
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescence and colorimetric analysis of β-estradiol based on aptamer assembled spherical nucleic acids†
Leyuan
Chen
abc,
Aijiao
Yuan
ac,
Dapeng
Zhang
 abc,
Wenjing
Xie
ac and
Hanyong
Peng
*ac
abc,
Wenjing
Xie
ac and
Hanyong
Peng
*ac
aState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China. E-mail: hypeng@rcees.ac.cn
bSchool of Environment, Hangzhou Institution for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 26th August 2024
Abstract
Detecting β-estradiol (E2) in environmental monitoring is a complex task due to its status as a significant environmental contaminant. The detection methods require precision, sensitivity, and the ability to be conducted on-site without expensive instrumentation. Herein, we developed a novel approach using E2 aptamer assembled spherical nucleic acids (SNAs), which combines the sensitivity of fluorescence and the simplicity of colorimetry. Initially, a fluorescein (FAM)-labeled DNA aptamer is attached to the surface of gold nanoparticles (AuNPs) through hybridization with thiol-labeled DNA, resulting in fluorescence quenching. However, when E2 is present, the aptamer specifically binds to it, displacing from the thiol-DNA and releasing from the AuNP's surface. This leads to the recovery of fluorescence, allowing for quantitative detection of E2 by measuring the increase in fluorescence signal. Additionally, E2 detection can also be achieved visually using ultraviolet light. For colorimetric analysis, we introduce another set of AuNPs modified with thiol-DNA complementary to the DNA remaining on the surface of the previous AuNPs. When E2 triggers the release of the aptamer, the DNA on both AuNPs hybridized to each other, causing the aggregation of AuNPs and resulting in a distinct color change from red to purple. The detection limits for fluorescence and colorimetric analyses are 1 nM and 5 nM, respectively. We successfully applied this biosensing strategy to determine E2 concentrations in tap water and serum samples. Furthermore, our assay exhibits high selectivity towards E2 over other estrogens. Overall, this innovative approach provides an effective and versatile method for convenient on-site monitoring of E2.
1 Introduction
Environmental estrogens, known as environmental endocrine disruptors (EDCs), are prevalent in our living environment and can interfere with hormone action.1–3 These EDCs, including natural and synthetic estrogens, as well as estrogen-like industrial chemicals,4–6 have the ability to imitate or hinder the actions of natural hormones in humans and animals.7,8 Among the natural estrogens, β-estradiol (E2) is particularly potent and can have negative effects on the endocrine systems of wildlife and humans, even at low concentrations.9–12The detection of E2 is crucial to prevent potential harm to the human body.13–15 Currently, methods such as liquid chromatography-tandem mass spectrometry (LC-MS/MS), enzyme-linked immunosorbent assay (ELISA), and in vitro bioassays are available for E2 detection.16,17 However, these techniques require skilled technicians, expensive instruments, and laboratory settings, making them unsuitable for portable and real-time detection.18 Consequently, there is an urgent need for alternative detection methods.
Aptamers, which are artificial single-stranded nucleic acid ligands, offer a promising solution.19 They can be selected from large libraries and used to target various molecules, including small organic compounds like E2.20–22 Aptamers have several advantages over antibodies, such as enhanced stability, lower cost, and programmable properties.23–27 They can be combined with different signaling platforms, making them highly suitable for detecting small and medium-sized molecules in environmental samples.
The assembly of the aptamer onto gold nanoparticles (AuNPs) can lead to the formation of spherical nucleic acids, which have attracted significant interest in the field of biosensing due to their distinctive characteristics.28 They possess a high specific surface area, exhibit good biocompatibility, can be controlled in size, possess catalytic properties, and exhibit complex optical characteristics. By utilizing fluorescence resonance energy transfer (FRET), AuNPs can quench fluorescence signals.29,30 Moreover, their aggregation can induce color changes due to the distance-dependent localized surface plasmon resonance (LSPR) property, which appear in the absorption spectrum as a redshift of the absorption peak along with a decrease in intensity.31,32
In this study, we developed a novel approach to detect E2, an environmental endocrine disruptor, through the development of a dual-mode analytical method. Our method utilizes E2 aptamer assembled spherical nucleic acids (SNAs) and incorporates the conformational change of the E2 aptamer along with complementary DNA modified AuNPs. As a result, we achieve significant responses in both fluorescence and colorimetric analyses. This innovative method offers rapid and accurate detection of E2, with a low detection limit and a wide linear range of response. Additionally, the colorimetric detection can be conveniently carried out using a portable UV-visible spectrophotometer, facilitating on-site detection. Overall, this technique provides a simple and efficient approach for detecting E2, demonstrating its potential for environmental monitoring as well as biological fluids detection.
2 Materials and methods
2.1 Materials and reagents
DNA oligonucleotides (sequences provided in Table S1†) were procured from Zixi Biotechnology Co., Ltd (Beijing, China). Sigma-Aldrich (MI, USA) supplied sodium chloride, magnesium chloride, and mercaptoethanol. Sangon Biotech Co., Ltd (Shanghai, China) provided Tween 20, Tween 80, and tris (2- carboxyethyl) phosphine (TCEP). Dimethyl sulfoxide (DMSO) was purchased from Aladdin Reagent Co., Ltd (Shanghai, China). Tris–HCl buffer solution was obtained from Thermo Scientific (MA, USA), while 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) buffer solution was procured from Solarbio Science & Technology Co., Ltd (Beijing, China). BBI solution (WI, USA) supplied 20 nm AuNPs.The E2 standard was purchased from J&K Scientific Co., Ltd (Beijing, China), while Aladdin Reagent Co., Ltd (Shanghai, China) provided standards of E1, E3, EE2, and BPA. Analytes were dissolved in DMSO at a concentration of 5 mM to create stock solutions, which were stored at 4 °C in the absence of light. Prior to detection, working solutions with specific concentrations were prepared by diluting the stock solutions with DMSO to 25 μM and then further diluting them with the sensing buffer to achieve the desired target concentration. Purified water from a Milli-Q system (Millipore, Bedford, MA, USA) was used for all experiments.
