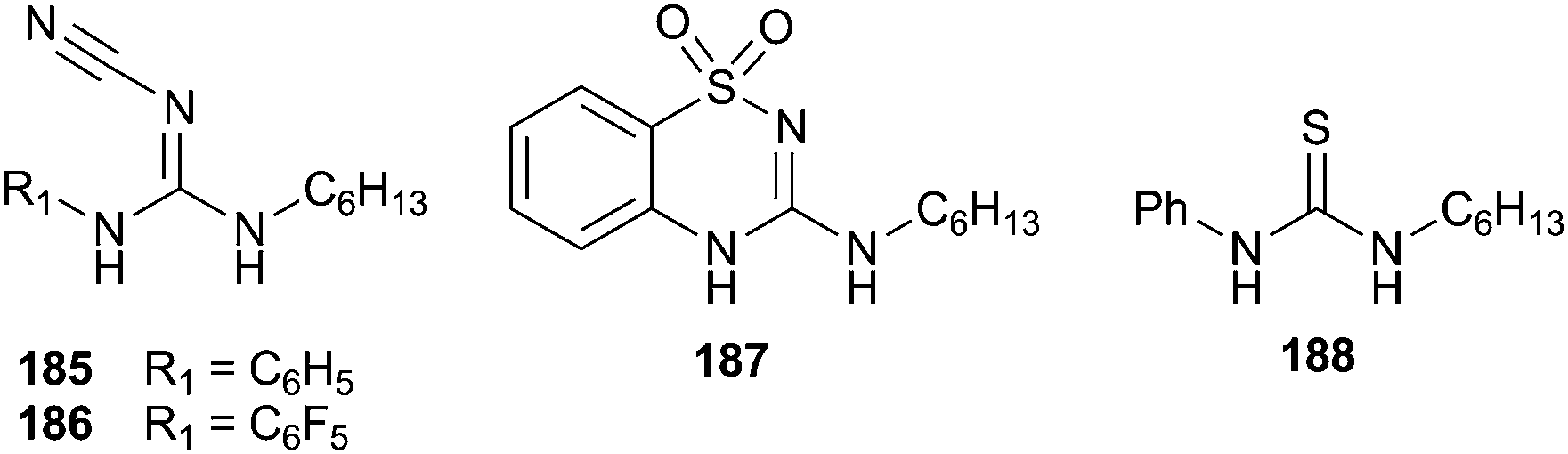
This indication of a dual host system was further investigated by the same group.103 Taking lead from work where separate anion and cation receptors can be used to extract metal salts from aqueous to organic media, they wondered if the same tactic could be used to co-transport metal salts across POPC lipid bilayers, utilising synergistically coupled cationic and anionic uniport processes (Scheme 22). Using lucigenin fluorescence assays they were able to determine that triazole-strapped calix[4]pyrroles 189 and 190, when added together with the known potassium cation transporter valinomycin, are able to significantly increase the internal vesicular concentration of chloride anions, more so than when valinomycin or the triazole-strapped calixpyrrole are added separately. This evidence supports the proposed K+/Cl− co-transport mechanism.
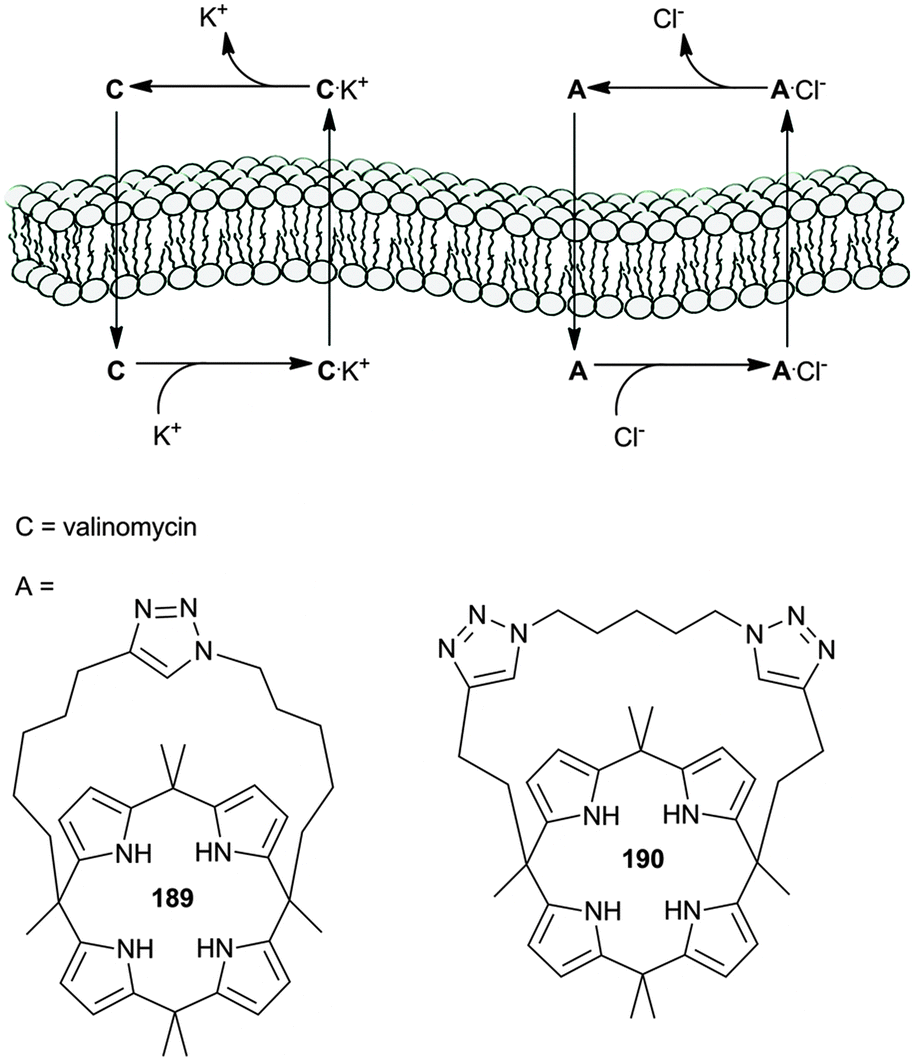 | ||
| Scheme 22 Design concept of the dual host approach to co-transport of KCl by cation transporter C (valinomycin) and anion transporter A (189 or 190). | ||
Gale and co-workers continued to improve on transport efficiency by small molecules and reported the first example of the use of squaramides 191–193 for transmembrane anion transport.104 This investigation showed that squaramide based compounds outperform urea and thiourea analogues when it comes to transmembrane anion transport by nearly one order of magnitude, presumably a result of the increased strength of the anion binding interaction. Receptor 193, for example, gave a chloride association constant of Ka = 643 M−1 and an EC50,270s value for chloride–nitrate antiport of 0.01 mol% with respect to lipid (compared to Ka = 41 M−1 and an EC50,270s = 0.16 mol% for the analogous thiourea), and was the most active transporter of the tested series. All transporters tested were shown to function by a mobile carrier mechanism, as opposed to channel formation, as indicated by U-tube experiments. Since the introduction of the squaramides motif reduces the receptors' lipophilicity, it seems that the squaramide motif is a good target for future development of potent anion transporters.
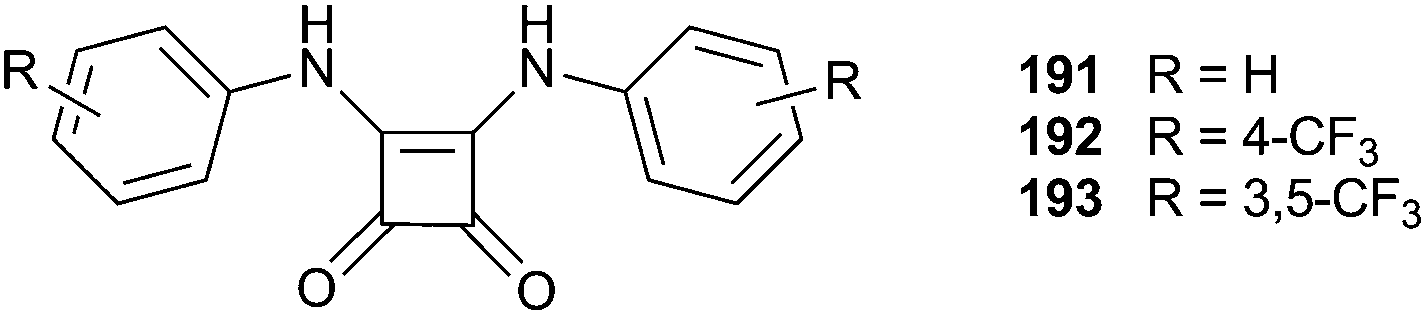
Bahmanjah, Zhang and Davis have investigated the use of monoacylglycerols 194–197 for chloride transport.105 It was suggested that the 1,2-diol unit would provide a good anion binding region whilst the C3-acyl chains would give the compound sufficient lipophilicity to pass through a phospholipid membrane. Furthermore, the addition of an extra hydrogen bond donor to the head group and/or fluorination of the tail group would improve transport capabilities. Lucigenin fluorescence tests on vesicles pre-incorporated with 194–197 showed that monoacylglycerol 194 is able to promote chloride influx, but when the 1,2-diol unit was substituted for additional acylglycerol units (195) transport activity was lost, confirming the importance of the diol for transport activity. Substituting an amide NH at the C3 position resulted in increased anion affinity, as confirmed by 1H NMR titrations in both CDCl3 and CD3CN, as well as increased transport efficiency (196, EC50 = 0.076 mM). This enhancement in transport efficiency was amplified even further upon fluorination of the acylglycerol tail (197, EC50 = 0.011 mM), and the group were able to confirm an antiport mechanism of transport for this class of compound.
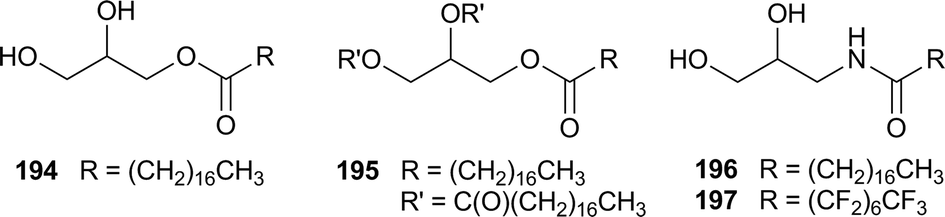
Matile and co-workers aimed to maximise atom efficiency of anion transporters by employing halogen bond functionalities (e.g.198–204).106 These are considered the hydrophobic equivalent of hydrogen bonds, and hence do not require the same level of overcompensation to counteract the hydrophilic binding sites. DFT calculations revealed that chloride anions could be encapsulated by up to 6 molecules of 199, forming a hydrophobic “shell”, and that the anion binding interactions reduce upon replacement of 199 with water, indicating that the binding process is reversible and useful for anion transport processes. Furthermore, defluorination of the receptor or replacement of the iodine for bromine gives weaker halogen bonding. The crystal structure of 199 in the presence of KCl and 18-crown-6 confirms that at least 2 halogen bonds form between a chloride anion and the receptors (Fig. 24). HPTS fluorescence assays were used to determine the chloride transport efficiency of 199 and revealed an EC50 = 21.6 μM with a Hill coefficient of n = 4.7, consistent with the computational results for the halogen bonded capsules. The longer chain receptor 200 (EC50 = 3.07 μM) was a better transporter due to its increased ability to fully encapsulate the anion. At the other end of the scale trifluoroiodomethane 198 is a relatively poor yet still detectable transporter, and with only one carbon is the smallest possible organic ion transporter.

 | ||
| Fig. 24 Section of the crystal structure of chloride bound to 2 molecules of 199 and potassium counterions bound to 18-crown-6; hydrogen atoms are omitted for clarity and halogen bonds are shown by dashed lines, with the anion in spacefill and the receptors in ball-and-stick. | ||
Matile and co-workers have also investigated a series of ditopic calix[4]arene receptors 205–208 with a single-point variation, allowing them to probe the contributions of hydrogen bonds, anion–π interactions and halogen bonds to anion transport.107 The benefit of ditopic receptors is that they can co-transport ion pairs thereby avoiding temporary separation of charge. These receptors bind TMA (tetramethylammonium) counter cations in the upper rim of the calix[4]arene, while substitutions at the lower rim allow for anion binding. The group used pH sensitive HPTS (8-hydroxy-1,3,6-pyrenetrisulfonate) assays to establish that this class of receptors is able to antiport chloride–hydroxide. The overall process occurs via the combination of chloride–TMA and hydroxide–TMA symport actions. Binding of TMA in the upper rim is essential to the function of these transporters, as in tests with different counter cations no transport was observed. Single mutation from 205 to 206 resulted in the total loss of transport activity, whilst mutation to 207 partially restored the transport activity. 19F NMR in dry acetone-d6 shows evidence of detectable anion binding in receptor 206 (but not in 205 and 207), hence it is suggested that the disruption in transport activity is a result of excessive binding by halogen bonding. Weakening of the halogen bond by mutation from 206 to 208 increased the transport ability more than 30 times, suggesting that 208 can transport chloride–hydroxide using halogen bonding if the halogen bond is weak enough.
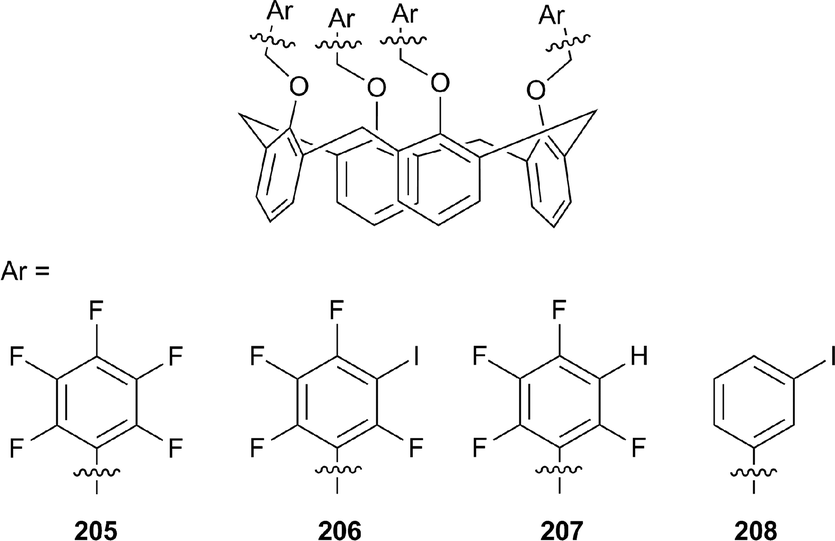
Matile and co-workers have also recently published work on self-sorting dimerisation.108 Dimerisation and self-sorting are known to be key events associated with the function of photosystems and anion transporters, and the group has therefore introduced cis-core naphthalenediimides (NDIs) with only one free chiral π-surface (e.g.209–211) as a means to direct self-assembly into dimers. NMR studies (CDCl3 at room temperature) provided evidence of dimerisation into heterodimers composed of pairs of enantiomers at high monomer concentrations. It was established that alternate self-sorting efficiency dropped with increasing π-acidity; a result of the increasing strength of π,π-contacts in the core. The most π-acidic NDIs (211) showed the highest level of self-sorting ability. These compounds showed a wide range of self-sorting ability, from complete self-sorting with less π-acidic species to no self-sorting with more π-acidic species, and alternate self-sorting somewhere in the middle. In terms of anion transport ability, the highest activity in HPTS transport assays was observed in systems where the π-acidity is intermediate and where the fewest number of dimers are detectable (various stereoisomers of 210). In the reported sulfoxide series the transport activities are reported to increase with increasing Hill coefficients.
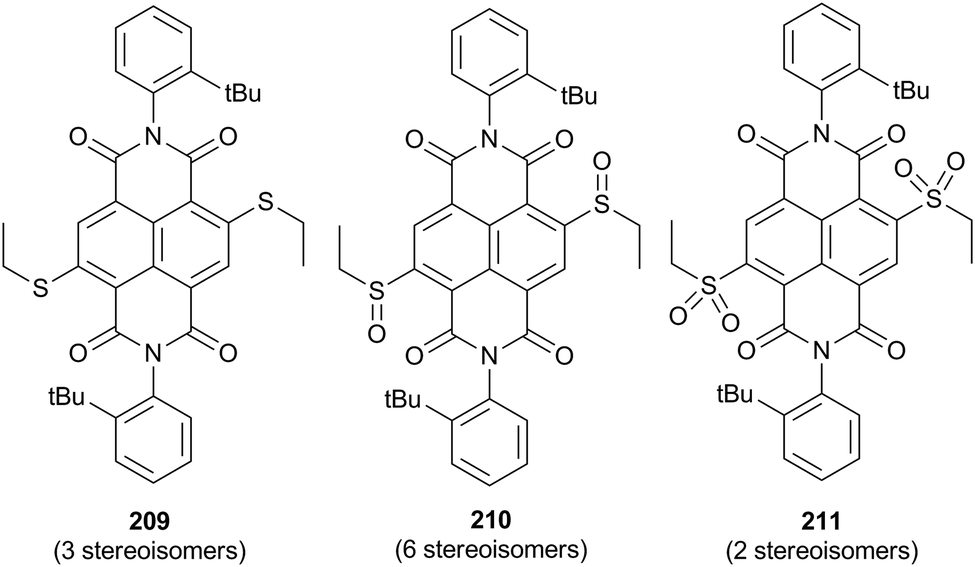
The use of small molecule mobile carriers is just one approach towards transmembrane anion transport. Gokel and co-workers reported the formation of lipid bilayer channels from branched-chain pyrogallol[4]arenes, which assemble into nanotubes or bilayers.109 Compound 212 crystallises as nanotubes formed by the stacking of two or more hexameric units interlocked by the branched pentyl side chains, while compound 213 crystallises as a head-to-head bilayer with a thickness of ∼17.6 Å.110 The authors postulate that when hexamers of 212 penetrate into the lipid bilayer, they reassemble into the nanotubular arrangement, encouraged by the interaction of the pentyl side chains and the fatty acid acyl chains of the lipid. It is suggested that two hexameric units stacked upon each other would form a channel and the lateral movement of these hexamer units of 212 over one another would “gate” the opening and closing of the nanotube, ready for ion transport. This gating was experimentally observed in an asolectin membrane by using planar lipid bilayer conductance studies, which also reveal a pore size of ∼17 Å in the membrane, corresponding well to the dimensions of the hexameric units seen in the crystal structure of 212 (18 Å × 20 Å).
Building on their work on the use of isophthalamides as anion transporters,111 Gokel and co-workers have also illustrated the use of tris-arenes based on isophthalic acid and 2,6-dipicolinic acid (214–221) for transport of plasmid DNA into ampicillin resistant E. coli.112 Transformation experiments using 2.6 kb plasmids showed isophthalamides with OCH3 (214) and H (215) substituents give an enhancement in transformation of 2.3- and 2-fold respectively compared with DMSO and water controls, while the Cl (216) and CN (217) substituted species were less effective than the controls. In fact the CN substituted compound appeared to totally suppress transformation. The results for the 2,6-dipicolinic acid derivatives (218–221) follow the same trend as for the isophthalic acid derivatives, with the only major difference being that the CN substituted compound shows reduced activity without total suppression of transformation. Tests using 20 kb plasmids illustrate how OCH3 and H substituted tris-arenes can dramatically enhance transformation up to 10- and 8-fold respectively, which is remarkable considering that increased plasmid size usually hinders transformation. The authors theorise that the tris-arenes bind individually to the phosphate residues of the DNA backbone, encapsulating the DNA in a hydrophobic “coat”, which can be transported across the lipid bilayer by passive diffusion.
Yao, Yang and colleagues have continued their work on the electrophysical properties and potential function of 222,113 which has previously been shown to form channels in liposomes.114 The group used single-channel patch clamp techniques to confirm the formation of channels in HEK 293 living cells and showed that 1 μmol of 222 is enough for the small molecule to self-assemble into chloride ion channels within the membrane. Subsequent whole-cell patch clamp recordings showed that compound 222 induces a large increase in current relative to the control experiments. By varying the size of the counter cation it was possible to confirm that the increase in whole-cell current was due to chloride channel formation. Channel selectivity was tested by measuring reverse cell potentials when the extracellular chloride was replaced with different anions, including fluoride, bromide, iodide and nitrate. The activity sequence was as follows F− < Cl− < Br− < I− < NO3−. The group were also able to show that compound 222, once pre-incorporated into plasma membranes of cystic fibrosis airway epithelial cells, can increase the chloride permeability of such cells with damaged CFTR (Cystic Fibrosis transmembrane conductance regulator) channels; a significant step towards therapeutic use of this compound in the treatment of channelopathies such as cystic fibrosis.
Conclusions
The years 2011 and 2012 have seen many advances in the area of supramolecular chemistry of anionic species and related subjects. The use of “unconventional” interactions to bind anions, such as CH hydrogen bonding, halogen bonding and anion⋯π interactions, has become increasingly popular. Additionally, more research has been devoted to employing the lessons learned from pure anion binding studies into real-world applications, such as sensing, catalysis and the medicinal use of transmembrane anion transport, and we can anticipate that this research field will continue to grow in the coming years.Acknowledgements
We thank the EPSRC for funding (CJEH (EP/J009687/1) and LEK). NB thanks the University of Southampton and A*STAR for a postgraduate scholarship. ILK thanks the University of Southampton for a teaching assistantship. PAG thanks the Royal Society and the Wolfson Foundation for a Royal Society Wolfson Research Merit Award.Notes and references
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC.
- P. A. Gale, Chem. Soc. Rev., 2010, 39, 3746–3771 RSC.
- P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC.
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC.
- C. Bazzicalupi, A. Bencini, C. Giorgi, B. Valtancoli, V. Lippolis and A. Perra, Inorg. Chem., 2011, 50, 7202–7216 CrossRef CAS PubMed.
- C. Bazzicalupi, A. Bencini, S. Puccioni, B. Valtancoli, P. Gratteri, A. Garau and V. Lippolis, Chem. Commun., 2012, 48, 139–141 RSC.
- A. Bencini, C. Coluccini, A. Garau, C. Giorgi, V. Lippolis, L. Messori, D. Pasini and S. Puccioni, Chem. Commun., 2012, 48, 10428–10430 RSC.
- M. A. Hossain, S. A. Kang, J. A. Kut, V. W. Day and K. Bowman-James, Inorg. Chem., 2012, 51, 4833–4840 CrossRef CAS PubMed.
- M. B. Nielsen, J. O. Jeppesen, J. Lau, C. Lomholt, D. Damgaard, J. P. Jacobsen, J. Becher and J. F. Stoddart, J. Org. Chem., 2001, 66, 3559–3563 CrossRef CAS PubMed.
- S. S. Andersen, M. Jensen, A. Sorensen, E. Miyazaki, K. Takimiya, B. W. Laursen, A. H. Flood and J. O. Jeppesen, Chem. Commun., 2012, 48, 5157–5159 RSC.
- C. J. Serpell, J. Cookson, D. Ozkaya and P. D. Beer, Nat. Chem., 2011, 3, 478–483 CAS.
- S. R. Beeren and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3804–3807 CrossRef CAS PubMed.
- C. N. Carroll, B. A. Coombs, S. P. McClintock, C. A. Johnson II, O. B. Berryman, D. W. Johnson and M. M. Haley, Chem. Commun., 2011, 47, 5539–5541 RSC.
- C. N. Carroll, O. B. Berryman, C. A. Johnson, L. N. Zakharov, M. M. Haley and D. W. Johnson, Chem. Commun., 2009, 2520–2522 RSC.
- J. M. Engle, C. N. Carroll, D. W. Johnson and M. M. Haley, Chem. Sci., 2012, 3, 1105–1110 RSC.
- Y. P. Zhou, M. Zhang, Y. H. Li, Q. R. Guan, F. Wang, Z. J. Lin, C. K. Lam, X. L. Feng and H. Y. Chao, Inorg. Chem., 2012, 51, 5099–5109 CrossRef CAS PubMed.
- D. M. Jessen, A. N. Wercholuk, B. Xiong, A. L. Sargent and W. E. Allen, J. Org. Chem., 2012, 77, 6615–6619 CrossRef CAS PubMed.
- M. J. Kim, H. W. Lee, D. Moon and K. S. Jeong, Org. Lett., 2012, 14, 5042–5045 CrossRef CAS PubMed.
- R. M. Duke, T. McCabe, W. Schmitt and T. Gunnlaugsson, J. Org. Chem., 2012, 77, 3115–3126 CrossRef CAS PubMed.
- J. Krishnamurthi, T. Ono, S. Amemori, H. Komatsu, S. Shinkai and K. Sada, Chem. Commun., 2011, 47, 1571–1573 RSC.
- Z. W. Yang, B. A. Wu, X. J. Huang, Y. Y. Liu, S. G. Li, Y. N. Xia, C. D. Jia and X. J. Yang, Chem. Commun., 2011, 47, 2880–2882 RSC.
- R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. H. Zhang, L. A. Watson, G. Van Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525–8534 CrossRef CAS PubMed.
- P. G. Young, J. K. Clegg, M. Bhadbhade and K. A. Jolliffe, Chem. Commun., 2011, 47, 463–465 RSC.
- C. D. Jia, B. A. Wu, S. G. Li, X. J. Huang, Q. L. Zhao, Q. S. Li and X. J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486–490 CrossRef CAS PubMed.
- S. G. Li, C. D. Jia, B. Wu, Q. Luo, X. J. Huang, Z. W. Yang, Q. S. Li and X. J. Yang, Angew. Chem., Int. Ed., 2011, 50, 5720–5723 Search PubMed.
- S. G. Li, M. Y. Wei, X. J. Huang, X. J. Yang and B. Wu, Chem. Commun., 2012, 48, 3097–3099 RSC.
- B. Wu, C. D. Jia, X. L. Wang, S. G. Li, X. J. Huang and X. J. Yang, Org. Lett., 2012, 14, 684–687 CrossRef CAS PubMed.
- A. Rostami, C. J. Wei, G. Guerin and M. S. Taylor, Angew. Chem., Int. Ed., 2011, 50, 2059–2062 CrossRef CAS PubMed.
- P. Sokkalingam, J. Yoo, H. Hwang, P. H. Lee, Y. M. Jung and C.-H. Lee, Eur. J. Org. Chem., 2011, 2911–2915 CrossRef CAS.
- P. Sokkalingam, D. S. Kim, H. Hwang, J. L. Sessler and C.-H. Lee, Chem. Sci., 2012, 3, 1819–1824 RSC.
- S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938–15941 CrossRef CAS PubMed.
- S. K. Kim, G. I. Vargas-Zúñiga, B. P. Hay, N. J. Young, L. H. Delmau, C. Masselin, C.-H. Lee, J. S. Kim, V. M. Lynch, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 1782–1792 CrossRef CAS PubMed.
- H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266–9269 CrossRef CAS PubMed.
- H. Maeda, K. Naritani, Y. Honsho and S. Seki, J. Am. Chem. Soc., 2011, 133, 8896–8899 CrossRef CAS PubMed.
- H. Maeda and Y. Terashima, Chem. Commun., 2011, 47, 7620–7622 RSC.
- H. Maeda, K. Kinoshita, K. Naritani and Y. Bando, Chem. Commun., 2011, 47, 8241–8243 RSC.
- J.-m. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938–13941 CrossRef CAS PubMed.
- J.-m. Suk, D. A. Kim and K.-S. Jeong, Org. Lett., 2012, 14, 5018–5021 CrossRef CAS PubMed.
- J.-i. Kim, H. Juwarker, X. Liu, M. S. Lah and K.-S. Jeong, Chem. Commun., 2010, 46, 764–766 RSC.
- Q.-Y. Cao, T. Pradhan, S. Kim and J. S. Kim, Org. Lett., 2011, 13, 4386–4389 CrossRef CAS PubMed.
- P. Das, A. K. Mandal, M. K. Kesharwani, E. Suresh, B. Ganguly and A. Das, Chem. Commun., 2011, 47, 7398–7400 RSC.
- N. Ahmed, B. Shirinfar, I. Geronimo and K. S. Kim, Org. Lett., 2011, 13, 5476–5479 CrossRef CAS PubMed.
- K. P. McDonald, Y. Hua, S. Lee and A. H. Flood, Chem. Commun., 2012, 48, 5065–5075 RSC.
- Y. Hua, R. O. Ramabhadran, J. A. Karty, K. Raghavachari and A. H. Flood, Chem. Commun., 2011, 47, 5979–5981 RSC.
- Y. Hua, R. O. Ramabhadran, E. O. Uduehi, J. A. Karty, K. Raghavachari and A. H. Flood, Chem.–Eur. J., 2011, 17, 312–321 CrossRef CAS PubMed.
- K. P. McDonald, R. O. Ramabhadran, S. Lee, K. Raghavachari and A. H. Flood, Org. Lett., 2011, 13, 6260–6263 CrossRef CAS PubMed.
- A. Caballero, N. G. White and P. D. Beer, Angew. Chem., Int. Ed., 2011, 50, 1845–1848 CrossRef CAS PubMed.
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed.
- M. G. Chudzinski, C. A. McClary and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 10559–10567 CrossRef CAS PubMed.
- M. Giese, M. Albrecht, T. Krappitz, M. Peters, V. Gossen, G. Raabe, A. Valkonen and K. Rissanen, Chem. Commun., 2011, 48, 9983–9985 RSC.
- S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS PubMed.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2011, 133, 7344–7347 CrossRef CAS PubMed.
- X. M. He and V. W. W. Yam, Org. Lett., 2011, 13, 2172–2175 CrossRef CAS PubMed.
- H. C. Gee, C. H. Lee, Y. H. Jeong and W. D. Jang, Chem. Commun., 2011, 47, 11963–11965 RSC.
- M. A. Tetilla, M. C. Aragoni, M. Arca, C. Caltagirone, C. Bazzicalupi, A. Bencini, A. Garau, F. Isaia, A. Laguna, V. Lippolis and V. Meli, Chem. Commun., 2011, 47, 3805–3807 RSC.
- M. Mameli, M. C. Aragoni, M. Arca, M. Atzori, A. Bencini, C. Bazzicalupi, A. J. Blake, C. Caltagirone, F. A. Devillanova, A. Garau, M. B. Hursthouse, F. Isaia, V. Lippolis and B. Valtancoli, Inorg. Chem., 2009, 48, 9236–9249 CrossRef CAS PubMed.
- C. R. K. Glasson, J. K. Clegg, J. C. McMurtrie, G. V. Meehan, L. F. Lindoy, C. A. Motti, B. Moubaraki, K. S. Murray and J. D. Cashion, Chem. Sci., 2011, 2, 540–543 RSC.
- C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, P. Turner, M. B. Duriska and R. Willis, Chem. Commun., 2008, 1190–1192 RSC.
- R. A. Bilbeisi, J. K. Clegg, N. Elgrishi, X. de Hatten, M. Devillard, B. Breiner, P. Mal and J. R. Nitschke, J. Am. Chem. Soc., 2012, 134, 5110–5119 CrossRef CAS PubMed.
- Y. R. Hristova, M. M. J. Smulders, J. K. Clegg, B. Breiner and J. R. Nitschke, Chem. Sci., 2011, 2, 638–641 RSC.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Nat. Chem., 2012, 4, 751–756 CrossRef CAS PubMed.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch, K. M. Keller and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 1526–1533 CrossRef CAS PubMed.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS PubMed.
- A. S. Singh and S.-S. Sun, Chem. Commun., 2011, 47, 8563–8565 RSC.
- L. M. Hancock, L. C. Gilday, N. L. Kilah, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 1725–1727 RSC.
- N. H. Evans, H. Rahman, A. V. Leontiev, N. D. Greenham, G. A. Orlowski, Q. Zeng, R. M. J. Jacobs, C. J. Serpell, N. L. Kilah, J. J. Davis and P. D. Beer, Chem. Sci., 2012, 3, 1080–1089 RSC.
- A. V. Leontiev, C. J. Serpell, N. G. White and P. D. Beer, Chem. Sci., 2011, 2, 922–927 RSC.
- M. Chas, G. Gil-Ramírez and P. Ballester, Org. Lett., 2011, 13, 3402–3405 CrossRef CAS PubMed.
- M. Chas and P. Ballester, Chem. Sci., 2012, 3, 186–191 RSC.
- V. Valderrey, E. C. Escudero-Adán and P. Ballester, J. Am. Chem. Soc., 2012, 134, 10733–10736 CrossRef CAS PubMed.
- J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed.
- H. Ito, M. Ikeda, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2011, 133, 3419–3432 CrossRef CAS PubMed.
- Y. Bao, B. Liu, H. Wang, J. Tian and R. Bai, Chem. Commun., 2011, 47, 3957–3959 RSC.
- R. M. Duke and T. Gunnlaugsson, Tetrahedron Lett., 2011, 52, 1503–1505 CrossRef CAS PubMed.
- P. Singh and M. Kaur, Chem. Commun., 2011, 47, 9122–9124 RSC.
- H. J. Kim, K. C. Ko, J. H. Lee, J. Y. Lee and J. S. Kim, Chem. Commun., 2011, 47, 2886–2888 RSC.
- P. Calero, M. Hecht, R. Martinez-Manez, F. Sancenon, J. Soto, J. L. Vivancos and K. Rurack, Chem. Commun., 2011, 47, 10599–10601 RSC.
- W. Chen, S. A. Elfeky, Y. Nonne, L. Male, K. Ahmed, C. Amiable, P. Axe, S. Yamada, T. D. James, S. D. Bull and J. S. Fossey, Chem. Commun., 2011, 47, 253–255 RSC.
- I. Richter, J. Minari, P. Axe, J. P. Lowe, T. D. James, K. Sakurai, S. D. Bull and J. S. Fossey, Chem. Commun., 2008, 1082–1084 RSC.
- Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC.
- J. A. Birrell, J.-N. Desrosiers and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 13872–13875 CrossRef CAS PubMed.
- S. Lin and E. N. Jacobsen, Nat. Chem., 2012, 4, 817 CrossRef CAS PubMed.
- N. Z. Burns, M. R. Witten and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS PubMed.
- K. Ohmatsu, M. Kiyokawa and T. Ooi, J. Am. Chem. Soc., 2011, 133, 1307–1309 CrossRef CAS PubMed.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- S. Matile, A. Vargas Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- G. W. Gokel and N. Barkey, New J. Chem., 2009, 33, 947–963 RSC.
- P. A. Gale, Acc. Chem. Res., 2011, 44, 216–226 CrossRef CAS PubMed.
- J. T. Davis, in Topics in Heterocyclic Chemistry, ed. P. A. Gale and W. Dehaen, Springer, New York, 2010, vol. 24, pp. 145–176 Search PubMed.
- J. L. Seganish and J. T. Davis, Chem. Commun., 2005, 5781–5783 RSC.
- R. Pérez-Tomás, B. Montaner, E. Llagostera and V. Soto-Cerrato, Biochem. Pharmacol., 2003, 66, 1447–1452 CrossRef.
- B. DíazdeGreñu, P. I. Hernández, M. Espona, D. Quiñonero, M. E. Light, T. Torroba, R. Pérez-Tomás and R. Quesada, Chem.–Eur. J., 2011, 17, 14074–14083 CrossRef PubMed.
- P. I. Hernandez, D. Moreno, A. A. Javier, T. Torroba, R. Perez-Tomas and R. Quesada, Chem. Commun., 2012, 48, 1556–1558 RSC.
- N. J. Andrews, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis, W. A. Harrell Jr and P. A. Gale, Chem. Sci., 2011, 2, 256–260 RSC.
- C. J. E. Haynes, S. J. Moore, J. R. Hiscock, I. Marques, P. J. Costa, V. Felix and P. A. Gale, Chem. Sci., 2012, 3, 1436–1444 RSC.
- P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633–3647 RSC.
- S. Hussain, P. R. Brotherhood, L. W. Judd and A. P. Davis, J. Am. Chem. Soc., 2011, 133, 1614–1617 CrossRef CAS PubMed.
- Y. R. Choi, M. K. Chae, D. Kim, M. S. Lah and K.-S. Jeong, Chem. Commun., 2012, 48, 10346–10348 RSC.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed.
- S. J. Moore, M. Wenzel, M. E. Light, R. Morley, S. J. Bradberry, P. Gomez-Iglesias, V. Soto-Cerrato, R. Perez-Tomas and P. A. Gale, Chem. Sci., 2012, 3, 2501–2509 RSC.
- M. Wenzel, M. E. Light, A. P. Davis and P. A. Gale, Chem. Commun., 2011, 47, 7641–7643 RSC.
- S. J. Moore, M. G. Fisher, M. Yano, C. C. Tong and P. A. Gale, Chem. Commun., 2011, 47, 689–691 RSC.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS PubMed.
- S. Bahmanjah, N. Zhang and J. T. Davis, Chem. Commun., 2012, 48, 4432–4434 RSC.
- A. V. Jentzsch, D. Emery, J. Mareda, S. K. Nayak, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, Nat. Commun., 2012, 3, 905 CrossRef PubMed.
- A. Vargas Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675–11678 CrossRef CAS PubMed.
- N.-T. Lin, A. Vargas Jentzsch, L. Guenee, J.-M. Neudorfl, S. Aziz, A. Berkessel, E. Orentas, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1121–1127 RSC.
- S. Negin, M. M. Daschbach, O. V. Kulikov, N. Rath and G. W. Gokel, J. Am. Chem. Soc., 2011, 133, 3234–3237 CrossRef CAS PubMed.
- O. V. Kulikov, M. M. Daschbach, C. R. Yamnitz, N. Rath and G. W. Gokel, Chem. Commun., 2009, 7497–7499 RSC.
- C. R. Yamnitz, S. Negin, I. A. Carasel, R. K. Winter and G. W. Gokel, Chem. Commun., 2010, 46, 2838–2840 RSC.
- J. L. Atkins, M. B. Patel, M. M. Daschbach, J. W. Meisel and G. W. Gokel, J. Am. Chem. Soc., 2012, 134, 13546–13549 CrossRef CAS PubMed.
- B. Shen, X. Li, F. Wang, X. Yao and D. Yang, PLoS One, 2012, 7, e34694 CAS.
- X. Li, B. Shen, X.-Q. Yao and D. Yang, J. Am. Chem. Soc., 2007, 129, 7264–7265 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |