
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalyzed asymmetric addition of imides to alkenes†
Kentaro
Yamakawa
,
Kana
Sakamoto
and
Takahiro
Nishimura
 *
*
Department of Chemistry, Graduate School of Science, Osaka MetropolitanUniversity, Sumiyoshi, Osaka 558-8585, Japan. E-mail: tnishi@omu.ac.jp
First published on 5th October 2023
Abstract
Enantioselective addition of an imide N–H bond to alkenes was realized by use of a cationic iridium catalyst. Bulky diphosphine ligands such as DTBM-segphos, DTBM-MeO-biphep, and DTBM-binap were indispensable for the reaction. A variety of styrene derivatives, allylsilanes, and norbornene were good substrates to give the corresponding chiral adducts with high enantioselectivity.
Chiral amines are essential motifs that appear in pharmaceuticals, agrochemicals, and natural products.1,2 In particular, chiral 1-arylethylamines exist as important pharmacophores and are known to exhibit bioactivities as shown in Fig. 1.3 Therefore, much effort has been devoted to developing efficient methods for the synthesis of enantiopure 1-arylethylamines, and a variety of approaches such as nucleophilic addition to imines,4 reductive amination,5 hydrogenation of imine, enamine, and enamides,6 and biocatalytic methods7 have been developed.
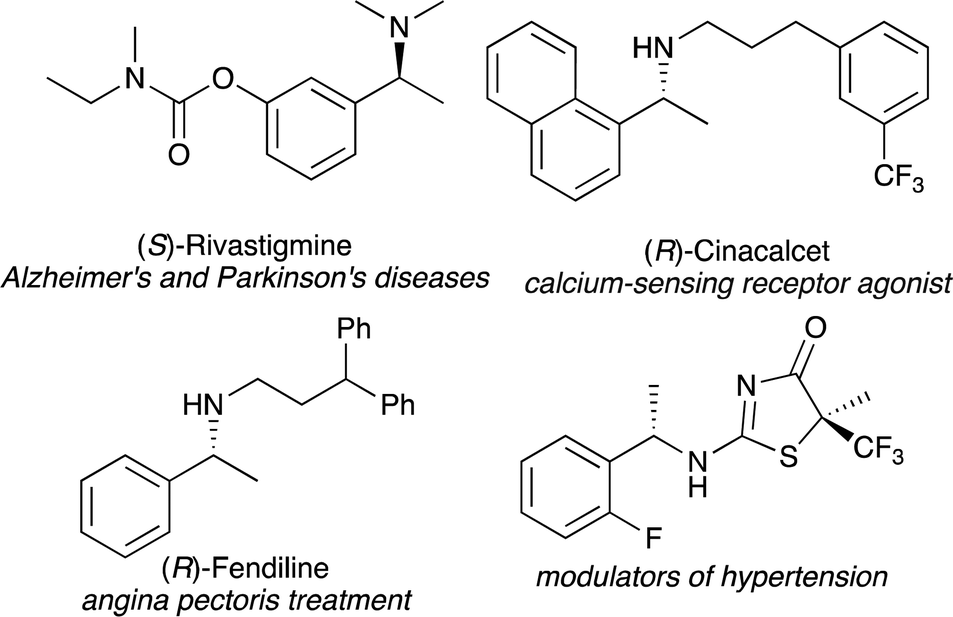 | ||
| Fig. 1 Selected examples of 1-arylethylamine pharmaceuticals and bioactive compounds. | ||
Transition-metal-catalysed asymmetric intermolecular hydroamination, which is the addition of N–H bond across unsaturated bonds, is the most straightforward, effective, and atom-economical methods for preparing chiral amines from alkenes.8 In this respect, Togni and co-workers reported the first enantioselective hydroamination of 2-norbornene with aniline catalysed by an iridium/diphosphine complex.9 It was revealed that the presence of fluoride anion was necessary for reactivity and enantioselectivity. Afterward, Hartwig and co-workers reported iridium-catalyzed hydroamination of norbornene derivatives with a series of anilines using potassium bis(trimethylsilyl)amide (KHMDS) as a base, instead of the fluoride anion.10 Thus far, several intermolecular enantioselective hydroamination of terminal alkenes,11 alkynes,12 allenes,13 and dienes14 have been developed.15 2-Aminopyridine derivatives have been often used as a nitrogen source in iridium-catalysed asymmetric hydroamination. Shibata and co-workers reported asymmetric hydroamination of styrene derivatives using C3-TUNEPHOS as a chiral ligand, where the pyridyl group assisted the efficient oxidative addition of the N–H bond to iridium, providing the Markovnikov's adducts in moderate enantioselectivities (Scheme 1a).16 Recently, Hartwig and co-workers demonstrated the enantioselective hydroamination of internal alkenes17 or terminal alkenes18 with 6-methyl-2-aminopyridine catalysed by iridium/(R)-TMS-SYNPHOS catalyst (Scheme 1b).
 | ||
| Scheme 1 Iridium-catalysed enantioselective hydroamination of alkenes with 2-aminopyridine derivatives. | ||
Phthalimide has been used to synthesize primary amines from alkyl halides in the Gabriel synthesis, where the intermediate N-alkylphthalimides are prepared by the substitution reaction.19 To the best of our knowledge, enantioselective addition of phthalimide to alkenes for the synthesis of chiral N-alkylphthalimides has not been achieved to date. Here we report iridium-catalyzed enantioselective intermolecular hydroamination of alkenes with phthalimide derivatives. The bulky chiral diphosphine ligand enabled the addition of the N–H bond achieving high enantioselectivities. Not only styrene derivatives but also allylsilanes were suitable for the present catalytic system.
Treatment of phthalimide (1a) with styrene (2a, 3 equiv.) in the presence of a cationic iridium catalyst, which is generated from [IrCl(cod)]2 (5 mol% Ir), (S)-DTBM-segphos (5 mol%), and NaBArF4 (10 mol%) (cod = 1,5-cyclooctadiene, ArF = 3,5-(CF3)2C6H3) gave the addition product 3aa in 85% yield with 93% ee (Table 1, entry 1).20 The use of other bulky diphosphine ligands, such as (S)-MeO-DTBM-binphep and (R)-DTBM-binap, also promoted the reaction (entries 2 and 3), and smaller dihedral angle of the ligands was found to increase the enantioselectivity (dihedral angles: segphos (67.2°), MeO-biphep (72.3°), binap (86.2°)).21 In sharp contrast, no addition products were formed by use of (S)-segphos or (R)-binap, which has diphenylphosphino groups (entries 4 and 5). The cationic iridium complex [Ir(cod)2]BArF4 as a catalyst precursor was less effective in catalysing the present reaction, thus giving 3aa in 13% yield (entry 6). After optimizing reaction conditions, such as solvent, temperature, reaction time, and so on, we selected the reaction conditions shown in entry 1 of Table 1 (see Table S1 in ESI†). The absolute configuration of 3aa obtained by use of (S)-DTBM-segphos was assigned to be S-(−) by correlation with the reported specific rotation values.22
|
|
|||
|---|---|---|---|
| Entry | Ligand | Yieldb (%) | Eec (%) |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.30 mmol), [IrCl(cod)]2 (5 mol% Ir), ligand (5 mol%), and NaBArF4 (10 mol%) in MCH (0.4 mL) at 140 °C for 48 h. MCH: methylcyclohexane. b Isolated yields. c Determined by HPLC analysis with a chiral stationary phase column: Chiralpak ID. d With 5 mol% of [Ir(cod)2]BArF4 complex instead of [IrCl(cod)]2 and NaBArF4. e Not determined. | |||
| 1 | (S)-DTBM-segphos | 85 | 93 |
| 2 | (S)-DTBM-MeO-biphep | 85 | 89 |
| 3 | (R)-DTBM-binap | 92 | 84 |
| 4 | (S)-segphos | 0 | — |
| 5 | (R)-binap | 0 | — |
| 6d | (S)-DTBM-segphos | 13 | N.De |

Scheme 2 summarizes the results obtained for the addition of several imides to styrene. Not only substituted phthalimides (1b and 1c) but also 3,4-diphenylmaleimide (2d) and succinimide (2e) underwent the hydroamination of styrene to give the corresponding products 3ba–3ea with high enantioselectivities (82–92% ee). 1-Methylhydantoin (2f) and 5,5-dimethylhydantoin (2g) were also good substrates, giving 3fa and 3ga in high yields with high enantioselectivity. The present catalytic system could not be applied to the addition of saccharin, six-membered piperidine-2,6-dione, or 1-methylimidazolidin-2-one.
 | ||
Scheme 2 Ir-catalysed asymmetric addition of an amide N–H bond to alkenes. Reaction conditions: 1 (0.10 mmol), 2a (0.30 mmol), [IrCl(cod)]2 (5 mol% Ir), (S)-DTBM-segphos (5 mol%), and NaBArF4 (10 mol%) in MCH (0.4 mL) at 140 °C for 48 h. Isolated yields were shown. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 mol% of Ir, 10 mol% of (S)-DTBM-MeO-biphep, and 20 mol% of NaBArF4 were used. 10 mol% of Ir, 10 mol% of (S)-DTBM-MeO-biphep, and 20 mol% of NaBArF4 were used. | ||
Under the catalysis of Ir+/(S)-DTBM-segphos, para-alkyl substituted alkenes 2b–2d effectively undergo the addition of phthalimide (1a) to give the corresponding adducts 3ab–3ad in high yields and enantioselectivity (79–93% yield, 90–93% ee, Scheme 3). In contrast, the reaction with 4-methoxystyrene (2e) did not give the adduct 3ae due to its intense polymerization in the presence of a cationic iridium complex.23p-Trifluoromethoxylstyrene (2f) participated in the reaction, but the yield of the adduct 3af was still low (20% yield) due to the polymerization. In the reaction of styrenes 2g (F), 2h (Cl), and 2i (CF3), which are substituted with electron-withdrawing groups, 3,4-diphenylmaleimide (1d) exhibited the higher reactivity than phthalimide (1a), thus giving the corresponding adducts 3ag–3ai (24–86% yields, 93% ee).24 The reaction of meta-, ortho-, and multiply substituted styrenes 2j–2p reacted with phthalimide (1a) or maleimide 2d to give the adducts with high enantioselectivity (74–93% ee). To our delight, non-conjugated alkenes such as allylsilanes were applicable to the reaction. Thus, the reaction of trimethyl- (2q), diphenylmethy- (2r), and triphenyl-allylsilane (2s) proceeded to give the desired products 3aq–3as with high enantioselectivity (80–86% ee). 2-Norbornene (2t) was also a good substrate to give exo-adduct 3at in high yield with high enantioselectivity, even in a 1.0 mmol scale reaction of 1a (94% yield, 96% ee). Unfortunately, however, the present catalytic system could not be applied to the addition to simple alkenes such as 1-hexene, (Z)-3-octene, cis-stilbene, and indene.
 | ||
| Scheme 3 Scope of alkenes 2. Reaction conditions: 1a or 1d (0.10 mmol), 2 (0.30 mmol), [IrCl(cod)]2 (5 mol% Ir), (S)-DTBM-segphos (5 mol%), and NaBArF4 (10 mol%) in MCH (0.4 mL) at 140 °C for 48 h. Isolated yields were shown. a10 mol% of Ir, 10 mol% of (S)-DTBM-segphos, and 20 mol% of NaBArF4 were used. bWith 5 equiv. of 2q. cAt 120 °C. d1.0 mmol scale. | ||
To gain some insight into the reaction mechanism, stoichiometric reactions of 1a with a cationic iridium complex were conducted. When the reaction of phthalimide (1a, 2 equiv.), [IrCl(coe)]2 (1 equiv.), (S)-DTBM-segphos (1 equiv.), and NaBArF4 (1 equiv.) in acetonitrile-d3 was carried out at 80 °C for 15 min in an NMR tube, two major hydride resonances (77% and 14% yields determined by 1H NMR using an internal standard) as well as three small resonances were observed. 1H NMR of the major two hydride resonances showed a triplet at −18.0 ppm (JP–H = 15 Hz) and a doublet of doublets at −20.0 ppm (JP–H = 23, 14 Hz), implying that hydrogen atoms on Ir are located at the cis-position to the two phosphorous atoms. The result indicates that the cationic iridium coordinated with DTBM-segphos readily undergoes oxidative addition of phthalimide to give the hydride complexes. We also conducted a deuterium-labeling experiment (Scheme 4). The reaction of 1a with deuterated styrene (2a-d3) under the standard reaction conditions gave the corresponding product 3aa-D in 40% yield, where H/D exchange was observed at both methine and methyl groups. The result indicates that the reversible insertion of the alkene moiety into the Ir–H occurs during the reaction, although the hydroamination products would be formed via aminometallation.17,18
 | ||
| Scheme 4 Deuterium-labelling experiment. | ||
In summary, we have developed the enantioselective addition of the N–H bond of phthalimide to alkenes. Diphosphine ligands possessing biaryl backbones with bulky aryl groups on the phosphorus atoms enabled smooth addition to give the adducts in high yield with high enantioselectivities. Styrene and allylsilane derivatives were good substrates to give the adducts with high enantioselectivities.
Kentaro Yamakawa: conceptualization; data curation; investigation; visualization; writing – original draft. Kana Sakamoto: conceptualization; data curation; funding acquisition; investigation; visualization; writing – review & editing. Takahiro Nishimura: conceptualization; data curation; funding acquisition; investigation; project administration; supervision; visualization; writing – review & editing.
This work was supported by JSPS KAKENHI Grant Number JP19H02721 and JP20J23499. K. S. thanks the JSPS for a research Fellowship for Young Scientists.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42 CrossRef; (b) C. E. Paul, M. R.-Mata, E. Busto, I. Lavandera, V. G.-Fernández, V. Gotor, S. G.-Cerrada, J. Mendiola, Ó. de Frutos and I. Collado, Org. Process Res. Dev., 2014, 18, 788 CrossRef CAS.
- (a) M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788 CrossRef CAS PubMed; (b) M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985 CrossRef CAS; (c) D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284 CrossRef CAS.
- D. J. St. Jean Jr., C. Yuan, E. A. Bercot, R. Cupples, M. Chen, J. Fretland, C. Hale, R. W. Hungate, R. Komorowski, M. Veniant, M. Wang, X. Zhang and C. Fotsch, J. Med. Chem., 2007, 50, 429 CrossRef PubMed.
- J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984 CrossRef CAS PubMed.
- (a) T. C. Nugenta and M. El-Shazlya, Adv. Synth. Catal., 2010, 352, 753 CrossRef; (b) O. l Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857 CrossRef CAS PubMed.
- (a) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (b) A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2022, 122, 269 CrossRef.
- (a) M. Hçhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42 CrossRef; (b) C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305 CrossRef CAS PubMed; (c) S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Ornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef PubMed; (b) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed; (c) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed.
- R. Dorta, P. Egli, F. Zürcher and A. Togni, J. Am. Chem. Soc., 1997, 119, 10857 CrossRef CAS.
- J. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 11960 Search PubMed.
- (a) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9546 CrossRef CAS; (b) A. Hu, M. Ogasawara, T. Sakamoto, A. Okada, K. Nakajima, T. Takahashi and W. Lina, Adv. Synth. Catal., 2006, 348, 2051 CrossRef CAS; (c) C. S. Z. Zhang, S. D. Lee and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 5372 CrossRef PubMed; (d) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed; (e) M. Otsuka, H. Yokoyama, K. Endo and T. Shibata, Org. Biomol. Chem., 2012, 10, 3815 RSC; (f) C. S. Sevov, J. S. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 11960 CrossRef CAS PubMed; (g) C. S. Sevov, J. S. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 3200 CrossRef CAS PubMed; (h) E. P. Vanable, J. L. Kennemur, L. A. Joyce, R. T. Ruck, D. M. Schultz and K. L. Hull, J. Am. Chem. Soc., 2019, 141, 739 CrossRef CAS PubMed.
- Q.-A. Chen, Z. Chen and V. M. Dong, J. Am. Chem. Soc., 2015, 137, 8392 CrossRef CAS PubMed.
- (a) R. L. LaLonde, B. D. Sherry, E. J. Kang and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2452 CrossRef CAS PubMed; (b) K. L. Butler, M. Tragni and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2012, 51, 5175 CrossRef CAS PubMed; (c) M. L. Cooke, K. Xu and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 10876 CrossRef CAS PubMed; (d) Y.-M. Wang, A. D. Lackner and F. D. Toste, Acc. Chem. Res., 2014, 47, 889 CrossRef CAS PubMed.
- (a) O. Löber, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4366 CrossRef PubMed; (b) S. Hong and T. J. Marks, J. Am. Chem. Soc., 2002, 124, 7886 CrossRef CAS PubMed.
- For recent reviews, see; (a) H. Liu, S. Saha and M. S. Eisen, Coord. Chem. Rev., 2023, 493, 215284 CrossRef CAS; (b) S. Ma and J. F. Hartwig, Acc. Chem. Res., 2023, 56, 1565 CrossRef CAS PubMed.
- S. Pan, K. Endo and T. Shibata, Org. Lett., 2012, 14, 780 CrossRef CAS PubMed.
- Y. Xi, S. Ma and J. F. Hartwig, Nature, 2020, 588, 254 CrossRef CAS PubMed.
- S. Ma, Y. Xi, H. Fan, S. Roediger and J. F. Hartwig, Chem, 2022, 8, 532 CAS.
- M. S. Gibson and R. W. Bradshaw, Angew. Chem., Int. Ed. Engl., 1968, 7, 919 CrossRef CAS.
- The cationic iridium complex [Ir((S)-DTBM-segphos)(cod)]BArF4 was in situ prepared by mixing [IrCl(cod)]2, (S)-DTBM-segphos, and NaBArF4 in CH2Cl2. After removing CH2Cl2 under vacuum, substrates and the solvent were added. This procedure contributed the reproducibility of the reaction.
- S. Jeulin, S. D. de Paule, V. Ratovelomanana-Vidal, J.-P. Genêt, N. Champion and P. Dellis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5799 CrossRef CAS PubMed.
- See the ESI† for details.
- The polymerization was separately confirmed in the reaction of 2e in the presence of the cationic iridium catalyst without 1a.
- For example, the reaction of phthalimide (1a) with p-chlorostyrene (2h) gave the corresponding adduct 3ah in 15% yield with 49% ee.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc04406h |
| This journal is © The Royal Society of Chemistry 2023 |