2.2 Preparation of spherical nucleic acids (SNAs)
This study employed two types of spherical nucleic acids. The first type involved functionalizing the AuNPs with double strand (ds) DNA. A fluorescein (FAM)-labeled DNA aptamer is attached to the surface of AuNPs through hybridization with thiol-labeled DNA. Initially, 100 μM E2 aptamer was annealed with 100 μM thiol labelled DNA in the annealing buffer (20 mM Tris–HCl + 100 mM NaCl + 20 mM MgCl2 + 0.1% Tween 20) to obtain 25 μL of 10 μM dsDNA. The obtained dsDNA was then incubated with 5 μL of 20 mM TCEP for 30 minutes. Subsequently, in a 1.5 mL test tube, 500 μL of 20 nM AuNPs, 50 μL of 4% Tween 80, 30 μL of 5 M NaCl, and 30 μL of the aforementioned dsDNA solution were added sequentially and incubated for 1 hour. The molar ratio of DNA to AuNP was approximately 1200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Next, 50 μL of 20% Tween 20 was added, followed by another 30 minutes incubation. Afterward, 350 μL of 5 M NaCl was added, and the solution was subjected to 3 hours of salt aging with intermittent ultrasound treatment every hour. After salt aging, the mixture underwent centrifugation at 13000 g for 20 minutes to remove the supernatant containing unconjugated DNA. The washing step, involving the addition of 500 μL of washing buffer (10 mM Tris–HCl + 100 mM NaCl + 0.05% Tween 20) to the precipitate, resuspension, centrifugation, and removal of the supernatant, was repeated three times. Finally, the functionalized AuNPs were resuspended in 250 μL of preserving buffer (20 mM HEPES + 100 mM NaCl) to achieve a final concentration of 2 nM.
:1. Next, 50 μL of 20% Tween 20 was added, followed by another 30 minutes incubation. Afterward, 350 μL of 5 M NaCl was added, and the solution was subjected to 3 hours of salt aging with intermittent ultrasound treatment every hour. After salt aging, the mixture underwent centrifugation at 13000 g for 20 minutes to remove the supernatant containing unconjugated DNA. The washing step, involving the addition of 500 μL of washing buffer (10 mM Tris–HCl + 100 mM NaCl + 0.05% Tween 20) to the precipitate, resuspension, centrifugation, and removal of the supernatant, was repeated three times. Finally, the functionalized AuNPs were resuspended in 250 μL of preserving buffer (20 mM HEPES + 100 mM NaCl) to achieve a final concentration of 2 nM.
The second type of SNA involved functionalizing the AuNPs with single strand (ss) DNA, a thiol-DNA complementary to the thiol-DNA on the first SNA. To achieve this, 500 μL of 20 nm AuNPs was combined with 6 μL of 100 μM ssDNA and incubated for 5 minutes in a 1.5 mL micro test tube. The molar ratio of DNA to AuNP was approximately 1200:1. Next, 50 μL of 20% Tween 20 was added, followed by another 30 minutes incubation. Subsequently, 350 μL of 5 M NaCl was added, and the solution was subjected to 3 hours of salt aging with intermittent ultrasound treatment every hour. After salt aging, the mixture underwent centrifugation at 13000 g for 20 minutes to remove the supernatant containing unconjugated DNA. This washing step, involving the addition of 500 μL of washing buffer (10 mM Tris–HCl + 100 mM NaCl + 0.05% Tween 20) to the precipitate, resuspension, centrifugation, and removal of the supernatant, was repeated three times. Finally, the functionalized AuNPs were resuspended in 250 μL of preserving buffer (20 mM HEPES + 100 mM NaCl) to achieve a final concentration of 2 nM.
To modulate the loading quantity of ssDNA on AuNPs, ssDNA was mixed with polyT at a ratio of 6:n, where n represents values of 2, 4, 6, 7, 8, 10, and 16. The resulting mixture was then added to AuNPs. Following a 3 hours salt aging process, the mixture underwent washing steps and was subsequently stored in a preserving buffer.
The confirmation of DNA loading on the surface of AuNPs was validated through transmission electron microscopy (TEM) and energy-dispersive X-ray (EDX) mapping analyses (Fig. S1†), by comparing absorption spectra between DNA-AuNPs and bare AuNPs (Fig. S2†), and by assessing changes in fluorescence intensity of DNA-AuNPs before and after the introduction of mercaptoethanol (ME) (Fig. S3†).
2.3 Polyacrylamide gel electrophoresis (PAGE)
A 10% nondenaturing polyacrylamide gel electrophoresis (PAGE) was prepared, and the electrophoresis conditions were set to 110 V for a duration of 40 minutes. Once the electrophoresis process was completed, a mixture of SYBR Gold dye (5×TBE: 25 mL, SYBR Gold: 2.5 μL) was added to the polyacrylamide gel, and the mixture was stained on a shaker for 15 minutes. Finally, the resulting products were examined using a ChampGel 6000 gel imager for characterization.2.4 Dual-mode detection of β-estradiol
A dual-mode sensing buffer comprising 20 mM HEPES, 200 mM NaCl, and 80 mM MgCl2 was prepared. DsDNA-AuNPs, ssDNA-AuNPs, and E2 were combined in the sensing buffer. Both dsDNA-AuNPs and ssDNA-AuNPs had a final concentration of 0.6 nM. The mixture was thoroughly mixed in a 200 μL micro test tube, and then 100 μL of the solution was transferred to the wells of a 96-well quartz glass microplate for colorimetric detection, with a duration of 60 minutes. The absorbance at 525 nm and 650 nm was simultaneously measured using a SpectraMax iD5 instrument (USA, Molecular Devices). For fluorescence detection, the mixture was loaded into the wells of a black 96-well NUNC microplate (USA, Thermofisher). Fluorescence intensity was measured using a SpectraMax iD5 instrument (USA, Molecular Devices) with an excitation wavelength of 485 nm and an emission wavelength of 535 nm after a reaction time of 60 minutes. Pictures were captured while illuminating the mixture in the micro test tube with an ultraviolet lamp, using an excitation wavelength of 472 nm and an emission wavelength of 595 nm.2.5 Detection of E2 in water samples
To assess the applicability of the sensing method in environmental samples, tap water samples were utilized to detect E2. The tap water samples were diluted tenfold using the fluorescence sensing buffer. Subsequently, E2 was added to the diluted tap water samples, and the mixture was subjected to fluorescent signal measurement for 60 minutes. The fluorescence sensing buffer employed in this analysis consisted of 20 mM HEPES, 250 mM NaCl, and 60 mM MgCl2. dsDNA-AuNPs and E2 were combined in the sensing buffer, with the final concentration of dsDNA-AuNPs set at 0.5 nM.3 Results and discussion
3.1 Principle of dual-mode biosensor
Scheme 1 illustrates the design and functioning of a dual-mode biosensor designed for the detection of E2. The biosensor consists of two SNA components: dsDNA-AuNPs and ssDNA-AuNPs. The dsDNA-AuNPs are composed of a target aptamer labeled with FAM (fluorescein) and contain a complementary fragment to the thiol-labeled ssDNA-A on AuNPs. On the other hand, the ssDNA-AuNPs are constructed on AuNPs coated with thiol-labeled ssDNA-B that is complementary to the ssDNA-A on dsDNA-AuNPs.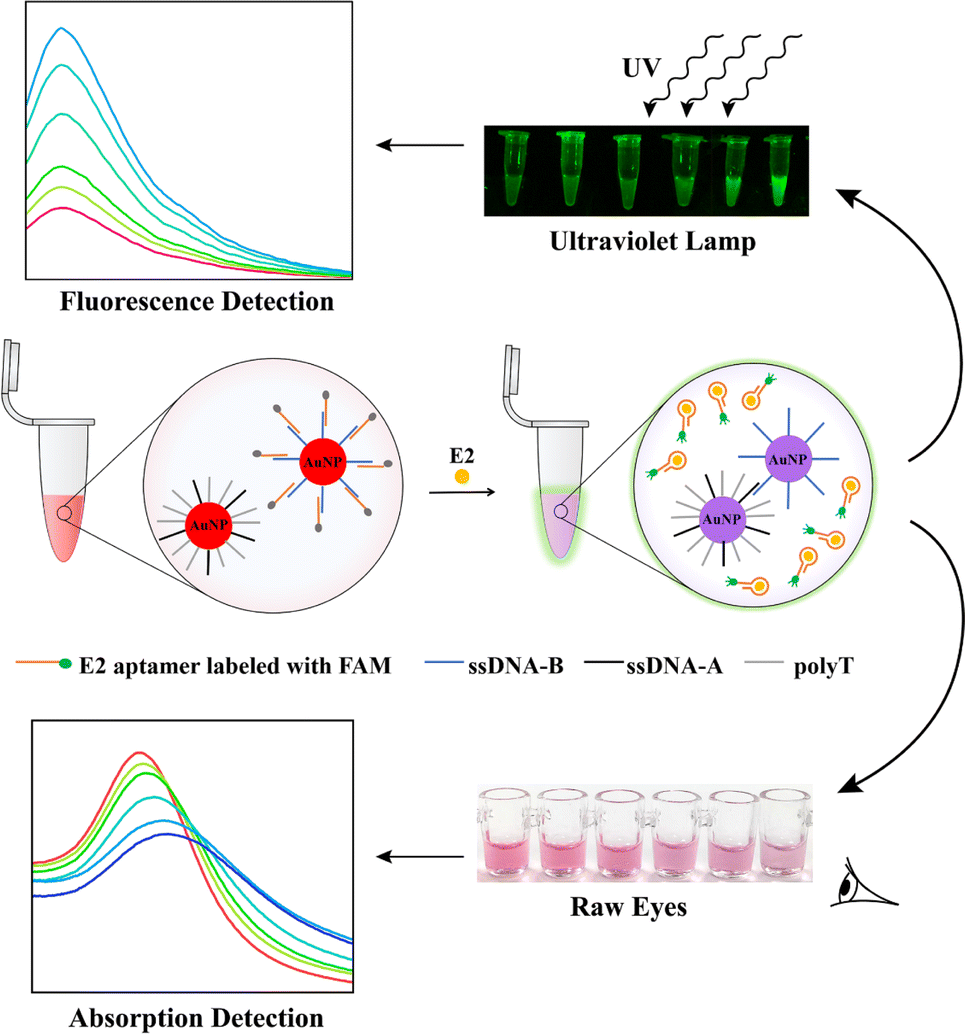 | ||
| Scheme 1 Schematic description of the dual-mode biosensor for E2 detection based on the aggregation of AuNPs and recovery of fluorescence signal induced by aptamer. | ||
Initially, the fluorescence of the FAM-labeled aptamer is quenched when it assembles on the AuNPs. However, in the presence of E2, the dual-mode biosensor becomes activated. The aptamer specifically binds to E2, initiating a strand displacement reaction that releases the FAM-labeled aptamer from the dsDNA-AuNPs. This release generates a fluorescence signal that can be monitored in real-time.
Simultaneously, the remaining ssDNA-A on the dsDNA-AuNPs hybridizes with its complementary DNA on the ssDNA-B-AuNPs. This hybridization causes the aggregation of AuNPs, resulting in a distinct solution colour change from red to purple. Therefore, the biosensor enables the detection of an increase both in fluorescence signal and the A650/A525 ratio (where A650 and A525 represent the absorbance intensity at 650 nm and 525 nm, respectively).
The developed colorimetric-fluorescence dual-mode biosensor effectively translates target-specific recognition into the aggregation of AuNPs as a reporter and the fluorescence signal resulting from the release of the FAM-labeled aptamer from the AuNPs' surface. This biosensor exhibits potential for various environmental applications.
3.2 Viability of the assay
In order to demonstrate the viability of the assay, the binding and separation capability of ssDNA on AuNPs were initially investigated. The binding interaction between ssDNA-A and ssDNA-B, ssDNA-B and the aptamer, as well as their optimal ratios, were assessed using polyacrylamide gel electrophoresis (PAGE). The results revealed the appearance of a hybrid band following the combination of ssDNA-B and the aptamer, indicating successful binding between the two strands (refer to Fig. S4†). As the molar ratio of the aptamer to ssDNA-B increased from 1:1.2 to 1.2:1, it was observed that the aptamer was slightly in excess. Additionally, the mixing of ssDNA-A and ssDNA-B resulted in the formation of a hybrid band representing dsDNA, signifying successful combination of the two strands with an optimal molar ratio of 1:1.
We proceeded to investigate the formation of AuNPs aggregates and the colorimetric reaction between ssDNA-A and ssDNA-B decorated AuNPs, based on the principle of base complementary pairing. To examine the impact of ionic strength, we varied the concentration of Mg2+ ions while maintaining the concentration of ssDNA-A-AuNPs and ssDNA-B-AuNPs at 2 nM. As depicted in Fig. 1A, the characteristic absorbance peak of AuNPs displayed a red shift and decreased intensity with increasing Mg2+ ion concentration from 5 mM to 45 mM. The colorimetric reaction between ssDNA-A and ssDNA-B was observed, leading to a change in the colour of the solution depending on the concentration of Mg2+ ions. Activation of the colorimetric reaction occurred when the Mg2+ ion concentration exceeded 20 mM.
 | ||
| Fig. 1 Confirmation of the aggregation effect of ssDNA-A-AuNPs and ssDNA-B-AuNPs, as well as the binding effect between E2 and the aptamer. (A) The UV-vis spectra and corresponding colour changes (inset: photographic images) were observed in mixtures of ssDNA-A-AuNPs and ssDNA-B-AuNPs with varying concentrations of Mg2+. Used ssDNA-A-AuNPs/ssDNA-B-AuNPs in the presence of Mg2+ at highest concentration (45 mM) as control. (B) The absorption peak position (represented by the green polyline), A650/A525 value (represented by the purple polyline), and colour changes (inset: photographic images) were analysed in mixtures of ssDNA-A-AuNPs and ssDNA-B-AuNPs with different proportions. (C) Fluorescence kinetic curves were recorded for mixtures of dsDNA-AuNPs with various concentrations of E2. | ||
Due to variations in the loading capacity of dsDNA and ssDNA on AuNPs, as well as incomplete reaction between E2 and dsDNA, the amount of ssDNA-B in the system was lower than that of ssDNA-A. To assess the performance of the colorimetric reaction between ssDNA-A-AuNPs and ssDNA-B-AuNPs, we utilized ssDNA-B-AuNPs with a lower proportion. The mixtures were subjected to a 30 minutes reaction in a buffer with a Mg2+ concentration of 60 mM, with the ssDNA-A-AuNPs solution serving as a negative control. As shown in Fig. 1B, the A650/A525 ratio and the colorimetric change were still distinguishable in the mixture, even when the concentration of ssDNA-B-AuNPs was only 0.1 times that of ssDNA-A-AuNPs. The position of the absorption peak and the A650/A525 ratio demonstrated a positive correlation with the amount of ssDNA-B-AuNPs in the mixture, indicating the feasibility of quantitative detection of E2 using the colorimetric method.
Furthermore, we explored the reaction efficiency of E2 and the aptamer by monitoring the fluorescence kinetics of the reaction between E2 and dsDNA-AuNPs. Upon the release of the FAM-labeled aptamer from dsDNA-AuNPs in the presence of E2, the fluorescence signal of FAM was restored (Fig. 1C). The intensity of fluorescence recovery increased with higher E2 concentrations. Changes in circular dichroic (CD) spectra of dsDNA after addition of estradiol also implied effective binding of E2 and aptamer (Fig. S5†).
Next, we focused on optimizing the reaction conditions for the detection of E2 using dsDNA-AuNPs. The efficiency of fluorescence intensity recovery in the mixture was monitored using the (F1 − F0)/F0 value, where F1 represents the fluorescence intensity of the test sample and F0 represents the fluorescence intensity of the negative control. To begin, we examined the influence of Mg2+ ion concentration on E2 detection. Fig. 2A demonstrates that the maximum recovery efficiency was achieved at a Mg2+ ion concentration of 60 mM. Conversely, the concentration of Na+ had minimal effect on the fluorescence intensity recovery efficiency within the range of 0 to 250 mM, as shown in Fig. 2B. Furthermore, we investigated the impact of different surfactants, such as Tween 20 and Tween 80, on the reaction (Fig. 2C). Interestingly, the addition of surfactants was found to have an adverse effect on the reaction. Hence, the inclusion of surfactants in the buffer was deemed unnecessary.
 | ||
| Fig. 2 The optimization of conditions for the binding of E2 and the aptamer involved the following parameters: (A) varying concentrations of Mg2+ were tested. (B) Different concentrations of Na+ were examined. (C) Various types and concentrations of surfactants were evaluated. (D) Different concentrations of AuNPs were investigated. | ||
Lastly, we explored the concentration of dsDNA-AuNPs within the range of 0.125 to 0.5 nM (Fig. 2D). It was observed that the recovery efficiency increased with higher concentrations of dsDNA-AuNPs. Based on these results, a concentration of 0.5 nM was determined to be the appropriate concentration for dsDNA-AuNPs.
3.3 Optimization of buffer conditions
To achieve optimal detection performance, we conducted optimization of several parameters, including the loading amount of ssDNA-A on ssDNA-A-AuNPs (Fig. S6† and 3A), the concentration of Mg2+ in the sensing buffer (Fig. 3B), and the concentration of Na+ in the sensing buffer (Fig. 3C).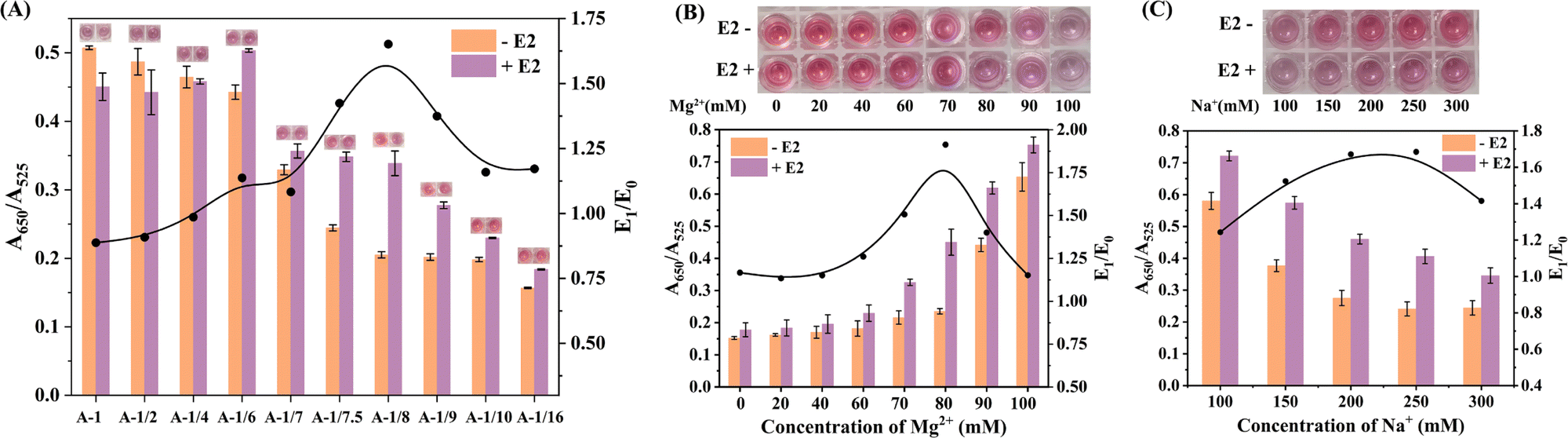 | ||
| Fig. 3 The optimization of conditions for E2 detection involved the following parameters: (A) the loading capacity of ssDNA-A-AuNPs was varied, where A-n represents ssDNA-A-AuNPs in the mixture with a load of 1/n of the original. (B) Different concentrations of Mg2+ in the sensing buffer were tested. (C) Various concentrations of Na+ in the sensing buffer were examined. | ||
Initially, we investigated the impact of DNA loading amount on AuNPs and its effect on the colorimetric reaction. By synthesizing dsDNA-AuNPs and ssDNA-A-AuNPs with varying DNA loading capacities, we evaluated their detection efficiency. Based on the fluorescence intensity results (Fig. S6†), we observed that reducing the DNA loading capacity of dsDNA-AuNPs had minimal effect on the fluorescence intensity recovery efficiency. However, when the loading capacity of ssDNA-A-AuNPs was reduced to 1/8, the fluorescence recovery efficiency increased to nearly double that of the original sample. Therefore, we prepared dsDNA-AuNPs with the highest achievable loading capacity and tested their colorimetric reaction performance against ssDNA-A-AuNPs with various loading capacities. Fig. 3A demonstrated that the A650/A525 ratio remained similar in samples with and without the presence of E2 when the loading capacity of ssDNA-A-AuNPs was reduced to 1/2 or 1/4. However, a noticeable difference in the A650/A525 ratio was observed between samples with and without E2 when the loading capacity of ssDNA-A-AuNPs was reduced to 1/8. Further reduction in the loading capacity to 1/10 or 1/16 led to significant decreases in the intensities of the A650/A525 ratio in samples with and without E2. To quantify this difference, we calculated the E1/E0 value, where E1 represents the A650/A525 value of the sample in the presence of E2, and E0 represents the A650/A525 value of the negative sample without E2. It was observed that the E1/E0 value increased initially and then decreased with decreasing loading capacity. When the ssDNA-A-AuNPs loading capacity was reduced to 1/8 of the original, the E1/E0 value reached a maximum value of 1.6.
Subsequently, we analyzed the optimal concentration of Mg2+ in the sensing buffer using AuNPs with optimized loading efficiency. The results showed that the addition of E2 did not significantly alter the solution color when the concentration of Mg2+ ranged from 0 to 60 mM (Fig. 3B). The solution color transitioned from pink to lavender when the Mg2+ concentration increased to 80 mM, and the biosensor exhibited good detection performance. However, both samples with and without the presence of E2 exhibited an evident color change as the concentration of Mg2+ continued to increase beyond 90 mM.
Furthermore, we investigated the effect of Na+ concentration in the sensing buffer. As depicted in Fig. 3C, both samples with and without the presence of E2 displayed a decrease in the A650/A525 ratio with increasing Na+ concentration. The E1/E0 value exhibited an increase within the range of Na+ concentration between 150 and 250 mM, indicating a significant difference between samples with and without the presence of E2. Based on these findings, we determined 200 mM to be the appropriate Na+ concentration.
3.4 Sensitivity of the assay
We conducted an evaluation of the established biosensor's linear range and limit of detection (LOD) for detecting E2 under optimized parameters. Various concentrations of E2 ranging from 0 nM to 10 μM were added to assess sensitivity. As depicted in Fig. 4B, the A650/A525 value exhibited a gradual increase with increasing E2 concentration within the tested range. When the added E2 concentration reached 10 μM, the A650/A525 value was approximately 0.58, representing a 205% increase compared to the control samples.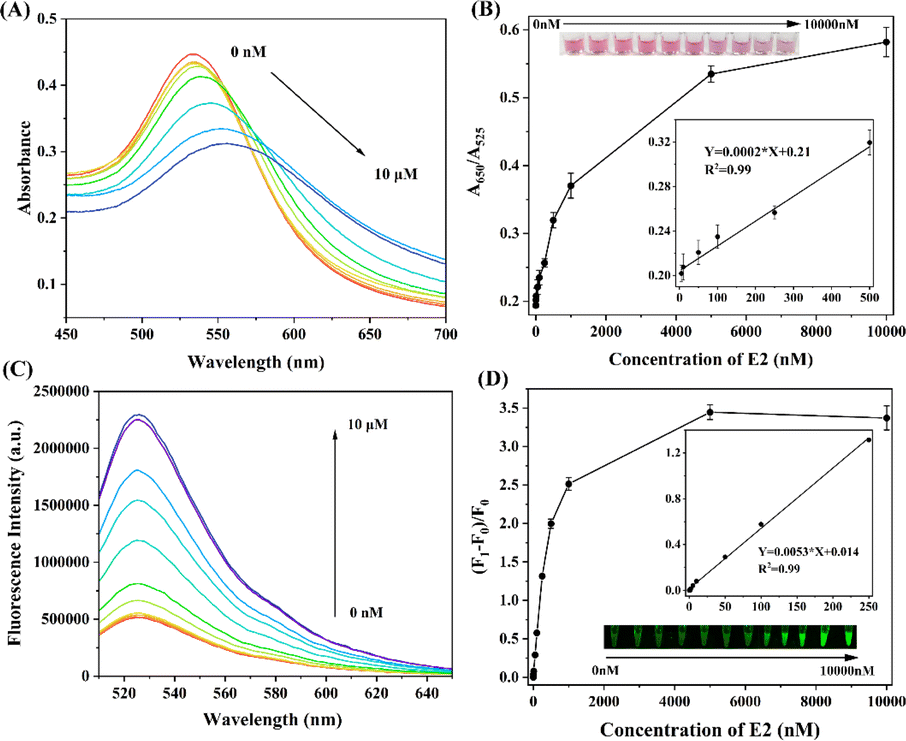 | ||
| Fig. 4 (A) Absorption spectra for mixtures of the biosensor with various concentrations of E2. (B) The correlation between the change in A650/A525 value and different concentrations of E2 is depicted. The bottom right inset displays the response of A650/A525 value to E2 concentrations ranging from 10 to 500 nM. Additionally, the top left inset illustrates the corresponding colour changes observed with varying E2 concentrations. (C) Fluorescence spectra for mixtures of the biosensor with various concentrations of E2. (D) The relationship between the change in (F1 − F0)/F0 value and different concentrations of E2 is presented. The inset demonstrates the response of (F1 − F0)/F0 value to E2 concentrations ranging from 0 to 250 nM. | ||
Furthermore, a noticeable color change from pink to light pink and eventually to purple was observed as the E2 concentration increased (inset of Fig. 4B), indicating a colorimetric response within the E2 concentration range of 0 nM to 10 μM. The limit of detection (LOD), which was determined when the signal change caused by E2 was three times the standard deviation of the control samples, was as low as 5 nM for E2. The dual-mode biosensor also exhibited a linear A650/A525 response within the E2 concentration range of 10 to 500 nM.
Subsequently, the fluorescence signal was monitored under the same conditions. As shown in Fig. 4B, a strong positive correlation was observed between the detected fluorescence signal and the added E2 concentrations within the linear range of 0 to 250 nM. The LOD was calculated to be 1 nM. A semi-quantitative detection of E2 was performed by monitoring the fluorescence change under ultraviolet lamp irradiation. As depicted in the inset of Fig. 4D, the solution displayed a bright green fluorescence at an E2 concentration of 10 μM. The green fluorescence of the solution remained observable as the E2 concentration decreased to 100 nM. However, when the E2 concentration dropped below 50 nM, it became challenging to discern the green fluorescence of the solution under ultraviolet irradiation. Comparing the results to other previously reported biosensors (Table S2†), both of these methods exhibited competitiveness in terms of sensitivity and efficiency for detecting E2.
3.5 Specificity for detection of E2
To assess the selectivity of the dual-mode biosensor for E2, we selected four other estrogens, namely E1, E3, EE2, and BPA, for investigation. In the colorimetric selectivity experiment, a concentration of 5 μM for E2 and its analogs was individually added to the system (Fig. 5A). Due to the higher sensitivity of the fluorescence signal, a lower concentration of 2 μM was used for E2 and its analogs in the fluorescence selectivity experiment (Fig. 5B). The results revealed that E3, EE2, and BPA exhibited negligible signal responses in both colorimetric and fluorescence detection, suggesting low cross-reactivity with the biosensor. On the other hand, E2 displayed a sensitive response in both colorimetric and fluorescence signals, indicating its high selectivity. Additionally, E1 also elicited a response signal, which is consistent with a previous study.21 | ||
| Fig. 5 (A) The selectivity of the proposed biosensor for colorimetric detection of E2 was examined in comparison to other estrogens. The concentrations of all estrogens, including E2, were set at 5 μM. (B) The selectivity of the proposed biosensor for fluorescence detection of E2 was investigated in comparison to other estrogens. The concentrations of all estrogens, including E2, were set at 2 μM. | ||
3.6 Detection of E2 in water and serum samples
To assess the performance of the dual-mode biosensor in real samples, we conducted an analysis using tap water and fetal bovine serum (FBS) samples. Spike and recovery experiments were performed to evaluate the practical applicability of the biosensor. As depicted in Fig. S7,† the biosensor exhibited a linear fluorescence response within the concentration range of 0 to 100 nM for E2, and the limit of detection (LOD) was determined to be 2 nM in a 10-fold diluted tap water sample. In a 20-fold dilution of FBS, the biosensor demonstrated a linear fluorescence response between 0 and 500 nM for E2, with a LOD of 2 nM (Fig. S8†).We proceeded with testing tap water and FBS samples spiked with E2 (Table 1). Our assay revealed recovery rates ranging between 98.1% and 107.0%, indicating the biosensor's precise detection of the spiked E2 levels. The relative standard deviations (RSD) for these measurements ranged from 3.8% to 6.2%, based on three replicates (n = 3). These results demonstrate the feasibility of the biosensor for the detection of E2 in complex sample matrices, such as environmental water and biological fluids. Furthermore, we conducted a 14 days stability assessment of the biosensor (Fig. S9†). The results demonstrated that there was no self-aggregation within the 14 days timeframe, and the fluorescence signal recovery efficiency remained consistently robust. This observation underscores the substantial practical application potential of the biosensor.
| Samples | Added (nM) | Found (nM) | RSD (%) | Recovery (%) |
|---|---|---|---|---|
| Tap water | 10 | 10.56 | 6.2 | 105.6 |
| 75 | 75.92 | 3.8 | 101.2 | |
| FBS | 60 | 64.18 | 6.2 | 107.0 |
| 400 | 392.56 | 4.3 | 98.1 |
4 Conclusion
In summary, we present a novel dual-mode biosensing approach for the detection of E2 in water samples using aptamer-induced gold nanoparticles aggregation, enabling rapid fluorescence and colorimetric readouts. By combining the high sensitivity of the fluorescence method with the interpretability of the colorimetric method, our strategy caters to the detection requirements of various scenarios. It is noteworthy to mention that colorimetric detection can be carried out either utilizing a portable ultraviolet-visible spectrophotometer or simply by visual observation, enabling on-site detection capabilities. Overall, the detection process is straightforward, reliable, and efficient, with results obtainable within 60 minutes. Furthermore, our methods exhibit strong selectivity towards E2 and maintain their detection performance even in complex sample matrices. This research provides valuable insights for the advancement of multi-mode biosensors, and the outcomes hold promising prospects for practical applications in E2 detection.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Key Research and Development Program of China (Grant no. 2023YFA0915102 and 2022YFC3701302), the National Natural Science Foundation of China (Grant no. 22276199), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0750100). Wenjing Xie were supported by the Youth Fund from National Natural Science Foundation of China (Grant no. 22306195).References
- W. Wang, Y. Peng, J. Wu, M. Zhang, Q. Li, Z. Zhao, M. Liu, J. Wang, G. Cao, J. Bai and Z. Gao, Anal. Chem., 2021, 93, 4488–4496 CrossRef CAS PubMed
.
- A. C. Gore, V. A. Chappell, S. E. Fenton, J. A. Flaws, A. Nadal, G. S. Prins, J. Toppari and R. T. Zoeller, Endocr. Rev., 2015, 36, E1–E150 CrossRef CAS PubMed
- H. S. Changa, K. H. Chooa, B. Lee and S. J. Choi, J. Hazard. Mater., 2009, 172, 1–12 CrossRef PubMed
- M. Adeel, X. Song, Y. Wang, D. Francis and Y. Yang, Environ. Int., 2017, 99, 107–119 CrossRef CAS PubMed
- M. Morgan, A. Deoraj, Q. Felty and D. Roy, Mol. Cell. Endocrinol., 2017, 457, 89–102 CrossRef CAS PubMed
- B. S. Rodriguez, M. A. Ramos, J. H. Borges, A. V. H. Herrera and M. A. R. Delgado, Trends Anal. Chem., 2013, 44, 58–77 CrossRef
- H. Hamid and C. Eskicioglu, Water Res., 2012, 46, 5813–5833 CrossRef CAS PubMed
- G. Ying, R. S. Kookana and Y. Ru, Environ. Int., 2002, 28, 545–551 CrossRef CAS PubMed
- Z. Liu, G. Lu, H. Yin, Z. Dang and B. Rittmann, Environ. Sci. Technol., 2015, 49, 5288–5300 CrossRef CAS PubMed
- S. Wang, Y. Li, M. Ding, X. Wu, J. Xu, R. Wang, Ti. Wen, W. Huang, P. Zhou, K. X. Ma Zhou and S. Du, J. Chromatogr. B, 2011, 879, 2595–2600 CrossRef CAS PubMed
- K. L. Thorpe, T. H. Hutchinson, M. J. Hetheridge, M. Scholze, J. P. Sumpter and C. R. Tyler, Environ. Sci. Technol., 2001, 35, 2476–2481 CrossRef CAS PubMed
- A. Amalraj and P. Perumal, Microchem. J., 2022, 173, 106971 CrossRef CAS
- Q. Zhang, J. Zhao, G. Ying, Y. Liu and C. Pan, Environ. Sci. Technol., 2014, 48, 7982–7992 CrossRef CAS PubMed
- N. Xu, Y. Xu, S. Xu, J. Li and H. Tao, Environ. Pollut., 2012, 165, 215–224 CrossRef CAS PubMed
- L. Barreiros, J. F. Queiroz, L. M. Magalhaes, A. M. T. Silva and M. A. Segundo, Microchem. J., 2016, 126, 243–262 CrossRef CAS
- K. M. S. Ana and M. P. Espino, Chemosphere, 2020, 256, 127122 CrossRef PubMed
- C. P. Silva, T. Carvalho, R. J. Schneider, V. I. Esteves and D. L. D. Lima, Anal. Methods, 2020, 12, 2517–2526 RSC
- B. I. Escher, M. Allinson, R. Altenburger, P. A. Bain, P. Balaguer, W. Busch, J. Crago, N. D. Denslow, E. Dopp, K. Hilscherova, A. R. Humpage, A. Kumar, M. Grimaldi, B. S. Jayasinghe, B. Jarosova, A. Jia, S. Makarov, K. A. Maruya, A. Medvedev, A. C. Mehinto, J. E. Mendez, A. Poulsen, E. Prochazka, J. Richard, A. Schifferli, D. Schlenk, S. Scholz, F. Shiraishi, S. Snyder, G. Su, J. Y. M. Tang, B. Burg, S. C. Linden, I. Werner, S. D. Westerheide, C. K. C. Wong, M. Yang, B. H. Y. Yeung, X. Zhang and F. D. L. Leusch, Environ. Sci. Technol., 2014, 48, 1940–1956 CrossRef CAS PubMed
- S. Tombelli, M. Mascini and C. Opin, Mol. Ther., 2009, 11(2), 179–188 CAS
- Y. S. Kim, N. H. A. Raston and M. B. Gu, Biosens. Bioelectron., 2016, 76, 2–19 CrossRef PubMed
- C. Niu, C. Zhang and J. Liu, Environ. Sci. Technol., 2022, 56, 17702–17711 CrossRef CAS PubMed
- Y. Li, L. Sun and Q. Zhao, Anal. Bioanal. Chem., 2018, 410, 6269–6277 CrossRef CAS PubMed
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2004, 126, 12298–12305 CrossRef CAS PubMed
- J. Liu and Y. Lu, Chem. Commun., 2007, 46, 4872–4874 RSC
- Y. He, F. Tian, J. Zhou, Q. Zhao, R. Fu and B. Jiao, J. Hazard. Mater., 2020, 388, 121758 CrossRef CAS PubMed
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS PubMed
- Y. Cao, J. Wu, B. Pang, H. Zhang and X. C. Le, Chem. Commun., 2021, 57, 6871–6874 RSC
- L. Guo, Y. Xu, A. R. Ferhan, G. Chen and D. Kim, J. Am. Chem. Soc., 2013, 135, 12338–12345 CrossRef CAS PubMed
- A. Amalraj, R. Pavadai, S. Subramanian and P. Perumal, Appl. Surf. Sci., 2022, 602, 154222 CrossRef CAS
- A. Amalraj, N. P. S. Chauhan and P. Perumal, New J. Chem., 2023, 47, 3956 RSC
- H. Aldewachi, T. Chalati, M. N. Woodroofe, N. Bricklebank, B. Sharrack and P. Gardiner, Nanoscale, 2018, 10, 18–33 RSC
- A. Nezami, R. Nosrati, B. Golichenari, R. Rezaee, G. I. Chatzidakis, A. M. Tsatsakis and G. Karimi, Trends Anal. Chem., 2017, 94, 95–105 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay01283f |
| This journal is © The Royal Society of Chemistry 2024 |