
Rhodium(II)-catalyzed intramolecular annulation of 1-sulfonyl-1,2,3-triazoles with indoles: facile synthesis of functionalized tetrahydro-β-carbolines†
Hai Shanga,
Yu Tiana,
Jun Luoa,
Lingyu Lia,
Yefeng Tang*b and
Zhongmei Zou*a
aInstitute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100193, P. R. China. E-mail: zmzou@implad.ac.cn
bComprehensive AIDS Research Center, Department of Pharmacology & Pharmaceutical Sciences, School of Medicine, Tsinghua University, Beijing 100084, China
First published on 17th March 2016
Abstract
A novel rhodium(II)-catalyzed intramolecular annulation of 1-sulfonyl-1,2,3-triazoles with indoles has been developed, which enables the facile synthesis of various 4-functionalized tetrahydro-β-carbolines as well as relevant scaffolds from readily available starting materials.
Tetrahydro-β-carbolines (THBCs) as well as relavent scaffolds (e.g. β-carbolines) are privileged structural motifs present in many natural products and medicinally valuable molecules,1 as represented by the compounds 1–5 outlined in Fig. 1.2 Their broad spectrum of biological and pharmacological profiles such as hypnotic, anxiolytic, antimicrobial, antiviral, anti-tumor, and anticonvulsant activities have stimulated significant interest in their synthesis.3 The most commonly used methods to access tetrahydro-β-carbolines rely on Pictet–Spengler condensations of tryptamine or tryptophan with aldehydes or ketones.4 Of note, such methods are oftenly applied to the synthesis of C1- or C3-functionalized THBCs. Comparably, obtaining 4-functionalized THBCs remains a challenging task, and multistep procedures are normally required. Recently, some new approaches have been developed to address this issue. For examples, Bandini and co-workers reported an efficient intramolecular cyclization reaction to give a variety of 4-functionalized THBCs.5 Roussi et al. established an elegant methods for the direct functionalization at the 4-position of THBCs.6 More recently, Wang et al. disclosed a novel I2-promoted cascade electrophilic cyclization for the rapid access of 4-iodomethyl substituted THBCs.7 Despite of such significant progresses, the development of a general synthetic method for the efficient synthesis of 4-substituted THBCs remains a considerable challenge.
 | ||
| Fig. 1 Representative molecules containing tetrahydro-β-carboline and β-carboline motifs. | ||
Recently, Rh(II)-azavinylcarbene, which is readily generated from 1-sulfonyl-1,2,3-triazole through denitrogenative reaction, has evolved into a capable intermediate for the synthesis of various nitrogen heterocycles.8 Along with the major contribution from Gevorgyan, Fokin, Davies, Murakami, Shi, and Lee's groups,9 we also show keen interest on the development of novel Rh-AVC-promoted cycloadditions or transannulations for the synthesis of various valuable heterocyclic scaffolds.10 In light of the importance of THBCs and relevant scaffolds in organic and medicinal chemistry, as well as the fact that only limited methods exist for their synthesis, we sought to develop a new entry to the aforementioned heterocycles based on the emerging Rh-AVC chemistry. As shown in Scheme 1, we envisioned that a substrate like 6 might readily undergo a denitrogenative reaction upon the treatment of Rh(II)-catalyst to afford the putative Rh(II)-azavinylcarbene species 7, which then induces an intramolecular cyclization with the nucleophilic attack of the C-3 position of indole to the electrophilic metal carbene center, thus furnishing the zwitterion 8. Intermediate 8 could rapid convert to 9 via rearomatization with the release of a proton, and the latter, upon protonation, would afford the enamide 10 as final product. Of note, the enamide moiety could serve as a handle for further functionality elaborations, which allows accessing some more synthetically useful 4-substituted tetrahydro-β-carbolines 11 as well as relevant scaffolds (e.g. β-carbolines).
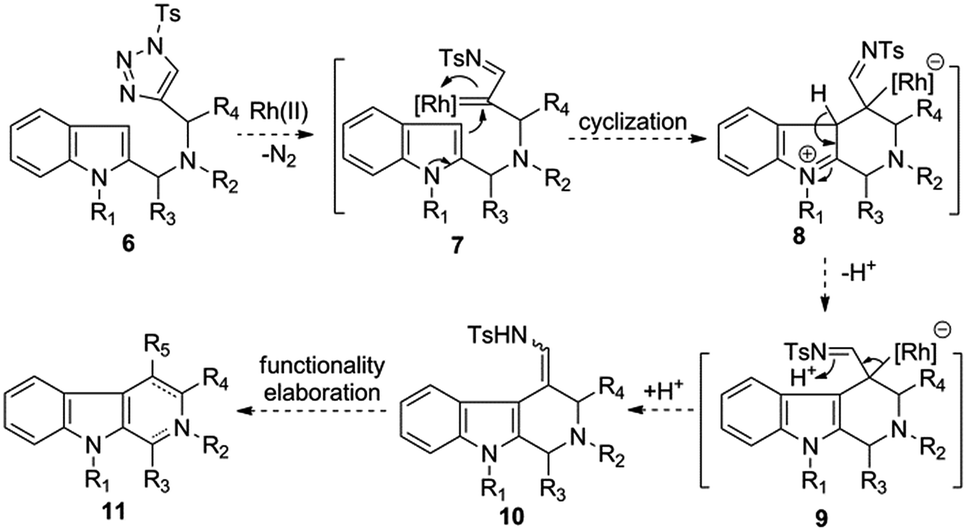 | ||
| Scheme 1 Working hypothesis of the Rh-AVC-promoted intramolecular annulation of 1-sulfonyl-1,2,3-triazoles with indoles. | ||
The readily available indole–triazole hybrid compound 6a was chosen as standard substrate in the initial study (Scheme 2). Thus, treatment of 6a with catalytic amount of Rh2(OOct)4 in 1,2-DCE at 80 °C for 15 min resulted in the complete consumption of the starting material, with the expected annulation product 10a obtained in 67% isolated yield. Of note, the enamide 10a proved to be a mixture of Z/E isomers, the ratio of which was 3.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Moreover, it was found that 10a was not very stable and slightly decomposed upon chromatographic purification. Thus, to obtain a single and stable product for structural determination, 10a was quickly reduced to the corresponding amide derivative 11a with NaBH3CN in 87% yield, whose structure was unambiguously confirmed by the X-ray crystallographic study.11 Furthermore, we found that the Rh(II)-catalyzed annulation and reduction could be conducted in an operationally simple one-pot manner, which allowed for the access of 11a directly from 6a with comparable efficiency.
:1. Moreover, it was found that 10a was not very stable and slightly decomposed upon chromatographic purification. Thus, to obtain a single and stable product for structural determination, 10a was quickly reduced to the corresponding amide derivative 11a with NaBH3CN in 87% yield, whose structure was unambiguously confirmed by the X-ray crystallographic study.11 Furthermore, we found that the Rh(II)-catalyzed annulation and reduction could be conducted in an operationally simple one-pot manner, which allowed for the access of 11a directly from 6a with comparable efficiency.
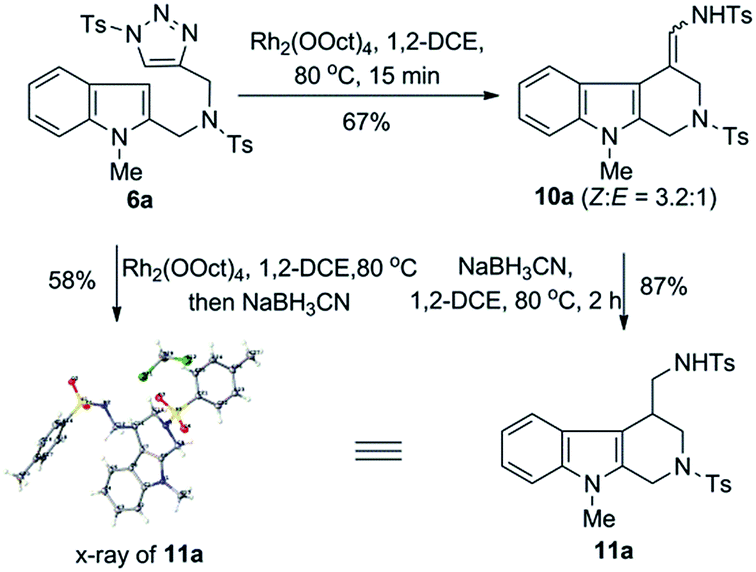 | ||
| Scheme 2 Initial result of Rh(II)-catalyzed intramolecular annulation of 1-sulfonyl-1,2,3-triazoles with indoles. | ||
To improve the efficiency of the above one-pot transformations, we performed an extensive condition screening (Table 1). First of all, it was found that increasing the reaction temperature was beneficial to the reaction (entries 2–4), with the best result (86%) obtained at 140 °C. Furthermore, it was shown that the nature of Rh-catalyst also had a notable influence on the outcome. While Rh2(OAc)4 and Rh2(S-DOSP)4 gave comparable results to Rh2(OOct)4 (entry 5 and 8), Rh2(S-PTAD)4 and Rh2(esp)2 displayed inferior reactivity (entries 6 and 7). In addition, the effect of solvents was also evaluated (entries 9–11). It turned out that the polar solvent such as CHCl3 was suitable choice for the reaction, and the less polar solvent PhMe and PhCl afforded decreased yield of product.
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent | T [°C] | Yieldb |
| a Reaction conditions: 6a (0.2 mmol), Rh2(OOct)4 (2.0 μmol), and 1,2-DCE (1.0 mL) in a sealed tube, then NaBH3CN (0.4 mmol).b Refers to isolated yield. 1,2-DCE = 1,2-dichloroethane, esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid, OOct = octanoate, (S)-ptad = N-phthaloyl-(S)-adamantylglycine, (S)-dosp = 4-(dodecyl-phenyl)-sulfonyl-(2S)-prolinate. | ||||
| 1 | Rh2(OOct)4 | DCE | 80 | 58% |
| 2 | Rh2(OOct)4 | DCE | 100 | 73% |
| 3 | Rh2(OOct)4 | DCE | 120 | 81% |
| 4 | Rh2(OOct)4 | DCE | 140 | 86% |
| 5 | Rh2(OAc)4 | DCE | 140 | 85% |
| 6 | Rh2(S-PTAD)4 | DCE | 140 | 30% |
| 7 | Rh2(esp)2 | DCE | 140 | 64% |
| 8 | Rh2(S-DOSP)4 | DCE | 140 | 83% |
| 9 | Rh2(OOct)4 | CHCl3 | 140 | 83% |
| 10 | Rh2(OOct)4 | PhMe | 140 | 60% |
| 11 | Rh2(OOct)4 | PhCl | 140 | 61% |
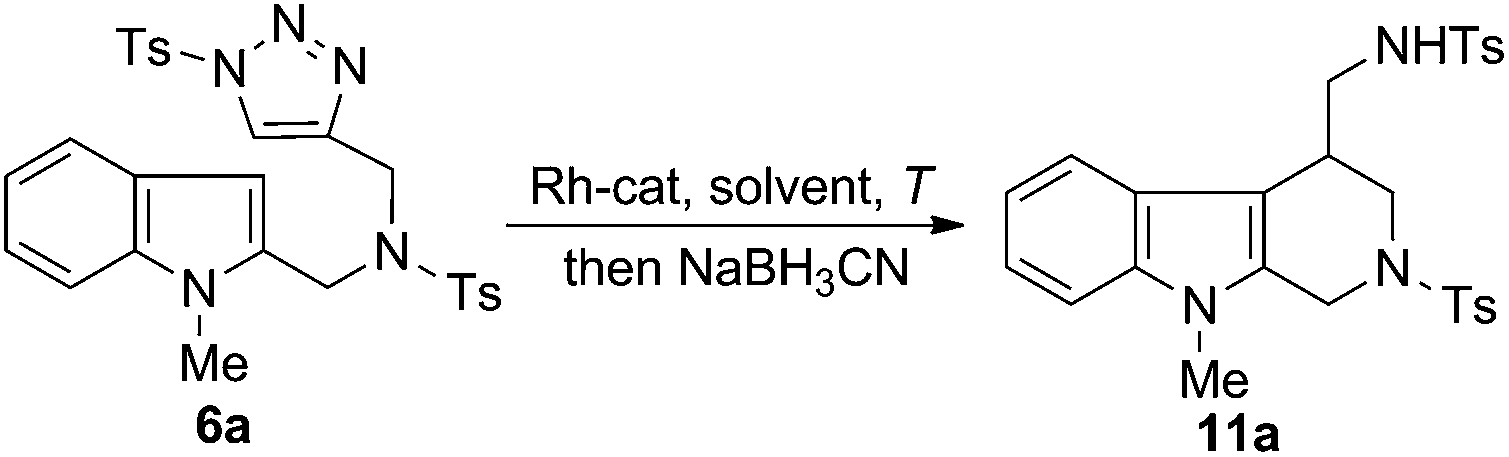
Having the optimal conditions in hands, we then turned to evaluate the generality of the above reaction (Table 2). First of all, the substituent variants on the aromatic ring of indoles were examined. A wide range of 5-substituted indole derivatives underwent the desired reactions smoothly to provide the corresponding products in good to excellent yields. Both electron-withdrawing (11b–d) and electron-donating (11e–g) substituents were well tolerated. Moreover, the reaction could also be applied to various other mono- or disubstituted indole derivatives (11h–m). As shown, the substrates bearing a substituent on the 6-position (11h and 11i) or 7-position (11j) showed comparable reactivity to the 5-substituted ones. Comparably, the substituent on the C-4 position of indoles seemed to have negative influence on the reaction outcome (11k and 11l), most likely due to the steric effect. Of note, the 4,6-disubstituted substrate was also amenable to the reaction, as proved by the case leading to 11m.
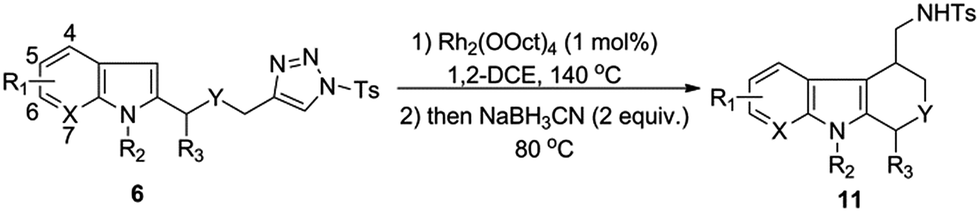
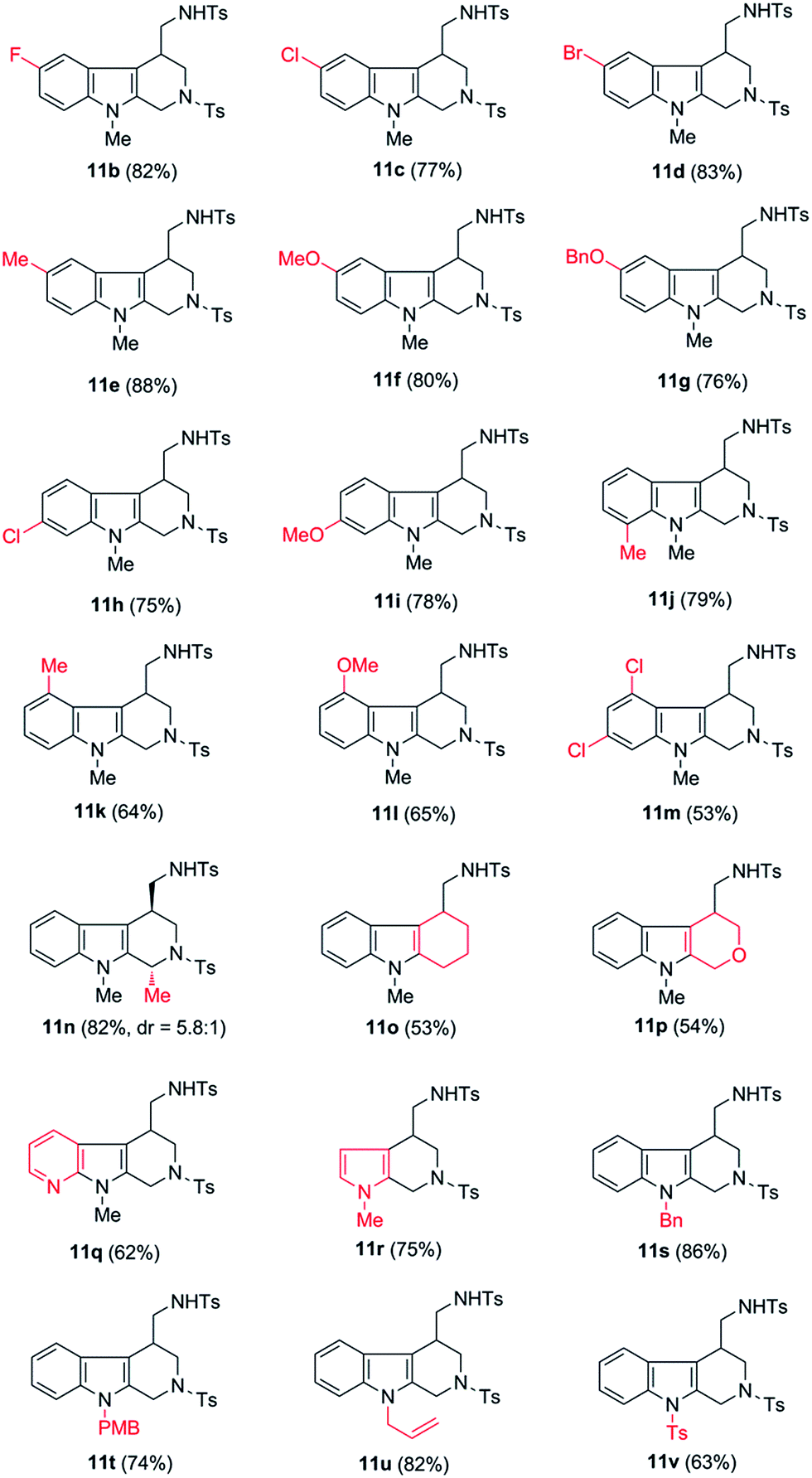
In addition to the substituent on the aromatic ring of indoles, some other structural variants were also evaluated. Gratifyingly, the transformation could be applied to the synthesis of 1,4-disubstituted tetrahydro-β-carboline, as shown by the case of 11n which was obtained as a mixture of diastereoisomers (dr = 5.8:1), favoring the 1,4-trans-product as major component. Moreover, besides the tetrahydro-β-carboline derivatives, some other relevant scaffolds (e.g. tetrahydrocarbazole derivative 11o or tetrahydropyrano[3,4-b]indole derivative 11p) could also be obtained from the corresponding substrates. Finally, it was found that the indole unit of the substrates could be replaced by some other heterocycles, such as 1H-pyrrolo[2,3-b]pyridine and pyrrole, as shown by the cases of 11q and 11r.
Notably, all of the above-examined substrates bear a N-methyl protecting group, the removal of which might be a challenging task in practice. To improve the synthetic utility of the transformations, we next examined several substrates bearing the removable protecting groups, such as Bn, PMB, and allyl groups. Fortunately, it was shown that all of these substrates provided the corresponding products in good yields (11s–11u). Interestingly, even the substrate bearing a strong electron-withdrawing Ts group on the N-1 position of indole could also tolerate in the transformation, with acceptable yield of the corresponding product obtained (11v).
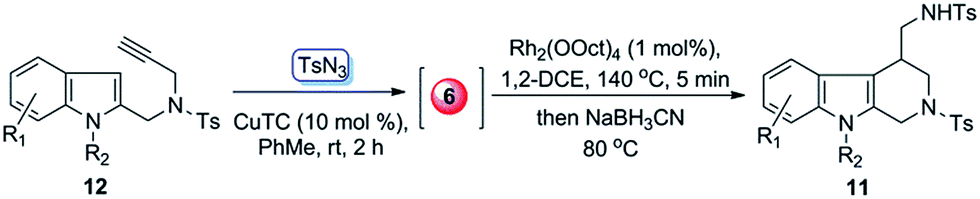
To further simply the operation of above transformations, we also developed a one-pot protocol for the direct synthesis of tetrahydro-β-carboline derivatives from terminal alkynes and tosyl azide. As proof-of-concept case, the alkyne 12a was treated with TsN3 in the presence of CuTC (10 mol%) in PhMe at room temperature for 2 h. Then the solvent was removed, and the resulting mixtures, without isolation, were redissolved in 1,2-DCE12 and sequentially treated with Rh2(OOct)4 (1 mol%) at 140 °C for 5 min and then NaBH3CN at 80 °C for another 3 h, which finally afforded 11a in 61% yield (entry 1, Table 3). This one-pot protocol was also applied to some other terminal alkynes (12b–c, 12e–f and 12s). Gratifyingly, all of them gave the corresponding products in acceptable yields (entries 2–6).
|
||
|---|---|---|
| Entry | Alkyne | Product: yieldb |
| a Reaction conditions: 12 (0.20 mmol), TsN3 (0.22 mmol) and CuTC (20.0 μmol) in PhMe (2.0 mL), then PhMe was replaced by 1,2-DCE (1.0 mL), Rh2(OOct)4 (2.0 μmol), and then NaBH3CN (0.40 mmol).b Refers to isolated yield. | ||
| 1 | 12a: R1 = H, R2 = Me | 11a: 61% |
| 2 | 12b: R1 = 5-F, R2 = Me | 11b: 63% |
| 3 | 12c: R1 = 5-Cl, R2 = Me | 11c: 54% |
| 4 | 12e: R1 = 5-Me, R2 = Me | 11e: 70% |
| 5 | 12f: R1 = 5-OMe, R2 = Me | 11f: 56% |
| 6 | 12s: R1 = H, R2 = Bn | 11s: 65% |
Lastly, the synthetic utility of the developed chemistry was demonstrated by its application to the formal synthesis of oxopropaline G (4), a naturally occurring β-carboline alkaloid (Scheme 3). Thus, starting from the readily available alkyne 12w, triazole 6w was synthesized through copper-catalyzed alkyne-azide cycloaddition in 87% yield. Treatment of 6w via Rh-catalyzed denitrogenative cyclization followed by reduction gave the key intermediate 11w in 77% yield. Serendipitously, we found that 11w readily underwent the deprotection followed by aromatization upon treatment with NaOH to afford the compound 13 in 61% yield,13 thereby completing the formal synthesis of oxopropaline G (4).14,15
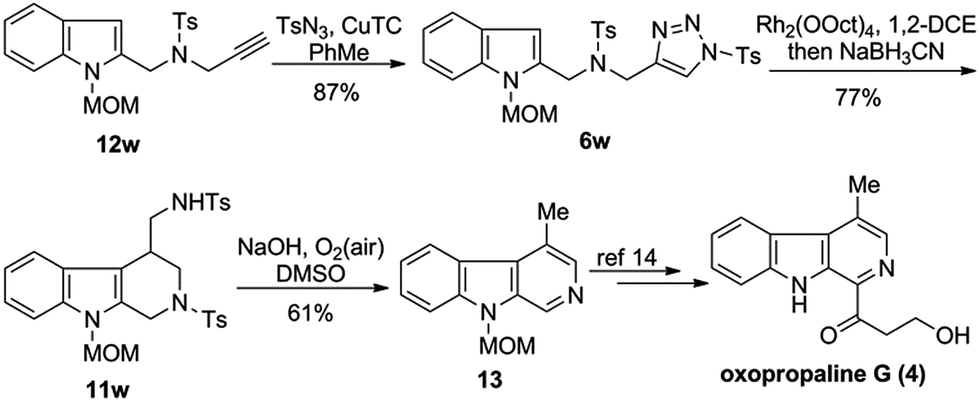 | ||
| Scheme 3 Formal synthesis of oxopropaline G. | ||
In summary, we have developed an efficient method for the construction of 4-substituted tetrahydro-β-carbolines through a novel Rh(II)-catalyzed intramolecular annulation of 1-sulfonyl-1,2,3-triazoles with indoles. An operationally simple one-pot three-steps protocol was also established, which enable the rapid access of the title compounds from readily available starting materials. The potential utility of the present chemistry was demonstrated by a short formal synthesis of oxopropaline G. It is anticipated that this method would provide an alternative to the current toolbox for the synthesis of highly functionalized tetrahydro-β-carbolines, and thus might find widespread applications in the organic and medicinal chemistry.
Acknowledgements
We gratefully acknowledge the financial supports from the NSFC (81502929).Notes and references
- For selected reviews, see: (a) C. R. Edwankar, R. V. Edwankar, O. A. Namjoshi, S. K. Rallapappi, J. Yang and J. M. Cook, Curr. Opin. Drug Disc., 2009, 12, 752 CAS; (b) J. E. Saxton, Nat. Prod. Rep., 1997, 559 RSC; (c) B. J. Magnier and Y. Langlois, Tetrahedron, 1998, 54, 6201 CrossRef; (d) G. Lazurjevski and I. Terentjeva, Heterocycles, 1976, 4, 1783 CrossRef.
- For selected examples, see: (a) J. Garnier, J. Mahuteau, M. Plat and C. Merienne, Phytochemistry, 1989, 28, 308 CrossRef CAS; (b) A. Itoh, T. Tanahashi, N. Nagakura and T. Nishi, Phytochemistry, 2003, 62, 359 CrossRef CAS PubMed; (c) P. Crews, J. J. Farias, R. Emrich and P. A. Keifer, J. Org. Chem., 1994, 59, 2932 CrossRef CAS; (d) N. Abe, Y. Nakakita, T. Nakamura, N. Enoki, H. Uchida, S. Takeo and M. Munekata, J. Antibiot., 1993, 46, 1672 CrossRef CAS PubMed.
- For selected examples, see: (a) H. R. Snyder, C. H. Hansch, L. Katz, S. M. Parmerter and E. C. Spaeth, J. Am. Chem. Soc., 1948, 70, 219 CrossRef CAS PubMed; (b) P. Molina, P. M. Fresneda and S. García-Zafra, Tetrahedron Lett., 1995, 36, 3581 CrossRef CAS; (c) K. Narayanan, L. Schindler and J. M. Cook, J. Org. Chem., 1991, 56, 359 CrossRef CAS; (d) Y. Ohta, S. Oishi and H. Ohno, Org. Lett., 2009, 11, 1979 CrossRef CAS PubMed; (e) M. Bandini, A. Alfonso, F. Piccinelli, R. Sinisi, S. Tommasi and A. Umani-Ronchi, J. Am. Chem. Soc., 2003, 128, 1424 CrossRef PubMed; (f) T. Arai, M. Wasai and N. Yokoyama, J. Org. Chem., 2011, 76, 2909 CrossRef CAS PubMed.
- For leading reviews, see: (a) E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797 CrossRef CAS; (b) J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538 CrossRef PubMed.
- M. Agnusdei, M. Bandini, A. Melloni and A. Umani-Ronchi, J. Org. Chem., 2003, 68, 7126 CrossRef CAS PubMed.
- C. Rannoux, F. Roussi, P. Retailleau and F. Gueritte, Org. Lett., 2010, 12, 1240 CrossRef CAS PubMed.
- H. J. Song, Y. X. Liu and Q. M. Wang, Org. Lett., 2013, 13, 3274 CrossRef PubMed.
- For leading reviews, see: (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862 CrossRef CAS PubMed; (b) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371 CrossRef CAS PubMed; (c) H. M. L. Davie and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC.
- For selected recent examples, see: (a) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034 CrossRef CAS PubMed; (b) N. Grimster, L. Zhang and V. V. Fokin, J. Am. Chem. Soc., 2010, 132, 2510 CrossRef CAS PubMed; (c) T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194 CrossRef CAS PubMed; (d) T. Miura, T. Tanaka, T. Biyajima, A. Yada and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 3883 CrossRef CAS PubMed; (e) S. Chuprakov, B. T. Worrell, N. Selander, R. K. Sit and V. V. Fokin, J. Am. Chem. Soc., 2014, 136, 195 CrossRef CAS PubMed; (f) E. E. Schultz and R. Sarpong, J. Am. Chem. Soc., 2013, 135, 4696 CrossRef CAS PubMed; (g) S. Chuprakov, S. W. Kwok and V. V. Fokin, J. Am. Chem. Soc., 2013, 135, 4652 CrossRef CAS PubMed; (h) J. S. Alford and H. M. L. Davies, Org. Lett., 2012, 14, 6020 CrossRef CAS PubMed; (i) B. T. Parr, S. A. Green and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 4716 CrossRef CAS PubMed; (j) J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6802 CrossRef CAS PubMed; (k) Y. Z. Zhao, H. B. Yang, X. Y. Tang and M. Shi, Chem.–Eur. J., 2015, 21, 3562 CrossRef CAS PubMed; (l) T. Ryu, Y. Baek and P. H. Lee, J. Org. Chem., 2015, 80, 2376 CrossRef CAS PubMed; (m) T. Miura, Y. Funakoshi and M. Murakami, J. Am. Chem. Soc., 2014, 136, 2272 CrossRef CAS PubMed; (n) S. Park, W. Yong, S. Kim and P. H. Lee, Org. Lett., 2014, 16, 4468 CrossRef CAS PubMed; (o) B. Seo, W. H. Jeon, J. Kim, S. Kim and P. H. Lee, J. Org. Chem., 2015, 80, 722 CrossRef CAS PubMed; (p) C. Kim, S. Park, D. Eom, B. Seo and P. H. Lee, Org. Lett., 2014, 16, 1900 CrossRef CAS PubMed; (q) E. Lee, T. Ryu, E. Shin, J. Son, W. Choi and P. H. Lee, Org. Lett., 2015, 17, 2470 CrossRef CAS PubMed; (r) C. Kim, Y. Park, S. Park and P. H. Lee, Adv. Synth. Catal., 2015, 357, 210 CrossRef CAS; (s) S. Kim, J. Mo, J. Kim, T. Ryu and P. H. Lee, Asian J. Org. Chem., 2014, 3, 926 CrossRef CAS; (t) J. M. Yang, C. Z. Zhu, X. Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 5142 CAS; (u) K. Chen, Z. Z. Zhu, Y. S. Zhang, X. Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 6645 CrossRef CAS PubMed; (v) Y. Shi and V. Gevorgyan, Org. Lett., 2013, 15, 5394 CrossRef CAS PubMed; (w) D. Yadagiri and P. Anbarasan, Org. Lett., 2014, 16, 2510 CrossRef CAS PubMed; (x) B. Rajagopal, C. Chou, C. Chung and P. Lin, Org. Lett., 2014, 16, 3752 CrossRef CAS PubMed; (y) D. J. Lee and E. J. Yoo, Org. Lett., 2015, 17, 1830 CrossRef CAS PubMed; (z) A. Boyer, Org. Lett., 2014, 16, 1660 CrossRef CAS PubMed.
- (a) H. Shang, Y. Wang, Y. Tian, J. Feng and Y. Tang, Angew. Chem., Int. Ed., 2014, 53, 5662 CrossRef CAS PubMed; (b) Y. Tian, Y. H. Wang, H. Shang, X. D. Xu and Y. F. Tang, Org. Biomol. Chem., 2015, 13, 612 RSC; (c) J. Feng, Y. H. Wang, Q. G. Li, R. W. Jiang and Y. F. Tang, Tetrahedron Lett., 2014, 55, 6455 CrossRef CAS; (d) Y. H. Wang, X. Q. Lei and Y. F. Tang, Synlett, 2015, 26, 2051 CrossRef; (e) Y. H. Wang, X. Q. Lei and Y. F. Tang, Chem. Commun., 2015, 51, 4507 RSC; (f) X. Q. Lei, L. B. Li, Y. P. He and Y. F. Tang, Org. Lett., 2015, 17, 5224 CrossRef CAS PubMed.
- CCDC 1448884 (11a).†.
- While the one-pot three-steps reaction of 12a could be conducted using 1,2-DCE as the sole solvent, a slightly inferior yield (53% yield) was obtained.
- J. Dong, X. X. Shi, J. J. Yan, J. Xing, Q. Zhang and S. Xiao, Eur. J. Org. Chem., 2010, 6987 CrossRef CAS.
- (a) T. Choshi, T. Kuwada, M. Fukui, Y. Matsuya, E. Sugino and S. Hibino, Chem. Pharm. Bull., 2000, 48, 108 CrossRef CAS PubMed; (b) T. Choshi, Y. Matsuya, M. Okita, K. Inada, E. Sugino and S. Hibino, Tetrahedron Lett., 1998, 39, 2341 CrossRef CAS.
- Of note, only 1H NMR data of 13 was provided in ref. 14. However, it seemed that the documented data was incorrect. The structure of 13 obtained in our hands was unambigously confirmed by extensive spectroscopic study as well as its transformation into other well-known compound (for details, see ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and spectral data for all products were given. CCDC 1448884. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra02923j |
| This journal is © The Royal Society of Chemistry 2016 |