
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic applications of waste derived materials
James A.
Bennett
*,
Karen
Wilson
and
Adam F.
Lee
 *
*
European Bioenergy Research Institute, Aston University, Aston Triangle, Birmingham B4 7ET, UK. E-mail: a.f.lee@aston.ac.uk; Tel: +44 (0)121 2044036
First published on 20th January 2016
Abstract
Sustainability has become a watchword and guiding principle for modern society, and with it a growing appreciation that anthropogenic ‘waste’, in all its manifold forms, can offer a valuable source of energy, construction materials, chemicals and high value functional products. In the context of chemical transformations, waste materials not only provide alternative renewable feedstocks, but also a resource from which to create catalysts. Such waste-derived heterogeneous catalysts serve to improve the overall energy and atom-efficiency of existing and novel chemical processes. This review outlines key chemical transformations for which waste-derived heterogeneous catalysts have been developed, spanning biomass conversion to environmental remediation, and their benefits and disadvantages relative to conventional catalytic technologies.
 James A. Bennett | Dr James Andrew Bennett obtained his Master and PhD at the University of Leicester, where he investigated the use of perfluoroalkyl moieties to allow heterogenisation of homogeneous catalysts over zirconium phosphonate supports. He then worked at the University of Birmingham, researching biogenic heterogeneous catalysts composed of transition metal nanoparticles supported on bacterial biomass, using waste sources of metals and biomass to produce “green” catalyst materials. He is currently working with Professors Karen Wilson and Adam Lee at the European Bioenergy Research Institute at Aston University, developing environmentally sustainable catalysts derived from industrial waste for pyrolysis oil upgrading. |
 Karen Wilson | Karen Wilson is Professor of Catalysis and Research Director of the European Bioenergy Research Institute at Aston University, where she holds a Royal Society Industry Fellowship. Her research interests lie in the design of heterogeneous catalysts for clean chemical synthesis, particularly the design of tunable porous materials for sustainable biofuels and chemicals production from renewable resources. She was educated at the Universities of Cambridge and Liverpool, and following postdoctoral research at Cambridge and the University of York, was appointed a Lecturer and subsequently Senior Lecturer at York, prior to appointment as a Reader in Physical Chemistry at Cardiff University. |
 Adam F. Lee | Adam Lee is Professor of Sustainable Chemistry and an EPSRC Leadership Fellow in the European Bioenergy Research Institute, Aston University. He holds a BA (Natural Sciences) and PhD from the University of Cambridge, and following postdoctoral research at Cambridge and Lecturer/Senior Lecturer roles at the Universities of Hull and York respectively, held Chair appointments at Cardiff, Warwick and Monash universities. His research addresses the rational design of nanoengineered materials for clean catalytic technologies, with particular focus on sustainable chemical processes and energy production, and the development of in situ methods to provide molecular insight into surface reactions, for which he was awarded the 2012 Beilby Medal and Prize by the Royal Society of Chemistry. |
1. Introduction
The field of catalysis has seen a recent resurgence of interest in the design and development of catalytic materials derived from earth abundant and low cost components, i.e. non-platinum group metals (PGMs), with a view to improving the overall sustainability of catalytic processes. In some processes, notably exhaust emission control systems, catalyst production can be the single largest monetary cost, and has driven significant efforts to minimise precious metal losses during operation and recover/recycle spent catalysts. However, in addition to their expense, PGMs are also subject to geographical security concerns or supply chain vulnerability, with over 70% of Pt and 36% of Pd located in South Africa alone,1 hence there is a driver to identify widely available catalysts amenable to low cost manufacturing routes.Waste biomass has been increasingly targeted as a renewable feedstock for the production of fuels2–4 and more recently platform chemicals.5 Although by definition required in small amounts, the cost of the catalyst in a given process is potentially one of the most significant expenses, particularly in a process using waste biomass as starting material.6 Using waste material to generate the catalyst in addition to the target product makes the system more cost effective and environmentally friendly. There are an increasing number of reports of the preparation of heterogeneous catalysts from waste materials. The nature of the waste source affects the properties of the resulting catalyst, and therefore catalysts for a given application are often made from similar groups of materials. This review will summarise the aforementioned reports from the literature, with the various catalysts being classified by application. The synthesis of heterogeneous catalysts from waste materials has become increasingly popular over the past two decades. Most reports utilise abundant waste streams generated on the millions to hundreds of millions of tonnes per annum scale, which is important if catalysts obtained from them are to be utilised in major industrial processes. Amongst the most abundant resources are waste biomass from agriculture (inedible crops7 and animal parts8), sewage,9 mining and metal fabrication,10 in particular the iron and steel industry.11 Aside from generating highly active, selective and stable catalysts, a suitable waste stream should be simple and safe to collect, and its use for the purpose of catalyst manufacture should raise no ecological concerns. Any collection or handling costs must be factored into catalyst production costs, and will reduce the benefits of such a waste derived product. Consumption of waste materials is highly advantageous where there are existing financial or environmental costs associated with their disposal, as these may be alleviated or negated by its diversion to a valuable catalyst product. Sustainability of the waste stream must also be considered, for example food waste is likely to be generated at ever rising levels due to global population growth,12–14 whereas future technological developments and changes in regulations/legislation (e.g. EU Waste Framework Directive15) may downscale certain process, diminishing the scale of waste they generate. Waste stream valorisation through utilisation as a raw material must not encourage or justify an increase in the rate of its generation, as this is clearly in conflict with the goals of green chemistry.
2. Esterification and transesterification
Biodiesel production, via low temperature esterification of free fatty acids (FFAs) or transesterification of triacyl glycerides (TAGs), is the most popular process for which waste-derived catalysts have been investigated; solid acid and base catalysts have been prepared from diverse waste streams including plant matter, algae, eggshell, animal bone, clay, ash and industrial waste/by-products. In general, solid acid catalysts have been employed for esterification, and based upon activated carbons produced by pyrolysis or carbonisation and subsequent sulfonation. Solid base catalysts used for transesterification principally comprise calcium oxides, typically derived from bone and the shell of molluscs or eggs. Waste-derived solid base catalysts for biodiesel production has been widely reported and is also reviewed elsewhere.16–202.1. Solid base catalysts from waste
Heterogeneous solid bases typically offer significantly higher rates of TAG transesterification than their solid acid counterparts. A wealth of literature exists on the application of CaO transesterification catalysts, due to the wide range of calcium-rich waste materials available. The majority of such catalysts are synthesised via the high temperature calcination (at typically ≥800 °C) of shells from shellfish,21–31 snails32–35 or eggs.26,27,36–40 This approach offers a cost-effective route to the disposal of maricultural/agricultural and concomitant creation of a value-added product for the conversion of other wastes. Fig. 1 illustrates the synthesis of a solid base transesterification catalyst from freshwater mussel shells.23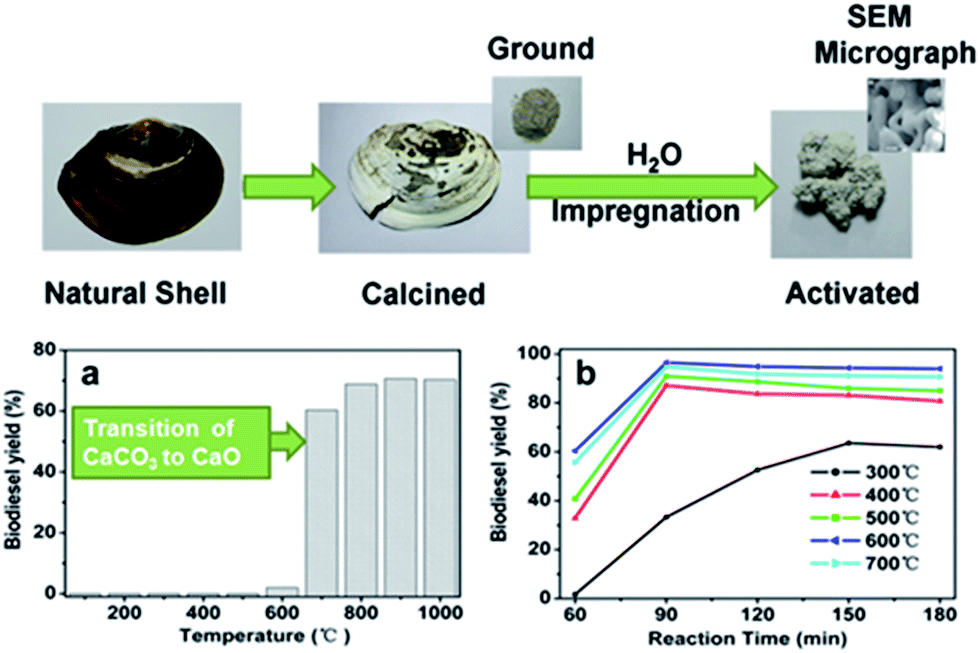 | ||
| Fig. 1 Preparation of transesterification catalysts derived from mussel shells and the effect of (a) calcination and (b) activation temperature. Adapted from ref. 23. Copyright 2011, with permission from Elsevier. | ||
Calcination temperature has emerged as a critical factor in the production of efficient solid base catalysts from mollusc/egg shells. These shells comprise mainly CaCO3, which TGA and XRD studies show decomposes to CaO at temperatures >700 °C,27,30,37,40 resulting in active transesterification catalysts, typically offering >90% conversion of palm, soybean and karanja oils, in 2–6 h at reaction temperatures as low as 60–70 °C, alcohol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oil molar ratios around 10, and catalyst loadings of 2–5 wt%.19 Regeneration of deactivated waste shell-based catalysts can be achieved through simple re-calcination at 900 °C.33 However, regeneration of the catalyst in this way has a high energy cost and may not be practical for industrial scale processes. Another major issue with these catalysts is leaching of Ca2+ into the alcohol phase of the reaction media,41 leading to a gradual loss in activity (which may be masked by homogeneous contributions), and saponification where impure or untreated oils are used. Also, activated CaO catalysts may be deactivated by poisoning upon exposure to air.42
:oil molar ratios around 10, and catalyst loadings of 2–5 wt%.19 Regeneration of deactivated waste shell-based catalysts can be achieved through simple re-calcination at 900 °C.33 However, regeneration of the catalyst in this way has a high energy cost and may not be practical for industrial scale processes. Another major issue with these catalysts is leaching of Ca2+ into the alcohol phase of the reaction media,41 leading to a gradual loss in activity (which may be masked by homogeneous contributions), and saponification where impure or untreated oils are used. Also, activated CaO catalysts may be deactivated by poisoning upon exposure to air.42
Active CaO-containing catalysts have also been generated at temperatures <700 °C by incomplete carbonisation and subsequent KF impregnation and thermal activation. Shrimp31 and turtle shells43 have been used to synthesis solid bases by this route via calcination at 450 and 500 °C respectively (Fig. 2). Such catalysts have comparable activity to the aforementioned CaO catalysts created at higher temperatures, but do not result in soap formation during transesterification via saponification, unlike some conventional solid bases.43 However, these catalysts suffer from deactivation when reused. The loss of activity was attributed to coating of the catalyst surface by products,31,43 but conversions still decreased when the used catalyst was washed with petroleum ether between each subsequent cycle. This leads to the speculation that some potassium may be lost from the catalyst surface during the reaction, although leaching studies were not reported.
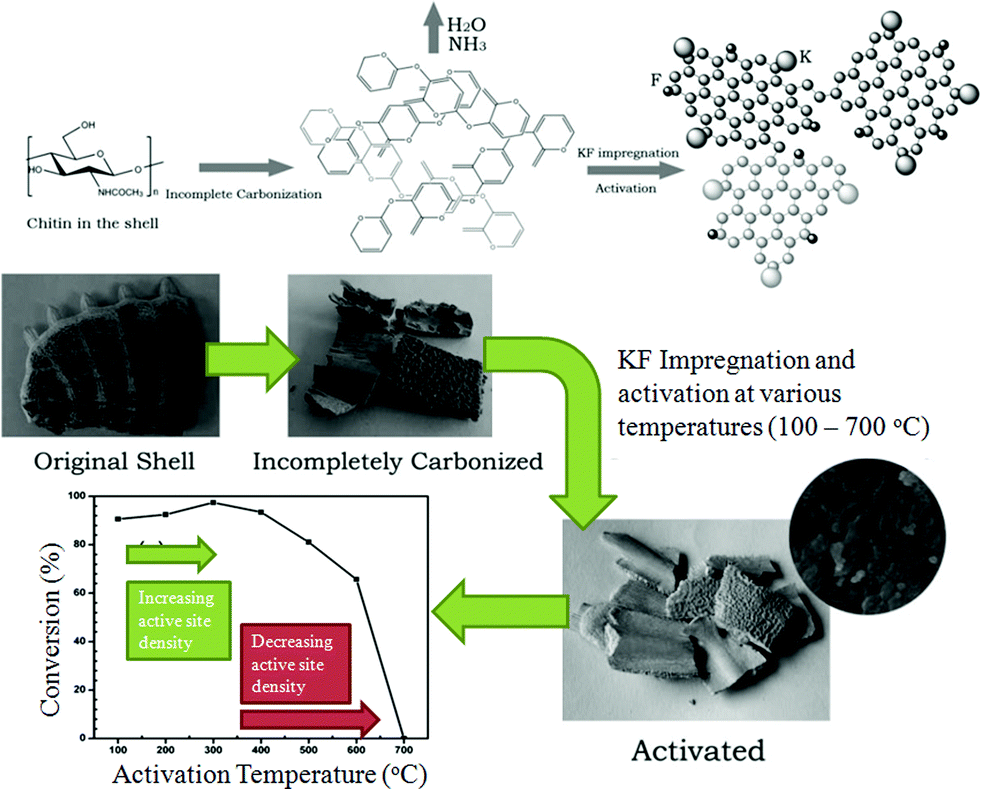 | ||
| Fig. 2 Preparation procedure of transesterification catalysts derived from turtle shells and the effect of activation temperature. Adapted from ref. 43. Copyright 2008, with permission from the Royal Society of Chemistry. | ||
Other calcium-containing catalysts from waste include those from coral44 bones,8,45 scales46 and calcite-containing rocks.47–52 Corals are marine polyps which possess a calcareous exoskeleton formed from aragonite, a crystalline form of CaCO3. Roschat et al. converted coral fragments to CaO by calcination at 700 °C, and applied the resulting material as a catalyst for the transesterification of various oils, including palm, soybean, rice bran and waste cooking oils, with methanol.44 Calcined coral fragments catalysed >98% FAME production in 2 h at 65 °C with a methanol:oil ratio of 15. However, since the coral fragments (0.5–1 cm in diameter) possessed a very low surface area and pore volume, they necessitated a high catalyst loading of 100 wt% compared with catalyst loadings of <5 wt% typical for conventional CaO materials.
At calcination temperatures >650 °C, calcium phosphate in animal bones and scales transforms into hydroxyapatite, which may be employed directly in biodiesel production. Under optimal conditions, bone-derived catalysts have been demonstrated to deliver conversions comparable to laboratory grade CaO;8,45 however high catalyst loadings (8–20 wt%) were again required, coupled with a higher reaction temperature (80 °C) and alcohol:oil ratio (18) versus CaO catalysts prepared from mollusc or egg shells. The impact of calcination temperature on catalyst surface area varies with the waste source. Smith et al. found no significant improvement in BET surface area upon calcining bovine bone >650 °C, with values remaining a mere 2–3 m2 g−1, reflecting crystallite sintering and loss of pore volume.8 However, a similar study by Obadiah et al. observed an order of magnitude increase in the surface area of calcined sheep bone with calcination temperature, rising from 6.8 m2 g−1 to 885 m2 g−1 between 600 and 800 °C.45 This may reflect different methods of bone preparation prior to calcination. Chakraborty and co-workers utilised a similar route to prepare calcium-based catalysts from fish scales.46 Calcination at 900 °C for 2 h yielded a fibrous, porous material composed mainly of β-tricalcium phosphate with a surface area of 39 m2 g−1, affording 98% FAME in the transesterification of soybean oil after 5 h at 70 °C with only 1 wt% catalyst and an excellent, low methanol:oil ratio of 6.
There are clear ecological and sustainability issues with the use of coral and turtle shells as a calcium source for the large scale production of solid base catalysts. Animal bones and seafood waste are the more viable feedstocks for calcium-based catalyst production, being readily available, abundant and continually produced. Worldwide mollusc, fishery and meat production are currently over 14,53 160 (ref. 53) and 300 million tonnes per annum respectively,54 with up to 25 wt% of fish and up to 28 wt% of the livestock ending as non-edible solid waste, including bone.55 Waste produced from shrimp, crab and lobster fishing is a much less abundant source, currently between 6 and 8 million tonnes per annum56
There are many natural calcium-rich minerals, some of which have been exploited as solid base catalysts for transesterification. A range of natural calcium sources were calcined at 800 °C by Ngamcharussrivichai et al. and screened for the transesterification of palm kernel oil at 60 °C using a high methanol:oil ratio of 30 and catalyst loading of 6 wt%.47 Calcite, a stable polymorph of CaCO3, was found superior to calcined cuttlebone and hydroxyapatite, with TAG conversions of 47, 24 and 3% respectively. However, dolomite, a calcium magnesium carbonate, CaMg(CO3)2, was the best such mineral tested, with 99% conversion to FAME under these reaction conditions, outperforming CaO, MgCO3 and MgO which gave 93, 1 and 1% conversion respectively. These results indicated a synergy between alkali earth metals. Dolomite was also utilised by Wilson and co-workers, who synthesised a high activity solid base catalyst comprising MgO nanocrystallites supported on CaO via prolonged calcination of dolomitic quarry waste at 900 °C.57 The catalyst was effective towards the transesterification of glyceryl tributyrate, glyceryl trioctanoate and olive oil with methanol, giving 100% conversion of C4 and C8 substrates within 3 h at 60 °C and low catalyst loadings of only 1.7 and 1.1 wt% respectively, with FAME selectivities >98%. However, transesterification rates were far slower for bulkier TAGs, possibly reflecting mass transport limitations in accessing base sites. Similar synergy between alkali earth metals was observed by Balakrishnan et al.,48 who used waste marble from construction sites as a CaO solid base catalyst for the transesterification of waste cooking oil. Marble was crushed and ground, impregnated with barium and then calcined at 830 °C, increasing its natural porosity and surface area from 40 to 56 m2 g−1. The resulting Ba/CaO catalyst gave slightly lower FAME yields than other waste CaO materials, with 88% TAG conversion after 3 h at 65 °C and 3 wt% catalyst, albeit with a modest MeOH:oil ratio of 9 and more challenging waste feedstock high in FFAs (which readily neutralise conventional solid bases58–61).
Red mud is a waste material formed during of the production of alumina from bauxite. Annual production of this waste residue has steadily increased over the last century, due to increasing worldwide demands for aluminium metal, and is currently generated at over 120 million tonnes per annum.62 Red mud comprises a complex mixture of metal oxides, notably iron oxides and small amounts of alkali earth metals. Liu et al. applied red mud, dried at 200 °C in air as a catalyst for soybean oil transesterification.49 A 4 wt% loading of red mud resulted in >94% biodiesel yield after 3 h at 65 °C and a MeOH:oil molar ratio of 24, although the authors noted that their red mud sample contained much higher levels of Ca than commonly found in this waste stream, with the major component being CaCO3 rather than iron oxides. Waste cement is composed of a similar mixture of metal oxides, principally calcium and iron oxides, with silica and alumina. Wang et al. used Portland cement as catalyst for the transesterification of soybean oil with methanol.50 Under optimal conditions, cement calcined at 650 °C gave 98% conversion after 3 h at 65 °C, but with an extremely high 32.8 wt% catalyst loading and methanol:soybean oil ratio of 24. While the catalyst loading was high, it must be recalled that a large proportion of the sample mass was fine aggregates of waste concrete from which the cement was recycled, and that this aggregate was itself inactive in transesterification. When using fresh Portland cement as a catalyst for soybean oil transesterification complete conversion was achieved with only a 4 wt% loading under identical conditions. Sea sand may also be used as calcium oxide source since it is rich in CaCO3. Calcined sea sand was therefore used by Muciño to transesterify oils for biodiesel.51 Conversions to FAME of 9 5%, 97% and 98% were obtained from waste cooking oil, safflower oil and soybean oil respectively after 6 h at 60 °C with a methanol:oil ratio of 12 and 7.5 wt% catalyst. High calcination temperatures of 800 °C were required to fully decompose the CaCO3 to CaO, and the resulting material, composed of 98.7% CaO and 1.3% MgO by EDS, was strongly basic (9.3 < H− < 15 via Hammett indicators) but possessed a low surface area and porosity of only 5 m2 g−1 and 0.001 cm3 g−1. MgO–CaO/SiO2 from calcined waste slag has also been utilised for rapeseed oil transesterification with methanol.52 Under optimum reaction conditions of 68 °C, methanol:oil ratio of 6.5 and 3.5 wt% catalyst, 98% oil conversion was achieved in 3.5 h. This material was superbasic with an H− of 22, and was as active as lab grade CaO but at a tenth of the cost.
Although CaO is the most widely studied solid base for transesterification due to the abundance of natural, calcium-rich waste sources, other solid bases have been developed. Waste plant biomass has been utilised to create solid base catalysts for biodiesel production. Chen et al. used Li-modified rice husk ash as a solid base for soybean oil the transesterification.7 Rice husk ash was chosen as an abundant, low cost source of amorphous silica, which was mixed with solid Li2CO3 and calcined at 900 °C. XRD showed the resulting material comprised mainly Li2SiO3, which afforded 99% oil conversion at 65 °C after 3 h and a methanol:oil ratio of 24 and 4 wt% catalyst. Though not as basic as pure CaO (H− of 15), this catalyst was air-insensitive, rendering it advantageous to CaO which rapidly loses basicity upon air exposure. Hindryawati et al. also used rice husk ash as a support for alkali metals to create a strong solid base for the transesterification of waste cooking oil.63 Catalysts were prepared by wet impregnation of the ash with aqueous alkali metal hydroxide solutions, dried and calcined between 200 °C and 700 °C. FTIR and XRD studies showed that the materials which were calcined ≥500 °C and composed of Li2SiO3, Na2SiO3 or K2SiO3 depending on the starting hydroxide. These materials gave >96% conversion to FAME after 3 h at 65 °C with a catalyst loading of 3 wt% and a methanol:oil ratio of 9. Catalysts were reused successively six times with only small conversion losses due to alkali leaching; the residual methanolic filtrate after exposure to catalysts for 1 h conferred 33–40% oil conversion. The same group prepared a similar material from waste sponge skeletons as a silica source.64 These were impregnated with aqueous NaOH at a Na:Si molar ratio of 2, and the resulting material dried and calcined at 500 °C to form predominantly amorphous SiO2 and Na2SiO3. This was also active in waste cooking oil transesterification, leading to 98% FAME yield after only 30 min at 55 °C for an identical reaction mixture. However, it is difficult to compare the activity of the latter catalyst to similar waste-derived transesterification catalysts since its rapid reaction was ultrasound-assisted. Nevertheless, the sponge-derived catalyst was tolerant of up to 8 wt% water in the oil, and recyclable up to seven times with a small drop in methyl ester yields whose origin was not discussed. A potassium-containing silica has also been prepared from the ash of waste aquatic plants by Chouhan et al.65 Plants were first dried and calcined at 550 °C, containing around 82% SiO2 and 11% K, with the balance Co, C, Pb, Cl and Na. The calcined ash was used without further processing for the transesterification of refined Jatropha curcas L. oil. With 5 wt% ash, 89% conversion to biodiesel was attained after 5 h at 65 °C and a methanol:oil ratio of 9. However, this catalyst exhibited significant deactivation, with oil conversion falling to 47% after only two re-uses, although the authors (contentiously) argued that this was compensated for by the minimal catalyst production cost.
The most abundant organic sources of CaO are fish and livestock solid wastes. These are available on sufficiently large scale for catalytic applications, and can be readily collected from slaughterhouses, fisheries or processing plants, and their use for this purpose would not raise ecological concerns. Calcium-rich minerals, such as dolomite, are potentially available on a much greater scale, require less processing/purification than organic Ca sources and often generate more active catalysts.
2.2. Solid acid catalysts from waste
Solid acid catalysts are not as widely reported for biodiesel production as their base counterparts, reflecting the lower intrinsic activity of the former. However, solid acids offer a significant practical advantage in their tolerance of free fatty acids (FFA), and hence potential for the simultaneous esterification of FFA impurities and transesterification of TAG components of challenging bio-oils. In contrast, solid bases require pretreatment of FFA-containing bio-oil to prevent in situ deactivation and post-reaction separation issues arising from saponification. The application of synthetic acid catalyst to biodiesel production has been extensively reviewed.66–69Waste polymers70 and clays71–74 have been used to produce solid acid biodiesel catalysts however the majority of such catalysts are produced from waste biomass, often by-products of biodiesel production. The manufacture of catalysts from waste seed cakes75,76 or microalgal residues77 following bio-oil extraction could enhance the cost-effectiveness of the overall biodiesel production process.
Solid acid catalysts have been prepared by pyrolysis and subsequent sulphonation of oil seed cakes. Dawodu et al. extracted oil from non-edible seeds of the evergreen tree Calophyllum inophyllum, which contained around 15% FFA, and then used the seed cake to prepare a catalyst for one-step conversion of the extracted oil into biodiesel.75 Residues were incompletely carbonised at 400 °C under N2 and then functionalised with –SO3H by heating with concentrated H2SO4 under N2. The same procedure was also used to produce catalysts from pure glucose. Resulting materials composed large, irregular carbon sheets of low BET surface areas (0.2–3.4 m2 g−1) and a sulphur content ranging from 0.1–3.6 wt%. Catalysts pyrolysed for shorter times afforded smaller carbon sheets and higher acid site density, resulting in superior FFA conversion and concomitant FAME production. However, these catalysts also suffered high degrees of sulphur leaching in situ. Catalysts derived from seed cakes gave good FFA conversions (>75%) but poor FAME yields (up to 36%). Glucose-derived catalysts performed more favourably, offering 99% FAME yield after 4–5 h with an excellent MeOH:oil ratio of 1.1:1, albeit requiring high catalyst loadings of 7.6 wt% and a high reaction temperature at of 180 °C, and hence autoclave operation at 10 bar to maintain a liquid phase.
Konwar and co-worker adopted a similar approach to create a solid acid catalyst from the seed cake of the Mesua ferrea tree.76 Residues were carbonised at 500 °C and sulphonated by covalent attachment of 4-benzenediazoniumsulfonate radicals to give catalysts with a sulphur content of 1–2 wt% and a high surface area (550–700 m2 g−1). At 80 °C, a 5 wt% catalyst loading provided >97% FFA conversion within Mesua ferrea oil after 8 h, however a very high MeOH:oil ratio of 43:1 was required to drive the equilibrium and lower FFA content below 2%. The transesterification activity of sulphonated catalysts was low when using oils of low FFA content, but surprisingly improved with increasing FFA content due to enhanced methanol miscibility in acidic oils. These catalysts were reused 5 times with minimal loss in FFA conversion (which was quantitative in sulphur leaching).
Oil for biodiesel production may also be harvested from microalgae. Fu et al. used the same approach as described above employing microalgae residue, which is often combusted as a fuel source post-oil extraction, to generate solid acid catalysts for oleic acid esterification and triglyceride transesterification with methanol.77 Partially-carbonised, sulphonated materials were composed of graphene sheets with a high density of –SO3H, –OH and –COOH functions. Despite an extremely poor surface area of only 1 m2 g−1, the catalyst exhibited superior performance to that of the widely used, commercial polymeric solid acid, Amberlyst-15, which was attributed to beneficial surface–reagent interactions with the carbon-based support. A 5 wt% loading of microalgae-derived catalyst resulted in 98% oleic acid conversion after 12 h at only 80 °C and was reusable 6 times with negligible activity loss. Under the same conditions a FAME yield of 24% was obtained in triolein transesterification. Although the results are promising, the methanol:oil ratio was not reported hampering comparison with literature catalysts.
A family of mesoporous materials, named Starbons®, have been prepared from starch78 (Fig. 3) with high surface areas, good mechanical stability and a range of hydrophilicities. While raw and heat or chemically modified starches are widely used as foods, and therefore should not be considered a waste resource, starch is also employed on the multimillion ton per annum scale within the papermaking industry, and as the basis of corrugated board adhesives. These applications result in vast quantities of entrained starch at the end of product lifetime, and hence are available as a waste resource for recycling into the original products alongside the direction of a small fraction into alternative higher value products such as catalysts. Solid acids were synthesised from carbonised starch via sulphonation for 4 h at 80 °C, and subsequently tested in the aqueous phase esterification of various organic diacids, including succinic, fumaric, itaconic and levulinic acids.79,80 Starbons® offered superior conversion and selectivity to commercial sulphonated carbons, despite their lower BET surface areas (386 vs. 585–626 m2 g−1).80 In the case of succinic acid, >99% conversion and diester selectivity were achieved after 500 min at 80 °C with a 30:1 molar ratio of EtOH:acid and (0.1 g catalyst and 0.124 g succinic acid) 85 wt% catalyst, compared to 72 and 81% conversion and 5 and 28% selectivity for NORIT® and DARCO® commercial solid acids respectively.52 These starch-based materials possessed acid site densities comparable to commercial acidic carbons (∼200 mmol g−1 (ref. 80)), and hence their improved activity was attributed to a unique combination of hydrophilic/hydrophobic functions and mixed micro- and mesoporosity. Starbons® were also able to catalyse simultaneously the esterification of free fatty acids present in waste cooking oil with methanol, and the transesterification of associated triglycerides to yield FAME and glycerol,81 and shown activity towards alkylation,52,82 acetylation52,82 and hydrogenation83 and even as photocatalytic supports.84
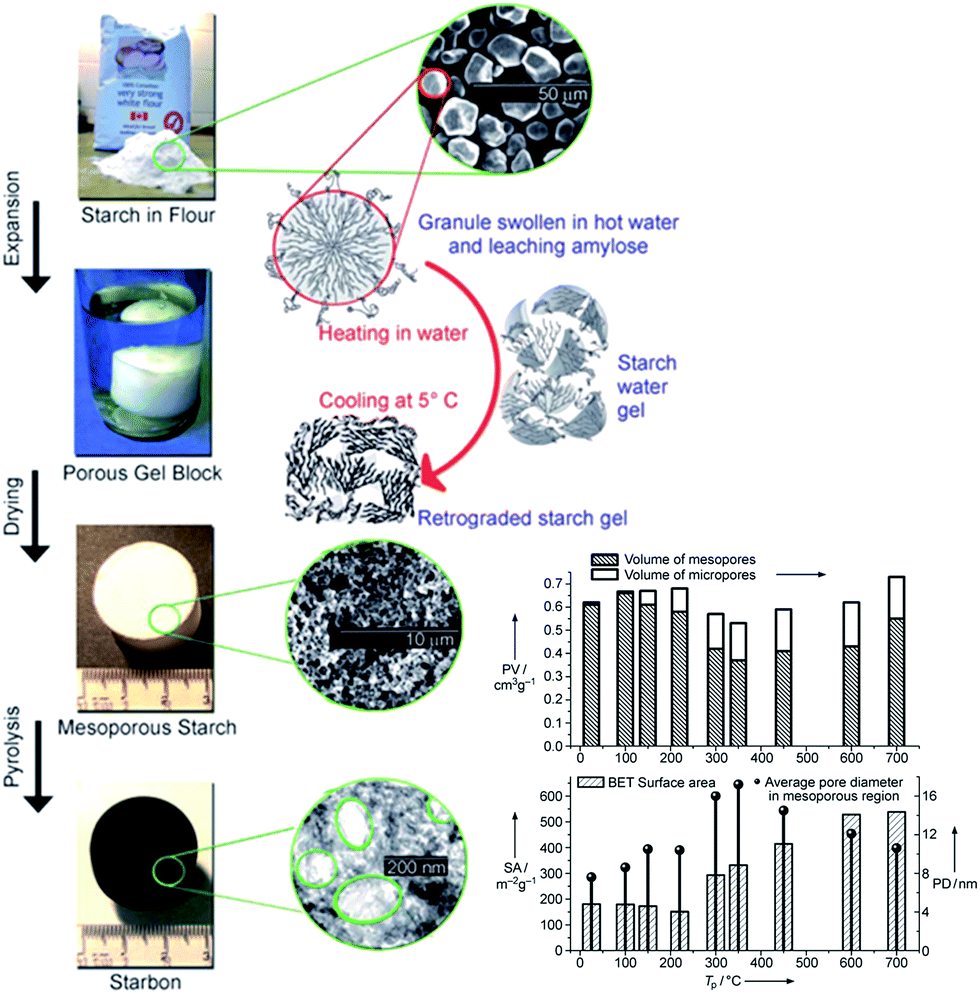 | ||
| Fig. 3 Preparation and surface properties of mesoporous Starbons®. Adapted from ref. 78. Copyright 2006, with permission from Wiley-VCH. | ||
Other approaches to synthesise carbonised and sulphonated biomass-derived catalysts include the use of glycerol,85 glucose,86 microcellulose,87 rice husk,88 vegetable oil89,90 and petroleum oil asphalts,90 cassava stillage residue from bioethanol production91 and pyrolysis biochar.92 A comparison of the catalytic performance of these catalysts and corresponding experimental conditions employed in their applications is provided in Table 1. In all cases, solid acid catalyst synthesis involves biomass carbonisation at 200–800 °C and functionalisation with –SO3H groups through heating with a sulphuric or other sulphur-containing acid. Resulting materials generally comprise non-porous aromatic carbon sheets of low surface area (>10 m2 g−1), although some high area exceptions exist.
