
Thermally activated delayed fluorescence tetradentate ligand-containing gold(III) complexes with preferential molecular orientation and their application in organic light-emitting devices†
Cathay Chai
Au-Yeung
 a,
Ming-Yi
Leung
ab,
Shiu-Lun
Lai
a,
Shun-Cheung
Cheng
c,
Lok-Kwan
Li
a,
Man-Chung
Tang
a,
Wing-Kei
Kwok
ab,
Chi-Chiu
Ko
c,
Mei-Yee
Chan
*ab and
Vivian Wing-Wah
Yam
*ab
a,
Ming-Yi
Leung
ab,
Shiu-Lun
Lai
a,
Shun-Cheung
Cheng
c,
Lok-Kwan
Li
a,
Man-Chung
Tang
a,
Wing-Kei
Kwok
ab,
Chi-Chiu
Ko
c,
Mei-Yee
Chan
*ab and
Vivian Wing-Wah
Yam
*ab
aInstitute of Molecular Functional Materials and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: wwyam@hku.hk; chanmym@hku.hk; Tel: +(852) 2859-2153 Tel: +(852) 2857-1586
bHong Kong Quantum AI Lab Limited, 17 Science Park West Avenue, Pak Shek Kok, Hong Kong, P. R. China
cDepartment of Chemistry, City University of Hong Kong, Kowloon, Hong Kong, P. R. China
First published on 27th October 2023
Abstract
A new class of thermally activated delayed fluorescence (TADF) pyridine-/pyrazine-containing tetradentate C^C^N^N gold(III) complexes have been designed and synthesized. Displaying photoluminescence quantum yields (PLQYs) of up to 0.77 in solid-state thin films, these complexes showed at-least a six-fold increase in the radiative decay rate constant (kr) in toluene upon increasing temperature from 210 to 360 K. Using variable-temperature (VT) ultrafast transient absorption (TA) spectroscopy, the reverse intersystem crossing (RISC) processes were directly observed and the activation parameters were determined, in line with the results of the Boltzmann two-level model fittings, in which the energy separation values between the lowest-lying singlet excited state (S1) and the lowest-lying triplet excited state (T1), ΔE(S1–T1), of these complexes were estimated to be in the range of 0.16–0.18 eV. Through strategic modification of the position of the electron-donating –tBu substituent in the cyclometalating ligand, the permanent dipole moments (PDMs) of these tetradentate gold(III) emitters could be manipulated to enhance their horizontal alignment in the emitting layer of organic light-emitting devices (OLEDs). Consequently, the resulting vacuum-deposited OLEDs demonstrated a 30% increase in the theoretical out-coupling efficiency (ηout), as well as promising electroluminescence (EL) performance with maximum external quantum efficiencies (EQEs) of up to 15.7%.
 Vivian Wing-Wah Yam | Our first Material Horizons article was published in the special issue in honour of Professor Seth Marder reporting a new class of robust C^C^N gold(III) complexes with a promising operational half-lifetime of more than 200 |
New conceptsIt is known that by promoting the horizontal orientation of the emitters, a larger fraction of generated photons can be dissipated as emission, leading to a higher ηout of the OLED devices. While many studies have investigated the influence of several parameters such as the aspect ratios or the glass-transition temperature of emitter/host molecules on the molecular orientation, the direct relationship between the PDMs of the emitters and their molecular orientation, however, has much less been explored. Here, we present the design and synthesis of horizontally oriented tetradentate C^C^N^N ligand-containing gold(III) complexes and the direct observation of the RISC processes and the determination of their activation parameters. This not only marks the first example to demonstrate green-emitting TADF gold(III) complexes with tetradentate C^C^N^N ligand frameworks, but also a successful demonstration of enhancing the horizontal alignment of emitters via a slight modification in the molecular design to boost the gold(III) OLED performance. The strategy of manipulating the PDMs of emitters towards the control of molecular orientations has never been demonstrated in the gold(III) system and is also scarcely reported in the widely studied iridium(III), platinum(II) and TADF systems. This work demonstrates the functionality of the tert-butyl substituents in realizing the horizontal-preferred molecular orientation as well as the enhancing operational stability, providing novel insights into the design of horizontally aligned gold(III) emitters. |
Introduction
Being a mainstream display technology for smartphones and televisions nowadays, organic light-emitting diodes (OLEDs) have been extensively studied in both industry and academia. To meet the standard for commercialization, high internal quantum efficiencies (ηint) and high out-coupling efficiencies (ηout) are required for OLEDs to achieve high external quantum efficiencies (EQEs), which are defined by ηext = γ·ηr·qeff·ηout ≡ ηint·ηout.1,2 Particularly, ηint is contributed by γ, the carrier balance of electrons and holes; ηr is the radiative exciton generation efficiency, and qeff is the emission efficiency, which is usually referred to as the photoluminescence quantum yield (PLQYs) of the emitting layer. With the aim of maximizing ηr, thermally activated delayed fluorescence (TADF) has been frequently featured in the emitter design,3,4 in which TADF can be enabled with a small energy gap between the lowest triplet excited state (T1) and the lower singlet excited state (S1), ΔE(S1–T1), to harvest both singlet and triplet excitons in the organic TADF emitters. Meanwhile, metal-containing TADF emitters have gained huge attention for their high PLQYs approaching 100% and excellent electroluminescence (EL) performances.5–7 The presence of the heavy metal atom leads to a strong spin–orbit coupling (SOC) effect, resulting in much faster intersystem crossing (ISC) and reverse-ISC (RISC) rate constants when compared to organic TADF emitters. Metal-TADF emitters generally afford high radiative decay rate constants (kr) of 105–106 s−1 and have been widely demonstrated in d10 metal complexes, e.g. copper(I) and gold(I) carbene-metal-amides (CMAs).6–8 Recently, several luminescent gold(III) complexes have also been found to display TADF properties and exhibit promising EL performance with maximum EQEs reaching >20%.9–17Apart from ηr, the PLQY of emitters is also vital for attaining high EQEs. To this end, several strategies could be applied to the emitter design to achieve higher PLQYs and thus higher ηint, for example, incorporating multidentate ligands into the molecular structure to reduce the non-radiative decay rate constants (knr), as a higher denticity and rigidity could suppress the conformational deformation upon the interconversions between the ground state and the emissive state, resulting in a more efficient radiative decay.17–19 Several tetradentate emitters with d6 iridium(III)20–22 or d8 platinum(II)23–27 metal center have shown high PLQYs. As for the d8 gold(III) system, Yam and co-workers first reported tetradentate gold(III) emitters in 2017, prepared through post-synthetic cyclization.28 These gold(III) complexes with a rigid C^N^C^C tetradentate scaffold displayed high PLQYs of up to 0.78 and afforded orange-red-emitting solution-processed OLEDs with EQEs of up to 11.1%.28 Since then, a lot of effort has been made to expand the family of tetradentate gold(III) complexes,15,17,28–32 including the one-pot two bond-forming approach for the preparation of fully π-conjugated tetradentate C^C^N^C- and N^C^N^C-cyclometalated gold(III) complexes reported by Yam and co-workers,31 and the ether linkage-containing C^N^C^C gold(III) complexes reported by Che and co-workers32 similar to the amine-linked C^N^C^C gold(III) system.28 Recently, inspired by the formerly reported highly efficient tridentate C^C^N gold(III) emitters,33 our group reported another series of tetradentate gold(III) complexes containing fully π-conjugated C^C^N^N ligands.29 These complexes exhibited not only high PLQYs of up to 0.70, but also a preferential horizontal alignment of transition dipole moment vectors (TDMVs) in the emissive host–guest matrix with a horizontal dipole ratio (Θh) of 0.87 in solid-state thin films.29 The restricted rotation of the carbazole moiety in these complexes is believed to render a higher robustness of the ligand framework and thus a better control of the intermolecular interactions with the host, leading to the high Θh.29 Given the attractiveness of the square-planar geometry of d8 gold(III) complexes where all TDMVs lie in the plane of the complex molecules, a more horizontally aligned TDMV with respect to the substrate surface resulting from the more horizontal orientation of the gold(III) complex molecules can reduce the intensity loss in waveguide and surface plasmon modes, and hence result in a higher ηout,29 as demonstrated in several vacuum-evaporated organic molecules and OLED applications.17,34–36 With a Θh of 0.87 and a high estimated-ηout of 30%, the green-emitting vacuum-deposited OLEDs fabricated with these tetradentate C^C^N^N gold(III) emitters demonstrated EQEs reaching 20.6%.29
In consideration of higher ηr, PLQYs and ηout towards the design of efficient gold(III) emitters, we present a new series of green-/red-emitting tetradentate C^C^N^N ligand-containing gold(III) complexes 1–6 (Scheme 1), in which the pyridine unit in 1–4 was replaced by the more electron-accepting pyrazine unit in 5–6 to extend the emission color range of the C^C^N^N gold(III) complexes from green to red. A rigid tetradentate framework has been adopted to reduce knr. Furthermore, a simplified design with one less phenyl ring at the 4-position of the central phenyl ring compared to the aforementioned C^C^N^N gold(III) emitters29 reduces the freedom of rotation in the molecules. As a result, these complexes demonstrated an improvement in the maximum PLQY by 10% from 0.7029 to 0.77 in doped 1,3-bis(N-carbazolyl)benzene (mCP) thin films. Despite the highly rigid structure under the tetradentate ligand framework, the presence of the twisted donor–acceptor-structure reduces the highest occupied molecular orbital (HOMO)–lowest occupied molecular orbital (LUMO) overlap to afford a smaller ΔE(S1–T1). Notably, TADF was observed in the variable-temperature (VT) emission studies, in which selected complexes showed more than a six-fold increase in kr in toluene with increasing intensity and a hypsochromic shift of 0.10–0.17 eV upon increasing temperature. Such an increase in the emission intensity and the spectral shift was also found in the doped mCP thin films. The ΔE(S1–T1) values of 1, 3 and 5 estimated by the Boltzmann model were found to be ∼0.16–0.18 eV, which further support the TADF properties in 1–6, in co-existence with phosphorescence (see below). VT femtosecond transient absorption (fs-TA) spectroscopy was conducted on 1, 3 and 5 to explore their excited state dynamics. The activation energy for the RISC process of these complexes was estimated to be 0.06–0.11 eV, which is generally in line with the estimation by the VT emission studies and the Boltzmann model. By changing the position of the electron-donating –tBu substituent in the molecular design, we could further enhance ηout by manipulating the permanent dipole moments (PDMs) of the gold(III) complexes to enhance their horizontal alignments in the OLEDs. Complexes 3 and 4 were specifically designed as the regioisomer of 1 and 2, respectively, which differ only in the position of the –tBu group at either R1 or R2 site of the C^C^N^N ligand (see Scheme 1). It is known that the orientation of TDMVs can be engineered by the host–dopant interaction.35–37 While the PDM of the complex molecules could be correlated with the horizontal orientation of the TDMVs of the dopants in the host–guest matrix,35–38 the change in the electrostatic interactions brought about by the manipulation of the PDMs might afford a more horizontally aligned TDMV of the emitters, and thus a higher ηout and EQE of the resulting devices. Consequently, complexes 3 and 4 demonstrated a more horizontal alignment of TDMVs in vacuum-deposited films than 1 and 2, and improvements in theoretical ηout by ∼30%, which is reflected in the higher EQEs of the vacuum-deposited devices based on the preferentially horizontal-aligned 3 and 4, i.e., ∼24 and ∼49% higher than those of 1 and 2. This work not only demonstrates the promising EL performances of the C^C^N^N ligand-containing TADF gold(III) complexes, but also shows that a slight structural modification of the emitters can effectively manipulate the molecular anisotropy, leading to a significant improvement in the EQEs of OLEDs.
 | ||
| Scheme 1 Molecular structures of the tetradentate C^C^N^N ligand-containing gold(III) complexes 1–6. | ||
Results and discussion
Synthesis, characterization and crystal packing
Complexes 1–6 were synthesized by treating the respective chlorogold(III) precursors with the corresponding boronic ester acids in the presence of a catalytic amount of palladium catalyst (Scheme S1, ESI†). The C^C^N cyclometalating ligand precursors were prepared by following procedures reported previously by us with slight modifications,29 followed by metalation and transmetalation to the gold(III) metal center to afford the tridentate ligand-containing chlorogold(III) precursors. The complexes were then purified using column chromatography, followed by recrystallization by diffusion of diethyl ether into a concentrated dichloromethane solution of the respective complexes.39–42 All the complexes were characterized by 1H, 13C{1H} nuclear magnetic resonance (NMR) spectroscopy, and high-resolution electrospray ionization (ESI) mass spectroscopy. The molecular structure and crystal packing of 5 were studied by X-ray crystallography as shown in Fig. S1 and S2 (ESI†) and the details are listed in Tables S1 and S2 (ESI†). Unlike the previously reported tetradentate C^C^N^N gold(III) complex,295 did not show a parallel packing between two molecules. The loose molecular packing and the absence of π–π stacking interactions between neighboring molecules are probably due to the bent molecular geometry led by the incorporation of the bulky –tBu groups and the restricted rotation of the C^C^N^N ligand.Electrochemistry
The electrochemical properties of 1–6 in dichloromethane (0.1 M nBu4NPF6) were investigated by cyclic voltammetry (Fig. S3, ESI†). Their electrochemical data and the estimated HOMO–LUMO energy gaps are listed in Table S3 (ESI†). In the reductive scans, 1–4 show an irreversible reduction process at ca. −1.70 V vs. SCE, while 5 and 6 show a quasi-reversible reduction couple at −1.27 V vs. SCE. With reference to previously reported tetradentate gold(III) complexes,29 these reduction waves are assigned to the ligand-centered reduction of the C^C^N^N ligand. The replacement of the pyridine moiety in 1–4 by a more electron-accepting pyrazine in 5–6 has led to a less negative reduction potential, attributed to the more stabilized π*(C^C^N^NPz) orbital. As for the oxidative scans, 1 and 3 show an irreversible first oxidation wave at +1.07 V vs. SCE, while 2, 4, 5 and 6 show a quasi-reversible oxidation couple at ca. +0.95 V vs. SCE. The attachment of –tBu groups onto the carbazolyl unit accounts for the less positive potentials for oxidation in 2, 4, 5 and 6.29 Therefore, these oxidation waves are assigned to the carbazolyl ligand-centered oxidations. The estimated HOMO–LUMO energy gaps display a decreasing trend, i.e., ∼2.78 (1 and 3) > ∼2.66 (2 and 4) > ∼2.23 eV (5 and 6), which is in agreement with the results from the emission studies and theoretical calculations (see below).Photophysical properties
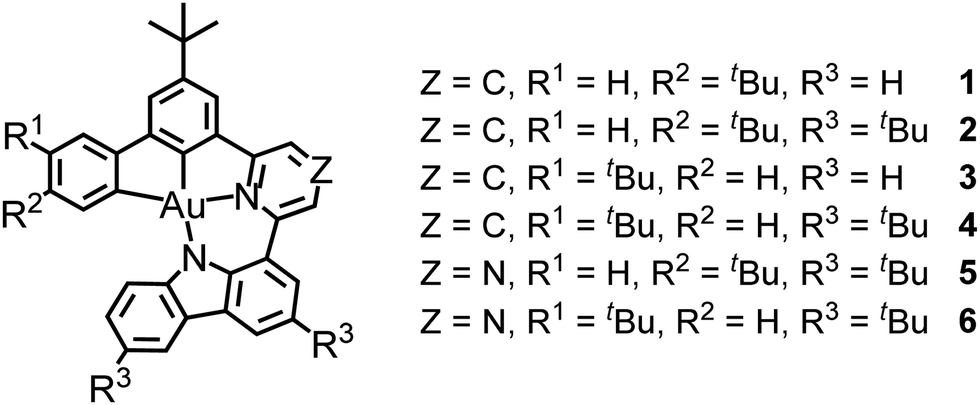
UV–vis absorption and emission studies were conducted on 1–6 in degassed toluene at 298 K (Fig. 1). Their electronic absorption and luminescence data are shown in Table S4 (ESI†) and Table 1, respectively. 1–6 show an intense absorption band at wavelength (λ) of ca. 300–400 nm and a moderately intense structureless band peaking at 462–528 nm, with extinction coefficient (ε) in the order of 104 mol−1 dm3 cm−1 (Fig. 1a). The higher-energy intense absorption bands at <400 nm can be mainly attributed to the spin-allowed intraligand (IL) [π → π*(C^C^N^N)] transition,29 while the intense lowest-energy absorption bands at >460 nm are attributed to the IL charge transfer (ILCT) [π(Cbz) → π*(Py/Pz)] transition. They are found to be dependent on the nature of the N-heterocycle moieties and the electron-donating strength of the carbazole moieties. Compared to 1 and 3 which comprise an unsubstituted carbazole, the incorporation of electron-donating –tBu groups into the carbazolyl unit in 2 and 4 has led to a slight bathochromic shift of the absorption bands, owing to the destabilization of HOMO that is mainly localized on the π(Cbz) orbital, while replacing the pyridine moiety by a pyrazine unit in 5 and 6 has led to a more stabilized π* orbital of the C^C^N^N ligand, with the lowest-energy absorption band further red-shifted to 528 nm. Overall, the trend of the absorption bands of 1–6 is found to be in good agreement with the electrochemical studies and the computational studies (see below).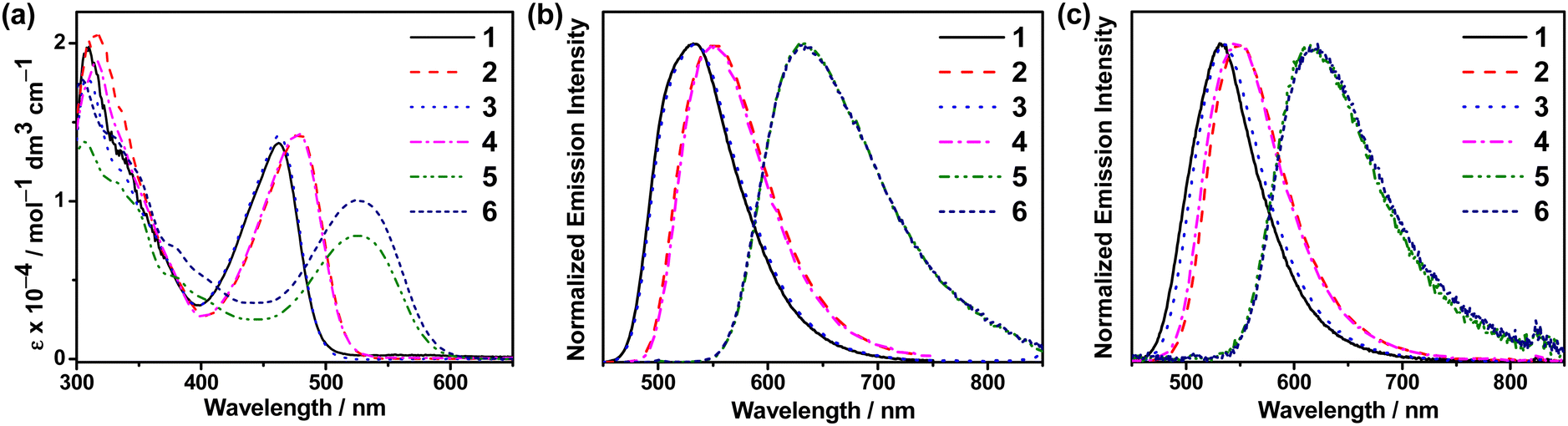 | ||
| Fig. 1 (a) UV–vis absorption spectra of 1–6 in toluene at 298 K. (b) Normalized emission spectra of 1–6 in (b) degassed toluene and (c) 10 wt%-doped mCP thin films at 298 K. | ||
| Complex | Medium (T/K) | Emission λmax/nm (τ0/μs) | Φ PL | k r /s−1 | k nr /s−1 |
|---|---|---|---|---|---|
a The relative luminescence quantum yields of the gold(III) complexes in solution were measured at room temperature using [Ru(bpy)3]Cl2 in degassed acetonitrile as the reference (excitation wavelength = 436 nm, Φlum = 0.060).
b Absolute luminescence quantum yield of the gold(III) complexes doped into mCP excited at a wavelength of 310 nm.
c Radiative decay rate constant determined from the equation kr = ΦPL/τ0; non-radiative decay rate constant determined from the equation knr = (1 − ΦPL)/τ0.
d Measured in EtOH–MeOH–CH2Cl2 (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:1, v/v).
e Prepared by spin-coating a blend of the complex:host in chloroform.
f Biexponential decay. :10:1, v/v).
e Prepared by spin-coating a blend of the complex:host in chloroform.
f Biexponential decay.
|
|||||
| 1 | Toluene (298) | 532 (8.1) | 0.23 | 2.8 × 104 | 9.5 × 104 |
| Solid (298) | 600 (0.2) | ||||
| Solid (77) | 559 (5.4, 38.9)f | ||||
| Glass (77)d | 540, 578 (262.4) | ||||
| Thin film (298) | |||||
| 5 wt% in mCPe | 530 (2.2, 21.6)f | 0.76 | |||
| 10 wt% in mCPe | 531 (1.9, 22.1)f | 0.77 | |||
| 15 wt% in mCPe | 533 (1.7, 17.3)f | 0.71 | |||
| 20 wt% in mCPe | 533 (1.6, 13.6)f | 0.68 | |||
| 2 | Toluene (298) | 550 (7.2) | 0.19 | 2.6 × 104 | 1.3 × 105 |
| Solid (298) | 530 (0.3) | ||||
| Solid (77) | 550, 583 (4.8, 31.7)f | ||||
| Glass (77)d | 525, 580 (253.5) | ||||
| Thin film (298) | |||||
| 5 wt% in mCPe | 546 (2.9, 18.8)f | 0.75 | |||
| 10 wt% in mCPe | 548 (2.8, 16.4)f | 0.74 | |||
| 15 wt% in mCPe | 550 (2.3, 15.4)f | 0.74 | |||
| 20 wt% in mCPe | 554 (2.2, 13.6)f | 0.68 | |||
| 3 | Toluene (298) | 534 (10.5) | 0.20 | 1.9 × 104 | 7.6 × 104 |
| Solid (298) | 575 (0.2) | ||||
| Solid (77) | 552, 581 (4.4, 36.8)f | ||||
| Glass (77)d | 517, 552 (243.7) | ||||
| Thin film (298) | |||||
| 5 wt% in mCPe | 534 (2.3, 20.6)f | 0.74 | |||
| 10 wt% in mCPe | 535 (1.9, 16.6)f | 0.72 | |||
| 15 wt% in mCPe | 540 (2.0, 14.7)f | 0.63 | |||
| 20 wt% in mCPe | 541 (2.1, 13.3)f | 0.63 | |||
| 4 | Toluene (298) | 549 (4.8) | 0.21 | 4.4 × 104 | 1.7 × 105 |
| Solid (298) | 548 (0.2) | ||||
| Solid (77) | 548 (4.0, 23.8)f | ||||
| Glass (77)d | 567, 617 (212.8) | ||||
| Thin film (298) | |||||
| 5 wt% in mCPe | 546 (2.4, 20.9)f | 0.67 | |||
| 10 wt% in mCPe | 548 (1.5, 16.9)f | 0.64 | |||
| 15 wt% in mCPe | 549 (1.4, 13.5)f | 0.59 | |||
| 20 wt% in mCPe | 554 (0.9, 9.3)f | 0.58 | |||
| 5 | Toluene (298) | 635 (5.3) | 0.09 | 1.7 × 104 | 1.7 × 105 |
| Solid (298) | 628 (0.1) | ||||
| Solid (77) | 627 (0.3, 20.1)f | ||||
| Glass (77)d | 620 (94.7) | ||||
| Thin film (298) | |||||
| 5 wt% in mCPe | 604 (1.6, 8.2)f | 0.30 | |||
| 10 wt% in mCPe | 614 (0.9, 5.1)f | 0.21 | |||
| 15 wt% in mCPe | 621 (0.6, 3.5)f | 0.16 | |||
| 20 wt% in mCPe | 627 (0.4, 2.0)f | 0.13 | |||
| 6 | Toluene (298) | 630 (5.6) | 0.11 | 2.0 × 104 | 1.6 × 105 |
| Solid (298) | 631 (0.3) | ||||
| Solid (77) | 629 (33.9) | ||||
| Glass (77)d | 652, 677, 711 (87.0) | ||||
| Thin film (298) | |||||
| 5 wt% in mCPe | 608 (2.4, 9.7)f | 0.24 | |||
| 10 wt% in mCPe | 615 (0.4, 3.9)f | 0.17 | |||
| 15 wt% in mCPe | 621 (0.3, 3.0)f | 0.15 | |||
| 20 wt% in mCPe | 630 (0.2, 1.4)f | 0.08 | |||
Upon excitation at λ > 400 nm, 1–6 show structureless emission bands with λem = 532 and 534 (1 and 3), 550 and 549 (2 and 4) and 635 and 630 nm (5 and 6), which are found to be sensitive to the nature of the N-heterocycles and the carbazolyl units (Fig. 1b). The Gaussian-shaped emission bands of 2 and 4 are ca. 15 nm red-shifted with respect to those of 1 and 3, owing to the more stabilized HOMO in 1 and 3 that is localized on the π(Cbz) orbital. On the other hand, the use of pyrazine in 5 and 6 has led to red emissions, owing to the more stabilized π*(Pz) orbital of the C^C^N^N ligand. The emission bands of this series of gold(III) C^C^N^N complexes are assigned as originating from the 3ILCT [π(Cbz) → π*(Py/Pz)] excited state. These complexes in toluene solution exhibit PLQYs of 0.09–0.23 and excited state lifetimes of 4.8–10.5 μs, with kr in the order of 104 s−1.
Solvent-dependent absorption and emission spectra were measured for 3 and 5 to investigate the excited state nature. The lowest-energy absorption bands of 3 and 5 show negative solvatochromism (Fig. S4, ESI†), which further supports the assignment of ILCT [π(Cbz) → π*(Py/Pz)] transitions.29 Upon increasing solvent polarities, the emission of 3 exhibits an insignificant spectral change (Fig. S5a, ESI†). This is because the emission of 3 is not predominantly of CT character but involves also an IL character, as it has a similar wavelength to the vibronic-structured emission of its chlorogold(III) precursor in toluene peaking at 486, 525 and 564 nm (Fig. S6, ESI†), which is originated from the IL [π → π*(C^C^N^N)] excited state. Nevertheless, 3 shows a positive slope of 6217 cm−1 in the Lippert–Mataga plot, indicating a mixing of a CT character of its excited states. On the other hand, the emission of 5 exhibits a significant spectral shift upon changing solvent polarities, with a more positive slope of 9767 cm−1 in the Lippert–Mataga plot (Fig. S5b, ESI†), suggesting a much larger dipole moment in the excited state than in the ground state and a greater CT character in the emission of 5.
In 10 wt%-doped mCP thin films (Fig. 1c), 1–4 show high PLQYs of up to 0.77 and structureless emissions with λem = 531–548 nm, which follow the trend observed in toluene. The pyrazine-containing 5 and 6 show Gaussian-shaped emissions with λem = 614–615 nm and PLQYs of up to 0.30. The lower PLQYs of 5 and 6 can be ascribed to the energy gap law, which suggests that a lower emission energy results in a faster non-radiative decay process. The emission spectra of the doped mCP thin films of 1–6 show bi-exponential decays with lifetimes of up to 22.1 μs. With the calculated kr in the range of 104 s−1, the emissions of 1–6 in mCP films are ascribed to be originating mainly from the 3ILCT [π(Cbz) → π*(Py/Pz)] excited state, possibly with some mixing of the 3IL [π → π*(C^C^N^N)] state and 1ILCT [π(Cbz) → π*(Py/Pz)] state (see below). The time-resolved emission of 3 in the 5 wt%-doped mCP film shows a more structured band at longer delay times (Fig. S7, ESI†), which probably originates from the 3IL state due to its longer lifetime, again revealing the possible mixing of the 3ILCT and 3IL excited states. Upon increasing dopant concentration (Fig. S8, ESI†), the emission spectra of 1–4 show almost no spectral shift, suggesting the absence of excimeric emission. The insensitivity of the emission energies towards the change in the matrix polarity also suggests the involvement of 3IL [π → π*(C^C^N^N)] state besides the 1/3ILCT [π(Cbz) → π*(Py/Pz)] emission origin. As for 5 and 6, the bathochromic shifts from ca. 604 to 630 nm upon increasing dopant concentration are due to a larger contribution of the 3ILCT excited state that becomes more stabilized at a higher local polarity of the host matrix.12
Variable-temperature (VT) studies
VT studies were conducted on selected C^C^N^N carbazolylgold(III) complexes to investigate their TADF properties. The emission spectra of 3 and 5 in 5 wt%-doped mCP thin films show increased intensities, shortened emission lifetimes, and a hypsochromic shift of λem of ∼0.05 eV (437 and 371 cm−1, respectively) upon increasing temperature (Fig. S9 and Table S5, ESI†), which are likely due to TADF.11,12,32,43 However, at low temperatures, 3 shows a vibronic-structured emission, which could be attributed to the reduced molecular motions and the more rigid environment especially in thin films.7,44 Although the emergence of such a 3IL state-originated emission at low temperatures is common for metal complexes, the presence of the IL state would complicate the TADF analysis, as the TADF process in metal complexes concerns mainly the 1/3CT states.5,7 Thus, VT studies were also performed in the solution state of the complexes to further investigate their TADF properties. Fig. 2 shows the VT emission spectra of 1, 3, 4 and 5 in toluene, which exhibit an overall enhancement in intensity and maintain a structureless band shape throughout the whole temperature range. As for the spectral changes, hypsochromic shifts of 24–25 nm (799–879 cm−1, ∼0.10 eV) for 1, 3 and 4, and 58 nm (1363 cm−1, ∼0.17 eV) for 5 are observed upon increasing temperature, indicating an up-conversion process to another CT state of higher energy. Apart from the enhanced intensities and hypsochromic shifts, the change in emission lifetimes has also been recorded for 1, 3 and 5, in which shortened emission lifetimes from 15.2 to 7.1 μs for 1, 16.8 to 4.8 μs for 3, and 5.4 to 4.0 μs for 5 are observed upon increasing temperature from 210 to 360 K (Table S6, ESI†). It implies that kr also increases upon increasing temperature. By fitting to the Boltzmann two-level model, ΔE(S1–T1) and the lifetime of the S1 and T1 states have been estimated using the formula:45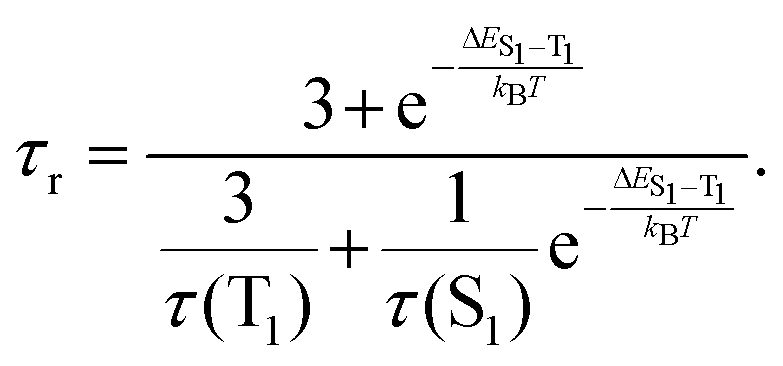 | (1) |
 | ||
| Fig. 2 Emission spectra of (a) 1, (b) 3, (c) 4 and (d) 5 in degassed toluene upon increasing temperature. | ||
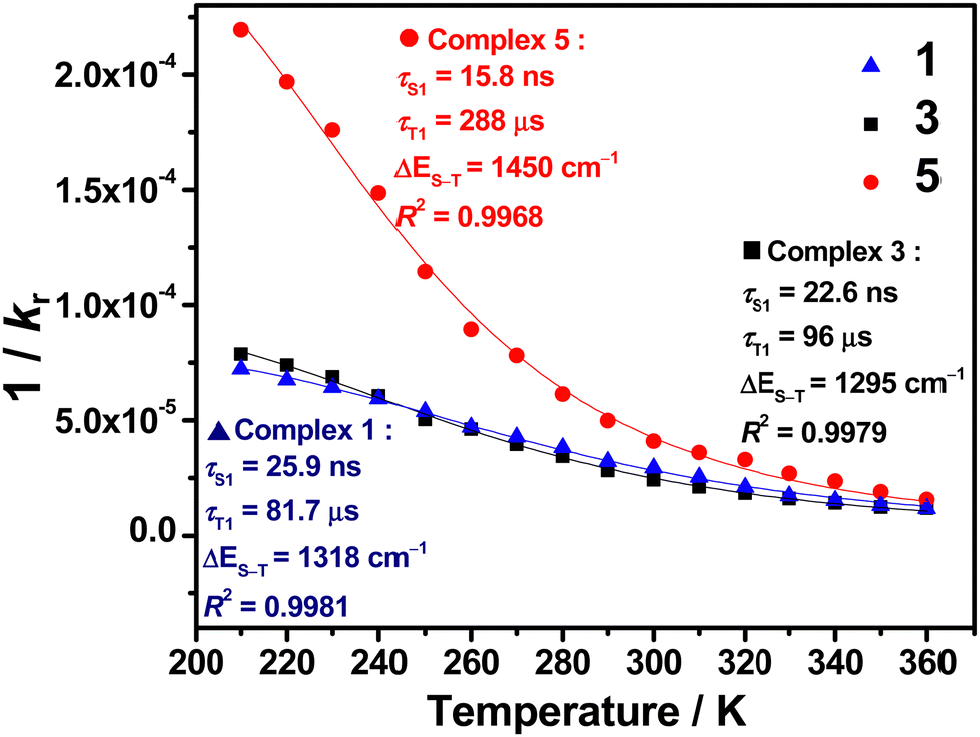 | ||
| Fig. 3 A plot of radiative lifetime vs. temperature of 1, 3 and 5 in degassed toluene monitored at λem and the fits of the temperature-dependent data to eqn (1). | ||
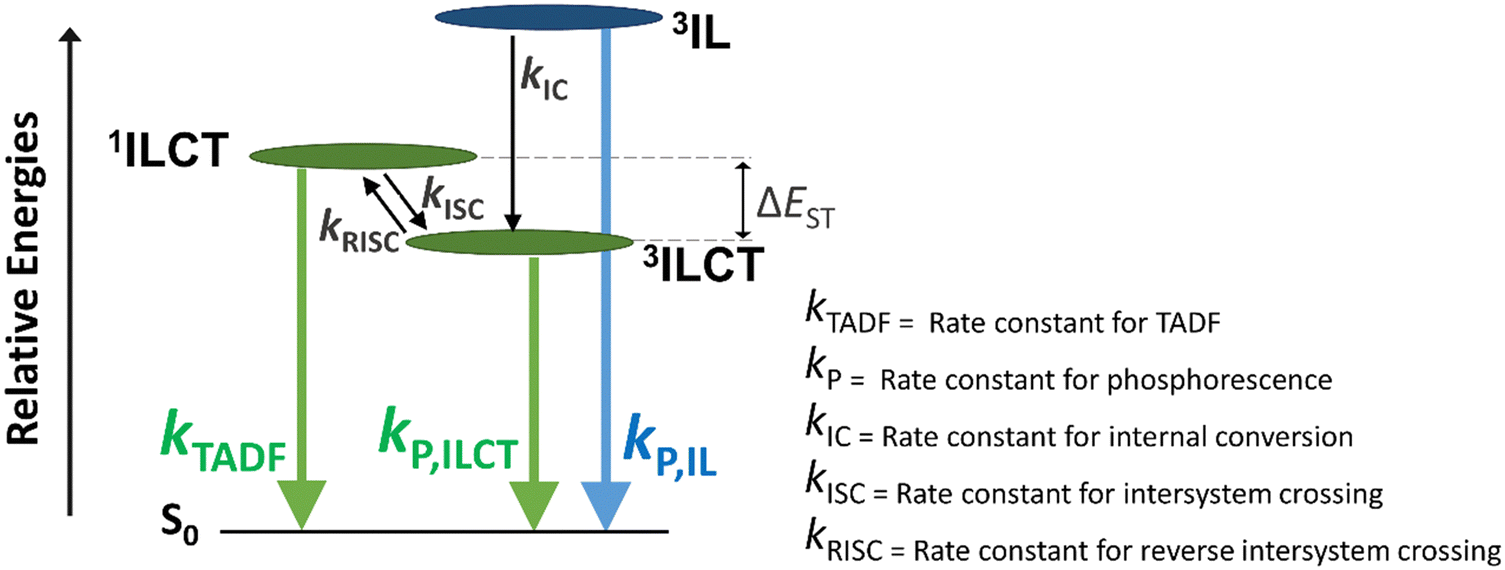 | ||
| Fig. 4 General schematic diagram illustrating the emission pathways of 1–6. kTADF, kP, kIC, kISC, and kRISC represent the rate constants for thermally activated delayed fluorescence, phosphorescence, internal conversion, intersystem crossing, and reverse intersystem crossing, respectively. | ||
Ultrafast transient absorption spectroscopy (TAS)
To elucidate the excited-state dynamics of the complexes, 1, 3 and 5 have been further investigated with femtosecond (fs) and picosecond (ps) TA measurements in toluene solution. The fs-TA difference spectra measured at 293 K at different pump–probe delay times are shown in Fig. 5 and Fig. S10, S11 (ESI†). In general, the kinetics of the TA spectral changes of 1 and 3 are similar and could be described using four major exponential components, whereas those of 5 can be described using five exponential components. With reference to the time constants of these components, selected TA spectra in the time ranges showing the major spectral changes of each component are separately shown in panels (a)–(d). Taking the TA spectra of 1 (Fig. 5) as an illustrative example, the four exponential components include (a) an evolution of the ground-state bleaching to a more negative ΔOD at ca. 460 nm with an initial rise of absorption at 550–750 nm (Fig. 5a) with the shortest time constant (τ1) of ca. 200 fs, (b) a growth of an absorption band at 500–525 nm (τ2 = ca. 1.0 ps) with a subtle change in the absorption band at the 650–750 nm region (Fig. 5b), (c) a further rise of the signal at 500–625 nm which likely overlaps with the two absorption bands at the 500–525 and 650–750 nm regions (Fig. 5c), and (d) a prolonged decay with spectra (Fig. 5d) similar to those observed in the ps-TA spectra (Fig. 5g). Since the fs-TA spectra are recorded within a 3 ns pump–probe delay, the time constants (>20 ns) for the prolonged decay cannot be determined accurately, and the isosbestic point observable in the ps-TA spectra (Fig. 5g) cannot be clearly identified in the long timescale fs-TA spectra (Fig. 5d). With reference to other luminescent gold(III) complexes,46 the initial rise (τ1) can be ascribed to the ISC from an initially populated singlet excited state to a triplet state. The ground-state bleaching at ca. 460 nm, which corresponds to the ILCT [π(Cbz) → π*(Py)] band of the respective complexes in the absorption spectra, and the similarity of the TA band beyond 500 nm in the prolonged decay to that of the carbazolyl radical cations31,47 are consistent with the population of the ILCT excited state. As for 5, which consists of different N-heterocyclic and carbazolyl moieties, its spectral changes (Fig. S11, ESI†) are dissimilar to those for 1 and 3. The bleaching observed at ca. 530 nm corresponding to the more red-shifted ILCT band in 5 is consistent with its ILCT [π(Cbz) → π*(Pz)] assignment. In addition, 5 shows an initial rise of two absorption bands at ca. 420 nm and ca. 685 nm with τ1 of ca. 350 fs, similar to those observed in 1 and 3, corresponding to the ISC. Despite the differences in the excited-state dynamics between 1/3 and 5 (see illustration in Fig. S11, ESI†), the assignments of the initial rise (τ1) component and the ground-state bleaching for 1, 3 and 5 are generally the same.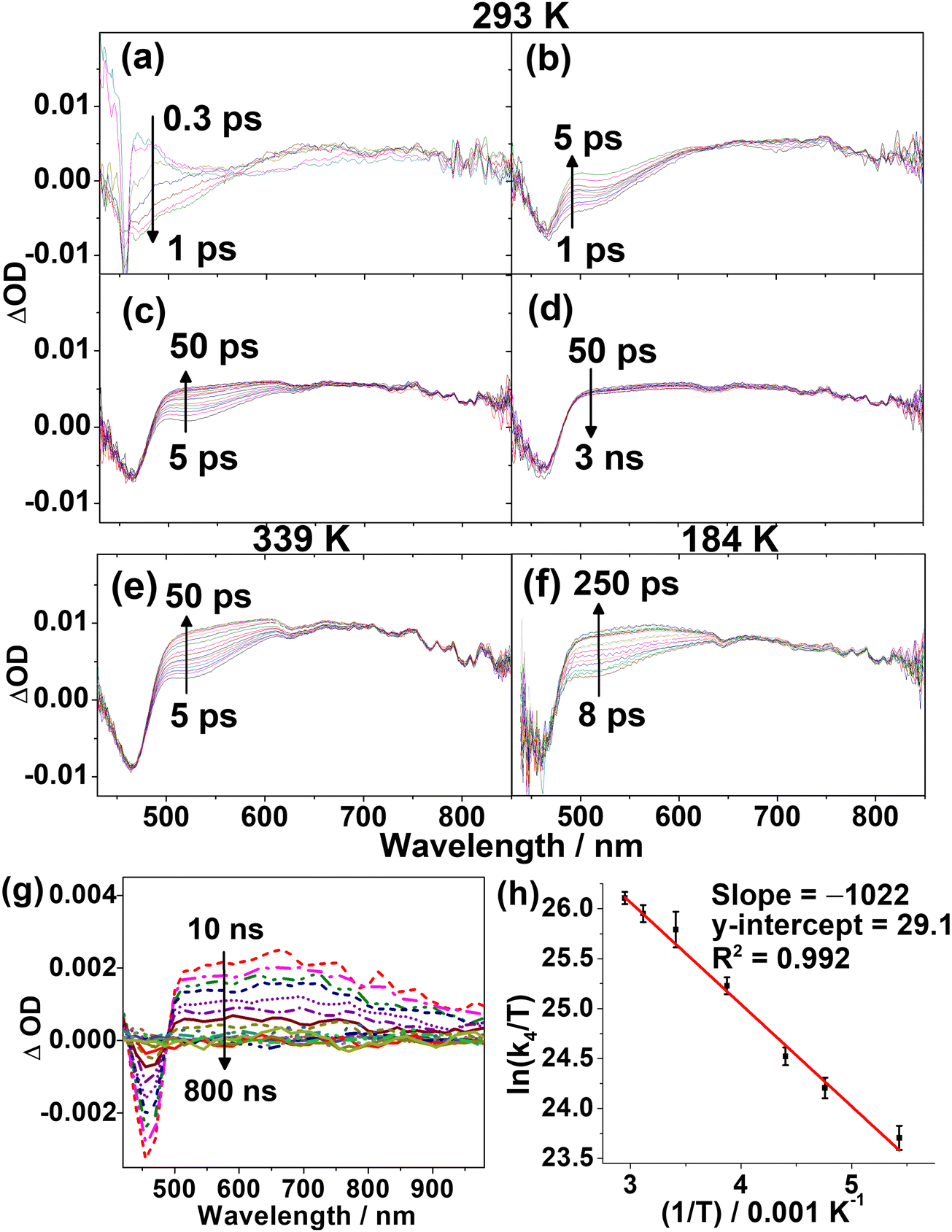 | ||
| Fig. 5 Selected fs-TA spectra of 1 in aerated toluene at 293 K recorded at (a) 0.3–1 ps, (b) 1–5 ps, (c) 5–50 ps, and (d) 50–3000 ps, and the spectra recorded at (e) 5–50 ps at 339 K and (f) 8–250 ps at 184 K after 400 nm fs-laser excitation. (g) ps-TA spectra of 1 in toluene at 298 K. (h) Arrhenius plot of k4(τ4) of 1. | ||
To gain insights into the absorption growth processes subsequent to the initial ISC (τ1), VT fs-TA spectra of 1, 3 and 5 (Fig. S12–S14, ESI†) from 184 to 339 K were analyzed (Table S7, ESI†). The close similarity of the fs-TA spectral changes in 3 at different temperatures (Fig. S10, S13 and Table S7, ESI†) to those observed in 1 is understandable due to their similar structures and also suggests that 1 and 3 have similar excited-state dynamics. As shown in the TA spectra of 1 recorded at 184 and 339 K (Fig. S12, ESI†), the ΔOD profiles are similar to those at 293 K (Fig. 5a–d). Notably, the kinetics of the further absorption growth at delay times beyond 5 ps (component τ4 = 4.6–50.7 ps) is found to be highly sensitive to temperature changes when compared to the traces recorded at 184 and 339 K (Fig. 5e and f), while the kinetics of the other processes (τ1 = ca. 200 fs; τ2 = ca. 1.0 ps) are almost temperature-independent (Table S7, ESI†). While τ1 is assigned to the ISC as described earlier, the initial spectral growth process (τ2, Fig. 5b) of ca. 1 ps is ascribed to vibrational cooling.32,46 On the other hand, the highly temperature-dependent component (τ4, Fig. 5c) is indicative of an activated process and is assigned to the RISC step. Hence, the activation energy (Ea) for the RISC process is determined from the Arrhenius plot. For 1 and 3, Ea values of +8.49 and +6.02 kJ mol−1 are obtained (Fig. 5h and Fig. S15, ESI†), corresponding to ΔE(S1–T1) of 0.09 and 0.06 eV, respectively (Table S8, ESI†). In contrast, the fs-TA spectra and excited-state dynamics of 5 appear to be less similar to those of 1 and 3. It is worth mentioning that the initial identification of the time constant component for vibrational cooling for 5 at 293 K is less obvious, given a temperature-dependent component that spans a time constant at a similar timescale as vibrational cooling (with references to 1 and 3) at room temperature and higher is observed. Thus, VT fs-TA spectra at lower temperatures were recorded, which indeed show the resolution and identification of an additional component, i.e., the excited-state dynamics for 5 contain two components (τ2 and τ3), with time constants of very similar orders of magnitudes at room temperature that can be resolved at low temperatures. With references to 1 and 3, the τ2 and τ3 components correspond to the vibrational cooling and RISC processes, respectively, and they are difficult to be determined separately by global fitting from the TA spectra of 5 at higher temperatures. Therefore, only the time constants of τ3 recorded at <273 K were used to construct the Arrhenius plot (Fig. S16, ESI†). Ea for 5 is estimated to be +10.70 kJ mol−1, corresponding to ΔE(S1–T1) of 0.11 eV (Table S8, ESI†). Overall, the Ea values estimated from VT fs-TAS are in trend with the estimated ΔE(S1–T1) by the emission spectral studies and the two-state kinetic model. It is also noteworthy that the activation parameters for 5 are values not of the highest precision given that the vibrational cooling and RISC processes are quite close in their time constants at room temperature and higher temperatures.
Computational studies
To further study the electronic structures and the nature of absorption and emission origins of these gold(III) complexes, density functional theory (DFT) and time-dependent DFT (TDDFT) calculations were conducted on 1–6. The optimized ground-state geometries of 1–6 are shown in Fig. S17 (ESI†). The first fifteen singlet excited states of 1–6 computed by TDDFT and the selected molecular orbitals involved in the transitions are shown in Table S9 and Fig. S18–S23 (ESI†), respectively. The simulated UV–vis spectra generated by Multiwfn are shown in Fig. S24 (ESI†). The S0 → S1 transitions computed at 433–508 nm correspond to the HOMO → LUMO excitation. The HOMOs of these complexes are predominantly the π orbitals localized on the carbazole units, and the LUMOs are the π* orbitals predominantly localized on the central phenyl ring and the pyridine/pyrazine moiety of the C^C^N^N ligand. The lowest-energy absorption band observed in the stimulated UV–vis spectra is attributed to the ILCT [π(Cbz) → π*(Py/Pz)] transition within the C^C^N^N ligand. The ILCT bands computed for 1–6 show a descending trend in energy, i.e., 1 and 3 (433 and 435 nm) > 2 and 4 (453 and 455 nm) > 5 and 6 (506 and 508 nm), which agrees with the trend observed in the experimental result. The higher-energy absorption bands of 1–6 computed at ca. 320 nm are predominantly attributed to the transition from the π orbital on the whole cyclometalating ligand to the π* orbital localized on the central phenyl ring and the pyridine moiety. Therefore, the higher-energy absorption bands of 1–6 found in the absorption spectra are assigned to the IL [π → π*(C^C^N^N)] transitions with some CT character from the phenyl rings to the pyridine, in line with the spectral assignments. The PDMs computed for 1–6 are summarized in Table S10 (ESI†). The orbital energy diagram of the frontier molecular orbitals of 1–4 is shown in Fig. S25 (ESI†). It is found that 2 and 4 possess higher HOMO energy levels (ca. −5.25 eV) than 1 and 3 (ca. −5.43 eV), owing to the destabilization of the HOMO with the incorporation of electron-donating 3,6-di-tert-butylcarbazolyl units in 2 and 4. The HOMO–LUMO energy gap computed for 1 (3.58 eV) and 3 (3.56 eV) are larger than those for 2 (3.43 eV) and 4 (3.41 eV), in good agreement with the results from absorption spectroscopy and cyclic voltammetry.To confirm the nature of the emissive states, the geometries of the T1 state of 1–4 in toluene have been optimized using the unrestricted UPBE0/CPCM method. The plots of the spin density of the optimized T1 excited states are shown in Fig. S26 (ESI†). The plots of the spin density show that 1–4 possess the spin density mainly localized on the carbazole and the pyridine moieties, supporting the predominant 3ILCT [π(Cbz) → π*(Py/Pz)] character of the triplet emissive states. The emission wavelengths of 1–4, approximated by the energy difference between the S0 and T1 states at the corresponding optimized geometries in toluene, are summarized in Table S11 (ESI†). In general, the emission wavelengths computed for 1 (527 nm) and 3 (528 nm) are higher in energy than those for 2 (553 nm) and 4 (548 nm), which agrees with the trend observed in the experimental emission spectra.
Analyses of the electrostatic potential were also performed on 1–4. As shown in Fig. S27 (ESI†), the isosurfaces of the electrostatic potential (ESP) of these complexes indicate the highly electron-deficient nature of the pyridine unit, comparable to the previously reported C^C^N^N gold(III) complex,29 revealing the potential of these parts interacting with the electron-rich regions of the m-CBP host molecules. Comparing 1–2 and 3–4 which differ mainly on the –tBu position on the R1/R2 site (see Scheme 1), however, no significant difference can be found among the ESP surfaces of 1–4. Based on the optimized excited state geometries, the TDMVs of 1 and 3 at S1 and T1 geometries were calculated using TDDFT with the Tamm–Dancoff approximation (TDA-DFT), as shown in Fig. S28 (ESI†). The natural transition orbital (NTO) pairs for the S1 and T1 excited states of 1 and 3 in their respective optimized structures are shown in Fig. S29 and S30 (ESI†). The TDMVs of the S1 and T1 states for 1 and 3 were found to be alike, lying near the C–Au–NCbz bond on the C^C^N^N plane.
Preferential molecular orientation
From the previous sections, 1 and 2 are found to share very similar photophysical properties with their regioisomers, 3 and 4, respectively, as the regioisomers differ only in the position of the electron-donating –tBu substituent, either on R1 or R2 site of the C^C^N^N ligand (see Scheme 1). By such a slight modification, the PDMs of the complexes are manipulated. The computed PDMs at the optimized S0 state (μg) are found to increase from 6.0 (1) to 6.3 D (3) and from 5.7 (2) to 5.9 D (4), respectively (Table S10, ESI†). It is known that the changes in PDM of the dopant molecules would affect the electrostatic interactions between the dopant and the host in the host–guest system, making horizontal alignment of the TDMVs of the dopants more preferred. Angular-dependent PL measurements of 1–4 in vacuum-deposited thin films were performed and analyzed. With the ability to adopt random orientations in a wide range of deposition temperatures, m-CBP was selected as a host material such that any preferential orientation in the thin films would solely be induced by the gold(III) complexes.29,37 The angular-dependent PL spectra of 1–4 doped at 11% v/v in m-CBP thin films are shown in Fig. 6. On the other hand, the PDMs of 5 and 6 are found to be 2.7 and 3.3 D (Table S10, ESI†), respectively, which are in general much lower when compared to those of 1–4 (5.7–6.3 D), and therefore, 5 and 6 were not selected for further studies of their preferential molecular orientation.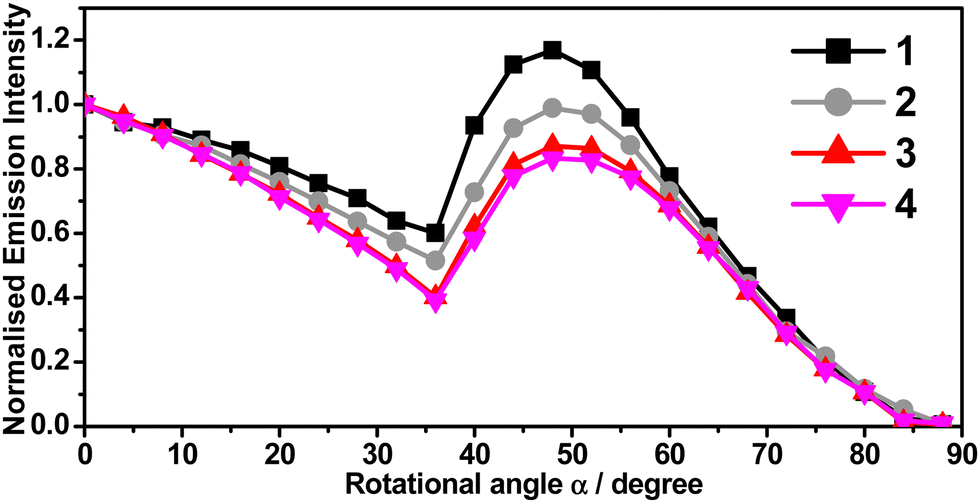 | ||
| Fig. 6 Angular-dependent PL intensities of the p-polarized light of 1–4 (11% v/v) in m-CBP thin films with 20 nm thickness. | ||
The TDMV anisotropies can be characterized by the orientation order parameter, S.48,49S = 0 implies a random orientation, while the S value varies from −0.50 for a completely horizontal orientation to +1.0 for a completely vertical orientation,49 From the angular-dependent measurements, S = 0.09, −0.06, −0.23 and −0.31 are found for 1–4, respectively. Table 2 summarizes the molecular orientation parameters of 1–4. Given that θ is the angle between the normal of a substrate and the TDMV, S = 0.09, −0.06, −0.23 and −0.31 correspond to Θh of 0.61, 0.71, 0.81 and 0.87, respectively (Table 2). These results suggest that 3 and 4 show a higher preference for the horizontal transition dipole orientation than their structural isomers, 1 and 2. This could be ascribed to the higher PDMs of 3 and 4, which would exhibit increased Coulombic attraction with the host, making the horizontal alignment more preferred. The –tBu position change has led to an increase in Θh by 32% from 0.61 (1) to 0.81 (3), and by 23% from 0.71 (2) to 0.87 (4). The Θh value reaching 0.87 is found to be comparable to that of many other reported highly efficient horizontal-aligned emitters (Table S16, ESI†). For the present C^C^N^N gold(III) system, a larger PDM of the complexes would be advantageous to the realization of the horizontal alignment of the TDMVs of the emitters. The findings here further confirm the functionality of the –tBu substituent, which assists a desired molecular anisotropy of the gold(III) emitters in the host matrix, resulting in an enhanced horizontal-aligned preference without altering the photophysical properties of the gold(III) complexes.
| Complex | PDM/Debye | Order parameter (S) | θ /° | Θ h | (h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :v)b :v)b |
|---|---|---|---|---|---|
a
θ represents the angle between the normal of a substrate and the transition dipole moment vector (TDMV) and is calculated using the equation, 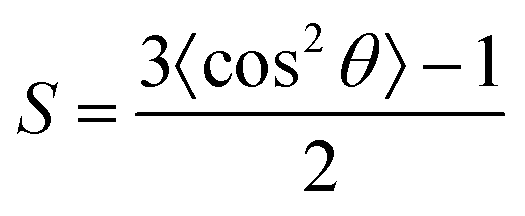 , with the bracketed values 〈… 〉 indicating an ensemble average of 〈cos2θ〉.49
b
Θ
h represents the ratio of the horizontal dipole to the total dipole of the emitters and is obtained by the equation Θh:Θv = 〈sin2θ〉:〈cos2θ〉 = h:v.49 , with the bracketed values 〈… 〉 indicating an ensemble average of 〈cos2θ〉.49
b
Θ
h represents the ratio of the horizontal dipole to the total dipole of the emitters and is obtained by the equation Θh:Θv = 〈sin2θ〉:〈cos2θ〉 = h:v.49
|
|||||
| 1 | 5.9659 | 0.09 | 51.2 | 0.61 | 0.61:0.39 |
| 2 | 5.6588 | −0.06 | 57.2 | 0.71 | 0.71:0.29 |
| 3 | 6.2510 | −0.23 | 64.4 | 0.81 | 0.81:0.19 |
| 4 | 5.8844 | −0.31 | 69.1 | 0.87 | 0.87:0.13 |
With a positive correlation found between the PDMs of the emitters and their molecular orientation, attempts have been made to explore an optimal molecular design for a more preferred molecular orientation by identifying the structure that gives the largest PDM on the current gold(III) C^C^N^N backbone with –tBu substituents at different positions. Based on this concept, twelve molecular structures of potential gold(III) C^C^N^N emitters have been proposed. The proposed structures, their optimized ground state structures, and the computed PDMs are summarized in Table S13 and Fig. S31 (ESI†). The PDMs of the complexes are found to be in the range of 5.0 to 6.2 D and are significantly affected by the position of the –tBu substituents. Particularly, only two of the proposed structures show PDMs of >6 D, similar to those of 3 and 4 which display more horizontally oriented TDMVs. As a significant increase in PDM is not observed, with 3 being the highest in the PDMs, the experimental confirmation of the physical properties of the proposed structures was not performed.
OLED fabrication and characterization
Complexes 1–5 were utilized to prepare vacuum-deposited OLEDs to examine their EL properties, while 5 and 6 have not been systematically studied due to their comparatively lower PDMs, low PLQYs and relatively poor EL performance (Table S13, ESI†). The key performances of the devices based on 1–4 are summarized in Table 3. Fig. 7 shows the normalized EL spectra of the vacuum-deposited devices based on these complexes, and the traces of their EQEs as a function of current density. The EL spectra of these devices exhibit Gaussian-shaped emission bands, which is in good agreement with the PL spectra of the respective complexes. Although 1 and 3 are regioisomers of each other and show similar PL properties, the EL performances of the devices made with 3 are much higher than those made with 1 (Table S14, ESI†). In particular, devices based on 1 afford maximum current efficiency (CE) and EQE of 43.9 cd A−1 and 12.6%, respectively. On the other hand, devices made with 3 reach CEs and power efficiencies (PEs) of up to 55.5 cd A−1 and 58.1 lm W−1, respectively. Notably, 3 affords a maximum EQE of 15.7%, that is ∼24% higher than that based on 1 (12.6%). Given that the PLQYs for the devices of 1 and 3 are 0.80 and 0.76, respectively, the estimated ηout for 3 is found to be 20.6%, which is ∼30% higher than that estimated from device made with 1 (ηout = 15.7%). Similar improvement is observed when comparing 2 and 4, in which the maximum EQE for 4 (15.4%) is ∼49% higher than that for 2 (10.3%). The enhancement in the EQEs and the theoretical ηout on 3 and 4 can be ascribed to their preferential molecular anisotropy, in which they are found to possess higher Θh of TDMVs (Θh = 0.81 for 3 and 0.87 for 4) in m-CBP thin films than those of 1 (Θh = 0.61) and 2 (Θh = 0.70). Meanwhile, red-emitting devices with a maximum CE of 10.3 cd A−1 and an EQE of 7.0% have been realized for devices based on 5. The relatively low EQEs of 5 could possibly be ascribed to the deep electron trapping in 5, owing to its low LUMO level energy of −3.07 eV (Table S3, ESI†).11| Complex (conc.) | Θ h | CEb/cd A−1 | PEc/lm W−1 | EQEd/% | λ max /nm | Φ PL | CIEf (x, y) |
|---|---|---|---|---|---|---|---|
| a Refer to Table 2. b CE represents the maximum current efficiency. c PE represents the maximum power efficiency. d EQE represents the maximum external quantum efficiency. e λ max represents the peak maximum. f CIE coordinates are taken at a luminance of 100 cd m−2. | |||||||
| 1 (14 v/v%) | 0.61 | 43.9 | 38.8 | 12.6 | 528 | 0.80 | 0.32, 0.61 |
| 2 (5 v/v%) | 0.71 | 34.8 | 31.2 | 10.3 | 552 | 0.85 | 0.42, 0.56 |
| 3 (14 v/v%) | 0.81 | 55.5 | 58.1 | 15.7 | 532 | 0.76 | 0.33, 0.61 |
| 4 (8 v/v%) | 0.87 | 53.0 | 41.6 | 15.4 | 544 | 0.82 | 0.39, 0.58 |
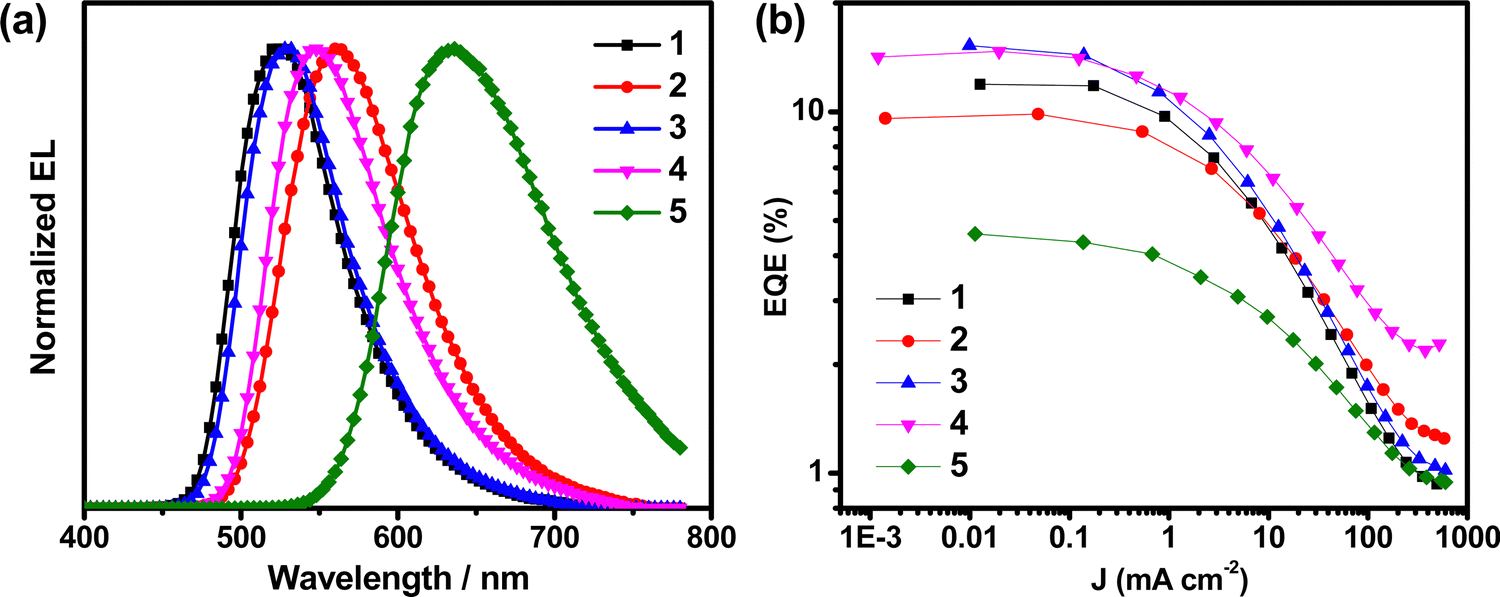 | ||
| Fig. 7 (a) Normalized EL spectra and (b) plots of EQEs vs. current density of the vacuum-deposited devices based on 1–5. | ||
The operational stabilities of the vacuum-deposited devices based on 1–6 were measured by accelerated tests at a constant driving current density of 20 mA cm−2. Fig. S33 and Table S15 (ESI†) depict the relative luminance (L/L0) of the devices based on 1–6 as a function of time and their key lifetime data, respectively. Generally, complexes with the presence of –tBu groups on the carbazole are found to afford better operational stabilities. For example, the operational half-lifetime (LT50) values of devices based on 2 and 4 at 100 cd m−2 are projected to be 27760 hours and 16316 hours, respectively, much longer than those of 1 (1358 hours) and 3 (6353 hours). This might be ascribed to the introduction of electron-donating –tBu groups on the carbazole, which would lead to a lower hole injection barrier from the host transporting layer to the emitting layer.29 Such longer operational lifetimes could also be explained by the increased steric hindrance in 2 and 4 with more –tBu substituents present, reducing the rate of Dexter-type triplet-charge quenching,50 and thus prolonging the OLEDs.
Conclusion
A new series of TADF tetradentate C^C^N^N ligand-containing gold(III) complexes have been designed and synthesized. Hypsochromic spectral shifts of ∼0.05–0.17 eV and drastically shortened emission lifetimes were found in the thermally enhanced emissions of these gold(III) complexes in toluene and doped mCP thin films, suggesting an increased kr upon increasing temperature due to TADF. The experimental variable-temperature kr in the solution state was fitted to the Boltzmann two-level model, which gave estimated ΔE(S1–T1) values for 1, 3 and 5 of 0.16–0.18 eV, respectively. The TADF properties have also been supported by temperature-dependent ultrafast transient absorption studies, with the direct observation of RISC and the determination of the activation parameters. This is the first time that green-emitting TADF gold(III) complexes with tetradentate C^C^N^N ligand frameworks are demonstrated. In light of the effects of the host–guest interactions on the molecular anisotropy, through the structural modification of the electron-donating –tBu substituent, the PDMs of the complexes were manipulated in 1–4 to realize an enhancement in Θh of the TDMVs of the emitters and ηout of the resulting OLEDs. The vacuum-deposited OLEDs based on the more horizontal-orientated complexes 3 and 4 showed improvements in ηout by ∼30%, compared to their regioisomers, 1 and 2. The introduction of a –tBu substituent to the carbazolyl moieties also improved the operational stability of the corresponding OLEDs. This work has demonstrated the functionality of the –tBu substituent in realizing the horizontal-preferred molecular orientation as well as the enhancing of operational stability, providing insights into optimization of the gold(III) C^C^N^N systems towards the development of efficient gold(III)-based OLEDs.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
V. W.-W. Y. acknowledges The University of Hong Kong. The work described in this paper was supported by a grant from Hong Kong Quantum AI Lab Ltd under the AIR@InnoHK administrated by the Innovation and Technology Commission (ITC), a grant from the Shenzhen-Hong Kong-Macau Technology Research Programme (Type C) (contract no. SGDX2020110309520101), the Collaborative Research Fund (CRF) (C7075-21G) from the Research Grant Council of the Hong Kong Special Administrative Region, P. R. China, and the Croucher-CAS Fund Scheme for Joint Laboratories on Molecular Functional Materials for Electronics, Switching and Sensing. We would like to thank Dr Keith Man-Chung Wong, Miss Shu-Nan Zhao and Miss Si-Ye Wu for their help in X-ray crystal structure data collection. Dr Liangliang Yan is acknowledged for his help in the collection and analysis of the crystal data. Ms Yan-Kiu Brigid-Bernadette Ng is acknowledged for her assistance in ligand synthesis. C. C. A.-Y. acknowledges the receipt of postgraduate studentships from HKU. The computations were performed using the HKU ITS research computing facilities.References
- T. D. Schmidt, T. Lampe, M. R. Daniel Sylvinson, P. I. Djurovich, M. E. Thompson and W. Brütting, Phys. Rev. Appl., 2017, 8, 037001 CrossRef
.
- K.-H. Kim and J.-J. Kim, Adv. Mater., 2018, 30, 1705600 CrossRef PubMed
- D.-H. Kim, A. D’Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed
- P. J. Conaghan, S. M. Menke, A. S. Romanov, S. T. E. Jones, A. J. Pearson, E. W. Evans, M. Bochmann, N. C. Greenham and D. Credgington, Adv. Mater., 2018, 30, 1802285 CrossRef PubMed
- J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold and M. Bochmann, Chem. Commun., 2015, 51, 16629–16632 RSC
- C.-Y. Wong, M.-C. Tang, L.-K. Li, M.-Y. Leung, W.-K. Tang, S.-L. Lai, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2022, 13, 10129–10140 RSC
- L.-K. Li, C. C. Au-Yeung, M.-C. Tang, S.-L. Lai, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W.-W. Yam, Mater. Horiz., 2022, 9, 281–293 RSC
- C. C. Au-Yeung, L.-K. Li, M.-C. Tang, S.-L. Lai, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2021, 12, 9516–9527 RSC
- L.-K. Li, W.-K. Kwok, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2021, 12, 14833–14844 RSC
- D. Zhou, W.-P. To, Y. Kwak, Y. Cho, G. Cheng, G. S. M. Tong and C.-M. Che, Adv. Sci., 2019, 6, 1802297 CrossRef CAS
- D. Zhou, G. S. M. Tong, G. Cheng, Y.-K. Tang, W. Liu, D. Ma, L. Du, J.-R. Chen and C.-M. Che, Adv. Mater., 2022, 34, 2206598 CrossRef CAS
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994–2015 CrossRef CAS PubMed
- M.-C. Tang, M.-Y. Chan and V. W.-W. Yam, Chem. Rev., 2021, 121, 7249–7279 CrossRef CAS PubMed
- T. Fleetham, Y. Ji, L. Huang, T. S. Fleetham and J. Li, Chem. Sci., 2017, 8, 7983–7990 RSC
- H.-H. Kuo, Z.-l Zhu, C.-S. Lee, Y.-K. Chen, S.-H. Liu, P.-T. Chou, A. K.-Y. Jen and Y. Chi, Adv. Sci., 2018, 5, 1800846 CrossRef PubMed
- J. H. Palmer, M. W. Day, A. D. Wilson, L. M. Henling, Z. Gross and H. B. Gray, J. Am. Chem. Soc., 2008, 130, 7786–7787 CrossRef CAS PubMed
- K. Koren, S. M. Borisov, R. Saf and I. Klimant, Eur. J. Inorg. Chem., 2011, 1531–1534 CrossRef CAS PubMed
- D. Chen, K. Li, X. Guan, G. Cheng, C. Yang and C.-M. Che, Organometallics, 2017, 36, 1331–1344 CrossRef CAS
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS
- H. Fukagawa, T. Shimizu, H. Hanashima, Y. Osada, M. Suzuki and H. Fujikake, Adv. Mater., 2012, 24, 5099–5103 CrossRef CAS PubMed
- G. Cheng, P.-K. Chow, S. C. F. Kui, C.-C. Kwok and C.-M. Che, Adv. Mater., 2013, 25, 6765–6770 CrossRef CAS PubMed
- X.-C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753–6756 CrossRef CAS PubMed
- T. Fleetham, G. Li, L. Wen and J. Li, Adv. Mater., 2014, 26, 7116–7121 CrossRef CAS PubMed
- B. Y.-W. Wong, H.-L. Wong, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2017, 56, 302–305 CrossRef CAS PubMed
- M.-C. Tang, L.-K. Li, S.-L. Lai, W.-L. Cheung, M. Ng, C.-Y. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2020, 59, 21023–21031 CrossRef CAS PubMed
- M.-C. Tang, A. K.-W. Chan, M.-Y. Chan and V. W.-W. Yam, Top. Curr. Chem., 2016, 374, 46 CrossRef PubMed
- C.-H. Lee, M.-C. Tang, F. K.-W. Kong, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2020, 142, 520–529 CrossRef CAS PubMed
- D. Zhou, W.-P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips and C.-M. Che, Angew. Chem., Int. Ed., 2020, 59, 6375–6382 CrossRef CAS PubMed
- L.-K. Li, M.-C. Tang, S.-L. Lai, M. Ng, W.-K. Kwok, M.-Y. Chan and V. W.-W. Yam, Nat. Photonics, 2019, 13, 185–191 CrossRef CAS
- S.-Y. Kim, W.-I. Jeong, C. Mayr, Y.-S. Park, K.-H. Kim, J.-H. Lee, C.-K. Moon, W. Brütting and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 3896–3900 CrossRef CAS
- H. J. Jang and J. Y. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 54907–54913 CrossRef CAS PubMed
- T. Lampe, T. D. Schmidt, M. J. Jurow, P. I. Djurovich, M. E. Thompson and W. Brütting, Chem. Mater., 2016, 28, 712–715 CrossRef CAS
- T. Komino, H. Tanaka and C. Adachi, Chem. Mater., 2014, 26, 3665–3671 CrossRef CAS
- F. Tenopala-Carmona, O. S. Lee, E. Crovini, A. M. Neferu, C. Murawski, Y. Olivier, E. Zysman-Colman and M. C. Gather, Adv. Mater., 2021, 33, 2100677 CrossRef CAS PubMed
- M.-C. Tang, C.-H. Lee, M. Ng, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2018, 57, 5463–5466 CrossRef CAS PubMed
- L.-K. Li, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M. Ng, C. K.-M. Chan, M.-Y. Chan and V. W.-W. Yam, Chem. Mater., 2019, 31, 6706–6714 CrossRef CAS
- M.-C. Tang, L. H.-Y. Lo, W.-L. Cheung, S.-L. Lai, M.-Y. Chan and V. W.-W. Yam, Chem. Commun., 2019, 55, 13844–13847 RSC
- C.-H. Lee, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2018, 9, 6228–6232 RSC
- W.-P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 14036–14041 CrossRef CAS PubMed
- D. J. Casadonte, Jr. and D. R. McMillin, J. Am. Chem. Soc., 1987, 109, 331–337 CrossRef
- R. Czerwieniec, J. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed
- W.-K. Kwok, L.-K. Li, S.-L. Lai, M.-Y. Leung, W. K. Tang, S.-C. Cheng, M.-C. Tang, W.-L. Cheung, C.-C. Ko, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2023, 145, 9584–9595 CrossRef CAS PubMed
- Y.-J. Cho, S.-Y. Kim, M. R. Son, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 20093–20100 RSC
- Y. Esaki, T. Matsushima and C. Adachi, Org. Electron., 2019, 67, 237–241 CrossRef CAS
- J.-S. Huh, K.-H. Kim, C.-K. Moon and J.-J. Kim, Org. Electron., 2017, 45, 279–284 CrossRef CAS
- C. Zhao, F. Zhao, K. Wang, H. Yu, T. Huang, R. Wang, C. Zhang, B. Hu and L. Duan, Phys. Rev. Appl., 2020, 14, 034059 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the gold(III) complexes; X-ray crystal structure of 5; electrochemical studies; photophysical studies; temperature-dependent lifetime data; Boltzmann fitting details; computational studies; EL performance of the corresponding OLEDs; physical measurements and instrumentation details. CCDC 2269449. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3mh00910f |
| This journal is © The Royal Society of Chemistry 2024 |