 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual protection of amino functions involving Boc
Ulf Ragnarsson*a and
Leif Grehnb
aDepartment of Chemistry-BMC, University of Uppsala, SE-751 23 Uppsala, Box 576, SE-751 23, Sweden. E-mail: ulf.ragnarsson@kemi.uu.se
bDepartment of Chemistry-BMC, University of Uppsala, SE-751 23 Uppsala, Box 576, SE-751 23, Sweden. E-mail: leif.grehn@bredband.net
First published on 17th July 2013
Abstract
Protecting groups play a pivotal role in the synthesis of multifunctional targets and as amino functions often occur in this context, issues related to their protection become prominent. Primary amines are unique because they can accommodate two such groups. This review highlights various aspects related to the synthesis, properties and applications of products containing one or two Boc-groups resulting from dual protection of amines and amides. Attention is directed towards cases of facilitated cleavage due to mutual interaction between two protecting groups on the same nitrogen.
1 Introduction
When in a synthetic project there is a need to protect an amino function, its conversion to tert-butyl carbamate is nowadays generally the first option, because of the attractive properties of the resulting so-called Boc-derivative. Boc-protection1 was introduced in the late fifties and was rapidly applied in the field of peptide synthesis. In that context Boc-derivatives nicely complemented the corresponding, then relatively novel, benzyl carbamates (Cbz-compounds) which could be deprotected by catalytic hydrogenolysis or sodium in liquid ammonia, conditions under which Boc is completely stable. Instead Boc can be cleaved by mild acidolysis, whereas benzyl carbamates require significantly stronger acidic conditions for their deprotection. Initially the preparation of Boc-amino acids was a bit cumbersome but gradually better reagents and methods became available. These and other developments and aspects related to Boc-protection in peptide synthesis have already been comprehensively documented.2 Besides, within a few years Boc-amino acids became commercially available which contributed to the fast success of this protecting group. It was also applied nearly from the outset of solid phase peptide synthesis for temporary protection of the α-amino group and continues to play an important role in this context in parallel with the Fmoc-group (Fmoc = 9-fluorenylmethyloxycarbonyl).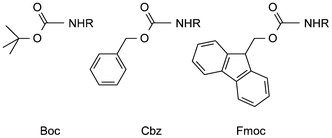
In retrospect it seems that outside the peptide field the break-through in the use of Boc came a bit later as judged from the first monographs devoted to protecting groups.3 More recently the preferences have changed and nowadays Boc is ranked as “one of the most commonly used protective groups for amines”.4 Other NH-compounds including various protected/acylated amines have also been substituted in this way that thereby give rise to bis-protected amino functions. The synthetic aspects leading to such compounds and the properties and applications of them are discussed in this paper. To the best of our knowledge this is the first review dealing with these topics.
2 Statistic perspectives
In addition to providing access to relevant previous work in the chemical literature, modern computerized chemical data banks can furnish surveys of related information available, otherwise difficult to foresee, search for or collect. This is illustrated in Table 1, in which figures on the occurrence of various classes of Boc-compounds, Boc-N-(GH)2 and Boc-N-G2, collected from Reaxys® are presented. To put things in a proper perspective, for comparison the corresponding figures for Cbz and Fmoc are also included.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000
000Presently, over 7.2 × 107 organic and inorganic compounds are described in the CAS® registry and as can be seen from Table 1 nearly 0.7% contain one of the three protecting groups mentioned. The figures further demonstrate that most of these substances, over 65%, are Boc-derivatives and that the majority contain one substituent in addition to hydrogen. The very large number of amino acid and peptide derivatives initially referred to belong to the subgroups on line 2, whereas many of the compounds with bis-protected amino functions discussed below are included among them in column 2 on the last line.
3 Synthetic background
Di-tert-butyl dicarbonate or tert-butyl pyrocarbonate, Boc2O, is an excellent reagent for the preparation of all sorts of Boc-derivatives.5 In connection with attempts to convert lactams to ω-amino acids, Grieco et al. reacted a few CO–NH-containing compounds with an excess of this reagent in the presence of stoichiometric amounts of 4-dimethylaminopyridine (DMAP) and Et3N in dichloromethane at room temperature and obtained the corresponding N-Boc-derivatives in high yields (Scheme 1).6 The CO–N(Boc) bonds could subsequently be cleaved by hydrolysis or methanolysis under remarkably mild conditions. From the acyclic substrates the corresponding acyl components were isolated.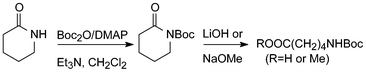
In parallel with the work of Grieco et al. the present authors performed structure–activity studies on compounds containing pyrrole 2-carboxylic acid and, to avoid the formation of cyclic dimers, attempted to protect pyrrole nitrogens by Boc.7 As the methods of Boc-protection of that time did not furnish the desired products we started searching for conditions that would allow substitution also of such non-basic NH-groups. This work resulted in a procedure also involving Boc2O/DMAP, but with catalytic amounts (10%) of DMAP, only a small excess of Boc2O and without additional base. This procedure later proved useful in a wider context.
DMAP was introduced as a catalyst in a synthetic context by Steglich and Höfle, who used it in conjunction with acetic anhydride for the esterification of sterically hindered alcohols and for the C-acylation of oxazolones.8 The effect of DMAP is rationalized in terms of increased nucleophilicity of an intermediary N-Boc-DMAP complex (Scheme 2), nucleophilic catalysis, compared with that of the anhydride.9 Simultaneously CO2 gas escapes driving the reaction towards completion. The isolation of the corresponding N-Boc-DMAP salts including the tetrafluoroborate was described by Guibé-Jampel and Wakselman10a and the spectroscopic identification of the related Boc2O/catalyst complex by Knölker et al.10b DMAP soon found wide application for catalysis of a variety of synthetic reactions and is nowadays a product of great technical and commercial interest.11
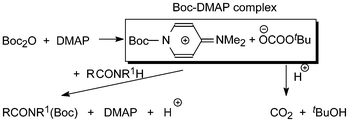 | ||
| Scheme 2 The Boc-DMAP complex and its reaction products with RCONR1H. | ||
4.1 Synthesis and properties of N-Boc-acylamides [RCONR1(Boc)]12
Formamides, acetamides and benzamides are quite stable compounds, and often require heating in strongly acidic or basic solution for their cleavage.4 Unless severely sterically hindered they generally react with a slight excess of Boc2O and catalytic amounts of DMAP under carbon dioxide evolution to the corresponding Boc-acylamides (acylcarbamates) within 1–20 h at room temperature in essentially quantitative yields.13 Their aminolysis was occasionally slow, whereas hydrazinolysis was much faster. In this way N-formyl, -acetyl and -benzoyl protecting groups could be cleaved under much milder conditions than before.14 Both steps could also be combined in a one-pot reaction procedure. Discrimination between amino functions of mixed primary/secondary amines via acylation and subsequent treatment with Boc2O/DMAP is feasible, because after the acylation step obviously only the primary groups can react. This was demonstrated by selective cleavage of N-acetyl from the primary amino groups of spermidine.15It is in the nature of amino-protecting groups that they are electron withdrawing. Substituting a hydrogen atom at an acyl nitrogen by an electron-withdrawing Boc-group can therefore be expected to decrease the electron density on nitrogen. In order to study the resulting bonding geometry, the 3D structure of two Boc-compounds of this type, one containing acetyl and one benzoyl, was determined by X-ray crystallography.16 Evidence for strong interaction of their acyl- and Boc-substituents with nitrogen was noticed in both molecules. Thus on Boc-substitution the acyl CO–N bond distances increased by 0.06–0.08 Å. Inversely, the acyl group increased the carbamate CO–N bond lengths by about the same amount compared with those in Boc-amino acids. Milder nucleophiles are therefore required to release the Boc-protected amine from acylcarbamates than for cleavage of the original amides. Similarly, strong interaction involving the benzoyl- and Boc-groups on nitrogen was also evident from cyclic voltammetry experiments.17 After Boc-substitution the electrolytic cleavage of benzoyl could be accomplished at a significantly less negative potential.
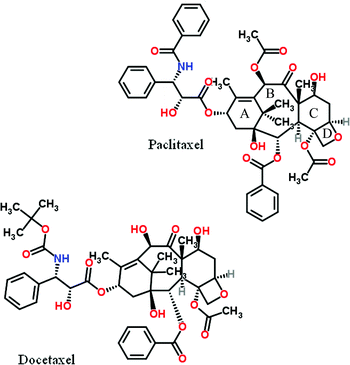
Paclitaxel is a drug of immense medicinal and commercial interest, originally isolated from the bark of the Pacific yew tree. It inhibits cell division and is therefore used in the treatment of various types of cancer. Structurally it is composed of a highly functionalized tetracyclic skeleton with one carbonyl and two free hydroxyls and another four esterified such, one of which in the A-ring notably in this context with a N-benzoylated β-amino acid sidechain. Several elegant total syntheses of the drug have been reported. A related drug of comparable importance is docetaxel in which one of the previously mentioned paclitaxel skeleton ester groups in the B-ring is replaced by a free hydroxyl and the sidechain N-benzoyl group is missing and instead substituted with a Boc-group.
A conversion of paclitaxel to docetaxel based on reaction with Boc2O/DMAP of relevance in this context has been reported but due to hydroxyl group interference rather elaborate protection of them including silylation was required prior to the N-debenzoylation step.18a Subsequently after desilylation the remaining acetate group in the B-ring that is absent in docetaxel could be cleaved.
Other relevant work dealing with the modification of a synthetic OH-protected precursor A of the paclitaxel sidechain has also been described (Scheme 3).18b After introduction of Boc on its nitrogen, first the ethoxyethoxy group of B was selectively cleaved by mild acidolysis and this was followed by benzoyl hydrazinolysis. Before oxidation of C to E could take place its free OH-group had to be acetylated.
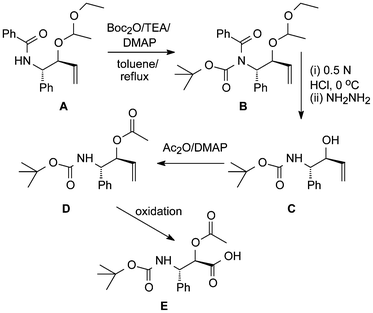 | ||
| Scheme 3 Modification of a paclitaxel sidechain precursor.18b | ||
Capitalizing on recent developments in the solid phase synthesis of peptides innumerable compounds have been prepared on polymeric supports including such of safety-catch type. To release protected, variably multisubstituted products with acyl links to a resin containing amine functions, their acyl-NH bonds were activated for cleavage by reaction with Boc2O/DMAP in order to allow subsequent hydrolysis or alcoholysis.19 After deprotection these primary products could be further modified.
Boc-derivatives of benzyl amides and amino acid esters, especially such containing benzoyl, on treatment with excess lithium diisopropylamide have been found to undergo fast acyl nitrogen to carbon migration at −78 °C to give the corresponding α-aminoketones (Scheme 4).20 An experiment with chiral N-Boc(Bz)-α-benzyl amine provided the product with 94% ee in 85% yield.
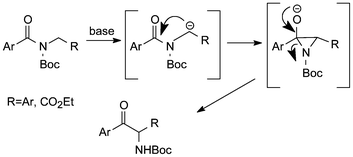 | ||
| Scheme 4 Rearrangement of N-Boc-acylamide derivatives under basic conditions.20 | ||
4.2 Synthesis and properties of N-Boc-toluene-sulfonamides [TsNR(Boc)]21
Also other amide NH-functions including more acidic ones such as those in tosylanilide and several N-benzyl benzene-sulfonamides react with Boc2O/DMAP.13,22 In fact it has been demonstrated that the reaction rate with this reagent mixture varies dramatically with the NH-acidity of the substrates and that quantitative yields can be reached in two minutes for the most acidic ones studied.23 A very useful reagent of Gabriel type for the synthesis of compounds of this kind from halides is TsNHBoc (Ts = 4-methylbenzenesulfonyl-).24 Its pKa has been determined in dimethyl sulfoxide and found to differ by about eight orders of magnitude from that of Boc2NH further discussed below.25 Therefore TsNHBoc can also be applied by alkylation with alcohols under Mitsunobu conditions.Sulfonamides are normally very stable compounds. Because of the demanding conditions required for their cleavage, for many years amino-protecting groups of this type fell into disrepute, although they could be cleaved under useful reductive conditions. Interestingly, Boc-substitution in Ts-compounds causes a considerable shift in the cleavage potentials to less negative values, indicating significant Boc/Ts interaction.17 This effect could be exploited for a rather mild, experimentally simple reductive cleavage of Boc-substituted arenesulfonamides by magnesium powder in dry methanol, only requiring sonication (Scheme 5).26 Conversion of stable Ts-NH into acid-labile Boc-NH functions in two convenient steps by treatment with Boc2O/DMAP followed by magnesium reduction is often preparatively useful.27
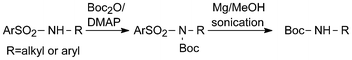 | ||
| Scheme 5 Reductive cleavage of N-Boc-arenesulfonamides with magnesium in anhydrous methanol. | ||
With the aim to develop practically useful, reductively more labile alternatives to tosyl for protection of amino functions, a number of N-arenesulfonyl-protected derivatives were prepared and studied by cyclic voltammetry.26b Among them were various 1- and 2-naphthalenesulfonamides, 1-Ns- and 2-Ns-amides, that all cleaved at significantly less negative potential than tosyl compounds. Actually both groups could be cleaved by Mg/MeOH without prior conversion to sulfonylcarbamate. It was also concluded that N-arenesulfonyl-protected derivatives that give CV peaks above −2.30 V can be cleaved by Mg/MeOH. Later the 2-Ns-analogue of the previously mentioned Gabriel reagent TsNHBoc was also made.26c
A case of debenzylation by catalytic hydrogenolysis at sulfonylated nitrogens after Boc-substitution has also been reported.28 Thus, TsNHBoc was obtained from TsNBn(Boc) in essentially quantitative yield, obviously also due to strong Boc/benzyl interaction in the substrate.
4.3 Boc2-amine derivatives [R-N(Boc)2]29
The simplest bis-Boc-derivative of this kind, di-tert-butyl imidocarbonate (Boc2NH), was made a long time ago but not until the last decades, by analogy with the text-book method of Gabriel, it has gradually replaced phthalimide as the reagent of choice in conjunction with alkyl halides.30a,b It has so far been used for the synthesis of amines in over 100 patents. Most conveniently it can be prepared in essentially quantitative yield with excess Boc2O and DMAP directly from NH4Cl via nitridotricarbonic acid tri-tert-butyl ester (Boc3N) because the compound is susceptible to nucleophiles and one Boc-group can be selectively cleaved by aminolysis.30c This procedure is ideal for the preparation of Boc215NH and has been used to make a complete set of 13C,15N-labelled Boc-glycines (Scheme 6).31 | ||
| Scheme 6 Synthesis of isotope-labelled Boc-glycines.31 | ||
By analogy with what is said in section 4.1 above, in Boc2N-derivatives one Boc-group is highly acid-labile and can be cleaved under very mild acidic conditions as exemplified by 1.5 equiv. trifluoroacetic acid32a or with catalytic amounts of a Lewis acid like Mg(ClO4)2.32b This reagent is believed to form a six-membered imidodicarbonate chelate that eliminates one molecule of isobutylene and then collapses. More recently CeCl3·7H2O,32c indium and zinc powder,32d montmorillonite K1032e and BiBr332f have also been used in this context. As Boc-derivatives of primary amines have previously been selectively alkylated, two different alkyl substituents can in principle be introduced stepwise into Boc2NH when used instead of phthalimide.33
Boc2N-compounds were originally prepared from urethanes with Boc2O/DMAP.13 For amine synthesis from halides alkylation of Boc2NH is rather convenient. Similarly, alkylation of Cbz(Boc)NH provided a product of type Cbz(Boc)N-R.34 Reacting an easily obtained Cbz-protected spermidine substrate with Boc2O/DMAP ultimately provided N8-Boc-N1-Cbz-spermidine via a derivate of this kind, i.e. full discrimination between two primary and one secondary amino functions was accomplished.35a In the same way Cbz-hydrazine served as a reagent for stepwise alkylation/acylation of hydrazine.35b,c Also chiral products of these and related types have been obtained in the presence of a proper ligand.36
The conversion of Boc-amino acids into Boc2-amino acids using Boc2O/DMAP has also been investigated.37 The procedure requires intermediary esterification but even with small ester groups occasionally the combined bulk of sidechain and protecting groups provides difficulties in the final steps. An enhanced tendency for hydantoin formation on carboxyl activation has also been noticed. In peptide synthesis the best results have been obtained in coupling reactions with the fluorides.38 This is presumably due both to their strong activation and small-size leaving group. The X-ray structure of Boc2-alanine has also been determined.39 The Boc–N bond lengths are 0.05–0.07 Å longer than in Boc-alanine and Boc-distances and bond angles deviate significantly from the normal tetrahedral values, indicating strong Boc/Boc electronic and steric interactions.
A variety of N-protected serine and threonine esters, including Boc-protected species, on treatment with two moles of Boc2O and catalytic amounts of DMAP gave rise to elimination and provided the corresponding pure N,N-diprotected dehydroamino acid esters in 87–99% yields.40 When substrates with a free carboxyl were used, the tert-butyl esters were formed in slightly lower yield (Scheme 7).
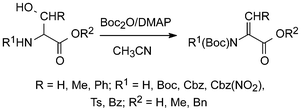 | ||
| Scheme 7 Synthesis of protected dehydroamino acids using Boc2O/DMAP.40 | ||
Like the N-Boc-N-acylamides described above also benzyl-substituted Boc2-derivatives rearrange easily in the presence of strong base providing the corresponding Boc-phenylglycine tert-butyl esters.41 With a chiral precursor products in up to 40% ee were formed dependent on the solvent.
4.4 N-Boc2-acylamides [R-CO-N(Boc)2]42
The synthesis of Boc2-acylamides (acylimidodicarbonates) was first undertaken with amino acid derivatives using Boc2O/ DMAP.43a To our surprise with 2.2 eq. Boc2O products seemed to form about as fast as the Boc2-glycine esters simultaneously studied. According to more recent work these reactions go to completion without the formation of any side-products within 24 h, thereby eliminating the need for chromatographic purification.43b These stable amide derivatives can be smoothly reduced to the corresponding alcohols with sodium borohydride (Scheme 8).43b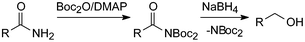
Like Boc3N above, Boc2-acylamides are susceptible to nucleophiles and they have therefore been investigated and successfully applied as acylating agents as well as cleaved by base under mild conditions (Scheme 9).44
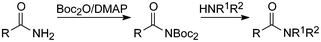
Up to now relatively few compounds of this type have been made.42
5 Scope of the Boc2O/DMAP reagent
Due to its excellent reactivity and specificity Boc2O is a superb reagent for the synthesis of derivatives of amines. Addition of the powerful nucleophilic catalyst DMAP highly increases its reactivity and is accompanied by decreases in selectivity.In addition to the products formed from amides including sulfonamides and urethanes on treatment with Boc2O/DMAP described above, isocyanates have been made from amines in this way.10b The best yields were obtained with substrates containing bulky substituents close to the amino functions that slowed down their further reaction with the tert-butanol simultaneously formed. Other reactive NH-functions include heterocyclic ones like those in pyrrole and indole.7
The reactions of Boc2O with amines and alcohols in the presence and absence of DMAP have been investigated by Basel and Hassner with respect to products and mechanisms under widely varying conditions.45 Different products were obtained depending on the ratio of reagents, polarity of the solvent and type of amine used. Without DMAP aliphatic amines gave Boc-derivatives in quantitative yields. In the presence of DMAP isocyanate formation was favoured at low temperatures, whereas otherwise in addition to Boc- and Boc2-amine also urea and carbamic-carbonic anhydrides and related substances could be isolated. It was suggested that carbamic acids could occasionally play a role as reaction intermediates. Aliphatic alcohols gave O-Boc derivatives and symmetrical carbonates in the presence of DMAP. From cysteine methyl ester in the presence of DMAP, the N-Boc-, N,S-Boc2- and N,N,S-Boc3-derivatives could all be isolated.
In a few cases Boc2O/DMAP has been reported also to give rise to products with Boc-groups bound to carbon.13,38a,46 Although occasionally forcing conditions have been used, this demonstrates the scope of the Boc2O/DMAP reagent. Bordwell et al. have investigated the reactivity of Boc2O/DMAP with substrates of different acidity.23 As already mentioned, for the most acidic substrates studied the reactions were very fast.
An early attempt to react acetanilide with dimethyl dicarbonate in the presence of DMAP was unsuccessful13 but with 4-thiazolidinone as substrate a methyloxycarbonyl derivative was obtained, although in lower yield than with Boc2O.23 The latter substrate with Cbz2O/DMAP also afforded the corresponding Cbz-protected product.23 Of other analogous reagents investigated only di-1-adamantyl dicarbonate with DMAP was preparatively useful.47
Recently, considerably more powerful catalysts than DMAP with the nitrogen(s) incorporated into rings annelated to pyridine have been developed.48 To our knowledge, however, they have not been used in conjunction with Boc2O so far.
6 Summary and conclusion
The highly increased reactivity of Boc2O/DMAP in comparison with that of Boc2O has allowed the substitution of nitrogen-bound hydrogens far beyond such in ordinary amino functions. Of particular relevance in this context are amides and carbamates, due to the fact that many amino-protecting groups are of these types. In this respect the application of Boc2O/DMAP has paved the way for dual protection of amino functions.Several classical amino-protecting groups require rather harsh conditions for their cleavage and are therefore nowadays often being abandoned in favour of more labile ones. Groups of acetyl-, benzoyl- and tosyl-type constitute typical examples. Amino functions protected in this way can all be further substituted with Boc2O/DMAP provided a further NH is present and bulky substituents in the vicinity of nitrogen do not set a limit. This usually also applies to carbamates. The products are generally crystalline solids and exhibit good stabilities with significantly increased sensitivity to nucleophiles and reducing agents, respectively, as a consequence of the additional electron-withdrawal effect of the Boc-group. Thus, at the cost of a simple additional step, selective cleavage of the original protecting group results in its replacement with Boc as demonstrated in several cases in this paper. The uniquely useful properties of this type of amine protection and its applicability in general was already outlined in the introduction.
The Boc2O/DMAP reagent has a considerable scope. Boc-substituents have also been introduced in this way on oxygen, sulphur as well as carbon atoms but so far mainly N-Boc-substitution has been explored preparatively.
Notes and references
- L. A. Carpino, J. Am. Chem. Soc., 1957, 79, 4427–4431 CrossRef CAS.
- For a review on the present state of the art of protecting groups in peptide synthesis, see: Protection of Functional Groups, in Synthesis of Peptides and Peptidomimetics, M. Goodman, A. Felix, L. Moroder and C. Toniolo, ed., Methods of Organic Synthesis (Houben-Weyl), Thieme: Stuttgart, 2002, vol. E 22a, pp. 39–423 Search PubMed.
- (a) J. F. W. McOmie, Protective groups. In Advances in Organic Chemistry. Methods and Results, R. A. Raphael, E. C. Taylor and H. Wynberg, ed., Interscience, New York, 1963, vol. 3, Chapter 4 Search PubMed; (b) J. W. Barton, Protection of N–H bonds and NR3. In Protective Groups in Organic Chemistry, J. F. W. McOmie, Ed., Plenum Press, London, 1973, Chapter 2 Search PubMed.
- P. G. M. Wuts and T. W. Greene, Greene’s Protective Groups in Organic Synthesis, 4th ed., Wiley, Hoboken, 2007, Chapter 7 Search PubMed.
- (a) Preparation of Boc2O: B. M. Pope, Y. Yamamoto and D. S. Tarbell, Org. Synth., Coll., 1988, VI, 418–421 Search PubMed and references therein; (b) For preparation of amino acid derivatives with Boc2O: L. Moroder, A. Hallett, E. Wünsch, O. Keller and G. Wersin, Hoppe-Seyler's Z. Physiol. Chem., 1976, 357, 1651–1653 CrossRef CAS; (c) Application of Boc2O: M. Wakselman, Di-tert-butyl dicarbonate. In Encyclopedia of Reagents for Organic Synthesis, L. A. Paquette, D. Crich, P. L. Fuchs and G. A. Molander, Eds; Wiley, Chichester, 2009, vol. 4, pp. 3199–3206 Search PubMed.
- D. L. Flynn, R. E. Zelle and P. A. Grieco, J. Org. Chem., 1983, 48, 2424–2426 CrossRef CAS.
- L. Grehn and U. Ragnarsson, Angew. Chem., 1984, 96, 291–292 (Angew. Chem., Int. Ed. Engl., 1984, 23, 296–301) CrossRef CAS.
- W. Steglich and G. Höfle, Angew. Chem., 1969, 81, 1001 (Angew. Chem., Int. Ed. Engl., 1969, 8, 981) CrossRef.
- G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., 1978, 90, 602–615 (Angew. Chem., Int. Ed. Engl., 1978, 17, 569–583) CrossRef and references therein.
- (a) E. Guibé-Jampel and M. Wakselman, Synthesis, 1977, 772 CrossRef; (b) H.-J. Knölker, T. Braxmeier and G. Schlechtingen, Angew. Chem., 1995, 107, 2746–2749 (Angew. Chem., Int. Ed. Engl., 1995, 34, 2497–2500) CrossRef.
- D. J. Berry, C. V. DiGiovanna, S. S. Metrick and R. Murugan, Arkivoc, 2001, 201–226 CAS.
- A Reaxys search (June 2013) for R-CO-N-R′(Boc), both R and R′ any group except hydrogen, corrected for R-N(Boc)2 below that are obviously included, gave 7800 − 5600 = 2200 hits.
- L. Grehn, K. Gunnarsson and U. Ragnarsson, Acta Chem. Scand., Ser. B, 1986, B40, 745–750 CrossRef CAS.
- L. Grehn, K. Gunnarsson and U. Ragnarsson, Acta Chem. Scand., Ser. B, 1987, B41, 18–23 CrossRef CAS and reference therein.
- M. L. S. Almeida, L. Grehn and U. Ragnarsson, Acta Chem. Scand., 1989, 43, 990–994 CrossRef CAS.
- J. Symersky, P. Malon, L. Grehn and U. Ragnarsson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 683–686 CrossRef.
- H. L. S. Maia, L. S. Monteiro, F. Degerbeck, L. Grehn and U. Ragnarsson, J. Chem. Soc., Perkin Trans. 2, 1993, 495–500 RSC.
- (a) P. G. Jagtap and D. G. I. Kingston, Tetrahedron Lett., 1999, 40, 189–192 CrossRef CAS; (b) S. Bhatia, S. Singh, R. Kumar, A. Kumar, C. E. Olsen and A. K. Prasad, Indian J. Chem., 2013, 52B, 379–386 Search PubMed.
- C. Hulme, J. Peng, G. Morton, J. M. Salvino, T. Herpin and R. Labaudiniere, Tetrahedron Lett., 1998, 39, 7227–7230 CrossRef CAS.
- O. Hara, M. Ito and Y. Hamada, Tetrahedron Lett., 1998, 39, 5537–5540 CrossRef CAS.
- A Reaxys search (June 2013) for Ts-N-R(Boc), R any group except hydrogen, gave 650 hits.
- B. Nyasse, L. Grehn, U. Ragnarsson, H. L. S. Maia, L. S. Monteiro, I. Koppel and J. Koppel, J. Chem. Soc., Perkin Trans. 1, 1995, 2025–2031 RSC.
- M. M. Hansen, A. R. Harkness, D. S. Coffey, F. G. Bordwell and Y. Zhao, Tetrahedron Lett., 1995, 36, 8949–8952 CrossRef CAS.
- J. R. Henry, L. R. Marcin, M. C. McIntosh, P. M. Scola, G. D. Harris Jr and S. M. Weinreb, Tetrahedron Lett., 1989, 30, 5709–5712 CrossRef CAS.
- I. Koppel, J. Koppel, F. Degerbeck, L. Grehn and U. Ragnarsson, J. Org. Chem., 1991, 56, 7172–7174 CrossRef CAS.
- (a) B. Nyasse, L. Grehn and U. Ragnarsson, Chem. Commun., 1997, 1017–1018 RSC; (b) B. Nyasse, L. Grehn, H. L. S. Maia, L. S. Monteiro and U. Ragnarsson, J. Org. Chem., 1999, 64, 7135–7139 CrossRef CAS; (c) L. Grehn and U. Ragnarsson, J. Org. Chem., 2002, 67, 6557–6559 CrossRef CAS.
- (a) K. Juhl, N. Gathergood and K. A. Jørgensen, Angew. Chem., 2001, 113, 3083–3085 (Angew. Chem., Int. Ed. Engl., 2001, 40, 2995–2997) CrossRef; (b) A. W. van Zijl, F. López, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2007, 72, 2558–2563 CrossRef CAS.
- D. C. Johnson II and T. S. Widlanski, Tetrahedron Lett., 2004, 45, 8483–8487 CrossRef.
- A Reaxys search (June 2013) for R-N(Boc)2, R any group except hydrogen, gave 5630 hits and for R-NCbz(Boc) 230 hits.
- (a) U. Ragnarsson and L. Grehn, Acc. Chem. Res., 1991, 24, 285–289 CrossRef CAS; (b) S. Neelamkavil, Di-tert-butyl-imidocarbonate. In Encyclopedia of Reagents for Organic Synthesis, L. A. Paquette, D. Crich, P. L. Fuchs and G. A. Molander, Eds, Wiley, Chichester, 2009, vol. 5, pp. 3226–3228 Search PubMed; (c) F. Degerbeck, L. Grehn and U. Ragnarsson, Acta Chem. Scand., 1993, 47, 896–898 CrossRef CAS.
- L. Grehn, T. Pehk and U. Ragnarsson, Acta Chem. Scand., 1993, 47, 1107–1111 CrossRef CAS.
- (a) R. D. Connell, T. Rein, B. Åkermark and P. Helquist, J. Org. Chem., 1988, 53, 3845–3849 CrossRef CAS; (b) J. A. Stafford, M. F. Brackeen, D. S. Karanewsky and N. L. Valvano, Tetrahedron Lett., 1993, 34, 7873–7876 CrossRef CAS; (c) J. S. Yadav, B. V. S. Reddy and K. S. Reddy, Synlett, 2002, 468–470 CrossRef CAS; (d) J. S. Yadav, B. V. S. Reddy, K. S. Reddy and K. B. Reddy, Tetrahedron Lett., 2002, 43, 1549–1551 CrossRef CAS; (e) J. N. Hernández, F. R. P. Crisóstomo, T. Martín and V. S. Martín, Eur. J. Org. Chem., 2007, 5050–5058 CrossRef; (f) J. Zheng, B. Yin, W. Huang, X. Li, H. Yao, Z. Liu, J. Zhang and S. Jiang, Tetrahedron Lett., 2009, 50, 5094–5097 CrossRef CAS.
- P. Dalla Croce, C. La Rosa and A. Ritieni, J. Chem. Res. (S), 1988, 346–347 CAS.
- L. Grehn and U. Ragnarsson, Collect. Czech. Chem. Commun., 1988, 53, 2778–2786 CrossRef CAS.
- (a) M. L. S. Almeida, L. Grehn and U. Ragnarsson, J. Chem. Soc., Perkin Trans. 1, 1988, 1905–1911 RSC; (b) U. Mäeorg, L. Grehn and U. Ragnarsson, Angew. Chem., 1996, 108, 2802–2803 (Angew. Chem., Int. Ed. Engl., 1996, 35, 2626–2627) CrossRef; (c) U. Mäeorg, T. Pehk and U. Ragnarsson, Acta Chem. Scand., 1999, 53, 1127–1133 CrossRef.
- O. V. Singh and H. Han, Tetrahedron Lett., 2007, 48, 7094–7098 CrossRef CAS.
- K. Gunnarsson and U. Ragnarsson, Acta Chem. Scand., 1990, 44, 944–951 CrossRef CAS.
- (a) J. Šavrda, L. Chartanova and M. Wakselman, Tetrahedron, 1994, 50, 5309–5322 CrossRef; (b) L. A. Carpino, E. M. E. Mansour and A. El-Faham, J. Org. Chem., 1993, 58, 4162–4164 CrossRef CAS.
- T. Gustafsson and K. Gunnarsson, Acta Chem. Scand., 1993, 47, 33–37 CrossRef CAS.
- P. M. T. Ferreira, H. L. S. Maia, L. S. Monteiro and J. Sacramento, J. Chem. Soc., Perkin Trans. 1, 1999, 3697–3703 RSC.
- N. Kise, H. Ozaki, H. Terui, K. Ohya and N. Ueda, Tetrahedron Lett., 2001, 42, 7637–7639 CrossRef CAS.
- A Reaxys search (June 2013) for R-CO–N(Boc)2, R any group except hydrogen, gave 85 hits.
- (a) L. Grehn and U. Ragnarsson, Angew. Chem., 1985, 97, 519–520 (Angew. Chem., Int. Ed. Engl., 1985, 24, 510–511) CrossRef CAS; (b) U. Ragnarsson, L. Grehn, L. S. Monteiro and H. L. S. Maia, Synlett, 2003, 2386–2388 CrossRef CAS.
- S. K. Davidsen, P. D. May and J. D. Summers, J. Org. Chem., 1991, 56, 5482–5485 CrossRef CAS.
- Y. Basel and A. Hassner, J. Org. Chem., 2000, 65, 6368–6380 CrossRef CAS.
- P. Hudhomme, Synlett, 2010, 1331–1332 CrossRef CAS.
- A. Könnecke, L. Grehn and U. Ragnarsson, Tetrahedron Lett., 1990, 31, 2697–2700 CrossRef.
- (a) M. R. Heinrich, H. S. Klisa, H. Mayr, W. Steglich and H. Zipse, Angew. Chem., 2003, 115, 4975–4977 (Angew. Chem., Int. Ed. Engl., 2003, 42, 4826–4828) CrossRef; (b) R. Tandon, T. Unzner, T. A. Nigst, N. De Rycke, P. Mayer, B. Wendt, O. R. P. David and H. Zipse, Chem.–Eur. J., 2013, 19, 6435–6442 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |