
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDispersion, solvent and metal effects in the binding of gold cations to alkynyl ligands: implications for Au(I) catalysis†
Luisa
Ciano
a,
Natalie
Fey
 *b,
Connor J. V.
Halliday
a,
Jason M.
Lynam
*a,
Lucy M.
Milner
a,
Nimesh
Mistry
a,
Natalie E.
Pridmore
a,
Nell S.
Townsend
b and
Adrian C.
Whitwood
a
*b,
Connor J. V.
Halliday
a,
Jason M.
Lynam
*a,
Lucy M.
Milner
a,
Nimesh
Mistry
a,
Natalie E.
Pridmore
a,
Nell S.
Townsend
b and
Adrian C.
Whitwood
a
aDepartment of Chemistry, University of York, York, YO10 5DD, UK. E-mail: jason.lynam@york.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: natalie.fey@bristol.ac.uk
First published on 15th May 2015
Abstract
The coordination modes of the [Au(PPh3)]+ cation to metal alkynyl complexes have been investigated. On addition to ruthenium, a vinylidene complex, [Ru(η5-C5H5)(PPh3)2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCPh{AuPPh3})]+, is obtained while addition to a gold(III) compound gives di- and trinuclear gold complexes depending on the conditions employed. In the trinuclear species, a gold(I) cation is sandwiched between two gold(III) alkynyl complexes, suggesting that coordination of multiple C–C triple bonds to gold is facile.
CCPh{AuPPh3})]+, is obtained while addition to a gold(III) compound gives di- and trinuclear gold complexes depending on the conditions employed. In the trinuclear species, a gold(I) cation is sandwiched between two gold(III) alkynyl complexes, suggesting that coordination of multiple C–C triple bonds to gold is facile.
Gold catalysts are effective reagents for activating multiple bonds towards intramolecular cycloaddition and nucleophilic attack. In addition to the growing number of synthetic applications of gold complexes in both the +1 and +3 oxidation states,1 there is clear evidence that binding of two gold atoms to the unsaturated substrate (diauration) plays an important role in catalysis.2 In the case of binding to arenes or alkenyl groups, two gold atoms may bind to form a “gem-diaurate” A,2a,3 terminal alkynes may be activated by formation of σ–π complex, B,4 and it has been proposed that vinylidene complexes, C,5 (Fig. 1) are key intermediates in the cyclisation of diynes. Indeed, all three binding modes have been proposed within a single catalytic cycle,6 illustrating the flexibility of digold coordination to support key intermediates in C–C bond forming reactions.
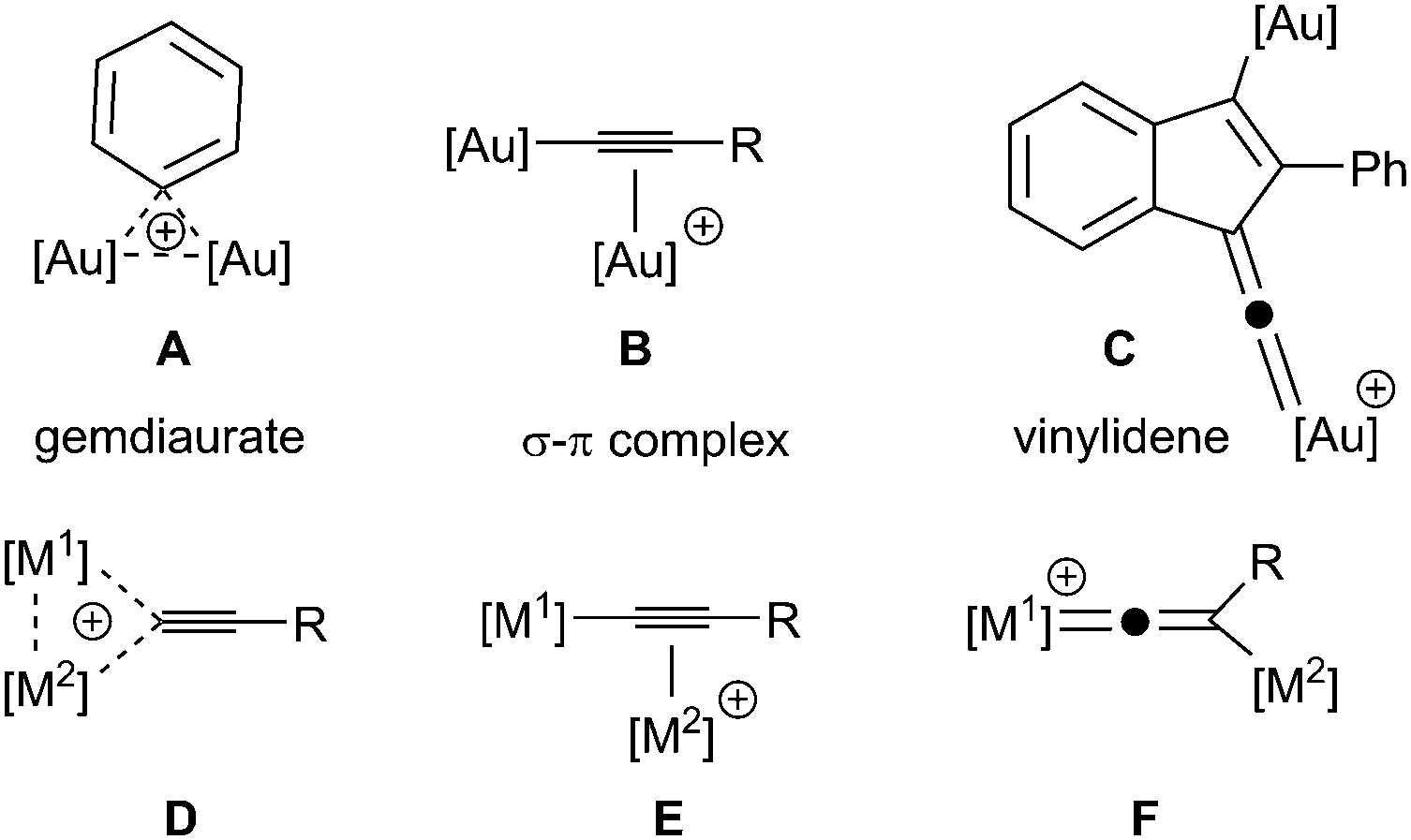 | ||
| Fig. 1 Binding modes of gold to unsaturated substrates. | ||
Vinylidene complexes are important intermediates in a number of transition metal-mediated processes,7 notably those involving the functionalisation of terminal alkynes, although typically these reactions have involved elements from groups 6–9.7,8 In these cases, the conversion of alkynes to their vinylidene tautomers has been shown to generate stable complexes, whereas this is not the case for gold.9 There is, however, a continuum in behaviour between gem-diaurate-type structures, D, dinuclear σ–π complexes, E, and vinylidenes, F, which differ only in terms of the relationship between the position of [M2] and the alkynyl system. In the case where [M1] = Au, no structures of type F have been isolated and in the solid state σ–π complexes E are observed and D is a probable transition state for the exchange of gold atoms.4f However, evidence for mono-nuclear gold vinylidene complexes as intermediates in metal-catalysed reactions has been presented recently.10 Insight into how a given combination of [M1] and [M2] affects relative stability of forms D, E and F will aid in the selection and optimisation of catalysts promoting reaction pathways involving these intermediates.
It was envisaged that by making the isolobal substitution of a proton by [Au(PPh3)]+ a vinylidene complex of type F with [M1] = Ru, [M2] = Au, [Ru(η5-C5H5)(PPh3)2(CCH{AuPPh3})]+ would be a viable target. As complexes [Ru(η5-C5H5)(PPh3)2(CCHR)]+ may be prepared from protonation of alkynyl complexes [Ru(η5-C5H5)(PPh3)2(–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)],11 a related reaction with “[Au(PPh3)]+” would provide a straightforward route to the desired complex.
CR)],11 a related reaction with “[Au(PPh3)]+” would provide a straightforward route to the desired complex.
Addition of the cationic species [Au(PPh3)]SbF6, [2]SbF6, (generated in situ from the reaction of [AuCl(PPh3)] with AgSbF6) to [Ru(η5-C5H5)(PPh3)2(–CCPh)], [1], in CH2Cl2 solution resulted in a colour change from yellow to orange (Scheme 1). Addition of Et2O gave an orange precipitate identified as [Ru(η5-C5H5)(PPh3)2(CCPh{AuPPh3})]SbF6, [3]SbF6. The 13C{1H} NMR spectrum of [3]+ exhibited resonances typical of a vinylidene ligand, with a triplet observed for the ruthenium-bound carbon atom at δ 354.1 (t, 2JPC = 15.7 Hz), the β-carbon atom was observed at δ 118.8 (d, 2JPC = 15.4 Hz) with the coupling presumably arising from the single PPh3 ligand bound to gold. A peak in the ESI-MS at m/z 1251.2225 with the correct isotope pattern confirmed the presence of a complex containing both ruthenium and gold atoms as well as the additional ancillary ligands.
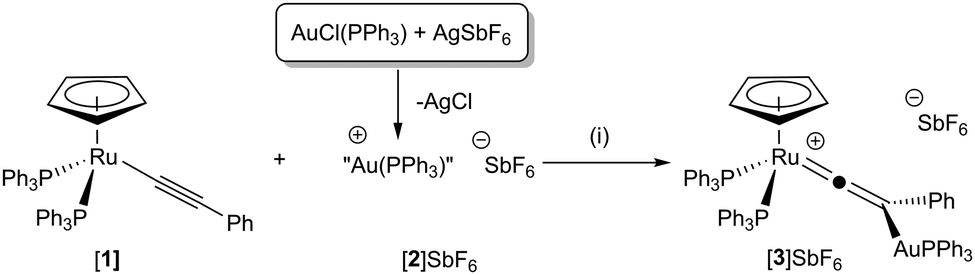 | ||
| Scheme 1 (i) CH2Cl2 solution, 1 h, r.t. | ||
The postulated structure of [3]+ was further supported by DFT calculations (see ESI† for computational details), finding geometry F lowest in energy, with E 19 kJ mol−1 higher in energy, while optimisation of D remained unsuccessful; with restraints, this lies 90 kJ mol−1 above F (Table 1). The formation of [3]SbF6 demonstrates that it is possible to form vinylidene complexes F with [M1] = Ru and [M2] = Au and that the reaction with [Au(PPh3)]+ is analogous to the addition of other electrophiles to [1].11,12
For gold(I) alkynyl complexes with coordinated gold cations, the σ–π binding of type E is observed most frequently in the solid state,4 and rapid exchange of environments through a low energy gem-diaurate species D has been proposed.4f Indeed, an examination of the structure of σ–π complexes, E, indicates that in most cases a distortion is observed, with the π-bound gold atom being closer to the gold-bound carbon of the alkynyl group (see Table S2.5†). Furthermore, our calculations (Table 1) indicate that when [M1] = [M2] = Au(PPh3) the gem-diaurate complex D is actually lower in energy than E. For this complex, powder X-ray diffraction experiments predict an Au–Au distance of 3.08 Å,13 in agreement with the calculated structure of D (3.09 Å); in DFT-calculated structure E it is predicted to be 3.24 Å.
The effect of the gold oxidation state on the relationship between D, E, and F has not been explored so far. Our recent calculations have indicated that d8 systems are better at stabilising vinylidene ligands than their d10 analogues, so gold(III) might be more effective at stabilising vinylidene ligands than gold(I).9a Further calculations, (Table S3.3†) using propyne as a model, also suggested that a smaller energy difference between alkyne and vinylidene tautomers could be achieved for gold(III) than for gold(I). However, the calculated predictions for other substitution patterns were less clear, with carbocationic species observed instead of alkynes (Table S3.3†). Comparing the three binding modes computationally for two gold complexes (Table 1), the simple gold(I) alkynyl favours D, while the gold(III) complex favours E, and F becomes comparatively less unfavourable. Experimental confirmation of the favoured form would provide validation for our computational predictions, as well as giving insight into the role of [M1] in determining the relative preference.
The cyclometallated gold(III) alkynyl complex [Au(tBuC^N^CtBu)(CCPh)], [4], previously prepared by Yam and co-workers, was selected as a precursor for this study (Scheme 2).14 Bochmann has demonstrated that this pincer ligand is effective at stabilising gold complexes with hydride15 and η2-alkene ligands.16 Tilset and co-workers have shown that cationic Au(III) alkene complexes may be prepared,17 and that a cyclometallated gold(III) complex may promote the insertion of ethene into a metal–oxygen bond.18
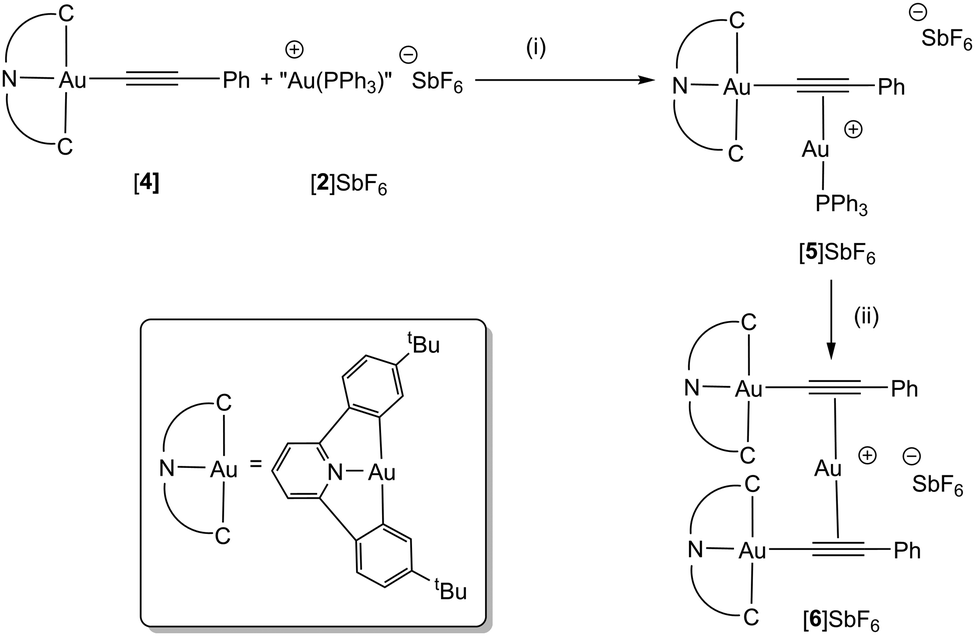 | ||
| Scheme 2 (i) Toluene 16 h, r.t., (ii) CH2Cl2, 16 h, r.t. – [Au(PPh3)2]SbF6. | ||
Reaction of a toluene solution of [4] with [2]SbF6 resulted in the formation of a yellow precipitate of the Au(III)–Au(I) σ–π complex, [5]SbF6 (Scheme 2). Although the poor solubility of [5]SbF6 in C6D6 and subsequent reactivity observed in CD2Cl2 (q.v.) precluded a detailed NMR study, the identity of the complex was confirmed by HR-MS (where a peak for the molecular ion was observed) and single crystal X-ray diffraction of the C6D6 (Fig. 2a) and C6H5CH3† solvates.
 | ||
| Fig. 2 X-ray structure of (a) the [5]+ cation and (b) the [6]+ cation. ADPs are shown at the 50% probability level, hydrogen atoms are omitted for clarity. | ||
Repeating the reaction but using CH2Cl2 as solvent for [4], or dissolving isolated [5]SbF6 in CD2Cl2, resulted in a colour change from yellow to green, indicating formation of a different product, identified as [6]SbF6 (Scheme 2). Slow diffusion of Et2O into these solutions afforded green crystals suitable for study by X-ray diffraction. The resulting structural determination (Fig. 2b) demonstrated that an unusual reaction had occurred to give a trimetallic complex, [6]SbF6, in which a single Au(I) cation was sandwiched between two Au(III) alkynyl groups. In the 13C{1H} NMR spectrum, resonances for the two alkynyl carbon atoms were observed at δ 92.8 (Au–C) and δ 114.8 (CC–Ph) that exhibited small coordination shifts (Δδ 0.1 and 13.4 respectively, identified with the aid of a 13C-enriched sample). A peak in the ESI-MS of [6]SbF6 at m/z 1475.4042, with the correct isotopic distribution pattern, confirmed the presence of the trinuclear framework. Analysis of the supernatant demonstrated that [Au(PPh3)2]+ has been formed, accounting for the mass balance of the reaction.
In the structure of [6]+ observed crystallographically, the Au(I) cation is bound asymmetrically to the two alkynyl ligands; in both cases the Au(I) is closer to the Au(III)-coordinated carbon atom, but one of these interaction is more marked than any example reported to date [Au(1)–C(1) 2.145(4) Å, Au(1)–C(2) 2.357(4) Å]. In [5]+ and [6]+ there are intermolecular π–π interactions between the C^N^C-pincer ligands, however, in the case of [6]+ there is also evidence for intramolecular interactions between the pyridyl and aryl groups (Table S2.4†). It seems likely that these intramolecular π–π interactions (along with the formation of [Au(PPh3)2]+) are favouring formation of the trinuclear gold complex.
To the best of our knowledge [6]+ is the first example of a species with three gold atoms in a “σ–π2–σ-complex”.19 A single gold(I) binding two alkyne ligands has been reported with the strained cyclooctyne,20 and when a preorganised supramolecular diyne is used.21 In addition, a structure in which a gold(I) cation is coordinated between two alkyne units has been shown to be a transition state for the exchange of gold between the two triple bonds.5d This, together with the analysis of complexes of form D and E, suggests that the potential energy surface for the binding of gold to alkynes is likely to be flat, with small perturbations determining whether a given species is a minimum or a transition state. Therefore, the factors controlling the coordination of gold to alkynes were investigated in more detail.
Calculations of simple fragment binding energies illustrate that the binding of [Au(PPh3)]+ to [4], giving [5]+, is more favourable than binding to benzene and PhCCH (Table S3.4†) reinforcing the notion that binding of gold cations to alkynyl ligands is more favourable than binding to alkynes.3a The calculations also show clear energy differences between the binding of benzene and CH2Cl2, helping to explain the observed experimental solvent effect.
The difference in the calculated energies of [5]+ and [6]+ is small and dispersion corrections favour [6]+ slightly (by 4–6 kJ mol−1), while [5]+ is favoured if such corrections are not included (Tables S3.5 and S3.6†). The effects of dispersion corrections on the thermodynamic preference for [6]+ support the hypothesis that intramolecular π–π interactions are responsible for favouring the formation of the trinuclear scaffold.
We have also explored the energy balance between the σ–π complex E (for some complexes, this optimises to the gem complex D, discussed in greater detail in the ESI†) and the trinuclear gold(I) complex H with a range of different substituents and ligands (Scheme 3, Table 2 and Table S3.8†). Remarkably, the calculations predict that for other compounds with gold in the +1 oxidation state, σ–π2–σ complexes H might also be accessible. Indeed, the predicted energy differences between these species and D/E, are small and probably within the limits of reliability of the theoretical method used. The calculations therefore predict that it should be possible to observe complexes of type H in other systems.
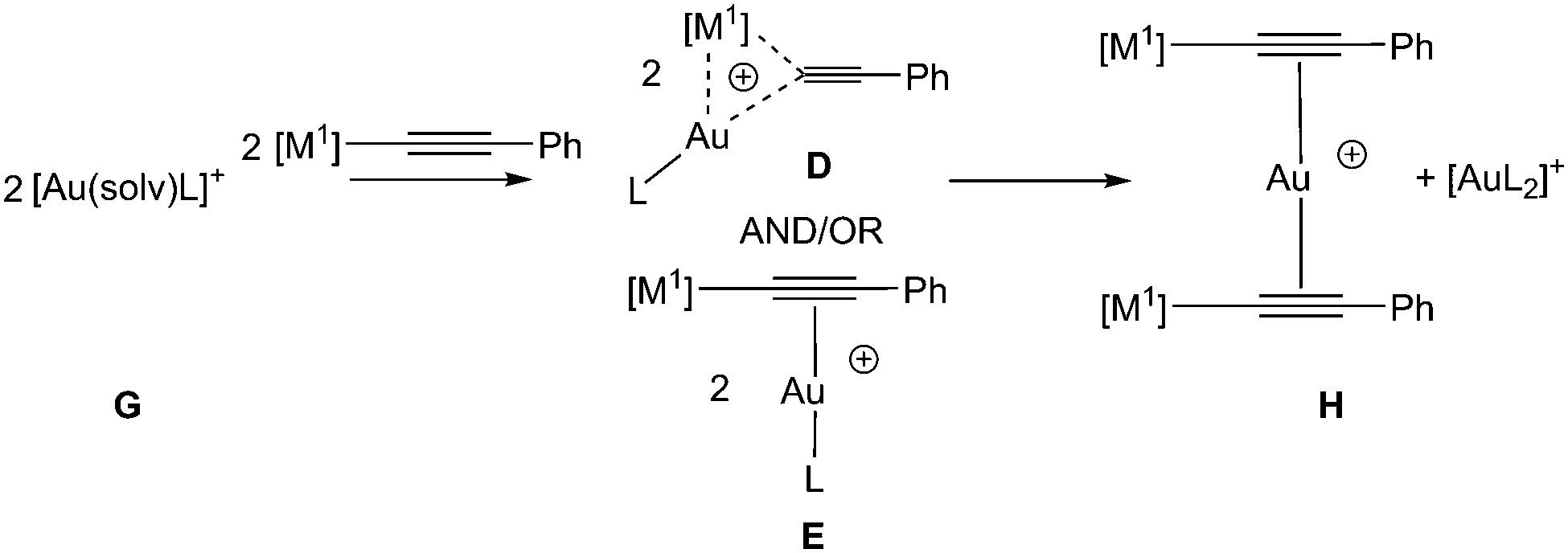 | ||
| Scheme 3 Model reaction used to explore preference for gold(I) complexes. | ||
| Solvent | L | [M1] | G | D/E | H | D/E–H |
|---|---|---|---|---|---|---|
| C6H6 | PPh3 | Au(tBuC^N^CtBu) | 0 | −207 | −211 | +4 |
| CH2Cl2 | PPh3 | Au(tBuC^N^CtBu) | 0 | −234 | −240 | +6 |
| C6H6 | PPh3 | H | 0 | −47 | −43 | −4 |
| CH2Cl2 | PPh3 | H | 0 | −100 | −99 | −1 |
| C6H6 | PPh3 | Au(PPh3) | 0 | −200 | −192 | −8 |
| CH2Cl2 | PPh3 | Au(PPh3) | 0 | −219 | −213 | −6 |
| C6H6 | PPh3 |
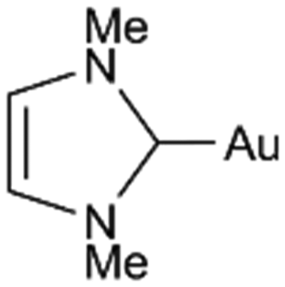
|
0 | −184 | −196 | +12 |
| CH2Cl2 | PPh3 |
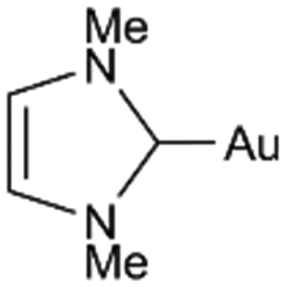
|
0 | −202 | −216 | +14 |
| C6H6 | PPh3 | Au(IPr) | 0 | −225 | −211 | −14 |
| CH2Cl2 | PPh3 | Au(IPr) | 0 | −233 | −223 | −10 |
To test this hypothesis, a sample of [Au(–CCPh)(PPh3)], [7], was treated with [2]SbF6 in CD2Cl2 solution. An ESI-MS spectrum of the reaction mixture confirmed the presence of [Au(PPh3)2]+ and an intense peak at m/z 1019.1556 was consistent with the formation of either D or E with [M1] = [M2] = Au(PPh3). A less intense peak at m/z 1317.1643 was assigned to the trinuclear complex H ([M1] = Au(I){PPh3}), supporting predictions from the DFT calculations that the trinuclear framework may not be limited to the gold(III) example. The 31P{1H} NMR spectrum exhibited a sharp resonance for [Au(PPh3)2]+ and a very broad feature at δ 37.5 which on cooling to 195 K decoalesced to a number of new resonances. Repeating the reaction with a 13C-enriched sample, [Au(–13CCPh)(PPh3)], [7-13C], resulted in a number of these resonances exhibiting P–C couplings (Section S1.8†). Although the NMR data do not unambiguously demonstrate the formation of a trinuclear complex, they do indicate that a number of gold alkynyl phosphine complexes are present and undergoing rapid exchange. A detailed investigation of this behaviour is currently underway.
These results have demonstrated that the key factor controlling the preference between isomers D, E or F is the nature of [M1]. Presumably, the more π-basic ruthenium(II) and crowded environment in [3]+ effectively stabilize the vinylidene ligand, whereas (at least in the systems explored to date) neither Au(I) nor Au(III) are suitably π-basic and/or bulky. The observation of trinuclear complexes for both gold(I) and gold(III) complexes raises the possibility that coordination by more than two gold atoms to a substrate may be involved in catalytic processes. In the case of the gold(III) complexes, dispersion interactions between the two pincer ligands ensure that the trinuclear complex is the lowest energy structure, and although these effects are not as pronounced in the gold(I) analogue, there is only a small difference in energy between complexes of type D/E and H. Furthermore, our data suggest that the solvent may play a key role in determining the nuclearity of gold-containing complexes and this should also be considered as an important factor in catalysis.
We thank the University of York (pump-prime funding) and the EPSRC (studentship to LMM) for funding and the Centre for Computational Chemistry, University of Bristol, for computing resources. We are grateful to Professors Paul Low and George Koutsantonis (University of Western Australia) for helpful discussions.
During the review of the manuscript, evidence for a gold(I) vinylidene has been reported.22
Notes and references
- (a) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592 CrossRef CAS PubMed; (b) G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112 CrossRef CAS PubMed; (c) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2013, 136, 254 CrossRef PubMed; (d) W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159 CrossRef CAS PubMed.
- (a) A. Gomez-Suarez and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 8156 CrossRef CAS PubMed; (b) D. Weber and M. R. Gagne, Chem. Sci., 2013, 4, 335 RSC; (c) I. Braun, A. M. Asiri and A. S. K. Hashmi, ACS Catal., 2013, 3, 1902 CrossRef CAS; (d) C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16 RSC; (e) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864 CrossRef CAS PubMed.
- (a) P. H.-Y. Cheong, P. Morganelli, M. R. Luzung, K. N. Houk and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 4517 CrossRef CAS PubMed; (b) S. Flugge, A. Anoop, R. Goddard, W. Thiel and A. Furstner, Chem. – Eur. J., 2009, 15, 8558 CrossRef PubMed; (c) D. Weber, M. A. Tarselli and M. R. Gagne, Angew. Chem., Int. Ed., 2009, 48, 5733 CrossRef CAS PubMed; (d) H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2 CrossRef CAS.
- (a) T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313 RSC; (b) A. Himmelspach, M. Finze and S. Raub, Angew. Chem., Int. Ed., 2011, 50, 2628 CrossRef CAS PubMed; (c) T. J. Brown and R. A. Widenhoefer, J. Organomet. Chem., 2011, 696, 1216 CrossRef CAS PubMed; (d) T. J. Brown and R. A. Widenhoefer, Organometallics, 2011, 30, 6003 CrossRef CAS; (e) A. Grirrane, H. Garcia, A. Corma and E. Álvarez, ACS Catal., 2011, 1, 1647 CrossRef CAS; (f) R. E. M. Brooner, T. J. Brown and R. A. Widenhoefer, Chem. – Eur. J., 2013, 19, 8276 CrossRef CAS PubMed; (g) R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714 CrossRef CAS PubMed; (h) A. S. K. Hashmi, T. Lauterbach, P. Noesel, M. H. Vilhelmsen, M. Rudolph and F. Rominger, Chem. – Eur. J., 2013, 19, 1058 CrossRef CAS PubMed; (i) C. Obradors and A. M. Echavarren, Chem. – Eur. J., 2013, 19, 3547 CrossRef CAS PubMed; (j) A. Gomez-Suarez, S. Dupuy, A. M. Z. Slawin and S. P. Nolan, Angew. Chem., Int. Ed., 2013, 52, 938 CrossRef CAS PubMed; (k) A. Grirrane, H. Garcia, A. Corma and E. Alvarez, Chem. – Eur. J., 2013, 19, 12239 CrossRef CAS PubMed; (l) L. Jasikova and J. Roithova, Organometallics, 2013, 32, 7025 CrossRef CAS; (m) A. Homs, C. Obradors, D. Lebœuf and A. M. Echavarren, Adv. Synth. Catal., 2014, 356, 221 CrossRef CAS PubMed; (n) A. Gimeno, A. B. Cuenca, S. Suarez-Pantiga, C. R. de Arellano, M. Medio-Simon and G. Asensio, Chem. – Eur. J., 2014, 20, 683 CrossRef CAS PubMed.
- (a) L. W. Ye, Y. Z. Wang, D. H. Aue and L. M. Zhang, J. Am. Chem. Soc., 2012, 134, 31 CrossRef CAS PubMed; (b) A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644 CrossRef CAS; (c) M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 2593 CrossRef CAS PubMed; (d) M. H. Vilhelmsen and A. S. K. Hashmi, Chem. – Eur. J., 2014, 20, 1901 CrossRef CAS PubMed; (e) J. Bucher, T. Stößer, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 1666 CrossRef CAS PubMed; (f) P. Nösel, V. Müller, S. Mader, S. Moghimi, M. Rudolph, I. Braun, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2015, 357, 500 CrossRef PubMed; (g) J. Bucher, T. Stosser, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 1666 CrossRef CAS PubMed.
- K. Graf, P. D. Hindenberg, Y. Tokimizu, S. Naoe, M. Rudolph, F. Rominger, H. Ohno and A. S. K. Hashmi, ChemCatChem, 2014, 6, 199 CrossRef CAS PubMed.
- (a) B. M. Trost and A. McClory, Chem. – Asian J., 2008, 3, 164 CrossRef CAS PubMed; (b) J. M. Lynam, Chem. – Eur. J., 2010, 16, 8238 CrossRef CAS PubMed.
- (a) B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630 CrossRef CAS PubMed; (b) C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2006, 45, 2176 CrossRef CAS PubMed; (c) D. G. Johnson, J. M. Lynam, N. S. Mistry, J. M. Slattery, R. J. Thatcher and A. C. Whitwood, J. Am. Chem. Soc., 2012, 135, 2222 CrossRef PubMed.
- (a) M. M. Hansmann, F. Rominger and A. S. K. Hashmi, Chem. Sci., 2013, 4, 1552 RSC; (b) O. J. S. Pickup, I. Khazal, E. J. Smith, A. C. Whitwood, J. M. Lynam, K. Bolaky, T. C. King, B. W. Rawe and N. Fey, Organometallics, 2014, 33, 1751 CrossRef CAS.
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569 CrossRef CAS PubMed.
- M. I. Bruce and R. C. Wallis, Aust. J. Chem., 1979, 32, 1471 CrossRef CAS.
- (a) M. I. Bruce, A. G. Swincer and R. C. Wallis, J. Organomet. Chem., 1979, 171, C5 CrossRef CAS; (b) M. I. Bruce and A. G. Swincer, Aust. J. Chem., 1980, 33, 1471 CrossRef CAS.
- V. L. Korsunsky, K. I. Grandberg, E. I. Smyslova and T. V. Baukova, J. Organomet. Chem., 1987, 335, 277 CrossRef.
- (a) K. M.-C. Wong, L.-L. Hung, W. H. Lam, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 4350 CrossRef CAS PubMed; (b) V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107 CrossRef CAS PubMed.
- D.-A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643 CrossRef PubMed.
- N. Savjani, D.-A. Roşca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874 CrossRef CAS PubMed.
- E. Langseth, M. L. Scheuermann, D. Balcells, W. Kaminsky, K. I. Goldberg, O. Eisenstein, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2013, 52, 1660 CrossRef CAS PubMed.
- E. Langseth, A. Nova, E. A. Tråseth, F. Rise, S. Øien, R. H. Heyn and M. Tilset, J. Am. Chem. Soc., 2014, 136, 10104 CrossRef CAS PubMed.
- Examples of gold bridging ruthenium and rhenium alkynyl complexes have been reported see C. S. Griffith and G. E. Koutsantonis, Aust. J. Chem., 2012, 65, 698 CrossRef CAS and G. A. Carriedo, D. Miguel, V. Riera and X. Solans, J. Chem. Soc., Dalton Trans., 1987, 2867 RSC respectively.
- (a) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940 CrossRef CAS PubMed; (b) A. Das, C. Dash, M. A. Celik, M. Yousufuddin, G. Frenking and H. V. R. Dias, Organometallics, 2013, 32, 3155 CrossRef.
- S.-K. Yip, E. C.-C. Cheng, L.-H. Yuan, N. Zhu and V. W.-W. Yam, Angew. Chem., Int. Ed., 2004, 43, 4957 CrossRef PubMed.
- R. J. Harris and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201501474.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, X-ray structure determinations including a comparison of structural metrics of this class of compounds, further discussion of computational data including a more complete evaluation of method effects and xyz coordinates of optimised geometries. CCDC 1032551–1032553. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc02629f |
| This journal is © The Royal Society of Chemistry 2015 |