| Catalyst material | FFA conversion/% | FAME yield/% | T/°C | MeOH:oil molar ratio |
Time/h | Loading/wt% |
|---|---|---|---|---|---|---|
| a Esterification in the absence of TG. | ||||||
| Seed cake75 | 75 | 25 | 150 | 1.1 | 5 | 7.6 |
| Seed cake76 | 97 | 71 | 80 | 43 | 8 | 5 |
| Microalgae77 | 98 | 24 | 80 | n.a. | 12 | 5 |
| Glycerol85 | >99 | >99 | 160 | 45 | 4 | 20 |
| Glucose86 | >95 | — | 80 | 10 | 8 | 10 |
| Cellulose87 | 89 | — | 60 | 3 | 3 | 7 |
| Rice husk88 | 98 | 88 | 110 | 20 | 15 | 5 |
| Vegetable oil asphalt53 | 95 | 81 | 220 | 17 | 4.5 | 0.2 |
| Petroleum oil asphalt90 | 96 | 88 | 220 | 21 | 3 | 0.3 |
| Cassava stillage91 | 93 | — | 80 | 18 | 6 | 5 |
| Pyrolysis biochar92 | — | 48 | 150 | 30 | 3 | 3 |
The temperatures of carbonisation and sulphonation strongly influence catalytic activity, with higher carbonisation temperatures conferring higher surface areas accompanying oxygen and hydrogen loss, and higher sulphonation temperatures enhancing acid site density. Table 2 shows the physical properties of the aforementioned carbon-based catalysts.
| Catalyst material | BET surface area/m2 g−1 | Acid site density/mmol g−1 | S loading/% | Pore size/nm |
|---|---|---|---|---|
| Seed cake75 | 0.2–3.4 | 0.6–2.8 | 2.4–3.6 | — |
| Seed cake76 | 550–700 | 2.2–2.4 | 1.2–2.4 | — |
| Microalgae77 | 1 | — | — | — |
| Glycerol85 | — | — | — | — |
| Glucose86 | 1–1300 | — | 0.5–2.3 | — |
| Cellulose87 | 105 | 2.5 | — | 1.3 |
| Rice husk88 | 4 | 2.5 | 2.8 | 7.7 |
| Vegetable oil asphalt53 | <10 | 2.2 | 7.1 | 44 |
| Petroleum oil asphalt90 | <10 | 1.1 | 3.6 | — |
| Cassava stillage91 | 4–155 | 1.1–2.7 | — | — |
| Pyrolysis biochar92 | 990 | 1.7–2.0 | 0.4 | 3.2–3.5 |
Waste polymer was utilised by Alonso-Fagúndez et al.70 who employed sulfonated polystyrene to prepare a soluble polystyrene sulphonic acid catalyst and a solid SiO2–polystyrene sulphonic acid nanocomposite via a sol–gel route from tetraethylorthosilicate. Despite promising acid site densities (4.9–5.4 mmol g−1) and activities of the soluble polystyrene catalysts, the insoluble SiO2 nanocomposite analogues were unsuitable for biodiesel preparation from Cynara cardunculus oil. For sulfonated polystyrene, the FFA content of the oil was rapidly reduced by esterification, giving a FAME yield of 80% after 7 h at 130 °C with 4 wt% catalyst and a MeOH:oil molar ratio of 24. However, the same conditions rapidly deactivated the heterogeneous catalyst.
Natural hydroxyapatite (NHA) derived from fish waste has also been employed to support acidic copper species for oleic acid esterification.93 Fish bones were washed, pulverised and the resulting NHA impregnated with copper via a Cu(NO3)2 precursor, and then calcined to yield a low surface area mesoporous material with large (31 nm) pore diameters. The Lewis acid site density of 11 mmol g−1 was higher than those derived from sulphonated carbonised biomass, resulting in a highly active catalyst with 92% FFA conversion after 1 h at 70 °C. Unfortunately the catalyst loading was unspecified prohibiting comparison with alternative solid acid catalysts.
Kaolin, or china clay, is a mineral clay of chemical formula Al2Si2O5(OH)4 used in the manufacture of china and porcelain. Waste flint kaolin has found application by Nascimento et al. for oleic acid esterification.71 The kaolin was calcined at 950 °C prior to activation at 90 °C for 1 h with sulphuric acid, then washed, dried and re-calcined at 400 °C. While this clay had a high surface area (406 m2 g−1), it only possessed a low acid site density of 0.2 mmol g−1, but still delivered >98% oleic acid conversion in 4 h with 5 wt% catalyst, though this necessitated an extremely high reaction temperature of 160 °C and MeOH:acid ratio of 60. Kaolin has also been used for the esterification of waste distillate from palm oil deodorisation into biodiesel,72 converting 93% of fatty acids within the oil residue (which contained 17 wt% FFA), and highlighting the potential of acidified flint kaolin as a viable, low-cost catalyst for biodiesel production. Dang et al.73 also developed a zeolite-like material through the hydrothermal transformation of kaolin. While the surface area of this LTA zeolite was much lower than related zeolites at only 7–9 m2 g−1 it gave high conversion in vegetable oil transesterification: a 10 wt% catalyst loading afforded 95% FAME yield after 4 h at 60 °C and a MeOH:oil ratio of 20 for palm oil, and a 55 wt% loading achieved 98% FAME yield after 2.5 h for soyabean oil under the same conditions. Such high catalyst loadings were partially mitigated by its recyclability (80% FAME yield after four successive reactions). Pires et al.74 employed waste kaolin to support 12-tungstophosphoric acid for the esterification of waste distillate derived from palm oil deodorisation with ethanol. A 25 wt% HPW/kaolin catalyst was highly active, but suffered unacceptable degrees of acid leaching due to the high solubility of the heteropolyacid and resulting homogeneous catalysis. A purification step to remove excess adsorbed HPW from the kaolin produced a more stable 14 wt% heteropolyacid/kaolin catalyst giving 83% palm oil distillate esterification with ethanol in 2 h for an ethanol:oil molar ratio of 10 but only under a forcing reaction temperature of 200 °C. Catalyst recyclability was poor, with FAME yields falling to 20% after three re-uses due to further heteropolyacid leaching.
3. Bio-oil upgrading catalysts
Fast pyrolysis of biomass, which is undertaken typically around 500 °C with retention times of 2 s, can generate high bio-oil yields of up to 75% for the subsequent production of drop-in gasoline and diesel transportation fuels. Unfortunately, the high oxygen content, acidity and poor thermochemical stability of pyrolysis bio-oils prohibits their direct utilization as a bio-fuel, necessitating additional chemical upgrading to deliver a commercial grade fuel.2 An important approach to bio-oil upgrading is the neutralisation of its constituent acids and concomitant lowering of oxygen content via ketonisation, the reaction between two carboxylic acid molecules to form a ketone, CO2 and H2O.94 Metal oxides are active catalysts for ketonisation.95 Waste material from the Bayer process, the refining of bauxite ore to alumina, was used by Karimi et al. for the ketonisation of several carboxylic acids,96 and the upgrading of pyrolysis bio-oils.97,98 This waste, commonly termed ‘red mud’, is a mixture of metal oxides such as Fe2O3, SiO2, Al2O3, SiO2 and alkali and alkali earth oxides such as CaO and Na2O.99 Red mud at a 10 mol% loading and the carboxylic acid were added to an autoclave which was flushed with H2 or inert, sealed and heated to 365 °C for 8 h. In the absence of H2 the catalyst achieved a high conversion of linear alkanoic acids (>89%) and ketone selectivity of 21–41%.96 While similar conversions were obtained under H2 pressures, ketone lower ketone selectivity was observed due to the initiation of additional upgrading reactions such as hydrodeoxygenation and hydrogenation; in situ molecular hydrogen generation is possible through the thermal decomposition of formic acid to produce H2O and CO, which can undergo the water–gas shift reaction over iron to yield H2 and CO2.100 Hemp seed pyrolysis oil was also successfully upgraded using 25 wt% red mud catalyst, with the resulting oil less viscous and more chemically stable.97 A more acidic pyrolysis oil from hardwood sawmill chips, was also successfully upgraded, increasing bio-oil heating value from 12 to 34 kJ g−1 due to deoxygenation.64 The authors noted that red mud was less active than platinum group metal catalysts such as Ru/C or Pt/Al2O3, however this disadvantage is at least partially offset by the low cost of red mud, which adds value to this otherwise alkaline, problematic wastestream.64,101 Due to the huge scale of red mud production, it offers great potential as a cost-effective and earth abundant catalyst/reagent, and has therefore also been employed for catalytic transesterification,49 hydrodechlorination and hydrogenation as reviewed elsewhere.1024. Oxidation catalysts
Metal waste from several industries has been explored in oxidation catalysis. Waste from the aluminium (red mud), tannery and electroplating industries were used as catalyst precursors by Sushil et al.103 At temperatures ≥350 °C, calcined red mud was active for the total oxidation of propane under continuous flow of 1 bar air and a modest gas hourly space velocity of 5000 h−1, although the composition and activity of red mud varied depending on the site is was sourced from, delivering 60–90% propane conversion by 500 °C. Addition of tannery shavings, a source of Cr2O3, to red mud catalysts increased their activity, enabling complete conversion at lower temperature. Pure tannery shavings were the most active, but due to their small particle size were unsuitable for high gas flow rates. Metal extracts obtain via acid leaching of electroplating waste sludge had poor dispersions and their use as additives decreased the activity of red mud catalysts. In all cases activities were lower than those of commercial catalysts e.g. Co3O4/CeO2 and Pt/Al2O3, which afford total propane conversion under similar conditions around 280 °C, although the low cost of such waste-based catalysts permits their use at higher catalyst loadings.Teixeira et al. demonstrated red mud as a catalyst for the conversion and storage of petroleum gas, a mixture of methane and other hydrocarbons produced and flared during offshore petroleum exploration.104 Metal oxides in red mud reacted with CH4 to form CO/CO2 and water, with the resulting reduced metals further reacting to produce hydrogen. The coked metal was then reacted with steam to generate syngas (Fig. 4). Methane decomposition was performed in a continuous flow reactor between 25–900 °C in a CH4/N2 stream, with significant conversion observed >650 °C. Oxidation of metal/carbon composites under steam occurred >650 °C, releasing H2, with CO produced >800 °C from oxidation of carbon deposits. Red mud provides new opportunities for the catalytic conversion, transport and use of associated petroleum gas.
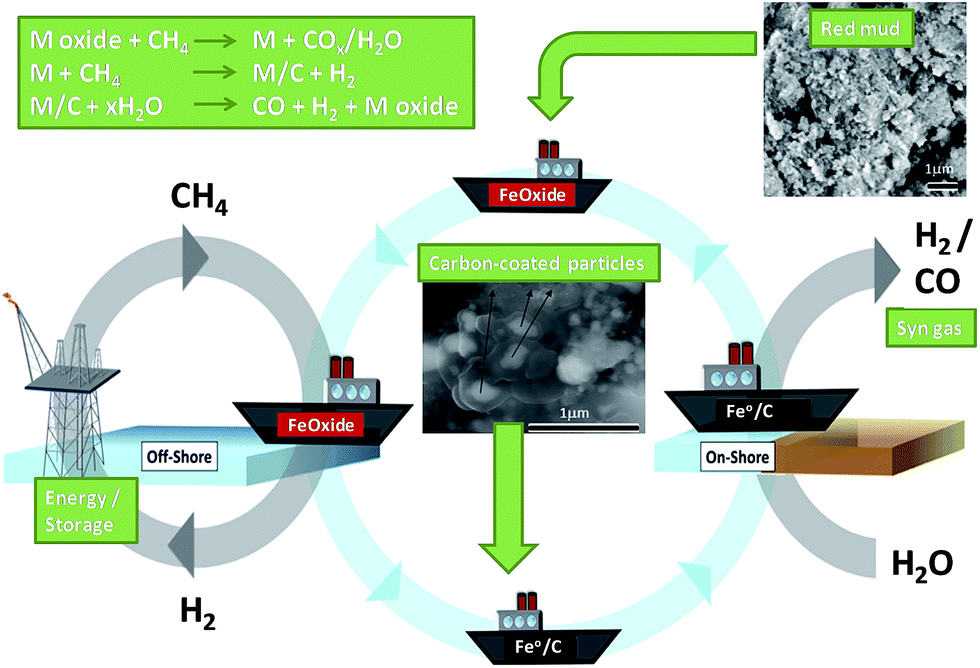 | ||
| Fig. 4 Two-step process of carbon deposition and steam oxidation using associated petroleum gas and red mud. Adapted from ref. 104. Copyright 2014, with permission from Elsevier. | ||
Metal oxides prepared from industrial metal waste via spark erosion have also been exploited.105 These materials comprised fine amorphous powders, containing ∼50 wt% Fe and trace Cr, Cu and other metals, with low surface areas of 12–70 m2 g−1 and were tested as fixed bed catalysts for the oxidation of volatile organic compounds (ethanol and xylene) in continuous flow between 250–300 °C. The highest conversion was obtained with an oxide catalyst containing 50 wt% Fe, 1 wt% Cr and <1 wt% transition and s-block elements calcined at 600 °C, with total ethanol (xylene) oxidation achieved at 300 °C (400 °C). Spark erosion thus appears a viable method to recover catalytically active metals from industrial waste (the source of which was lamentably unstated).
Liquid-phase naphthalene oxidation to aromatic alcohols, aldehydes and anhydrides, such as naphthols, phthaldialdehyde and phthalic anhydride, was also undertaken over a (silica-supported) iron based catalyst, with hydrogen peroxide as the oxidant.106 The silica support was extracted from rice husk and impregnated with 5–20 wt% Fe from a ferric nitrate precursor. The resulting Fe/SiO2 iron catalyst contained Lewis and Brönsted acid sites and good surface areas spanning 96–282 m2 g−1. The most active catalyst 10 wt% Fe catalyst gave 49% naphthalene conversion after 4 h at 70 °C in acetonitrile for a modest catalyst loading and 0.5 equivalents of peroxide. Activity fell on re-use, accompanied by iron leaching, despite which the authors concluded that the reaction was truly heterogeneous. Iron-based catalysts for dye degradation via a Fenton-like oxidation process have also been prepared from goldmine waste,107 blast furnace dust,108 sewage sludge,9 printed circuit boards109 and grinding wheel ash.110 These waste streams are produced in sufficiently high quantities for large scale catalyst production, with steel industry waste the most abundant. For every tonne of steel, 2 to 4 tonnes of waste is produced,111 with blast furnace slag and dust comprising ∼400 kg t−1 and 28 kg t−1 of the waste respectively. Millions of tonnes of blast furnace dust is produced each year,108 tens of millions of dry tonnes of sewage sludge,9 and electronic waste is estimated at around 25 million tonnes annually.112
A catalyst derived from goldmine waste was a fine powder comprising calcium, iron, copper and zinc oxides. Modification with iron nanoparticles resulted in a material able to lower the dye content of wastewater by 69% via Fenton-like oxidation with H2O2, albeit using a high loading. Blast furnace dust contained a mix of metal oxides, including SiO2, Al2O3, CaO and 54 wt% Fe in various phases. Despite its low surface area (3.4 m2 g−1) this dust was highly active for azo dye degradation, in the presence of both H2O2 and UV irradiation, the latter108 claimed to promote free hydroxyl radical formation and increase the efficiency of the Fenton reaction via Fe3+ photoreduction to Fe2+. A mesoporous, iron-rich catalyst was also prepared from wastewater sewage sludge, treated with iron sulfate, for the photo-degradation of azo-dyes and thermally stable organic pollutants (Fig. 5).9 Though possessing a low area (∼7 m2 g−1), this catalysts, consisting primarily of quartz and hematite, was active in the photo-Fenton reaction under UV and visible light, and was resistant to metal leaching.
 | ||
| Fig. 5 Waste derived catalyst SS-Fe-350 and the mechanism of the photo-Fenton reaction for degradation of Rhodamine B and p-nitrophenol under visible light irradiation. Adapted from ref. 9. Copyright 2014, with permission from Elsevier. | ||
Waste printed circuit boards are generated at ever increasing scale due to consumer desire to replace old technology with next-generation electric and electronic equipment. Li et al. demonstrated a potential second life for waste circuit boards as catalysts for Fenton-like oxidation of wastewater organic pollutants.109 The waste comprised 51 wt% SiO2, small amounts of Cu, Sn, Al, Pb, Ba, Fe and Ni metals and 30 wt% organics. Catalyst preparation was achieved by shredding, grinding and sieving the waste circuit boards. Resulting catalysts were active for pyridine degradation at room temperature with H2O2, giving comparable rates to pure Cu powder, although requiring a large excess of catalyst (4 g L−1) to achieve 90% conversion of a 0.1 g L−1 pyridine solution after 14 h. It is unlikely that the catalytic reaction was truly heterogeneous since a substantial fraction of metal ions were leached in situ, notably toxic Cu and Pb which raises concerns over the utility of such materials for wastewater depollution.
