Dextrin cross linked with poly(HEMA): a novel hydrogel for colon specific delivery of ornidazole†
Dipankar
Das
a,
Raghunath
Das
a,
Paulomi
Ghosh
b,
Santanu
Dhara
b,
Asit Baran
Panda
c and
Sagar
Pal
*a
aPolymer Chemistry Laboratory, Department of Applied Chemistry, Indian School of Mines, Dhanbad-826004, India. E-mail: sagarpal1@hotmail.com; Fax: +91-326-2296615; Tel: +91-326-2235769
bSchool of Medical Science & Technology, Indian Institute of Technology, Kharagpur – 721302, India
cDiscipline of Inorganic Materials and Catalysis, Central salt and Marine Chemicals Research Institute (CSIR), Bhavnagar-364002, Gujarat, India
First published on 18th September 2013
Abstract
We report on the synthesis and characterization of a novel hydrogel based on dextrin grafted with poly(2-hydroxyethyl methacrylate) by embedding N,N′-methylene bis acrylamide (MBA) as cross linker, into a polymeric network in the presence of potassium persulphate (KPS) initiator for colon specific delivery of ornidazole. Various grades of hydrogels [Dxt-g-p(HEMA)] have been synthesized by altering the reaction parameters and the best one optimized. The developed hydrogel has been characterized using FTIR spectra, 13C NMR spectra, elemental analysis, XRD study, SEM analysis, TGA analysis, swelling study and cell viability study. The equilibrium swelling ratio of the hydrogels has been recorded in different media and found to be at a maximum at pH 7.4. A cell viability study indicates that the hydrogel is non-cytotoxic in nature. The drug delivery results demonstrate that Dxt-g-p(HEMA) delivers ornidazole successfully in the colonic region in a controlled way and is a good candidate for an orally administered drug delivery system. The release mechanism and kinetics of ornidazole from various hydrogels have been determined using different linear and nonlinear mathematical models, which confirm that ornidazole release from hydrogel follows first order kinetics and a non-Fickian diffusion mechanism.
1. Introduction
In recent years, hydrogels have drawn significant attention because of their numerous applications in the pharmaceutical and biomedical fields.1 Hydrogels are physically or chemically cross linked natural or synthetic three dimensional polymer networks, which are capable of absorbing large amounts of water while maintaining their structure. Remarkable attempts have been made to design and develop novel hydrogels with combined characteristics like, tuneable chemical and three-dimensional physical structure,2 desired mechanical properties,3 high water content capability and biocompatibility,2 all of which are essential for applications in tissue engineering, biomedical implants, drug delivery, and nano-biotechnology.4More recently, natural polymer-based hydrogels have attracted ample interest in tissue engineering and drug delivery applications.5 They can be obtained with various desired properties similar to their synthetic counter-parts and hence hydrogels are employed alone or with their modified forms in pharmaceutical and biomedical fields as they are biodegradable, abundant in nature, renewable, non-toxic, and relatively cheap. However, natural polymers have certain drawbacks, like uncontrolled rate of hydration, microbial contamination, and drop in viscosity on storing. Fortunately, several strategies have already been developed to overcome these drawbacks by suitable chemical modification. One of the most significant ways to modify natural polymers is grafting of synthetic polymers onto their backbones. The insoluble nature of grafted hydrogels is mainly because of the chemical and/or physical cross links. It is also easy to tune the physico-chemical properties of grafted hydrogels, which are sensitive towards various stimuli present in our body like pH, ionic strength, temperature etc., owing to flexible synthetic methods.6 Thus, use of these graft copolymer based hydrogels, specifically for drug delivery applications is advantageous over other developed biomaterials based on hydroxyapatite, collagen, chitosan, alginate and nanocomposites,7 as the drug release behaviour of these hydrogels not only depends on the extent of cross linking but also on various other factors, including swelling properties, pH of the release media and finally for their biocompatibility. In particular, for colon specific drug delivery, pH-sensitive hydrogels have attracted increasing attention in a stimuli-responsive manner based on pH changes in GI tract. In recent years, several natural polymer based hydrogels have been reported for colon specific delivery, but their release characteristics as well as drug stability are not satisfactory from a pharmacological point of view.8
Further, several antibiotics, such as ciprofloxacin,9 metronidazole,10 ornidazole,8a tetracycline hydrochloride11 have been used for treatment of colon related disorders. Out of these ornidazole is the preferred drug, mostly used in the treatment of severe hepatic and intestinal amoebiasis, as it has antiprotozoal and antibacterial properties against anaerobic bacteria.8a,11,12 It is used in the treatment and prophylaxis of susceptible anaerobic infections in gastric surgery.12 However, due to the complete and prompt absorption after oral administration and short biological half life of ornidazole,8a it is essential to deliver the drug in a more sustained way to provide longer bioavailability and hence its administration in tablet form may provide a minimum amount of ornidazole for local action in the colon.8a In spite of the development of various drug delivery systems, like carboxymethyl chitosan hydrogel, hydrogel based on sterculia gum–polyHEMA–polyacrylic acid, hydrogel derived from sterculia gum and poly(vinyl pyrrolidone), HPMC, and ethyl cellulose, to investigate the colon specific release behaviour of ornidazole, it is essential to develop new colon specific delivery systems for ornidazole to overcome important shortcomings of developed hydrogels, like specificity, biodegradability, biocompatibility, drug stability, and most importantly controlled release properties.8a–d For example, Singh et al.8b reported that ∼65% of ornidazole drug was released from sterculia-cl-poly(HEMA-co-AAc) hydrogel in 5 h, Vaghani et al.8a reported ∼100% ornidazole release in 12 h from the carboxymethyl chitosan hydrogel system. Singh et al.8d also reported the controlled release behaviour of ornidazole from sterculia-cl-poly(NVP) hydrogel. Further, the non-cytotoxicity of the materials and the stability of the drug in that matrix are also the vital factors for a material to be a novel and useful drug delivery system. Unfortunately, based on our knowledge, there are no literature reports on the toxicity level as well as the ornidazole stability in these matrices, except that Patel et al.8c studied the ornidazole release from HPMC based coated system, which released the drug ∼65% in 12 h and the stability of drug loaded tablet after 2 months for 12 h is ∼74–78% and for 24 h is ∼96–98% at 40 ± 2 °C/75 ± 5% RH. In view of the potential pharmaceutical importance of ornidazole and the lack of literature on the non-cytotoxic and compatible matrix for oral administration of ornidazole in a sustainable way through prolonged control release, it is essential to develop a novel natural polymer based hydrogel, which is non-toxic, compatible with the drug and shows a more controlled/sustained ornidazole release behaviour.
Out of various natural polymers, pure/modified polysaccharide-based hydrogels are the most interesting and the most used because of their high content of functional groups, like hydroxyl, amino, carboxylic acid groups, which are utilized for cross linking with additional functional cross linkers as well as further bioconjugation with cell targeting agents.13 In recent years, polysaccharide based graft copolymer hydrogels have been a new entrance in the field of controlled drug delivery by solving the shortcomings as mentioned above for natural polymers.14 Dextrin, one of the most promising natural polysaccharides, is enzymatically degradable and hydrophilic in nature. It is water soluble, optically active, low viscosity, non-toxic and an ideal polymer to form hydrogels. Previously, it has been reported that various synthetic polymers such as polymethyl acrylate, polyvinyl acrylate, polyacrylamide and so on have been introduced on the dextrin backbone for their application in different fields.15 It has also been reported that cross linking density is very crucial for desirable polymeric network formation and its application in the biomedical field.16 On the other hand, poly-2-hydroxy ethyl methacrylate [poly(HEMA)] based hydrogels recently received great attention in biomedical applications, such as corneal implants, cardiovascular implants, contact lenses, tissue repair surgery, dental applications as well as in the controlled release of antibiotic drugs, anti cancer drugs, peptides, proteins.17 HEMA can be polymerized either by the homo-polymerization or copolymerization technique.18 Because of its biocompatibility, poly(HEMA) can act as an ideal component for various biomedical and pharmaceutical applications. Although, HEMA–different polysaccharide based hydrogels have already been developed for other purposes, to the best of our knowledge there is no report on a hydrogel based on poly(HEMA) and dextrin i.e. Dxt-g-p(HEMA). Thus, it is presumed that the grafted hydrogel derived from dextrin and poly(HEMA) may be a probable potential candidate for colon specific drug delivery of ornidazole.
Considering all these assertions and in order to improve the effectiveness of dextrin as a recipient for a colon specific drug delivery system, poly(HEMA) has been grafted on the dextrin backbone in the presence of N,N′-methylene bis acrylamide (MBA) as a cross linker. The developed hydrogel showed excellent pH dependent swelling behaviour, which favours its applicability as a colon targeted drug delivery agent. Further, the developed hydrogel is non-cytotoxic, released the enclosed ornidazole drug in a more sustained way and most importantly the drug loaded tablet is stable (∼98%) up to 3 months. To the best of our knowledge, this is the first investigation on the preparation of a dextrin and poly(HEMA) based hydrogel and on its use as carrier for colon specific ornidazole delivery.
2. Experimental
2.1 Chemicals
Dextrin was purchased from Fluka, Switzerland. 2-Hydroxyethyl methacrylate (HEMA) was procured from Alfa Aesar, Lancaster, UK. N,N′-Methylene bis acrylamide (MBA) was acquired from Loba Chemie Pvt. Ltd., Mumbai, India. Potassium persulphate (KPS) was supplied by Glaxo Smith Kline Pharmaceuticals Ltd., Mumbai, India. Acetone was obtained from E-Mark (I) Pvt. Ltd., Mumbai, India. Ornidazole was a gift sample from Endoc Pharma, Rajkot, Gujarat, India. Polyvinyl pyrrolidone (PVP-K30) was supplied by Spectrochem Pvt. Ltd., Mumbai, India. Double distilled water was used in all experimental works.2.2 Synthesis
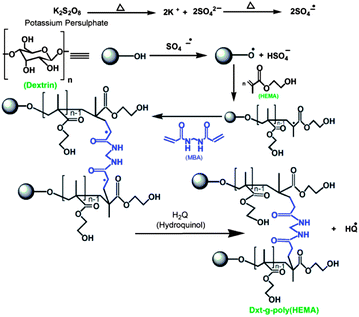 | ||
| Scheme 1 Schematic representation of the synthesis of Dxt-g-p(HEMA) hydrogel. | ||
| Hydrogel | Amount of dextrin = 0.0062 mol (i.e. 1 g) for all hydrogel syntheses | ||||||
|---|---|---|---|---|---|---|---|
| Initiator conc. (mole × 10−6) | Cross linker conc. (mole × 10−3) | Monomer conc. (mole × 10−2) | % Cross linking | Equilibrium swelling (%) | |||
| pH 1.4 | pH 6.8 | pH 7.4 | |||||
| Dxt-g-p(HEMA) 1 | 0.36992 | 0.6486 | 7.684 | 61.97 | 230 ± 7 | 255 ± 7 | 280 ± 3 |
| Dxt-g-p(HEMA) 2 | 0.92480 | 0.6486 | 7.684 | 83.42 | 175 ± 4 | 189 ± 6 | 194 ± 3 |
| Dxt-g-p(HEMA) 3 | 1.84960 | 0.6486 | 7.684 | 71.37 | 194 ± 7 | 205 ± 7 | 224 ± 7 |
| Dxt-g-p(HEMA) 4 | 0.92480 | 0.9729 | 7.684 | 87.67 | 165 ± 2 | 180 ± 1 | 186 ± 1 |
| Dxt-g-p(HEMA) 5 | 0.92480 | 1.2972 | 7.684 | 91.08 | 150 ± 3 | 174 ± 4 | 184 ± 1 |
| Dxt-g-p(HEMA) 6 | 0.92480 | 1.6215 | 7.684 | 82.08 | 181 ± 5 | 200 ± 8 | 210 ± 3 |
| Dxt-g-p(HEMA) 7 | 0.92480 | 1.2972 | 9.605 | 80.08 | 187 ± 3 | 195 ± 3 | 215 ± 2 |
2.3 Characterization
FTIR spectra of the dextrin, Dxt-g-p(HEMA) 5 hydrogel, ornidazole drug and the triturated form of the tablet were recorded using the KBr pellet method (Model IR-Perkin Elmer, Spectrum 2000). The scan range was 400 and 4000 cm−1.C, H, N analysis was performed using an Elemental Analyzer (Vario EL III, Elementar, Germany). The C, H, N analysis was carried out by the combustion process. In this procedure, the sample was burned in presence of excess oxygen, and various traps collect the combustion products: carbon dioxide, water, and nitric oxide. The masses of these combustion products can be used to calculate the composition of the unknown sample.
Solid state 13C nuclear magnetic resonance (NMR) spectra of dextrin and the hydrogel were recorded at 500 MHz on a Bruker Advance II-500NMR spectrophotometer.
Wide angle X-ray diffraction (XRD) behaviour of the hydrogel, ornidazole drug and triturated form of tablet were recorded using a Bruker X-ray diffractometer (Bruker, D8-Fecus, Germany).
The surface morphologies of neat dextrin and the hydrogel were investigated using scanning electron microscopy. After complete drying, the polysaccharide and the hydrogel were sputter-coated with gold, and the surface morphologies were analyzed by SEM (Model: S-3400N, HITACHI, Japan).
Thermogravimetric analysis of the native dextrin and the hydrogel was carried out with a TGA analyzer (Model – DTG 60, Shimadzu, Japan) in the presence of an inert atmosphere of nitrogen. The heating rate was uniform in both cases i.e. 5 °C min−1.
2.4 Measurements of equilibrium swelling capacity and swelling kinetics
The equilibrium swelling ratio (ESR) of hydrogels was assessed in pH 1.2, 6.8 and 7.4 at 37 °C. A known amount (0.5 g) of dried hydrogel was immersed in buffer and then left to swell for 10 h. The swollen hydrogel was withdrawn after every 1 h and the excess water was blotted off carefully using tissue paper and then reweighed. The equilibrium swelling was attained at ∼5 h. The % ESR has been calculated using eqn (1):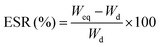 | (1) |
For swelling kinetics, water absorption of the hydrogels has been measured at consecutive time intervals until and unless equilibrium was achieved. The pH-sensitivity of the hydrogel was determined using the equilibrium swelling ratio in various buffer media. The Voigt model (eqn (2)) is used to find out the swelling rate of the hydrogel.16,19
| St = Se(1 − e−t/τ) | (2) |
2.5 In vitro cell compatibility
The powdered samples were made into pellets for assessing cell viability and proliferation. The pellets were sterilized using 70% alcohol and UV followed by washing with sterilized PBS, pH 7.4. HaCat cell line (NCCS, Pune) was cultured in 5% CO2 atmosphere at 37 °C (Heracell 150i, Thermo, USA) in DMEM (Himedia) supplemented with 10% foetal bovine serum, 1% antibiotics, 3.7% sodium bicarbonate, and 1% L-glutamine (all Himedia). The cells were harvested from tissue culture flasks using 0.25% trypsin in 1 mM EDTA (Himedia) and plated onto pellets. To seed similar cell density, cell suspension was counted using a Countess (Invitrogen, USA). 104 cells were plated on both samples and wells of a tissue culture plate (TCP).2.6 Preparation of hydrogel based tablets and in vitro drug release study
The Dxt-g-p(HEMA) hydrogel (450 mg) was finely ground in a blender, with a binder, PVP-K30 (50 mg) and ornidazole drug (500 mg). The mixture was wet with ethanol and mixed further. The paste was dried at 40 °C to a constant weight. Then a mixture of silicon dioxide and magnesium stearate (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratios) was added as a lubricant, in an amount not exceeding 3% of the ground powder. After mixing and sieving (20 meshes), tablets of 1 g each were prepared by compression in a tablet punching machine at a pressure of 2–3 t per cm2.
:1 ratios) was added as a lubricant, in an amount not exceeding 3% of the ground powder. After mixing and sieving (20 meshes), tablets of 1 g each were prepared by compression in a tablet punching machine at a pressure of 2–3 t per cm2.
During drug release, some tablets disintegrated partially. The degree of erosion (D) was calculated using eqn (3), based on the difference between the initial dry weight of the tablet (Wi) and the dry weight of the tablet (Wd(t)) at time t, considering the initial amount of drug in tablet (Wd) and fraction of drug release at time t (Mt/M∞),
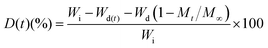 | (3) |
The in vitro release of entrapped drug (i.e. ornidazole) from the hydrogels was determined using standard Dissolution Test Apparatus (Lab India, Model: DS 8000), under a constant rotation of 60 rpm at 37 ± 0.5 °C using 900 mL simulated gastric fluid (SGF, pH 1.2) for 2 h, phosphate buffer having pH 6.8 for the next 3 h and subsequently in 900 mL of simulated intestinal fluid (SIF, pH 7.4) for a further 13 h. Aliquots were withdrawn from the release media after every 60 min and replaced by an equal volume at each sampling time. The amount of drug release was measured with the help of UV-Visible spectrophotometer (Shimadzu, Japan; Model – UV 1800).
To determine the release kinetics and mechanism of the drug release, various mathematical models were used such as the zero order kinetic model, first order kinetic model, Korsemeyar–Peppas model, Higuchi model, Hixson–Crowell model and nonlinear Kopcha model.
3. Results and discussions
3.1 Synthesis of Dxt-g-poly(HEMA) based hydrogel
Poly(HEMA) has been grafted onto dextrin backbone in the presence of KPS as initiator and MBA as cross linker in an inert atmosphere of nitrogen. An expected explanation for the formation of the graft copolymer hydrogel is based on the assumption that KPS generates free radical sites on dextrin. In the presence of cross linker MBA, because of its poly-functionality, a new macro-radical was formed that has four reactive sites20 and these sites can be linked with the radical generated on dextrin grafted poly(HEMA) backbone as shown in Scheme 1. This results in the formation of a three dimensional cross linked hydrogel. In order to observe the effect of reaction parameters, several grades of hydrogels have been prepared (Table 1) and the best one has been selected with respect to its higher % cross linking (% CE) and lower equilibrium swelling (% ESR).The cross linker concentration was varied from 0.6486 × 10−3 mol to 1.6215 × 10−3 mol (Table 1) and maximum cross linking was obtained (i.e. minimum % equilibrium swelling) with a MBA concentration of 1.2972 × 10−3 mol. This is because the higher the amount of cross linker incorporated in the hydrogel structure, the higher will be the cross linking ratio. For this reason, the hydrogel structure will be more rigid and will swell less compared to the same hydrogel with a lower cross linking ratio.14
It has been found that with an increase in monomer conc. (i.e. HEMA) from 7.684 × 10−2 mol to 9.605 × 10−3 mol, the % ESR increased, which implied that the amount of cross linking is less (Table 1). This may be because of the formation of more homopolymer (which has been separated from the hydrogel during the synthesis process) with higher monomer concentration.
