
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient carbene transfer reactivity mediated by Fe(II) complexes supported by bulky alkoxides†
Lakshani W.
Kulathungage
a,
Sudheer S.
Kurup
a,
Edison A.
Browne‡
b,
Gabriel H.
Spalink‡
b,
Cassandra L.
Ward
c,
Richard L.
Lord
 *b and
Stanislav
Groysman
*a
*b and
Stanislav
Groysman
*a
aDepartment of Chemistry, Wayne State University, 5101 Cass Ave., Detroit, MI 48202, USA. E-mail: groysman@chem.wayne.edu
bDepartment of Chemistry, Grand Valley State University, 1 Campus Dr, Allendale, MI 49401, USA. E-mail: lordri@gvsu.edu
cLumigen Instrument Center, Wayne State University, 5101 Cass Avenue, Detroit, MI 48202, USA
First published on 14th June 2024
Abstract
Herein we describe the stoichiometric and catalytic carbene-transfer reactivity of iron(II) alkoxide complexes with iodonium ylide precursors. Treatment of PhIC(CO2Me)2 with styrene in the presence of catalytic amounts of several different Fe(OR)2(THF)2 precursors results in efficient cyclopropanation for a variety of styrenes. Computational and reactivity studies suggest a novel remote metallocarbene/vinyl radical intermediate, Fe(OR)2(κ2-(O![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C(OMe))2C), which could be responsible for the reactive nature of the catalyst.
C(OMe))2C), which could be responsible for the reactive nature of the catalyst.
There is long-standing interest in the chemistry of metallocarbenes, one of the most important functionalities in organometallic chemistry.1–15 The reactivity of a metallocarbene is determined by its electronic structure.2,15 Common types of metallocarbenes include nucleophilic Schrock carbenes,2,3 electrophilic Fischer carbenes,2,4 and carbene radicals.6–8,15 The reactivity difference between different types of metallocarbenes is most convincingly illustrated via their reactions with olefins. Whereas nucleophilic carbenes usually demonstrate olefin metathesis,2 both electrophilic and radical carbenes catalyze cyclopropanation.4,7,13–15 However, while electrophilic carbenes usually demonstrate two-electron concerted reactivity with olefins, radical carbenes typically exhibit one-electron stepwise reactivity.15 In addition to their reactions with olefins, metallocarbenes have been previously shown to react with isocyanides to form ketenimines, although this reactivity is generally stoichiometric.16–19
We previously reported the synthesis and group-transfer reactivity of middle and late 3d metal complexes in weak-field bis(alkoxide) ligand environments.20–30 The reaction of Co(OR)2(THF)2 (OROCtBu2Ph) with diphenyldiazomethane formed high-valent, low-spin cobalt-carbene Co(OR)2(CPh2) with an electronic structure intermediate between cobalt(IV)-alkylidene and carbene(III)-carbene radical.22 One-electron reduction of this compound produced high-spin Co(II) weakly coupled with a carbene radical.27 Both complexes exhibited carbene transfer reactivity with isocyanides.24 In contrast, no substantial carbene transfer reactivity with olefins was observed for Co(OR)2(CPh2). The lack of catalytic cyclopropanation reactivity prompted us to turn to the corresponding iron complexes.26 However, no carbene formation via N2 release was observed with Fe(OR)2(THF)2. Instead, iron alkoxide complexes reductively coupled diazo compounds through the terminal nitrogens.26 We hypothesized that a different carbene precursor is needed for the formation and carbene transfer reactivity with iron. Herein we describe the reactivity of Fe(OR)2(THF)2 and related iron(II) alkoxide complexes with iodonium ylide precursors,30 which are known to serve as precursors for metallocarbene-catalyzed cyclopropanation.31 We demonstrate facile carbene transfer to olefins to form cyclopropanes. Mechanistic computational studies suggest the formation of a remote radical carbene Fe(OR)2(κ2-(OC(OMe))2C) in which the carbene is coordinated to the metal via two ester carbonyls, with the reactive functionality facing away from the metal.
Addition of PhIC(CO2Me)232 to a pale yellow solution of Fe(OR)2(THF)2 (1) in THF at room temperature led to a color change to orange. 1H NMR analysis of the reaction mixture in the presence of an internal standard indicated formation of (MeO2C)2CC(CO2Me)233 (5) in 78% yield (Fig. 1); formation of PhI was also observed. The formation of 5 was further confirmed by recrystallization (Fig. 1 and ESI†); a closely related structure exhibiting somewhat different cell parameters was recently reported.34 Similarly, the reaction of PhIC(CO2Ph)2 (see ESI†) with 1 led to the formation of (PhO2C)2CC(CO2Ph)2 (6, 74%) and PhI. Albeit small amounts of the dimerized products ((RO2C)2CC(CO2R)2) are typically observed in the solutions of the corresponding iodonium ylides, no formation of significant amounts of 5 or 6 was observed in the absence of 1 under identical reaction conditions.
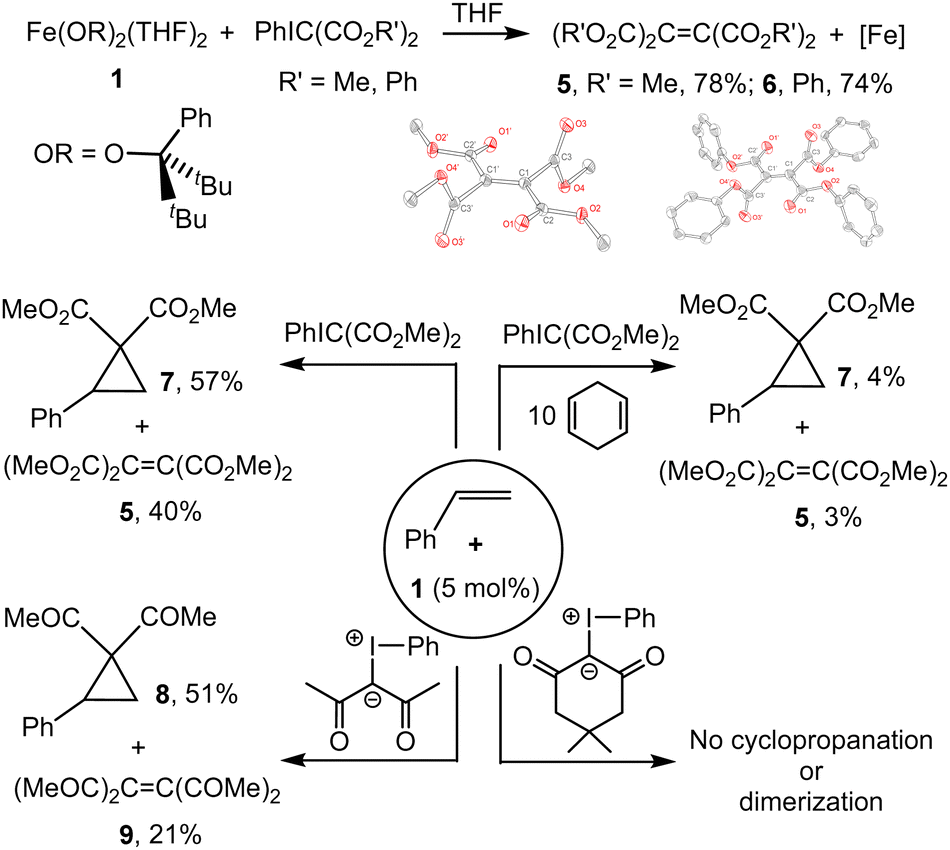 | ||
| Fig. 1 Reactions between 1 and various iodonium ylides, in the presence/absence of styrene and other reagents. | ||
Addition of PhIC(CO2Me)2 to a yellow solution of Fe(OR)2(THF)2 (1) and styrene led to formation of the corresponding cyclopropane 7 in 63% yield; the formation of (MeO2C)2CC(CO2Me)2 (5) was also observed (37%). Combining equimolar amounts of PhIC(CO2Me)2 and styrene under catalytic conditions (5 mol% of 1, C6D6, 24 h, RT) leads to formation of cyclopropane 7 in 41% yield. Increasing the amount of styrene to 2 equiv. decreases the yield to 26%. In contrast, increasing the amount of ylide to 2 equiv. increases the yield to 57%. Notably, the nature of the iron-alkoxide catalyst has a significant effect on the yield. We previously described the synthesis of three different iron(II) bis(alkoxide) complexes differing primarily in the steric effect around the metal (1–3, Fig. 2), and reported diverging reactivity of 1–3 in the catalytic dimerization of aryl nitrenes to form azoarenes.21,23,25 Conducting cyclopropanation of styrene under the optimized conditions (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PhIC(CO2Me)2:styrene) with 2 and 3 led to yields of 69% and 95%, respectively, indicating dependence on the nature of the catalyst.
