
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical hydrogenative coupling of nitrobenzene into azobenzene over a mesoporous palladium–sulfur cathode†
Jie
Xiao
,
Yanzhi
Wang
,
Bo
Xiao
* and
Ben
Liu
 *
*
Key Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, China. E-mail: xiaobo_chem@scu.edu.cn; ben.liu@scu.edu.cn
First published on 15th April 2025
Abstract
Azobenzene (AZO) and its derivatives are of great importance in the dyestuff and pharmaceutical industries; however, their sustainable synthesis is much slower than expected due to the lack of high-performance catalysts. In this work, we report a robust yet highly efficient catalyst of PdS mesoporous nanospheres (MNSs) with confined mesostructures and binary elemental composition that achieved sustainable electrosynthesis of value-added AZO by selective hydrogenative coupling of nitrobenzene (NB) feedstocks in H2O under ambient conditions. Using a renewable electricity source and H2O, binary PdS MNSs exhibited a remarkable NB conversion of 95.4%, impressive AZO selectivity of 93.4%, and good cycling stability in selective NB hydrogenation reaction (NBHR) electrocatalysis. Detailed mechanism studies revealed that the confined mesoporous microenvironment of PdS MNSs facilitated the hydrogenative coupling of key intermediates (nitrosobenzene and phenylhydroxylamine) into AZO and/or azoxybenzene (AOB), while their electron-deficient S sites stabilized the Pd-spillovered active H* and inhibited the over-hydrogenation of AZO/AOB into AN. By coupling with the anodic methanol oxidation reaction (MOR), the (−)NBHR‖MOR(+) two-electrode system exhibits much better NB-to-AZO performance in a sustainable and energy-efficient manner. This work thus paves the way for designing functional mesoporous metal alloy electrocatalysts applied in the sustainable electrosynthesis of industrial value-added chemicals.
Introduction
Azobenzene (AZO) and its derivatives are important organic chemicals, which have been widely utilized as building blocks in the dyestuff and pharmaceutical industries, as well as in the photonics field, because of the excellent optical and photochromic properties of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/NN–O moieties.1–6 Selective hydrogenation of nitrobenzene (NB) feedstocks has been extensively used to synthesize AZO and its derivatives. Of the various routes available, the thermochemical reduction of NB is recognized as the most popular route for the industry-scale production of these chemicals.7–11 However, this route involves the utilization of pressurized hydrogen gas and/or other costly hydrogen sources at high temperature, resulting in serious safety risks and environmental concerns. Alternatively, photocatalytic coupling of NB has been investigated to produce AZO and its derivatives by direct or transfer hydrogenation routes.12–16 Unfortunately, the selectivity depends strongly on the irradiation wavelengths and photocatalysts used, remarkably restricting their wide practical application (Fig. 1). Therefore, it is of great importance to develop a sustainable route for selective and efficient AZO synthesis from NB hydrogenation under mild conditions.
N/NN–O moieties.1–6 Selective hydrogenation of nitrobenzene (NB) feedstocks has been extensively used to synthesize AZO and its derivatives. Of the various routes available, the thermochemical reduction of NB is recognized as the most popular route for the industry-scale production of these chemicals.7–11 However, this route involves the utilization of pressurized hydrogen gas and/or other costly hydrogen sources at high temperature, resulting in serious safety risks and environmental concerns. Alternatively, photocatalytic coupling of NB has been investigated to produce AZO and its derivatives by direct or transfer hydrogenation routes.12–16 Unfortunately, the selectivity depends strongly on the irradiation wavelengths and photocatalysts used, remarkably restricting their wide practical application (Fig. 1). Therefore, it is of great importance to develop a sustainable route for selective and efficient AZO synthesis from NB hydrogenation under mild conditions.
 | ||
| Fig. 1 Comparison of the routes for AZO synthesis from NB. Schematic illustrations of reported routes (including chemical reduction and photocatalytic coupling) and the newly developed electrocatalytic route for AZO synthesis from NB. | ||
Recently, electrochemical transformation in H2O has been proposed as a green and appealing route in synthetic chemistry for the electrosynthesis of valued-added chemicals.17–19 In contrast to other methods, H2O as a sustainable hydrogen source can be electrochemically reduced into active H*, which can further participate in chemical hydrogenation reactions to produce a variety of products.20–22 However, the chemisorption of active H* heavily depends on the electrocatalyst materials used, which determine the product selectivity of the route, especially considering the competitive hydrogen evolution reaction (HER) at the cathode.23,24 In the past five years, some important electrocatalysts have been designed to engineer active H* and exhibit promising performance in electrochemical alkene semi-hydrogenation and CO2 reductions.19,25–29 Despite some impressive progress, the achievements in electrochemical NB hydrogenation reaction (NBHR) for selective AZO electrosynthesis by engineering active H* in H2O are still in their infancy.30–33 This is likely because of the complicated multi-electron synthesis of thermodynamically unfavorable nitrosobenzene and phenylhydroxylamine intermediates as well as their further hydrogenative coupling, in which undesired electrocatalysts enable thermodynamically favorable aniline (AN) electrosynthesis (Fig. S1†).34–37
In this work, we report that the synergy of a mesoporous structure and Pd-nonmetal alloy efficiently promotes sustainable AZO electrosynthesis from NB in H2O under ambient conditions, without the over-hydrogenation of AZO into thermodynamically favorable AN. The best PdS mesoporous nanospheres (MNSs) exhibited impressive NB conversion (95.4%), remarkable AZO selectivity (93.4%), and high cycling selectivity in selective NB-to-AZO electrocatalysis. We proposed that the confined mesoporous microenvironment of PdS MNSs optimized the hydrogenative coupling of nitrosobenzene and phenylhydroxylamine intermediates into AZO and/or azoxybenzene (AOB), while their adjacent S–Pd–S sites stabilized the active H* and inhibited competitive HER and over-hydrogenation of AZO/AOB. Moreover, PdS MNSs performed well as bifunctional electrocatalysts in the two-electrode coupling system, in which anodic methanol oxidation reaction (MOR) further promoted cathodic NB-to-AZO electrocatalysis, further demonstrating the sustainable electrosynthesis of AZO in an energy-efficient manner.
Results and discussion
Electrochemical NBHR studies were performed at room temperature (25 °C) in 1.5 mM NB with 1.0 M KOH as the electrolyte and dimethylsulfoxide (DMSO) as the co-solvent. Three products, including AZO, AOB, and AN, were evaluated here by high-performance liquid chromatography (HPLC) and 1H nuclear magnetic resonance (NMR) for selective NBHR electrocatalysis (Fig. 2a and S2–S4†). Linear sweep voltammetry (LSV) curves were first recorded in 1.0 M KOH with and without NB (Fig. S5†). In comparison to the one collected without NB, the LSV curve of PdS MNSs increased remarkably in the presence of NB, suggesting the promotion of favorable NBHR and the inhibition of competitive HER at the cathode. Further, the long-term chronoamperometry test was performed to investigate the selectivity and conversion of NBHR electrocatalysis in the potential range of −0.7 to −1.0 V (vs. Ag/AgCl) (Fig. 2b). At the higher potential (−0.7 V), 20.2% of NB was converted with electrocatalysis by PdS MNSs for 6 h. Meanwhile, all three products were monitored, and 33.7% AZO, 45.3% AOB, and 21.0% AN were recorded. Interestingly, with the increase in the potential, both NB conversion and AZO selectivity gradually increased and reached the best values at the potential of −0.9 V. Specifically, PdS MNSs exhibited the best NB conversion of 95.4% and the remarkable AZO selectivity of 93.4% with the reaction time of 6 h. AZO selectivity was slightly decreased under the higher potentials (−1.0 V), while NB conversion was still higher than 95%. Then, we studied the electrochemical NBHR performance at the potential of −0.9 V under different reaction times (Fig. 2c). In the initial stage (1 h), 44.5% of NB was electrochemically hydrogenated with the major product being AOB (75.8%) and the minor product AZO (22.4%). As the reaction proceeded, NB conversion gradually increased and reached 95.4% at 6 h. Meanwhile, the AOB intermediate was further electroreduced into AZO with a superior selectivity of 93.4%. With longer reaction times (for example 12 h), AOB cannot be fully electroreduced into AN and retained a high AZO selectivity of >93%. The results clearly highlighted the high potential of PdS MNSs as an efficient electrocatalyst for the selective electrosynthesis of high value-added AZO by electrochemical hydrogenative coupling reaction of NB in H2O (as the hydrogen source). | ||
| Fig. 2 Electrochemical performance. (a) Reaction products of electrochemical NBHR in this work. Electrochemical selectivity and conversion of NBHR (b) under different potentials (for 6 h) and (c) with different reaction times (at −0.9 V) over PdS MNSs. (d) AZO selectivity of NBHR over PdS MNSs, Pd MNSs, Pd NPs, and PdS NPs. (e) Electrochemical selectivity and conversion of NBHR over PdS MNSs, PdP MNSs, PdH MNSs, PdB MNSs, and PdN MNSs. (f) Electrochemical cycling stability of PdS MNSs in NBHR. (g) Electrochemical selectivity and conversion of AZOHR over PdS MNSs. (h) Electrochemical selectivity and conversion of AOBHR over PdS MNSs. | ||
To further reveal the intrinsic nature of PdS MNSs in selective NBHR electrocatalysis, some important reference electrocatalysts were carefully designed and studied for clear comparisons. First, monometallic Pd MNSs, binary PdS nanoparticles (NPs), and Pd NPs were used for selective NB-to-AZO electrocatalysis under the same conditions (Fig. 2d and S6–S11†). As expected, the PdS MNSs exhibited the best electrochemical NB-to-AZO selectivity at all potentials (from −0.7 to −1.0 V). In comparison, the Pd MNSs exhibited slightly lower AZO selectivity at the lower potentials (−1.0 V and −0.95 V), while PdS NPs exhibited lower AZO selectivity at the higher potentials (from −0.7 to −0.8 V). Second, four other Pd-nonmetal MNSs, PdP, PdH, PdB, and PdN, were also studied for selective NBHR electrocatalysis (Fig. S12–S16†). In comparison to PdS MNSs, PdP, PdH, and PdB MNSs exhibited slightly lower NB conversion (from 80% to 90%) and poorer AZO selectivity (from 78% to 90%) (Fig. 2e). Binary PdN MNSs exhibited the lowest NB-to-AZO performance. These results clearly confirmed that the synergy of the mesoporous structure and binary PdS composition of the PdS MNSs promoted NBHR electrocatalysis and facilitated selective AZO electrosynthesis in H2O under ambient conditions.
PdS MNSs demonstrated not only superior NB-to-AZO conversion and selectivity but also remarkable catalytic stability. After being performed for six consecutive cycles, both NB conversion (>90%) and AZO selectivity (90%) were retained well with a reaction time of 6 h (Fig. 2f). More impressively, the electrocatalytic performance slightly decreased, after 20 cycles, indicating high cycling stability of the PdS MNSs in NB-to-AZO electrocatalysis (Fig. S17†). Meanwhile, the mesoporous structure and PdS composition were maintained perfectly after NBHR electrocatalysis, further confirming the high stability of the PdS MNSs (Fig. S18†). Then, the catalytic substrate was changed from NB to AZO. Very interestingly, AZO cannot be further converted electrochemically into over-hydrogenated AN, even when the reaction is carried out for 12 h (Fig. 2g). This clearly suggested the reactive inertness of PdS MNSs for selective AZO electrocatalysis in H2O. In comparison, AOB as the substrate was quickly converted electrochemically into AZO with superior selectivity of ∼100% and high conversion of 85%, further indicating the high reactivity of AOB electrocatalysis for selective AZO electrosynthesis over PdS MNSs (Fig. 2h).
The physicochemical properties of the binary PdS MNSs were then characterized and compared to reveal the nature of their high performance in selective NB-to-AZO electrocatalysis. Transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) images showed that the products were highly uniform and homogeneous with a nearly spherical morphology (Fig. 3a and S19†). The size of the PdS MNSs was estimated to be in the range of 35–55 nm. The high-magnification HAADF-STEM image further disclosed the highly penetrated mesoporous channels that were radially extended from the center of the MNSs. The average mesopore size and framework thickness were measured to be 3.7 and 4.2 nm, respectively. The high-resolution TEM image further showed the mesoporous structure of the PdS MNSs (Fig. 3b). Meanwhile, two clear lattice fringes with interplanar distances of 0.24 nm and 0.19 nm were observed, which were precisely assigned to the (111) and (100) planes of face-centered cubic (fcc) crystals. In comparison to pure Pd, slightly smaller lattice distances confirmed that S atoms randomly substituted Pd atoms and thus formed binary PdS nanocrystals (see X-ray diffraction (XRD) patterns in Fig. S20†). Meanwhile, HAADF-STEM energy dispersive X-ray (EDX) mapping images also disclosed homogeneously dispersed Pd and S in PdS MNSs with a Pd/S ratio of 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 (Fig. 3c). A lack of phase-separated elemental compositions confirmed that the PdS MNSs were compositionally alloyed, rather than having other atomic structures. These physicochemical characterizations clearly indicated the successful synthesis of PdS MNSs with abundant mesoporous channels and binary elemental compositions.
:12 (Fig. 3c). A lack of phase-separated elemental compositions confirmed that the PdS MNSs were compositionally alloyed, rather than having other atomic structures. These physicochemical characterizations clearly indicated the successful synthesis of PdS MNSs with abundant mesoporous channels and binary elemental compositions.
 | ||
| Fig. 3 Physicochemical properties of PdS MNS cathode. (a) HAADF-STEM image, (b) high-resolution TEM image, and (c) HAADF-STEM EDX mapping images of binary PdS MNSs. Inset in (a): high-magnification HAADF-STEM image. (d) High-resolution XPS Pd 3d spectra and (e) electrochemical CO stripping curves of PdS MNSs and Pd MNSs. (f) Pd K-edge XANES spectra, (g) FT-EXAFS spectra, and (h) two-dimensional WT contour plots of PdS MNSs and Pd MNSs (PdO and Pd foils were used as the counterparts). (i) Proposed electronic structures and coordination environments of PdS MNSs around mesopores. | ||
The surface electronic structures of the PdS MNSs were then analyzed by X-ray photoelectron spectroscopy (XPS) and electrochemical CO stripping experiments. High-resolution XPS spectra revealed that, in comparison to Pd MNSs, all the binding energies of Pd 3d in the PdS MNSs positively shifted about 0.35 eV (Fig. 3d), indicating electron-deficient Pd sites (Pdδ+) due to the stronger Pd–S binding interactions (see high-resolution S 2p spectra in Fig. S21†).38 Similar electron-deficient Pd sites of PdS MNSs were also seen from electrochemical CO stripping experiments, where a negative shift of 63 mV was observed when comparing peak potentials of PdS MNSs and Pd MNSs (Fig. 3e). Moreover, X-ray adsorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) experiments were conducted and compared to reveal the electronic structure and coordination environment of PdS MNSs and Pd MNSs. As shown in Fig. 3f, the absorption edge positions of PdS and Pd MNSs were located in between those of Pd and PdO foils, demonstrating the partially oxidized states of Pd sites in the samples. In comparison to pure Pd MNSs, in particular, a higher oxidized state of Pd sites was seen for binary PdS MNSs that was mostly induced by the electron-rich S (Sδ−) sites. The Fourier transform EXAFS (FT-EXAFS) spectra of the PdS MNSs further disclosed the weak peak at ∼2.5 Å that corresponded to the Pd–Pd coordination structure. In comparison, a strong peak appeared at ∼1.8 Å, which was slightly larger than that at ∼1.6 Å (Pd–O bonds), further indicating the formation of a binary Pd–S coordination shell (Fig. 3g).38,39 Wavelet transform (WT) contour plots also confirmed the co-existence of major Pd–S bonds and minor Pd–Pd bonds in the PdS MNSs (Fig. 3h and S22†). These results clearly highlighted that alloying electron-rich S (Sδ−) changed the electronic structure of Pd and thus introduced the formation of electron-deficient Pd sites (Pdδ+) through the strong Sδ−–Pdδ+–Sδ− binding interactions in binary PdS MNSs (Fig. 3i). Such electronic structures of the PdS MNSs were thus expected to optimize the chemisorption properties of the intermediates (especially active H*) and thus enable selective NBHR electrocatalysis for AZO electrosynthesis with H2O as the H* source.
During NBHR electrocatalysis, controlling the production of active H* and inducing the subsequent hydrogenative coupling of nitrosobenzene and phenylhydroxylamine intermediates is of great importance for selective AOB electrosynthesis from NB in H2O. We thus designed a series of controlled experiments to reveal the kinetics of active H* produced from H2O during NBHR electrocatalysis. First, we conducted cyclic voltammetry (CV) studies of PdS MNSs in KOH with and without NB (Fig. 4a). There was a strong peak at 0.53 V in KOH that was assigned to active hydrogen desorption, indicating the high activity of PdS MNSs for producing active H* from H2O.40 After the addition of NB, the peak decreased remarkably, suggesting that active H* produced from H2O was dramatically consumed for NBHR electrocatalysis. Then, D2O was used instead of H2O as the solvent for NBHR electrocatalysis (Fig. S23†). Both current density and onset potential decreased significantly in D2O compared to H2O. This was attributed to the stronger and stable D–O bonds in D2O, which make the oxidative cleavage of D2O into active D* more difficult (in comparison to H*).41–44 The cations of the base electrode are discussed in the context of the selective NB-to-AZO electrocatalysis (Fig. S24†). As summarized in Fig. 4b, the highest NB conversion and AZO selectivity were achieved in the presence of KOH, and a trend of KOH > NaOH > LiOH was followed. This originated from a smaller hydrated K+ (K+(H2O)7) that favored the formation of a hybrid network of Sδ−–K+(H2O)7 on the surface of the PdS MNSs.40 Such a network optimized the oxidative cleavage of H2O for the formation of active H* and thus facilitated selective NB-to-AZO electrocatalysis. The results further highlighted the critical role of active H* produced from H2O on PdS MNSs in the electrocatalytic hydrogenative coupling of NB into value-added AZO.
 | ||
| Fig. 4 Electrocatalytic mechanism. (a) CV curves of PdS MNSs without NB and with NB collected in 1.0 M KOH. (b) Electrochemical selectivity and conversion of NBHR over PdS MNSs recorded in different base solutions. (c) Chronoamperometry measurements of PdS MNSs and Pd MNSs collected at the reduction (adsorption) and oxidation (desorption) potentials. (d) Electrochemical selectivity and conversion of NBHR over PdS MNSs and Pd MNSs in the presence of Zn(NO3)2. (e) Electrochemical in situ FTIR spectra of PdS MNSs for selective NBHR electrocatalysis with different reaction times. (f) Schematic illustration of the proposed mechanism of selective NB-to-AZO electrocatalysis in H2O over the PdS MNSs cathode. | ||
More mechanistic studies were focused on the effect of alloyed electron-rich S sites (Sδ−) in promoting selective NB-to-AZO electrocatalysis over PdS MNSs. First, we compared the amounts of active H* produced during NBHR electrocatalysis over PdS and Pd MNSs using a coulometric stripping experiment (Fig. 4c).45 Both PdS and Pd MNSs exhibit constant reductive potentials until hydrogen adsorption  reaches equilibration (Fig. S25†). After being applied at a constant oxidative potential,
reaches equilibration (Fig. S25†). After being applied at a constant oxidative potential, 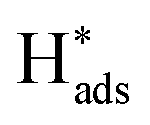 was desorbed on the PdS and Pd MNSs. Both adsorption and desorption experiments revealed that, in comparison to Pd MNSs, PdS MNSs exhibited a lower amount of
was desorbed on the PdS and Pd MNSs. Both adsorption and desorption experiments revealed that, in comparison to Pd MNSs, PdS MNSs exhibited a lower amount of 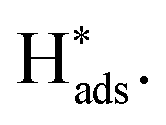 This was mostly because active
This was mostly because active 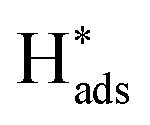 was partially transferred from Pd to S (similar to H spillover)46–48 that not only disfavored the HER but also inhibited the over-hydrogenation of AZO to AN. Second, we injected Zn(NO3)2 into the electrolyte to selectively inhibit S sites, as Zn is a sulfophilic metal that forms of stable Zn–S bonds, thereby covering S sites on the surface of the PdS MNSs.49 LSV curves of the PdS MNSs showed that, in the presence of Zn(NO3)2, the current density remarkably decreased and the onset potential shifted negatively, compared to those recorded without Zn(NO3)2 (Fig. S26†). However, almost no change in both current density and onset potential can be observed in Pd MNSs. We further compared the electrochemical NB-to-AZO performance of PdS MNSs with and without Zn(NO3)2 (Fig. 4d). After the addition of Zn(NO3)2, both NB conversion and AZO selectivity were decreased dramatically over PdS MNSs. In comparison, the Pd MNSs retained almost the NB conversion and AZO selectivity with and without Zn(NO3)2. Clearly, the decreased NB-to-AZO performance originated from the covering of the S sites with extra Zn2+ inhibitor. The results clearly indicated the role of electron-rich S sites (Sδ−) in optimizing selective NB-to-AZO electrocatalysis on PdS MNSs. To demonstrate how S sites in the PdS MNSs effectively stabilized H* and further suppressed the HER, we further calculated the adsorption energies of H* at the Pd sites and S sites in both Pd MNSs and PdS MNSs (Fig. S27†). The adsorption energies of H* were −0.16 eV and −0.08 eV on the Pd sites of Pd and PdS, respectively. In comparison, the adsorption energy of H* was −0.39 eV on the S sites of PdS. The results clearly demonstrated that H* was more effectively stabilized at the S sites, and the adsorption capability of H* at the Pd sites was reduced. This all led to the spillover of H* from Pd sites to S sites in the PdS MNSs. Meanwhile, the strong adsorption of H* at the S sites suppressed the coupling of H* to form H2, enabling the H* produced by H2O splitting to participate in the NBHR.
was partially transferred from Pd to S (similar to H spillover)46–48 that not only disfavored the HER but also inhibited the over-hydrogenation of AZO to AN. Second, we injected Zn(NO3)2 into the electrolyte to selectively inhibit S sites, as Zn is a sulfophilic metal that forms of stable Zn–S bonds, thereby covering S sites on the surface of the PdS MNSs.49 LSV curves of the PdS MNSs showed that, in the presence of Zn(NO3)2, the current density remarkably decreased and the onset potential shifted negatively, compared to those recorded without Zn(NO3)2 (Fig. S26†). However, almost no change in both current density and onset potential can be observed in Pd MNSs. We further compared the electrochemical NB-to-AZO performance of PdS MNSs with and without Zn(NO3)2 (Fig. 4d). After the addition of Zn(NO3)2, both NB conversion and AZO selectivity were decreased dramatically over PdS MNSs. In comparison, the Pd MNSs retained almost the NB conversion and AZO selectivity with and without Zn(NO3)2. Clearly, the decreased NB-to-AZO performance originated from the covering of the S sites with extra Zn2+ inhibitor. The results clearly indicated the role of electron-rich S sites (Sδ−) in optimizing selective NB-to-AZO electrocatalysis on PdS MNSs. To demonstrate how S sites in the PdS MNSs effectively stabilized H* and further suppressed the HER, we further calculated the adsorption energies of H* at the Pd sites and S sites in both Pd MNSs and PdS MNSs (Fig. S27†). The adsorption energies of H* were −0.16 eV and −0.08 eV on the Pd sites of Pd and PdS, respectively. In comparison, the adsorption energy of H* was −0.39 eV on the S sites of PdS. The results clearly demonstrated that H* was more effectively stabilized at the S sites, and the adsorption capability of H* at the Pd sites was reduced. This all led to the spillover of H* from Pd sites to S sites in the PdS MNSs. Meanwhile, the strong adsorption of H* at the S sites suppressed the coupling of H* to form H2, enabling the H* produced by H2O splitting to participate in the NBHR.
In situ Fourier-transform infrared spectroscopy (FTIR) was also carried out to reveal the intermediates and products of NBHR electrocatalysis at different times. As presented in Fig. 4e, –NO2 in NB was gradually adsorbed on the PdS MNSs and quickly reached a constant amount with a reaction time of 40 s. In comparison, –NO and N–H appeared with a reaction time of 70 s, indicating the prompt formation of nitrosobenzene and phenylhydroxylamine intermediates. More importantly, some new peaks assigned to –NN– also appeared at the same time (70 s), suggesting the hydrogenative coupling of nitrosobenzene and phenylhydroxylamine into AOB and/or AZO. Based on the above discussion, we proposed a Haber mechanism of PdS MNSs for electrochemical hydrogenative coupling of NB into AZO with H2O as the H* source (Fig. 4f). First, H2O as a solvent was electroreduced into active H* on electron-deficient Pdδ+ sites. Then, H* was quickly shifted to adjacent electron-rich Sδ− sites and further stabilized without direct hydrogenation into H2 (HER). After H* was reacted with NB, nitrosobenzene was immediately formed and further hydrogenated into phenylhydroxylamine. Thanks to concave mesopores and a confined microenvironment, –NO of nitrosobenzene was coupled with –NHOH of phenylhydroxylamine to form –N+(O−)N–(AOB). After being reacted further, –N+(O−)N– of AOB was finally hydrogenated to –NN–(AZO). Owing to the weaker hydrogenation ability of active H* on electron-rich Sδ− sites, AZO was not further over-hydrogenated into AN, thus disclosing high selectivity of AZO for selective NBHR electrocatalysis in H2O over PdS MNSs. In our controlled experiments, both Pd MNSs and PdS NPs exhibit lower AZO selectivity, further highlighting the synergy of the confined mesoporous microenvironment and binary PdS composition, promoting selective NB-to-AZO electrocatalysis.22,50,51 Moreover, the weaker adsorption ability of AZO on the PdS MNSs also confirmed that AZO favored the electrochemical desorption from PdS MNSs and thus resulted in high AZO selectivity (Fig. S28†).
Considering the sluggish kinetics and high onset potential of the anode water oxidation reaction (WOR), we designed a kinetically favorable electrochemical oxidation reaction in H2O, such as MOR, to couple with cathode NBHR in a two-electrode system using PdS MNSs as a bifunctional electrocatalyst (Fig. 5a). In comparison to traditional (−)NBHR‖WOR(+), our PdS MNSs exhibited a remarkably lower onset potential in (−)NBHRMOR(+), highlighting its high performance in the two-electrode coupling system (Fig. 5b). Specifically, the (−)NBHR‖MOR(+) coupling system required only 0.69 and 1.40 V at current densities of 5 and 10 mA cm−2, which were 1.17 and 1.26 V lower than the (−)NBHR‖WOR(+) system, respectively (Fig. 5c). More impressively, over a wide cell voltage range from −1.8 to −1.4 V, the PdS MNSs exhibit remarkable NB conversion of >98% and AZO selectivity of >92% in the (−)NBHR‖MOR(+) coupling system, both of which are much better than those in the (−)NBHR‖WOR(+) system (Fig. 5d). Meanwhile, the electrocatalytic rate of the cathode NBHR was also remarkably accelerated. As shown in Fig. 5e, with a reaction time of only 2 h, 90.3% of NB was electroreduced into AZO (91.4% selectivity) in the (−)NBHR‖MOR(+) coupling system. Meanwhile, the AZO selectivity was retained even upon reaction for 10 h. In comparison, the (−)NBHR‖WOR(+) system required at least 6 h for electroreducing ∼90% of NB under the same conditions. The results clearly highlighted the high performance of the (−)NBHR‖MOR(+) coupling system for selective AZO electrosynthesis with a coupled electrochemical oxidation reaction with faster kinetics and lower onset potential. Additionally, the (−)NBHR‖MOR(+) coupling system of the PdS MNSs was electrochemically stable, with only a slight delay in both NB conversion and AZO selectivity after 6 consecutive cycles (Fig. 5f).
 | ||
| Fig. 5 Two-electrode coupling system. (a) A scheme illustrating the two-electrode coupling system with cathode NBHR and anode MOR. (b) LSV curves of PdS MNSs in (−)NBHR‖MOR(+) and (−)NBHR‖WOR(+). (c) Cell voltages of PdS MNSs at different current densities in (−)NBHR‖MOR(+) and (−)NBHR‖WOR(+). (d) Electrochemical selectivity and conversion of PdS MNSs at different current densities in (−)NBHR‖MOR(+). (e) Time-dependent electrochemical selectivity and conversion of PdS MNSs in (−)NBHR‖MOR(+) (−1.5 V). (f) Electrochemical stability of PdS MNSs in (−)NBHR‖MOR(+) (−1.5 V). | ||
Conclusion
In conclusion, this work proposed that the synergy of a confined mesoporous microenvironment and binary PdS composition of an efficient electrocatalyst, such as PdS MNSs, can optimize the reaction pathways and promote the hydrogenative coupling of NB for value-added AZO electrosynthesis under ambient conditions in H2O (as an H* source). Mechanistic studies revealed that active H* on electron-deficient S sites (spilt over from Pd sites) inhibited electrocatalytic over-hydrogenation of NB into AN, while the confined mesoporous microenvironment facilitated the hydrogenative coupling of nitrosobenzene and phenylhydroxylamine intermediates for AZO electrosynthesis. The resultant PdS MNSs thus exhibited high NB conversion of 95.4%, remarkable AZO selectivity of 93.4%, and superior cycling stability in selective NB-to-AZO electrocatalysis in H2O. Moreover, when further coupling with kinetically favorable MOR as the anode reaction instead of sluggish WOR, the two-electrode (−)NBHR‖MOR(+) system with PdS MNSs as a bifunctional electrocatalyst exhibits much better NB-to-AZO performance with a higher reaction rate and AZO selectivity. Our findings in synergistically engineering active H* and stabilizing/coupling key intermediates are thus expected to provide a new strategy in designing new functional mesoporous binary electrocatalysts applied in the selective electrosynthesis of industrially value-added chemicals.Data availability
Additional data supporting this article have been included as part of the ESI.†Author contributions
B. L. conceived and supervised the project. J. X. and Y. W. conducted the experiments, performed the structural characterizations, and analyzed the data. B. X. provided important comments on the project. All the authors discussed the results and co-wrote the paper.Conflicts of interest
We have filed a patent on the invention.Acknowledgements
B. L. kindly acknowledges the financial supports from the National Key Research and Development Program of China (2023YFC3905002) and the Fundamental Research Funds for the Central Universities.References
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Advances and opportunities in the exciting world of azobenzenes, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef PubMed.
- S. Mehrparvar, Z. N. Scheller, C. Wölper and G. Haberhauer, Design of Azobenzene beyond Simple On–Off Behavior, J. Am. Chem. Soc., 2021, 143, 19856–19864 CrossRef CAS PubMed.
- J. Gemen, J. R. Church, T.-P. Ruoko, N. Durandin, M. J. Białek, M. Weißenfels, M. Feller, M. Kazes, M. Odaybat and V. A. Borin, et al., Disequilibrating azobenzenes by visible-light sensitization under confinement, Science, 2023, 381, 1357–1363 CrossRef CAS PubMed.
- S. Crespi, N. A. Simeth and B. König, Heteroaryl azo dyes as molecular photoswitches, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- F. Corrado, U. Bruno, M. Prato, A. Carella, V. Criscuolo, A. Massaro, M. Pavone, A. B. Muñoz-García, S. Forti and C. Coletti, et al., Azobenzene-based optoelectronic transistors for neurohybrid building blocks, Nat. Commun., 2023, 14, 6760 CrossRef CAS PubMed.
- Y. Li, B. Xue, J. Yang, J. Jiang, J. Liu, Y. Zhou, J. Zhang, M. Wu, Y. Yuan and Z. Zhu, et al., Azobenzene as a photoswitchable mechanophore, Nat. Chem., 2024, 16, 446–455 CrossRef CAS PubMed.
- J. Li, S. Song, Y. Long, L. Wu, X. Wang, Y. Xing, R. Jin, X. Liu and H. Zhang, Investigating the Hybrid-Structure-Effect of CeO2-Encapsulated Au Nanostructures on the Transfer Coupling of Nitrobenzene, Adv. Mater., 2018, 30, 1704416 CrossRef PubMed.
- X. Liu, H.-Q. Li, S. Ye, Y.-M. Liu, H.-Y. He and Y. Cao, Gold-Catalyzed Direct Hydrogenative Coupling of Nitroarenes to Synthesize Aromatic Azo Compounds, Angew. Chem., Int. Ed., 2014, 53, 7624–7628 CrossRef CAS PubMed.
- Z. Yuan, L. Huang, Y. Liu, Y. Sun, G. Wang, X. Li, J. A. Lercher and Z. Zhang, Synergy of Oxygen Vacancies and Base Sites for Transfer Hydrogenation of Nitroarenes on Ceria Nanorods, Angew. Chem., Int. Ed., 2024, 63, e202317339 CrossRef CAS PubMed.
- D. He, T. Li, X. Dai, S. Liu, X. Cui and F. Shi, Construction of Highly Active and Selective Molecular Imprinting Catalyst for Hydrogenation, J. Am. Chem. Soc., 2023, 145, 20813–20824 CrossRef CAS PubMed.
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang and Z. Zhao, et al., Interfacial electronic effects control the reaction selectivity of platinum catalysts, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed.
- J. Song, Z.-F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J.-J. Zou, Review on selective hydrogenation of nitroarene by catalytic, photocatalytic and electrocatalytic reactions, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS.
- B. Wang, Z. Deng and Z. Li, Efficient chemoselective hydrogenation of nitrobenzene to aniline, azoxybenzene and azobenzene over CQDs/ZnIn2S4 nanocomposites under visible light, J. Catal., 2020, 389, 241–246 CrossRef CAS.
- X. Guo, C. Hao, G. Jin, H.-Y. Zhu and X.-Y. Guo, Copper Nanoparticles on Graphene Support: An Efficient Photocatalyst for Coupling of Nitroaromatics in Visible Light, Angew. Chem., Int. Ed., 2014, 53, 1973–1977 CrossRef CAS PubMed.
- F. Xu, K. Meng, S. Cao, C. Jiang, T. Chen, J. Xu and J. Yu, Step-by-Step Mechanism Insights into the TiO2/Ce2S3 S-Scheme Photocatalyst for Enhanced Aniline Production with Water as a Proton Source, ACS Catal., 2022, 12, 164–172 CrossRef CAS.
- Y. Dai, C. Li, Y. Shen, T. Lim, J. Xu, Y. Li, H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, Light-tuned selective photosynthesis of azo- and azoxy-aromatics using graphitic C3N4, Nat. Commun., 2018, 9, 60 CrossRef PubMed.
- C. Liu, F. Chen, B.-H. Zhao, Y. Wu and B. Zhang, Electrochemical hydrogenation and oxidation of organic species involving water, Nat. Rev. Chem., 2024, 8, 277–293 CrossRef CAS PubMed.
- C. Liu, Y. Wu, B. Zhao and B. Zhang, Designed Nanomaterials for Electrocatalytic Organic Hydrogenation Using Water as the Hydrogen Source, Acc. Chem. Res., 2023, 56, 1872–1883 CrossRef CAS PubMed.
- Y. Zhao, J. Xu, K. Huang, W. Ge, Z. Liu, C. Lian, H. Liu, H. Jiang and C. Li, Dopant- and Surfactant-Tuned Electrode–Electrolyte Interface Enabling Efficient Alkynol Semi-Hydrogenation, J. Am. Chem. Soc., 2023, 145, 6516–6525 CrossRef CAS PubMed.
- G. Han, G. Li and Y. Sun, Electrocatalytic dual hydrogenation of organic substrates with a Faradaic efficiency approaching 200%, Nat. Catal., 2023, 6, 224–233 CrossRef CAS.
- H. Xu, G. Xu, B. Huang, J. Yan, M. Wang, L. Chen and J. Shi, Zn-Organic Batteries for the Semi-Hydrogenation of Biomass Aldehyde Derivatives and Concurrently Enhanced Power Output, Angew. Chem., Int. Ed., 2023, 62, e202218603 Search PubMed.
- L. Sun, H. Yao, F. Jia, Y. Wang and B. Liu, Intermediate Confinement for Selective Ammonia Electrosynthesis from Nitrate on Robust Mesoporous Metal Catalysts, Adv. Energy Mater., 2023, 13, 2302274 Search PubMed.
- Y. Wang, H. Lv, L. Sun, F. Jia and B. Liu, Ordered Mesoporous Intermetallic Trimetals for Efficient and pH-Universal Hydrogen Evolution Electrocatalysis, Adv. Energy Mater., 2022, 12, 2201478 CrossRef CAS.
- L. Sun, H. Yao, Y. Wang, C. Zheng and B. Liu, Mesostructures Engineering to Promote Selective Nitrate-to-Ammonia Electroreduction, Adv. Energy Mater., 2023, 13, 2303054 CrossRef CAS.
- Y. Wu, C. Liu, C. Wang, S. Lu and B. Zhang, Selective Transfer Semihydrogenation of Alkynes with H2O (D2O) as the H (D) Source over a Pd-P Cathode, Angew. Chem., Int. Ed., 2020, 59, 21170–21175 CrossRef CAS PubMed.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Promoting electrocatalytic CO2 reduction to formate via sulfur-boosting water activation on indium surfaces, Nat. Commun., 2019, 10, 892 CrossRef PubMed.
- X. Zhang, X. Xie and Y. Liu, Nickel-Catalyzed Highly Regioselective Hydrocyanation of Terminal Alkynes with Zn(CN)2 Using Water as the Hydrogen Source, J. Am. Chem. Soc., 2018, 140, 7385–7389 Search PubMed.
- S. Mukhopadhyay, M. S. Naeem, G. Shiva Shanker, A. Ghatak, A. R. Kottaichamy, R. Shimoni, L. Avram, I. Liberman, R. Balilty and R. Ifraemov, et al., Local CO2 reservoir layer promotes rapid and selective electrochemical CO2 reduction, Nat. Commun., 2024, 15, 3397 CrossRef CAS PubMed.
- A. M. Sheta, S. Fernández, C. Liu, G. C. Dubed-Bandomo and J. Lloret-Fillol, An Electrocatalytic Cascade Reaction for the Synthesis of Ketones Using CO2 as a CO Surrogate, Angew. Chem., Int. Ed., 2024, 63, e202403674 CrossRef CAS PubMed.
- W. Gong, X. Mao, J. Zhang, Y. Lin, H. Zhang, A. Du, Y. Xiong and H. Zhao, Ni–Co Alloy Nanoparticles Catalyze Selective Electrochemical Coupling of Nitroarenes into Azoxybenzene Compounds in Aqueous Electrolyte, ACS Nano, 2023, 17, 3984–3995 CrossRef CAS PubMed.
- X. Chong, C. Liu, Y. Huang, C. Huang and B. Zhang, Potential-tuned selective electrosynthesis of azoxy-, azo- and amino-aromatics over a CoP nanosheet cathode, Natl. Sci. Rev., 2020, 7, 285–295 CrossRef CAS PubMed.
- M. Jin, Y. Liu, X. Zhang, J. Wang, S. Zhang, G. Wang, Y. Zhang, H. Yin, H. Zhang and H. Zhao, Selective electrocatalytic hydrogenation of nitrobenzene over copper-platinum alloying catalysts: Experimental and theoretical studies, Appl. Catal., B, 2021, 298, 120545 CrossRef CAS.
- W. Qiao, I. Waseem, G. Shang, D. Wang, Y. Li, F. Besenbacher, H. Niemantsverdriet, C. Yan and R. Su, Paired Electrochemical N–N Coupling Employing a Surface-Hydroxylated Ni3Fe-MOF-OH Bifunctional Electrocatalyst with Enhanced Adsorption of Nitroarenes and Anilines, ACS Catal., 2021, 11, 13510–13518 CrossRef CAS.
- Q. Mao, X. Mu, W. Wang, K. Deng, H. Yu, Z. Wang, Y. Xu, L. Wang and H. Wang, Atomically dispersed Cu coordinated Rh metallene arrays for simultaneously electrochemical aniline synthesis and biomass upgrading, Nat. Commun., 2023, 14, 5679 CrossRef CAS PubMed.
- Y. Zhao, T. Xu, X. Bai, Y. Jia, Y. Pan, X. Shi, H. Zheng and L. Zheng, Selective electrochemical hydrogenation of nitrobenzene to aniline coupled with efficient 5-hydroxymethylfurfural oxidation in aqueous electrolyte using a broccoli-like CuNi catalyst, Chem. Eng. J., 2024, 482, 149054 CrossRef CAS.
- Q. Mao, X. Mu, K. Deng, H. Yu, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Sulfur Vacancy-Rich Amorphous Rh Metallene Sulfide for Electrocatalytic Selective Synthesis of Aniline Coupled with Efficient Sulfion Degradation, ACS Nano, 2023, 17, 790–800 CrossRef CAS PubMed.
- J. Ma, Z. Wang, T. Majima and G. Zhao, Role of Ni in PtNi Alloy for Modulating the Proton–Electron Transfer of Electrocatalytic Hydrogenation Revealed by the In Situ Raman–Rotating Disk Electrode Method, ACS Catal., 2022, 12, 14062–14071 Search PubMed.
- H. Li, Y. Gao, Y. Wu, C. Liu, C. Cheng, F. Chen, Y. Shi and B. Zhang, σ-Alkynyl Adsorption Enables Electrocatalytic Semihydrogenation of Terminal Alkynes with Easy-Reducible/Passivated Groups over Amorphous PdSx Nanocapsules, J. Am. Chem. Soc., 2022, 144, 19456–19465 CrossRef CAS PubMed.
- Z. Wang, J. Zhu, X. Zu, Y. Wu, S. Shang, P. Ling, P. Qiao, C. Liu, J. Hu and Y. Pan, et al., Selective CO2 Photoreduction to CH4 via Pdδ+-Assisted Hydrodeoxygenation over CeO2 Nanosheets, Angew. Chem., Int. Ed., 2022, 61, e202203249 CrossRef CAS PubMed.
- Y. Gao, R. Yang, C. Wang, C. Liu, Y. Wu, H. Li and B. Zhang, Field-induced reagent concentration and sulfur adsorption enable efficient electrocatalytic semihydrogenation of alkynes, Sci. Adv., 2022, 8, eabm9477 Search PubMed.
- J. Sheng and X. Cheng, Electrochemical Mono-Deuterodefluorination of Trifluoromethyl Aromatic Compounds with Deuterium Oxide, CCS Chem., 2023, 6, 230–240 Search PubMed.
- T. Jia, D. Meng, R. Duan, H. Ji, H. Sheng, C. Chen, J. Li, W. Song and J. Zhao, Single-Atom Nickel on Carbon Nitride Photocatalyst Achieves Semihydrogenation of Alkynes with Water Protons via Monovalent Nickel, Angew. Chem., Int. Ed., 2023, 62, e202216511 CrossRef CAS PubMed.
- S. P. Cummings, T.-N. Le, G. E. Fernandez, L. G. Quiambao and B. J. Stokes, Tetrahydroxydiboron-Mediated Palladium-Catalyzed Transfer Hydrogenation and Deuteriation of Alkenes and Alkynes Using Water as the Stoichiometric H or D Atom Donor, J. Am. Chem. Soc., 2016, 138, 6107–6110 CrossRef CAS PubMed.
- C.-Q. Zhao, Y.-G. Chen, H. Qiu, L. Wei, P. Fang and T.-S. Mei, Water as a Hydrogenating Agent: Stereodivergent Pd-Catalyzed Semihydrogenation of Alkynes, Org. Lett., 2019, 21, 1412–1416 CrossRef CAS PubMed.
- R. S. Sherbo, M. Moreno-Gonzalez, N. J. J. Johnson, D. J. Dvorak, D. K. Fork and C. P. Berlinguette, Accurate Coulometric Quantification of Hydrogen Absorption in Palladium Nanoparticles and Thin Films, Chem. Mater., 2018, 30, 3963–3970 CrossRef CAS.
- L. Jiang, K. Liu, S.-F. Hung, L. Zhou, R. Qin, Q. Zhang, P. Liu, L. Gu, H. M. Chen and G. Fu, et al., Facet engineering accelerates spillover hydrogenation on highly diluted metal nanocatalysts, Nat. Nanotechnol., 2020, 15, 848–853 Search PubMed.
- Y. Zhang, S. Zhan, K. Liu, M. Qiao, N. Liu, R. Qin, L. Xiao, P. You, W. Jing and N. Zheng, Heterogeneous Hydrogenation with Hydrogen Spillover Enabled by Nitrogen Vacancies on Boron Nitride-Supported Pd Nanoparticles, Angew. Chem., Int. Ed., 2023, 62, e202217191 CrossRef CAS PubMed.
- M. D. Marcinkowski, A. D. Jewell, M. Stamatakis, M. B. Boucher, E. A. Lewis, C. J. Murphy, G. Kyriakou and E. C. H. Sykes, Controlling a spillover pathway with the molecular cork effect, Nat. Mater., 2013, 12, 523–528 CrossRef CAS PubMed.
- Y. Wu, C. Liu, C. Wang, Y. Yu, Y. Shi and B. Zhang, Converting copper sulfide to copper with surface sulfur for electrocatalytic alkyne semi-hydrogenation with water, Nat. Commun., 2021, 12, 3881 CrossRef CAS PubMed.
- L. Sun and B. Liu, Mesoporous PdN Alloy Nanocubes for Efficient Electrochemical Nitrate Reduction to Ammonia, Adv. Mater., 2023, 35, 2207305 CrossRef CAS PubMed.
- H. Lv, L. Sun, D. Xu, W. Li, B. Huang and B. Liu, Precise Synthesis of Hollow Mesoporous Palladium–Sulfur Alloy Nanoparticles for Selective Catalytic Hydrogenation, CCS Chem., 2022, 4, 2854–2863 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08608b |
| This journal is © The Royal Society of Chemistry 2025 |