3.2 Characterization of hydrogel
From the FTIR spectrum of dextrin (Fig. 1a), it has been observed that the peak at 3393 cm−1 is due to the stretching vibrations of O–H, a small peak at 2928 cm−1 is attributed to the C–H stretching vibrations. The peaks at 1026 and 931 cm−1 are assigned to the C–O–C stretching vibrations. Fig. 1b shows the FTIR spectrum of the synthesised hydrogel [Dxt-g-p(HEMA) 5]. The O–H stretching band of dextrin and the N–H stretching band of the cross linker (i.e. MBA) overlap with each other and lead to a broad peak appearing at 3464 cm−1. Two intense peaks at 1735 and 1153 cm−1 are attributed to the stretching frequency of the ester group (i.e. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O) and the peak 2952 cm−1 is due to C–H stretching of the –CH3 group, which indicates the presence of poly(HEMA) on the dextrin backbone. Two peaks at 1642 and 1491 cm−1 are due to the amide-I and amide-II bands of MBA and one peak at 1397 cm−1 indicates the presence of C–N group which verifies the existence of the cross linker (i.e. MBA) in the polymer matrix.
O and C–O) and the peak 2952 cm−1 is due to C–H stretching of the –CH3 group, which indicates the presence of poly(HEMA) on the dextrin backbone. Two peaks at 1642 and 1491 cm−1 are due to the amide-I and amide-II bands of MBA and one peak at 1397 cm−1 indicates the presence of C–N group which verifies the existence of the cross linker (i.e. MBA) in the polymer matrix.
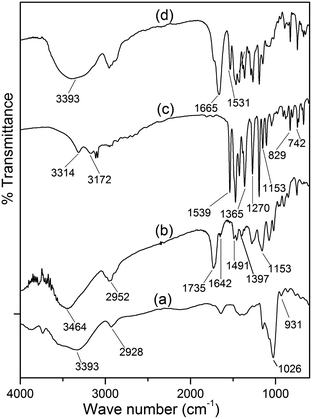 | ||
| Fig. 1 FTIR spectra of (a) dextrin, (b) Dxt-g-p(HEMA) 5 hydrogel, (c) ornidazole, and (d) triturated form of ornidazole tablet. | ||
In the FTIR spectrum of ornidazole (Fig. 1c), peaks at 3314 and 3172 cm−1 are due to the –O–H stretching mode and C–H stretching mode respectively. For asymmetric NO2 stretching, the peak appears at 1539 cm−1. The peaks at 1365–1270 cm−1 are due to the symmetric stretching mode of the NO2 group. The peak at 1153 cm−1 is due to the C–O stretching vibration. The peaks at 829 and 742 cm−1 are attributed to C–N i.e. the carbon atom connected with an NO2 group and stretching frequency of C–Cl bond vibration respectively. In the spectrum of the triturated form of the ornidazole tablet (Fig. 1d), the carbonyl group stretching frequency shifted from 1735 cm−1 to 1665 cm−1, amide-I from 1642 cm−1 to 1531 cm−1 and N–H stretching shifts from 3464 cm−1 to 3393 cm−1 which signify that the interaction between hydrogel and drug is through H-bonding as shown in Fig. 2.
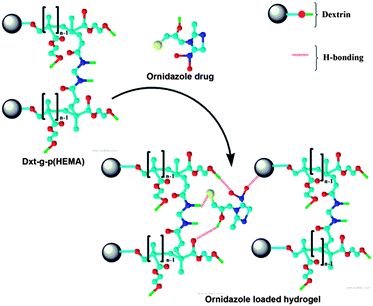 | ||
| Fig. 2 Interaction between ornidazole drug and the hydrogel. | ||
XRD analysis was carried out to investigate the drug's polymorphism after tablet formation. XRD profiles of cross linked hydrogel, ornidazole and triturated form of ornidazole tablet are shown in Fig. 3. In the case of the cross linked hydrogel (Fig. 3a), a broad characteristic peak appeared in 2θ = 18° which indicates that cross linked hydrogel is amorphous in nature. Ornidazole (Fig. 3b) showed peaks at 2θ = 13.5°, 16.2°, 21.1°, 23.2°, 24.4°, 25.8°, 27.6°, 30.7°, 32.6° because of its close molecular packing and regular crystallization structure. From the X-ray diffraction pattern of the ornidazole loaded tablet (Fig. 3c), it has been observed that the peak of cross linked hydrogel shifted towards a lower 2θ value (i.e. to 13.7°). This shifting indicates the molecular level dispersion of ornidazole drug in the hydrogel matrix.21 Also, the XRD of the tablet formulation emphasizes the existence of good compatibility between the drug and the excipients.21
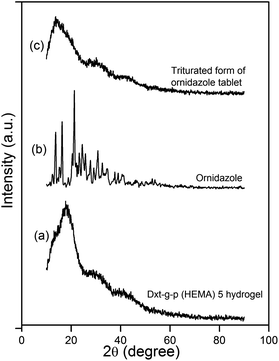 | ||
| Fig. 3 XRD profile of (a) Dxt-g-p(HEMA) 5 hydrogel, (b) ornidazole and (c) triturated form of ornidazole tablet. | ||
The CHN analyses of dextrin (% C – 39.65, % H – 6.83, % N – 0.00) and Dxt-g-p(HEMA) 5 (% C – 52.81, % H – 8.61, % N – 0.37) depict that there is substantial enhancement of amount of carbon, hydrogen and nitrogen in the hydrogel. The increase in % C and % H is because of the presence of poly(HEMA) chains in the hydrogel network. However, an increase in % N corroborates the presence of MBA as cross linker in the hydrogel structure.
The 13C NMR spectra of dextrin and Dxt-g-p(HEMA) 5 are shown in Fig. 4. The NMR spectrum of dextrin (Fig. 4a) has four characteristics peaks at δ = 111.4, 88.5, 79.4, and 68.1 ppm, which are attributed to the anomeric carbon atom, carbon atom of CH2OH group, carbon atom connected by –OH group in the six member ring except for the anomeric carbon atom (i.e. C2–C4) and C5 carbon atom respectively. In comparison to dextrin, the cross linked hydrogel has few additional peaks. The peak at 179.2 ppm correlates to the carbonyl carbons, which originate from the cross linker as well as from poly(HEMA). Peaks at 55.9 and 17.1 ppm are assigned to the carbon atom connected with –OH and methyl carbon of poly(HEMA). Another peak at 45.8 ppm can be assigned to sp3 hybridized carbon atom, which has been formed during polymerization of HEMA (i.e. two sp2 hybridized carbon atoms of HEMA monomer shifted to sp3 hybridized carbon atoms during polymerization). Thus, the NMR result confirms that poly(HEMA) has been grafted onto the dextrin backbone in the presence of MBA as cross linker.
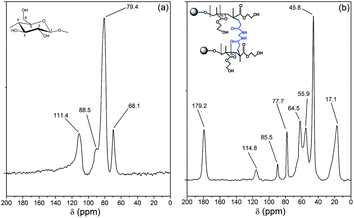 | ||
| Fig. 4 13C NMR spectra of (a) dextrin, (b) Dxt-g-p(HEMA) 5 hydrogel. | ||
The surface morphologies of dextrin and Dxt-g-p(HEMA) 5 are shown in Fig. 5. Dextrin (Fig. 5a) has a fine oval shaped granular morphology. However, after modification, the granular appearance of dextrin is distorted and the morphology of the hydrogel became porous (Fig. 5b). The incorporation of poly(HEMA) and cross linker onto dextrin backbone affects the cross link ratio as well as the structure of the hydrogel, leading to a change of microstructure of modified dextrin based hydrogel. Due to numerous interconnected pores in the hydrogel network, water molecules can easily spread and hence affect the rate of swelling. It is also expected that these pores are the regions of water permeation and interaction sites of external stimuli like pH, temperature, ionic strength etc.
 | ||
| Fig. 5 SEM micrograph (magnification × 800) of (a) dextrin, (b) Dxt-g-p(HEMA) 5 hydrogel. | ||
Thermogravimetric analysis of dextrin (Fig. 6a) and Dxt-g-p(HEMA) 5 (Fig. 6b) hydrogel explain the different thermal decomposition mechanisms. Dextrin has two different zones of weight loss. The initial decomposition may be because of the traces of moisture present. The second weight loss region (258–330 °C) is due to the degradation of the polysaccharide backbone. In contrast to dextrin, the hydrogel has two additional weight loss regions; one is in between 205 and 230 °C, due to the degradation of the presence of cross linker in the hydrogel structure and another extra zone is in the region of 350–440 °C, which is owing to the degradation of the poly(HEMA) present in the hydrogel structure. The presence of these extra zones of weight loss supports the presence of MBA as well as poly(HEMA) in the hydrogel structure.
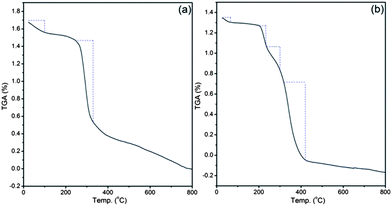 | ||
| Fig. 6 Thermogravimetric analysis of (a) dextrin, (b) Dxt-g-p(HEMA) 5 hydrogel. | ||
Cell attachment, viability and proliferation indicate cellular compatibility towards a material. An MMT reduction assay was used to determine cell viability after 1, 3, 5 days (Fig. 7). The Dxt-g-p(HEMA) 5 hydrogel as well as tissue culture plate (TCP) maintained a viable population of HaCaT cells throughout the study period (Fig. 7a). No significant differences were seen on the 1st day of cell seeding. However, after 3 days of culture, the hydrogel showed significantly higher viability than TCP.
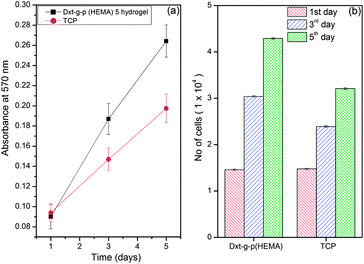 | ||
| Fig. 7 Cell viability study of Dxt-g-p(HEMA) 5 hydrogel. | ||
Thus, the synthesized material is not toxic for HaCaT cells. The absorbance values were further converted to rate of cell proliferation using a standard curve. After 5 days, the no. of cells on Dxt-g-p(HEMA) 5 hydrogel and TCP were 4.29 ± 0.18 × 104 and 3.21 ± 0.19 × 104, respectively (Fig. 7b). This may be because the hydrogel being hydrophilic in nature, swells to form a three dimensional structure that provides more surface area for cells to attach and proliferate whereas cell proliferation is restricted on a two dimensional surface like TCP.
3.3 Swelling characteristics of hydrogel
The equilibrium swelling properties of dextrin and various hydrogels were investigated at pH 1.2, 6.8 and 7.4 buffer solutions at 37 °C temperature. It has been observed that dextrin shows a declining trend of swelling with time which is because of its solubility in aqueous solution. The hydrogel attained its equilibrium swelling at ∼5 h (Fig. 8). It is obvious that the hydrogels demonstrate a faster swelling rate. This observation can be explained by the fact that the presence of poly(HEMA) and MBA in the hydrogel structure enhances the hydrophilicity of the network and facilitates the hydration as well as expansion of the network. In addition to that, the porous morphology of the hydrogel also enhances the diffusion of water into the hydrogel network. However, out of various hydrogels, Dxt-g-p(HEMA) 5 shows the lowest equilibrium swelling (Table 1 & Fig. 8) because of its maximum cross linking ratio (Table 1), which will make the hydrogel structure more rigid and hence will swell less as compared to same hydrogel having a lower cross linking ratio.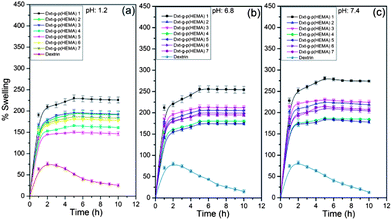 | ||
| Fig. 8 Swelling characteristic of dextrin and various hydrogels at (a) pH 1.2 (b) pH 6.8 and (c) pH 7.4 (results represented are mean ± SD, n = 3). | ||
As obvious from Fig. 8, all hydrogels show pH dependent swelling behaviour. This may be because of the fact that, at acidic pH (i.e. pH 1.2), the hydrophilic groups present in the hydrogel network get protonated which hindered the formation of H-bonding with water. This will result in less swelling compared to pH 7.4. However, at pH 7.4, all the hydrophilic groups remain free and thus able to form more H-bonding with the media. This would facile the penetration of a large no of water molecules into the hydrogel network, making the swelling higher.