Grinding wheel ash is a high iron-content waste from automobile production. Dissolution, precipitation and ageing of such ash was used to create a catalyst for methylene blue degradation110 containing 2 wt% Si and 1 wt% Al on iron oxide (74 wt% goethite, α-FeOOH) which was active for the Fenton-like dye oxidation. An aqueous solution of 1.0 g L−1 Si–Al/α-FeOOH catalyst gave 100% conversion of a 20 mg L−1 methylene blue solution in only 60 min with H2O2 under neutral or basic conditions. While silica was found to be beneficial to the rate whereas the aluminium had little or no effect on dye degradation but could reduce the degree of iron leaching.
Fly ash, an alumino-silicate containing waste product of coal combustion, has been used to support vanadium to create heterogeneous catalysts for the partial oxidation of toluene in a continuous flow microreactor.113 A series of low area V/fly ash catalysts containing an active monolayer of Lewis/Brönsted acidic vanadia gave 83% toluene conversion at 260 °C, under air but was not selective, producing a mixture of benzaldehyde and benzoic acid. High loadings suppressed activity due to the formation of V2O5 crystallites.
Stainless steel slags were used as raw materials for silica and silica-alumina supports for Pt-based, VOC oxidation catalysts.114 Slags were subjected to acid digestion and hydrothermal treatment resulting in high area (290 m2 g−1), largely amorphous silicas prior to impregnation with 1 wt% platinum to yield 9–20 nm diameter nanoparticles. Pt/SiO2 catalysts were efficiacious for both CO and toluene oxidation in flow under 21 vol% O2, giving 95% conversion at temperatures between 200–500 °C. The superior performance of these slag-derived catalysts for CO oxidation relative to commercial Pt/SiO2 was attributed to additional transition metal promoters in the waste-derived support, however toluene activity was poorer possibly a result of the lower Pt dispersion over the waste support. Stainless steel slags and dusts were also employed to create reticulated ceramic foams (termed Type III monoliths in which the foam was modified by sequential coatings of a silica, steel dust and slag mixture) for supporting Au/Al2O3 or Au/CeO2 oxidation catalysts (Fig. 6).115,116 Polyurethane foam was used to template these materials, which were subsequently impregnated with alumina or ceria and a gold salt. The final supports were rich in SiO2 (17–35 wt%), CaO (46–59 wt%) and other alkali, alkaline earth and transition metal oxides. Washcoating of the monoliths increased the mesoporous foam supports from <1 m2 g−1 to 10–23 m2 g−1. The Au/Al2O3 monolith gave 100% conversion of CO and 2-propanol at 350 and 300 °C respectively; Au/CeO2 showed superior activity with complete conversion at 150 and 225 °C. The higher ceria activity was attributed to its redox properties and excellent oxygen storage capacity. Despite their promising performance, these catalysts involve complex syntheses, with only the foam support sourced from waste, and hence offer limited environmental benefits.
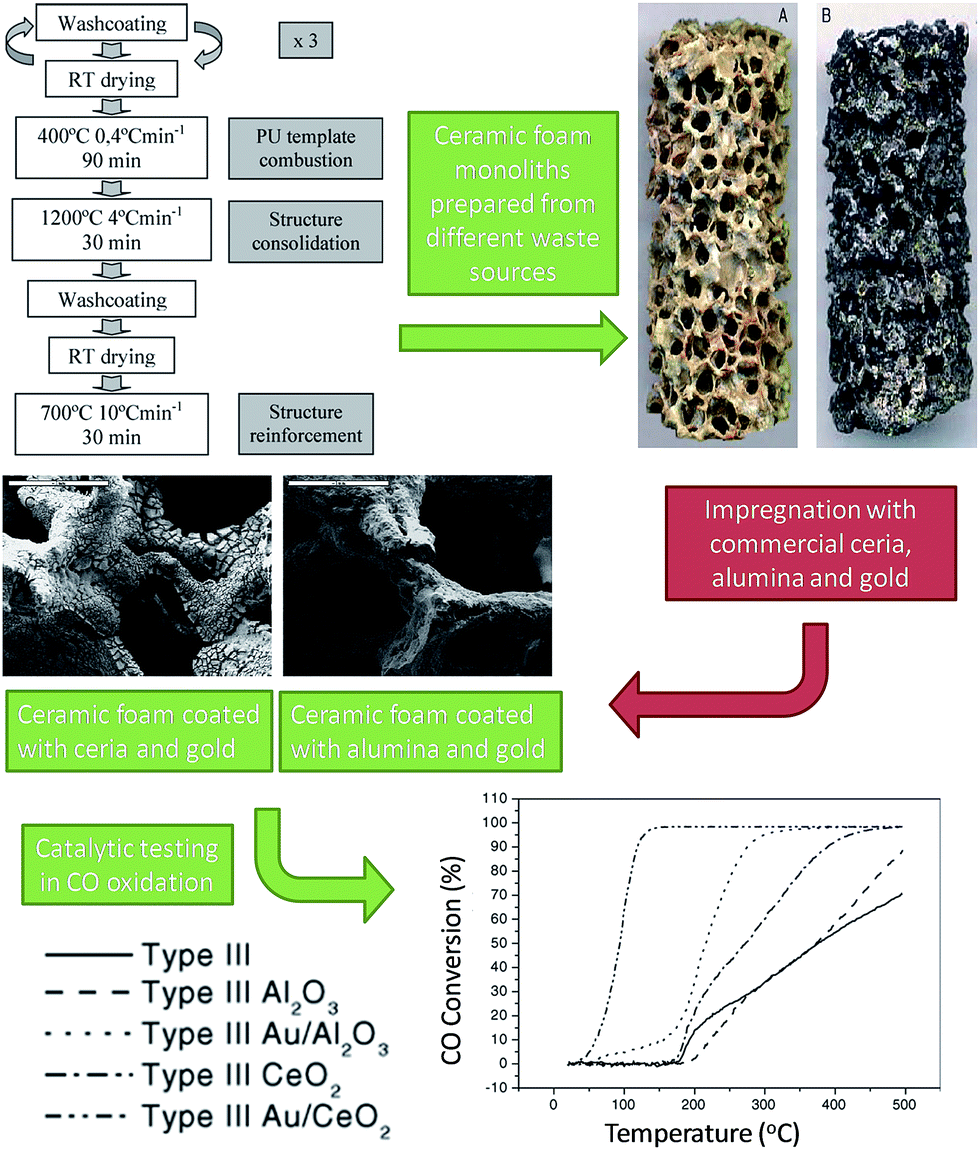 | ||
| Fig. 6 Preparation of Au-coated ceramic foam monoliths from waste for catalytic oxidation. Adapted from ref. 115 and 116. Copyright 2006 and 2007, with permission from Elsevier. | ||
Wastewater sludge from electroplating, tannery and textile dyeing industries were used as sources of transition metals for hydrocarbon oxidation by Klose et al.,117 who aimed to not only to produce catalysts from waste, but also to minimise the degree and cost of waste pretreatment. Wastes were simply dried, crushed and calcined at 1000 °C to remove organic residue, a standard procedure in waste sludge certification. The resulting materials all contained significant Fe (36 wt% for textile dyeing sludge), Cr (47 wt% for tannery sludge) and Cu (20 wt% for electrical appliance waste). Some of these wastestreams performed comparably to commercial catalysts for propane oxidation at temperatures from 200 to 600 °C. The most active catalysts were those sludges calcined at 550 °C which exhibited porous structures with surface areas of 50–100 m2 g−1 which thermally stable. A significant organic waste content was proposed as important to yield mechanically robust, high surface area mesoporous particles, with high iron contents conveying mechanical stability.
While the preceding examples delivered active oxidation catalysts, in many cases only the inert support was derived from waste, with the active phase generated from commercial chemicals. Despite this, supports are the dominant component by mass of most heterogeneous catalysts, and hence producing supports for oxidation catalysis from waste sources would inevitably reduce catalyst costs and the quantity of waste for e.g. landfill disposal or incineration. This is significant for waste materials whose large scale storage poses environmental risks, as seen in the Hungarian red mud spill disaster in Ajka.118 Nevertheless, the active phase of a catalyst is often the most expensive, earth scarce component, and hence exploiting waste sources to produce active components remains both an academic and economic challenge, with an ideal waste catalyst being one comprised entirely from recycled elements, necessitating minimal additives and/or costly processing. Spent catalysts, no longer fit for their intended purpose, are already commonly recycled for the same or a new application. This approach is common for PGM catalysts wherein refining has strong economic drivers, but has also been adopted for the re-use of waste catalysts from the petrochemical industry for e.g. sulphide removal from seawater by oxidation with air.119 Herein, a range of spent catalysts including NiO, ZnO, a CoO/MoO3/Al2O3 sulphur hydrogenation catalyst and a CuO/ZnO/Al2O3 CO-reduction catalyst were assessed, and while minimal efforts were made to characterise these spent materials, all were active for sulphide removal. Best results were obtained for a packed bed of CuO/ZnO/Al2O3, which gave 100% conversion of a 2 ppm aqueous sulphide solution in 40 min in a recycling batch reactor, indicating that the direct recycling of waste catalysts in an alternative catalytic process is may be an economically feasible strategy to circumvent costly waste catalyst handling and disposal. This re-apportioning of a catalyst to a new reaction once its performance in the original function becomes unacceptable could significantly improve life cycle assessments and alter industrial perspectives on what constitutes a ‘spent’ catalyst.
5. Pyrolysis and cracking catalysts
Another area where the re-use of spent catalysts has been explored is in the catalytic cracking of polymers for waste plastics recycling, in which spent fluid catalytic cracking (FCC) catalysts have been evaluated.120,121 Salmiaton compared fresh, steam regenerated and deactivated FCC catalysts in the cracking of high density polyethylene (HDPE) to volatile hydrocarbons in a fluidised bed reactor.120 The catalysts constituted low loadings of rare earth oxides (0.8–1.6 wt%) and sodium oxide (0.2 wt%) supported on 125–180 μm alumina or silica particles; deactivated catalysts also contained 100–6500 ppm Ni and V poisons. As expected, fresh catalysts were the most active, but the activity of spent/steam regenerated catalysts was roughly equal to that of the poisoned deactivated catalyst, giving between 70–90% conversion of the feed at a catalyst:polymer mass ratio of 6 at 450 °C. The poisoned deactivated catalyst gave a similar yield of gaseous products to fresh catalysts (61 and 64%) but a higher alkene:alkane ratio (1:1 versus 4:1). Such deactivated catalysts would typically be disposed of by FCC process operators, but clearly remained active for the catalytic cracking of waste plastics at temperature substantially lower than those used in incineration (500–900 °C). Further studies showed that regeneration of the deactivated catalyst was unnecessary for this application, offering little improvement in activity or selectivity.122 Similar work by Lee used spent FCC catalysts for waste plastic degradation, including high and low density polyethylene, polystyrene (PS) and polypropylene,121,123,124 in which spent catalysts were steam-regenerated at 400 °C. Resulting catalysts comprised an alumino-silicate support loaded with transition metals (0.3–1.7 wt% Ti, Ni, Fe, Zn and Cu) and residual coke (0.13 wt%).121 At 10 wt% catalyst, i.e. a much lower than that employed by Salmiaton, gave complete conversion of a waste plastic feed at 400 °C in a stirred semi-batch reactor within 40 min. Product distributions depended on reaction time and feed composition with PS selective to aromatic products, HDPE giving 80% olefins, and mixtures of these two plastics producing aromatics, olefins, parafins and napthenes, offering a route to tunable gasoline or diesel range hydrocarbons. Similar observations were reported for polyethylene pyrolysis over spent FCC catalysts in a semi-batch reactor at 300–500 °C.125
Lin et al. demonstrated the use of spent FCC catalysts for pyrolysis of waste plastic feeds of increasing complexity in a fluidised bed reactor.126–128 Detailed studies of the product distribution from polypropylene degradation over high loadings of spent FCC catalysts, revealed an 88% yield of volatile hydrocarbon products (C1–C9) after 20 min pyrolysis at 390 °C, with the main products being C4–C6 olefins. The degree of mixing of different types of waste plastic was found to affect not only conversion of the waste plastic but also the resulting product distribution. Similar results were obtained for a spent FCC catalyst using a 2:1 polyethylene:polypropylene blend92 and commingled polymer waste (polyethylene, polypropylene, polystyrene and polyvinylchloride),93 wherein yields of 82% and 83% were achieved in 30 and 20 min respectively under the same reaction conditions.
Biomass has also been investigated as a waste source for pyrolysis catalyst synthesis. Shen et al. prepared such catalysts from rice husk char impregnated with Fe or Ni.129 The rice husk was calcined at 700 °C to produce a thermally stable char with a surface area of 117 m2 g−1 and high Si content, and subsequently impregnated with iron or nickel nitrate, and tested either pre-calcined at 600 °C or uncalcined for in situ tar removal during biomass pyrolysis. Metallic Ni and Fe appeared the active sites for tar conversion, with the Ni-based catalysts more active, attributed to superior activation of C–H and C–C bonds in adsorbed organics. An uncalcined bimetallic char catalyst gave the greatest tar reduction (92%) and consequent highest proportion of desirable syngas with minimal CO2. The carbon of the SiO2-containing char support itself played a role in promoting reduction of the metal oxides and concomitant CO2, metal ion and tar poisoning.
Red mud has shown promise as a catalyst for waste plastic pyrolysis (Fig. 7),130 employing a feed of 40 wt% polyethylene, 35 wt% polypropylene, 18 wt% PS, 4 wt% poly(ethylene terephthalate) and 3 wt% poly(vinyl chloride). Although red mud was inactive below 440 °C, it achieved a 98% yield of volatile hydrocarbon products at 500 °C, delivering higher gas yields and lower viscosity bio-oils than a ZSM-5 zeolite catalyst. The different product selectivity (less C3–C6 hydrocarbons and increased ethane, methane and CO2) obtained from Fe2O3 and TiO2 in the red mud relative to ZSM-5 was ascribed to the occurrence of additional hydrogenation and hydrocracking reactions over the transition metal oxides. Despite its far lower surface area (28 m2 g−1vs. 400 m2 g−1 for ZSM-5), the red mud appears a promising solid acid catalyst for waste plastic recycling for bio-fuels production.
