
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive photoswitch–actinide binding: a match made in MOFs†
Kyoung Chul
Park
a,
Jaewoong
Lim
a,
Grace C.
Thaggard
a,
Buddhima K. P.
Maldeni Kankanamalage
a,
Ingrid
Lehman-Andino
 b,
Yuan
Liu
c,
Jennii M.
Burrell
a,
Corey R.
Martin
b,
An T.
Ta
c,
Andrew B.
Greytak
a,
Jake W.
Amoroso
b,
David D.
DiPrete
b,
Mark D.
Smith
a,
Simon R.
Phillpot
c and
Natalia B.
Shustova
*a
b,
Yuan
Liu
c,
Jennii M.
Burrell
a,
Corey R.
Martin
b,
An T.
Ta
c,
Andrew B.
Greytak
a,
Jake W.
Amoroso
b,
David D.
DiPrete
b,
Mark D.
Smith
a,
Simon R.
Phillpot
c and
Natalia B.
Shustova
*a
aUniversity of South Carolina, Department of Chemistry and Biochemistry, Columbia, South Carolina 29208, USA. E-mail: shustova@sc.edu
bSavannah River National Laboratory, Aiken, South Carolina 29808, USA
cUniversity of Florida, Department of Materials Science and Engineering, Gainesville, Florida 32611, USA
First published on 17th June 2025
Abstract
The fundamentals of switchable actinide–ligand binding modes are central for designing new platforms for addressing challenges associated with, for example, isolation of pure radiological daughters for nuclear medicine or methods for efficient nuclear stockpile recycling. This study is the first to report actinide binding modes controlled by an external stimulus via photochromic moieties, realized through probing thermodynamics and kinetics aspects, including changes in photoswitch isomerization constants upon metal coordination and enthalpies associated with the synergistic actinide–switch photochromic processes. A comprehensive analysis of the presented concept was executed through evaluation of data acquired through a multivariate strategy involving isothermal titration calorimetry, crystallography, spectroscopy, and theoretical modeling on the example of actinide-containing compounds based on thorium(IV)-, and uranium(IV, VI), as well as transuranic elements such as plutonium(IV) in solution and within a metal–organic framework (MOF) matrix for the first time. Overall, the presented concept could usher in an alternative direction in stimuli-responsive actinide-based platforms that could be adapted to confront current and upcoming challenges in f-block chemistry.
Introduction
Stimuli-responsive platforms that take advantage of cooperative interactions between molecular “switches” and metal cations could allow for the development of the next generation of on-demand catalysts, optoelectronic devices, or multivariable sensors.1–13 These applications take advantage of significant changes in molecular properties of photochromic metal–ligand interactions (e.g., metal–photoswitch binding strength or optical profile) upon isomerization of a stimuli-responsive fragment between two (or more) distinct states.14–18 For instance, the class of spiropyran derivatives is known to exhibit stimuli-responsive cation binding associated with isomerization from the neutral spiropyran isomer to the zwitterionic merocyanine form (Scheme 1).15,19 Even though the described advantages of cation binding and release via external stimuli could be expanded to f-block elements, there are currently no studies that target stimuli-responsive actinide (including transuranics) coordination and/or demetallation. For example, key thermodynamic and kinetics aspects that govern the behavior of photochromic actinide–switch platforms are lacking in the literature (Scheme 1). In particular, the binding strength between photochromic compounds and actinides dictates the selectivity and reversibility of actinide–switch coordination.20–22 To the best of our knowledge, there are no reports of binding constants for actinide–photochromic molecule interactions. Moreover, there are only three reports on binding constants describing photochromic molecules' interactions with non-radioactive metals in general23–25 while the kinetics of actinide–switch systems have never been reported before (Fig. 1). | ||
| Scheme 1 (Top) photoisomerization kinetics of a stimuli-responsive spiropyran derivative in the absence (blue) and presence (red) of actinides studied by UV-vis spectroscopy. (Bottom) thermogram obtained through isothermal titration calorimetry used in this work for estimation of Gibbs free energy, enthalpy, and entropy of stimuli-responsive actinide binding. | ||
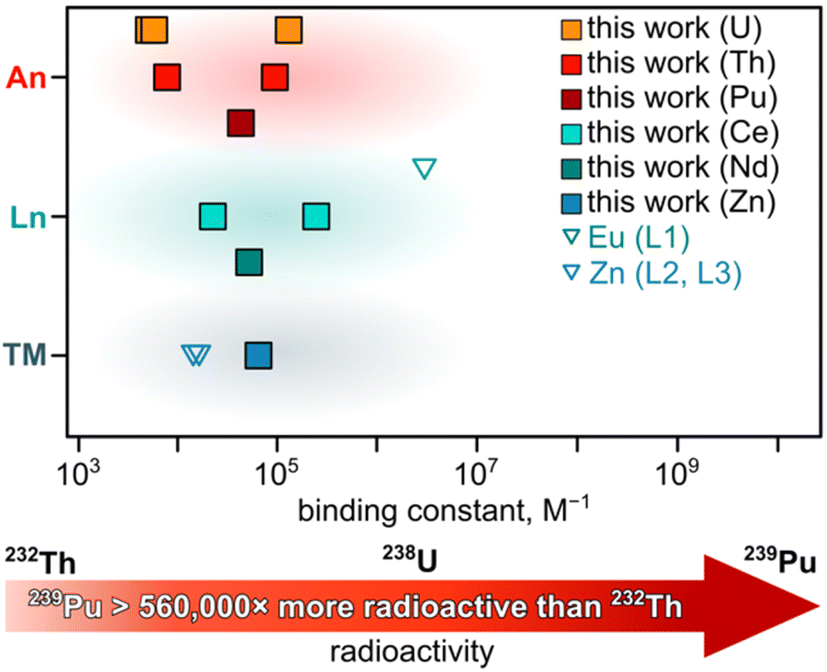 | ||
| Fig. 1 Analysis of all binding constants obtained in this work (filled squares) and literature (unfilled triangles) for photochromic molecules and actinides (red), lanthanides (green), and transition metals (blue). The specific values and ligands are presented in Table 1. The red arrow demonstrates the relative radioactivity of the specific isotopes used in the presented work. | ||
This study represents the first report of actinide binding modes controlled by an external stimulus via photochromic moieties, realized through probing thermodynamics and kinetics aspects, including changes in photoswitch isomerization constants upon metal coordination or enthalpies associated with the synergistic actinide–switch photochromic processes. We also report the first X-ray crystal structure demonstrating binding between a photochromic molecule and actinide. Moreover, we build a conceptual understanding of the thermodynamic and kinetic aspects underlying photoswitch-directed actinide binding and release modes (Scheme 1) through a combination of isothermal titration calorimetry (ITC) experiments and UV-vis spectroscopic studies, as well as X-ray crystallography and theoretical modeling. We report the first thermodynamic parameters, including Gibbs free energy, enthalpy, and entropy of actinide–photoswitch binding as fundamental characteristics of the interactions between photochromic molecules and actinides (e.g., U and Th) evaluated through ITC. Furthermore, we probe the binding constants between spiropyran derivatives and transuranic Pu(IV) species spectroscopically, providing a method to monitor both aspects, actinide binding and spiropyran photoisomerization, simultaneously. We evaluate the photoisomerization kinetics of spiropyran–actinide complexes, effectively shedding light on the impact of metal cation coordination on the binding and demetallation dynamics driven by stimuli-responsive ligands. Importantly, this report is currently the only analysis of both the thermodynamics and kinetics of stimuli-responsive actinide binding, which are two inseparable and equally important factors that govern the feasibility of photochromic material design for the nuclear sector. Finally, we translate the fundamental knowledge regarding thermodynamic and kinetics aspects of discrete molecular systems acquired in solution to the design of stimuli-responsive extended structures (e.g., metal–organic frameworks (MOFs)26–49) for on-demand actinide coordination, providing a pathway for transitioning from solution-based fundamental studies of actinide capture toward strategic design of functional materials.
Overall, these studies lay the conceptual groundwork for understanding the thermodynamic and kinetic patterns that could dictate future applications involving actinide–photoswitch binding modes, for instance, for the development of versatile and modular platforms that could control actinide-related processes on demand.
Results and discussion
To gain the necessary fundamental thermodynamic and kinetic insights on stimuli-responsive platforms capable of “on-demand” coordination and release of actinides, we first explored photoswitch–metal cation interactions that included commonly accessible actinides, non-radioactive surrogates, as well as highly radioactive transuranic species.The selection of the specific photochromic moiety allowing for control of metal coordination via an external stimulus was based on its ability to (i) rapidly and reversibly photoisomerize between discrete states that (ii) possess significantly different binding affinities for metal cations. As a result, we focus on spiropyran derivatives that can undergo rapid photoisomerization to a charge-separated merocyanine form upon exposure to UV irradiation, thereby satisfying criterion (i). At the same time, the zwitterionic merocyanine isomer can form strong coordination bonds with metal cations, as previously shown on the examples of its interactions with some transition metals15,19 and, therefore, satisfies the second criterion (ii). Thus, photoisomerization under UV excitation results in the formation of a zwitterionic merocyanine form, capable of metal coordination (Fig. 2) while the release of corresponding metal cations could be achieved upon merocyanine-to-spiropyran photoisomerization under visible light.15,19 Overall, the rapid transitions between spiropyran and merocyanine states and the ability to bind metal cations provide the rationale for the exploration of the thermodynamic and kinetic aspects associated with spiropyran–actinide systems (vide infra).
 | ||
| Fig. 2 (Top) schematic representation of spiropyran derivative photoisomerization within a UiO-67 MOF upon exposure to UV or visible light. The structures of the corresponding organic linker (gray rod) and metal node (white sphere) are shown. The blue polyhedra, as well as the gray and red spheres, represent zirconium, carbon, and oxygen atoms, respectively. (Bottom) X-ray crystal structures of SP–COOH and MC–COOH, demonstrating the change in the length of the molecular backbone upon photoisomerization. The gray, red, and blue ellipsoids represent carbon, oxygen, and nitrogen atoms, respectively. Hydrogen atoms have been omitted for clarity. Displacement ellipsoids are drawn at the 80% probability level. | ||
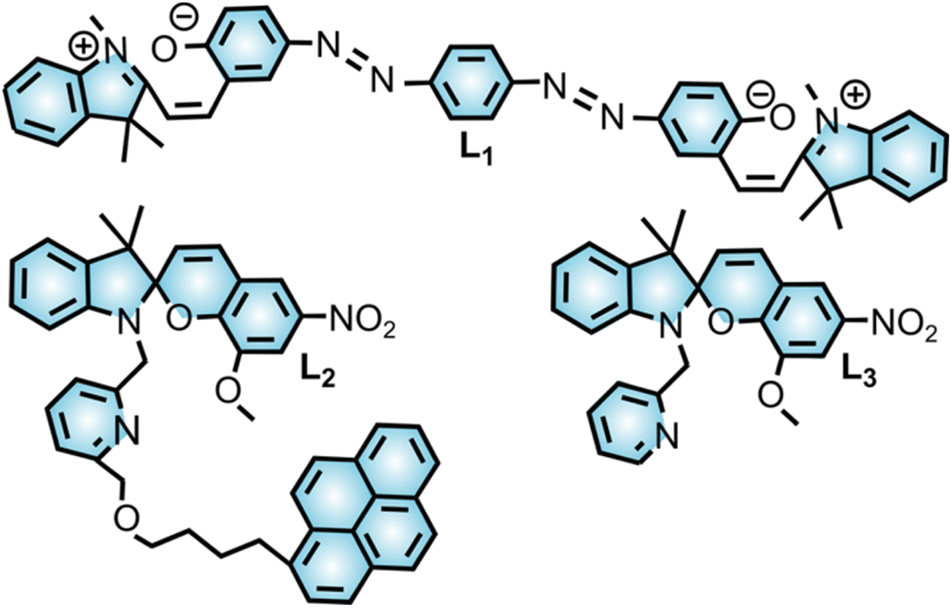
Spiropyran derivatives (1′,3′,3′-trimethyl-6-nitrospiro[chromene-2,2′-indoline] (SP) and 1′,3′,3′-trimethyl-6-nitrospiro[chromene-2,2′-indoline]-5′-carboxylic acid (SP–COOH); Scheme 1 and Fig. 2 and 3) were employed for probing their stimuli-responsive behavior toward actinide cations. In particular, SP (Fig. 3) was used to probe actinide coordination in solution while SP–COOH (Fig. 2), containing a carboxylic acid in its structure, was selected for promoting the anchoring of the photochromic unit to unsaturated metal nodes of the MOF extended structure, i.e., for studies of actinide binding to photochromic derivatives in the solid state.18,50
 | ||
| Fig. 3 (Top) the single-crystal X-ray structure of MC coordinated to a uranyl cation. The presence of NO3− is due to the UO2(NO3)2 salt used for crystal growth. The gray, blue, red, and orange ellipsoids represent carbon, nitrogen, oxygen, and uranium atoms, respectively. Hydrogen atoms are omitted for clarity. Displacement ellipsoids are drawn at the 80% probability level. (Bottom) baseline-corrected ITC thermograms and integrated heat responses resulting from titration of a metal salt (UCl4 or ThCl4) solution (0.25 mM, 2.5 μL injection in EtOH) into a pre-irradiated MC solution (1 mM, 350 μL). The inset shows the calculated thermodynamic parameters associated with the best-fit curves shown. | ||
Synthesis of selected SP–COOH was carried out using a reported literature procedure.18 For SP–COOH integration, a Zr-based MOF, Zr6O4(OH)4(BPDC)6 (BPDC2− = 4,4′-biphenyldicarboxylate; UiO-67, UiO = University of Oslo), was selected due to (i) appropriate pore size (12.7 × 16.2 × 19.7 (diagonal dimension) Å, Fig. S36†)51 suitable for accommodation of SP–COOH (13 × 2.7 Å),18 (ii) relatively broad chemical and thermal stability,52 and (iii) presence of defect sites (i.e., missing linkers resulting in unsaturated metal nodes, Fig. 2) suitable for SP–COOH coordination.53,54
Preparation of UiO-67 was carried out using a literature procedure,55 and corresponding photochromic UiO-67 + SP–COOH was synthesized by heating a sample of parent UiO-67 in an N,N-dimethylformamide (DMF) solution of SP–COOH (60 mM) at 75 °C for three days to promote anchoring of SP–COOH to the defect sites of UiO-67 (see more details of synthesis and material characterization in the ESI†). After the UiO-67 + SP–COOH synthesis, a thorough washing procedure using a Soxhlet apparatus was employed to remove any uncoordinated photochromic molecules from the MOF pores and surface prior to further characterization.
The prepared UiO-67 + SP–COOH samples were analyzed by powder X-ray diffraction (PXRD, Fig. S4†) to ensure that the sample maintained its crystallinity for the duration of the experimental procedures. Further, the amount of SP–COOH installed in the MOF was analyzed by 1H nuclear magnetic resonance (NMR) spectroscopy performed on the digested (destroyed in the presence of acid) MOF samples (Fig. S5†). More detailed information is provided in the ESI.†
Isothermal titration calorimetry
As a starting point for thermodynamic studies of merocyanine (MC)–actinide systems, we evaluated Gibbs free energy (as well as the binding constants) for MC–actinide (MC–Mn+) interactions through ITC studies (Fig. 3). ITC has emerged as a powerful technique for studying the thermodynamic aspects of molecular interactions, including metal–ligand binding, over the past several decades.56–60 More importantly, ITC allows for direct quantification of the heat exchange during molecular-level interactions, offering comprehensive insights into the thermodynamics of molecular associations involving proteins, nucleic acids, and small molecules, unlike UV-vis spectroscopy which can indirectly infer binding parameters.61–63 Despite the advantageous data that could be acquired through ITC studies, there is a large gap in the study of binding interactions between photochromic molecules and metal cations using ITC experiments. For example, only very few studies64–67 have implemented ITC to evaluate parameters for just photochromic molecules themselves. However, there are no reports of utilizing ITC to probe interactions between photochromic molecules and any metals to date. We envisioned that the use of ITC experiments could be significantly expanded as a valuable technique to measure the binding constants, ΔH, and ΔS of MC–actinide cation interactions, which are critical parameters for a fundamental understanding of the role of photochromic molecules in controlled binding and release of actinides, and these studies are a first step toward this direction.The experimental setup for ITC experiments (explained in more detail in the ESI) involved the titration of metal species in ethanol (EtOH) into a sample cell containing a 1 mM EtOH solution of SP. The choice of a polar protic solvent such as EtOH for the ITC studies was dictated by two factors: (i) the solubility of the actinide cations and (ii) the necessity of stabilizing the desired MC isomer for actinide coordination via solvent–photoswitch electrostatic interactions (e.g., hydrogen bonding between EtOH and MC) to promote MC–actinide binding.68,69
To induce the formation of MC and facilitate MC–Mn+ interactions, the SP solution was irradiated for 10 minutes at 365 nm before the measurements were taken, and the subsequent titrations were performed in the dark in a closed sample cell to prevent exposure to visible light for the duration of the ITC experiments. A solution of, for instance, UCl4 (0.25 mM) was prepared in a titration syringe for incremental injection into the sample cell. To ensure that the heat changes measured during the ITC experiments were solely due to the binding of the metal cation to the photoswitch, we conducted control titration experiments that involved studies of EtOH⋯EtOH, metal salt⋯EtOH, and EtOH⋯MC interactions. Indeed, the latter experiments did not show pronounced heat exchange effects above the background level (Fig. S6–S9†).
The thermograms obtained from the titration of MC with the prepared UCl4 solution revealed negative peaks, indicating that the binding process is exothermic (Fig. 3). As a result of the independent model thermogram fitting based on the Langmuir model describing binding at identical independent sites, we were able to evaluate the MC–Mn+ binding constants, Gibbs free energy, enthalpy, and entropy for these processes for the first time. The relatively high ITC-derived binding constant of 1.9 × 105 M−1 for MC–U(IV) indicates favorable binding between MC and uranium cations (Table S2†).
To confirm the validity of the acquired data, we also carried out a number of control experiments. For instance, we considered that exposure of UCl4 to air could potentially result in oxidation of U(IV) to U(VI), resulting in uranyl cation formation.70 To address this possibility, we monitored changes in the absorbance profile of the used UCl4 solution (0.25 mM in EtOH) over time by UV-vis spectroscopy. In particular, we monitored the potential changes in absorbance at 424 nm corresponding to π–π* transitions of uranyl cations, i.e., associated with uranyl cation formation.70 After mimicking the experimental conditions by exposing the U(IV) solution to air for two and even four hours, no changes in the absorbance profile of UCl4 in the solution were detected. This allowed us to conclude that the oxidation state of U(IV) was most likely preserved during the timeline of the ITC experiments.
In the next step, performing similar titrations of ThCl4 with an MC solution in EtOH yielded a binding constant of 9.1 × 104 M−1 (Fig. 3). The binding constant obtained in the presented studies is consistent with the results of the other ITC measurements performed to analyze interactions between thorium cations and non-photochromic compounds.71 For instance, the binding constant of Th4+ to a commercial-grade solvent extraction reagent (Cyanex 301) estimated in a micelle solution was found to be 1.9 × 104 M−1.71 Overall, the reported values of binding constants estimated for coordination of Th(IV) and U(IV) to photochromic MC species estimated using ITC measurements are in agreement with the values reported for the actinides across both synthetic and biological systems.71,72 The ITC experiments also allowed us to evaluate the contributions of ΔH and ΔS to ΔG. Uranium(IV) cation coordination to MC is spontaneous based on an estimated negative value for ΔG (−30.08 kJ mol−1), and the binding interaction is driven by a negative ΔH value (−98.45 kJ mol−1). The slightly lower binding constant determined for ThCl4 compared to UCl4 is due to its less favorable enthalpy of the binding process (ΔH = −56.47 kJ mol−1). In the case of MC–Th(IV) interactions, the less negative enthalpy is incompletely compensated by a less negative entropy, leading to a less negative ΔG = −28.31 kJ mol−1. A comprehensive summary of all thermodynamic parameters derived from the presented ITC experiments is given in Table S2.†
Photoisomerization and metal coordination/demetallation kinetics via UV-vis spectroscopy
As a next step, we evaluated both the kinetics and thermodynamics of the discussed MC–Mn+ interactions using UV-vis spectroscopy studies to allow for simultaneous monitoring of two subsequent processes: (i) the merocyanine-to-spiropyran photoisomerization and (ii) the metal cation chelation to the in situ generated merocyanine isomer. Real-time monitoring of both processes (i and ii) is plausible since spiropyran, merocyanine, and merocyanine-based metal complexes exhibit significantly different absorbance profiles that could be distinguished through spectroscopic analysis.73,74 As a result, UV-vis spectroscopic data can provide support for the formation of coordination bonds between the negatively charged oxygen of MC and metal cations, as well as shed light on the kinetics of the corresponding processes (Fig. 4). Both of these aspects cannot be proven directly by ITC experiments. Moreover, the binding constants determined through UV-vis spectroscopic methods could serve as a reference point (i.e., control experiments) that allows for placing the experimental values determined in this work in perspective with other binding constants reported in the literature, for instance, for some transition metals.73,74 Notably, none of the published binding constants relate to interactions between actinides and stimuli-responsive (photochromic) molecules, and therefore, we used non-radioactive metals (e.g., zinc) in our control experiments. Thus, the selected series of metal salts include radioactive compounds (e.g., ThCl4, UCl4, and UO2(NO3)2) for evaluation of the binding constants between merocyanine and the related actinide as well as non-radioactive cations, such as Zn(II), Ce(III), and Nd(III) as a reference using UV-vis spectroscopy.24,25,54,75,76 | ||
| Fig. 4 (Top) a schematic representation of a stimuli-responsive spiropyran derivative before and after binding to a radioactive metal cation. (Middle left) UV-vis absorbance spectra of SP (3 mM in EtOH) upon exposure to a 365 nm excitation wavelength for 30 s to promote MC formation followed by exposure to white light (λex = 400–900 nm). (Middle right) UV-vis absorbance spectra of SP (3 mM in EtOH) with UO2(NO3)2 (3 mM in EtOH) upon exposure to a 365 nm excitation wavelength for 30 s followed by exposure to white light (λex = 400–900 nm). The hypsochromic shift in the absorbance profile is consistent with complex formation. (Bottom left) plot demonstrating the change in absorbance of SP after exposure to a 365 nm excitation wavelength for 30 s followed by exposure to white light (λex = 400–900 nm). Data fitting (blue line) with a first-order exponential decay equation resulted in a photoisomerization rate constant (k) of 0.25 s−1. (Bottom right) plot demonstrating the change in absorbance of MC with UO2(NO3)2 after exposure to a 365 nm excitation wavelength for 30 s followed by exposure to white light (λex = 400–900 nm). Data fitting (red line) resulted in k = 0.11 s−1 for the MC–UO22+ complex. | ||
As a starting point, we evaluated the photoisomerization kinetics of a selected photochromic unit in the presence and absence of the metal cations since it correlates with the metal coordination/demetallation rates. For instance, isomerization from neutral spiropyran to zwitterionic merocyanine is the first step for the MC–Mn+ formation. Similarly, the rates related to photoisomerization from merocyanine to spiropyran are associated with the reverse demetallation process.15,19,54 Based on the relatively high binding constants determined for MC–actinide complexes via ITC (Ka = 9.1 × 104 and 1.9 × 105 M−1, vide supra), we hypothesized that strong coordination bonds between MC and metal cations would stabilize the zwitterionic MC isomer and, as a result, decrease the rate of MC-to-SP photoisomerization.18,50 Therefore, we evaluated the photoisomerization rate of “free” (i.e., uncoordinated) SP in solution in comparison with MC–Mn+ complexes (where Mn+ = Th4+, U4+, or UO22+). For this, a solution of either SP or a SP/metal salt mixture in EtOH was first exposed to a 365 nm excitation wavelength for 30 s to promote SP-to-MC conversion and subsequent MC–Mn+ complex formation. Both processes were supported by the formation of strong absorbance bands centered at 563 nm and 430 nm, corresponding to “free” MC and the MC–Mn+ complex, respectively. The reverse processes (i.e., demetallation of MC–Mn+ and MC-to-SP photoisomerization) were monitored in real-time through attenuation of the absorbance band at either 563 nm or 430 nm upon exposure to visible light, corresponding to MC-to-SP photoisomerization and MC–Mn+ demetallation, respectively (Fig. 4). Fitting the acquired time-dependent absorbance data with a first-order exponential decay equation allowed us to estimate photoisomerization rate constants (k) for unbound MC and MC in MC–Mn+ complexes.18,50 As a result, a photoisomerization rate constant of k = 0.25 s−1 was estimated for unbound MC in ethanol, which was decreased by a factor of 2.3 in the presence of UO2(NO3)2 (k = 0.11 s−1, Fig. 8). Similarly, the photoisomerization rate constants for MC in MC–Th4+ and MC–U4+ were found to be k = 0.16 and 0.13 s−1, respectively (Fig. S34 and S35†). As anticipated, these examples illustrate that the formation of MC–Mn+ complexes reduces the switching rate of the photochromic unit, which is associated with the stabilization of the zwitterionic MC isomer upon the formation of coordination bonds.
As a next step in our studies, we evaluated the binding constants for MC–Mn+ complexes via UV-vis absorbance spectroscopy (Fig. 5). For that, we prepared 10 mM solutions of SP in three different solvents: EtOH, DMF, and acetonitrile (Fig. S11–S28†). The choice of these experimental conditions was dictated by the intent to compare the acquired data with the existing literature reports on non-radioactive metal binding.25 The prepared SP solutions were then transferred into a quartz cuvette for further analysis using UV-vis spectroscopy. Each sample was then exposed to a 365 nm excitation wavelength for 180 s to promote the formation of the MC photoisomer prior to collecting the initial absorbance spectrum. As shown in Fig. 5, a strong absorbance feature centered at 539 nm could be attributed to the presence of the colored MC isomer.73 Next, aliquots of each metal salt with varying equivalents of metal with respect to SP (0.1–10000 equivalents) were added to the same cuvette, and the absorbance spectra were recollected. As a result, we detected a decrease in the absorbance band at 539 nm upon the addition of metal salt to the MC solution, which corresponds to the chelation of the metal cations by MC (Fig. 5).73 Plotting the absorbance data as a function of the modified Benesi–Hildebrand equation  (ΔA = AC − AMC, where Ac represents the absorbance at a specific metal concentration, and AMC is the absorbance of merocyanine in the absence of metal cations)77 allowed us to estimate the binding constants for MC with each metal cation in different solvents. This procedure was adopted from the previous reports25 and Table 1 summarizes the acquired results.
(ΔA = AC − AMC, where Ac represents the absorbance at a specific metal concentration, and AMC is the absorbance of merocyanine in the absence of metal cations)77 allowed us to estimate the binding constants for MC with each metal cation in different solvents. This procedure was adopted from the previous reports25 and Table 1 summarizes the acquired results.
 | ||
| Fig. 5 (Top) changes in absorbance spectra of spiropyran (5.0 × 10−5 M in EtOH) upon exposure to a 365 nm excitation wavelength for 180 s, followed by the addition of 0.10, 0.15, 0.20, 0.30, 0.40, and 0.50 equivalents of ThCl4 (from top to bottom). Prior to the collection of each spectrum, the solution was irradiated with a 365 nm excitation wavelength for 180 s to promote the binding of Th cations by MC. The photographs show a 10 mM MC solution in EtOH, as well as upon addition of a 10 mM solution of ThCl4 in EtOH, and after mixing the two solutions, from top to bottom. (Bottom) plot demonstrating the ratio [A∞ − AMC]/[AC − AMC] as a function of 1/[Th4+] for binding constant determination of MC (5.0 × 10−5 M in EtOH) toward Th4+, measured through absorbance changes at 539 nm upon addition of ThCl4 (Ka = 8.8 ± 3.4 × 104 M−1, R2 = 0.99). | ||
| Metal ion | Ligand | Solvent | K a, M−1 | Ref. |
|---|---|---|---|---|
| Zn2+ | MC | MeCN | 6.5 × 104 | This work |
| Ce3+ | MC | MeCN | 2.3 × 105 | This work |
| Ce3+ | MC | DMF | 2.1 × 104 | This work |
| Nd3+ | MC | DMF | 5.2 × 104 | This work |
| Th4+ | MC | EtOH | 8.8 × 104 | This work |
| Th4+ | MC | DMF | 7.3 × 103 | This work |
| U4+ | MC | EtOH | 1.3 × 105 | This work |
| U4+ | MC | DMF | 5.8 × 103 | This work |
| UO22+ | MC | DMF | 5.3 × 103 | This work |
| Pu4+ | MC | DMF | 4.3 × 104 | This work |
| Eu3+ | L1 | MeCN | 3.0 × 106 | 23 |
| Zn2+ | L2 | MeCN | 1.4 × 104 | 24 |
| Zn2+ | L3 | MeCN | 1.6 × 104 | 25 |
|
||||
As a reference, we evaluated the binding constant for the MC–Zn2+ complex in acetonitrile for comparison with the previously reported one for zinc cations and methoxy-functionalized spiropyran in acetonitrile.24 Indeed, the binding constant of 6.5 × 104 M−1 estimated in the presented studies is in line with the value, 1.4 × 104 M−1,24 previously reported for chelation of zinc cations by methoxy-functionalized merocyanine (Scheme 1 and Table 1). In addition, we also probed the binding constants of non-radioactive trivalent cerium and neodymium cations. Applying the described UV-vis spectroscopic analysis for evaluation of MC–Ce3+ and MC–Nd3+ binding constants, we were able to estimate the MC–Nd3+ binding constant in DMF as 5.2 × 104 M−1 which is similar to that of the Nd(III) coordinated to serum albumins (5.7 × 104 M−1).78 The MC–Ce3+ binding constant in DMF was found to be 2.1 × 104 M−1 using Ce(NO3)3·6H2O salt for coordination.
After confirming the feasibility of the selected methodology and experimental setup on the example of binding constants for non-radioactive MC–Zn2+, MC–Ce3+, and MC–Nd3+, we transitioned toward studies of radioactive U- and Th-based salts such as ThCl4, UCl4, and UO2(NO3)2 for which the corresponding binding constants were evaluated in DMF and EtOH. In particular, the MC–Th4+ binding constant in EtOH was estimated to be 8.8 × 104 M−1 which is in line with the data previously reported for the binding, for instance, of thorium cations to other organic non-photochromic molecules, including pyrene-based fluorophores (4.5 × 104 M−1).79 In a similar vein, the estimated MC–U(VI) binding constant in DMF (5.3 × 103 M−1) was in the same order magnitude as the binding constant reported for uranyl ions coordinated to cross-linked hydrogel networks (1.2 × 103 M−1).80 As shown in Tables 1 and S2,† the binding constants obtained by UV-vis spectroscopic analysis are in agreement with the ITC-derived ones (e.g., 9.1 × 104 M−1 for ThCl4 in EtOH based on ITC data versus 8.8 × 104 M−1 based on UV-vis spectroscopy). Likewise, UV-vis spectroscopic analysis of the MC–U(IV) interactions in EtOH revealed a binding constant of 1.3 × 105 M−1, which is in line with the value obtained through ITC experiments (1.9 × 105 M−1).
The detailed information regarding UV-vis spectra and data fitting analysis for estimating the reported binding constants can be found in Fig. S11–S28.† Thus, to place them in perspective, the estimated binding constants for MC with the selected cations (e.g., Zn2+, Ce3+, Nd3+, Th4+, U4+, and UO22+), which are in the range of 5.3 × 103 to 2.3 × 105 M−1 (Table 1), are consistent with the values reported for actinide capture by sorbent materials (3.4 × 103 to 4.5 × 104 M−1)72,78,79 or chelation by fluorophores (e.g., 6.0 × 103 M−1 for Th(IV)).81 However, when comparing binding constants reported herein and in the broader scientific literature, it is important to note that many variables, including solution concentration, solvent, oxidation state of the metal cation, and counterion, may impact the formation of spiropyran-based complexes. Systematic studies of the correlation between the oxidation states of actinides and binding constants in photochromic actinide-base complexes are underway.
In addition to estimating the binding constants for MC–metal cation interactions, we also analyzed the obtained UV-vis spectroscopic data using Job's plots to evaluate the binding stoichiometry for the MC–Mn+ complexes (Fig. 6).82 For this, the absorbance profiles of a series of solutions containing varying equivalents of SP and metal salts (e.g., Ce(NO3)3, NdCl3, UCl4, UO2(NO3)2, and ThCl4) were analyzed spectroscopically. The absorbance band centered around the 433 nm region (the exact λmax is metal-salt-dependent) corresponding to the MC–metal complex was monitored as a function of the MC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn+ molar ratio (Fig. 4 and S29–S33†).82 Next, the molar ratio, at which the maximum absorbance of the complex was obtained, was taken to be the preferred binding stoichiometry based on previous literature reports (Fig. 6).82,83 For example, we determined a ratio of 3:1 for MC–Nd3+ and MC–Ce3+ complexes, while a binding ratio of 4:1 was determined for MC–U4+ and MC–Th4+ complexes. Finally, a ratio of 2:1 was determined for the MC–UO22+ complex (Fig. S29–S33†). As a result, the determined stoichiometry for each of the studied complexes was consistent with the charge of the metal cation.
:Mn+ molar ratio (Fig. 4 and S29–S33†).82 Next, the molar ratio, at which the maximum absorbance of the complex was obtained, was taken to be the preferred binding stoichiometry based on previous literature reports (Fig. 6).82,83 For example, we determined a ratio of 3:1 for MC–Nd3+ and MC–Ce3+ complexes, while a binding ratio of 4:1 was determined for MC–U4+ and MC–Th4+ complexes. Finally, a ratio of 2:1 was determined for the MC–UO22+ complex (Fig. S29–S33†). As a result, the determined stoichiometry for each of the studied complexes was consistent with the charge of the metal cation.
 | ||
| Fig. 6 The Job's plot analysis of changes in the absorbance profile of an MC solution at 433 nm as a function of the mole fraction of U4+ (gray squares), Ce3+ (blue triangles), and UO22+ (red circles) added, resulting in a complex stoichiometry of 4:1 (MC–U4+), 3:1 (MC–Ce3+), and 2:1 (MC–UO22+). Specific details regarding solution preparation for Job's plot analysis can be found in the ESI. The Job's plot analyses for the other MC–Mn+ complexes can be found in Fig. S29–S33.† | ||
Finally, we expanded our studies toward 239Pu(IV), as a representative example of transuranic elements central to the nuclear fuel cycle,84–86 using Pu(NO3)4 that was prepared and handled by following all safety protocols at the Savannah River National Laboratory (SRNL) in a designated transuranics lab (Fig. 7). Using the procedure outlined above for UV-vis spectroscopic analysis of MC–Mn+ interactions, we estimated the binding constant of Pu(IV) to MC in DMF to be 4.3 × 104 M−1, which is 5.9 and 7.4-fold higher in comparison with those of Th4+ (7.3 × 103 M−1) and U4+ (5.8 × 103 M−1), respectively, measured in the same solvent system (Table 1). The estimated binding constant for the MC–Pu(IV) pair in DMF is in the same order of magnitude as the interactions between actinides (U and Th) and biomolecules, such as hemoglobin in buffered saline (logKd = 22.5).87 However, the evaluation of 239Pu(IV) binding to the photochromic spiropyran derivatives was studied for the first time in the presented work.
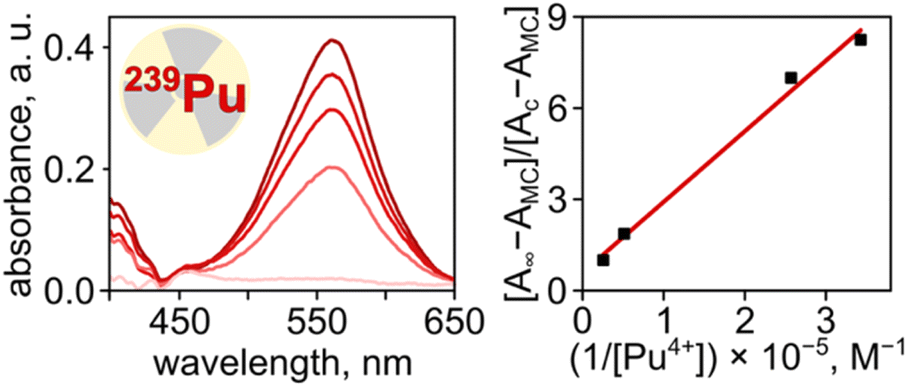 | ||
| Fig. 7 (Left) changes in absorbance spectra of spiropyran (1.43 × 10−5 M in DMF) upon exposure to a 365 nm excitation wavelength for 180 s (top trace), followed by the addition of 0.3, 1.0, 1.4, and 2.7 equivalents of Pu(NO3)4 (from top to bottom). Prior to the collection of each spectrum, the solution was irradiated with a 365 nm excitation wavelength for 180 s to promote the binding of Pu(IV) cations by merocyanine. The absorbance centered at 550 nm corresponds to uncoordinated spiropyran molecules. (Right) plot demonstrating the ratio [A∞ − AMC]/[AC − AMC] as a function of the known value of 1/[Pu4+] for binding constant determination of MC (1.43 × 10−5 M in DMF) toward Pu4+, measured through absorbance changes at 565 nm upon addition of Pu(NO3)4 (Ka = 4.3 × 104 M−1, R2 = 0.99). | ||
Theoretical modeling
To support our experimental studies of the thermodynamic parameters governing MC⋯Mn+ binding, we employed theoretical modeling using the experimentally obtained X-ray crystal structure of MC bound to UO2(NO3)2 as a starting point (Fig. 3). We first modeled the interaction between MC and either uranyl or thorium cations in DMF, which coincides with the conditions used for the previously described UV-vis spectroscopic studies (Fig. 4 and 6). Density functional theory (DFT; TPSSh functional and 6-311G* basis set) was employed to model the binding interactions between spiropyran and either uranyl, thorium, or plutonium(IV) at room temperature while incorporating the solvation effects of DMF (Fig. 8). The method used to calculate the binding energy, along with detailed parameters such as basis sets and pseudopotentials, are provided in the ESI. | ||
| Fig. 8 (Left) the optimized structure of MC coordinated to the plutonium(IV) cation. The gray, blue, red, green, and orange represent carbon, nitrogen, oxygen, chloride, and plutonium atoms, respectively. Hydrogen atoms are omitted for clarity. (Right) plot demonstrating the experimentally determined MC⋯Mn+ binding constants (turquoise color) for U(VI), Th(IV), and Pu(IV) in DMF and estimated binding energy (in DMF) based on theoretical calculations (gray). | ||
As a result, the theoretical binding energies were estimated to be −77 kJ mol−1 (UO22+), −106 kJ mol−1 (Th4+), and −132 kJ mol−1 (Pu4+) in DMF, respectively, using chloride anions to balance the charge (Fig. 8). The found trend demonstrating changes in binding constants is in line with the experimentally determined values based on UV-vis or ITC data (Fig. 4 and 6 and Tables 1 and S2†). We also expanded our theoretical studies toward EtOH as a solvent to mimic the conditions used in the previously described ITC experiments (Fig. 3). In this solvent, our findings indicated that the binding energy changes in the following order of Pu(IV) (−130 kJ mol−1) > Th(IV) (−108 kJ mol−1) > U(VI) (−50 kJ mol−1). The calculations performed considering an EtOH solvate system show a consistent trend with our experimental values.
Photochromic MOF-promoted actinide binding
Based on the outcomes of the aforementioned thermodynamic and kinetics studies demonstrating the coordination of actinide cations to a merocyanine derivative, we probed the feasibility of utilizing the spiropyran derivatives grafted to a solid-state matrix with the assumption that the integrated spiropyran derivatives will undergo reversible SP ⇄ MC isomerization upon exposure to an external stimulus such as light. For that, we integrated SP–COOH (Fig. 2) into a porous host matrix, resulting in the formation of UiO-67 + SP–COOH, as discussed earlier (Fig. 2 and 10).As a next step, we evaluated the photochromic behavior by probing the photoisomerization of integrated SP–COOH units through time-resolved diffuse reflectance spectroscopy (Fig. 9). As expected, based on previous reports18 and the acquired data for SP in solution herein, an increase of the band centered at 590 nm was detected upon irradiation of UiO-67 + SP–COOH with 365 nm light, corresponding to SP–COOH–to-MC–COOH isomerization. Subsequent exposure to white light (λex = 400–900 nm) resulted in a decrease in the sample absorbance (λmax = 590 nm), indicating successful photoisomerization from the merocyanine to the spiropyran photoisomer (Fig. 9). Thus, our studies confirmed that coordinatively-integrated SP–COOH could exhibit successful isomerization within a porous MOF. Based on crystallographic analysis of the UiO-67, SP–COOH, and MC–COOH structures (Fig. S37 and S39†) and subsequent spectroscopic characterizations, SP–COOH photoisomerization occurs within the framework voids.
 | ||
| Fig. 9 (Left) diffuse reflectance spectra of UiO-67 + SP–COOH upon exposure to a 365 nm excitation wavelength for 30 s followed by exposure to white light (λex = 400–900 nm). The gradient from orange to yellow indicates photoisomerization from the merocyanine to spiropyran isomers. (Right) plot demonstrating the change in absorbance of UiO-67 + SP–COOH after exposure to a 365 nm excitation wavelength for 30 s followed by exposure to white light (λex = 400–900 nm). Data fitting (orange line) with a first-order exponential decay equation resulted in a photoisomerization rate constant (k) of 0.04 s−1. | ||
As a proof of concept, we also probed whether the integrated SP–COOH is still capable of actinide coordination upon photoisomerization to an MC-photoisomer when integrated within a solid-state matrix. In contrast to solution studies where both photochromic ligands and metal cations could “move freely”, a porous scaffold such as a MOF could slow down the metal cation diffusion. To account for that, we allowed the UiO-67 + SP–COOH suspension in an actinide solution to equilibrate for an extended time (e.g., at least 24 hours). A sufficient equilibration time was determined based on spectroscopic analysis of changes in the metal cation concentration (outside the MOF matrix).
As a starting point, we performed a control experiment to demonstrate that exposure of parent, non-photochromic UiO-67 MOF to actinide cations does not result in an alternation in actinide concentration as a function of excitation with different wavelengths. For that, we utilized Th4+ in DMF as a model system since we confirmed its facile coordination to MC-isomer in solution (vide supra). As expected, no detectable alternation in thorium concentration in the presence of UiO-67 was detected upon exposure to UV or visible light (Fig. 10). We have also explored different actinide concentrations using thorium cations as an example, but for all studied concentrations, no significant changes were detected spectrophotometrically. In contrast to parent UiO-67, the use of photochromic UiO-67 + SP–COOH resulted in changes in thorium concentration upon light exposure as shown in Fig. 10. Photochromic UiO-67 + SP–COOH was exposed to a 20 mM solution of Th(NO3)4 in DMF for 120 h at 75 °C (i.e., similar conditions used for the control experiment). After 120 h of equilibration time, we determined that ∼45% of Th4+ was adsorbed in the framework pores by monitoring the concentration of the Th4+ in the supernatant spectrophotometrically (see more details in the ESI; Fig. S10†). In particular, aliquots of the Th@UiO-67 + SP–COOH suspension were diluted with a complexing solution of arsenazo(III), and the changes in the absorbance band centered at 667 nm, which corresponds to the arsenazo(III)–Th complex, were monitored to study the capture and release of Th4+ from the UiO-67 + SP–COOH framework (Fig. 10). After collecting the initial absorbance profile, the Th@UiO-67 + SP–COOH suspension was irradiated with a 365 nm excitation wavelength for 10 minutes to facilitate the formation of the MC-photoisomer (MC-photoisomer formation occurs under this irradiation time based on control experiments).
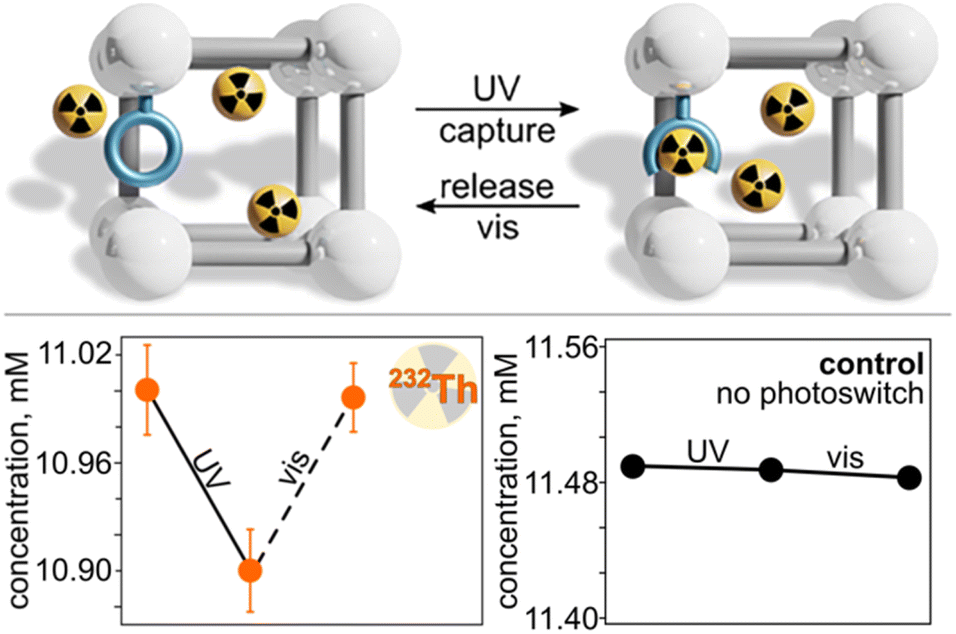 | ||
| Fig. 10 (Top) schematic representation of on-demand actinide capture and release using UiO-67 + SP–COOH upon exposure to UV (365 nm) or visible light (590 nm). (Bottom left) plot demonstrating the change in Th(IV) concentration in the UiO-67 + SP–COOH suspension as a function of excitation wavelength. (Bottom right) plot demonstrating no change in Th(IV) concentration in a non-photochromic UiO-67 suspension as a function of excitation wavelength. | ||
As shown for studies in solution, the MC-photoisomer could coordinate to the thorium cations (vide supra). The Th@UiO-67 + SP–COOH suspension was then allowed to equilibrate at room temperature for 24 h to accommodate for slow diffusion of thorium cations throughout the bulk material. Similar to the control experiments, the equilibration time was chosen based on the absence of changes in thorium concentration in the supernatant solution. As a result, we detected a decrease in thorium concentration in the solution outside the MOF matrix based on UV light exposure due to the MC-photoisomer formation and subsequent Th4+ coordination. Notably, the decrease in thorium concentration in the supernatant corresponds to coordination of Th cations by approximately 33% of available SP–COOH units.
As a next step, the suspension was irradiated with visible light (λex = 590 nm) for one hour to test the hypothesis that photoisomerization from MC–COOH to SP–COOH (integrated within the MOF) would result in demetallation and diffusion of thorium cations out of the MOF pores into the supernatant. Similar to samples after UV irradiation, the MOF suspension was allowed to equilibrate at room temperature over the course of one day prior to analysis. As expected, the absorbance of the band centered at 667 nm, which corresponded to the arsenazo(III)–Th complex, increased after visible light exposure, indicating demetallation of thorium cations followed by diffusion out of the MOF pores. Notably, these studies are a proof-of-principle for actinide coordination/demetallation, and optimization of benchmark materials with desirable characteristics is underway. Thus, as indicated by a combination of our spectroscopic analysis with the results of the control experiments, the coordination and demetallation of thorium cations to spiropyran-based compounds integrated into a MOF could be controlled through the alternation of an excitation wavelength (Fig. 10).
Conclusions
This study represents the first report of actinide binding modes controlled by an external stimulus via photochromic moieties, realized through probing thermodynamics and kinetics aspects and provides quantitative information, such as binding constants and binding energy, for the coordination of actinides to photochromic molecules, including the first X-ray crystal structure; demonstrating the possibility of coordination of actinides to photochromic molecules on the example of the MC–UO22+ system. This work also includes novel thermodynamic studies of highly radioactive transuranic 239Pu(IV) species which were carried out at the U.S. Savannah River National Laboratory according to the appropriate safety protocols. In situ spectroscopic analysis was employed to simultaneously evaluate the photoisomerization kinetics of the spiropyran derivative in the presence of actinide cations while monitoring the metal coordination to MC-photoisomer in solution and upon its integration within the solid support such as a MOF. We probed the complex stoichiometry for non-radioactive surrogates and radioactive cations using Job's plot analysis. At the same time, ITC experiments allowed us to quantify critical thermodynamic values, including Gibbs free energy, enthalpy, and entropy changes associated with actinide binding by spiropyran derivatives. As a result, we highlighted the coordination strength between actinide cations and merocyanine derivatives in general, including the MC–Pu(IV) interactions for the first time. Finally, we applied the fundamental principles found in the solution studies toward functional material design on the example of a spiropyran-functionalized MOF. As a result, we report a photochromic framework capable of photo-triggered binding and release of actinide cations as a function of excitation wavelength, demonstrating how the acquired thermodynamic and photoisomerization kinetics information can be used to guide functional material design. In summary, these studies are a critical first step for addressing urgent fundamental questions concerning thermodynamic, spectroscopic, and structural aspects of photoswitch–actinide interactions, demonstrating the proof-of-concept toward actinide binding on demand.Data availability
The data that support the findings of this study are available in the ESI† of this article. Crystallographic data for MC–COOH and the MC–UO22+ complex have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2454510 and 2409152.Author contributions
K. C. P.: investigation (lead), data curation (lead), methodology (lead), visualization (lead), writing – original draft preparation (lead). J. L.: investigation (supporting), visualization (supporting), writing – review & editing (supporting). G. C. T.: investigation (supporting), visualization (supporting), writing – original draft preparation (lead). B. K. P. M. K.: visualization (supporting), writing – review & editing (supporting). I. L.-A.: investigation (supporting), writing – review & editing (supporting). Y. L.: investigation (supporting), formal analysis (lead), methodology (supporting). J. M. B.: investigation (supporting), formal analysis (supporting), methodology (supporting). C. R. M.: investigation (supporting), writing – review & editing (supporting). A. T. T.: investigation (supporting), formal analysis (lead), methodology (supporting). A. B. G.: supervision (supporting), writing – review & editing (supporting). J. W. A.: supervision (supporting), funding acquisition (supporting). D. D. P.: supervision (supporting), funding acquisition (supporting). M. D. S.: formal analysis (supporting), investigation (supporting), visualization (supporting). S. R. P.: supervision (supporting), writing – review & editing (supporting). N. B. S.: conceptualization (lead), project administration (lead), funding acquisition (lead), writing – original draft preparation (lead), writing – review & editing (lead).Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported as part of the Center for Hierarchical Wasteform Materials (CHWM), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science under award DE-SC0016574. N. B. S. acknowledges support from the Camille Dreyfus Teaching-Scholar Award supported by the Camille and Henry Dreyfus Foundation. The synthesis of photoresponsive materials and corresponding photochromic linkers is supported by the National Science Foundation (DMR-2405868). N. B. S. is also grateful for support from the Friedrich Wilhelm Bessel Research Award. G. C. T. is supported by the National Science Foundation's Graduate Research Fellowship under grant no. DGE-2034711. Y. L., A. T. T., and S. R. P. acknowledge the University of Florida Research Computing for providing computational resources and support that have contributed to the results reported in this publication. A. B. G. and J. M. B. are supported by the US NSF under grant numbers CHE-2109064 and CHE-2404405. J. M. B. is additionally supported by a SPARC Graduate Research Grant from the Office of the Vice President for Research at the University of South Carolina.References
- Q. Qiu, Z. Sun, D. Joubran, X. Li, J. Wan, K. Schmidt-Rohr and G. G. D. Han, Angew. Chem., Int. Ed., 2023, 62, e202300723 CrossRef CAS PubMed.
- W. Fu, Y. Pi, M. Gao, W. Wang, C. Li, R. Tan and D. Yin, Chem. Commun., 2020, 56, 5993–5996 RSC.
- R. G. DiNardi, S. Rasheed, S. S. Capomolla, M. H. Chak, I. A. Middleton, L. K. Macreadie, J. P. Violi, W. A. Donald, P. J. Lusby and J. E. Beves, J. Am. Chem. Soc., 2024, 146, 21196–21202 CrossRef PubMed.
- G. C. Thaggard, J. Haimerl, R. A. Fischer, K. C. Park and N. B. Shustova, Angew. Chem., Int. Ed., 2023, 62, e202302859 CrossRef CAS.
- C. R. Martin, G. A. Leith, P. Kittikhunnatham, K. C. Park, O. A. Ejegbavwo, A. Mathur, C. R. Callahan, S. L. Desmond, M. R. Keener, F. Ahmed, S. Pandey, M. D. Smith, S. R. Phillpot, A. B. Greytak and N. B. Shustova, Angew. Chem., Int. Ed., 2021, 60, 8072–8080 CrossRef CAS PubMed.
- D. Wang, F. Schellenberger, J. T. Pham, H.-J. Butt and S. Wu, Chem. Commun., 2018, 54, 3403–3406 RSC.
- G. A. Leith, C. R. Martin, A. Mathur, P. Kittikhunnatham, K. C. Park and N. B. Shustova, Adv. Energy Mater., 2022, 12, 2100441 CrossRef CAS.
- A. Iagatti, B. Shao, A. Credi, B. Ventura, I. Aprahamian and M. D. Donato, Beilstein J. Org. Chem., 2019, 15, 2438–2446 CrossRef CAS.
- G. C. Thaggard, B. K. P. Maldeni Kankanamalage, K. C. Park, J. Haimerl, R. A. Fischer and N. B. Shustova, Chem. Phys. Rev., 2024, 5, 011305 CrossRef CAS.
- M. Qin, Y. Huang, F. Li and Y. Song, J. Mater. Chem. C, 2015, 3, 9265–9275 RSC.
- M. Mandal, D. Banik, A. Karak, S. K. Manna and A. K. Mahapatra, ACS Omega, 2022, 7, 36988–37007 CrossRef CAS.
- Y. Huang, F. Li, C. Ye, M. Qin, W. Ran and Y. Song, Sci. Rep., 2015, 5, 9724 CrossRef CAS.
- S. Pattaweepaiboon, W. Foytong, N. Phiromphu, T. Nanok, N. Kaewchangwat, K. Suttisintong and W. Sirisaksoontorn, ACS Omega, 2022, 7, 18671–18680 CrossRef CAS.
- N. Kobayashi, S. Sato, K. Takazawa, K. Ikeda and R. Hirohashi, Electrochim. Acta, 1995, 40, 2309–2311 CrossRef CAS.
- A. Radu, S. Scarmagnani, R. Byrne, C. Slater, K. T. Lau and D. Diamond, J. Phys. D: Appl. Phys., 2007, 40, 7238 CrossRef CAS.
- C. Raju, H. P. Q. Nguyen and G. G. D. Han, Chem. Sci., 2024, 15, 17273–17283 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- G. C. Thaggard, K. C. Park, J. Lim, B. K. P. Maldeni Kankanamalage, J. Haimerl, G. R. Wilson, M. K. McBride, K. L. Forrester, E. R. Adelson, V. S. Arnold, S. T. Wetthasinghe, V. A. Rassolov, M. D. Smith, D. Sosnin, I. Aprahamian, M. Karmakar, S. K. Bag, A. Thakur, M. Zhang, B. Z. Tang, J. A. Castaño, M. N. Chaur, M. M. Lerch, R. A. Fischer, J. Aizenberg, R. Herges, J.-M. Lehn and N. B. Shustova, Nat. Commun., 2023, 14, 7556 CrossRef CAS PubMed.
- P. K. Kundu, G. L. Olsen, V. Kiss and R. Klajn, Nat. Commun., 2014, 5, 3588 CrossRef PubMed.
- Z. Lin, W. Li, Q. Chen, L. Chen, C. Zhang and J. Zhang, J. Mater. Chem. C, 2022, 10, 1672–1680 RSC.
- G. C. Thaggard, J. Haimerl, K. C. Park, J. Lim, R. A. Fischer, B. K. P. Maldeni Kankanamalage, B. J. Yarbrough, G. R. Wilson and N. B. Shustova, J. Am. Chem. Soc., 2022, 144, 23249–23263 CrossRef CAS.
- Y.-D. Li, L.-F. Ma, G.-P. Yang and Y.-Y. Wang, Angew. Chem., Int. Ed., 2025, 64, e202421744 CrossRef CAS.
- F. Nourmohammadian, M. Ghahari and M. D. Gholami, J. Appl. Spectrosc., 2015, 82, 561–566 CrossRef CAS.
- A. Perry, S. J. Green, D. W. Horsell, S. M. Hornett and M. E. Wood, Tetrahedron, 2015, 71, 6776–6783 CrossRef CAS.
- M. Natali, L. Soldi and S. Giordani, Tetrahedron, 2010, 66, 7612–7617 CrossRef CAS.
- P. Kittikhunnatham, G. A. Leith, A. Mathur, J. K. Naglic, C. R. Martin, K. C. Park, K. McCullough, H. D. A. C Jayaweera, R. E. Corkill, J. Lauterbach, S. G. Karakalos, M. D. Smith, S. Garashchuk, D. A. Chen and N. B. Shustova, Angew. Chem., Int. Ed., 2022, 61, e202113909 CrossRef CAS.
- A. M. Rice, G. A. Leith, O. A. Ejegbavwo, E. A. Dolgopolova and N. B. Shustova, ACS Energy Lett., 2019, 4, 1938–1946 CrossRef CAS.
- H. Lin, Y. Yang, B. G. Diamond, T.-H. Yan, V. I. Bakhmutov, K. W. Festus, P. Cai, Z. Xiao, M. Leng, I. Afolabi, G. S. Day, L. Fang, C. H. Hendon and H.-C. Zhou, J. Am. Chem. Soc., 2024, 146, 1491–1500 CrossRef CAS.
- R. Wang, K. Shi, J. Liu, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2023, 145, 13979–13988 CrossRef CAS.
- D. Wang, S. Ostresh, D. Streater, P. He, J. Nyakuchena, Q. Ma, X. Zhang, J. Neu, G. W. Brudvig and J. Huang, Angew. Chem., Int. Ed., 2023, 62, e202309505 CrossRef CAS PubMed.
- H. Yang, F. Peng, A. N. Hong, Y. Wang, X. Bu and P. Feng, J. Am. Chem. Soc., 2021, 143, 14470–14474 CrossRef CAS PubMed.
- H. Yang, Y. Chen, C. Dang, A. N. Hong, P. Feng and X. Bu, J. Am. Chem. Soc., 2022, 144, 20221–20226 CrossRef CAS PubMed.
- J. Chen, K. Zhou, J. Li, G. Xu, X. Hei and J. Li, Chem. Sci., 2025, 16, 1106–1114 RSC.
- M. Kim, H.-S. Lee, D.-H. Seo, S. J. Cho, E.-C. Jeon and H. R. Moon, Nat. Commun., 2024, 15, 1174 CrossRef CAS PubMed.
- S.-M. Jung, S. Kim, J. An, K.-S. Kim, M. Kim, J. Kwon, H. Kim, Y.-T. Kim and S. S. Park, ACS Catal., 2025, 15, 5568–5576 CrossRef.
- F. A. Son, O. J. Bailey, T. Islamoglu and O. K. Farha, ACS Appl. Mater. Interfaces, 2024, 16, 31798–31806 CrossRef CAS PubMed.
- R. E. Sikma and S. M. Cohen, Angew. Chem., Int. Ed., 2022, 61, e202115454 CrossRef CAS PubMed.
- J. Y. Choi, B. Check, X. Fang, S. Blum, H. T. B. Pham, K. Tayman and J. Park, J. Am. Chem. Soc., 2024, 146, 11319–11327 CAS.
- G. Skorupskii, K. N. Le, D. L. M. Cordova, L. Yang, T. Chen, C. H. Hendon, M. Q. Arguilla and M. Dincă, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2205127119 CrossRef CAS PubMed.
- G. O. N. Ndjawa, M. R. Tchalala, O. Shekhah, J. I. Khan, A. E. Mansour, J. Czaban-Jóźwiak, L. J. Weselinski, H. A. Ahsaine, A. Amassian and M. Eddaoudi, Materials, 2019, 12, 2457 CrossRef CAS PubMed.
- Z. Xiao, H. F. Drake, G. S. Day, J. E. Kuszynski, H. Lin, H. Xie, P. Cai, M. R. Ryder and H.-C. Zhou, Cell Rep. Phys. Sci., 2022, 3, 101074 CrossRef CAS.
- H. Nagatomi, L. C. Gallington, S. Goswami, J. Duan, K. W. Chapman, N. Yanai, N. Kimizuka, O. K. Farha and J. T. Hupp, ACS Omega, 2020, 5, 30299–30305 CrossRef CAS PubMed.
- J. Nyakuchena, S. Ostresh, D. Streater, B. Pattengale, J. Neu, C. Fiankor, W. Hu, E. D. Kinigstein, J. Zhang, X. Zhang, C. A. Schmuttenmaer and J. Huang, J. Am. Chem. Soc., 2020, 142, 21050–21058 CrossRef CAS.
- E. Velasco, G. Zhang, S. J. Teat, K. Tan, S. Ullah, T. Thonhauser and J. Li, Inorg. Chem., 2023, 62, 16435–16442 CrossRef CAS.
- S. Cho, C. Park, M. Jeon, J. H. Lee, O. Kwon, S. Seong, J. Kim, I.-D. Kim and H. R. Moon, Chem. Eng. J., 2022, 449, 137780 CrossRef CAS.
- G. Park, M. C. Demuth, C. H. Hendon and S. S. Park, J. Am. Chem. Soc., 2024, 146, 11493–11499 CAS.
- X. Wang, K. Ma, T. Goh, M. R. Mian, H. Xie, H. Mao, J. Duan, K. O. Kirlikovali, A. E. B. S. Stone, D. Ray, M. R. Wasielewski, L. Gagliardi and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 12192–12201 CrossRef CAS PubMed.
- J. Karges, R. W. Stokes and S. M. Cohen, Dalton Trans., 2021, 50, 2757–2765 RSC.
- C. Lu, J. Y. Choi, B. Check, X. Fang, S. Spotts, D. Nuñez and J. Park, J. Am. Chem. Soc., 2024, 146, 26313–26319 CrossRef CAS PubMed.
- G. C. Thaggard, G. R. Wilson, M. Naik, M. A. Quetel, J. Lim, B. K. P. Maldeni Kankanamalage, M. D. Smith and N. B. Shustova, J. Am. Chem. Soc., 2024, 146, 31746–31756 CrossRef CAS.
- N. Ko, J. Hong, S. Sung, K. E. Cordova, H. J. Park, J. K. Yang and J. Kim, Dalton Trans., 2015, 44, 2047–2051 RSC.
- J. Lim, K. C. Park, G. C. Thaggard, Y. Liu, B. K. P. Maldeni Kankanamalage, D. J. Toler, A. T. Ta, P. Kittikhunnatham, M. D. Smith, S. R. Phillpot and N. B. Shustova, J. Am. Chem. Soc., 2024, 146, 12155–12166 CrossRef CAS PubMed.
- G. Kaur, S. Øien-Ødegaard, A. Lazzarini, S. M. Chavan, S. Bordiga, K. P. Lillerud and U. Olsbye, Cryst. Growth Des., 2019, 19, 4246–4251 CrossRef CAS.
- K. C. Park, C. R. Martin, G. A. Leith, G. C. Thaggard, G. R. Wilson, B. J. Yarbrough, B. K. P. Maldeni Kankanamalage, P. Kittikhunnatham, A. Mathur, I. Jatoi, M. A. Manzi, J. Lim, I. Lehman-Andino, A. Hernandez-Jimenez, J. W. Amoroso, D. P. DiPrete, Y. Liu, J. Schaeperkoetter, S. T. Misture, S. R. Phillpot, S. Hu, Y. Li, A. Leydier, V. Proust, A. Grandjean, M. D. Smith and N. B. Shustova, J. Am. Chem. Soc., 2022, 144, 16139–16149 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- R. J. Drout, S. Kato, H. Chen, F. A. Son, K.-I. Otake, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 12357–12366 CrossRef CAS PubMed.
- S. Kato, R. J. Drout and O. K. Farha, Cell Rep. Phys. Sci., 2020, 1, 100006 CrossRef.
- A. B. Greytak, S. L. Abiodun, J. M. Burrell, E. N. Cook, N. P. Jayaweera, M. M. Islam and A. E. Shaker, Chem. Commun., 2022, 58, 13037–13058 RSC.
- M. Bastos, O. Abian, C. M. Johnson, F. Ferreira-da-Silva, S. Vega, A. Jimenez-Alesanco, D. Ortega-Alarcon and A. Velazquez-Campoy, Nat. Rev. Methods Primers, 2023, 3, 17 CrossRef CAS.
- W. R. Archer and M. D. Schulz, Soft Matter, 2020, 16, 8760–8774 RSC.
- D. Prozeller, S. Morsbach and K. Landfester, Nanoscale, 2019, 11, 19265–19273 RSC.
- A. Thanassoulas, P. Barthélémy, L. Navailles and G. Sigaud, J. Phys. Chem. B, 2014, 118, 6570–6585 CrossRef CAS PubMed.
- Y. Ding and J. Liu, ChemBioChem, 2024, 25, e202400225 CrossRef CAS.
- R. Migliore, M. N. Zavalishin, G. A. Gamov, T. R. Usacheva, V. A. Sharnin, G. I. Grasso and C. Sgarlata, J. Therm. Anal. Calorim., 2022, 147, 5483–5490 CrossRef CAS.
- U. Osswald, J. Boneberg and V. Wittmann, Chem.–Eur. J., 2022, 28, e202200267 CrossRef CAS PubMed.
- F. Höglsperger, F. A. Larik, C. Bai, M. D. Seyfried, C. Daniliuc, H. Klaasen, P. Thordarson, J. E. Beves and B. J. Ravoo, Chem.–Eur. J., 2023, 29, e202302069 CrossRef PubMed.
- J. S. Boruah and D. Chowdhury, Appl. Nanosci., 2022, 12, 4005–4017 CrossRef CAS.
- E. B. Gaeva, V. Pimienta, A. V. Metelitsa, N. A. Voloshin, V. I. Minkin and J. C. Micheau, J. Phys. Org. Chem., 2005, 18, 315–320 CrossRef CAS.
- R. F. Khairutdinov, K. Giertz, J. K. Hurst, E. N. Voloshina, N. A. Voloshin and V. I. Minkin, J. Am. Chem. Soc., 1998, 120, 12707–12713 CrossRef CAS.
- K. C. Park, P. Kittikhunnatham, J. Lim, G. C. Thaggard, Y. Liu, C. R. Martin, G. A. Leith, D. J. Toler, A. T. Ta, N. Birkner, I. Lehman-Andino, A. Hernandez-Jimenez, G. Morrison, J. W. Amoroso, H.-C. zur Loye, D. P. DiPrete, M. D. Smith, K. S. Brinkman, S. R. Phillpot and N. B. Shustova, Angew. Chem., Int. Ed., 2023, 62, e202216349 CrossRef CAS PubMed.
- H. Liang, Q. Chen, C. Xu and X. Shen, Sep. Purif. Technol., 2019, 210, 835–842 CrossRef CAS.
- A. Kumar, M. Ali, R. S. Ningthoujam, P. Gaikwad, M. Kumar, B. B. Nath and B. N. Pandey, J. Hazard. Mater., 2016, 307, 281–293 CrossRef CAS PubMed.
- J. Meng, H. Xu, Z. Li, S. Xu and C. Yao, Tetrahedron, 2017, 73, 6637–6643 CrossRef CAS.
- C. R. Martin, K. C. Park, G. A. Leith, J. Yu, A. Mathur, G. R. Wilson, G. B. Gange, E. L. Barth, R. T. Ly, O. M. Manley, K. L. Forrester, S. G. Karakalos, M. D. Smith, T. M. Makris, A. K. Vannucci, D. V. Peryshkov and N. B. Shustova, J. Am. Chem. Soc., 2022, 144, 4457–4468 CrossRef CAS PubMed.
- E. J. Watkinson, R. M. Ambrosi, H. R. Williams, M. J. Sarsfield, K. Stephenson, D. P. Weston, N. Marsh and C. Haidon, J. Nucl. Mater., 2017, 486, 308–322 CrossRef CAS.
- M. Tribet, C. Jégou, S. Miro, J. Delrieu, F. Doreau and S. Peuget, J. Nucl. Mater., 2023, 585, 154634 CrossRef CAS.
- I. D. Kuntz, F. P. Gasparro, M. D. Johnston and R. P. Taylor, J. Am. Chem. Soc., 1968, 90, 4778–4781 CrossRef CAS.
- R. Karmakar and K. Sen, J. Radioanal. Nucl. Chem., 2019, 322, 57–66 CrossRef CAS.
- B. K. Rani and S. A. John, New J. Chem., 2017, 41, 12131–12138 RSC.
- D. Saraydin, D. Şolpan, Y. Işıkver, S. Ekici and O. Güven, J. Macromol. Sci., Part A: Pure Appl. Chem., 2002, 39, 969–990 CrossRef.
- R. Selva Kumar and S. K. Ashok Kumar, Dalton Trans., 2019, 48, 12607–12614 RSC.
- D. Samanta, D. Galaktionova, J. Gemen, L. J. W. Shimon, Y. Diskin-Posner, L. Avram, P. Král and R. Klajn, Nat. Commun., 2018, 9, 641 CrossRef PubMed.
- H. Zhao, J. Huai, C. Weng and H. Han, J. Mol. Struct., 2022, 1263, 133146 CrossRef CAS.
- T. Taiwo and R. Wigeland, Ann. Nucl. Energy, 2021, 156, 108182 CrossRef CAS.
- L. W. Cooper, J. M. Kelley, L. A. Bond, K. A. Orlandini and J. M. Grebmeier, Mar. Chem., 2000, 69, 253–276 CrossRef CAS.
- B. M. Rotermund, J. M. Sperling, G. P. Horne, N. B. Beck, H. B. Wineinger, Z. Bai, C. Celis-Barros, D. G. Martinez and T. E. Albrecht-Schönzart, Inorg. Chem., 2023, 62, 12905–12912 CrossRef CAS PubMed.
- S. Sauge-Merle, D. Lemaire, R. W. Evans, C. Berthomieu and J. Aupiais, Dalton Trans., 2017, 46, 1389–1396 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, experimental procedures, UV-vis absorbance spectra, and powder X-ray diffraction patterns. CCDC 2409152 and 2454510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03171k |
| This journal is © The Royal Society of Chemistry 2025 |