| Polymer | Value of rate parameter | ||
|---|---|---|---|
| pH 1.2 | pH 6.8 | pH 7.4 | |
| Dxt-g-p(HEMA) 1 | 143.06 | 109.64 | 78.24 |
| Dxt-g-p(HEMA) 2 | 153.84 | 115.47 | 83.68 |
| Dxt-g-p(HEMA) 3 | 145.56 | 112.48 | 83.19 |
| Dxt-g-p(HEMA) 4 | 187.26 | 123.45 | 105.59 |
| Dxt-g-p(HEMA) 5 | 259.74 | 123.76 | 106.04 |
| Dxt-g-p(HEMA) 6 | 185.87 | 123.30 | 104.71 |
| Dxt-g-p(HEMA) 7 | 164.47 | 120.91 | 99.30 |
3.4 In vitro drug release studies
The cumulative percentage of ornidazole release from various hydrogels as well as from dextrin as a function of time is shown in Fig. 9.The chemical structure of the hydrogel affects the drug release behaviour.22 The release rate of the drug is directly proportional to the equilibrium swelling ratio of the hydrogels.12 The higher swelling ratio of hydrogels creates a larger surface area for diffusion of the drug from the inside of the hydrogel to the environment.12 In particular, the release of water soluble drugs from hydrogels occurs only after penetration of water into the hydrogel network, which will swell and dissolve the drug, followed by drug diffusion.12 It is obvious from Fig. 9 that the rate of drug release is higher at pH 6.8 and pH 7.4 than that at pH 1.2. This may be because of the fact that at pH 1.2, the network hydrogel is in a collapsed state, resulting in a lower rate of swelling and hence the drug was unable to diffuse from the hydrogels. However, at higher pH, the hydrogels swelled rapidly, leading to an increase in drug diffusion from the matrix. Out of various hydrogels, Dxt-g-p(HEMA) 5 showed more sustained drug release (∼68.7% release after 18 h) behaviour. Further, it has been observed that the rate of erosion (Table 3) of different hydrogels is much lower in comparison with dextrin, which also has a direct impact on the rate of drug release.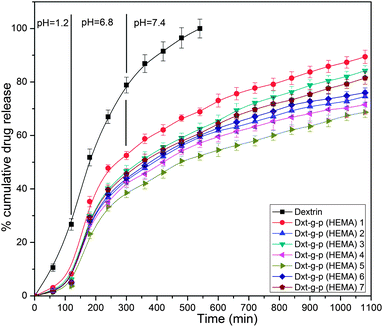 | ||
| Fig. 9 Drug release profile of dextrin and various hydrogels at pH 1.2, 6.8 and 7.4 for 2 h, 3 h and 13 h respectively (results represented are mean ± SD, n = 3). | ||
| Hydrogel | % Erosion (ornidazole tablet) | |
|---|---|---|
| pH 6.8 | pH 7.4 | |
| Dextrin (Dxt) | 76.12 ± 4.23 | 80.35 ± 5.25 |
| Dxt-g-p(HEMA) 1 | 24.01 ± 2.34 | 27.16 ± 3.20 |
| Dxt-g-p(HEMA) 2 | 19.81 ± 1.18 | 21.70 ± 3.25 |
| Dxt-g-p(HEMA) 3 | 20.06 ± 1.02 | 21.79 ± 3.12 |
| Dxt-g-p(HEMA) 4 | 16.11 ± 1.14 | 18.09 ± 2.25 |
| Dxt-g-p(HEMA) 5 | 12.61 ± 2.88 | 15.54 ± 4.32 |
| Dxt-g-p(HEMA) 6 | 17.00 ± 1.19 | 18.99 ± 2.80 |
| Dxt-g-p(HEMA) 7 | 17.89 ± 3.86 | 19.57 ± 4.25 |
Thus, the results suggested that ornidazole was released in a controlled manner from modified dextrin based hydrogels. This observation indicates that the presence of poly(HEMA) as well MBA in the hydrogel structure enhances the hydrophilicity of the network and thus causing the release of the enclosed drug in a more sustained way.
| Qt = Q0 + K0t | (4) |
On the other hand, the first order kinetic model (eqn (5)) describes the release from systems containing water soluble drugs in porous matrices, where the drug release is proportional to the amount of drug remaining in the interior of the matrix.24
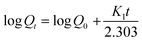 | (5) |
Here, the in vitro release of ornidazole from hydrogels was executed in various buffer solutions (Fig. 9). It has been observed that the release follows a first order kinetic model than that of zero order kinetic model (based on higher R2 value, Table S1, see ESI†).
The Korsemeyar–Peppas model (eqn (6)) is very crucial to find out the mechanism of drug release from a polymer matrix.27 This model applies up to 60% of the total amount of drug released.
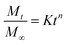 | (6) |
The Higuchi model (eqn (7)) illustrates the release of drugs as a diffusion process from an insoluble matrix based on Fick's law.28
| Qt = Q0 + KHt1/2 | (7) |
The Hixson–Crowell cube root law (eqn (8)) exemplifies the release from systems when there is a change in surface area and diameter of particles or tablets, which supports that the rate of erosion of the matrix is the main principle of drug release.29
| W1/30 − W1/3t = KHCt | (8) |
The Kopcha model (eqn (9)) is also used to quantify the relative contributions of diffusion and polymer relaxation to drug release.30
| Qt = At1/2 + Bt | (9) |
To understand the mechanism of drug release, we have used the Korsemeyar–Peppas model in both media (i.e. pH 6.8 and pH 7.4). It has been found that the ‘n’ value lies between 0.45 and 0.89 (Table S1, see ESI†), which clearly indicates that the drug release from Dxt-g-p(HEMA) based hydrogels follows a non-Fickian diffusion mechanism i.e. the drug release depends on both a diffusion process as well as a polymer relaxation i.e. erosion process.
Further it has also been observed that comparable linearity was found in both media in the case of the Higuchi model (R2 = 0.9810–0.9968 in pH 6.8, R2 = 0.9824–0.9960 in pH 7.4) (Table S1, see ESI†). Consequently release follows the Higuchi model which supports that the drug release is controlled by a diffusion process.
The release mechanism was further confirmed by calculating the diffusion exponent (A) and erosion exponent (B) derived from the nonlinear fitted Kopcha model. The calculated value of ‘A’ and ‘B’ indicates both factors (diffusion and erosion) are responsible for drug release (Table S1, see ESI†). In pH 6.8, the value of ‘A’ is greater than ‘B’ which states that in this medium drug release is quantitatively controlled by a diffusion process rather than an erosion process. On the other hand at pH 7.4, the value of both ‘A’ and ‘B’ are comparable which clarifies that the drug release is controlled by both diffusion as well as polymer relaxation.
Considering all the shortcomings of reported hydrogels as well as colon specific ornidazole delivery systems regarding biodegrability, cytotoxicity, compatibility and stability (as mentioned in introduction section), the developed Dxt-g-p(HEMA) hydrogel is novel. Use of Dxt-g-p(HEMA) for colon specific ornidazole delivery is advantageous over reported systems due to (a) excellent swelling properties, (b) non-cytotoxicity, (c) release in a more sustained way (∼68.7% even after 18 h). The last one is the most important prerequisite for pharmaceutical application, as slow release of ornidazole may prevent any severe side effects. Moreover, % of drug stability using Dxt-g-p(HEMA) hydrogel as carrier for ornidazole is ∼98% up to 3 months, which is quite promising and restricts the degradation of the drug in the stomach region.
4. Conclusions
A series of cross linked hydrogels, composed of dextrin and poly(HEMA), were synthesized via free radical polymerization technique. Through introducing poly(HEMA) onto a dextrin backbone, the porous structure of a hydrogel was achieved and the hydrophilicity was increased. Compared with pure dextrin, the hydrogels exhibited enhanced thermal stability and faster swelling rates. The results confirm that the prepared hydrogel shows good biocompatibility, being also able to effectively stimulate cell proliferation. Further, the hydrogel demonstrates excellent potential as a novel matrix for controlled release of ornidazole in colonic region in a controlled and sustained way. The drug release from dissolution media follows a first order kinetic model and non-Fickian type diffusion mechanism. Finally, the developed hydrogel is probably a better candidate for pharmaceutical applications, in particular for colon specific ornidazole release.Acknowledgements
The first author wishes to acknowledge University Grant Commission, New Delhi, India (Ref. no.19-06/2011(i) EU-IV; Sr. no. 2061110303, Dated: 30.11.2011) for providing financial assistance Under Junior Research Fellowship Scheme. The authors acknowledge the financial support from Department of Science and Technology, New Delhi, India in form of a research grant (NO:SR/FT/CS-094/2009) to carry out the reported investigation. The authors also acknowledge the kind help of Dr Animesh Ghosh, Department of Pharmaceutical Sciences, BIT, Mesra, Ranchi, India for providing drug stability study.Notes and references
- (a) T. Coviello, P. Matricardi, C. Marianecci and F. Alhaigue, J. Controlled Release, 2007, 119, 5–24 CrossRef CAS PubMed; (b) L. Pescosolido, T. Piro, T. Vermonden, T. Coviello and F. Allhaique, Carbohydr. Polym., 2011, 86, 208–213 CrossRef CAS PubMed; (c) A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- (a) P. Kurian and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1209–1217 CrossRef CAS; (b) A. Finne and A. C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1296–1305 CrossRef CAS; (c) B. V. Slaughter, S. S. Khurshid and O. Z. Fisher, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed; (d) N. A. Peppas, J. Z. Hilt and A. Khademhosseini, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- (a) M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed; (b) E. Soussan, S. Cassel and M. Blanzat, Angew. Chem., Int. Ed., 2009, 48, 274–288 CrossRef CAS PubMed; (c) L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed; (d) L. Tang, T. M. Fan, L. B. Borst and J. Cheng, ACS Nano, 2012, 6, 3954–3966 CrossRef CAS PubMed.
- (a) J. A. Killion, L. M. Geever, D. M. Devine, J. E. Kennedy and C. L. Higginbotham, J. Mech. Behav. Biomed. Mater., 2011, 4, 1219–1227 CrossRef CAS PubMed; (b) S. G. Lee, G. F. Brunello, S. S. Jang and D. G. Bucknall, Biomaterials, 2009, 30, 6130–6141 CrossRef CAS PubMed; (c) H. Shin, B. D. Olsen and A. Khademhosseini, Biomaterials, 2012, 33, 3143–3152 CrossRef CAS PubMed.
- (a) Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1652 CrossRef CAS PubMed; (b) S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5–28 CrossRef CAS PubMed; (c) B. K. Nanjawade, F. V. Manvi and A. S. Manjappa, J. Controlled Release, 2007, 122, 119–134 CrossRef CAS PubMed; (d) M. D. M. Ortega, C. A. Lorenzo, A. Concheiro and T. Loftsson, Int. J. Pharm., 2012, 428, 152–163 CrossRef PubMed; (e) C. He, L. Yin, C. Tang and C. Yin, Biomaterials, 2013, 34, 2843–2854 CrossRef CAS PubMed.
- (a) J. O. Karlsson and P. Gatenholm, Polymer, 1997, 38, 4727–4731 CrossRef CAS; (b) K. Pal, A. K. Banthia and D. K. Majumdar, Des. Monomers Polym., 2009, 12, 197–220 CrossRef CAS PubMed; (c) N. A. Peppas, P. Bures, W. Leobandung and H. Ichi, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS; (d) Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- (a) W. F. Zambuzzi, C. V. Ferreira, J. M. Granjeiro and H. Aoyama, J. Biomed. Mater. Res., Part A, 2011, 97, 375–382 Search PubMed; (b) L. Zhang, J. Rodriguez, J. Raez, A. J. Myles, H. Fenniri and T. J. Webster, Nanotechnology, 2009, 20, 175101 CrossRef PubMed.
- (a) S. S. Vaghani, M. M. Patel and C. S. Satish, Carbohydr. Res., 2012, 347, 76–82 CrossRef CAS PubMed; (b) B. Singh and M. Vashistha, Nucl. Instrum. Methods Phys. Res., Sect. B, 2008, 266, 2009–2020 CrossRef CAS PubMed; (c) P. Patel, A. Roy, V. K. SM and M. Kulkarni, Int. J. Drug Dev. Res., 2011, 3, 52–61 CAS; (d) B. Singh and N. Sharma, Colloids Surf., B, 2011, 82, 325–332 CrossRef CAS PubMed; (e) M. R. Saboktakin, R. M. Tabatabaie, A. Maharramov and M. A. Ramazanov, Int. J. Biol. Macromol., 2011, 48, 381–385 CrossRef CAS PubMed.
- D. A. Talan, K. G. Naber, J. Palou and D. Elkharrat, Int. J. Antimicrob. Agents, 2004, 23, 54–66 CrossRef PubMed.
- Y. S. R. Krishnaiah, P. R. Bhaskar Reddy, V. Satyanarayana and R. S. Karthikeyan, Int. J. Pharm., 2002, 236, 43–55 CrossRef CAS.
- T. S. Anirudhan, S. Sandeep and P. L. Divya, RSC Adv., 2012, 2, 9555–9564 RSC.
- P. Singh, R. Mittal, G. C. Sharma, S. Singh, A. Singh, In profiles of Drug Substances, and Excipients, and Related Methodology, ed. H. G. Brittain, Academic Press: New York, 2006, vol. 30, pp. 123–184 Search PubMed.
- J. K. Oh, D. I. Lee and J. M. Park, Prog. Polym. Sci., 2009, 34, 1261–1282 CrossRef CAS PubMed.
- (a) T. Asoh, T. Kaneko, M. Matsusaki and M. Akashi, J. Controlled Release, 2006, 110, 387–394 CrossRef CAS PubMed; (b) S. Geresh, J. Peisahov, J. Voorspoels, J. P. Remon and J. Kost, Biomed. Health Res., 1998, 16, 192–196 CAS; (c) S. Geresh, G. Y. Gdalevsky, I. Gilboa, J. Voorspoels, J. P. Remon and J. Kost, J. Controlled Release, 2004, 94, 391–399 CrossRef CAS PubMed.
- (a) H. Garcia, A. S. Barros, C. Gonçalves, F. M. Gama and A. M. Gil, Eur. Polym. J., 2008, 44, 2318–2329 CrossRef CAS PubMed; (b) J. Carvalho, C. Gonçalves, A. M. Gil and F. M. Gama, Eur. Polym. J., 2007, 43, 3050–3059 CrossRef CAS PubMed; (c) J. M. Carvalho, M. A. Coimbra and F. M. Gama, Eur. Polym. J., 2009, 75, 322–327 CAS; (d) E. F. Okieimen and F. Egharevba, Eur. Polym. J., 1992, 28, 415–417 CrossRef CAS; (e) S. Pal, T. Nasim, A. Patra, S. Ghosh and A. B. Panda, Int. J. Biol. Macromol., 2010, 47, 623–631 CrossRef CAS PubMed.
- V. Rana, P. Rai, A. K. Tiwari, R. S. Singh, J. F. Kennedy and C. J. Knill, Carbohydr. Polym., 2011, 83, 1031–1047 CrossRef CAS PubMed.
- (a) J. S. Belkas, C. A. Munro, M. S. Shoichet, M. Johnston and R. Midha, Biomaterials, 2005, 26, 1741–1749 CrossRef CAS PubMed; (b) O. Hossein, P. Kinam and K. U. Jose, J. Bioact. Compat. Polym., 2010, 25, 483–497 CrossRef PubMed; (c) F. M. Veronese, G. Ceriotti, G. Keller, S. Lora and M. Carenza, Int. J. Radiat. Appl. Instrum., Part C, 1990, 35, 88–96 CAS; (d) M. Carenza, S. Lora, P. Caliceti, O. Schiavon and F. M. Veronese, Radiat. Phys. Chem., 1993, 42, 897–901 CrossRef CAS; (e) B. Tasdelen, N. K. Apohan, O. Guven and B. M. Baysal, Radiat. Phys. Chem., 2005, 73, 340–345 CrossRef CAS PubMed.
- (a) G. Mabilleau, M. F. Moreau, R. Filmon, M. F. Basle and D. Chappard, Biomaterials, 2004, 25, 5155–5162 CrossRef CAS PubMed; (b) B. Vazquez, M. Gurruchaga and I. Goni, Polymer, 1995, 36, 2311–2314 CrossRef CAS.
- H. Omidian, S. A. Hashemi, P. G. Sammes and I. Meldrum, Polymer, 1998, 39, 816–823 Search PubMed.
- B. Singh, N. Sharma and N. Chauhan, Carbohydr. Polym., 2007, 69, 631–643 CrossRef CAS PubMed.
- (a) Q. Wang, Z. Dong, Y. Du and J. F. Kennedy, Carbohydr. Polym., 2007, 69, 336–343 CrossRef CAS PubMed; (b) K. Ganguly, T. M. Aminabhavi and A. R. Kulkarni, Ind. Eng. Chem. Res., 2011, 50, 11797–11807 CrossRef CAS.
- G. R. Bardajee, A. Pourjavadi and R. Soleyman, Colloids Surf., A, 2011, 392, 16–24 CrossRef CAS PubMed.
- M. Donbrow and Y. Samuelov, J. Pharm. Pharmacol., 1980, 32, 463–470 CrossRef CAS.
- M. Gibaldi and S. Feldman, J. Pharm. Sci., 1967, 56, 1238–1242 CrossRef CAS.
- S. Kiortsis, K. Kachrimanis, T. Broussali and S. Malamataris, Eur. J. Pharm. Biopharm., 2005, 59, 73–83 CrossRef CAS PubMed.
- N. A. Peppas, Pharm. Acta Helv., 1985, 60, 110–111 CAS.
- R. W. Korsemeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef.
- T. Higuchi, J. Pharm. Sci., 1961, 50, 874–875 CrossRef CAS.
- A. W. Hixson and J. H. Crowell, Ind. Eng. Chem., 1931, 23, 923–931 CrossRef CAS.
- M. Kopcha, N. G. Lordi and K. J. Toja, J. Pharm. Pharmacol., 1991, 43, 382–387 CrossRef CAS.
- WHO, Technical Report Series, no. 953, Annex. 2, 2009.
Footnote |
| † Electronic supplementary information (ESI) available: Table containing drug release kinetics parameters, stability study result. See DOI: 10.1039/c3ra44716b |
| This journal is © The Royal Society of Chemistry 2013 |