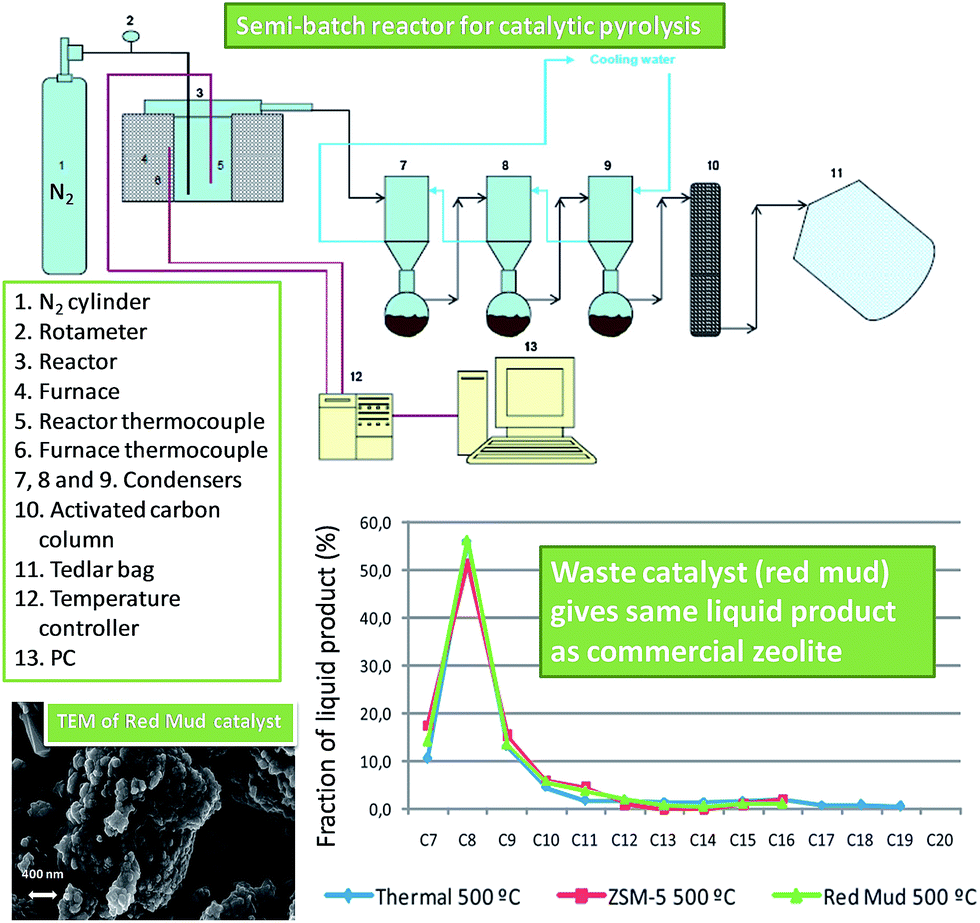 | ||
| Fig. 7 Reactor design and liquid fraction composition in the catalytic pyrolysis of waste plastics using red mud and ZSM5. Adapted from ref. 130. Copyright 2014, with permission from Elsevier. | ||
6. Catalysts for hydrogen production
Hydrogen has great potential as an energy carrier as it compatible with fuel cell technology, where its use does not lead to CO2 or toxic emissions, with water being the only byproduct of its consumption. It also possesses a higher energy density than natural gas or diesel.131,132 Although hydrogen can be produced from conventional fossil fuels or nuclear energy, these routes are at odd with green chemistry principles and therefore there is a great need for environmentally benign, cost-effective hydrogen production from renewable and sustainable sources, such as biomass.133,134 A number of waste-derived catalysts for hydrogen production from various precursors have been reported.In addition to its application in transesterification, ketonisation and oxidation, red mud has also been utilised as a catalyst for methane decomposition to generate H2 and Fe3C as a coproduct.10 Favourable hydrogen production rates of 3.80 × 10−5 mol g−1 s−1 were achieved employing 0.4 g of red mud in a continuous flow microreactor between 600–700 °C under a 1:4 flow of N2:CH4, comparable to literature rates for high loading Fe2O3/Al2O3 catalysts. This application offers multiple benefits, valorising Bayer process waste and mitigating the emissions of a potent greenhouse gas while producing a low carbon fuel. A modified mesoporous red mud was used by Cao et al. for hydrogen production from ammonia.135 Crystallinity and surface properties of the red mud were altered by acid digestion and base re-precipitation, which removed the majority of Na and Ca from the waste material. The processed red mud was subsequently impregnated with Ni at loadings between 0–15 wt%, ground and calcined prior to catalytic testing. Digestion increased the surface area from 8 to 225 m2 g−1 and pore volume from 0.08 to 0.39 cm3 g−1, while calcination at 600 °C transformed metal hydroxides to oxides. Ammonia decomposition in a continuous flow microreactor between 350–700 °C proceeded with 96% conversion over a 12 wt% Ni/red mud catalyst, achieving 5.33 × 10−4 mol g−1 s−1 of H2, this high activity attributed to a synergy between Ni and Fe species in the red mud support.
Kanamori et al. used battery waste as a source of Ni for preparing catalysts for H2 production via methane dry reforming.136 Waste from the Ni-metal hydride batteries contained Ni alongside La, Ce, Co, Mn and Nd. These metals were extracted by acid leaching, followed by base precipitation and final calcination. The major crystalline components, NiO, CeO2 and LaCoO3, were subsequently separated by three different extraction processes. The NiO phase was 97% pure, while ceria and lanthana both contained substantial metal impurities, although all three oxides exhibited low surface areas <10 m2 g−1. NiO proved the most active material for methane dry reforming in a fixed bed reactor at 780 °C under a CH4, CO2 and Ar mixture, giving 100% CH4 conversion for 9 days on-stream; battery extracted NiO was more active than commercial NiO which deactivated rapidly after 3 days operation. The active component was determined to be metallic nickel, demonstrating that toxic battery waste could be transformed into active and stable catalysts for syngas (H2 and CO) generation from a greenhouse gas.
Methanol is considered a potential hydrogen fuel carrier, due to the number of well-developed processes for its production (e.g. from biomass) and established pathways for its reforming into H2. Methanol decomposition over Co/Fe/C catalysts synthesised from waste biomass was explored by Genova et al.137 Activated carbon was prepared from peach stones by heating to 650 °C in the presence of water vapour. The resulting carbon was impregnated with 8 wt% Fe, Co or a mixture of both metals using nitrate precursors, and exhibited a high area 820 m2 g−1 micro–mesoporous structure. Iron and cobalt were present as metal oxides as Fe3O4 in the pure iron material, mixed Co3O4 and CoO in the pure cobalt material, and a CoFe2O4 phase in the bimetallic catalyst. Monometallic materials were active for methanol decomposition, giving higher conversion >330 °C than analogous SBA-15 catalysts, but were less selective to CO production. While the bimetallic catalyst was less active than either monometallic counterpart, it offered superior CO selectivity. These waste-derived, carbon supported catalysts provided higher yields than mesoporous silica supported counterparts for hydrogen production via methanol decomposition. Waste leaves have also been demonstrated as an abundant and low cost biomass source for preparing activated carbon supports.138 These high area and density supports were impregnated with 10 wt% Ni or 1 wt% Pt and tested as catalysts for electrocatalytic water hydrolysis via linear sweep and cyclic voltammetry, with a working electrode coated with the electrocatalyst. Ni and Pt materials were both active for H2 generation, with the precious metal affording higher productivity. Unfortunately, conversion of leaf waste into activated carbon supports necessitated significant processing steps and reagents, notably washing, drying and thermal treatment cycles and the addition of a stoichiometric amount of ZnCl2, weakening the environmental credentials of waste-derived electrocatalysts.
Methylcyclohexane, another proposed H2 carrier, was used to evaluate dehydrogenation catalysts prepared from waste tires139 which were pyrolysed, purified by toluene reflux and H2SO4 washing, followed by at 850 °C steam-activation. The micro- and mesoporous carbon support had an extremely high area >1000 m2 g−1, facilitating the genesis of highly dispersed Pt nanoparticles upon wet impregnation with low metal loadings (particle sizes of 2.5 nm). Catalysts were active for methylcyclohexane dehydrogenation at 300 °C in a flow reactor, with a maximal TON for 0.4 wt% Pt. Methylcyclohexane conversions of 95% were achieved in N2 flow for up to 52 h without deactivation and while retaining ∼100% selectivity to hydrogen and toluene; hydrogen production rates of 342 mmol gPt−1 min−1 were higher than for commercial 1 wt% Pt/C. However, the financial and environmental implications of the multiple processing steps and solvent/stoichiometric reagent utilisation must be balanced against this enhanced reactivity.
7. Photodegradation catalysts
Titania is a common catalyst for photocatalytic oxidations, notably photo-Fenton systems wherein UV and/or visible light is used to generate hydroxyl radicals in situ from hydrogen peroxide. A number of such TiO2 catalysts have been prepared from waste sources.140–143Transition metal-doping of TiO2 confers several benefits to its photocatalytic properties144,145 such as increased light harvesting and/or retardation of undesirable electron–hole charge carrier recombination. Cu-doped TiO2 has received particular attention.146–149 A Cu2O/TiO2 catalyst was prepared by Xiu et al. from waste printed circuit boards.143 The electronic equipment waste was subjected to supercritical water oxidation and then processed in an electrokinetic system with a TiO2 P25 suspension as the catholyte (the electrolyte on the cathode side of the electrochemical cell), resulting in the selective leaching of copper from the circuit boards. An electrokinetic process uses direct current as a “cleaning agent” where metal ions are leached from a surface then migrate to the cathode or anode where they may be reduced and the metal recovered. This method yielded nanocrystalline copper oxide supported on titania with a surface area of ∼50 m2 g−1 and Cu loadings spanning 0.7–9.1 wt%. Resulting catalysts showed promise in methylene blue degradation under UV irradiation, albeit under extremely high photocatalyst loadings of 2000 wt% relative to the dye. Cu-doped titanias were more active than undoped P25, with the best 4.5 wt% Cu giving complete conversion of 10 ppm methylene blue in 60 min. Despite the poor activity, protracted pretreatment steps, and use of commercial titania, this study offers tentative steps towards catalysts incorporating components selectively extraction from complex mixed waste sources.
Liou et al. utilised electrical and electronic waste as a source of silica support for TiO2 photocatalysts.140 Waste resin was ground, calcined and leached with HCl to remove unwanted metals prior to washing and drying (producing copious contaminated aqueous waste that itself requires remediation/recycling). Sodium silicate was prepared from the resin using NaOH solution and this was then used to synthesis mesoporous silica MCM-48 using cetyl trimethylammonium bromide (CTAB). MCM-48 was subsequently impregnated with a TiO2 suspension to help prevent aggregation of titania nanoparticles. The support had a high surface area (upto 1153 m2 g−1) and was able to stabilise TiO2 particles that were upto 10 times smaller than commercial bulk TiO2 (∼2.4 nm for 30 wt% TiO2/MCM-48 vs. 32.4 nm for pure titania). The optimal catalyst was a 30 wt% TiO2/MCM-48 calcined at 700 °C, which gave 75% conversion of 50 μM methylene blue after 3 h UV irradiation at a catalyst loading of 500 wt% (0.080 g).
Colemanite ore waste from boron mining has also been applied to synthesise supports for TiO2 photocatalysts (Fig. 8).141 This mining waste comprised alkali, alkali earth and transition metal oxides, largely SiO2, CaO and B2O3. Titania nanoparticles of 5–30 nm diameter were deposited on this waste oxide mix from titanium isopropoxide. The band gap of the supported TiO2 was 3.08 eV, comparable to the band gaps of pure anatase and rutile phases of 3.1 and 2.9 eV respectively.150 The supported titania was tested for the photodegradation of pesticides in a model wastewater stream, with a 1 g L−1 catalyst suspension able to remove 80% of atrazine from a 10 mg L−1 solution after 70 min UV irradiation. This catalyst was reused four times with minimal deactivation.
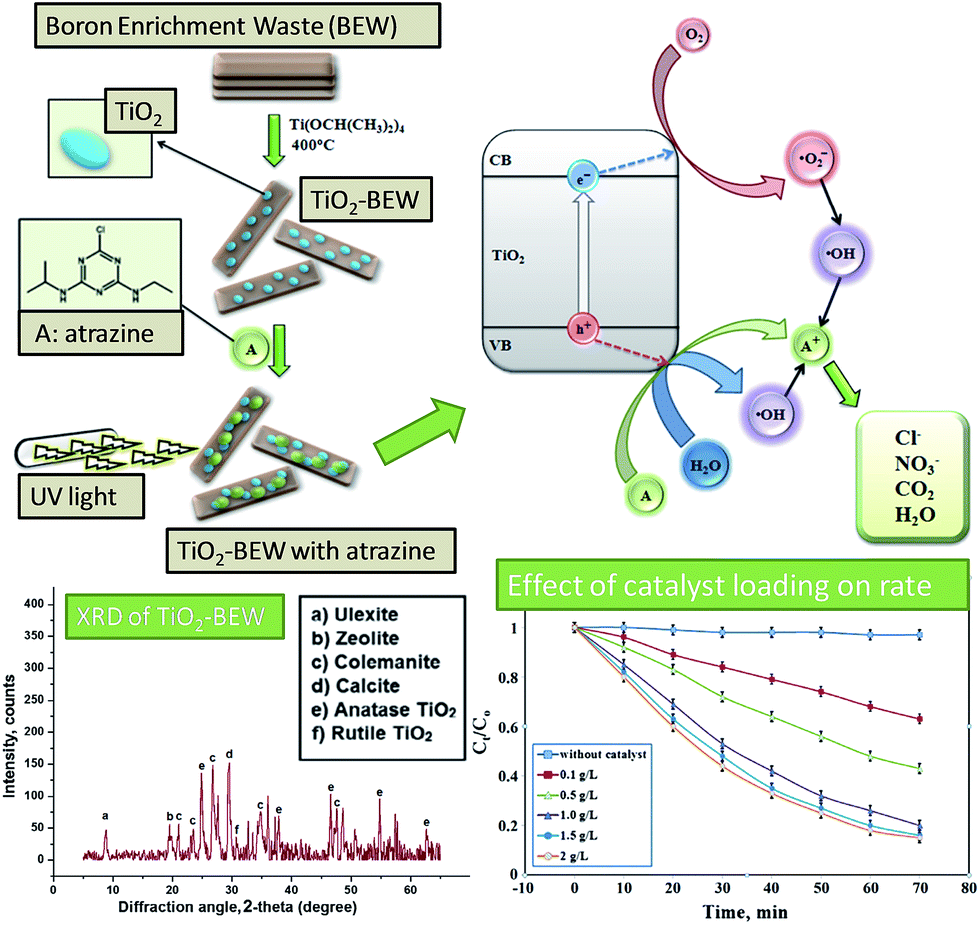 | ||
| Fig. 8 TiO2 photocatalyst supported on boron enriched waste and its use in atrazine photodegradation. Adapted from ref. 141. Copyright 2014, with permission from Elsevier. | ||
Da Silva et al. used a range of industrial residues supported on silica as catalysts for the photodegradation of Rhodamine B dye under UV or visible light142 which contained Zn, Ti, Ag, Cr or Cu depending on the waste origin (galvanic bath residues, polymerisation catalyst slurries and photography solutions). Metal loadings and surface areas ranged from 0.01–0.6 wt% and 96–280 m2 g−1 respectively. The most active photocatalyst was Ti-rich, affording gave 61% and 67% conversion of a 20 mg L−1 Rhodamine B solution in 60 min for a catalyst loading of 3500 wt% under visible versus UV light respectively. Although this catalyst was less active than pure P25 under UV irradiation, it was significantly more active under visible light, attributed to the presence of Mg, which narrowed the Ti band gap to 1.78 eV and may also have hampered electron–hole recombination.
Other photocatalytic materials have been synthesised from waste. For example, Sapiña et al. prepared a catalyst from electric arc furnace dust from the steel industry.151 The material was 33 wt% ZnO and 33 wt% Fe2O3, together with various alkali, alkali earth and transition metal oxides, including 0.1 wt% TiO2. This material was active for NO photodegradation to HNO3 (though less so than pure P25) and the photodegradation of Rhodamine B under UV and visible light. Higher Rhodamine B conversion was observed with UV light, wherein 70% conversion of 1 mg L−1 aqueous RhB solution was achieved with 750 wt% catalyst after 140 min. However, uncatalysed photolysis led to ∼20% conversion under the same conditions, while adsorption onto the catalyst in the absence of light artificially lowered the concentration of Rhodamine B by around 40%, due to the high catalyst loading. Thus, the contribution from photocatalysis by the electric furnace dust may be similar to or less than the rate of degradation by uncatalysed UV photolysis.
An obvious limitation of all these photocatalysts is the, in some cases extraordinarily, high catalyst:substrate ratios employed, in addition to the strong sensitivity of photocatalysts to minor compositional variations and hence site-to-site differences. Since photocatalysts are invariably employed in aqueous or polar solutions, the leaching of toxic metals such as lead during reactions may also be problematic, particularly for applications in wastewater depollution.
8. Catalysts for emission control
Nitrogen oxides (NOx) are a major air pollutant and contributor to environmental problems such as acid rain and tropospheric ozone depletion. Singh et al. used waste biomass from the cotton industry to prepare carbonaceous supports for selective catalytic reduction (SCR) deNOx catalysts.152 SCR processes utilise NH3 to convert NO and/or NO2 into N2 and H2O. Cotton stalks were converted to chars via either pyrolysis at 600 °C, chemical activation with H3PO4 and pyrolysis at 800 °C or co-activation in steam/H3PO4 at 800 °C. These chars were co-impregnated with Ce and Mn nitrates (2:1 molar ratio) to give a 4 wt% metal loading. Co-activated carbons gave the highest surface area materials (1300–1600 vs. 800–1200 m2 g−1 for chemically activated carbons), however NOx conversion levels were similar over both series between 50–66% at 280 °C, and lower than literature fully synthetic Mn or Ce catalysts, attributed to the much higher metal loadings (10–50 wt%) of the latter. It was proposed that the alkali content of the biomass waste source may neutralise catalytically active Brönsted acid sites however methods to reduce the alkali content of the cotton stalk were not explored, and it is hard to conceive how phosphoric acid treated chars could retain any residual basicity.
Furnace waste from the iron and steel industry with high titania contents (<20 wt%) have also been used to prepare deNOx catalysts.11 TiO2 was extracted with ∼75% efficiency from blast furnace slag by acid leaching, alongside silica and trace Al2O3 and Fe2O3; the associated slag residue contained CaSO3, SiO2 and TiO2 which it was proposed could serve as a construction material. Slag-extracted TiO2 was consistent with 20 nm anatase, but with a much higher surface area than a commercial analogue due to amorphous impurities. Both waste and commercial titanias were to prepare V–W–TiO2 catalysts for the denitration of flue gas (Fig. 9), wherein the waste-derived catalyst gave a superior conversion of 77% conversion of 600 ppm NO in an NH3 (480 ppm) and O2 (3 vol%) stream at 250 °C, possibly reflecting the presence of impurity Fe2O3 promoter. This study is a rare example wherein catalyst supports obtained from mixed waste sources offer enhanced reactivity to pure commercial counterparts.
 | ||
| Fig. 9 Preparation flow diagram of low-cost deNOx catalyst from blast furnace slag and denitration results. Adapted from ref. 11. Copyright 2014, with permission from Elsevier. | ||
Carbon dioxide is the most well-known and topical greenhouse gas, produced on a huge scale as a by-product of numerous industries, notably steel production through blast furnace operation. In an effort to improve carbon emissions from this highly polluting sector, Kuwahara et al. prepared a CO2-fixing catalyst from blast furnace slag,153 thereby offering a route to manage and valorise this solid waste stream. Slag was utilised as a source of Ca for the synthesis of layered double hydroxides (LDHs), an important class of solid base catalysts, and tested in the cycloaddition of epoxides and CO2 to form five-membered cyclic carbonates. The LDH catalyst was prepared by dissolving blast furnace slag in 3 M HCl at 100 °C, filtration of the residual SiO2 (which may also be used as a raw material for e.g. zeolite synthesis), and final precipitation of the acid leachate with 2 M NaOH. The latter step is problematic from the perspective of solid base catalysis, since alkali-free precipitation routes to LDHs must be employed in order to eliminate their contamination by alkali residues and concomitant leaching and homogeneous catalysis in polar media.154–158 The dried precipitates were calcined between 400–800 °C, yielding materials with structures similar to Ca2Al(OH)6Cl·2H2O (hydrocalumite), with trace Mg, Fe, Mn, Si and Ti.
These impurities were hypothesis to enhance the slag LDH surface area relative to pure hydrocalumite, with slag area decreasing with calcination temperature and accompanied by CaO, MgO, CaCO3 and Ca12Al14O33 mixed metal oxide formation. Cyclic carbonates are precursors to a range of plastics, pharmaceuticals and fine chemicals, and there is great interest in their direct synthesis from CO2 and epoxides.159–161 Slag LDH catalysts gave higher conversion and selectivity in the cycloaddition of styrene oxide with atmospheric carbon dioxide than pure hydrocalumite LDH, with conversion increasing with calcination temperature due to a higher basic strength and site density. Pure CaO and CaO/Al2O3 afforded low conversions due to the lack neighbouring acid and base sites required in the reaction mechanism. Optimum results were attained for slag LDH calcined at 800 °C using a 100 wt% catalyst loading, which yielded 85% styrene oxide conversion after 24 h at 100 °C under atmospheric CO2, and with 90% selectivity to styrene carbonate. Similar results were observed for a range of epoxides under the same reaction conditions.
Reports on blast furnace slag neatly illustrate how one waste source may be used to successfully prepare different catalysts, one Ti-rich and one Ca-rich, for different chemical transformations through appropriate extraction/separation procedures, highlighting the importance of evaluating the potential of all components of waste, both extract and residues.
9. Catalysts for waste biomass valorisation
Biodiesel production generates large volumes of glycerol by-product. With a view to develop routes to manage and valorise this major biomass waste source, Gonçalves et al. reported the preparation of carbon catalysts from glycerol and their application in the etherification of glycerol to generate fuel additives (Fig. 10).162 An acidic carbon was prepared by hydrothermal treatment of waste glycerol with sulphuric acid under reflux at 150 °C for 24 h, possessing an acid site density and S content of 3.5 mmol H+ per g and 3.5 wt% respectively. Under optimised conditions this waste carbon catalyst gave superior activity and selectivity than commercial Amberlyst-15 (which despite its high acid loading exhibits poor acid site accessibility), with a 5 wt% catalyst loading giving a 74% yield of desired ethers after 6 h at 120 °C with a t-butyl alcohol:glycerol molar ratio of 4. Although sulphonic acid leaching from similar solid acid catalysts has been commonly reported, this glycerol-derived carbon was reused eight times with negligible loss of product yield, although no characterisation of the spent catalyst was undertaken, or hot filtration tests and S analysis of the filtrate, hence stability was unproven. A similar study was undertaken by Sánchez et al., who created solid acid carbon catalysts via sulphonation of carbonised sucrose for the esterification of glycerol with acetic acid.163 These materials exhibited lower acid site densities, ranging from 0.1–1.4 mmol g−1, but contained micro- and mesopores with far higher surface areas reaching 800 m2 g−1 following carbonisation at 850 °C. All catalysts were active for glycerol esterification, with a 5 wt% loading giving >99% conversion after 4 h at 160 °C and acetic acid:glycerol molar ratio of 9. Selectivity to the desired triacetylglycerol product of 50% was highest over the catalyst carbonised at 600 °C with the highest S and acid site density (4.3 wt% and 1.4 mmol g−1). Again, commercial Amberlyst-15 performed poorly, with only 22% selectivity to triacetylglycerol under identical reaction conditions.
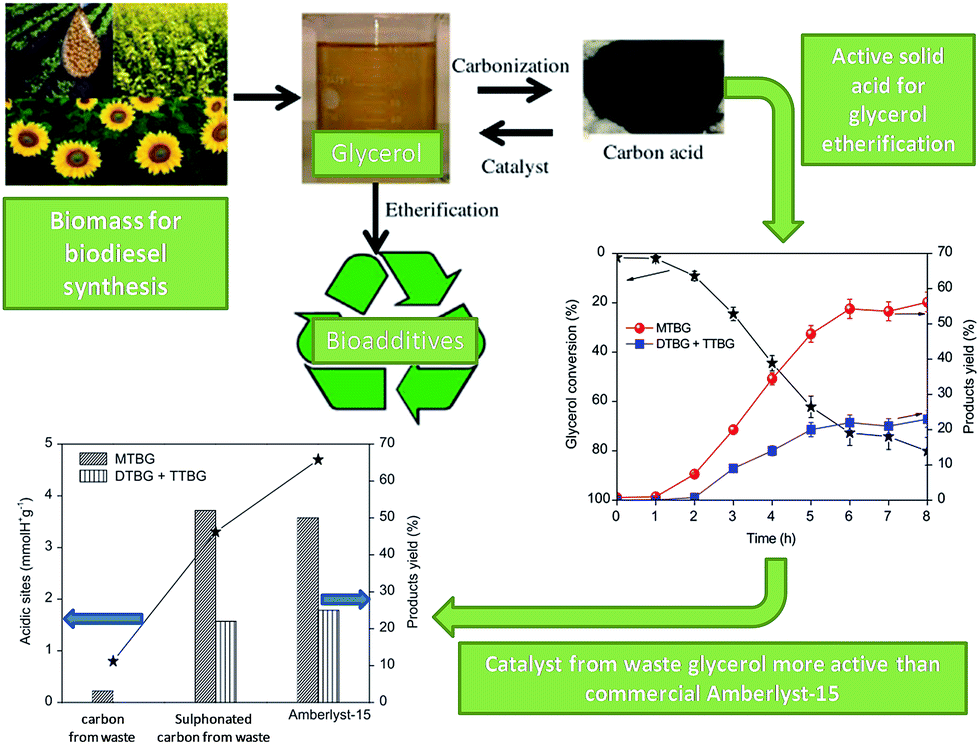 | ||
| Fig. 10 Preparation of a sulfonic acid functionalised carbon from waste glycerol for glycerol etherification. Adapted from ref. 162. Copyright 2014, with permission from Elsevier. | ||
Glycerol waste may also be transformed to value-added chemicals by hydrogenolysis to propanediols, used as solvents, antifreeze agents, food additives and as precursors to polyesters. Rode et al. prepared hydrogenolysis catalysts from fly ash waste obtained from coal-fired power stations.164 Fly ash, comprising largely SiO2, Al2O3 and trace Fe2O3, TiO2, CaO, MgO, K2O, and Na2O, was processed by solid state alkali metal hydroxide fusion and acid pretreatment. The resulting support was then impregnated with Cu and calcined. Surface areas of the 70 wt% silica-based catalysts varied from 31–113 m2 g−1, with Cu loadings varied as a function of the Al:Cu ratio. A 1:1 Al:Cu variant offered the highest glycerol conversion and selectivity to propanediol, with a 5 wt% catalyst loading achieving 57% conversion in 5 h at 220 °C under 52 bar hydrogen and 95% selectivity to the desired diol. A high copper dispersion and loading were critical to optimising hydrogenolysis, although the high hydrogen pressures require a renewable H2 source to improve the overall life cycle economics and environmental impact.
Milk permeate is a major waste product of the dairy industry and source of lactose. Lactose valorisation was proposed by Calzado et al.,165 who synthesised carbonaceous solid base catalysts for the isomerisation of lactose to lactulose, both widely used in the pharmaceutical and food sectors. Olive stone waste was first washed, ground and pyrolysed at 500 °C. The resulting char was then activated chemically or physically. Chemical activation involved impregnation of the char with 1:1 NaOH and Ba(OH)2 followed by carbonisation at 800 °C under N2, and then HCl and water washes, drying and grinding. Physical activation required carbonisation in N2 at 800 °C followed by high temperature gasification under CO2, and Ca impregnation. The solid bases were all microporous, with widely ranging surface areas from 4–1960 m2 g−1. A 5 wt% Ca/C catalyst gasified at 900 °C with the highest base site density gave the highest degree of isomerisation; 28% conversion of aqueous lactose was achieved after 3 h at 100 °C using 10 wt% of this solid base. Selectivity was high (∼80%), resulting in a 22% yield of lactulose, although leaching studies identified the loss of 1.4% of calcium after a single reaction, raising questions over even its short-term stability.
10. Miscellaneous applications
In addition to the preceding reaction classes, waste heterogeneous catalysts are reported for epoxidation,166 dehydration,167 ethanolysis,168 condensation169 and acylation.170Pyrolysed waste tires were used to derive a support for chiral Mn–salen complexes for the asymmetric epoxidation of unfunctionalised olefins.166 This thermally stable microporous support had a surface area of 85 m2 g−1, but lost 3% of its Mn content due to leaching over three successive recycles. Enantioselectivities achieved with the waste-supported catalysts were higher than those for analogous homogeneous complexes (62% vs. 50% for 1-phenylcyclohexene epoxidation).
Al and Fe-rich industrial wastes were used as sources of boehmite, γ-AlO(OH), and hematite (α-Fe2O3) respectively to generate iron oxide/alumina catalysts for ethanol dehydration,167 an important bio-alcohol for either transportation fuel or bulk chemical synthesis. Product selectivity could be switched from diethyl ether to ethene by varying the ratio of the waste feeds used in the catalyst preparation, with boehmite favouring diethyl ether formation and FeOx favouring ethane formation.
A highly active solid base catalyst containing calcium and magnesium oxides, was prepared from steel industry slag.168 This waste-derived catalyst gave higher rates for the ethanolysis of ethyl carbamate to diethyl carbonate than a more costly ZnO/PbO catalyst, and could be reused up to eight times without loss of activity or selectivity. Solid base and solid acid catalysts for respective Knoevenagel condensation and Friedel–Crafts acylation were synthesised from fly ash by amino-functionalisation169 and impregnation with scandium triflate170 respectively. A solid base catalyst composed of 10 wt% aminopropyl groups on fly ash demonstrated high activity in the synthesis of ethyl(cyclohexylidene) cyanoacetate (an intermediate of the epilepsy drug gabapentin) from ethyl cyanoacetate and cyclohexanone, giving a 92% yield.169 The catalyst was reused in 4 cycles and high yields were maintained, falling to 87% by the fourth run. The authors stated that the FTIR spectra of the fresh and used catalysts were the same, indicating no substantial loss of active sites. However, quantitative studies of the number of base sites were not reported for the fresh or spent catalysts so the stability of the catalyst could not be confirmed. A solid acid catalyst composed of fly ash-supported scandium triflate was showed superior activity than zeolite beta and La3+ ion exchanged zeolite beta in the Friedel–Crafts acylation of 2-methoxynaphthalene to form 2-acetyle-6-methoxynaphthalene, a naproxen precursor.170 The authors suggested that the fly ash catalyst could replace zeolite beta in industrial applications, such as the preparation of the anti-inflammatory drug naproxen, due to its superior activity and selectivity. The fly ash catalyst was shown to be active when reused in four reaction cycles, where conversion fell from 84 to 77%. Hot filtration leaching studies and FTIR studies of the fresh and used catalysts were carried out to demonstrate the stability of the catalyst. However, carbonaceous deposits were reported on the catalyst surface, a likely cause of the observed deactivation.
All the preceding materials in this review were prepared via chemical and/or physical processing of waste. However, microbiological approaches have also been developed to exploit the intrinsic metal-fixing properties of certain bacteria, which are able to extract valuable metals from waste. In some instances the resulting metallated bacteria have been processed for use as carbon-based catalysts. Macaskie et al. adapted this idea for a range of target metals and waste sources,171 extracting precious metals such as Pd, Pt and Au from industrial wastewater172,173 and metal leachates of electronic scrap,174 for testing in the catalytic reduction of heavy metals pollutants (carcinogenic and mutagenic H2CrO4 to noncarcinogenic Cr2O3). The same group developed a series of catalysts using various bacterial strains and active metals for wide-ranging applications including semi-hydrogenation of alkynes,175–177 carbon–carbon coupling,178,179 dehalogenation,180 oxidation of aromatic alcohols181,182 and fuel cells.183,184 However, many of these catalysts were actually prepared using solutions of commercial metal salts rather than the actual waste sources used in preliminary studies. The bioremediation approach to simultaneous metal recovery and catalyst preparation has been investigated by other researchers,185–187 with the use of biogenic Pd catalysts reviewed elsewhere.188 Waste bacteria or biomass from other industrial processes, such as brewing, rather than specifically grown cultures, makes such approaches especially appealing from economic and green chemistry perspectives. For example, Dimitriadis et al. employed yeast from distillery waste to scavenge platinum metal from a solution of commercial H2PtCl6 in order to prepare fuel cell anodes.189 While microorganisms can provide a highly selective means to recover specific metals from waste, and hence themselves constitute novel catalyst precursors, scale-up of such metal extraction and subsequent catalyst genesis has yet to be proven. Ideally, both the final active catalyst component and the intertwined carbonaceous support (i.e. microorganism) should be sourced from current waste streams.
11. Summary and concluding remarks
This review has explored the breadth of waste materials amenable to use as raw materials for the synthesis of heterogeneous catalysts, and the array of chemical transformations to which such catalysts may be applied. In many cases catalyst preparation is underpinned by diverse waste treatment processes, some highly complex while others are more facile, necessary to extract and isolate the desired components, be they inert/functional supports and/or active metals or metal oxides.Synthetic routes to effective catalysts can employ either a waste material alone, or their combination with additional commercial components. To date, the majority of catalysts are either transition metal oxides from industrial waste or carbonaceous and alkaline earth oxides from biomass waste. Common waste materials and their derived catalysts and applications are summarised in Table 3. The most intensive research has targeted biodiesel production via esterification and transesterification over solid acids/bases. CaO is an excellent base catalyst for triacyl glyceride transesterification (though debate remains concerning its solubility in methanol and resultant homogeneous catalytic contributions), predominantly obtainable from waste shells, but also bone and mining and construction wastes. Other popular catalyst targets are those containing iron, silicon, aluminium or titanium oxides which all occur in varying concentrations within red mud, fly ash, furnace slag and electronic circuit boards. Dissolved iron is also common in waste water from the electroplating, tannery and textile dyeing industries. Silica, a common inert support material in heterogeneous catalysis, may be extracted from organic waste, with rice husk an important source. Arguably, the most effective way of generating catalysts from waste is to recycle spent catalyst appropriated from other sectors which are unable to deliver the desired performance for their original purpose. Second lifetimes may be for less demanding reactions or those in which preceding deactivation may actually prove beneficial (e.g. phase or textural changes, or surface coking). This approach has been successfully employed with spent fluid catalytic cracking catalysts.
| Waste | Material produced | Active phase | Application | Properties |
|---|---|---|---|---|
| Furnace slag114 | High surface area SiO2 | Pt from commercial Pt(NH3)4(OH)2 | Oxidation of CO and volatile organic carbons | Inert support 290 m2 g−1 |
| Furnace slag11 | High surface area TiO2 | V2O5 and WO3 from commercial oxides | Denitration (deNOx) | Inert support 289 m2 g−1 |
| Furnace slag153 | Ca-based layered double hydroxide | Ca2Al(OH)6Cl, hydrocalumite | Cycloaddition of CO2 to epoxides | Solid base 21 m2 g−1 |
| Furnace slag168 | Mixed oxide | MgO | Ethanolysis of ethyl carbamate | Solid base upto 298 m2 g−1 |
| Furnace slag52 | MgO–CaO/SiO2 | MgO and CaO | Transesterification | Solid base |
| Red mud10 | Mixed oxide | Iron | H2 production | Mixed oxide upto 15 m2 g−1 |
| Red mud49 | Mixed oxide | Alkali oxides | Transesterification | Solid base upto 27 m2 g−1 |
| Red mud97,130 | Mixed oxide | Metal oxides | Catalytic pyrolysis | Solid acid/base upto 27 m2 g−1 |
| Red mud96 | Mixed oxide | Fe2O3 and other metal oxides | Ketonisation | Solid acid/base |
| Red mud104 | Mixed oxide | Fe2O3 | H2 carrier/production | Solid base |
| Fly ash39 | Mixed oxide | CaO from commercial Ca(OH)2 | Transesterification | Metal oxide support < 1 m2 g−1 |
| Fly ash113 | Mixed oxide | V2O5 from commercial vanadium salt | Oxidation of toluene | Metal oxide support upto 17 m2 g−1 |
| Fly ash164 | Mixed oxide | Cu from commercial salt | Hydrogenolysis of glycerol | Metal oxide support upto 113 m2 g−1 |
| Fly ash169 | Mixed oxide | Amine functionalisations from commercial APTMS | Knoevenagel condensation | Solid base < 1 m2 g−1 |
| Fly ash170 | Mixed oxide | Commercial scandium triflate | Friedel–Crafts acylation | Metal oxide support < 1 m2 g−1 |
Significant improvements are still necessary in the design of waste catalyst to minimise the need for commercially sourced components, and to reduce the length/number and cost of processing steps necessary to create the desired final catalyst. Environmental benefits of waste derived catalysts may be negated if their synthesis utilises large quantities of solvent or harmful/toxic chemicals, energy-intensive thermal or pressure regimes, or expensive commercial additives. However, despite these issues, the underlying ideology of seeking value from currently underutilised waste streams aligns perfectly with the pursuit of sustainable chemistry, and will become increasingly important in the modern world facing the combined challenges of shrinking natural resources, increasing air, land, and water pollution, and uncertain energy security. Holistic catalyst design strategies hold the promise of cleaner catalytic technologies from waste without compromising performance.
Acknowledgements
We thank the EPSRC (EP/K036548/1; EP/K014749/1; EP/K014706/1) for financial support.References
- P. J. Loferski, ed. U. S. D. o. t. Interior, 2013.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- M. Tabatabaei, K. Karimi, I. Sárvári Horváth and R. Kumar, Biofuel Res. J., 2015, 2, 258–267 CrossRef.
- H. Chen, D. Zhou, G. Luo, S. Zhang and J. Chen, Renewable Sustainable Energy Rev., 2015, 47, 427–437 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- T. R. Brown, Y. Zhang, G. Hu and R. C. Brown, Biofuels, Bioprod. Biorefin., 2012, 6, 73–87 CrossRef CAS.
- K.-T. Chen, J.-X. Wang, Y.-M. Dai, P.-H. Wang, C.-Y. Liou, C.-W. Nien, J.-S. Wu and C.-C. Chen, J. Taiwan Inst. Chem. Eng., 2013, 44, 622–629 CrossRef CAS.
- S. M. Smith, C. Oopathum, V. Weeramongkhonlert, C. B. Smith, S. Chaveanghong, P. Ketwong and S. Boonyuen, Bioresour. Technol., 2013, 143, 686–690 CrossRef CAS PubMed.
- S.-J. Yuan and X.-H. Dai, Appl. Catal., B, 2014, 154–155, 252–258 CrossRef CAS.
- M. Balakrishnan, V. S. Batra, J. S. J. Hargreaves, A. Monaghan, I. D. Pulford, J. L. Rico and S. Sushil, Green Chem., 2009, 11, 42 RSC.
- J. Yang, S. Lei, J. Yu and G. Xu, J. Environ. Chem. Eng., 2014, 2, 1007–1010 CrossRef CAS.
- P. R. Bureau, 2015 World Population Data Sheet, 2015 Search PubMed.
- G. Merino, M. Barange, J. L. Blanchard, J. Harle, R. Holmes, I. Allen, E. H. Allison, M. C. Badjeck, N. K. Dulvy, J. Holt, S. Jennings, C. Mullon and L. D. Rodwell, Global Environ. Change, 2012, 22, 795–806 CrossRef.
- P. K. Thornton, Philos. Trans. R. Soc. London, Ser. B, 2010, 365, 2853–2867 CrossRef PubMed.
- The European Parliament and the Council of the European Union, Official Journal of the European Union, 2008, 312, 3–30 Search PubMed.
- M. E. Borges and L. Díaz, Renewable Sustainable Energy Rev., 2012, 16, 2839–2849 CrossRef CAS.
- J. Boro, D. Deka and A. J. Thakur, Renewable Sustainable Energy Rev., 2012, 16, 904–910 CrossRef CAS.
- A. P. S. Chouhan and A. K. Sarma, Renewable Sustainable Energy Rev., 2011, 15, 4378–4399 CrossRef CAS.
- I. Nurfitri, G. P. Maniam, N. Hindryawati, M. M. Yusoff and S. Ganesan, Energy Convers. Manage., 2013, 74, 395–402 CrossRef CAS.
- D. A. Oliveira, P. Benelli and E. R. Amante, J. Cleaner Prod., 2013, 46, 42–47 CrossRef CAS.
- P. L. Boey, G. P. Maniam and S. A. Hamid, Bioresour. Technol., 2009, 100, 6362–6368 CrossRef CAS PubMed.
- R. Rezaei, M. Mohadesi and G. R. Moradi, Fuel, 2013, 109, 534–541 CrossRef CAS.
- S. Hu, Y. Wang and H. Han, Biomass Bioenergy, 2011, 35, 3627–3635 CrossRef CAS.
- N. Girish, S. P. Niju, K. M. Meera Sheriffa Begum and N. Anantharaman, Fuel, 2013, 111, 653–658 CrossRef CAS.
- P. Nair, B. Singh, S. N. Upadhyay and Y. C. Sharma, J. Cleaner Prod., 2012, 29–30, 82–90 CrossRef CAS.
- N. Viriya-empikul, P. Krasae, W. Nualpaeng, B. Yoosuk and K. Faungnawakij, Fuel, 2012, 92, 239–244 CrossRef CAS.
- N. Viriya-Empikul, P. Krasae, B. Puttasawat, B. Yoosuk, N. Chollacoop and K. Faungnawakij, Bioresour. Technol., 2010, 101, 3765–3767 CrossRef CAS PubMed.
- W. Suryaputra, I. Winata, N. Indraswati and S. Ismadji, Renewable Energy, 2013, 50, 795–799 CrossRef CAS.
- P.-L. Boey, S. Ganesan, G. P. Maniam and M. Khairuddean, Catal. Today, 2012, 190, 117–121 CrossRef CAS.
- N. Nakatani, H. Takamori, K. Takeda and H. Sakugawa, Bioresour. Technol., 2009, 100, 1510–1513 CrossRef CAS PubMed.
- L. Yang, A. Zhang and X. Zheng, Energy Fuels, 2009, 23, 3859–3865 CrossRef CAS.
- A. Birla, B. Singh, S. N. Upadhyay and Y. C. Sharma, Bioresour. Technol., 2012, 106, 95–100 CrossRef CAS PubMed.
- J. Boro, A. J. Thakur and D. Deka, Fuel Process. Technol., 2011, 92, 2061–2067 CrossRef CAS.
- J. Boro, L. J. Konwar, A. J. Thakur and D. Deka, Fuel, 2014, 129, 182–187 CrossRef CAS.
- S. Agrawal, B. Singh and Y. C. Sharma, Ind. Eng. Chem. Res., 2012, 51, 11875–11880 CrossRef CAS.
- Y. Gao and C. Xu, Catal. Today, 2012, 190, 107–111 CrossRef CAS.
- Z. Wei, C. Xu and B. Li, Bioresour. Technol., 2009, 100, 2883–2885 CrossRef CAS PubMed.
- P. Khemthong, C. Luadthong, W. Nualpaeng, P. Changsuwan, P. Tongprem, N. Viriya-empikul and K. Faungnawakij, Catal. Today, 2012, 190, 112–116 CrossRef CAS.
- R. Chakraborty, S. Bepari and A. Banerjee, Chem. Eng. J., 2010, 165, 798–805 CrossRef CAS.
- Y. C. Sharma, B. Singh and J. Korstad, Energy Fuels, 2010, 24, 3223–3231 CrossRef CAS.
- M. López Granados, D. Martín Alonso, A. C. Alba-Rubio, R. Mariscal, M. Ojeda and P. Brettes, Energy Fuels, 2009, 23, 2259–2263 CrossRef.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaría and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317–326 CrossRef CAS.
- J. Xie, X. Zheng, A. Dong, Z. Xiao and J. Zhang, Green Chem., 2009, 11, 355–364 RSC.
- W. Roschat, M. Kacha, B. Yoosuk, T. Sudyoadsuk and V. Promarak, Fuel, 2012, 98, 194–202 CrossRef CAS.
- A. Obadiah, G. A. Swaroopa, S. V. Kumar, K. R. Jeganathan and A. Ramasubbu, Bioresour. Technol., 2012, 116, 512–516 CrossRef CAS PubMed.
- R. Chakraborty, S. Bepari and A. Banerjee, Bioresour. Technol., 2011, 102, 3610–3618 CrossRef CAS PubMed.
- C. Ngamcharussrivichai, P. Nunthasanti, S. Tanachai and K. Bunyakiat, Fuel Process. Technol., 2010, 91, 1409–1415 CrossRef CAS.
- K. Balakrishnan, M. A. Olutoye and B. H. Hameed, Bioresour. Technol., 2013, 128, 788–791 CrossRef CAS PubMed.
- Q. Liu, R. Xin, C. Li, C. Xu and J. Yang, J. Environ. Sci., 2013, 25, 823–829 CrossRef CAS.
- J.-X. Wang, K.-T. Chen, B.-Z. Wen, Y.-H. B. Liao and C.-C. Chen, J. Taiwan Inst. Chem. Eng., 2012, 43, 215–219 CrossRef CAS.
- G. G. Muciño, R. Romero, A. Ramírez, S. L. Martínez, R. Baeza-Jiménez and R. Natividad, Fuel, 2014, 143–148 CrossRef.
- X. Zhang and W. Huang, J. Nat. Gas Chem., 2011, 20, 299–302 CrossRef CAS.
- FAO, FAO Handbook, Fishery and Aquaculture Statistics, 2012 Search PubMed.
- FAO, Food and Agriculture Organization of the United Nations, Statistics Division, http://www.faostat3.fao.org/browse/Q/QL/E.
- K. Jayathilakan, K. Sultana, K. Radhakrishna and A. S. Bawa, J. Food Sci. Technol., 2012, 49, 278–293 CrossRef CAS PubMed.
- FAO, The State of World Fisheries and Aquaculture, 2014 Search PubMed.
- K. Wilson, C. Hardacre, A. F. Lee, J. M. Montero and L. Shellard, Green Chem., 2008, 10, 654 RSC.
- A. P. S. Dias, J. Puna, M. J. Neiva Correia, I. Nogueira, J. Gomes and J. Bordado, Fuel Process. Technol., 2013, 116, 94–100 CrossRef.
- M. Kouzu, T. Kasuno, M. Tajika, Y. Sugimoto, S. Yamanaka and J. Hidaka, Fuel, 2008, 87, 2798–2806 CrossRef CAS.
- I. Haruna, M. Fatima and V. Ndam, International Journal of Scientific & Technology Research, 2015, 4, 186–189 Search PubMed.
- J. F. Puna, M. J. N. Correia, A. P. S. Dias, J. Gomes and J. Bordado, React. Kinet., Mech. Catal., 2013, 109, 405–415 CrossRef CAS.
- G. Power, M. Gräfe and C. Klauber, Hydrometallurgy, 2011, 108, 33–45 CrossRef CAS.
- N. Hindryawati, G. P. Maniam, M. R. Karim and K. F. Chong, Int. J. Eng. Sci. Res. Technol., 2014, 17, 95–103 Search PubMed.
- N. Hindryawati and G. P. Maniam, Ultrason. Sonochem., 2015, 22, 454–462 CrossRef CAS PubMed.
- A. P. S. Chouhan and A. K. Sarma, Biomass Bioenergy, 2013, 55, 386–389 CrossRef CAS.
- E. Lotero, Y. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. J. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- Y. M. Sani, W. M. A. W. Daud and A. R. Abdul Aziz, Appl. Catal., A, 2014, 470, 140–161 CrossRef CAS.
- F. Su and Y. Guo, Green Chem., 2014, 16, 2934 RSC.
- N. Alonso-Fagúndez, V. Laserna, A. C. Alba-Rubio, M. Mengibar, A. Heras, R. Mariscal and M. L. Granados, Catal. Today, 2014, 234, 285–294 CrossRef.
- L. A. S. do Nascimento, L. M. Z. Tito, R. S. Angélica, C. E. F. da Costa, J. R. Zamian and G. N. da Rocha Filho, Appl. Catal., B, 2011, 101, 495–503 CrossRef CAS.
- L. A. do Nascimento, R. S. Angelica, C. E. da Costa, J. R. Zamian and G. N. da Rocha Filho, Bioresour. Technol., 2011, 102, 8314–8317 CrossRef CAS PubMed.
- T. H. Dang, B. H. Chen and D. J. Lee, Bioresour. Technol., 2013, 145, 175–181 CrossRef CAS PubMed.
- L. H. O. Pires, A. N. de Oliveira, O. V. Monteiro Jr, R. S. Angélica, C. E. F. d. Costa, J. R. Zamian, L. A. S. d. Nascimento and G. N. R. Filho, Appl. Catal., B, 2014, 160–161, 122–128 CrossRef CAS.
- F. A. Dawodu, O. Ayodele, J. Xin, S. Zhang and D. Yan, Appl. Energy, 2014, 114, 819–826 CrossRef CAS.
- L. J. Konwar, R. Das, A. J. Thakur, E. Salminen, P. Mäki-Arvela, N. Kumar, J.-P. Mikkola and D. Deka, J. Mol. Catal. A: Chem., 2014, 388–389, 167–176 CrossRef CAS.
- X. Fu, D. Li, J. Chen, Y. Zhang, W. Huang, Y. Zhu, J. Yang and C. Zhang, Bioresour. Technol., 2013, 146, 767–770 CrossRef CAS PubMed.
- V. Budarin, J. H. Clark, J. J. E. Hardy, R. Luque, K. Milkowski, S. J. Tavener and A. J. Wilson, Angew. Chem., 2006, 118, 3866–3870 CrossRef.
- V. L. Budarin, J. H. Clark, R. Luque and D. J. Macquarrie, Chem. Commun., 2007, 634–636 RSC.
- J. Clark, V. Budarin, T. Dugmore, R. Luque, D. Macquarrie and V. Strelko, Catal. Commun., 2008, 9, 1709–1714 CrossRef CAS.
- R. Luque and J. H. Clark, ChemCatChem, 2011, 3, 594–597 CrossRef CAS.
- R. Luque, V. Budarin, J. H. Clark, P. Shuttleworth and R. J. White, Catal. Commun., 2011, 12, 1471–1476 CrossRef CAS.
- R. Luque and J. H. Clark, Catal. Commun., 2010, 11, 928–931 CrossRef CAS.
- J. C. Colmenares, P. Lisowski and D. Łomot, RSC Adv., 2013, 3, 20186 RSC.
- B. L. P. Devi, T. V. K. Reddy, K. V. Lakshmi and R. B. Prasad, Bioresour. Technol., 2014, 153, 370–373 CrossRef PubMed.
- C. A. Deshmane, M. W. Wright, A. Lachgar, M. Rohlfing, Z. Liu, J. Le and B. E. Hanson, Bioresour. Technol., 2013, 147, 597–604 CrossRef CAS PubMed.
- D. D. Chabukswar, P. K. K. S. Heer and V. G. Gaikar, Ind. Eng. Chem. Res., 2013, 52, 7316–7326 CrossRef CAS.
- M. Li, Y. Zheng, Y. Chen and X. Zhu, Bioresour. Technol., 2014, 154, 345–348 CrossRef CAS PubMed.
- Q. Shu, J. Gao, Z. Nawaz, Y. Liao, D. Wang and J. Wang, Appl. Energy, 2010, 87, 2589–2596 CrossRef CAS.
- Q. Shu, Z. Nawaz, J. Gao, Y. Liao, Q. Zhang, D. Wang and J. Wang, Bioresour. Technol., 2010, 101, 5374–5384 CrossRef CAS PubMed.
- L. Wang, X. Dong, H. Jiang, G. Li and M. Zhang, Bioresour. Technol., 2014, 158, 392–395 CrossRef CAS PubMed.
- A. M. Dehkhoda and N. Ellis, Catal. Today, 2013, 207, 86–92 CrossRef CAS.
- R. Chakraborty and D. RoyChowdhury, Chem. Eng. J., 2013, 215–216, 491–499 CrossRef CAS.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- M. Gliński, G. Zalewski, E. Burno and A. Jerzak, Appl. Catal., A, 2014, 470, 278–284 CrossRef.
- E. Karimi, I. F. Teixeira, L. P. Ribeiro, A. Gomez, R. M. Lago, G. Penner, S. W. Kycia and M. Schlaf, Catal. Today, 2012, 190, 73–88 CrossRef CAS.
- E. Karimi, C. Briens, F. Berruti, S. Moloodi, T. Tzanetakis, M. J. Thomson and M. Schlaf, Energy Fuels, 2010, 24, 6586–6600 CrossRef CAS.
- E. Karimi, I. F. Teixeira, A. Gomez, E. de Resende, C. Gissane, J. Leitch, V. Jollet, I. Aigner, F. Berruti, C. Briens, P. Fransham, B. Hoff, N. Schrier, R. M. Lago, S. W. Kycia, R. Heck and M. Schlaf, Appl. Catal., B, 2014, 145, 187–196 CrossRef CAS.
- C. Klauber, M. Gräfe and G. Power, Hydrometallurgy, 2011, 108, 11–32 CrossRef CAS.
- E. Karimi, A. Gomez, S. W. Kycia and M. Schlaf, Energy Fuels, 2010, 24, 2747–2757 CrossRef CAS.
- V. Jollet, C. Gissane and M. Schlaf, Energy Environ. Sci., 2014, 7, 1125 CAS.
- S. Sushil and V. S. Batra, Appl. Catal., B, 2008, 81, 64–77 CrossRef CAS.
- S. Sushil, P. Scholz, K. Pollok, B. Ondruschka and V. S. Batra, Chem. Eng. J., 2011, 166, 568–578 CrossRef CAS.
- I. F. Teixeira, T. P. V. Medeiros, P. E. Freitas, M. G. Rosmaninho, J. D. Ardisson and R. M. Lago, Fuel, 2014, 124, 7–13 CrossRef CAS.
- O. E. Lebedeva and A. G. Sarmurzina, Appl. Catal., B, 2000, 26, L1–L3 CrossRef CAS.
- J. Andas, F. Adam, I. A. Rahman and Y. H. Taufiq-Yap, Chem. Eng. J., 2014, 252, 382–392 CrossRef CAS.
- M. Huang, K. Zhao, M. Zhang, S. Chen, W. Xu, Y. Ye, W. Fu, Y. Wei, Z. Qiu and F. Sun, Procedia Eng., 2011, 18, 358–362 CrossRef CAS.
- C. C. Amorim, M. M. D. Leão, R. F. P. M. Moreira, J. D. Fabris and A. B. Henriques, Chem. Eng. J., 2013, 224, 59–66 CrossRef CAS.
- N. Li, X. Lu and S. Zhang, Chem. Eng. J., 2014, 257, 253–261 CrossRef CAS.
- B. Yuan, J. Xu, X. Li and M.-L. Fu, Chem. Eng. J., 2013, 226, 181–188 CrossRef CAS.
- B. Das, S. Prakash, P. S. R. Reddy and V. N. Misra, Resour., Conserv. Recycl., 2007, 50, 40–57 CrossRef.
- B. H. Robinson, Sci. Total Environ., 2009, 408, 183–191 CrossRef CAS PubMed.
- K. Srivastava, V. Devra and A. Rani, Fuel Process. Technol., 2014, 121, 1–8 CrossRef CAS.
- M. I. Domínguez, I. Barrio, M. Sánchez, M. Á. Centeno, M. Montes and J. A. Odriozola, Catal. Today, 2008, 133–135, 467–474 CrossRef.
- M. I. Domínguez, M. Sánchez, M. A. Centeno, M. Montes and J. A. Odriozola, Appl. Catal., A, 2006, 302, 96–103 CrossRef.
- M. I. Domínguez, M. Sánchez, M. A. Centeno, M. Montes and J. A. Odriozola, J. Mol. Catal. A: Chem., 2007, 277, 145–154 CrossRef.
- F. Klose, P. Scholz, G. Kreisel, B. Ondruschka, R. Kneise and U. Knopf, Appl. Catal., B, 2000, 28, 209–221 CrossRef CAS.
- A. D. Anton, O. Klebercz, A. Magyar, I. T. Burke, A. P. Jarvis, K. Gruiz and W. M. Mayes, Environ. Sci.: Processes Impacts, 2014, 16, 2677–2685 CAS.
- J. N. Alhajji and M. R. Reda, Water Res., 1994, 28, 2377–2381 CrossRef CAS.
- A. Salmiaton and A. Garforth, Waste Manag, 2007, 27, 1891–1896 CrossRef CAS PubMed.
- K. H. Lee, Polym. Degrad. Stab., 2008, 93, 1284–1289 CrossRef CAS.
- A. Salmiaton and A. A. Garforth, Waste Manag, 2011, 31, 1139–1145 CrossRef CAS PubMed.
- K.-H. Lee, N.-S. Noh, D.-H. Shin and Y. Seo, Polym. Degrad. Stab., 2002, 78, 539–544 CrossRef CAS.
- K.-H. Lee, D.-H. Shin and Y.-H. Seo, Polym. Degrad. Stab., 2004, 84, 123–127 CrossRef CAS.
- K. M. Park, T. Y. Kim, H. B. Kim, S. J. Kim and S. Y. Cho, in Studies in Surface Science and Catalysis, ed. I.-S. N. Hyun-Ku Rhee and P. Jong Moon, Elsevier, 2006, vol. 159, pp. 429–432 Search PubMed.
- Y. H. Lin and M. H. Yang, Polym. Degrad. Stab., 2007, 92, 813–821 CrossRef CAS.
- Y. H. Lin and M. H. Yang, Appl. Catal., A, 2007, 328, 132–139 CrossRef CAS.
- Y. Lin and M. Yang, Appl. Catal., B, 2007, 69, 145–153 CrossRef CAS.
- Y. Shen, P. Zhao, Q. Shao, D. Ma, F. Takahashi and K. Yoshikawa, Appl. Catal., B, 2014, 152–153, 140–151 CrossRef CAS.
- A. López, I. de Marco, B. M. Caballero, M. F. Laresgoiti, A. Adrados and A. Aranzabal, Appl. Catal., B, 2011, 104, 211–219 CrossRef.
- S. Sharma and S. K. Ghoshal, Renewable Sustainable Energy Rev., 2015, 43, 1151–1158 CrossRef CAS.
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2015, 40, 11094–11111 CrossRef CAS.
- S. E. Hosseini, M. A. Wahid, M. M. Jamil, A. A. M. Azli and M. F. Misbah, Int. J. Energy Res., 2015, 39, 1597–1615 CrossRef CAS.
- S. E. Hosseini, M. A. Wahid and A. Ganjehkaviri, Energy Convers. Manage., 2015, 94, 415–429 CrossRef CAS.
- J.-L. Cao, Z.-L. Yan, Q.-F. Deng, Y. Wang, Z.-Y. Yuan, G. Sun, T.-K. Jia, X.-D. Wang, H. Bala and Z.-Y. Zhang, Int. J. Hydrogen Energy, 2014, 39, 5747–5755 CrossRef CAS.
- T. Kanamori, M. Matsuda and M. Miyake, J. Hazard. Mater., 2009, 169, 240–245 CrossRef CAS PubMed.
- I. Genova, T. Tsoncheva, M. Dimitrov, D. Paneva, B. Tsyntsarski, R. Ivanova, Z. Cherkezova-Zheleva, T. Budinova, D. Kovacheva, I. Mitov and N. Petrov, Catal. Commun., 2014, 55, 43–48 CrossRef CAS.
- K. Palanichamy and A. Ariharaputhiran, Int. J. Hydrogen Energy, 2013, 38, 2263–2270 CrossRef CAS.
- C. Zhang, X. Liang and S. Liu, Int. J. Hydrogen Energy, 2011, 36, 8902–8907 CrossRef CAS.
- T.-H. Liou and B.-C. Lai, Appl. Catal., B, 2012, 115–116, 138–148 CrossRef CAS.
- M. L. Yola, T. Eren and N. Atar, Chem. Eng. J., 2014, 250, 288–294 CrossRef CAS.
- W. L. da Silva, M. A. Lansarin, F. C. Stedile and J. H. Z. dos Santos, J. Mol. Catal. A: Chem., 2014, 393, 125–133 CrossRef.
- F. R. Xiu and F. S. Zhang, J. Hazard. Mater., 2009, 172, 1458–1463 CrossRef CAS PubMed.
- E. Bae and W. Choi, Environ. Sci. Technol., 2002, 37, 147–152 CrossRef.
- B. Pal, S. Ikeda, H. Kominami, Y. Kera and B. Ohtani, J. Catal., 2003, 217, 152–159 CAS.
- J. Bandara, C. P. Udawatta and C. S. Rajapakse, Photochem. Photobiol. Sci., 2005, 4, 857–861 CAS.
- Y.-h. Xu, D.-h. Liang, M.-l. Liu and D.-z. Liu, Mater. Res. Bull., 2008, 43, 3474–3482 CrossRef CAS.
- J. Li, L. Liu, Y. Yu, Y. Tang, H. Li and F. Du, Electrochem. Commun., 2004, 6, 940–943 CrossRef CAS.
- K. Chiang, R. Amal and T. Tran, Adv. Environ. Res., 2002, 6, 471–485 CrossRef CAS.
- H. Tang, F. Lévy, H. Berger and P. Schmid, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 7771–7774 CrossRef CAS.
- M. Sapiña, E. Jimenez-Relinque and M. Castellote, Environ. Sci. Pollut. Res. Int., 2014, 21, 12091–12098 CrossRef PubMed.
- S. Singh, M. A. Nahil, X. Sun, C. Wu, J. Chen, B. Shen and P. T. Williams, Fuel, 2013, 105, 585–594 CrossRef CAS.
- Y. Kuwahara and H. Yamashita, J. CO2 Util., 2013, 1, 50–59 CrossRef CAS.
- K. Narasimharao, D. Brown, A. Lee, A. Newman, P. Siril, S. Tavener and K. Wilson, J. Catal., 2007, 248, 226–234 CrossRef CAS.
- L. Pesaresi, D. R. Brown, A. F. Lee, J. M. Montero, H. Williams and K. Wilson, Appl. Catal., A, 2009, 360, 50–58 CrossRef CAS.
- A. Osatiashtiani, A. F. Lee, M. Granollers, D. R. Brown, L. Olivi, G. Morales, J. A. Melero and K. Wilson, ACS Catal., 2015, 5, 4345–4352 CrossRef CAS.
- A. Osatiashtiani, A. F. Lee, D. R. Brown, J. A. Melero, G. Morales and K. Wilson, Catal. Sci. Technol., 2014, 4, 333–342 CAS.
- G. Morales, A. Osatiashtiani, B. Hernandez, J. Iglesias, J. A. Melero, M. Paniagua, D. Robert Brown, M. Granollers, A. F. Lee and K. Wilson, Chem. Commun., 2014, 50, 11742–11745 RSC.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- M. Gonçalves, M. Mantovani, W. A. Carvalho, R. Rodrigues, D. Mandelli and J. Silvestre Albero, Chem. Eng. J., 2014, 256, 468–474 CrossRef.
- J. A. Sánchez, D. L. Hernández, J. A. Moreno, F. Mondragón and J. J. Fernández, Appl. Catal., A, 2011, 405, 55–60 CrossRef.
- C. V. Rode, R. B. Mane, A. S. Potdar, P. B. Patil, P. S. Niphadkar and P. N. Joshi, Catal. Today, 2012, 190, 31–37 CrossRef CAS.
- M. Calzado, M. J. Valero-Romero, P. Garriga, A. Chica, M. O. Guerrero-Pérez, J. Rodríguez-Mirasol and T. Cordero, Catal. Today, 2015, 257, 229–236 CrossRef CAS.
- R. Ji, K. Yu, L.-L. Lou, C. Zhang, Y. Han, S. Pan and S. Liu, Inorg. Chem. Commun., 2012, 25, 65–69 CrossRef CAS.
- E. A. El-Katatny, S. A. Halawy, M. A. Mohamed and M. I. Zaki, Appl. Catal., A, 2000, 199, 83–92 CrossRef CAS.
- L. Wang, H. Li, S. Xin and F. Li, Catal. Commun., 2014, 50, 49–53 CrossRef CAS.
- D. Jain, M. Mishra and A. Rani, Fuel Process. Technol., 2012, 95, 119–126 CrossRef CAS.
- A. Rani, C. Khatri and R. Hada, Fuel Process. Technol., 2013, 116, 366–373 CrossRef CAS.
- L. E. Macaskie, I. P. Mikheenko, P. Yong, K. Deplanche, A. J. Murray, M. Paterson-Beedle, V. S. Coker, C. I. Pearce, R. Cutting, R. A. D. Pattrick, D. Vaughan, G. van der Laan and J. R. Lloyd, Hydrometallurgy, 2010, 104, 483–487 CrossRef CAS.
- A. N. Mabbett, D. Sanyahumbi, P. Yong and L. E. Macaskie, Environ. Sci. Technol., 2005, 40, 1015–1021 CrossRef.
- A. J. Murray, I. P. Mikheenko, E. Goralska, N. A. Rowson and L. E. Macaskie, in Biohydrometallury: From the Single Cell to the Environment, ed. A. Schippers, W. Sand, F. Glombitza and S. Willscher, Trans Tech Publications Ltd, Stafa-Zurich, 2007, vol. 20–21, pp. 651–654 Search PubMed.
- N. J. Creamer, V. S. Baxter-Plant, J. Henderson, M. Potter and L. E. Macaskie, Biotechnol. Lett., 2006, 28, 1475–1484 CrossRef CAS PubMed.
- G. A. Attard, J. A. Bennett, I. Mikheenko, P. Jenkins, S. Guan, L. E. Macaskie, J. Wood and A. J. Wain, Faraday Discuss., 2013, 162, 57 RSC.
- J. A. Bennett, G. A. Attard, K. Deplanche, M. Casadesus, S. E. Huxter, L. E. Macaskie and J. Wood, ACS Catal., 2012, 2, 504–511 CrossRef CAS.
- J. A. Bennett, N. J. Creamer, K. Deplanche, L. E. Macaskie, I. J. Shannon and J. Wood, Chem. Eng. Sci., 2010, 65, 282–290 CrossRef CAS.
- K. Deplanche, J. A. Bennett, I. P. Mikheenko, J. Omajali, A. S. Wells, R. E. Meadows, J. Wood and L. E. Macaskie, Appl. Catal., B, 2014, 147, 651–665 CrossRef CAS.
- J. A. Bennett, I. P. Mikheenko, K. Deplanche, I. J. Shannon, J. Wood and L. E. Macaskie, Appl. Catal., B, 2013, 140–141, 700–707 CrossRef CAS.
- L. E. Macaskie, A. C. Humphries, I. P. Mikheenko, V. S. Baxter-Plant, K. Deplanche, M. D. Redwood, J. A. Bennett and J. Wood, J. Chem. Technol. Biotechnol., 2012, 87, 1430–1435 CrossRef CAS.
- K. Deplanche, I. P. Mikheenko, J. A. Bennett, M. Merroun, H. Mounzer, J. Wood and L. E. Macaskie, Top. Catal., 2011, 54, 1110–1114 CrossRef CAS.
- K. Deplanche, M. L. Merroun, M. Casadesus, D. T. Tran, I. P. Mikheenko, J. A. Bennett, J. Zhu, I. P. Jones, G. A. Attard, J. Wood, S. Selenska-Pobell and L. E. Macaskie, J. R. Soc., Interface, 2012, 9, 1705–1712 CrossRef CAS PubMed.
- P. Yong, I. P. Mikheenko, K. Deplanche, M. D. Redwood and L. E. Macaskie, Biotechnol. Lett., 2010, 32, 1821–1828 CrossRef CAS PubMed.
- R. L. Orozco, M. D. Redwood, P. Yong, I. Caldelari, F. Sargent and L. E. Macaskie, Biotechnol. Lett., 2010, 32, 1837–1845 CrossRef CAS PubMed.
- L. S. Sobjerg, D. Gauthier, A. T. Lindhardt, M. Bunge, K. Finster, R. L. Meyer and T. Skrydstrup, Green Chem., 2009, 11, 2041–2046 RSC.
- D. Gauthier, L. S. Søbjerg, K. M. Jensen, A. T. Lindhardt, M. Bunge, K. Finster, R. L. Meyer and T. Skrydstrup, ChemSusChem, 2010, 3, 1036–1039 CrossRef CAS PubMed.
- M. Schlüter, T. Hentzel, C. Suarez, M. Koch, W. G. Lorenz, L. Böhm, R.-A. Düring, K. A. Koinig and M. Bunge, Chemosphere, 2014, 117, 462–470 CrossRef PubMed.
- S. De Corte, T. Hennebel, B. De Gusseme, W. Verstraete and N. Boon, Microb. Biotechnol., 2012, 5, 5–17 CrossRef CAS PubMed.
- S. Dimitriadis, N. Nomikou and A. P. McHale, Biotechnol. Lett., 2007, 29, 545–551 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |