
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst durability in electrocatalytic H2O2 production: key factors and challenges†
Ji Sik
Choi
ab,
Guilherme V.
Fortunato
 *abc,
Daniele C.
Jung
a,
Julio C.
Lourenço
bc,
Marcos R. V.
Lanza
c and
Marc
Ledendecker
*bd
*abc,
Daniele C.
Jung
a,
Julio C.
Lourenço
bc,
Marcos R. V.
Lanza
c and
Marc
Ledendecker
*bd
aDepartment of Technical Chemistry, Technical University of Darmstadt, Peter-Grünberg-Straße 8, 64287 Darmstadt, Germany. E-mail: g.fortunato@tum.de
bSustainable Energy Materials, Technical University Munich, Campus Straubing, Schulgasse 22, 94315 Straubing, Germany. E-mail: marc.ledendecker@tum.de
cSão Carlos Institute of Chemistry, University of São Paulo, Avenida Trabalhador São-Carlense 400, São Carlos, SP 13566-590, Brazil
dHelmholtz Institute Erlangen-Nürnberg for Renewable Energy (IEK-11), Forschungszentrum Jülich GmbH, Cauerstr. 1, 91058 Erlangen, Germany
First published on 29th May 2024
Abstract
On-demand electrocatalytic hydrogen peroxide (H2O2) production is a significant technological advancement that offers a promising alternative to the traditional anthraquinone process. This approach leverages electrocatalysts for the selective reduction of oxygen through a two-electron transfer mechanism (ORR-2e−), holding great promise for delivering a sustainable and economically efficient means of H2O2 production. However, the harsh operating conditions during the electrochemical H2O2 production lead to the degradation of both structural integrity and catalytic efficacy in these materials. Here, we systematically examine the design strategies and materials typically utilized in the electroproduction of H2O2 in acidic environments. We delve into the prevalent reactor conditions and scrutinize the factors contributing to catalyst deactivation. Additionally, we propose standardised benchmarking protocols aimed at evaluating catalyst stability under such rigorous conditions. To this end, we advocate for the adoption of three distinct accelerated stress tests to comprehensively assess catalyst performance and durability.
1. Introduction
Electrochemical devices powered by sustainable energy sources offer certain advantages over traditionally fossil fuel-based chemical production methods as they can be operated on-demand and in a decentralized manner.1,2 At the heart of the technology are electrocatalysts facilitating electrochemical reactions by providing pathways for electrons to move between electrodes and reactants.3 Seen as a significant technological advancement for many industrial, commercial and domestic end-users, the on-demand electrocatalytic hydrogen peroxide (H2O2) production has gained significant attention as a promising alternative to the traditional anthraquinone process. The former process involves the selective 2-electron oxygen-reduction (ORR-2e−) and a multitude of active and selective catalysts have been proposed in literature enabling on-demand H2O2 electrosynthesis.4,5 However, the critical aspect of stability, crucial for industrial viability, is frequently overlooked or inadequately assessed, if considered at all.Catalyst performance and degradation processes can significantly vary based on the reaction environment and the specific catalyst used particularly due to the low pH values, negative applied potentials and current densities.6 Here, we methodically explore commonly employed materials and design strategies for the electroproduction of H2O2 under low pH conditions. In order to understand the conditions, the catalysts have to overcome, we delve into currently discussed reactor configurations and analyse the factors that lead to catalyst deactivation. By doing so, we present standardised benchmarking protocols intended to evaluate catalyst stability in these demanding environments.
2. Electrocatalysts for H2O2 production
Given the extensive array of electrocatalysts available for hydrogen peroxide production, our initial focus is on introducing the catalysts most frequently utilised in the acidic oxygen reduction reaction for generating H2O2. The most commonly used catalyst materials are displayed in Fig. 1(B) and (C). As these catalysts exhibit diverse intrinsic properties, the prevalence of distinct degradation mechanisms is strongly influenced by the specific catalytic material in use. In general, the main classes of electrocatalysts for H2O2 production include metal-free carbon-based materials and transition metal-based materials that are usually supported on carbon. In acidic environments, platinum group metal electrocatalysts in particular Pt and Pd based, demonstrated the highest ORR exchange current densities.5 However, extended surfaces cleave the O–O bond, following a 4e− pathway toward water production as shown in Fig. 1(A). To achieve selective ORR-2e−, active sites have been isolated, preventing neighbouring sites from splitting the O–O bond. One approach to create these isolated active sites involves blending active metals, such as Pt and Pd, with metals possessing low oxygen affinity, such as Hg, Au, or Ag.4,7,8 Even though the specific nature of metal/metal interactions is occasionally ambiguous. In the case of the Pt/Hg system, Hg is electrodeposited on the Pt surface while in other cases solid solutions are formed. In all cases, the employed metal composition is rather poor in the active metal such as Pt or Pd in order to prevent overreduction to H2O.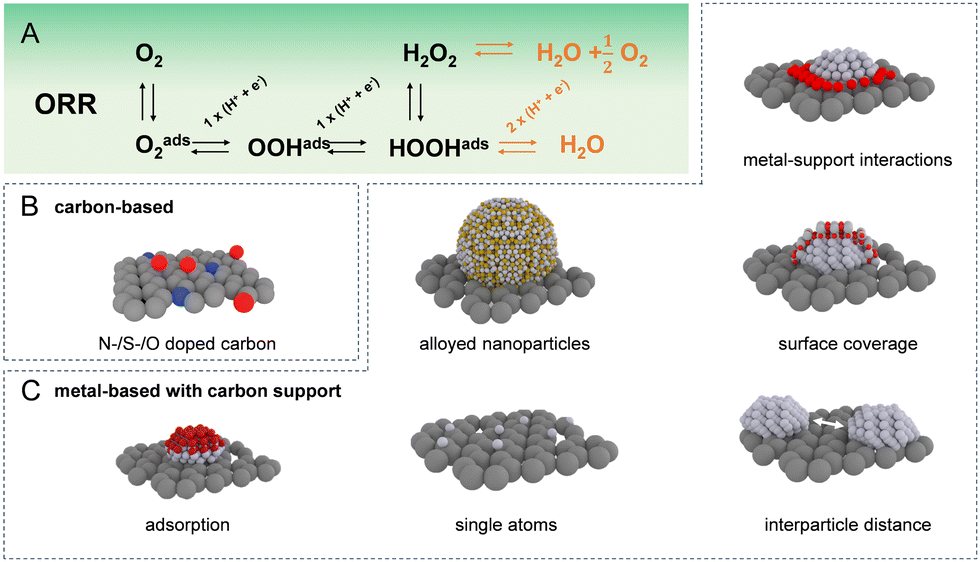 | ||
| Fig. 1 Catalysts and strategies for oxygen reduction reaction optimisation. (A) Mechanism of oxygen reduction reaction leading to the formation of both H2O2 and H2O. Commonly utilized catalysts include (B) carbon-based catalysts, which consist of (doped) carbonaceous materials, and (C) metal-based materials typically supported on high surface area carbon substrates. The different strategies to mitigate overreduction to H2O encompass site blocking by adsorption or surface coverage, altering the interparticle distance, using single atoms and alloyed nanoparticles as well as altering the binding energies by metal-support interactions. | ||
Active suppression of H2O2 re-adsorption through control of overall metal loading9–13 and interparticle distance12 has resulted in selectivities exceeding 90% in a rotating ring disk electrode configuration. Another approach is blocking Pt or Pd active sites using less active materials, for example, amorphous carbon,14 polymers,15,16 oxides,17 and polyatomic ions18 to achieve similar isolation of active sites. Equally, atomically dispersed metal sites, known as single-atom catalysts (SACs), supported on inert substrates have been proposed, where the coordination environment of the support matrix can tune the catalytic activity and selectivity.19–24 The trend towards downsizing metal catalysts from extended surfaces to isolated single-atom sites necessitates a meticulous re-evaluation of the metal-support interaction. As particle size shrinks, the interface between the metal and the support becomes increasingly dominant, significantly impacting catalyst activity and stability.25 This is critical since lower-coordinated metal atoms in a nanoparticle exhibit increased surface energy, making them more susceptible to degradation mechanisms such as dissolution and Ostwald ripening.26–28 For SACs, precisely controlling the interaction between the isolated metal atoms and the supporting material is paramount. This strong metal-support interaction directly governs the stability of the catalyst, the nature of its active sites, and ultimately the types of reactions it can facilitate.
When doped with nitrogen, sulfur, or oxygen, carbon supports enhance the physicochemical properties of catalysts by improving nanoparticle dispersion and stabilising single atoms during durability tests.21,29–31 These functional groups enable both, noble and non-noble metals such as Co, Ni, or Fe to remain stable in acidic media.32–34 For instance, Jiajun et al. maintained about 70% selectivity for O2 to H2O2 conversion with a Pt-S-CNT catalyst over 500 minutes of continuous electrolysis at 10 mA cm−2, with 0.4 ppb Pt leaching as confirmed by ICP-MS.35 The role of CoN4 coordination structures in the ORR pathway continues to be a subject of debate.34,36,37 Chen et al. found that pyrrole-type CoN4 primarily supports the ORR-2e−, maintaining stability for 90 hours at −50 mA in a flow cell, while pyridine-type CoN4 catalyzes the ORR-4e−. After stability testing, X-ray diffraction and electron microscopy analyses detected no Co nanoparticle aggregation.38 Huang et al. enhanced the long-term stability of a diatomic cobalt catalyst by integrating a second metal into cobalt's coordination sphere, achieving stability for 100 h at current densities of 400 mA cm−2 in a flow cell, optimising the binding strength of critical H2O2 intermediates.39
Bare carbon-based materials have been reported to be highly selective for ORR-2e−,4,7,40–42 though their activity is low, likely due to the weak interaction between carbon and *OOH. To boost the activity of carbon catalysts, additional treatments are effective, such as incorporating anthraquinone molecules,43,44 trying to emulate the anthraquinone process, or functionalizing carbon with nitrogen or oxygen groups.45–48 Oxidized or nitrogen-doped versions of carbon materials such as carbon black, carbon nanotubes, mesoporous carbon, and graphene, distinguished by their large surface areas and defect-rich structures, exhibit enhanced oxygen adsorption and activation, significantly boosting their catalytic efficacy compared with undoped carbon materials. While designated as “metal-free” catalysts, there is a high likelihood that these entities serve as anchor sites for prevalent transition metals such as iron, likely contributing to the overall activity.
It becomes clear that there is a plethora of material combinations that react to the applied reaction conditions. In electrocatalysis, degradation mechanisms are influenced by the applied conditions and the specific catalyst employed. Often, a mix of various degradation phenomena contributes to the observed overall deactivation, causing a loss in both catalytic activity and selectivity.6 The degradation mechanisms thereby heavily rely on the conditions the catalyst material faces which vary based on the electrochemical device being used. For more comprehensive insights on degradation pathways influenced by operating conditions and intrinsic material properties, readers may refer to other reviews.6,49–52 Operando techniques paired with a modified scanning flow cell system, including methods like ICP-MS, DEMS, Mössbauer spectroscopy, and Raman spectroscopy, can significantly enhance our understanding of the factors that influence catalyst degradation.53–59 In the subsequent section, we will present various configurations documented in literature and explore the potential conditions that the catalysts must withstand.
3. Identifying driving forces for catalyst deactivation in devices used for electrosynthesis of hydrogen peroxide
The electroproduction of H2O2 can be achieved in a variety of environments ranging from acidic, neutral, and alkaline media. However, the production in high pH media is less favourable, as the deprotonated form of hydrogen peroxide (HO2−) must be utilized immediately, limiting its storage capabilities and reducing its applicability.60,61 In the following discussion, we will focus on acidic media. Cation exchange membrane H2O2 fuel cells (H2O2-FC) stand out as a leading electrochemical cell, fulfilling a dual role: generating electricity while also yielding H2O2 as a valuable by-product.62,63 Initially employing O2-saturated liquid electrolytes on the cathode side as illustrated in Fig. 2(A), these cell designs face limitations such as low current densities restricted to a few mA cm−2, resulting in modest H2O2 accumulation at the μM per hour-scale.62 Efficient cell designs, which minimise the catalyst's exposure to generated peroxide, play a pivotal role in preventing unwanted side reactions that may consume the produced H2O2 and in extending the catalyst's lifespan, as will be shown later. It is feasible to generate significant amounts of H2O2 employing solid electrolytes and gas diffusion electrodes (Fig. 2(B)), with current densities reaching hundreds of mA cm−2.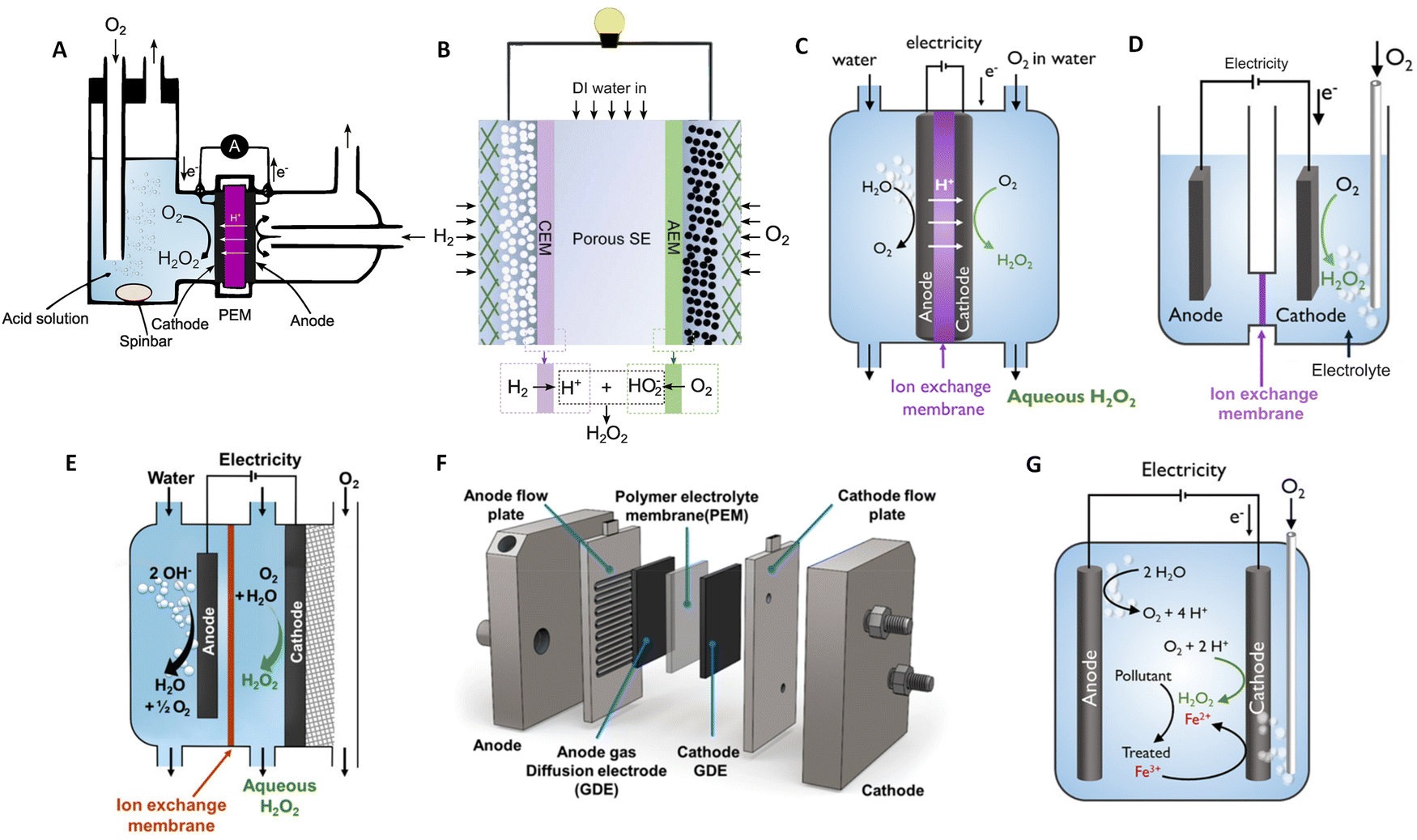 | ||
| Fig. 2 Schematic illustrations of H2O2-fuel cell and H2O2-electrolyser devices. (A) Liquid electrolyte-containing fuel cell. (B) Solid–electrolyte fuel cell with double membrane. (C) PEM electrolyser with submerged membrane electrode assembly (MEA). (D) H-cell with submerged electrodes. (E) Continuous flow reactor utilizing a gas diffusion electrode (GDE). (F) MEA-based flow reactor with a GDE anode and GDE cathode. (G) Non-dividing cell reactor for the electro-Fenton process. Panel A is adapted from ref. 62, Copyright 1990, with permission from Elsevier. Panel B is adapted from ref. 64. Copyright 2019, American Association for the Advancement of Science. Panels C, D, and G are adapted with permission from ref. 61, Copyright 2018, American Chemical Society. Panel E and F are adapted with permission from ref. 65, Copyright 2020, American Chemical Society. | ||
The potential applied poses a significant challenge to the catalyst's stability, particularly during start-stop. The operational potential, which must theoretically be lower than 0.7 VRHE, depends on the catalyst's activity,66 whereas the open circuit potential (OCP) can reach potentials around 0.9 VRHE, particularly for Pt- and Pd-based catalysts.67,68 The oscillation between operational potentials and OCP over time is expected to degrade the catalyst leading to electrocatalytic losses or changes in selectivity due to processes such as (transient) dissolution or leaching of the catalytic metal sites, support dissolution, detachment, or particle agglomeration.69–72 Non-noble metals such as Ni, Co, Mo, or W have been reported to be prone to dissolution at OCP upon uncontrolled immersion into the electrolyte.73
Potential challenges may encompass catalyst poisoning, which can be intensified depending on the electrochemical device used, for example, when another oxidation reaction on the anode side is used such as e.g., methanol or formic acid oxidation instead of HOR. Here, fuel crossover or contaminants such as sulphur or CO can adsorb onto the catalyst surface, blocking active sites and reducing the catalyst's activity or completely inhibiting its function.6,74
Elevated temperatures of up to 80 °C increase the rate of deactivation, leading to higher increased catalyst and/or support dissolution rates and particle detachment. This can result in a diminished activity and a reduced lifespan for the catalyst.71,72,75,76
An alternative approach for prominent production method involves the synthesis of H2O2 through an electrolytic cell as shown in Fig. 2(C)–(G). As an example, within a polymer electrolyte membrane (PEM) electrolyser, the anode and cathode catalyst layer directly interface with a solid electrolyte membrane. Flow-by-reactors, which facilitate continuous H2O2 extraction to limit the exposure of the catalytic layer to the generated peroxide (Fig. 2(E) and (F)) have been conceptualised and made their way into commercial applications.77 Non-divided cell reactors have been proposed for the Electro-Fenton process for wastewater treatment (Fig. 2(G)). With a non-divided cell configuration, both the anode and cathode can, directly and indirectly, contribute to the degradation of pollutant molecules, offering a promising solution for the environmentally friendly treatment of wastewater contaminants.
Regarding the applied potentials, the catalyst in an electrolyser is exposed to lower applied potentials (below 0 VRHE) compared to a H2O2-FC, especially when high H2O2-generation current density is required. Considering only Butler–Volmer kinetics, potentials below 0 VRHE are expected from Tafel extrapolations.6475,78–80 Conversely, the upper potential limit at the cathode should remain around OCP, similar to the conditions experienced by H2O2-FC systems. Higher potentials due to local fuel starvation will not be considered as mitigation strategies on the device level could potentially be employed.81–83 Consequently, the catalyst utilized in an H2O2 electrolyser is subjected to analogous potential-driven degradation mechanisms as mentioned earlier for H2O2-FC. If the applied potential surpasses the reduction potential of the Mx+/M redox couple, it is likely to induce a change in the valence state of surface atoms at sufficiently high overpotentials.84–86 These conditions can trigger structural reconstruction of the catalyst based on metals in a positive oxidation state, such as oxides or SACs, potentially leading to transformations of active sites, including the reduction to its metallic state followed by dissolution or agglomeration/cluster formation.29,87
A vital factor is the presence of H2O2 in close vicinity to the catalyst material as it may act as a degradation agent itself. H2O2 is known as an oxidising agent for organic matter and is used as bleaching agent, disinfectant, and antiseptic.5,88,89 The oxidative nature of H2O2 (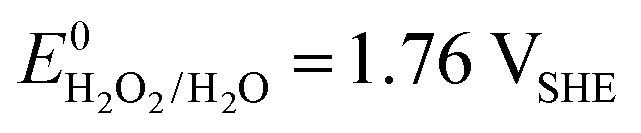 ) challenges the longevity of electrocatalysts employed in its production.90 Of particular concern is the potential formation of stronger or more reactive oxygen species (ROS) during H2O2 decomposition, such as hydroxyl (˙OH,
) challenges the longevity of electrocatalysts employed in its production.90 Of particular concern is the potential formation of stronger or more reactive oxygen species (ROS) during H2O2 decomposition, such as hydroxyl (˙OH, 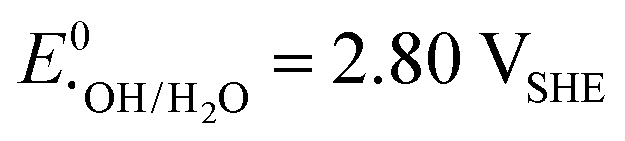 ) and hydroperoxyl (˙OOH,
) and hydroperoxyl (˙OOH, 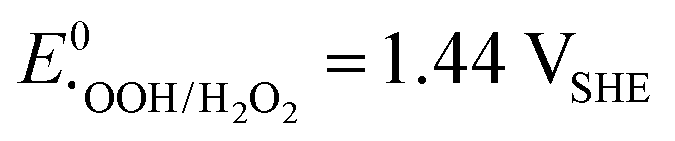 ) radicals in the presence of UV radiation or dissolved transition-metal ions.90–92 Effectively addressing the impact of ROS is imperative for enhancing the durability and sustained performance of electrocatalysts employed in the H2O2 production. In the previous section, it became clear that the unifying element in nearly all described catalyst materials lies in the utilization of carbon, either directly or as a supporting material, to achieve a high dispersion of the active catalyst. The generated ROS, especially the hydroxyl radical, renowned as the second strongest oxidizing agent after fluorine, can rapidly induce changes in the carbon matrix such as bond breakage ultimately leading to transformation into CO or CO2.90,93–96
) radicals in the presence of UV radiation or dissolved transition-metal ions.90–92 Effectively addressing the impact of ROS is imperative for enhancing the durability and sustained performance of electrocatalysts employed in the H2O2 production. In the previous section, it became clear that the unifying element in nearly all described catalyst materials lies in the utilization of carbon, either directly or as a supporting material, to achieve a high dispersion of the active catalyst. The generated ROS, especially the hydroxyl radical, renowned as the second strongest oxidizing agent after fluorine, can rapidly induce changes in the carbon matrix such as bond breakage ultimately leading to transformation into CO or CO2.90,93–96
For example, ˙OH can be generated by the gradual homolysis of H2O2. A reported rate constant of 1.2 × 10−7 s−1 at room temperature and low pH emphasizes the controlled formation of these radicals.97 However, ˙OH is also chemically generated much faster through the classical Fenton reaction (reaction rate of ca. 60 M−1 s−1),90,98 involving a mixture of diluted H2O2 solution and a Fe2+ species in acidic media (eqn (1)), following rather a heterolytic than a homolytic pathway.
The efficient homogeneous generation of hydroxyl radicals can be attributed to the high electron transfer cycle rate of the Fe3+/Fe2+ redox couple, particularly in solutions with a pH around 3.90 Even small amounts of Fe2+ are sufficient due to their ability to be regenerated from the homogeneous Fenton-like reaction (eqn (2)).99 The Fenton-like reaction is expected to be approximately four orders of magnitude slower than the classical Fenton reaction, eqn (1) (ca. 2 × 10−3 M−1 s−1).98 However, the consumed Fe2+ can be regenerated more rapidly (ca. 2 × 106 M−1 s−1) through the reduction of Fe3+ with ˙OOH (eqn (3)).98 Furthermore, the electrochemical regeneration of Fe2+ at the cathode is possible (eqn (4)).100
| Fe2+ + H2O2 + H+ → Fe3+ + H2O + ˙OH | (1) |
| Fe3+ + H2O2 → Fe2+ + ˙OOH + H+ | (2) |
| Fe3+ + ˙OOH → Fe2+ + O2 + H+ | (3) |
| Fe3+ + e− → Fe2+ | (4) |
Numerous studies especially in the context of advanced oxidation processes and as well as in durability of low temperature PEM-FC-4e− on various heterogeneous catalysts have explored their effectiveness in the Fenton and Fenton-like reaction (when the reaction is based on Fe3+ or on another reactant). These reactions involve diverse metals and are not limited to Fe as classical Fenton and Fenton-like catalysts including metals such as Cu, Mn, Ce, Cr, Co, Ru, Pd, Ag, and Au.97,99,101–106 In principle, several metals can generate radicals from H2O2 heterogeneously or through partly homogeneous Fenton-like chemistry. The unifying element is the prevalence of varying oxidation states. These oxidation states must remain stable over a wide pH range to prevent catalyst loss through leaching, and the metallic species should resist hydration forces and remain insoluble.
Despite the fact that most of these metals are less effective in Fenton and Fenton-like reactions than homogeneous Fe2+, these metals facilitate ROS production, typically through electron-transfer steps that alter the oxidation state of the metal, leading to the generation of ˙OH and ˙OOH as schematically shown in Fig. 3(A).107 Conversely, metals resistant to oxidation state changes, like Al, Ga, In, Zr, and Ti, also play a role in generating ROS from H2O2 through a metal hydroperoxo species, [M]-OOH, without direct electron transfer involvement.107 Additionally, ROS production can occur from the reaction of adsorbed protons with H2O2 on noble metals such as Ag, Pt, Rh, or Pd.108–110 Generally, all these processes of ROS generation from H2O2 exhibit slower rates compared to the homogeneous Fenton reaction. This difference is attributed to the additional mass transport barrier for H2O2 to access the solid-phase catalyst for Fenton and Fenton-like reactions.
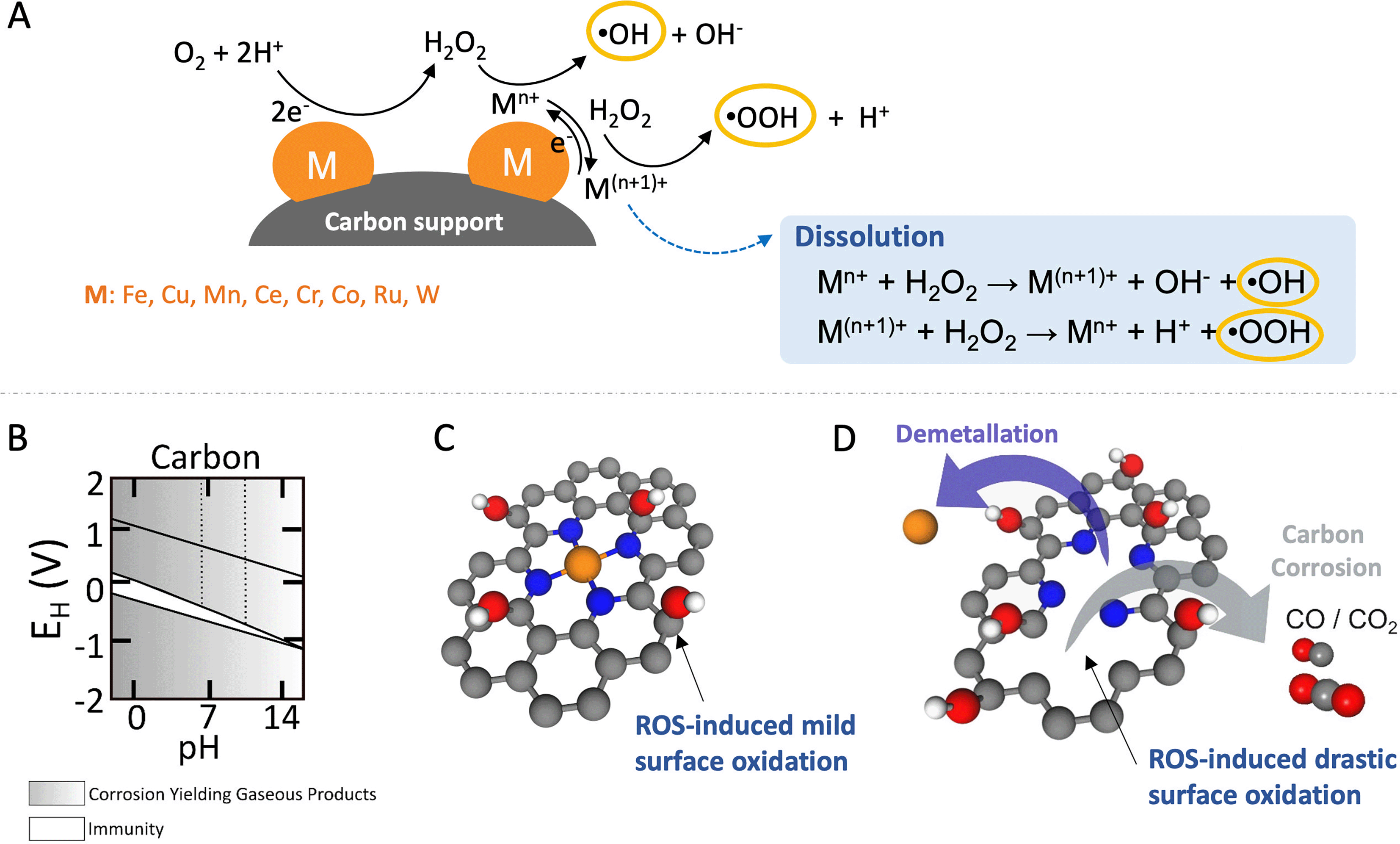 | ||
| Fig. 3 Schematic illustration depicting homogeneous and heterogeneous Fenton and Fenton-like reactions, showcasing their influence on carbon-based catalysts during the ORR. (A) Depiction of homogeneous and heterogeneous Fenton and Fenton-like reactions. (B) Potential–pH diagram for carbon in aqueous solutions. (C) Mild and reversible oxidation of carbon supports. (D) Irreversible oxidation or corrosion of carbon supports along with the demetallation. Color coding for the components: metal active sites, orange; C, gray; O, red; N, blue; and H, white. Panel B is adapted with permission from ref. 111 Copyright 1973 Springer Nature. | ||
In heterogeneous Fenton or Fenton-like catalysis, reducing particle size is expected to enhance the process efficiency. This is because a smaller particle size translates to a larger active surface area. This increased surface area provides more sites for the adsorption of reactants (hydrogen peroxide and target pollutants) and the generation of hydroxyl radicals, ultimately leading to faster reaction rates and potentially improved degradation of contaminants. Furthermore, SACs typically exhibit greater atom utilisation than NPs, they can show higher mass normalised Fenton or Fenton-like activity, as well as catalytic activity comparable or superior to homogeneous catalysts.112,113
The choice of support material significantly influences the stability and longevity of the catalyst as secondary degradation effects, such as nanoparticle agglomeration/coalescence or detachment are mitigated.114–118 Carbon-based materials, for instance, primarily degrade due to carbon oxidation at sufficiently high potentials.50,119 Thermodynamically, carbon exhibits a small immunity region in aqueous solutions as illustrated in Fig. 3(B), and can oxidize to CO2 and CO at 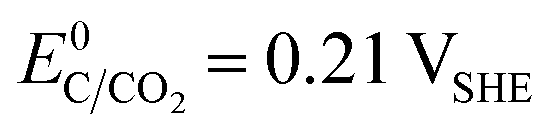 and
and 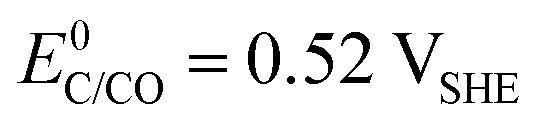 , respectively. These values significantly differ from
, respectively. These values significantly differ from 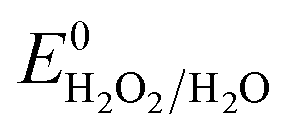 ,
, 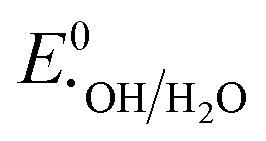 , or
, or 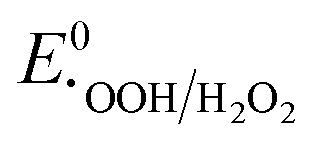 , thereby facilitating the thermodynamic oxidation of carbon materials by hydrogen peroxide.120
, thereby facilitating the thermodynamic oxidation of carbon materials by hydrogen peroxide.120
Kumar et al. demonstrated that subjecting a non-noble based catalyst to an electrochemical accelerated stress test (AST) in oxygen-containing electrolyte leads to a notable loss of iron within the FeNx sites of the catalyst. This loss is caused by the corrosion of the catalyst's carbon, facilitated either by H2O2 or radical species derived from H2O2.121 The same holds true for noble metal based catalysts. The presence of oxygen notably affects the degradation of Pt/C in the potential range of 0.05 to 0.5 VRHE. In this range, the two-electron ORR pathway generates H2O2-derived radical species which, in turn, inflicts damage on the carbon supports.122 Chemical oxidative stress can be intensified in Fe- and Co-based non-noble metal catalysts supported on carbon-based supports, as the chemical disproportionation of H2O2 on these catalysts generates a significant amount of ROS.50,123 It has been demonstrated that the Fe–N–C structure undergoes stronger degradation, along with a loss in ORR activity, when subjected to load cycling and start/stop testing in RDE and PEMFC,124 and potential hold in PEMFC125 in O2- vs. Ar-saturated acidic electrolyte. In addition, after enduring 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles from 0.6 to 1.0 VRHE and 100 hours hold at a potential of 0.7 VRHE in O2-purged electrolyte, Co–N–C catalysts exhibited superior durability compared to Fe–N–C.126 The enhanced stability is attributed to the lower reactivity of Co ions in Fenton-like reactions that generate radicals from H2O2. Complementing this finding, Laconti et al. suggested a hierarchy for the rate of chemical degradation by certain contaminants: Fe2+ impacts degradation rate the most, followed by Cu2+, TiO2+, Co2+, Pt2+, and Ni2+.127 The potential involvement of H2O2 and ROS species in catalytic processes was highlighted by Lefèvre et al.128 They observed that exposing Fe-based catalysts to a solution of 5 vol% H2O2 in 1 M H2SO4 solution for 5 hours – mimicking amounts of produced H2O2 similar to those found in ORR with a 2 wt% Pt/C catalyst led to a significant ORR activity loss of approximately 180 mV. This loss in catalytic efficiency was linearly correlated with the Fe loss within the catalyst induced by H2O2.
000 potential cycles from 0.6 to 1.0 VRHE and 100 hours hold at a potential of 0.7 VRHE in O2-purged electrolyte, Co–N–C catalysts exhibited superior durability compared to Fe–N–C.126 The enhanced stability is attributed to the lower reactivity of Co ions in Fenton-like reactions that generate radicals from H2O2. Complementing this finding, Laconti et al. suggested a hierarchy for the rate of chemical degradation by certain contaminants: Fe2+ impacts degradation rate the most, followed by Cu2+, TiO2+, Co2+, Pt2+, and Ni2+.127 The potential involvement of H2O2 and ROS species in catalytic processes was highlighted by Lefèvre et al.128 They observed that exposing Fe-based catalysts to a solution of 5 vol% H2O2 in 1 M H2SO4 solution for 5 hours – mimicking amounts of produced H2O2 similar to those found in ORR with a 2 wt% Pt/C catalyst led to a significant ORR activity loss of approximately 180 mV. This loss in catalytic efficiency was linearly correlated with the Fe loss within the catalyst induced by H2O2.
The efficiency of the Fenton reaction is influenced by temperature, pH, as well as H2O2, and iron concentration, with temperature being the most impactful, followed by Fe2+ and H2O2 concentration.127,129 Higher temperatures tend to increase the rate of the Fenton reaction, but they also increase the disproportionation of H2O2 into O2 and H2O.130
In the absence of iron, ˙OH is produced by homolytic dissociation of H2O2 (H2O2 → 2 ˙OH) on Pt catalyst. At an H2O2 concentration of 1 M, Gubler et al. reported that 45% of ˙OH are created via ˙OOH formation (˙OH + H2O2 → ˙OOH + H2O, ˙OOH + H2O2 → ˙OH + H2O + O2).101 Without Fe-ion impurities, ˙OOH primarily dismutates to H2O2 and O2, yet there remains a possibility for it to contribute to chemical oxidation of e.g. carbon.122 However, with typical Fe-ion impurities found in fuel cell membranes (around 1 ppm) and concentrations up to several millimolar H2O2, Fe ions catalyse ˙OH generation. The rate of ˙OH formation via the Fenton reaction is eight times higher than by homolysis, becoming the predominant source when Fe impurity levels exceed 40 ppm (1.1 mM).131
Carbon nanostructures exhibit varying levels and types of metallic impurities, which depend on the specific preparation methods used.132 For instance, analyses using ICP-MS reveal that bare graphite typically contains about 4 ppm of Fe. In graphene oxide, the metallic impurities were found to be different, comprising 3800 ppm of Mn, 120 ppm of Cu, 53 ppm of Fe, and 4 ppm of Ni. In a similar vein, carbon nanotubes display varied impurity profiles, with concentrations of 1200 ppm for Ni, 90 ppm for Fe, 11 ppm for Mn, and 3 ppm for Cu.133 Consequently, the influence of Fe and other metal impurities on the conversion of H2O2 into ROS in ostensibly metal-free catalysts is a critical consideration for carbon corrosion that cannot be overlooked.
The corrosion rate of carbon is notably affected by material properties, including surface area, porosity, degree of graphitisation, and heteroatom doping, all of which play pivotal roles in determining the overall durability of the catalyst system.134–136 ROS, generated through the decomposition of H2O2, readily interacts with unsaturated aliphatic or aromatic carbon compounds mainly at the edges, leading to the formation of oxygen-containing functional groups like –COOH, and –COH on the surface, as illustrated in Fig. 3(C). Ultimately, this process, detailed in Fig. 3(D), facilitates the conversion of volatile carbon corrosion products such as CO or CO2 and leads to metal dissolution and/or detachment (demetallation).121,123,128 In more applied H2O2 production devices using MEA or GDE (or both) configurations, the generation of hydrophilic groups by oxidation during operation causes flooding of the catalyst layer leading to mass transport related losses.137 In the case of catalysts containing active centers or organic ligands, such as catalysts based on organic molecules, organo-metallic complexes, and SACs, the impact on catalytic activity and selectivity can be even more pronounced.125
The intrinsic non-polarity of carbon makes it challenging to exhibit robust metal-support interactions without having anchoring functional groups present.138 The incorporation of oxygen functional groups onto carbon surfaces not only augments the number of anchoring sites for deposited particles/clusters/single atoms but also improves the catalytic activity and selectivity. The presence of carboxyl, carbonyl, and quinone functional groups on the carbon surface plays a vital role in enhancing the Fenton reaction. These groups expedite the conversion of Fe3+ to Fe2+, which is a pivotal step in the catalytic iron cycle. This conversion is particularly significant as it constitutes the rate-limiting step, profoundly influencing the efficiency of the reaction.103 Consequently, they can promote carbon oxidation and metal detachment/dissolution by increasing the ROS production rate. Resistance to carbon corrosion can be notably improved through graphitisation. Graphitised carbon supports exhibit remarkable long-term stability compared to disordered amorphous carbon supports.139,140 Enhancing ORR activity can be achieved by incorporating heteroatoms like nitrogen into their structure, which increases the negative charge density on adjacent carbon atoms. H2O2 and ROS, generated in situ at FeNx sites during ORR, induce mild surface oxidation, as previously reported in ex situ treatments of Fe–N–C with H2O2.141 However, they also trigger irreversible carbon corrosion. This corrosion forms volatile CO and CO2, creating new pores and increasing carbon surface area, while simultaneously disintegrating metal-N sites, resulting in demetallation and a decrease in catalytic activity.124
The decay of active sites and the oxidation of the carbon matrix are driven by a combination of electrochemical and chemical oxidation processes, with H2O2/ROS notably accelerating the chemical oxidation. Considering that H2O2 is the predominant product in ORR-2e−, closely examining its release rate during durability and stability tests becomes crucial. Simultaneously, standardised protocols play a crucial role in facilitating consistent comparisons of catalytic performance among various research groups.
4. Key considerations in unravelling catalyst degradation
While some degradation assessment protocols exist for specific electrochemical reactions, such as ORR-4e−,142–144 there is an absence of benchmarked protocols for the ORR-2e−. Accelerated stress tests can be utilized to induce and study catalyst (and cell) aging processes, as well as evaluate failure modes under real-world conditions. In the following discussion, our focus will be solely on catalyst-related aspects, excluding other potential failure modes. Establishing suitable degradation protocols and reliable measurement configurations at an early stage of electrocatalyst development is a fundamental task in this context. In light of the considerations mentioned earlier, a key objective is to design AST protocols capable of simulating specific scenarios that mirror the most challenging conditions imposed on the catalyst material. This includes scenarios such as start-stop cycling, continuous operation under H2O2 production, and extended contact with H2O2 at OCP.For PEM-FCs, AST protocols are capable of simulating the degradation of the catalysts at start-stop conditions. Typically, the catalysts are subjected to square wave voltammetry, using an operation potential, e.g. 0.6 VRHE and OCP, usually ≤1.0 VRHE for Pt and Pd-based catalysts. In Fig. 4(A), we exemplify a possible potential square wave-based AST for ORR-2e− catalysts, where the upper and lower potential limits are the OCP and the voltage recorded when forcing a certain H2O2-generation current density (J), respectively. Both, upper and lower potentials depend strongly on the catalysts employed and have to be measured for each catalyst system.
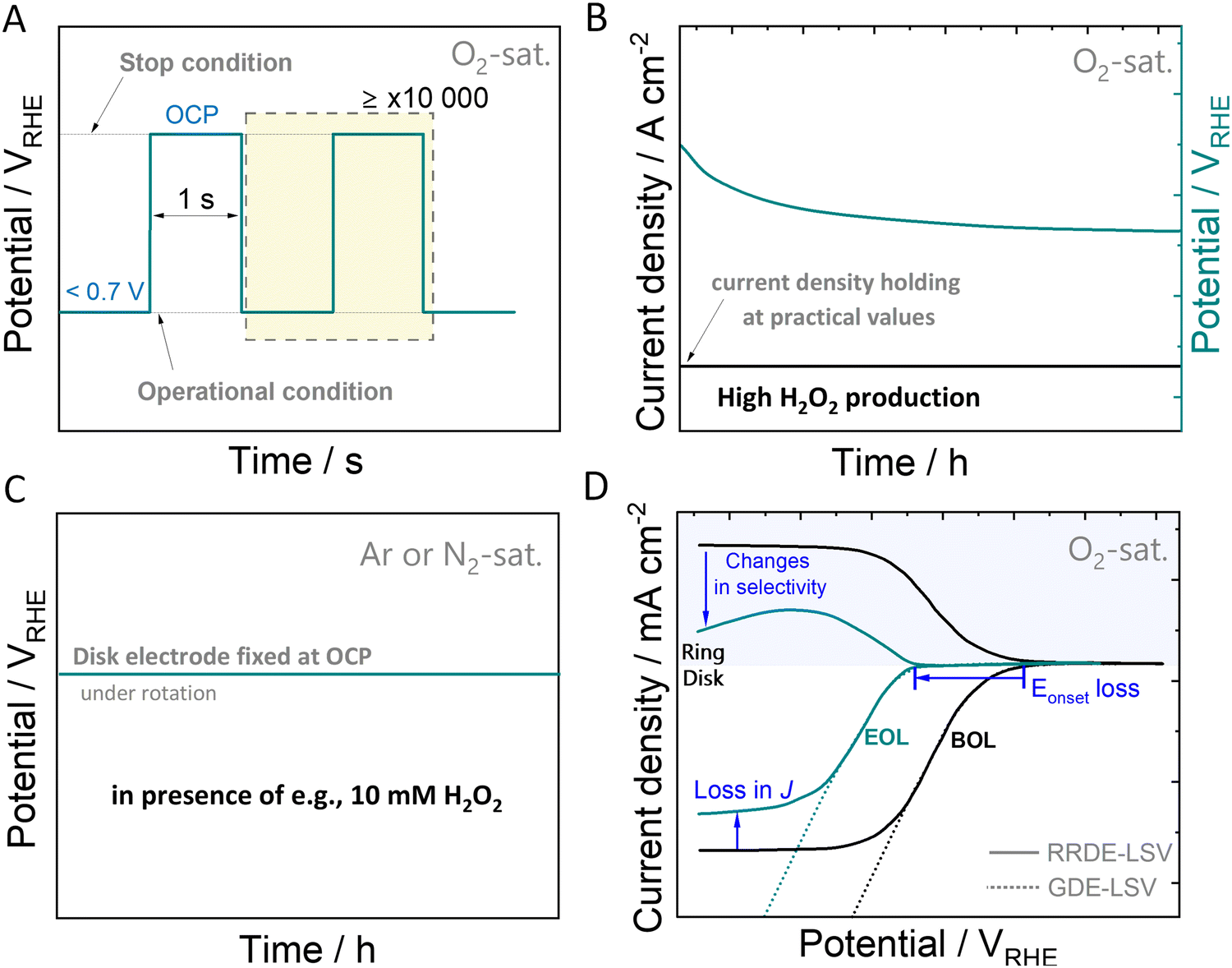 | ||
| Fig. 4 Schematic representation of the process for standardising degradation assessment protocols. Illustration of accelerated stress test protocol profiles simulating: (A) start-stop regimes, (B) practical electrolysis operating conditions, and (C) influence of the contact of H2O2 with the catalyst. Schematic responses for (D) RRDE- and GDE–LSV curves for ORR study. | ||
As H2O2 itself can inflict damage to the catalyst, evaluating its stability involves testing under practical electrolysis operating conditions and realistic H2O2 concentration in direct contact with the catalyst. Rather than relying solely on the applied potential, chronopotentiometric methods (Fig. 4(B)) allow quantitative insights into the potential contribution of generated H2O2 to the degradation process. When operated at high current densities of hundreds of mA cm−2, the applied potential becomes more and more negative creating a reductive environment. These reductive conditions, particularly relevant for catalysts composed of metals with positive oxidation states such as in oxides or single-atom catalysts, have the potential to induce structural changes, potentially leading to oxidation state changes, as discussed in Section 3.
During early-stage catalytic testing, conventional laboratory-scale electrochemical setups, such as rotating ring-disk electrode setups (RRDE) are commonly employed. However, limitations in O2 mass transport may hinder the attainment of practical H2O2-generation current densities, thereby preventing continuous exposure of the catalyst to realistic H2O2 concentrations. Acknowledging this challenge, we propose a supplementary protocol that aligns with the previously discussed protocols. This method involves conducting ASTs in an electrolyte solution containing H2O2, as illustrated in Fig. 4(C). Here, the catalyst is directly immersed in an H2O2-containing electrolyte solution such as e.g., 10 mM H2O2 for an extended duration. Thereby, a comparative analysis of chemical decomposition rates can be obtained. By conducting these three protocols, catalysts can be evaluated for their stability and compared. Further adaptation of these protocols will have to follow, depending on the catalyst used and the involved degradation mechanism of the specific catalyst material. The disparity in catalytic performance observed in polarisation curves, such as linear sweep voltammetry (LSV) curves, taken at the initiation and the end of the protocol, referred to as beginning of life (BOL) and end of life (EOL), provides valuable insights into the degradation induced by the accelerated stress test (Fig. 4(D)). This comparison aids in understanding changes in terms of ORR activity and selectivity, facilitating the evaluation of the catalyst's stability. To monitor variations in parameters such as onset potential, faradaic efficiency, mass activity, or losses of the electrochemical surface area should be considered.
5. Challenges and limitations
In the supporting information, we provide a comprehensive discussion of the obstacles and constraints associated with the performance of the catalyst in order to successfully implement the electrochemical H2O2 production. The efficiency of catalysts plays a crucial role in the cost-effectiveness of electrochemical technologies, underscoring the importance of enhancing catalyst efficiency and durability for substantial cost reduction.For practical applications, it is crucial that electrodes maintain high stability for H2O2 production across long-term operations and under varying conditions. Supporting substrates that hold nanoparticles or single atoms may chemically or physically degrade, significantly impacting performance. Currently, carbon materials are the preferred support for ORR catalysts because of their low cost and beneficial properties such as conductivity, variability in changing pore size and pore volume, chemical resistance and non-toxicity. However, carbon corrosion, which occurs at high electrode potentials and is exacerbated by ROS from H2O2, can lead to the detachment/agglomeration of metal particles/atoms from the support and subsequent performance degradation, as exemplified in Section 3. Arguably, future research should aim to enhance the stability of carbon-supported SACs in acidic conditions, possibly by modifying these supports with functional groups that may act as anchor point for the active metal. For instance, Chen et al. observed that thiolated carbon supports (Pt/SH-CNTs) significantly enhance the resistance against Pt dissolution at high potentials.145 More recently, Wang et al. demonstrated that graphitic N-doped CoN4C (CoN4C-N) catalysts achieve 82% Faraday efficiency for H2O2 in a flow cell, delivering a yield of 0.096 mmol cm−2 h−1 over 200 h at 0.358 VRHE, highlighting the important role of graphitic nitrogen.37
To our knowledge, little research has focused on catalyst stability in the context of H2O2 electrogeneration. We argue that for industrial applications, a thorough understanding of catalytic performance and degradation is crucial. In practical scenarios, high concentrations of H2O2 accumulating at the solid/liquid interface may decompose, leading to accelerated corrosion of the electrode and degradation of the catalyst.146 This decomposition can intensify the H2O2-induced oxidation process, causing the catalyst to detach from the support, agglomerate or dissolve into the electrolyte.
6. Summary and outlook
In summary, we have shown the crucial significance of catalyst stability in the electrocatalytic production of H2O2, emphasising essential considerations for designing and testing catalysts tailored to this application. Our analysis identifies several primary factors contributing to catalyst degradation, thereby leading to diminished H2O2 electrogeneration efficiency in devices. These factors include low pH values, potential-driven degradation, and the susceptibility of catalyst degradation induced by reactive oxygen species. Notably, H2O2 itself may act as a degradation agent. The oxidative nature of H2O2, coupled with the rapid kinetics of generated radicals, significantly impacts the longevity of electrocatalysts utilized in its production. Given that carbon serves as a unifying element in nearly all described ORR-2e− catalyst materials, whether directly or as a supporting material, it is imperative to effectively address the impact of ROS for enhancing the durability and sustained performance of electrocatalysts used in H2O2 production. Furthermore, considering the current absence of benchmarked AST protocols for ORR-2e−, and the need for the development and adoption of comprehensive standardised testing methodologies, we have made a first attempt to present three AST protocols capable of simulating specific scenarios that mirror the most challenging conditions imposed on the catalyst material during H2O2 production. By bridging gaps in our understanding of catalyst behaviour under realistic conditions, we can pave the way for the development of robust catalysts capable of withstanding the challenges posed by H2O2 electrogeneration.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. S. C. and M. L. are grateful to the Federal Ministry of Education and Research (BMBF) for the financial support received in the framework of NanoMatFutur (SynKat, FK: 03XP0265). G. V. F., J. C. L., and M. R. V. L. are grateful for the financial support received from the São Paulo Research Foundation (FAPESP – grants #2017/10118-0, #2023/04230-2, #2021/12053-8, #2019/04421-7, and # 2021/14194-8) and Brazilian National Council for Scientific and Technological Development (CNPq – grant #303943/2021-1). M. L. gratefully acknowledges funding within the framework of Fe-Upgraded from the “Deutsche Forschungsgemeinschaft” (DFG, German Research Foundation) – Project-ID 443703006 – CRC 1487.References
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- R. Xia, S. Overa and F. Jiao, JACS Au, 2022, 2, 1054–1070 CrossRef CAS.
- N. Dyantyi, J. C. Calderón Gómez, L. Mekuto, P. Bujlo and G. Pattrick, Electrocatalysts: Selectivity and utilization, INC, 2021 Search PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- G. V. Fortunato, E. Pizzutilo, I. Katsounaros, D. Göhl, R. J. Lewis, K. J. J. Mayrhofer, G. J. Hutchings, S. J. Freakley and M. Ledendecker, Nat. Commun., 2022, 13, 1–7 Search PubMed.
- E. Kolle-Görgen, G. Fortunato and M. Ledendecker, Chem. Mater., 2022, 34, 10223–10236 CrossRef.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed.
- J. S. Jirkovsk, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schi, J. Am. Chem. Soc., 2011, 14, 19432–19441 CrossRef PubMed.
- M. Inaba, H. Yamada, J. Tokunaga and A. Tasaka, Electrochem. Solid-State Lett., 2004, 7, 474–476 CrossRef.
- E. Fabbri, S. Taylor, A. Rabis, P. Levecque, O. Conrad, R. Kötz and T. J. Schmidt, ChemCatChem, 2014, 6, 1410–1418 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2016, 181, 298–313 CrossRef CAS.
- M. Nesselberger, M. Roefzaad, R. Fayçal Hamou, P. Ulrich Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, Nat. Mater., 2013, 12, 919–924 CrossRef CAS PubMed.
- G. V. Fortunato, E. Pizzutilo, A. M. Mingers, O. Kasian, S. Cherevko, E. S. F. Cardoso, K. J. J. Mayrhofer, G. Maia and M. Ledendecker, J. Phys. Chem. C, 2018, 122, 15878–15885 CrossRef CAS.
- C. H. Choi, H. C. Kwon, S. Yook, H. Shin, H. Kim and M. Choi, J. Phys. Chem. C, 2014, 118, 30063–30070 CrossRef CAS.
- A. Ohma, K. Fushinobu and K. Okazaki, Electrochim. Acta, 2010, 55, 8829–8838 CrossRef CAS.
- J. Chlistunoff and B. Pivovar, ECS Meet. Abstr., 2007, MA2007–02, 506 CrossRef.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS.
- D. He, L. Zhong, S. Gan, J. Xie, W. Wang, Z. Liu, W. Guo, X. Yang and L. Niu, Electrochim. Acta, 2021, 371, 137721 CrossRef CAS.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed.
- S. Yang, Y. J. Tak, J. Kim, A. Soon and H. Lee, ACS Catal., 2017, 7, 1301–1307 CrossRef CAS.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H. T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- M. Ledendecker, E. Pizzutilo, G. Malta, G. V. Fortunato, K. J. J. Mayrhofer, G. J. Hutchings and S. J. Freakley, ACS Catal., 2020, 10, 5928–5938 CrossRef CAS.
- Q. Chang, P. Zhang, A. H. B. Mostaghimi, X. Zhao, S. R. Denny, J. H. Lee, H. Gao, Y. Zhang, H. L. Xin, S. Siahrostami, J. G. Chen and Z. Chen, Nat. Commun., 2020, 11, 1–9 CrossRef.
- J. S. Choi, S. Yoo, E. S. Koh, R. Aymerich-Armengol, C. Scheu, G. V. Fortunato, M. R. V. Lanza, Y. J. Hwang and M. Ledendecker, Adv. Mater. Interfaces, 2023, 10, 1–7 Search PubMed.
- X. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- J. C. Meier, C. Galeano, I. Katsounaros, J. Witte, H. J. Bongard, A. A. Topalov, C. Baldizzone, S. Mezzavilla, F. Schüth and K. J. J. Mayrhofer, Beilstein J. Nanotechnol., 2014, 5, 44–67 CrossRef PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- F. D. Speck, M. T. Y. Paul, F. Ruiz-Zepeda, M. Gatalo, H. Kim, H. C. Kwon, K. J. J. Mayrhofer, M. Choi, C. H. Choi, N. Hodnik and S. Cherevko, J. Am. Chem. Soc., 2020, 142, 15496–15504 CrossRef CAS PubMed.
- H. E. Kim, I. H. Lee, J. Cho, S. Shin, H. C. Ham, J. Y. Kim and H. Lee, ChemElectroChem, 2019, 6, 4757–4764 CrossRef CAS.
- J. Xi, S. Yang, L. Silvioli, S. Cao, P. Liu, Q. Chen, Y. Zhao, H. Sun, J. N. Hansen, J. P. B. Haraldsted, J. Kibsgaard, J. Rossmeisl, S. Bals, S. Wang and I. Chorkendorff, J. Catal., 2021, 393, 313–323 CrossRef CAS.
- M. Campos, W. Siriwatcharapiboon, R. J. Potter and S. L. Horswell, Catal. Today, 2013, 202, 135–143 CrossRef CAS.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS.
- J. Gao, H. bin Yang, X. Huang, S. F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- Z. Jiajun, F. Cehuang, Y. Ke, L. Zheng, J. Fangling, S. Shuiyun, Z. Xiaoran, M. Lu, S. Zulipiya, W. Xiaoming, Z. Junliang and J. Kun, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- F. Wu, C. Pan, C. T. He, Y. Han, W. Ma, H. Wei, W. Ji, W. Chen, J. Mao, P. Yu, D. Wang, L. Mao and Y. Li, J. Am. Chem. Soc., 2020, 142, 16861–16867 CrossRef CAS PubMed.
- W. Wang, Y. Hu, P. Li, Y. Liu and S. Chen, ACS Catal., 2024, 14, 5961–5971 CrossRef CAS.
- S. Chen, T. Luo, X. Li, K. Chen, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li, Y. Chen, C. Ma, L. Zhu, Y. R. Lu, T. S. Chan, M. Zhu, E. Cortés and M. Liu, J. Am. Chem. Soc., 2022, 144, 14505–14516 CrossRef CAS PubMed.
- H. Huang, M. Sun, S. Li, S. Zhang, Y. Lee, Z. Li, J. Fang, C. Chen, Y.-X. Zhang, Y. Wu, Y. Che, S. Qian, W. Zhu, C. Tang, Z. Zhuang, L. Zhang and Z. Niu, J. Am. Chem. Soc., 2024, 146, 9434–9443 CrossRef CAS PubMed.
- T. P. Fellinger, F. Hasché, P. Strasser and M. Antonietti, J. Am. Chem. Soc., 2012, 134, 4072–4075 CrossRef CAS.
- S. K. Singh, K. Takeyasu and J. Nakamura, Adv. Mater., 2019, 31, 1–17 CrossRef.
- G. F. Han, F. Li, W. Zou, M. Karamad, J. P. Jeon, S. W. Kim, S. J. Kim, Y. Bu, Z. Fu, Y. Lu, S. Siahrostami and J. B. Baek, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- A. Wang, A. Bonakdarpour, D. P. Wilkinson and E. Gyenge, Electrochim. Acta, 2012, 66, 222–229 CrossRef CAS.
- R. B. Valim, R. M. Reis, P. S. Castro, A. S. Lima, R. S. Rocha, M. Bertotti and M. R. V. Lanza, Carbon N. Y., 2013, 61, 236–244 CrossRef CAS.
- P. Cui, L. Zhao, Y. Long, L. Dai and C. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218269 CrossRef CAS PubMed.
- K. Mamtani and U. S. Ozkan, Catal. Lett., 2015, 145, 436–450 CrossRef CAS.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS.
- Z. H. Sun, X. Zhang, X. D. Yang, W. N. Shi, Y. Q. Huang, Y. L. Men, J. Yang and Z. Y. Zhou, Chem. Commun., 2022, 58, 8998–9001 RSC.
- H. Singh, S. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon N. Y., 2019, 151, 160–174 CrossRef CAS.
- D. Banham, S. Ye, K. Pei, J. I. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- J. Zhang, Y. Yuan, L. Gao, G. Zeng, M. Li and H. Huang, Adv. Mater., 2021, 33, 1–23 Search PubMed.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 RSC.
- C. H. Choi, C. Baldizzone, J. P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed.
- M. Sun, S. Gong, Y. X. Zhang and Z. Niu, J. Energy Chem., 2022, 67, 250–254 CrossRef CAS.
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, 1–11 CrossRef PubMed.
- S. J. Ashton and M. Arenz, J. Power Sources, 2012, 217, 392–399 CrossRef CAS.
- Y. Yang, Y. Xiong, R. Zeng, X. Lu, M. Krumov, X. Huang, W. Xu, H. Wang, F. J. DiSalvo, J. D. Brock, D. A. Muller and H. D. Abruña, ACS Catal., 2021, 11, 1136–1178 CrossRef CAS.
- C. Long, J. Han, J. Guo, C. Yang, S. Liu and Z. Tang, Chem. Catal., 2021, 1, 509–522 CrossRef CAS.
- L. Liu, W. Li, X. He, J. Yang and N. Liu, Small, 2022, 18, 2104205 CrossRef CAS PubMed.
- W. D. Nicoll and A. F. Smith, Ind. Eng. Chem., 1955, 47, 2548–2554 CrossRef CAS.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- K. Otsuka and I. Yamanaka, Electrochim. Acta, 1990, 35, 319–322 CrossRef CAS.
- W. Li, A. Bonakdarpour, E. Gyenge and D. P. Wilkinson, ChemSusChem, 2013, 6, 2137–2143 CrossRef CAS PubMed.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- E. Jung, H. Shin, W. Hooch Antink, Y. E. Sung and T. Hyeon, ACS Energy Lett., 2020, 5, 1881–1892 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casdevall, M. Karamad, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, ECS Trans., 2013, 58, 53–62 CrossRef.
- P. J. Ferreira, G. J. la O’, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256 CrossRef.
- S. Cherevko, G. P. Keeley, S. Geiger, A. R. Zeradjanin, N. Hodnik, N. Kulyk and K. J. J. Mayrhofer, ChemElectroChem, 2015, 2, 1471–1478 CrossRef CAS PubMed.
- N. Seselj, S. M. Alfaro, E. Bompolaki, L. N. Cleemann, T. Torres and K. Azizi, Adv. Mater., 2023, 35, 1–28 CrossRef PubMed.
- F. D. Speck, J. H. Kim, G. Bae, S. H. Joo, K. J. J. Mayrhofer, C. H. Choi and S. Cherevko, JACS Au, 2021, 1, 1086–1100 CrossRef CAS PubMed.
- J. Zhao, X. Huang, H. Chang, S. Hwa Chan and Z. Tu, Energy Convers. Manage.: X, 2021, 10, 100087 CAS.
- T. Engl, L. Gubler and T. J. Schmidt, Energy Technol., 2016, 4, 65–74 CrossRef CAS.
- M. Ledendecker, J. S. Mondschein, O. Kasian, S. Geiger, D. Göhl, M. Schalenbach, A. Zeradjanin, S. Cherevko, R. E. Schaak and K. Mayrhofer, Angew. Chem., Int. Ed., 2017, 56, 9767–9771 CrossRef CAS PubMed.
- H. Cheng, J. Wang, C. Wu and Z. Liu, Energy Mater. Adv., 2023, 4, 1–26 Search PubMed.
- T. Imhof, R. K. F. Della Bella, B. M. Stühmeier, H. A. Gasteiger and M. Ledendecker, Phys. Chem. Chem. Phys., 2023, 25, 20533–20545 RSC.
- T. Lochner, R. M. Kluge, J. Fichtner, H. A. El-Sayed, B. Garlyyev and A. S. Bandarenka, ChemElectroChem, 2020, 7, 3545–3568 CrossRef CAS.
- A. V. Casadevall and R. Frydendal, US Pat., 20190071783A1, 2019 Search PubMed.
- M. S. Kronka, G. V. Fortunato, L. Mira, A. J. dos Santos and M. R. V. Lanza, Chem. Eng. J., 2023, 452, 139598 CrossRef CAS.
- X. Zhou, Y. Min, C. Zhao, C. Chen, M.-K. Ke, S.-L. Xu, J.-J. Chen, Y. Wu and H.-Q. Yu, Nat. Commun., 2024, 15, 193 CrossRef PubMed.
- T. Iwasaki, Y. Masuda, H. Ogihara and I. Yamanaka, Electrocatalysis, 2018, 9, 236–242 CrossRef CAS.
- F. Du, J. A. Hirschfeld, X. Huang, K. Jozwiak, T. A. Dao, A. Bauer, T. J. Schmidt and A. Orfanidi, J. Electrochem. Soc., 2021, 168, 074504 CrossRef CAS.
- X. Bai, Q. Jian, B. Huang, L. Luo and Y. Chen, J. Power Sources, 2022, 520, 230809 CrossRef CAS.
- Z. Xiong, B. Wen, D. Banham, S. H. Chan, Z. Xie, Y. Liang and S. Liao, Front. Energy Res., 2022, 10, 1–14 Search PubMed.
- S. H. Lee, J. C. Lin, M. Farmand, A. T. Landers, J. T. Feaster, J. E. Avilés Acosta, J. W. Beeman, Y. Ye, J. Yano, A. Mehta, R. C. Davis, T. F. Jaramillo, C. Hahn and W. S. Drisdell, J. Am. Chem. Soc., 2021, 143, 588–592 CrossRef CAS PubMed.
- W. Zhang, Q. Fu, Q. Luo, L. Sheng and J. Yang, JACS Au, 2021, 1, 2130–2145 CrossRef CAS PubMed.
- X. Liu, J. Meng, J. Zhu, M. Huang, B. Wen, R. Guo and L. Mai, Adv. Mater., 2021, 33, 1–34 Search PubMed.
- E. S. Koh, S. Geiger, A. Gunnarson, T. Imhof, G. M. Meyer, P. Paciok, B. J. M. Etzold, M. Rose, F. Schüth and M. Ledendecker, ChemElectroChem, 2023, 10, e202200924 CrossRef CAS.
- M. Abdollahi and A. Hosseini, Encyclopedia of Toxicology, Elsevier, 3rd edn, 2014, vol. 2, pp. 967–970 Search PubMed.
- J. Leifeld and I. Kögel-Knabner, Soil Biol. Biochem., 2001, 33, 2155–2158 CrossRef CAS.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- E. Brillas, Chemosphere, 2020, 250, 126198 CrossRef CAS PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, CRC Press, UK, 1985 Search PubMed.
- J. Weinstein and B. H. J. Bielski, J. Am. Chem. Soc., 1979, 101, 58–62 CrossRef CAS.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- W. Xing, G. Lalwani, I. Rusakova and B. Sitharaman, Part. Part. Syst. Charact., 2014, 31, 745–750 CrossRef CAS.
- T. Du, A. S. Adeleye, T. Zhang, N. Yang, R. Hao, Y. Li, W. Song and W. Chen, Environ. Sci.: Nano, 2019, 6, 2484–2494 RSC.
- E. Kuhnert, V. Hacker and M. Bodner, Int. J. Energy Res., 2023, 2023, 3183108 CrossRef.
- C. K. Duesterberg, S. E. Mylon and T. D. Waite, Environ. Sci. Technol., 2008, 42, 8522–8527 CrossRef CAS PubMed.
- S. A. Walling, W. Um, C. L. Corkhill and N. C. Hyatt, npj Mater. Degrad., 2021, 5, 1–20 CrossRef.
- S. Qiu, D. He, J. Ma, T. Liu and T. D. Waite, Electrochim. Acta, 2015, 176, 51–58 CrossRef CAS.
- L. Gubler, S. M. Dockheer and W. H. Koppenol, ECS Meet. Abstr., 2011, MA2011–02, 1141 CrossRef.
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Adv. Mater., 2019, 31, 1–20 Search PubMed.
- E. Fajardo-Puerto, A. Elmouwahidi, E. Bailón-García, A. F. Pérez-Cadenas and F. Carrasco-Marín, Catalysts, 2023, 13, 674 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- J. Feng, C. Chu and Z. Ma, Electrochem. Commun., 2021, 125, 106970 CrossRef CAS.
- S. Hussain, E. Aneggi and D. Goi, Environ. Chem. Lett., 2021, 19, 2405–2424 CrossRef CAS.
- N. V. Maksimchuk, J. Puiggalí-Jou, O. V. Zalomaeva, K. P. Larionov, V. Y. Evtushok, I. E. Soshnikov, A. Solé-Daura, O. A. Kholdeeva, J. M. Poblet and J. J. Carbó, ACS Catal., 2023, 13, 10324–10339 CrossRef CAS PubMed.
- Y. Li, C. J. Miller, L. Wu and T. D. Waite, Environ. Sci. Technol., 2022, 56, 5820–5829 CrossRef CAS PubMed.
- H. Zeng, G. Zhang, Q. Ji, H. Liu, X. Hua, H. Xia, M. Sillanpää and J. Qu, Environ. Sci. Technol., 2020, 54, 14725–14731 CrossRef CAS PubMed.
- W. Zheng, Y. Liu, F. Liu, Y. Wang, N. Ren and S. You, Water Res., 2022, 223, 118994 CrossRef CAS PubMed.
- M. Pourbaix, Lectures on Electrochemical Corrosion, Springer, US, 1973 Search PubMed.
- P. Cao, X. Quan, K. Zhao, S. Chen, H. Yu and Y. Su, Environ. Sci. Technol., 2020, 54, 12662–12672 CrossRef CAS PubMed.
- S. Weon, D. Huang, K. Rigby, C. Chu, X. Wu and J.-H. Kim, ACS ES&T Eng., 2021, 1, 157–172 Search PubMed.
- J. Bak, H. Kim, S. J. Lee, M. J. Kim, E. J. Kim, J. H. Roh, J. W. Shin, C. H. Choi and E. A. Cho, ACS Catal., 2020, 10, 12300–12309 CrossRef CAS.
- D. Böhm, M. Beetz, C. Gebauer, M. Bernt, J. Schröter, M. Kornherr, F. Zoller, T. Bein and D. Fattakhova-Rohlfing, Appl. Mater. Today, 2021, 24, 101134 CrossRef.
- L. Dubau, L. Castanheira, F. Maillard, M. Chatenet, O. Lottin, G. Maranzana, J. Dillet, A. Lamibrac, J. C. Perrin, E. Moukheiber, A. Elkaddouri, G. De Moor, C. Bas, L. Flandin and N. Caqué, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 540–560 CAS.
- Y. Yu, H. Li, H. Wang, X. Z. Yuan, G. Wang and M. Pan, J. Power Sources, 2012, 205, 10–23 CrossRef CAS.
- X. Zhang, Y. Yang, L. Guo and H. Liu, Int. J. Hydrogen Energy, 2017, 42, 4699–4705 CrossRef CAS.
- V. Goellner, C. Baldizzone, A. Schuppert, M. T. Sougrati, K. Mayrhofer and F. Jaouen, Phys. Chem. Chem. Phys., 2014, 16, 18454–18462 RSC.
- T. Kinumoto, K. Takai, Y. Iriyama, T. Abe, M. Inaba and Z. Ogumi, J. Electrochem. Soc., 2006, 153, A58 CrossRef CAS.
- K. Kumar, L. Dubau, M. Mermoux, J. Li, A. Zitolo, J. Nelayah, F. Jaouen and F. Maillard, Angew. Chem., Int. Ed., 2020, 59, 3235–3243 CrossRef CAS PubMed.
- L. Dubau, L. Castanheira, G. Berthomé and F. Maillard, Electrochim. Acta, 2013, 110, 273–281 CrossRef CAS.
- V. Goellner, V. Armel, A. Zitolo, E. Fonda and F. Jaouen, J. Electrochem. Soc., 2015, 162, H403–H414 CrossRef CAS.
- K. Kumar, L. Dubau, M. Mermoux, J. Li, A. Zitolo, J. Nelayah, F. Jaouen and F. Maillard, Angew. Chem., Int. Ed., 2020, 132, 3261–3269 CrossRef.
- S. Ünsal, R. Girod, C. Appel, D. Karpov, M. Mermoux, F. Maillard, V. A. Saveleva, V. Tileli, T. J. Schmidt and J. Herranz, J. Am. Chem. Soc., 2023, 145, 7845–7858 CrossRef PubMed.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- A. Laconti, H. Liu, C. Mittelsteadt and R. McDonald, ECS Trans., 2006, 1, 199–219 CrossRef CAS.
- M. Lefèvre and J. P. Dodelet, Electrochim. Acta, 2003, 48, 2749–2760 CrossRef.
- G. Zhang, R. Chenitz, M. Lefèvre, S. Sun and J. P. Dodelet, Nano Energy, 2016, 29, 111–125 CrossRef CAS.
- C. W. Jones, in Applications of Hydrogen Peroxide and Derivatives, ed. J. H. Clark and M. J. Braithwaite, The Royal Society of Chemistry, 1999, pp.1–36 Search PubMed.
- L. Gubler, S. M. Dockheer and W. H. Koppenol, J. Electrochem. Soc., 2011, 158, B755–B769 CrossRef CAS.
- W. Kiciński and S. Dyjak, Carbon N. Y., 2020, 168, 748–845 CrossRef.
- Y. Lum, Y. Kwon, P. Lobaccaro, L. Chen, E. L. Clark, A. T. Bell and J. W. Ager, ACS Catal., 2016, 6, 202–209 CrossRef CAS.
- P. T. Yu, W. Gu, J. Zhang, R. Makharia, F. T. Wagner and H. A. Gasteiger, in Polymer Electrolyte Fuel Cell Durability, ed. F. N. Büchi, M. Inaba and T. J. Schmidt, Springer, New York, New York, NY, 2009, pp.29–53 Search PubMed.
- W. Shi, K. H. Wu, J. Xu, Q. Zhang, B. Zhang and D. S. Su, Chem. Mater., 2017, 29, 8670–8678 CrossRef CAS.
- C. Poidevin and A. A. Auer, Carbon N. Y., 2021, 171, 618–633 CrossRef CAS.
- K. Ono, Y. Yasuda, K. Sekizawa, N. Takeuchi, T. Yoshida and M. Sudoh, Electrochim. Acta, 2013, 97, 58–65 CrossRef CAS.
- C. Poidevin, P. Paciok, M. Heggen and A. A. Auer, J. Chem. Phys., 2019, 150, 041705 CrossRef PubMed.
- Y. Qi, Y. Huang, Z. Gao, C. H. Chen, A. Perego, H. Yildirim, M. Odgaard, T. Asset, P. Atanassov and I. V. Zenyuk, J. Power Sources, 2022, 551, 232209 CrossRef CAS.
- S. Vinod Selvaganesh, G. Selvarani, P. Sridhar, S. Pitchumani and A. K. Shukla, Fuel Cells, 2011, 11, 372–384 CrossRef CAS.
- C. H. Choi, H. K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M. T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- Y. Hashimasa, T. Numata, K. Moriya and S. Watanabe, J. Power Sources, 2006, 155, 182–189 CrossRef CAS.
- J. S. Spendelow and D. C. Papageorgopoulos, Fuel Cells, 2011, 11, 775–786 CrossRef CAS.
- S. Stariha, N. Macauley, B. T. Sneed, D. Langlois, K. L. More, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F492–F501 CrossRef CAS.
- S. Chen, Z. Wei, L. Guo, W. Ding, L. Dong, P. Shen, X. Qi and L. Li, Chem. Commun., 2011, 47, 10984–10986 RSC.
- X. Huang, M. Song, J. Zhang, T. Shen, G. Luo and D. Wang, Nano-Micro Lett., 2023, 15, 86 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00109e |
| This journal is © The Royal Society of Chemistry 2024 |