Pseudomonas putida KT2440 response to nickel or cobalt induced stress by quantitative proteomics†
Prasun Rayab, Vincent Girardab, Manon Gaultab, Claudette Jobab, Marc Bonneuc, Marie-Andrée Mandrand-Berthelotab, Surya S. Singhd, Dominique Jobab and Agnès Rodrigue*ab
aUniversité de Lyon, Lyon, F-69003, INSA Lyon, Université Lyon 1, Villeurbanne, F-69621, France
bMicrobiologie, Adaptation et Pathogénie, UMR 5240 CNRS-UCBL-INSA-BCS, Bat. Lwoff, 10 rue Dubois, Villeurbanne Cedex, F-69622, France. E-mail: agnes.rodrigue@insa-lyon.fr; Fax: +33 472431584; Tel: +33 472447980
cPlate-forme Protéomique, Université Bordeaux 2, Bordeaux, France
dDepartment of Biochemistry, Osmania University, Hyderabad, India
First published on 28th November 2012
Abstract
Nickel and cobalt are obligate nutrients for the gammaproteobacteria but when present at high concentrations they display toxic effects. These two metals are present in the environment, their origin being either from natural sources or from industrial use. In this study, the effect of inhibitory concentrations of Ni or Co was assessed on the soil bacterium Pseudomonas putida KT2440 using a proteomic approach. The identification of more than 400 spots resulted in the quantification of 160 proteins that underwent significant variations in cells exposed to Co and Ni. This analysis allowed us to depict the cellular response of P. putida cells toward metallic stress. More precisely, the parallel comparison of the two proteomes showed distinct responses of P. putida to Ni or Co toxicity. The most striking effect of Co was revealed by the accumulation of several proteins involved in the defense against oxidative damage, which include proteins involved in the detoxification of the reactive oxygen species, superoxides and peroxides. The up-regulation of the genes encoding these enzymes was confirmed using qRT-PCR. Interestingly, in the Ni-treated samples, sodB, encoding superoxide dismutase, was up-regulated, indicating the apparition of superoxide radicals due to the presence of Ni. However, the most striking effect of Ni was the accumulation of several proteins involved in the synthesis of amino acids. The measurement of the amount of amino acids in Ni-treated cells revealed a strong accumulation of glutamate.
Introduction
Nickel and cobalt are trace elements widely utilized by organisms. In gammaproteobacteria, comparative genomic analyses showed that the majority of sequenced bacteria of this phylum are equipped with genes involved in the metabolism of Ni or Co.1 Ni enzymes fulfill nine different biological processes, the best characterized ones being hydrogenase and urease.2 Co is either a co-factor in itself or utilized by enzymes in the form of cobalamin (vitamin B12).3 A set of accessory proteins is necessary to ensure proper metal concentration within the cell. These are specific uptake systems, chaperones, regulatory proteins and efflux systems. Uptake systems as well as efflux pumps often transport both Ni and Co. These are for example the importer proteins of the NiCoT family exemplified by HoxN4 or the efflux pumps of the RND or RcnA families.5,6 By reason of their close chemical properties Ni and Co are often bound by the same protein. Ni and Co are present in the natural environment. Ni is found as a major constituent of serpentine soils for instance. It is admitted that both metals occur jointly in the environment.7 They are used in the metallurgical industry for the production of stainless steel for instance. Due to their extensive use, considerable amounts of Ni or Co are present in the aquatic environment or in biological wastewater treatment plants and are retrieved in sewage sludge or solid waste composts.8Pseudomonas putida is a ubiquitous gram-negative soil bacterium belonging to the gamma class of Proteobacteria. Genomic analysis revealed that P. putida KT2440 harbours a large variety of genes predicted to be involved in metal homeostasis or resistance9 with a great proportion of genes being present in genomic islands,10 inferring that this bacterium thrives in heavy metal contaminated environments. However, the molecular mechanisms of metal tolerance or resistance are poorly described in this bacterium.
The harmful effects of Ni and Co in prokaryotes have been understudied and still need to be investigated. The deleterious effect of Co seems to be due mainly to the inactivation of iron–sulfur clusters present in enzymes and to the inhibition of the Fe–S cluster assembly process.11 The aim of the present study was to analyze the cell response to inhibitory concentrations of Ni or Co in P. putida KT2440 by quantitative proteomics. The parallel investigation of the effect of the two metals allowed us to unveil the specific response to each metal.
Material and methods
Bacterial strain and culture conditions
P. putida KT2440 was cultivated in M63 minimal medium supplemented with 0.4% succinate as the carbon source. For the estimation of bacterial doubling time cells were grown in 250 mL Erlenmeyer flasks with 25 mL medium without metal or amended with 75 μM NiSO4 or 35 μM CoCl2 with orbital shaking (150 rpm) at 30 °C. Cell growth was monitored based on culture optical density at 600 nm (A600). The doubling time was estimated by the equation: g = (log10Nt − log10N0)/log102, where Nt = natural log of the number of cells (A600) at time t and N0 = natural log of the number of cells (A600) at time zero.Protein sample preparation
Bacteria were grown under the above conditions and were harvested in the logarithmic growth phase (A600 ≈ 2.00). Cell pellets were obtained by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 15 min). The pellets were briefly washed in 10 mM tris-HCl (pH 8.0) and were lysed by three passages in a French press at 1000 psi. The debris were removed by centrifugation (10000 × g for 15 min), and the lysate was centrifuged at 30000 × g for 2 h to separate the soluble and membrane fractions. The soluble proteins were quantified by the standard Bradford assay (BioRad). Bovine serum albumin was used as a standard. The protein samples were precipitated (TCA/acetone) and stored frozen at −80 °C.
000 × g for 15 min). The pellets were briefly washed in 10 mM tris-HCl (pH 8.0) and were lysed by three passages in a French press at 1000 psi. The debris were removed by centrifugation (10000 × g for 15 min), and the lysate was centrifuged at 30000 × g for 2 h to separate the soluble and membrane fractions. The soluble proteins were quantified by the standard Bradford assay (BioRad). Bovine serum albumin was used as a standard. The protein samples were precipitated (TCA/acetone) and stored frozen at −80 °C.Two-dimensional gel electrophoresis
An amount of 25 μg of protein was used for analytical gels, while an amount of 40 μg of protein was used for the preparative gels. Isoelectric focusing (IEF) of protein extracts in rehydratation buffer (8 M urea, 4% [w/v] CHAPS, 0.5% [v/v] IPG buffer, pH 3–10, 40 mM DTT, and 0.01% [w/v] bromophenol blue) was run using 24 cm immobilized pH gradient (3–10 nonlinear) IPG strips on the PROTEAN® IEF system (BIORAD). Strips were rehydrated for 20 h at 22 °C with the thiourea/urea lysis buffer containing 40 mM DTT and the protein extracts. Isoelectrofocusing was performed at 20 °C for 30 min at 100 V, 30 min at 250 V, 30 min at 1000 V, and 5 h at 7000 V.12 IEF strips were equilibrated for 2 × 20 min in 2 × 100 ml of equilibration solution containing 6 M urea, 37.5% (w/v) glycerol, 2.5% (w/v) SDS and tris-HCl (0.04 M pH 8.8). DTT 2% (w/v) was added to the first equilibration solution and iodoacetamide 2.5% (w/v) was added to the second.Equilibrated gel strips were placed on top of vertical 12% polyacrylamide gels. A denaturing solution containing 1% (w/v) low-melting agarose, 0.03% (w/v) Trizma-base, 1.4% (w/v) glycine, and 0.25% [w/v] SDS was loaded on gel strips. After agarose solidification, electrophoresis was performed at 10 °C in Laemmli buffer (pH 8.3), for 1 h at 35 V and 110 V overnight. Ten gels were run in parallel (Isodalt system from Amersham Pharmacia Biotech).12 For each condition, analyzed 2-D gels were made at least in triplicate and for a minimum of three independent extractions.
Protein staining and gel analyses
2-D gels were stained with silver nitrate13 using the Hoefer Automated Gel stainer apparatus from Amersham Pharmacia Biotech. Analytical silver-stained gels were scanned with the Sharp JX-330 scanner equipped with the Labscan version 3.00 from Amersham Pharmacia Biotech. After spot detection and background subtraction (mode: average on boundary), 2D gels were compared, matched, and the quantitative determination of the spot volumes was performed (mode: total spot volume normalization). For each analysis, statistical data showed a high level of reproducibility between normalized spot volumes of gels produced in triplicate. Data were subjected to statistical analysis by using Student's t test (α = 0.05).12The quantitative results herein described have been obtained from 494 spots for which a systematic quantification was performed, based on spot intensity, by comparing different conditions (without metal or amended with 75 μM Ni or 35 μM Co) with three gels per condition. For calibrating molecular weight and pI scales of 2D gels, commercial standard (GE, Serva and Pierce) was used.
In-gel digestion of proteins and sample preparation for MS analysis: data acquisition and database searching
For protein identification, 379 spots showing different levels of intensity were excised from 2D gels manually. Gel pieces were destained using the PROTSIL2 kit (Sigma-Aldrich, St. Louis, MO). Spots were rinsed twice in ultrapure water and shrunk in acetonitrile (ACN) for 10 min. After ACN removal, gel pieces were dried in a vacuum centrifuge and rehydrated in 10 ng μL−1 trypsin solution (Sigma-Aldrich) in 50 mM ammonium bicarbonate and incubated overnight at 37 °C. Hydrophilic peptides were extracted with 40 mM ammonium bicarbonate containing 10% (v/v) ACN at room temperature for 10 min. Hydrophobic peptides were extracted with 47.0% ACN, 5% (v/v) formic acid and the extraction step was repeated twice. All three supernatants were pooled together, concentrated in a vacuum centrifuge, and acidified with 0.1% formic acid.14 For MALDI TOF MS/MS analyses, a volume of 0.5 μL of each sample was spotted onto a Prespotted AnchorChip target plate (Part No. 231968, Bruker) according to the manufacturer's instructions. Mass spectrometry analyses were performed on an Ultraflex III TOF/TOF (Bruker, Bremen, Germany) in positive ion reflector mode.14 Mass spectra were acquired over the m/z range 450–5000 and were subsequently externally calibrated against adjacent spots prespotted with the peptide calibration mixture (PAC_PeptideCalibStandard, Bruker). 1500 and 300 shots were usually accumulated for MS and MS/MS spectra, respectively. MS/MS precursor ions were selected depending on Peptide Mass Fingerprint results (see below): in the case of PMF success, three out of the most intense ions were selected for MS/MS confirmation and five unidentified ions were selected for MS/MS exploration. In the case of PMF failure, MS/MS experiments were performed on the eight most intense ions from the MS spectra. For LC-ESI-MS/MS analyses, peptide mixtures were analyzed using an on-line capillary nano-HPLC (LC Packings, Amsterdam, Netherlands) coupled to a nanospray LCQ Deca XP ion trap mass spectrometer14 (ThermoFinnigan, San Jose, CA, USA). Peptide digests (10 μL) were loaded onto a 300 μm inner diameter × 5 mm C18 PepMapTM trap column (LC Packings, Amsterdam, Netherlands) at a flow rate of 30 μL min−1. The peptides were eluted from the trap column onto an analytical 75 μm inner diameter × 15 cm C18 PepMapTM column (LC Packings, Amsterdam, Netherlands) with a 5–40% linear gradient of solvent B in 30 min (solvent A was 0.1% formic acid in 5% ACN, and solvent B was 0.1% formic acid in 80% ACN). The separation flow rate was set at 200 nL min−1.14 The mass spectrometer operated in positive ion mode at a 2 kV needle voltage and a 3 V capillary voltage. Data acquisition was performed in a data-dependent mode alternating in a single run, a MS scan survey over the m/z range 150–2000, a zoom scan and a MS/MS scan of the most intense ion in the survey scan. MS/MS spectra were acquired using a 2 m/z unit ion isolation window at 35% relative collision energy and 0.5 min dynamic exclusion duration.PMF spectra were searched with MASCOT 2.2 software (http://www.matrixscience.com) against the P. putida KT2440 JCVI database (5437 entries). Monoisotopic mass tolerance was set at 50 ppm, one trypsin-missed cleavage was allowed, and oxidation of methionine was considered as a variable modification and carbamido-methylation of cysteine as a fixed modification. A minimal Mascot score of 54 was set for protein identity validation. A tolerance of 0.7 Da in MS/MS mode was also considered for MS/MS spectra searches. Proteins were validated as soon as two different peptides were validated (individual ion scores >20).14 Peptides from LC-MS/MS spectra were identified with the SEQUEST algorithm through Bioworks 3.3.1 interface (Thermo-Finnigan, Torrence, CA, USA) against the same P. putida KT2440 database used in MASCOT. DTA generation allowed the averaging of several MS/MS spectra corresponding to the same precursor ion with a tolerance of 1.4 Da. Spectra from precursor ions higher than 3500 Da or lower than 600 Da were rejected. The search parameters are as follows: mass accuracy of the peptide precursor and peptide fragments was set to 2 and 1 Da, respectively. Only b- and y-ions were considered for mass calculation. Oxidation of methionines and carbamidomethylation of cysteines were considered as differential modifications. Two missed trypsin cleavages were allowed. Trypsic peptides were validated using the following criteria (DeltaCN ≥ 0.1, Xcorr ≥ 1.9 (single charge), 2.2 (double charge), 3.75 (triple charge), peptide probability ≤ 0.001). A minimum of two different peptides was considered for protein validation.14
Amino acids assay
We used the Agilent method for HPLC analysis of amino acids.15 Chromatographic analysis was achieved with an HPLC 1100 series equipped with a quaternary pump module (G1311A), an automatic sampler (G1329A) and a Diode Array Detector (DAD G1315B) (Agilent Technologies). Before injection, 1 μl of samples was online-derivatized using orthophtalaldehyde (OPA) and 9-fluorenylmethyl chloroformate (FMOC). The separation was carried out at room temperature using a Zorbax Eclipse-AAA Column (4.6 × 150 mm, 3.5 μm, Agilent Technologies). The column was eluted at 2 ml min−1, with an optimized gradient established using solvents A (40 mM Na2HPO4 pH 7.8) and B (acetonitrile:methanol:water (45:45:10, v/v/v)). The used gradient was an increase of solvent B (0% to 57% in 8.8 min), then an increase of solvent B (57% to 100% in 0.2 min), and then isocratic conditions for 2 min. 3D data were recorded and specific wavelengths at 262 nm (secondary amino acids) and 338 nm (primary amino acids) were chosen for processing. The Chemstation Agilent software was used for integration and comparison of chromatograms. Each amino acid was quantified at an appropriate wavelength with a specific calibration curve.cDNA synthesis and real-time PCR
Total RNA were isolated with the cold phenol method. Briefly, the bacteria were grown in M63 minimal medium or supplemented with 75 μM NiCl2 or 35 μM CoCl2. Cells were broken mechanically in the presence of glass beads in a Fast Prep (MP) apparatus. RNA was extracted twice using a (25:24:1) mix of phenol/chloroform/isoamyl alcohol. RNA was ethanol precipitated and treated twice with DNase (Ambion) for 30 min at 37 °C. The absence of DNA contamination was verified by direct PCR. RNA was quantified by measuring the optical density at 260 nm (OD260), and its integrity was confirmed by agarose gel electrophoresis. RNA was reverse-transcribed using the RevertAid kit (Fermentas). Real-time PCR experiments were performed using the MasterPLUS SYBRGreen I kit (Roche Applied Science). The 16S RNA gene was chosen as reference gene for data normalization. Amplification and detection of the specific products were carried out with the LightCycler System, and data analysis was performed with the Lightcycler Relative Quantification software (Roche Applied Science). Relative expression was calculated as the ratio of the normalized value of each sample relative to that of the corresponding untreated cells according to ref. 16.Results and discussion
Ni or Co effect on the growth of P. putida
To assess the biological impact of challenging concentrations of Ni or Co on the growth of P. putida KT2440, the bacteria were grown in succinate minimal medium supplemented with increasing concentrations of one or the other metal added from the start of the culture. As expected, increasing concentrations of metals led to decreased cell growth. Under these conditions, Minimal Inhibitory Concentrations of 200 μM and 100 μM were obtained for Ni and Co respectively (Fig. S1, ESI†). For the present study, the chosen concentrations of Ni and Co were the ones resulting in an increase of about 30% of the average doubling time as compared to the growth in the absence of metal. In succinate supplemented minimal medium, the average doubling time measured was 100 min (Fig. 1). When 75 μM Ni or 35 μM Co were present, doubling times of 130 min and 140 min were measured respectively (Fig. 1). These doses, which are moderately cytotoxic, might activate both the specific cellular response as well as more general stress pathways. Interestingly, the parallel analysis of Ni or Co-treated proteome should give access to both types of information.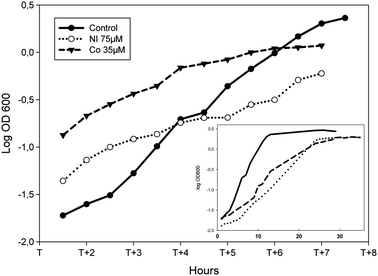 | ||
| Fig. 1 Effect of Ni or Co on the growth of P. putida KT2440. Logarithmic representation of growth curves during the logarithmic phase of cells grown in minimal medium supplemented with 0.4% succinate (closed dots, plain line), amended with 75 μM NiCl2 (open dots, dotted line) or 35 μM CoCl2 (closed triangles, dashed line). X axis: T corresponds to the time the bacteria enter into logarithmic growth phase. Inset: the same growth curves are shown from the start of the culture to the late stationary phase. | ||
2-DE
The bacteria were grown in the above-referred concentrations, 75 μM Ni or 35 μM Co, until late logarithmic phase (OD600 ≈ 1.00) and the soluble fraction of the total proteome was collected and analyzed by 2-DE. 2D-gels of protein extracts from cells grown in the absence of metal were compared with those from cells grown in the presence of 75 μM NiCl2 or 35 μM CoCl2. According to the cumulative analysis of various gels, the software Labscan determined approximately 580 spots (Fig. 2). After PMF or LC-MS analysis, 483 proteins were identified.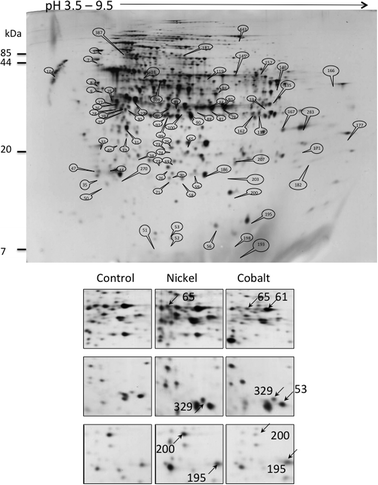 | ||
| Fig. 2 2-DE analysis of the P. putida proteome. Upper panel: total gel image of a 12 h culture soluble proteome of P. putida KT2440 grown in minimal medium supplemented with succinate. Lower panel: proteome comparison after growth in the presence of 75 μM Ni or 35 μM Co. Examples of spots variation are shown. Spots 61: PP_0018, 65: PP_4185, 53: PP_4730, 195: PP_5156, 200: PP_4981, 329: PP_5414. | ||
Protein identification and annotation
Finally, the quantification of 160 different proteins was achieved that correspond to the analysis of the spots containing only one protein. 41 up-regulated and 72 down-regulated proteins in the Co-treated samples and 54 up-regulated proteins and 46 down-regulated proteins in the Ni-treated proteome constituted the dataset (Fig. 3, Tables 1 and 2). Quite interestingly, the set of commonly regulated proteins included 17 up-regulated and 36 down-regulated proteins (Table 3). Protein identification was made according to the JCVI P. putida KT2440 database (version 1.2). Annotation was achieved using a combination of information from Swissprot (and linked databases) and additional elements from the literature. The proteins were sorted manually into 11 major functional classes (Fig. 3 and Tables 1–3).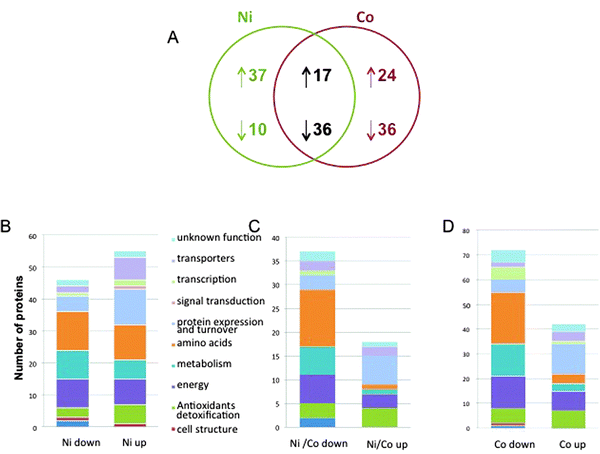 | ||
| Fig. 3 P. putida proteome response to Ni and Co. (A) Venn diagram of common and unique sets of proteins expressed in cells exposed to Ni and Co. The diagram indicates the number of proteins whose synthesis was increased (upward arrow) or decreased (downward arrow) after metal exposure. (B) Functional classification of the Ni-specific proteome. (C) Functional classification of the Ni and Co common proteome. (D) Functional classification of the Co-specific proteome. | ||
| Spot number | Co/Ctrla | Accession no | Protein (gene) [EC]b | Functional classification | PI | MW, kDa |
|---|---|---|---|---|---|---|
| a Ratio of protein abundance in the Co-treated cells to that in cells without metal.b Where available, the name of putative homologous gene is indicated in parentheses and the enzyme commission number is given in square brackets.c Transporters class: in this study the soluble proteome of P. putida was analyzed, which explains why transporter proteins were not identified apart from Periplasmic Binding Proteins (PBP) of the ABC transporter family that are soluble proteins. | ||||||
| 305 | 0.43 | PP_1210 | Ferritin Dps family protein | Antioxidants, detoxification | 5.2 | 17.8 |
| 352 | 3.72 | PP_0915 | Fe superoxide dismutase (sodB) [1.15.1.1] | Antioxidants, detoxification | 5.5 | 21.9 |
| 357 | 12.51 | PP_1859 | Organic hydroperoxide resistance protein (ohr) | Antioxidants, detoxification | 6.28 | 14.8 |
| 354 | 16.34 | PP_5215 | Thioredoxin (trx-2) | Antioxidants, detoxification | 4.9 | 11.7 |
| 82 | 3.10 | PP_0842 | Cysteine desulfurase (iscS-1) [2.8.1.7] | Antioxidants, detoxification | 5.85 | 44.4 |
| 213 | 0.10 | PP_0786 | Thioredoxin reductase (trxB) [1.8.1.9] | Antioxidants, detoxification | 5.22 | 34.0 |
| 37 | 2.44 | PP_1638 | Ferredoxin-NADP reductase (fpr) [1.18.1.2] | Antioxidants, detoxification | 5.22 | 29.7 |
| 75 | 0.36 | PP_4646 | Ferredoxin-NADP reductase (fprB) | Antioxidants, detoxification | 5.38 | 28.9 |
| 113_337 | 1.47 | PP_5415 | ATP synthase F1. Alpha subunit (atpA) [3.6.3.14] | Energy | 5.38 | 55.5 |
| 324 | 0.22 | PP_0072 | Quinone oxidoreductase (qor-1) [1.6.5.5] | Energy: E-transfer | 8.8 | 34.2 |
| 24 | 1.99 | PP_5085 | Malic enzyme (maeB) [1.1.1.40] | Energy: glyoxylate cycle | 5.09 | 45.4 |
| 148 | 0.45 | PP_1379 | 3-Carboxy-cis.cis-muconate cycloisomerase (pcaB) [5.5.1.2] | Energy: protocatechuate degradation | 6.08 | 48.4 |
| 341 | 4.79 | PP_2334 | Methylisocitrate lyase (prpB) [4.1.3.30] | Energy: TCA pathway | 5.4 | 31.8 |
| 349 | 1.82 | PP_2335 | Methylcitrate synthase [2.3.3.5] | Energy: TCA pathway | 6.3 | 42.0 |
| 125 | 0.11 | PP_0944 | Fumarate hydratase. Class II (fumC-1) [4.2.1.2] | Energy: TCA pathway | 5.65 | 48.6 |
| 246_509 | 0.51 | PP_4012 | Isocitrate dehydrogenase, NADP-dependent, monomeric-type [1.1.1.42] | Energy: TCA pathway | 5.44 | 80.1 |
| 132 | 0.61 | PP_4187 | 2-Oxoglutarate dehydrogenase, lipoamide dehydrogenase component (lpdG) [1.8.1.4] | Energy: TCA pathway | 5.93 | 50.1 |
| 284 | 0.16 | PP_4186 | Succinyl-CoA synthetase, beta subunit (sucC) [6.2.1.5] | Energy: TCA pathway | 5.83 | 41.5 |
| 312 | 0.36 | PP_3365 | Acetolactate synthase, catabolic | Metabolism: energy/amino acids/nucleotides | 5.75 | 60.3 |
| 85 | 0.33 | PP_0671 | Serine hydroxymethyltransferase (glyA-2) [2.1.2.1] | Metabolism: amino acids | 5.85 | 45.1 |
| 276 | 0.08 | PP_1471 | Threonine synthase (thrC) [4.2.99.2] | Metabolism: amino acids | 5.86 | 52.0 |
| 320 | 0.22 | PP_0817 | Aminotransferase, Class I | Metabolism: amino acids | 8.7 | 45.0 |
| 212 | 0.21 | PP_1237 | Dihydrodipicolinate synthase (dapA) [4.2.1.52] | Metabolism: amino acids | 5.73 | 31.9 |
| 433 | 0.25 | PP_2371 | Sulphite reductase hemoprotein, beta subunit (cysI) [1.8.1.2] | Metabolism: amino acids | 5.5 | 62.2 |
| 321 | 0.52 | PP_4473 | Aspartate kinase (lysC) [2.7.2.4] | Metabolism: amino acids | 5.25 | 44.7 |
| 211 | 0.37 | PP_4571 | Cysteine synthase A (cysK) [4.2.99.8] | Metabolism: amino acids | 5.55 | 34.5 |
| 269 | 0.20 | PP_4678 | Ketol-acid reductoisomerase (ilvC) [1.1.1.86] | Metabolism: amino acids | 5.48 | 36.6 |
| 314 | 0.22 | PP_4680 | Acetolactate synthase. Large subunit. Biosynthetic type (ilvB) [4.1.3.18] | Metabolism: amino acids | — | 62.8 |
| 342 | 8.16 | PP_2036 | Dihydrodipicolinate synthase | Metabolism: amino acids | 5.3 | 31.3 |
| 353 | 4.03 | PP_3778 | Pyrroline-5-carboxylate reductase | Metabolism: amino acids | 5.8 | 27.7 |
| 345 | 3.77 | PP_5155 | D-3-phosphoglycerate dehydrogenase (serA) [1.1.1.95] | Metabolism: amino acids | 5.9 | 44.3 |
| 147 | 0.32 | PP_1362 | Pyruvate kinase II (pykA) [2.7.1.40] | Metabolism: glycolysis | 6.22 | 52.0 |
| 223 | 0.28 | PP_1915 | Acyl carrier protein (acpP) | Metabolism: lipid and sterol | 4.11 | 8.7 |
| 234 | 0.20 | PP_0558 | Acetyl-CoA carboxylase, biotin carboxylase (accC-1) [6.4.1.2] | Metabolism: lipid and sterol | 6.09 | 49.5 |
| 328 | 0.24 | PP_4175 | 3-Oxoacyl-(acyl-carrier-protein) synthase I (fabB) [2.3.1.41] | Metabolism: lipid and sterol | 5.3 | 43.2 |
| 471 | 0.56 | PP_1457 | Phosphoribosylglycinamide formyltransferase 2 (purT) [2.1.2.-] | Metabolism: nucleotides | 5.61 | 42.8 |
| 264 | 0.24 | PP_1610 | CTP synthase (pyrG) [6.3.4.2] | Metabolism: nucleotides | 5.61 | 59.8 |
| 418 | 2.15 | PP_1179 | Ribonucleoside reductase, alpha subunit (nrdA) [1.17.4.1] | Metabolism: nucleotides | 5.5 | 106.9 |
| 225 | 0.24 | PP_0776 | Peptidyl-prolyl cis-trans isomerase, FKBP-type (slyD) | Protein destination and storage: folding and stability | — | 17.2 |
| 47 | 1.89 | PP_0684 | Peptidyl-prolyl cis-trans isomerase, FKBP-type (fklB-1) [5.2.1.8] | Protein destination and storage: folding and stability | 4.64 | 21.7 |
| 12_13 | 1.31 | PP_1361 | Chaperonin, 60 kDa (groEL) | Protein destination and storage: folding and stability | 4.97 | 56.7 |
| 8 | 1.92 | PP_2299 | Trigger factor (tig) [5.2.1.8] | Protein destination and storage: folding and stability | 4.81 | 48.5 |
| 329 | 0.32 | PP_4874 | Ribosomal protein L9 (rplI) | Protein synthesis: ribosomal proteins | 5.3 | 15.5 |
| 515 | 4.00 | PP_0469 | Ribosomal protein L6 (rplF) | Protein synthesis: ribosomal proteins | 9.73 | 19.1 |
| 559 | 2.83 | PP_1594 | Ribosome recycling factor (frr) | Protein synthesis: translation factors | 7.88 | 20.1 |
| 250_335 | 1.59 | PP_1213 | Aspartyl-tRNA synthetase (aspS) [6.1.1.12] | Protein synthesis: tRNA synthetases | 5.34 | 66.9 |
| 226 | 0.21 | PP_2084 | Regulator of ribonuclease activity A (rraA) | Transcription: RNA fate | — | 17.6 |
| 372_374 | 0.69 | PP_4708 | Polyribonucleotide nucleotidyltransferase (pnp) [2.7.7.8] | Transcription: mRNA processing | 5 | 75.0 |
| 406–412 | 0.26 | PP_0447 | DNA-directed RNA polymerase, beta subunit (rpoB) [2.7.7.6] | Transcription: mRNA synthesis | 5.57 | 151.5 |
| 308 | 3.40 | PP_0479 | DNA-directed RNA polymerase, alpha subunit (rpoA) [2.7.7.6] | Transcription: mRNA synthesis | 4.91 | 36.7 |
| 53 | 0.22 | PP_4730 | Transcriptional regulator Fur (fur) | Transcription: specific TFs | 5.43 | 15.2 |
| 207 | 4.02 | PP_0112 | ABC transporter, periplasmic binding protein, putative | Transporters: ABC-typec | 6.92 | 27.7 |
| 360 | 2.23 | PP_1137 | Branched-chain amino acid ABC transporter, ATP-binding protein (braG) | Transporters: ABC-type | 7.7 | 25.5 |
| 282 | 0.48 | PP_1826 | Isochorismatase hydrolase | Unclear classification | 7.05 | 21.5 |
| 171 | 0.15 | PP_3249 | Aldo/keto reductase | Unclear classification | 7.81 | 29.6 |
| 61 | 0.07 | PP_0018 | Hypothetical protein | Unknown function | 5.72 | 31.2 |
| 166 | 0.39 | PP_0766 | Hypothetical protein | Unknown function | 9.17 | 50.5 |
| 195 | 0.53 | PP_5156 | Hypothetical protein | Unknown function | 6.64 | 16.7 |
| 182 | 2.17 | PP_3611 | Hypothetical protein | Unknown function | 9.07 | 20.8 |
| 198 | 4.65 | PP_0998 | Hypothetical protein | Unknown function | 6.12 | 16.0 |
| Spot number | Ni/Ctrl | Accession no | Protein (gene) [EC] | Functional classification | PI | Mass |
|---|---|---|---|---|---|---|
| 220 | 0.25 | PP_1702 | Recombination-associated protein (rdgC) | Cell growth/division | 4.87 | 34.4 |
| 99 | 2.22 | PP_1799 | GDP-mannose 4.6 dehydratase (gmd) [4.2.1.47] | Cell structure: cell envelope | 6 | 40.1 |
| 305 | 2.14 | PP_1210 | Ferritin Dps family protein | Antioxidants, detoxification | 5.2 | 17.8 |
| 37 | 0.40 | PP_1638 | Ferredoxin-NADP reductase (fpr) [1.18.1.2] | Antioxidants, detoxification | 5.22 | 29.7 |
| 33 | 2.46 | PP_0538 | Inorganic pyrophosphatase (ppa) [3.6.1.1] | Energy | 4.77 | 19.2 |
| 324 | 4.05 | PP_0072 | Quinone oxidoreductase (qor-1) [1.6.5.5] | Energy: E-transfer | 8.8 | 34.2 |
| 349 | 0.20 | PP_2335 | Methylcitrate synthase [2.3.3.5] | Energy: TCA pathway | 6.3 | 42 |
| 460 | 0.42 | PP_2337 | Probable AcnD-accessory protein PrpF (prpF) | Energy: TCA pathway | 5.31 | 41.3 |
| 246_509 | 2.63 | PP_4012 | Isocitrate dehydrogenase, NADP-dependent [1.1.1.42] | Energy: TCA pathway | 5.44 | 80.1 |
| 132 | 1.28 | PP_4187 | 2-Oxoglutarate dehydrogenase, lipoamide dehydrogenase component (lpdG) [1.8.1.4] | Energy: TCA pathway | 5.93 | 50.1 |
| 441_504 | 2.02 | PP_4189 | 2-Oxoglutarate dehydrogenase, E1 component (kgdA) [1.2.4.2] | Energy: TCA pathway | 6.1 | 106.9 |
| 38 | 0.51 | PP_1389 | Oxaloacetate decarboxylase [4.1.3.3] | Energy: TCA pathway | 5.18 | 31.8 |
| 312 | 2.51 | PP_3365 | Acetolactate synthase, catabolic | Metabolism: energy/amino acids/nucleotides | 5.75 | 60.3 |
| 85 | 2.40 | PP_0671 | Serine hydroxymethyltransferase (glyA-2) [2.1.2.1] | Metabolism: amino acids | 5.85 | 45.1 |
| 533 | 7.73 | PP_0675 | Glutamate dehydrogenase (gdhA) [1.4.1.4] | Metabolism: amino acids | 6.12 | 49.3 |
| 320 | 4.62 | PP_0817 | Aminotransferase, Class I | Metabolism: amino acids | 8.7 | 45 |
| 212 | 2.80 | PP_1237 | Dihydrodipicolinate synthase (dapA) [4.2.1.52] | Metabolism: amino acids | 5.73 | 31.9 |
| 433 | 1.91 | PP_2371 | Sulphite reductase hemoprotein, beta subunit (cysI) [1.8.1.2] | Metabolism: amino acids | 5.5 | 62.2 |
| 321 | 14.70 | PP_4473 | Aspartokinase (lysC) [2.7.2.4] | Metabolism: amino acids | 5.25 | 44.7 |
| 211 | 2.35 | PP_4571 | Cysteine synthase A (cysK) [4.2.99.8] | Metabolism: amino acids | 5.55 | 34.5 |
| 269 | 2.15 | PP_4678 | Ketol-acid reductoisomerase (ilvC) [1.1.1.86] | Metabolism: amino acids | 5.48 | 36.6 |
| 314 | 2.40 | PP_4680 | Acetolactate synthase, large subunit (ilvB) [4.1.3.18] | Metabolism: amino acids | — | 62.8 |
| 133 | 3.96 | PP_3511 | Branched-chain amino acid aminotransferase (ilvE) [2.6.1.42] | Metabolism: amino acids | 6.72 | 36.8 |
| 147 | 2.14 | PP_1362 | Pyruvate kinase II (pykA) [2.7.1.40] | Metabolism: glycolysis | 6.22 | 52.05 |
| 234 | 2.10 | PP_0558 | Acetyl-CoA carboxylase, biotin carboxylase (accC-1) [6.4.1.2] | Metabolism: lipid and sterol | 6.09 | 49.4 |
| 445 | 1.70 | PP_4487 | Acetyl-CoA synthetase 1 (acsA1) [6.2.1.1] | Metabolism: lipid and sterol | 5.9 | 71.8 |
| 162 | 0.49 | PP_4169 | Glycerol-3-phosphate dehydrogenase (gpsA) [1.1.1.8] | Metabolism: lipid and sterol | 6.2 | 36.5 |
| 375 | 2.91 | PP_1031 | Inosine-5-monophosphate dehydrogenase (guaB) [1.1.1.205] | Metabolism: nucleotides | 6.5 | 51.8 |
| 274 | 0.41 | PP_4889 | Adenylosuccinate synthetase (purA) [6.3.4.4] | Metabolism: nucleotides | 5.56 | 47.1 |
| 225 | 1.89 | PP_0776 | Peptidyl-prolyl cis-trans isomerase, FKBP-type (slyD) | Protein destination and storage: folding and stability | — | 17.2 |
| 378 | 4.41 | PP_4727 | dnaK protein (dnaK) | Protein destination and storage: folding and stability | 4.83 | 68.8 |
| 8 | 0.41 | PP_2299 | Trigger factor (tig) [5.2.1.8] | Protein destination and storage: folding and stability | 4.81 | 48.5 |
| 329 | 3.48 | PP_4874 | Ribosomal protein L9 (rplI) | Protein synthesis: ribosomal proteins | 5.3 | 15.5 |
| 42 | 3.65 | PP_1592 | Translation elongation factor Ts (tsf) | Protein synthesis: translation factors | 5.14 | 30.4 |
| 260 | 0.53 | PP_5044 | GTP-binding protein TypA/BipA | Protein synthesis: translation factors | 5.4 | 67.3 |
| 261 | 1.85 | PP_2465 | Threonyl-tRNA synthetase (thrS) [6.1.1.3] | Protein synthesis: tRNA synthetases | 5.64 | 73.2 |
| 310 | 2.45 | PP_0397 | Putative serine protein kinase, PrkA | Signal transduction: kinases | 7.74 | 74.0 |
| 226 | 2.15 | PP_2084 | Regulator of ribonuclease activity A (rraA) | Transcription: RNA fate | — | 17.6 |
| 319 | 6.00 | PP_5214 | Transcription termination factor Rho (rho) | Transcription: general TFs | 7.71 | 47.1 |
| 327 | 3.78 | PP_0227 | Amino acid ABC transporter, Periplasmic binding protein | Transporters: ABC-type | — | 28.8 |
| 315 | 2.21 | PP_0885 | Dipeptide ABC transporter, periplasmic peptide-binding protein | Transporters: ABC-type | 6.64 | 60.5 |
| 137 | 2.44 | PP_3801 | Cation ABC transporter, periplasmic cation-binding protein, putative | Transporters: ABC-type | 6.67 | 32.8 |
| 89 | 2.31 | PP_3558 | Amino acid transporter, periplasmic amino acid-binding protein, putative | Transporters: ABC-type | 5.6 | 36.9 |
| 170 | 2.37 | PP_0824 | Phosphate ABC transporter, periplasmic phosphate-binding protein, putative | Transporters: ABC-type | 8.35 | 31.1 |
| 282 | 1.69 | PP_1826 | Isochorismatase hydrolase | Unclear classification | 7.05 | 21.5 |
| 195 | 3.84 | PP_5156 | Hypothetical protein | Unknown function | 6.64 | 16.7 |
| 198 | 0.55 | PP_0998 | Hypothetical protein | Unknown function | 6.12 | 16.1 |
| Spot number | Ni/Ctrl | Co/Ctrl | Accession no | Protein (gene) [EC] | Functional classification | PI | MW, kDa |
|---|---|---|---|---|---|---|---|
| 118 | 0.46 | 0.10 | PP_0011 | DNA polymerase III, beta subunit (dnaN) [2.7.7.7] | Cell growth/division | 5 | 40.7 |
| 10 | 0.24 | 0.29 | PP_4378 | Flagellin fliC (fliC) | Cell structure: motility | 4.39 | 67.80 |
| 362 | 1.78 | 6.75 | PP_3587 | Thiol peroxidase (tpx) [1.11.1.-] | Antioxidants, detoxification | 4.99 | 17.6 |
| 203 | 5.01 | 1.88 | PP_4506 | Nitroreductase family protein | Antioxidants, detoxification | 6.16 | 20.0 |
| 270–297 | 1.76 | 3.73 | PP_1084 | Alkyl hydroperoxide reductase, AhpC/Tsa family | Antioxidants, detoxification | 5.06 | 21.9 |
| 52 | 2.40 | 4.94 | PP_2463 | Cold shock protein CspA (cspA-2) | Antioxidants, detoxification | 6.56 | 7.7 |
| 44 | 0.23 | 0.52 | PP_0336 | Peptide methionine sulfoxide reductase (msrA) [1.8.4.6] | Antioxidants, detoxification | — | — |
| 196 | 0.37 | 0.23 | PP_2132 | Universal stress protein family UspA | Antioxidants, detoxification | 6.14 | 16.4 |
| 332 | 5.69 | 4.46 | PP_5414 | ATP synthase F1, gamma subunit (atpG) [3.6.3.14] | Energy | 9.5 | 31.4 |
| 222 | 0.21 | 0.11 | PP_5413 | ATP synthase F1, beta subunit (atpD) [3.6.3.14] | Energy | 4.88 | 49.4 |
| 18_107 | 0.33 | 0.20 | PP_4116 | Isocitrate lyase (aceA) [4.1.3.1] | Energy: glyoxylate cycle | 5.38 | 48.6 |
| 239 | 0.26 | 0.17 | PP_4965 | Transketolase (tktA) [2.2.1.1] | Energy: pentose phosphate pathway | 5.2 | 72.8 |
| 552 | 3.53 | 6.22 | PP_0339 | Pyruvate dehydrogenase, E1 component (aceE) [1.2.4.1] | Energy: TCA pathway | 5.56 | 99.7 |
| 393_567 | 2.00 | 1.67 | PP_2339 | Aconitate hydratase 2 (acnB) [4.2.1.3] | Energy: TCA pathway | 5.18 | 94.2 |
| 105 | 0.49 | 0.36 | PP_4011 | Isocitrate dehydrogenase, NADP-dependent, prokaryotic type (icd) [1.1.1.42] | Energy: TCA pathway | 5.4 | 45.7 |
| 65 | 0.40 | 0.27 | PP_4185 | Succinyl-CoA synthetase, alpha subunit (sucD) [6.2.1.5] | Energy: TCA pathway | 5.89 | 30.1 |
| 285 | 0.45 | 0.24 | PP_4194 | Citrate synthase (gltA) [4.1.3.7] | Energy: TCA pathway | 6.5 | 47.9 |
| 259 | 0.33 | 0.30 | PP_1304 | Sulfate adenylyltransferase, subunit 1/adenylylsulfate kinase (cysNC) [2.7.7.4] | Metabolism: energy/amino acids/nucleotides | 5.51 | 69.2 |
| 163 | 0.33 | 0.37 | PP_0432 | N-Acetylglutamate semialdehyde dehydrogenase (argC1) [1.2.1.38] | Metabolism: amino acids | 6.3 | 36.3 |
| 78 | 0.29 | 0.28 | PP_1000 | Ornithine carbamoyltransferase, catabolic (argI) [2.1.3.3] | Metabolism: amino acids | 5.92 | 37.9 |
| 106 | 0.45 | 0.20 | PP_1001 | Arginine deiminase (arcA) [3.5.3.6] | Metabolism: amino acids | 5.5 | 46.4 |
| 28 | 0.23 | 0.42 | PP_1988 | 3-Isopropylmalate dehydrogenase (leuB) [1.1.1.85] | Metabolism: amino acids | 5.03 | 38.9 |
| 149 | 0.28 | 0.40 | PP_2528 | O-acetylhomoserine sulfhydrylase (metY) [2.3.1.31] | Metabolism: amino acids | 6 | 45.2 |
| 135 | 0.20 | 0.21 | PP_4223 | Diaminobutyrate-2-oxoglutarate transaminase [2.6.1.76] | Metabolism: amino acids | 6.54 | 48.8 |
| 236 | 0.31 | 0.35 | PP_4477 | N-succinylarginine dihydrolase (astB) | Metabolism: amino acids | 6.03 | 49.2 |
| 122 | 0.11 | 0.31 | PP_4667 | Methylmalonate semialdehyde dehydrogenase (mmsA2) [1.2.1.27] | Metabolism: amino acids | 5.7 | 54.3 |
| 14 | 0.28 | 0.19 | PP_5046 | Glutamine synthetase, type I (glnA) [6.3.1.2] | Metabolism: amino acids | 5.21 | 51.7 |
| 40 | 0.18 | 0.26 | PP_5095 | Delta-1 pyrroline-5-carboxylate reductase (proC) [1.5.1.2] | Metabolism: amino acids | 4.98 | 28.2 |
| 67 | 0.05 | 0.12 | PP_5289 | Acetylglutamate kinase (argB) [2.7.2.8] | Metabolism: amino acids | 5.57 | 31.9 |
| 129 | 0.47 | 0.19 | PP_5338 | Aspartate ammonia-lyase (aspA) [4.3.1.1] | Metabolism: amino acids | 5.68 | 51.5 |
| 208 | 2.39 | 1.60 | PP_4869 | NH(3)-dependent NAD(+) synthetase (nadE) [6.3.5.1] | Metabolism: cofactors | 5.52 | 29.4 |
| 62 | 0.46 | 0.29 | PP_5104 | Thiazole synthase (thiG) | Metabolism: cofactors | 5.74 | 29.2 |
| 123 | 0.23 | 0.30 | PP_0763 | Medium-chain-fatty-acid CoA ligase | Metabolism: lipid and sterol | 5.47 | 62.1 |
| 74 | 0.53 | 0.33 | PP_2217 | Enoyl-CoA hydratase/isomerase FadB1x (fadB1x) | Metabolism: lipid and sterol | 5.43 | 27.7 |
| 73 | 0.19 | 0.33 | PP_1240 | Phosphoribosylaminoimidazole-succinocarboxamide synthase (purC) [6.3.2.6] | Metabolism: nucleotides | 5.37 | 26.9 |
| 127 | 0.41 | 0.26 | PP_4016 | Adenylosuccinate lyase (purB) [4.3.2.2] | Metabolism: nucleotides | 5.69 | 50.5 |
| 242 | 0.18 | 0.18 | PP_4179 | Heat shock protein HtpG (htpG) | Protein destination and storage: folding and stability | 5.21 | 71.5 |
| 386 | 2.51 | 1.69 | PP_2017 | Aminopeptidase N (pepN) [3.4.11.2] | Protein destination and storage: proteolysis | 4.9 | 99.5 |
| 58 | 0.11 | 0.09 | PP_2300 | ATP-dependent Clp protease, proteolytic subunit ClpP (clpP) [3.4.21.92] | Protein destination and storage: proteolysis | 5.53 | 23.5 |
| 177 | 2.41 | 4.95 | PP_0444 | Ribosomal protein L1 (rplA) | Protein synthesis: ribosomal proteins | 9.5 | 24.2 |
| 272 | 4.22 | 7.28 | PP_0721 | Ribosomal 5S rRNA E-loop binding protein Ctc/L25/TL5 | Protein synthesis: ribosomal proteins | 6 | 23.3 |
| 7 | 1.91 | 2.25 | PP_1772 | Ribosomal protein S1 (rpsA) | Protein synthesis: ribosomal proteins | 4.83 | 63.5 |
| 23 | 1.67 | 4.12 | PP_0440 | Translation elongation factor Tu (tuf-1) | Protein synthesis: translation factors | 5.22 | 43.8 |
| 43 | 1.29 | 2.16 | PP_1858 | Translation elongation factor P (efp) | Protein synthesis: translation factors | 4.73 | 21.3 |
| 219 | 0.29 | 0.37 | PP_0061 | Glycyl-tRNA synthetase, tetrameric type, alpha subunit (glyQ) [6.1.1.14] | Protein synthesis: tRNA synthetases | 5 | 36.1 |
| 50 | 0.23 | 0.20 | PP_4722 | Transcription elongation factor GreA (greA) | Transcription: specific TFs | 4.85 | 17.6 |
| 167 | 1.69 | 2.53 | PP_1071 | Amino acid ABC transporter, periplasmic amino acid-binding protein | Transporters: ABC type | 8.61 | 33.4 |
| 31 | 0.38 | 0.11 | PP_0282 | Amino acid ABC transporter, periplasmic amino acid-binding protein | Transporters: ABC type | 5.46 | 27.9 |
| 502 | 2.66 | 3.26 | PP_3828 | Molybdate ABC transporter, periplasmic molybdate-binding protein (modA) | Transporters: ABC type | 9 | 27.0 |
| 102 | 0.47 | 0.39 | PP_5196 | Iron ABC transporter, periplasmic iron-binding protein, putative | Transporters: ABC type | 5.6 | 36.8 |
| 69 | 0.17 | 0.32 | PP_0711 | Isochorismatase hydrolase | Unclear classification | 5.43 | 23.0 |
| 200 | 3.03 | 1.46 | PP_4981 | YceI1 family protein | Unknown function | 7.89 | 22.1 |
| 256 | 0.43 | 0.33 | PP_4448 | Hypothetical protein | Unknown function | 5.74 | 66.5 |
Comparison of Ni and Co effects unveils a common core of metal stress responsive proteins
The specific response to cobalt
Another strongly enhanced protein was Ohr (organic hydroperoxide resistance protein, PP_1859), a hydroperoxidase with a clear preference for organic peroxides. The in vivo substrates are unknown but are thought to include lipid peroxides.28 The anti-oxidative response is completed by Fe-superoxide dismutase (sodB, PP_0915) whose function is to scavenge superoxide O2−. To depict a more complete picture of the thiol-redox biology in P. putida under metallic stress, the expression of ohr and tpx (peroxiredoxin), trx-2 (thioredoxin), and trxB (thioredoxin reductase) was monitored at the mRNA level (see below).
Finally, the effect of Co on the stability of mRNAs can be hypothesized. The synthesis of RraA, ribonuclease E inhibitor protein A (PP_2084), decreased. This protein inhibits the activity of RNaseE that plays a central role in mRNA decay, the processing of tRNA, the tmRNA-mediated mRNA quality control.32 RraA binds to RNaseE and inhibits its activity leading to a global change in the half-life and abundance of mRNAs.33 Down-regulation of RraA is thus expected to increase RNA decay by RNaseE. In E. coli, over-production of RraA affected mRNA levels more notably involved in anaerobic metabolism and cell envelope biosynthesis. Another RNaseE interacting protein is Pnp, poly RNA nucleotidyltransferase (PP_4708). Pnp is involved in mRNA degradation by hydrolyzing single-stranded polyribonucleotides from 3′ to 5′ and is part of the RNA degradosome.34 Pnp can also function alone in the degradation of small RNAs. In E. coli it has been shown that Pnp plays a role for instance in the control of outer membrane protein (OMP) levels.35
The specific response to nickel
Conversion of aspartate to L-lysine via dihydrodipicolinate synthase DapA (PP_1237) and conversion of asparte to L-homoserine via aspartokinase LysC (PP_4473) seem to be favored by Ni. The conversion of serine into glycine via serine hydroxymethyltransferase GlyA2 (PP_0671) was favored in Ni-treated samples not in Co-ones, but the conversion of 3-phosphoglycerate to serine also increased in Co-treated samples via SerA (PP_5155) (Table 1). Finally, the synthesis of cysteine via the sulfate assimilation pathway was examined. Conversion of sulfate to adenosine-5′-phosphosulfate (APS) by CysNC (PP_1304) was lowered by both Co and Ni (Table 3). However, the conversion of sulfite to sulfide by CysI (PP_2371) and the condensation of sulfide with O-acetyl-L-serine by cysteine synthase (CysK, PP_4571) to yield cysteine were enhanced in Ni-treated samples (Table 2). Quite surprisingly, the level of these enzymes was reduced by Co (Table 1) suggesting that cysteine biosynthesis could not be supported by sulfate assimilation. Rather it could result from the conversion of cysteinylglycine into L-Cys and Gly via aminopeptidase PepN (PP_2017). Indeed, the level of PepN increased in Co and Ni samples (Table 3).
Quantification of the oxidative stress response at the mRNA level
The proteomic data revealed that the effect of Co on cellular stress leads to the accumulation of several proteins involved in the detoxification of peroxides (thioredoxin reductase, thioredoxin, organic hydroperoxide resistance protein and thiol peroxidase) as well as detoxification of superoxides (superoxide dismutase SodB). Concerning the effect of Ni, the effect on oxidative stress was less clear at the protein level. To address the involvement of defense mechanisms towards oxidative stress, the level of expression of the genes encoding the above-mentioned proteins was measured in samples prepared under the same conditions as the ones used for the proteomic analysis.In the presence of Co, ohr encoding organic hydroperoxide resistance protein and tpx encoding thiol peroxidase were up-regulated, indicating that under these conditions organic peroxides accumulate in the cell (Table 4). Similarly, the increase of trxB (thioredoxin reductase) and trx2 (thioredoxin) indicates the need for the cell to reduce oxidized thiols that can arise from the oxidation of peroxiredoxins or the direct damage of thiol-containing proteins.36 Finally, the most induced gene was sodB encoding superoxide dismutase. This enzyme is involved in the scavenging of superoxide ions to prevent their oxidative deleterious effect.37
| Gene | Protein | Ratio | |
|---|---|---|---|
| Ni | Co | ||
| sodB | Superoxide dismutase | 15.7 (0.5) | 45.3 (0.4) |
| trxB | Thioredoxin reductase | −2.5 (0.1) | 4.2 (2.2) |
| trx2 | Thioredoxin | 1.3 (0.4) | 1.9 (0.1) |
| ohr | Organic hydroperoxide resistance protein | 1.1 (0.1) | 1.8 (0.3) |
| tpx | Thiol peroxidase | 1.4 (0.0) | 7.6 (0.3) |
Interestingly, the situation in the presence of Ni was different. Indeed, trx2, ohr and tpx were only very slightly induced, and trxB was even repressed (Table 4). These regulations suggest that Ni, in contrast to Co, does not cause the apparition of peroxides and that thiol-damage is not more pronounced than under the control conditions. Still, oxidative stress is present in the Ni-treated samples as sodB, encoding superoxide dismutase, was strongly up-regulated, indicating the presence of superoxide radicals.
Modification of the amino acid pools by the addition of Ni or Co
The proteomics data revealed that the expression of a large set of proteins involved in amino acid biosynthesis was modified upon the addition of Ni or Co. This suggests that the level of amino acids might be changed upon the addition of these two metals. To assess the possible effect of these metals on the cellular pools of amino acids, their level in bacteria was assayed after growth either in minimal medium supplemented with 0.4% succinate or in the same medium supplemented with 75 μM NiCl2 or 35 μM CoCl2. In the absence of metal, the most abundant amino acids were glutamate (7.63 μg per mg of total protein), alanine (4.68 μg per mg of total protein), aspartate (3.95 μg per mg of total protein) and glycine (3.79 μg per mg of total protein) (Fig. 4). In the presence of Co, the level of serine increased 1.6 fold and the levels of branched chain amino acids valine, isoleucine and leucine increased twice, while the amount of arginine decreased twice (Fig. 4). It was previously reported that, in E. coli, the level of branched chain amino acids increased under paraquat-induced oxidative stress38 and in a tellurite-hyperresistant strain.39 A role for these amino acids in the defense against oxidative stress was thus proposed. In the presence of Ni, the level of alanine increased twice, the levels of isoleucine and leucine increased 1.3 fold while the level of aspartate decreased 1.6 fold. However, very striking was the level of glutamate that increased by a four-fold ratio (Fig. 4). Glutamate plays many roles in the cell. It can be converted to citric acid intermediates to provide energy for the cell. However, synthesis of proteins involved in this conversion was found to be reduced in this study (see above). Moreover, as glutamate accumulates here, its role as a precursor is not likely. Glutamate was also shown to enhance the stability of several proteins.40 Increased levels of glutamate might also reflect the adaptive response of cells to an increase of the osmolarity of the medium.41 It was shown that accumulation of glutamate has an effect on the transcription of several E. coli genes by acting differentially on RNA polymerase.42 The data of amino acid amounts measured here are in agreement with the proteomic data and this confirms that the amino acids play a role in the bacterial adaptation to noxious conditions caused by excess concentrations of Ni or Co.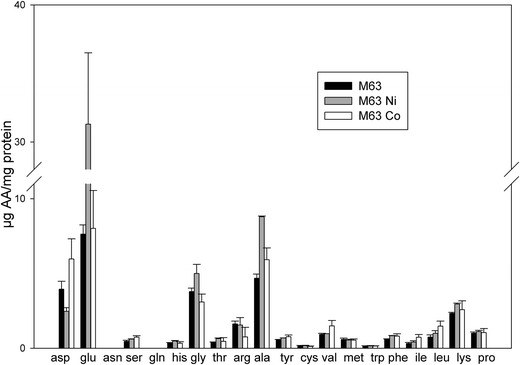 | ||
| Fig. 4 Concentrations of amino acids present in treated bacteria. The amount of amino acids was measured in whole cells after growth in 0.4% succinate M63 medium (black bars), in the same medium supplemented with 75 μM NiCl2 (grey bars) or 35 μM CoCl2 (white bars). The results are expressed in μg of amino acid per mg of total protein. Error bars indicate standard deviations. | ||
Concluding remarks
This study, by reason of a large number of proteins (160) that were identified and whose accumulation level was quantified, allowed us to perform a thorough investigation of the bacterial cell response to Ni or Co toxicity. The concentrations of these two metals we used influence the bacterial growth and this effect is obvious at the protein level where accumulation of components of the protein synthesis machinery was observed, suggesting the requirement for the cell to repair damaged macromolecules. We also observed a decrease in the level of proteins involved in the central metabolism or amino-acids metabolism. In the Co treated samples, several proteins involved in the defense against oxidative stress were shown to accumulate. Most of the anti-oxidant systems identified participate in the cellular thiol-disulfide homeostasis but interestingly, we also found proteins, such as superoxide dismutase, which are required for the detoxification of ROS. These data argue strongly in favor of Co causing the apparition of ROS, which is considered as controversial in the literature. These results were fully confirmed at the transcriptional level by the up-regulation of genes involved in the detoxification of superoxide radicals, peroxides and in maintaining the thiol-disulfide homeostasis. These observations were not retrieved in the Ni-treated samples, even if response to oxidative stress was also noticed at the protein level and was confirmed at the RNA level where sodB, encoding superoxide dismutase, was strongly up-regulated, indicating that Ni causes the apparition of superoxide radicals. This effect has never been described in the literature yet, however the mechanism by which superoxide ions appear is still to be uncovered. The superoxide radicals are known to damage iron–sulfur cluster containing enzymes leading to the release of iron that in turn can produce ROS through the Fenton reaction.37 The main signature of the effect of Ni was the accumulation of proteins involved in the biosynthesis of amino acids, more particularly, proteins involved in the synthesis of glutamate. When directly assayed in the Ni treated samples, the level of several amino acids changed, the most striking result being the increased accumulation of glutamate, a compound that plays a crucial role in cell survival. Further work is needed to decipher the precise role of amino acids in the defense against Ni toxicity.Acknowledgements
This work was supported by IFCPAR, grant 3709. We thank Floriant Bellvert and the CESN platform, UCBL, for the amino acids assay. This manuscript is dedicated to the memory of P. Maruthi Mohan, Department of Biochemistry, Osmania University.References
- Y. Zhang and V. N. Gladyshev, J. Biol. Chem., 2010, 285, 3393–3405 CrossRef CAS.
- S. B. Mulrooney and R. P. Hausinger, FEMS Microbiol. Rev., 2003, 27, 239–261 CrossRef CAS.
- M. Kobayashi and S. Shimizu, Eur. J. Biochem., 1999, 261, 1–9 CrossRef CAS.
- T. Eitinger, J. Suhr, L. Moore and J. A. Smith, Biometals, 2005, 18, 399–405 CrossRef CAS.
- D. Nies, FEMS Microbiol. Rev., 2003, 27, 313–339 CrossRef CAS.
- A. Rodrigue, G. Effantin and M. A. Mandrand-Berthelot, J. Bacteriol., 2005, 187, 2912–2916 CrossRef CAS.
- Z. L. He, X. E. Yang and P. J. Stoffella, J. Trace Elem. Med. Biol., 2005, 19, 125–140 CAS.
- S. Smith, Environ. Int., 2009, 35, 142–156 CrossRef CAS.
- D. Canovas, I. Cases and V. de Lorenzo, Environ. Microbiol., 2003, 5, 1242–1256 CrossRef CAS.
- A. Haritha, A. Rodrigue and P. M. Mohan, BMC Res. Notes, 2008, 1, 88 CrossRef.
- F. Barras and M. Fontecave, Metallomics, 2011, 3, 1130–1134 RSC.
- J. Catusse, J. M. Strub, C. Job, A. Van Dorsselaer and D. Job, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10262–10267 CrossRef CAS.
- A. Shevchenko, M. Wilm, O. Vorm and M. Mann, Anal. Chem., 1996, 68, 850–858 CrossRef CAS.
- C. Deytieux, L. Geny, D. Lapaillerie, S. Claverol, M. Bonneu and B. Doneche, J. Exp. Bot., 2007, 58, 1851–1862 CrossRef CAS.
- J. W. Henderson, R. D. Ricker, B. A. Bidlingmeyer and C. Woodward, Agilent application note, 2000 Search PubMed.
- M. W. Pfaffl, Nucleic Acids Res., 2001, 29, e45 CrossRef CAS.
- Z. A. Wood, E. Schroder, J. Robin Harris and L. B. Poole, Trends Biochem. Sci., 2003, 28, 32–40 CrossRef CAS.
- D. F. Ackerley, C. F. Gonzalez, M. Keyhan, R. Blake and A. Matin, Environ. Microbiol., 2004, 6, 851–860 CrossRef CAS.
- G. Horn, R. Hofweber, W. Kremer and H. Kalbitzer, Cell. Mol. Life Sci., 2007, 64, 1457–1470 CrossRef CAS.
- C. Loepfe, E. Raimann, R. Stephan and T. Tasara, Foodborne Pathog. Dis., 2010, 7, 775–783 CrossRef CAS.
- K. E. Nelson, C. Weinel, I. T. Paulsen, R. J. Dodson, H. Hilbert, V. A. P. Martins dos Santos, D. E. Fouts, S. R. Gill, M. Pop, M. Holmes, L. Brinkac, M. Beanan, R. T. DeBoy, S. Daugherty, J. Kolonay, R. Madupu, W. Nelson, O. White, J. Peterson, H. Khouri, I. Hance, P. C. Lee, E. Holtzapple, D. Scanlan, K. Tran, A. Moazzez, T. Utterback, M. Rizzo, K. Lee, D. Kosack, D. Moestl, H. Wedler, J. Lauber, D. Stjepandic, J. Hoheisel, M. Straetz, S. Heim, C. Kiewitz, J. Eisen, K. N. Timmis, A. Düsterhöft, B. Tümmler and C. M. Fraser, Environ. Microbiol., 2002, 4, 799–808 CrossRef CAS.
- J. J. Harrison, V. Tremaroli, M. A. Stan, C. S. Chan, C. Vacchi-Suzzi, B. J. Heyne, M. R. Parsek, H. Ceri and R. J. Turner, Environ. Microbiol., 2009, 11, 2491–2509 CrossRef CAS.
- Z. Cheng, Y. Y. Wei, W. W. Sung, B. R. Glick and B. J. McConkey, Proteome Sci., 2009, 7, 18 CrossRef.
- M. A. Molina-Henares, J. De La Torre, A. García-Salamanca, A. J. Molina-Henares, M. C. Herrera, J. L. Ramos and E. Duque, Environ. Microbiol., 2010, 12, 1468–1485 CAS.
- C. Tricot, V. Stalon and C. Legrain, J. Gen. Microbiol., 1991, 137, 2911–2918 CrossRef CAS.
- Y. Meyer, B. B. Buchanan, F. Vignols and J.-P. Reichheld, Annu. Rev. Genet., 2009, 43, 335–367 CrossRef CAS.
- S. Yeom, J. Yeom and W. Park, Microbiology, 2010, 156, 1487–1496 CrossRef CAS.
- J. A. Dubbs and S. Mongkolsuk, Subcell. Biochem., 2007, 44, 143–193 CrossRef.
- S. Nair and S. E. Finkel, J. Bacteriol., 2004, 186, 4192–4198 CrossRef CAS.
- J. R. Fantino, B. Py, M. Fontecave and F. Barras, Environ. Microbiol., 2010, 12, 2846–2857 CAS.
- K. Y. Choi and H. Zalkin, J. Bacteriol., 1992, 174, 6207–6214 CAS.
- D. A. Steege, RNA, 2000, 6, 1079–1090 CrossRef CAS.
- K. Lee, X. Zhan, J. Gao, J. Qiu, Y. Feng, R. Meganathan, S. N. Cohen and G. Georgiou, Cell, 2003, 114, 623–634 CrossRef CAS.
- V. Kaberdin, D. Singh and S. Lin-Chao, J. Biomed. Sci., 2011, 18, 23 CrossRef CAS.
- J. M. Andrade and C. l. M. Arraiano, RNA, 2008, 14, 543–551 CrossRef CAS.
- B. D'Autreaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813–824 CrossRef CAS.
- J. A. Imlay, Annu. Rev. Biochem., 2008, 77, 755–776 CrossRef CAS.
- H. Tweeddale, L. Notley-McRobb and T. Ferenci, Redox Rep., 1999, 4, 237–241 CrossRef CAS.
- V. Tremaroli, M. L. Workentine, A. M. Weljie, H. J. Vogel, H. Ceri, C. Viti, E. Tatti, P. Zhang, A. P. Hynes, R. J. Turner and D. Zannoni, Appl. Environ. Microbiol., 2009, 75, 719–728 CrossRef CAS.
- A. K. Mandal, S. Samaddar, R. Banerjee, S. Lahiri, A. Bhattacharyya and S. Roy, J. Biol. Chem., 2003, 278, 36077–36084 CrossRef CAS.
- L. N. Csonka, Microbiol. Rev., 1989, 53, 121–147 CAS.
- J. D. Gralla and D. R. Vargas, EMBO J., 2006, 25, 1515–1521 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2mt20147j |
| This journal is © The Royal Society of Chemistry 2013 |