:1 PhIC(CO2Me)2:styrene) with 2 and 3 led to yields of 69% and 95%, respectively, indicating dependence on the nature of the catalyst.
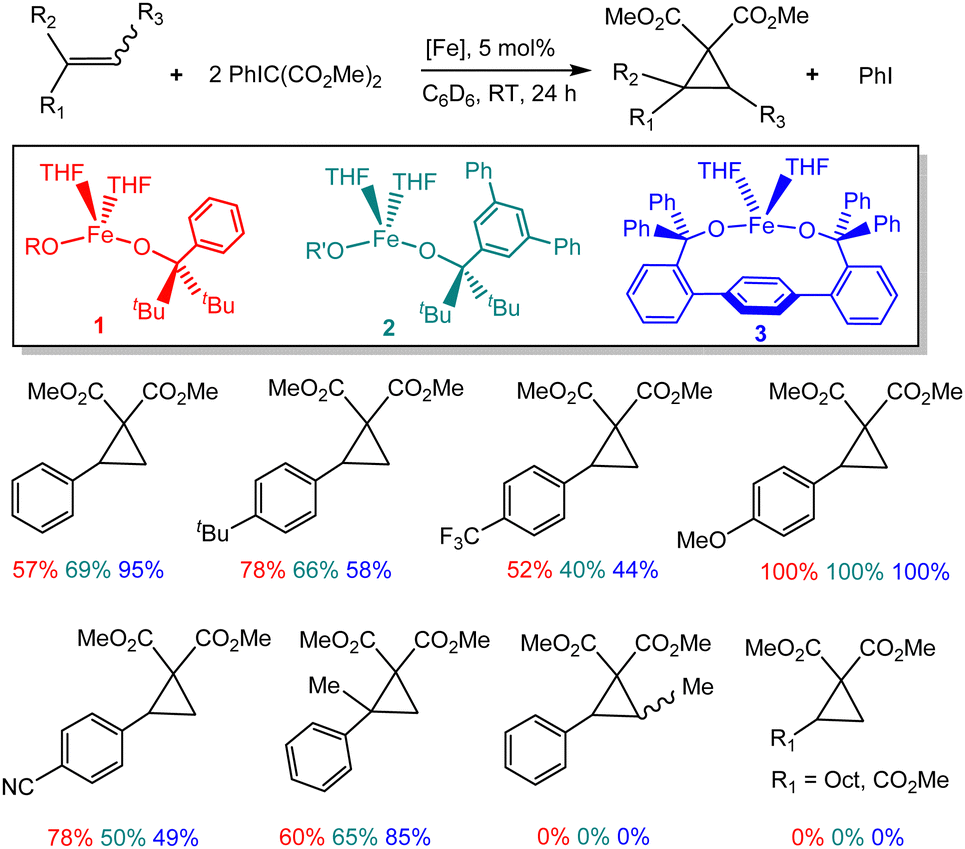 | ||
| Fig. 2 Catalytic reactivity of complexes 1–3 in cyclopropanation. | ||
Next, we investigated a series of different olefin precursors (Fig. 2), including styrenes with various electron-donating or electron-withdrawing groups in the para position, α- and β-methylstyrenes (cis and trans isomers), as well as 1-decene and methyl acrylate. For most of the para-substituted styrenes, moderate to excellent yields are observed; good to excellent yields are also observed for the α-methylstyrene. For some styrenes (4-methoxo, 4-trifluoromethyl), catalysts 1–3 generally exhibit similar reactivity. In contrast, some variability is observed for other styrenes (unsubstituted or 4-cyano). There appears to be higher reactivity for the electron-rich (4-tert-butyl, 4-methoxy) vs. electron-poor (4-trifluoromethyl, 4-cyano) substrates. No cyclopropanation was observed for β-methylstyrene, 1-decene, or methyl acrylate.
Attempts to isolate the reactive iron-carbene intermediate invariably resulted in formation of 5 and PhI. Thus, we turned to DFT studies of 3 to obtain insight into the reaction mechanism.35 Optimization of the putative carbon-bound carbene favored the quintet state by 2–39 kcal mol−1 (see ESI†) over the singlet, triplet, and septet states. This quintet corresponds to high-spin FeIII antiferromagnetically coupled to carbene radical anion. Significant ligand radical character is consistent with our earlier work on Co carbene,27 Fe/Mn nitrenes,21,29 and Fe azide/diazoester complexes in this ligand environment.20,26 Upon addition of styrene, either directly or through attempted coordination to Fe, significant rearrangement of the carbene during geometry optimization resulted in one or both of the esters coordinated to Fe and the carbene uncoordinated. We postulated that κ2 coordination through both ester carbonyls (reminiscent of acac) could be feasible for PhIC(CO2Me)2 (Fig. 3, 4-IPh). Significant C–I bond activation is observed with bond elongation from 2.074 Å in the free ylide to 2.259 Å in 4-IPh; this species already shows significant FeIII character suggesting oxidative addition is concurrent with binding (see ESI†). Dissociation of PhI to form 4 was barrierless and exergonic by 8 kcal mol−1. This new remote radical carbene 4 is lower in energy than the carbon-bound quintet carbenes by 2–9 kcal mol−1.
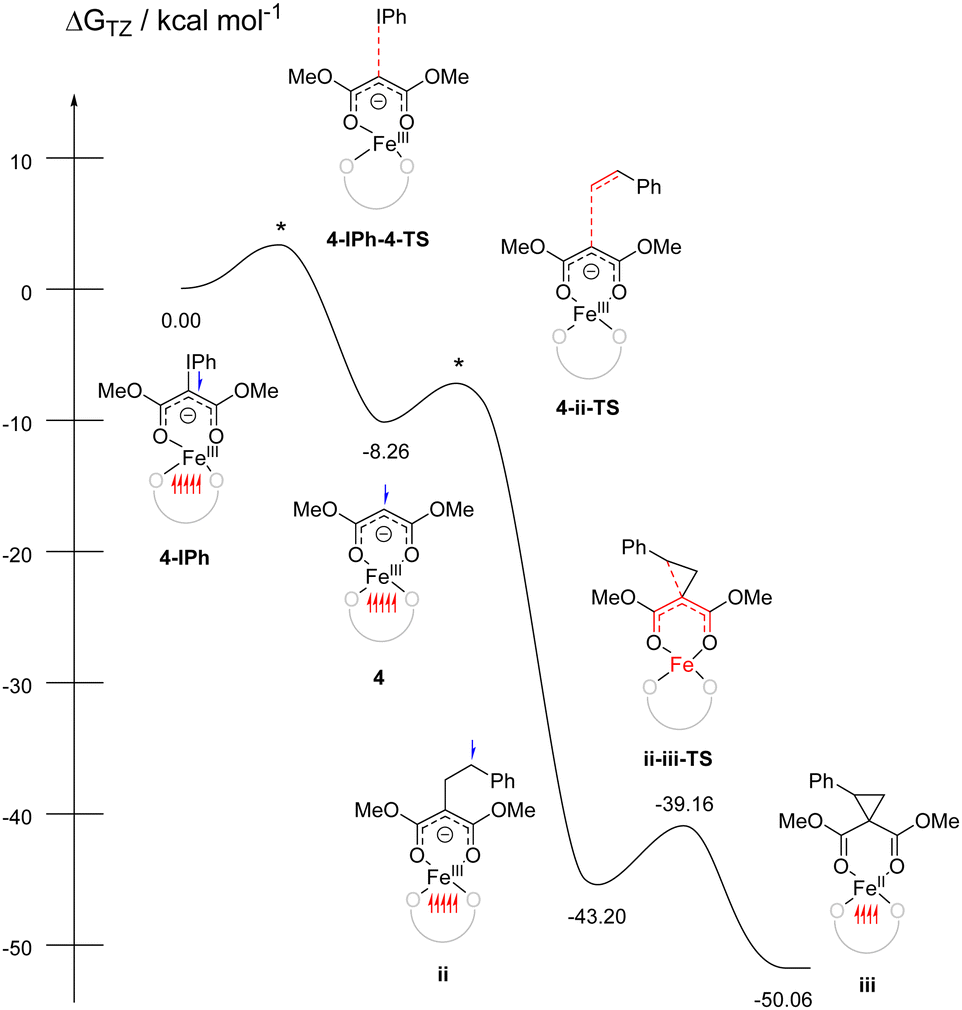 | ||
| Fig. 3 Free energy diagram for cyclopropanation. * indicates transition states that were not located. Unpaired spin up (red) and down (blue) electrons are shown for each intermediate. | ||
Reaction of 4 with styrene to form ii (only lowest energy regioisomer energy shown, see ESI†) was also barrierless, plausible given that this new coordination mode makes the carbene a carbon-based radical with little to no interaction with Fe. The new C–C bond makes this step exergonic by 35 kcal mol−1. Ring closing of ii to form iii, which concurrently reduces FeIII to FeII, is further exergonic by 7 kcal mol−1 with a low barrier of 5 kcal mol−1. This proposed reactivity is summarized in Fig. 3.
To provide experimental support to the computational predictions, we conducted additional experiments (Fig. 1); a summary of the observed reactivity and proposed mechanism is given in Fig. 4. The key intermediate proposed by DFT calculations is the remote carbene/vinyl radical 4. It is postulated that 4 forms due to the redox non-innocent nature of Fe(OR)2-carbene, which is facilitated by the stability of the delocalized form of the κ2-coordinated diester. However, the coordinative unsaturation of the metal center in 4 likely also plays an important role in preventing precedented κ1-C coordinated carbene radical derived from PhIC(CO2Me)2.36 Betley and coworkers also proposed the formation of a related vinyl radical derived from α-diazo-β-ketoesters, which led to proximal C–H bond activation followed by C–O bond formation (C–H alkoxylation).37 Remote carbenes were also reported for metals with non-coordinating N-heterocyclic carbenes.38–41
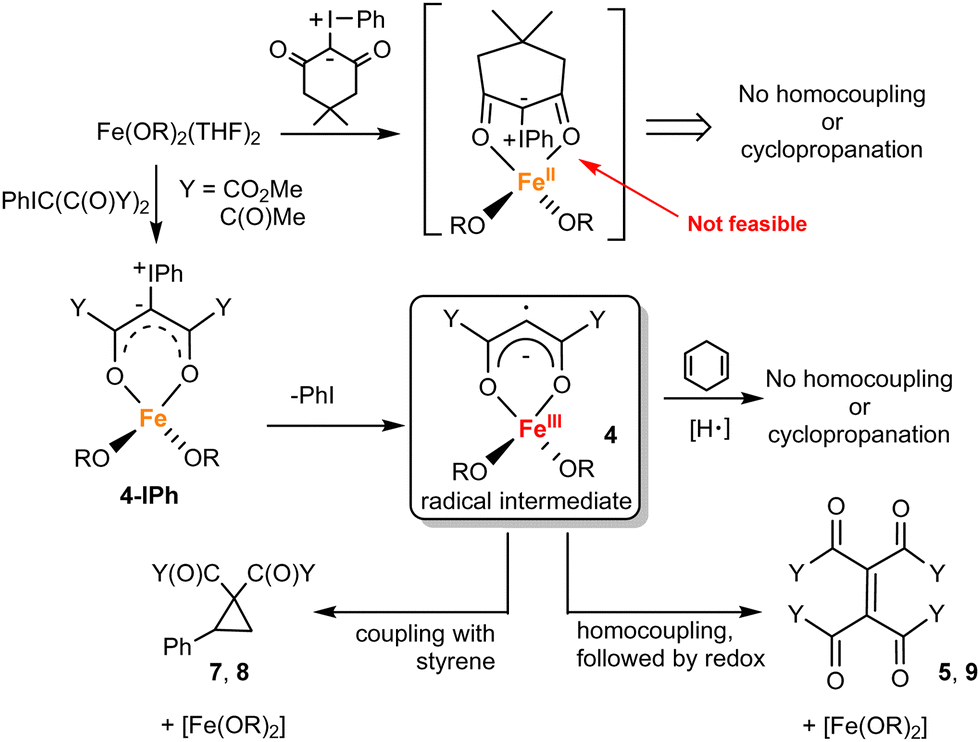 | ||
| Fig. 4 Formation and reactions of the postulated intermediate 4. | ||
The reactive radical nature of 4 is likely responsible for facile formation of olefins (R′O2C)2CC(CO2R′)2 or cyclopropanation (in the presence of styrene). H-atom donors such as cyclohexadiene or 9,10-dihydroanthracene are known to chemically probe ligand-localized unpaired spin density.42 These additions to the reaction of 1 with ylide and styrene shut down reactivity (trace products are observed), consistent with the expected catalyst's sensitivity to H-atom donors. The necessity and generality of the κ2 coordination of the dicarbonyl precursor (prior to PhI elimination) was probed by use of two additional ylides: 2,4-pentanedione-derived (i.e. acac-derived) ylide43 and dimedone-derived ylide.44 Acac-derived ylide is expected to coordinate to the metal like the diester-derived ylide and therefore should exhibit similar reactivity. As anticipated, the reaction between 2,4-pentanedione-derived ylide, styrene, and 1 (5 mol%) exhibited similar performance, producing the corresponding cyclopropane and olefin.45 Due to steric constraints, dimedone-derived ylide is unlikely to coordinate κ2 to the metal (Fig. 4). No reaction was observed between 1, dimedone ylide, and styrene (Fig. 1 and ESI†).
The reactivity between 1, PhIC(CO2Me)2, and isocyanides was also investigated (Fig. 5). No reaction is observed for PhIC(CO2Me)2 and xylyl isocyanide CNXyl (Xyl = 2,6-Me2C6H3). Adding the mixture to a solution of Fe(OR)2(THF)2 (1 equiv.) produced a color change to reddish-orange. 1H NMR suggested that no significant transformation of PhIC(CO2Me)2 took place. We previously reported formation of Fe(OR)2(CNXyl)2 (10);21 it is possible that its stability prevents turnover, which requires substitution of both isocyanides by the ylide. We also previously demonstrated notable differences in the reactivity between aromatic and aliphatic isocyanides; while no turnover (in ketenimine formation) was observed for Co(OR)2(CNXyl)2, catalytic reactivity was observed for Co(OR)2(CNAd)2 (Ad = adamantyl).24 Thus, we independently synthesized 1021 and Fe(OR)2(CNAd)2 (11). As 11 has not been previously reported, it was characterized by X-ray crystallography, NMR and IR spectroscopy, and elemental analysis. Both complexes lacked reactivity with PhIC(CO2Me)2, confirming that the lack of turnover in this reaction is due to the relative stability of the isocyanide complexes.
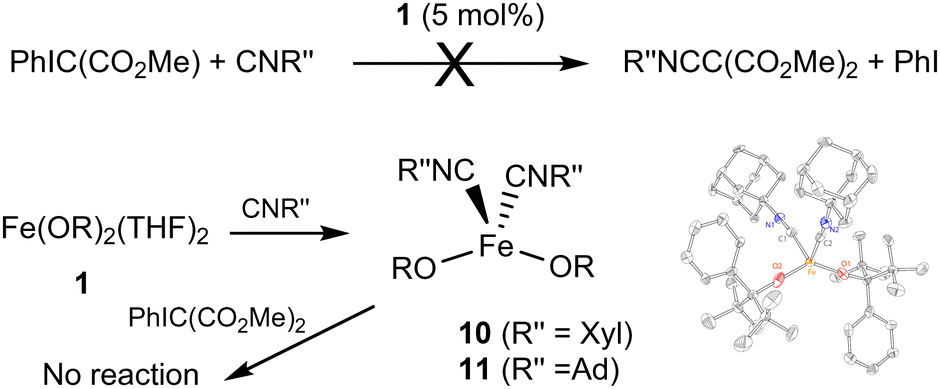 | ||
| Fig. 5 Reactions between 1, isocyanides, and PhIC(CO2Me)2. | ||
In summary, we described an efficient cyclopropanation reactivity between ylides and styrenes catalyzed by iron complexes in bulky alkoxide ligand environments. Mechanistic studies suggest that the reaction is mediated by a novel remote carbene/vinyl radical intermediate, whose formation is facilitated due to (1) the ability of [Fe(OR)2] to coordinate an iodonium ylide precursor in κ2-O,O-coordination mode, and (2) the propensity of [Fe(OR)2] to undergo oxidation to Fe(III). Future studies will focus on further mechanistic investigations, and additional carbene transfer reactions.
S. G. and R. L. L. are grateful to the National Science Foundation (NSF) for current support through CHE-2348382. Experimental characterization was carried out at Lumigen Instrument Center of Wayne State University. Lakshani W. Kulathungage is a Rumble Fellow. This work made use of the single-crystal XRD that was partially funded by the National Institutes of Health supplement grant #3R01EB027103-02S1.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- R. A. Moss and M. P. Doyle, Contemporary Carbene Chemistry, Wiley, Hoboken, New Jersey, 2013, 1118730267 Search PubMed.
- T. Strassner, Top. Organomet. Chem., 2004, 13, 1–20 CrossRef CAS.
- (a) R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS; (b) R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS.
- K. H. Doetz and J. Stendel, Chem. Rev., 2009, 109, 3227–3274 CrossRef CAS.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- H.-J. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 CrossRef CAS.
- W. I. Dzik, X. P. Zhang and B. de Bruin, Inorg. Chem., 2011, 50, 9896–9903 CrossRef CAS.
- S. K. Russell, J. M. Hoyt, S. C. Bart, C. Milsmann, S. C. E. Stieber, S. P. Semproni, S. DeBeer and P. J. Chirik, Chem. Sci., 2014, 5, 1168–1174 RSC.
- M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS.
- R. R. Schrock and C. Coperet, Organometallics, 2017, 36, 1884–1892 CrossRef CAS.
- A. Chirila, M. B. Brands and B. de Bruin, J. Catal., 2018, 361, 347–360 CrossRef CAS.
- A. Feliciano, J. L. Vázquez, L. J. Benítez-Puebla, I. Velazco-Cabral, D. C. Cruz, F. Delgado and M. A. Vázquez, Chem. – Eur. J., 2021, 27, 8233–8251 CrossRef CAS.
- W.-C. C. Lee, D.-S. Wang, Y.-L. Zhu and X. P. Zhang, Nat. Chem., 2023, 15, 1569–1580 CrossRef CAS.
- W.-C. C. Lee and X. P. Zhang, Trends Chem., 2022, 4, 850–851 CrossRef CAS.
- R. F. J. Epping, D. Vesseur, M. Zhou and B. de Bruin, ACS Catal., 2023, 13, 5428–5448 CrossRef CAS.
- R. Aumann and E. O. Fischer, Angew. Chem., Int. Ed. Engl., 1967, 6, 879–880 CrossRef CAS.
- I. Fernández, F. P. Cossío and M. A. Sierra, Organometallics, 2007, 26, 3010–3017 CrossRef.
- P. Lu and Y. Wang, Chem. Soc. Rev., 2012, 41, 5687–5705 RSC.
- Z. Tang, S. Mandal, N. D. Paul, M. Lutz, J. I. van der Vlugt and B. de Bruin, Org. Chem. Front., 2015, 2, 1561–1577 RSC.
- J. A. Bellow, P. D. Martin, R. L. Lord and S. Groysman, Inorg. Chem., 2013, 52, 12335–12337 CrossRef CAS.
- J. A. Bellow, M. Yousif, A. C. Cabelof, R. L. Lord and S. Groysman, Organometallics, 2015, 34, 2917–2923 CrossRef CAS.
- J. A. Bellow, S. A. Stoian, J. Van Tol, A. Ozarowski, R. L. Lord and S. Groysman, J. Am. Chem. Soc., 2016, 138, 5531–5534 CrossRef CAS.
- M. Yousif, D. Wannipurage, C. D. Huizenga, E. Washnock-Schmid, N. J. Peraino, A. Ozarowski, S. A. Stoian, R. L. Lord and S. Groysman, Inorg. Chem., 2018, 57, 9425–9438 CrossRef CAS.
- A. Grass, N. S. Dewey, R. L. Lord and S. Groysman, Organometallics, 2019, 38, 962–972 CrossRef CAS.
- S. S. Kurup, D. Wannipurage, R. L. Lord and S. Groysman, Chem. Commun., 2019, 55, 10780–10783 RSC.
- A. Grass, S. A. Stoian, R. L. Lord and S. Groysman, Chem. Commun., 2019, 55, 8458–8461 RSC.
- A. Grass, J. A. Bellow, G. Morrison, H.-C. zur Loye, R. L. Lord and S. Groysman, Chem. Commun., 2020, 56, 8416–8419 RSC.
- S. S. Kurup and S. Groysman, Dalton Trans., 2022, 51, 4577–4589 RSC.
- S. S. Kurup, N. M. Woodland, R. L. Lord and S. Groysman, Molecules, 2022, 27, 5751 CrossRef CAS.
- M. S. Yusubov and V. V. Zhdankin, Curr. Org. Synth., 2012, 9, 247–272 CrossRef CAS.
- C. Zhu, A. Yoshimura, L. Ji, Y. Wei, V. N. Nemykin and V. V. Zhdankin, Org. Lett., 2012, 14, 3170–3173 CrossRef CAS.
- S. R. Goudreau, D. Marcoux and A. B. Charette, Org. Synth., 2010, 87, 115–125 CrossRef CAS.
- (a) M. Barfield, T. Gotoh and H. K. Hall, Magn. Reson. Chem., 1985, 23, 705–709 CrossRef CAS; (b) H. K. Hall and R. C. Daly, Macromolecules, 1975, 8, 22–31 CrossRef CAS.
- I. Litvinov, Experimental Crystal Structure Determination 2022, CCDC 2152148† DOI: 10.5517/ccdc.csd.cc2b7h47.
- B3LYP-D3(BJ)/def2-TZVP/SMD(benzene)//BP86-D3(BJ)/def2-SVP. See ESI† for full computational details.
- R. F. J. Epping, M. M. Hoeksma, E. O. Bobylev, S. Mathew and B. de Bruin, Nat. Chem., 2022, 14, 550–557 CrossRef CAS.
- Y. Dong, A. T. Wrobel, G. P. Porter, J. J. Kim, J. Z. Essman, S.-L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2021, 143, 7480–7489 CrossRef CAS.
- A. J. Arduengo, D. Tapu and W. J. Marshall, Angew. Chem., Int. Ed., 2005, 44, 7240–7244 CrossRef CAS.
- D. M. Khramov, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2008, 47, 2267–2270 CrossRef CAS.
- B. Hildebrandt, W. Frank and C. Ganter, Organometallics, 2011, 30, 3483–3486 CrossRef CAS.
- J.-F. Lefebvre, M. Lo, D. Leclercq and S. Richeter, Chem. Commun., 2011, 47, 2976–2978 RSC.
- J. A. Valdez-Moreira, D. C. Wannipurage, M. Pink, V. Carta, P. Moënne-Loccoz, J. Telser and J. M. Smith, Chem, 2023, 9, 2601–2609 CAS.
- C. A. Montgomery, I. Jameel, F. Cuzzucoli, T. Chidley, W. S. Hopkins and G. K. Murphy, Chem. – Eur. J., 2022, 28, e202202029 CrossRef.
- T. A. Gazis, B. A. J. M. Thaker, D. Willcox, D. M. C. Ould, J. Wenz, J. M. Rawson, M. S. Hill, T. Wirthe and R. L. Melen, Chem. Commun., 2020, 56, 3345–3348 RSC.
- G. Adembri, F. De Sio, R. Nesi and M. Scotton, J. Chem. Soc. C, 1970, 1536–1540 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, X-ray data collection/refinement details, NMR, IR and GC-MS data, and computational details. CCDC 2351176–2351178. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02108h |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |