
Exploring the antimicrobial potential of isoniazid loaded Cu-based metal–organic frameworks as a novel strategy for effective killing of Mycobacterium tuberculosis†
Pawan
Kumar‡
a,
Ananyaashree
Behera‡
b,
Pranav
Tiwari
a,
Sibi
Karthik
c,
Mainak
Biswas
b,
Avinash
Sonawane
bc and
Shaikh M.
Mobin
 *acde
*acde
aDepartment of Chemistry, Indian Institute of Technology, Indore, Simrol, Madhya Pradesh, India
bSchool of Biotechnology, KIIT Deemed to be University, Bhubaneswar, Odisha, India. E-mail: xray@iiti.ac.in; Tel: +91-731-2438752
cDepartment of Biosciences and Biomedical Engineering, Indian Institute of Technology, Indore, Simrol, Madhya Pradesh, India
dCenter for Advance Electronic (CAE), Indian Institute of Technology, Indore, Simrol, Madhya Pradesh, India
eCenter for Electric Vehicle and Intelligent Transport Systems, Indian Institute of Technology, Indore, Simrol, Madhya Pradesh, India
First published on 24th October 2023
Abstract
Tuberculosis (TB) remains one of the most infectious pathogens with the highest human mortality and morbidity. Biofilm formation during Mycobacterium tuberculosis (Mtb) infection is responsible for bacterial growth, communication, and, most essentially, increased resistance/tolerance to antibiotics leading to higher bacterial persistence. Thus, biofilm growth is presently considered a key virulence factor in the case of chronic disease. Metal–Organic Frameworks (MOFs) have recently emerged as a highly efficient system to improve existing antibiotics' therapeutic efficacy and reduce adverse effects. In this regard, we have synthesized Cu-MOF (IITI-3) using a solvothermal approach. IITI-3 was well characterized by various spectroscopic techniques. Herein, IITI-3 was first encapsulated with isoniazid (INH) to form INH@IITI-3 with 10 wt% loading within 1 hour. INH@IITI-3 was well characterized by PXRD, TGA, FTIR, and BET surface area analysis. Furthermore, the drug release kinetics studies of INH@IITI-3 have been performed at pH 5.8 and 7.4 to mimic the small intestine and blood pH, respectively. The results show that drug release follows first-order kinetics. Furthermore, the antimycobacterial activity of INH@IITI-3 demonstrated significant bacterial killing and altered the structural morphology of the bacteria. Moreover, INH@IITI-3 was able to inhibit the mycobacterial biofilm formation upon treatment and showed less cytotoxicity toward the murine RAW264.7 macrophages. Thus, this work significantly opens up new possibilities for the applications of INH@IITI-3 in biofilm infections in Mtb and further contributes to TB therapeutics.
1. Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of human tuberculosis (TB), remains one of the deadliest pathogens, infecting approximately 10 million people globally with 1.3 million death cases annually.1 Primarily, TB affects the lungs, known as pulmonary TB, and the treatment regime involves six months long administration of multiple antibiotics where patients in the initial two months intensive phase are given four first-line antibiotics, i.e., isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA) followed by a four-month continuation period where patients are given INH and RIF.1–3 Multidrug resistant (MDR) TB continues to be a public health emergency with severe health safety threat. The continuous and frequent administration of multiple drugs leads to low or non-adherence towards the therapy; moreover, toxic side effects such as gastrointestinal (GI) upset, fever, rash, etc. due to multiple dosing along with a longer duration of treatment also lead to defiance towards the therapy resulting in the emergence of drug-resistant strains among non-compliance patients.4,5 Partial/incomplete clearance of tubercle bacilli often results in reversion of the disease thereby recurrence of persistent drug-tolerant bacteria. Thus, both drug resistance and drug persistence are primary concerns in the case of TB and are also associated with biofilm formation.6Many pathogens, including Mtb, Staphylococcus aureus, and Pseudomonas aeruginosa, are known to form biofilms.6,7 Biofilms are functionally heterogeneous clusters of microcolonies or single cells of replicating and non-replicating bacteria, swathed in a milieu of autogenous extracellular polymeric molecules attached to organic or abiotic surfaces or to each other.7,8 Mycobacterial biofilms and persistent cells are mostly formed because of stress and quorum sensing. Biofilm development starts with bacterial adherence, followed by various stages of surface attachment, sessile growth, matrix synthesis, and dispersal.9 Biofilm formation is an ingrained mechanism of antibiotic resistance, among others, where it provides protection against antibiotics to several bacterial species that are typically effective against identical bacteria in a planktonic state.9,10 Studies suggest that the minimum bactericidal concentration (MBC) of antibiotics for biofilm eradication is usually higher than the planktonic bacterial cells, thus making it difficult for biofilm treatment by conventional antibiotic administrations due to the toxicity and side effects of higher doses of antibiotics.10 Various mechanisms have been involved in this antibiotic resistance, such as permeability, persisted cells, activation of resistance genes, and metabolic states.11 Moreover, biofilm formations are associated with various infections, which may result in treatment failures and other medical complications, as antibiotic therapy is solely incompetent in eradicating sessile organisms.12,13 To overcome these complications, there is a requirement for the development of new drug targets as well as potential therapeutic applications, which harbor higher efficacy to penetrate and destruct the biofilms and enter intracellular targets, thereby eliminating the bacteria.
A regulated and sustained drug delivery system can support reducing adverse effects and improving the therapeutic efficacy.14 Control over guest release profiles can be found by selecting different types of functional groups of the linker and fine-tuning the pore size.15 Furthermore, it can inhibit the establishment of drug-resistant species since changes in the drug content will leave bacteria with little time to adapt. Several advanced biomaterials, including biopolymer, silica, and lipid-based materials, have been created to improve the effectiveness of drug delivery systems.16–19 Despite the increasing advancement of biomaterials for drug delivery systems, inadequate drug loading, and controlled drug delivery due to the material's tiny pore volume remains an unacceptable aspect.20
A large pore volume material was developed recently known as Metal–Organic Frameworks (MOFs) to address these issues.21–25 MOFs are porous materials with topologies composed of inorganic single ion or ion cluster nodes connected by organic linkers via covalent, non-covalent, and different types of non-covalent interactions.26–34 They have a large surface area, tunable pore size, and are biocompatible as well as hemocompatible.35–39 MOFs have been used in a wide range of applications such as catalyst, gas adsorption/separation, sorbet for the preparation of different analytical samples, solid phase extraction of hydrocarbons, dye degradation, solar cells, etc.40–47 MOFs' bioapplication mainly include drug delivery, biosensing, bioimaging, phototherapy, wound healing, antibiotic detection in food, etc.48–54 Drugs are loaded within the pores and surface of the MOFs via some covalent, non-covalent interactions (in the case of neutral drugs) and some electrostatic hydrophilic/hydrophobic interactions (in the case of ionic drugs).55,56 In 2019, Simon et al.57 performed isoniazid drug delivery using MIL-100(Fe) MOFs for the first time. Similarly, Gaweł et al.58 and Acharya et al.59 performed the isoniazid drug delivery using Fe-MIL-101-NH2 and BIO-MOF-100 but did not determine the antibacterial activity of the released isoniazid. A study by Sun et al., 2017 demonstrates the application of mixed ligand Cu-MOFs (MOFs-2) as a drug delivery vehicle for the release of an anthracycline antibiotic, Doxorubicin hydrochloride (DOX). MOFs-2 exhibited the highest efficiency in DOX loading without affecting the structure of MOFs-2 and complete DOX release from DOX@MOFs-2 was observed in acidic solution (pH 3.8).60 DOX is also known to be a potent inhibitor of Mtb DnaG and demonstrates effective antibacterial activity against Msm and Mtb.61 Thus, DOX encapsulated MOFs could be potentially useful as a therapeutic agent for sustained drug delivery and antibacterial activity against drug resistant TB.
In the present study, we have synthesized a Cu(II) based MOF named IITI-3 using our previously reported method,62 which has a large surface area (1258 m2 g−1) and is mesoporous in nature (having a pore diameter of 3.420 nm).63 Encouraged by our recent work, which showcases controlled insulin delivery by IITI-3, prompted us to explore this towards TB therapeutics. The in vitro cytotoxicity investigations of IITI-3 validated its safety, and the accumulation detected in the cell cytoplasm suggests potential utility in the treatment of pulmonary tuberculosis. After that, we incorporated IITI-3 with the INH drug and performed drug release kinetic studies under different conditions. Next, we demonstrated that upon treatment of M. smegmatis (Msm) and M. bovis BCG (BCG), the non-pathogenic laboratory strains of Mtb with INH@IITI-3, there was significant reduction in the bacteria count and altered the bacterial morphology. INH@IITI-3 treatment inhibited biofilm formation in Msm and the pellicle formed between air–liquid interfaces were loose and detached. However, no difference was observed in the biofilm disruption assay. Additionally, comparative cytotoxicity analysis was checked and INH@IITI-3 exhibited a less cytotoxic effect on the host RAW 264.7 macrophages. Together, these observations indicate that INH@IITI-3 could have significant implications in eradicating mycobacterial biofilms by preventing biofilm development and it could be an intriguing substitute for the standard TB therapy. Nevertheless, further studies of INH@IITI-3 against the pathogenic strains of Mtb are essential for better understanding the activity and functionality of drug-based MOFs.
2. Materials and methods
2.1 Materials
All reagents were commercially procured and were used without any further purification. Cu(NO3)2·3H2O (98%), 5-hydroxyisophthalic acid, 2,6-bis(bromomethyl)pyridine, DMF, HCl, EtOAc, Na2SO4, K2CO3, H2SO4, and isoniazid were purchased from Sigma-Aldrich, Loba Chemie and S. D. Fine Chemicals (India), respectively.2.2 Synthesis of H4L
H4L was synthesized by the previously reported method.62 In the first step, a simple substitution reaction takes place between 5-hydroxyisophthale and 2, 6-bis(bromomethyl)pyridine to form diethyl 5-((6-((3-(ethoxycarbonyl)-5-(propionyloxy)phenoxy)methyl)pyridin-2-yl)methoxy)isophthalate in the presence of potassium carbonate and acetonitrile solvent under a N2 atmosphere. In the second step, the basic hydrolysis takes place, resulting in the formation of 5,5′-((pyridine-2,6-diylbis(methylene))bis(oxy))diisophthalic acid (H4L). The detailed synthetic procedure with characterization (Fig. S1–S6) is given in the ESI.†2.3 Synthesis of IITI-3
The preparation method of IITI-3 has been taken from our previously reported method.62 In this method, H4L (0.040 g, 0.043 mmol), Cu(NO3)2·3H2O (0.112 g, 0.23 mmol), and 0.2 mL of 1 M HCL solution were combined with DMF (4.0 mL) and water (2.0 mL). This mixture was then agitated for 48 hours under high pressure in a Teflon-lined autoclave, and after completion of the reaction, it was cooled to room temperature at a rate of 1 °C min−1. The IITI-3 blue crystals were made. These crystals were dried in a vacuum oven after being constantly cleaned with water and acetone. (Yield = 79.8%) (Scheme 1). | ||
| Scheme 1 Schematic synthesis of IITI-3. | ||
2.4 Activation of IITI-3
IITI-3 was synthesized in a DMF/water solvent system, so there is a high probability that these solvent molecules may be present within the framework and occupy some space within the pores. So, to create a solvent-free and highly porous framework, before utilizing it as the drug delivery carrier, we have activated IITI-3. The synthetic material was activated using a solvent exchange experiment.64,65 In this experiment, we have taken acetone as a solvent because acetone does not disturb the structural integrity of the framework. This experiment took place over seven consecutive days and twice a day.Further heating of the solvent-exchanged sample at 130 °C for 6 hours under extreme vacuum resulted in the formation of additional pores and a solvent-free framework confirmed by the color changes of IITI-3 from blue to dark blue, resulting in the loss of metal-bound water molecules (Fig. S7, ESI†). Activated IITI-3 maintained the framework through the activation, which is confirmed by PXRD. PXRD data of activated IITI-3 have all the significant peaks present in as synthesized IITI-3 (Fig. S8, ESI†).
2.5 INH encapsulation within THE IITI-3 framework
To encapsulate the isoniazid, the crystals of activated IITI-3 were treated with the INH solution (DI water, 100 μg mL−1 at pH = 7.0) for continuous stirring at 200 rpm for 1 h at room temperature. The resulting solid composite was filtered, and the supernatant was collected. The excess INH molecules adhered to the surface of the IITI-3 crystals and were removed by washing the solid composite with DI water three times.2.6 In vitro INH release studies
A drug delivery experiment was performed using the dialysis technique. A definite amount (10 mg) of INH@IITI-3 was placed in a culture tube and submerged in 10 mL of a solution with pH values of 5.8 and 7.4, continuously stirring at 200 rpm at room temperature. 2 mL of sample was taken at every finite time interval. The same volume of the new solution with the same pH was added to keep the volume constant. A UV-Vis spectrophotometer was used to quantify how much drug had been released (for INH, λ = 263 nm).Using a linearity plot (Fig. S8, ESI†) and UV-Vis spectrometry, the loading and release characteristics of INH from INH@IITI-3 were identified, and the drug loading content (DLC) was calculated according to the following formula,66
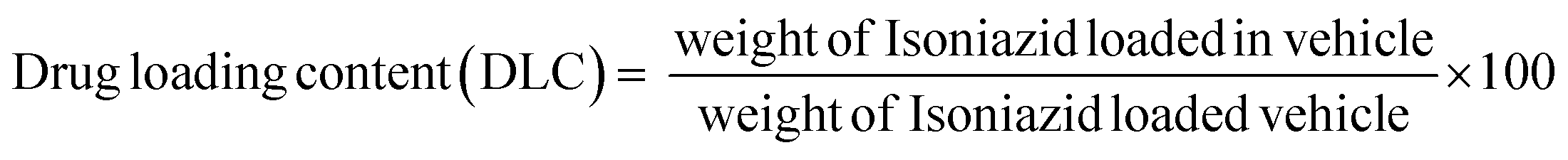 | (1) |
2.7 Bacterial strains and reagents
Mycobacterium smegmatis mc2155 (Msm) and Mycobacterium bovis BCG (BCG) were grown in Middlebrook's 7H9 broth medium (271310, Difco, New Jersey, USA) containing 0.05% Tween80 at 37 °C at 120 rpm. M. bovis BCG was supplemented with 10% OADC (oleic acid-albumin-dextrose-catalase) (FD018, Himedia, India). Mouse macrophage RAW264.7 cells were maintained in DMEM (Dulbecco's modified Eagle's medium) (10566016, Gibco, Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS) (10270106, Gibco, Invitrogen, USA) at 37 °C. Cell Counting Kit-8 (CCK-8) (CK04-13) was procured from Dojindo Molecular Technologies (Japan). Sauton's Fluid medium base (M1276, Himedia, India) was used to grow a mycobacterial biofilm. Crystal violet (28376) staining was obtained from Sisco Research Laboratories (SRL), India. Acridine orange (MB116) was procured from Himedia, India.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (7:3) solution for 1 hour, followed by spectrophotometric quantification at 595 nm.
:ethanol (7:3) solution for 1 hour, followed by spectrophotometric quantification at 595 nm.
3. Results and discussions
3.1 Preparation and characterization of IITI-3, INH@IITI-3
The preparation process of IITI-3, INH@IITI-3, is depicted in Schemes 1 and 2, respectively. | ||
| Scheme 2 Systematic scheme of INH encapsulation and release from INH@IITI-3. | ||
First, the IITI-3 has synthesized using solvothermal approach by combining H4L and Cu(NO3)2·3H2O in DMF and water (2:1) under acidic conditions and then heating to 90 °C for 48 hours. Blue-colored crystals were produced. The produced crystals of IITI-3's structure were evaluated using a variety of characterization techniques.
A powder X-ray diffractometer (PXRD) has been recorded and compared with the previously reported IITI-3 MOF to check the phase purity of as-synthesized material.62 Most of the peaks were matched with the as-synthesized material. Some different, less intense peaks are present due to the solvent molecules in the lattice. After solvent exchange, activation, and INH encapsulation, the framework maintained its crystallinity, as confirmed by PXRD in Fig. 1(a). To evaluate the functional group within IITI-3, Fourier Transfer Infrared (FT-IR) spectra have been recorded. In the FT-IR spectra of IITI-3, one low intense peak at 3451 cm−1 corresponds to the water molecule present in the framework, and two peaks at 1631 and 1377 cm−1 correspond to asymmetric and symmetric vibrations of the carboxylate ion.69 Two peaks around 1577 and 774 cm−1 confirm the presence of a pyridyl ring within IITI-3.70–72 After INH encapsulation, all the significant peaks present in INH@IITI-3 exactly match with the IITI-3, confirming that after encapsulation IITI-3 maintained their functionality (Fig. 1b). MOFs are well known for their enormous surface area, and BET analysis has been performed for this. Results show that IITI-3 has a high surface area of 1258 m2 g−1 and is mesoporous with a pore diameter of 3.420 nm.63,73,74 After encapsulation, INH@IITI-3 has less surface area of 803.21 m2 g−1 and a decrease in pore diameter of 3.346 nm compared to IITI-3, confirming that INH is present within the pore as well as on the surface of IITI-3 (Fig. 1c and d).
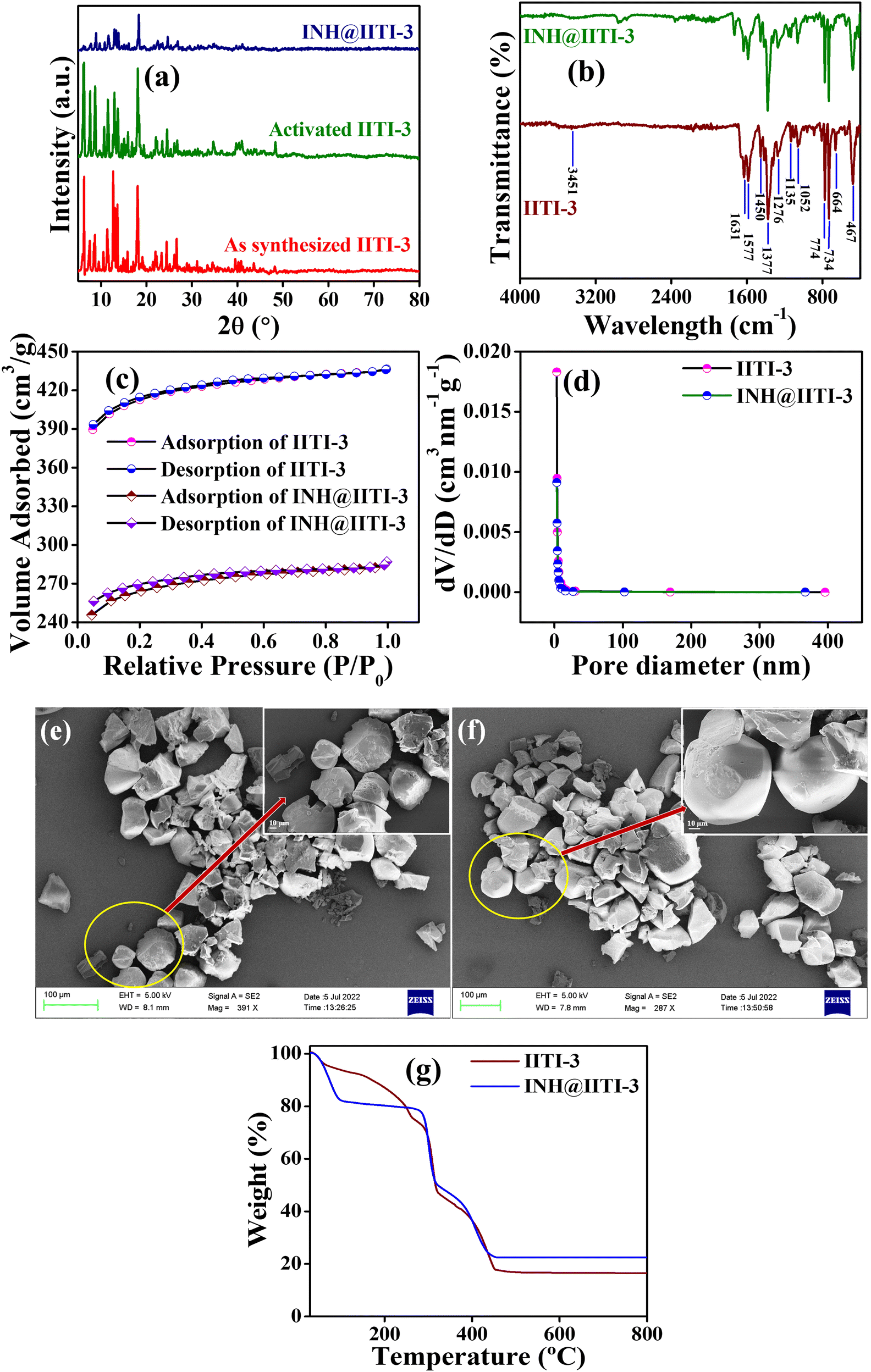 | ||
| Fig. 1 (a) PXRD of as synthesized IITI-3, activated IITI-3 and INH@IITI-3, (b) FT-IR data of IITI-3 and INH@IITI-3, (c) BET surface area analysis of IITI-3 and INH@IITI-3, (d) BJH pore size distribution of IITI-3 and INH@IITI-3, (e) and (f) SEM analysis of IITI-3 and INH@IITI-3 at 100 and 10 μm magnification respectively, and (g) TGA of IITI-3 and INH@IITI-3. | ||
Field emission surface electron microscopy (FE-SEM) has been performed to know the surface morphology of IITI-3. The results show that the IITI-3 has an irregular cuboid-type of shape. After INH encapsulation, INH@IITI-3 has a similar surface morphology as IITI-3, revealing no morphology changes after INH encapsulation (Fig. 1e and f). The thermal stability of the framework IITI-3 is confirm by Thermogravimetric analysis (TGA). The TGA graph shows a 25% decrease in weight percent from 70 to 260 °C, corresponding to the removal of guest molecules (water/DMF) from the lattice. From 296 to 320 °C the framework starts breaking, and from 320 to 458 °C framework ultimately reaches elemental form. And beyond 458 °C, after increasing the temperature, no peak is observed, confirming that the framework completely goes into elemental state. After INH encapsulation, there is an increase in weight percent in the TGA graph of INH@IITI-3, confirming that INH is successfully encapsulated within the framework of IITI-3 (Fig. 1g). From Dynamic light scattering (DLS), the average hydrodynamic size of INH@IITI-3 is calculated at 689.4 nm, which is increased from 392.1 nm due to INH encapsulation (Fig. S10, ESI†).
3.2 Drug loading content and in vitro drug release kinetics
A 10 wt % INH loading is achieved within 1 h of encapsulation, calculated from eqn (1). The drug is encapsulated within the pore and on the surface of the framework, confirm by the BET analysis. After INH loading within IITI-3, INH goes to inside the pore and some amount is also present on the surface of the IITI-3. IITI-3 upheld the INHvia various interactions like Electrostatic (hydrophilic and hydrophobic), hydrogen bonding, van der Waals, coordination bonds and some π–π interaction.75–77 As INH is a neutral small molecule it possibly goes inside the pore and will be held via covalent, π–π interaction and some hydrogen bond interaction. After successfully calculating the loading percentage, in vitro drug release studies from INH@IITI-3 have been performed at pH 5.8 and 7.4 to mimic the small intestine and blood pH, respectively.78 The drug release from INH@IITI-3 was due to the diffusion process.58 In this process the INH comes out from the higher concentration region to the lower concentration region.79,80 When we performed the drug delivery experiment, we suspended the 10 mg of INH@IITI-3 in the 10 mL of solution. INH started to come out from INH@IITI-3 to the solution due to the concentration difference. As in the dialysis method, the same amount (in our case, 2 mL) of the solution is taken out after a fixed time and maintained the same volume by fresh solution resulting the more concentration difference and more drug release.The drug release experiments have performed by gentle stirring at 200 rpm. The drug release model is described mainly from three types of models Higuchi (eqn (2)),81 Zero order (eqn (3)),81 and First order (eqn (4)),82 which are as follows:
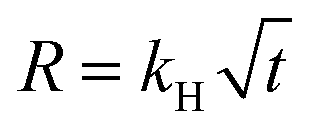 | (2) |
| R = q0 + k0t | (3) |
| R = Re(1 − e−kt) | (4) |
The model fitting into the drug release profile from INH@IITI-3 is shown in Fig. 2, Fig. S11 (ESI†). And parameter for the fitting of different models is given in Table 1. We have fitted in the Higuchi kinetic model, zero order, and first-order model, but the fitting parameter and analysis clearly show that the drug release follows 1st order kinetics which corresponds to the drug release from the porous matrices.14,84 This suggests that the drug is released continuously and at different rates.87 The experimental cumulative drug release (R) value after 24 hours is 39.08% and 43.47% at pH 5.8 and 7.4, respectively. From the first-order kinetic model, it is calculated from 35.91% and 39.33%, which is well matched with the calculated one.
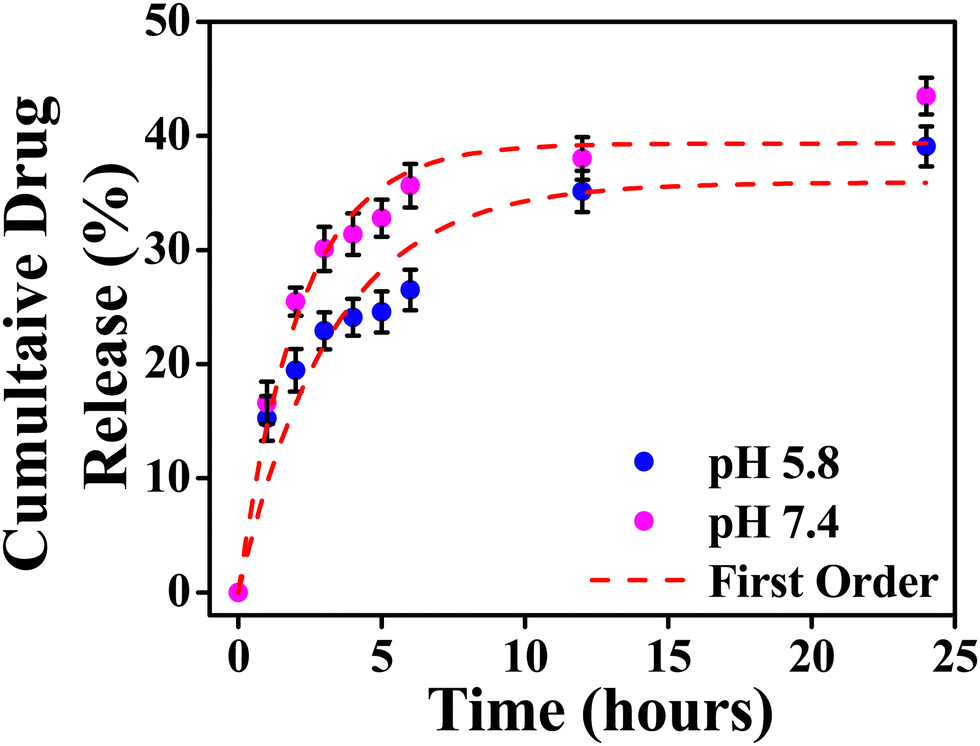 | ||
| Fig. 2 Cumulative drug release from INH@IITI-3 at pH 5.8 and 7.4 with first order kinetics respectively. | ||
| Model | Parameter | pH 5.8 | pH 7.4 |
|---|---|---|---|
| Higuchi | k H | 9.88922 ± 0.67901 | 11.80515 ± 1.10224 |
| R 2 | 0.79465 | 0.59184 | |
| Zero order | k 0 | 1.12194 ± 0.33423 | 1.2345 ± 0.46172 |
| R e, % | 15.26069 ± 3.17272 | 20.3483 ± 4.38294 | |
| R 2 | 0.6072 | 0.43458 | |
| First order | k | 0.30763 ± 0.0584 | 0.46443 ± 0.05691 |
| R e, % | 35.9153 ± 2.56969 | 39.33457 ± 1.46521 | |
| R 2 | 0.90805 | 0.96826 | |
The drug release was comparably high in alkaline pH, because at higher pH, after releasing the INH, positively charged sites are vacant and the negatively charged OH− from the alkaline solution stabilizes the IITI-3 framework.57 This is confirmed by the zeta potential value of INH@IITI-3, −23.5, and −25.6 at pH 5.8 and 7.4, respectively (Fig. S12, ESI†). There is a slight increase in zeta potential leading to a slight increase in drug delivery. The increased negative value of zeta potential will cause more positively charged sites present on the surface.88 And at pH 7.4, a greater number of positively charged sites are stabilized in comparison to lower pH 5.8. This concludes that at 7.4 pH, INH release is comparatively higher with respect to pH 5.8, leading to higher zeta potential.89 This will cause interference in interactions between INH molecules and the active surface of IITI-3.
3.3 Evaluation of antimycobacterial efficiency of the compounds
The synthesized IITI-3, INH@IITI-3, and free INH were tested against Msm and BCG to assess their antimycobacterial activity. The results obtained against the assayed Msm indicate a significant decrease in the antibacterial activity of IITI-3, INH@IITI-3, and free INH when compared with the buffer control after 24 hours of treatment (Fig. 3A). Significant killing of Msm when treated with IITI-3 alone indicates that MOFs, especially Cu-MOFs, can also release metal ions, which can destruct the cellular integrity, thereby killing the bacteria.90 Several studies have shown the synergistic effect of antibiotics when incorporated in MOFs. The release of metal ions from the MOFs concurrent with the antibiotic effect on the bacteria synergistically augments the antibacterial activity,91,92 which herein, with the treatment of INH@IIT-3, substantial Msm killing was observed (Fig. 3A). The cell wall of mycobacteria is crucial for its viability and virulence. There are two probable mechanisms for MOFs to incapacitate/eradicate bacteria. Leaching of metal ions from the MOFs or the interactions between the active sites of MOF surfaces and the bacterial surface to peroxidise bacterial membrane lipids, thus disrupting the membrane.93 However, interestingly, we did not observe a significant reduction in the bacterial count when BCG was treated with only IITI-3 (Fig. 3B). This might be because of the several virulence factors of BCG and its distinctive cell envelope that is not affected only by the Cu(II) ions. However, further studies are a prerequisite to establishing the MOF mechanism of action against BCG. Next, we observed significant killing of BCG with increasing concentration of INH@IITI-3 when compared with IITI-3 and free INH (Fig. 3B). The probable reason for significant bacterial reduction might be the synergistic effect of INH together with the Cu(II) ions. However, there is a necessity for detailed evaluation regarding the MOF activity against BCG for a better understanding of the mechanism.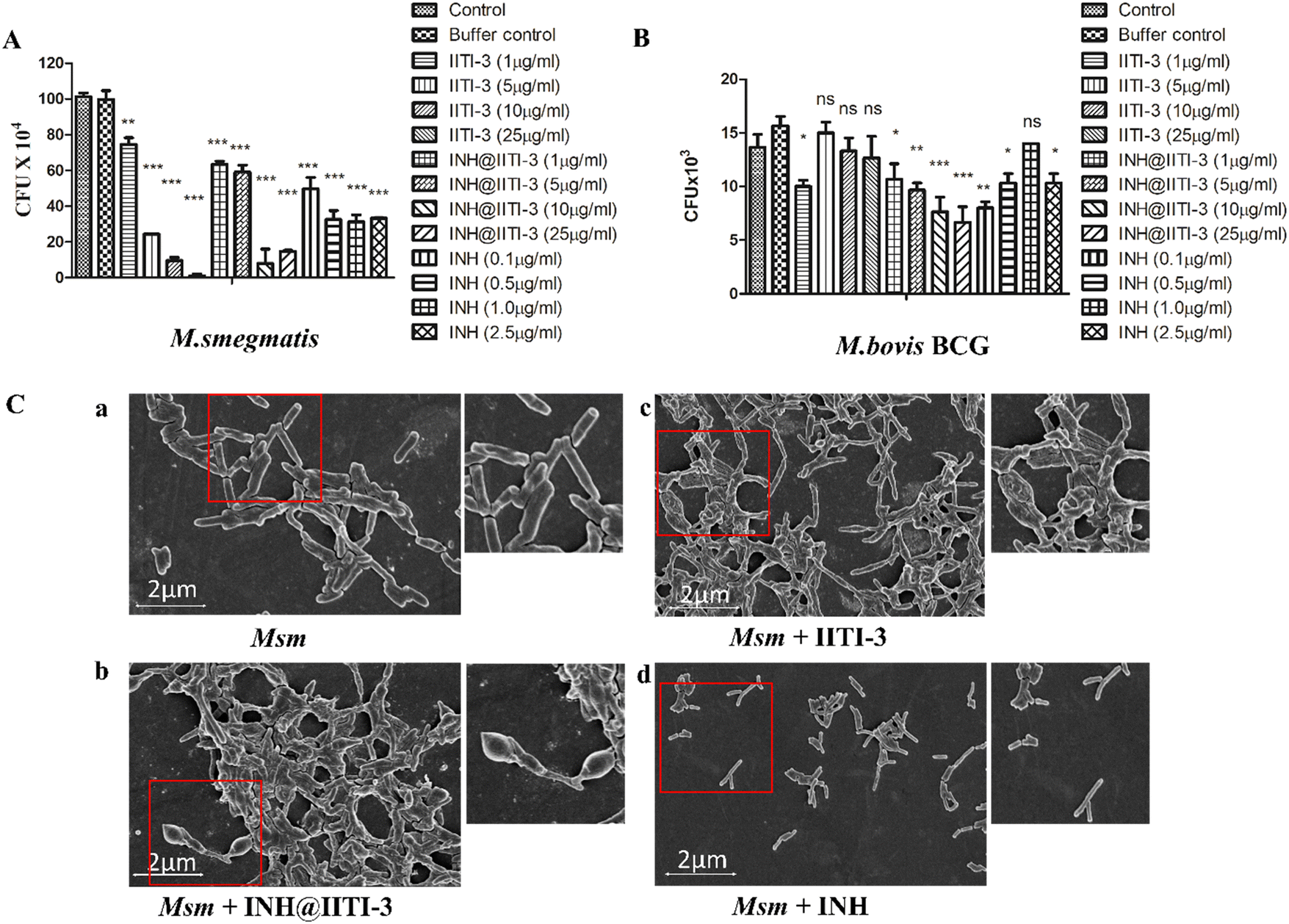 | ||
| Fig. 3 Antibacterial activity of INH@IITI3 was determined against (A) M. smegmatis and (B) M. bovis BCG. Bacteria were incubated with different concentrations of IITI-3, INH@IITI-3 and INH for the indicated time point and bacterial survival was determined by the CFU assay. (C) FE-SEM of M. smegmatis: (a) Control, (b) with INH@IITI-3 treatment, (c) with IITI-3 treatment, and (d) with INH treatment. Msm was analysed for morphological variations with different treatments of the compounds. | ||
Furthermore, we visualized the effects of IITI-3 and INH@IITI-3 on the morphology of Msm using a field emission scanning electron microscope (FE-SEM). The exposure of Msm to 25 μg mL−1IITI-3 or INH@IITI-3 for 24 hours resulted in morphological abnormalities of their cell shape. Untreated Msm demonstrated smooth, uniform, and slightly wrinkled cell structure (Fig. 3C-a). After treatment with IITI-3 or INH@IITI-3, the bacterial cells displayed a loss in cellular integrity. The cells were irregular, longer and bulbous with a rough and wrinkled surface when exposed to INH@IITI-3 as shown in (Fig. 3C-b). Likewise, IITI-3 treated Msm displayed irregular, wrinkled, and destructed cell wall (Fig. 3C-c). We did not observe any change in the bacterial structure when treated with INH (Fig. 3C-d). These results suggest that INH-incorporated IITI-3 has the potency to destruct the Msm cell wall, thereby effectively killing the bacteria.
3.4 Inhibition of biofilm formation by INH-encapsulated IITI-3
Biofilms are associated with several microbial infections leading to serious clinical complications.9,94 The emergent properties of microbial biofilms, especially mycobacterial biofilms, include increased resistance to antibiotics and antimicrobial agents that may result in treatment failure.95,96 Thus, a comparative anti-biofilm potential of newly synthesized INH@IITI-3 and INH was performed against Msm. As the bound crystal violet was measured after 5 days of incubation, Fig. 4A shows a significant reduction in the biofilm formation in INH@IITI-3 treated Msm as compared with IITI-3 and buffer control treated Msm. There was a marked inhibition of biofilm formation in INH-treated Msm. However, the measurement of bound crystal violet for dispersed biofilm quantification showed no significant reduction in the biofilms after 5 days of treatment, indicating no substantial difference in the biofilm disruption Fig. 4B. From the above result, it can be suggested that INH@IITI-3 has potential anti-biofilm activity against Msm.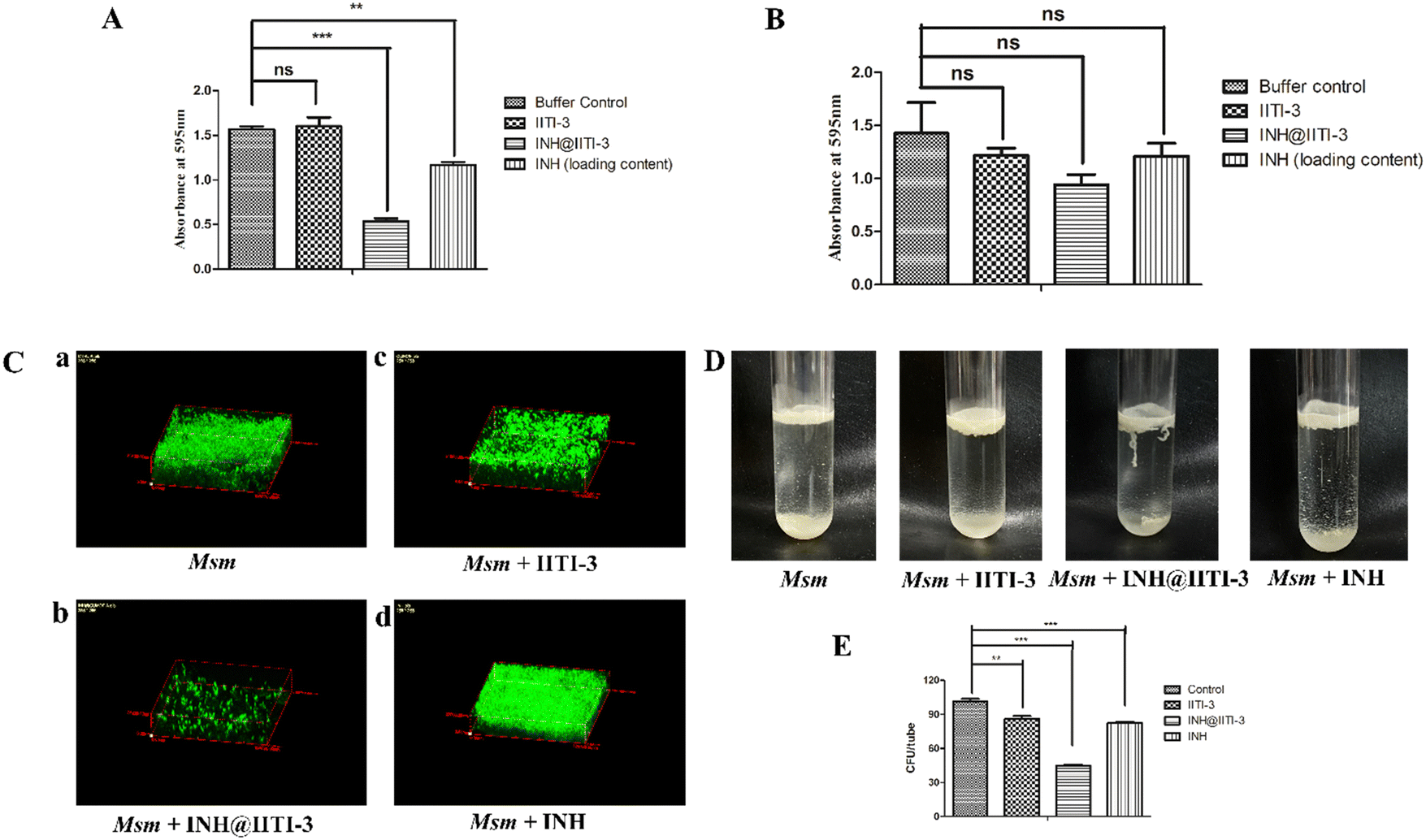 | ||
| Fig. 4 Biofilm assay of M. smegmatis using IITI-3, INH@IITI-3 and INH to determine (A) biofilm formation and (B) biofilm disruption, by crystal violet staining. (C) Acridine orange staining to determine the biofilm formation with various treatments using Confocal Laser Scanning Microscopy (CLSM). (D) Pellicle formation assay to determine the bacterial growth at the air–liquid interface with IITI-3, INH@IITI-3 and INH treatment. (E) Determination of bacterial count post pellicle formation using the CFU assay. | ||
Next, to corroborate the inhibition in biofilm formation, Acridine orange staining was performed. Microscopic analysis revealed significantly reduced, thin and scattered biofilms of INH@IITI-3 (Fig. 4C-b) treated Msm as compared to the highly dense, compact, and well-structured biofilms of only Msm control (Fig. 4C-a) and INH (Fig. 4C-d) treated Msm (Fig. 4C). Moreover, lesser dense and slightly scattered biofilms were observed in IITI-3-treated Msm (Fig. 4C-c). From the above data, it can be suggested that INH@IITI-3 has the potential to destruct the formed biofilms, thus indicating its possibility to exterminate biofilm infections during TB treatment.
Mycobacteria have the ability to form floating biofilms at the interface between air and liquid, known as the pellicle. Bacterial cells present within biofilm or pellicle are more resistant to antibiotic therapies and undergo horizontal transfer of antibiotic resistance genes among themselves.97 Studies suggest that isolates and mutants of Gram-negative bacteria such as Salmonella enterica and Pseudomonas aeruginosa demonstrate a strong relationship between pellicle formation and biofilm forming ability.98,99 A study by Chen et al., 2006 reports M. smegmatis lsr2 mutant was unable to form pellicles and was also defective in biofilm formation.100 Similarly, overexpression of rv1288 in M. smegmatis resulted in enhanced pellicle growth and aggregate formation indicating its link with the changed lipid composition of the cell wall.101 Herein, we studied the effect of the synthesized compounds on the Msm pellicle. We observed thick, intact, and enhanced pellicles for control Msm, IITI-3-treated Msm, and INH-treated Msm (Fig. 4D). Additionally, the pellicle of INH@IITI-3 treated Msm exhibited reduced capability to remain attached and was visualized to be loose and easily disturbed (Fig. 4D). Next, bacterial enumeration was performed by CFU assay, where we observed significantly reduced bacterial count in INH@IITI-3 treated Msm. Bacterial enumeration of IITI-3/INH treated Msm also indicated reduced bacterial count as compared to control Msm pellicle (Fig. 4E). Together, these results demonstrate that INH@IITI-3 is effective against Msm biofilms and pellicles, and with further studies, it can be effectively used in TB therapeutics.
3.5 Cytotoxicity study on mouse macrophages
Nanomaterials have a large surface area, making them better agents for various biological applications.102 Thus, determining the toxicity of synthesized nanomaterials is very critical. Despite the extensive use of INH for TB treatment, there are several toxic side effects associated with INH.103,104 Here, a comparative analysis of the cytotoxic action of IITI-3, INH@IITI-3, and INH has been performed against murine macrophages (RAW 246.7). After 24 hours of incubation with various concentrations of each molecule, cell viability has been determined. Fig. 5A indicates that cells treated with IITI-3 and INH alone did not show any toxic effects; however, with the increase in the concentration (25 μg mL−1) of INH@IITI-3, the viability of cells has decreased. The obtained data suggests that a higher concentration of INH@IITI-3 leads to a slight decrease in the cell viability. To further validate the effect of the synthesized and drug incorporated MOFs on macrophage morphology, cells were visualized under the light microscope. As shown in Fig. 5B, IITI-3 and INH alone treated RAW 264.7 macrophages showed negligible cytotoxic effect with no damage to the cell morphology.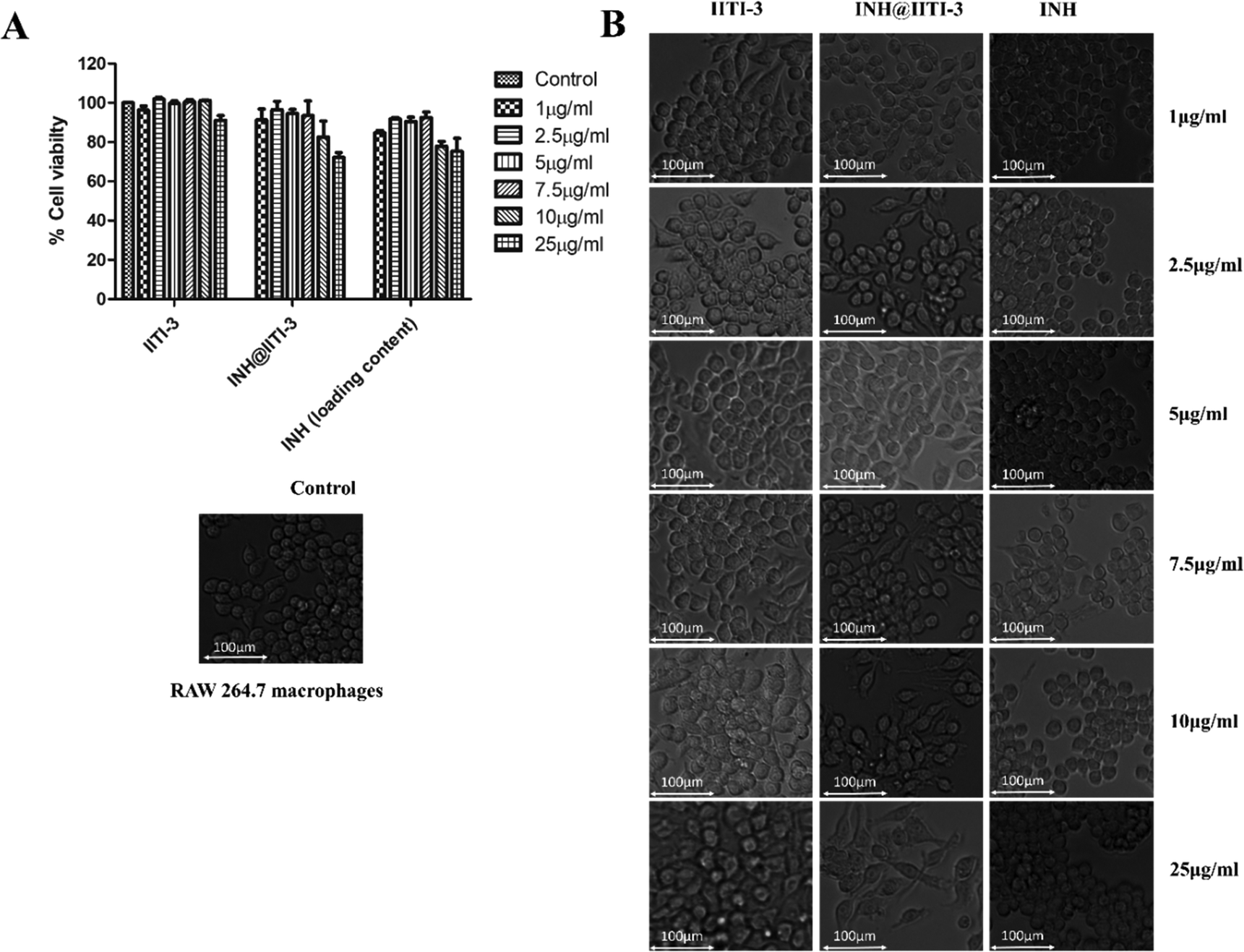 | ||
| Fig. 5 In vitro cytotoxicity of IITI-3, INH@IITI-3 and INH was determined in (A) RAW 264.7 murine macrophages. Cells were treated with different concentrations of compounds for 24 hours and CCK-8 analysis was performed to check the viability. (B) Bright-field microscopy images of macrophages after treatment with different concentrations of the compounds. | ||
However, with an increase in the concentration of INH@IITI-3, macrophages exhibited few pseudopodia formation which could be due to the effect of the compound on the cells. These results imply that INH@IITI-3 could be a promising candidate for therapeutic applications.
4. Conclusion
In this research work, IITI-3 has been synthesized successfully. IITI-3 has a large surface area and is mesoporous in nature. Furthermore, IITI-3 was incorporated with INH to form INH@IITI-3. The prepared INH@IITI-3 is well characterized by different physiochemical methods. The drug loading content is 10 wt% within 1 hour of time. Further drug release kinetics has been studied at two different pH values mimicking the small intestine and blood pH conditions, and the results showed that drug release behaviour follows first-order kinetics with an R2 value of 0.9 which corresponds to drug release continuously with different rates. Furthermore, INH@IITI-3 presented significant antimicrobial properties against Msm and BCG that were evident from the in vitro studies and morphological analysis. The study provides an overview of potential INH@IITI-3 that holds promise in inhibiting mycobacterial biofilms and pellicles. A better understanding of INH@IITI-3 and its underlying mechanism on the pathogenic TB strains could play an important role in developing more active, less toxic TB therapy approaches and in lowering the prevalence of Mtb treatment failures.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. M. M. acknowledges BRNS (project no. 58/14/17/2020-BRNS/37215), and IIT Indore for financial support. P. K. would like to thank the Ministry of Education, Government of India, for providing a research fellowship. This work was supported by an Indo-Russian joint research grant (INT/RUS/RFBR/380) from the Department of Science and Technology, Government of India, to A. S. P. T. thanks BRNS (project no. 58/14/17/2020-BRNS/37215). The authors gratefully acknowledge Dr Ravinder, Mr Kinny Pandey, Mr Ghanashyam Bhavsar, and Dr Nitin Upadhyay (Sophisticated Instrumentation Centre, IIT Indore) for their help during characterization studies. The authors are also thankful to Material Research Centre, MNIT Jaipur, for proving the DLS and zeta potential facility. The authors are also thankful to Mr Ravindra Vishwakarma and Dr Tridib K. Sharma, Department of Chemistry IIT Indore, for providing DLS and Zetasizer facility. P. K. also thanks Dr Diptangshu Datta Mal, Dr Navpreet Kaur, and Praveen Kumar for their help with manuscript preparation. The authors would also like to thank the members of the A. S. lab and S. M. M. lab for fruitful suggestions and discussions.References
- Global tuberculosis report 2021, https://www.who.int/publications-detail-redirect/9789240037021, (accessed August 28, 2022).
- G. Sotgiu, R. Centis, L. D’ambrosio and G. B. Migliori, Cold Spring Harbor Perspect. Med., 2015, 5, a017822 CrossRef PubMed.
- R. E. Maphasa, M. Meyer and A. Dube, Front. Cell. Infect. Microbiol., 2021, 10, 618414 CrossRef PubMed.
- Host-directed therapies for bacterial and viral infections|Nature Reviews Drug Discovery, https://www.nature.com/articles/nrd.2017.162, (accessed April 24, 2023).
- M. Badrinath and S. John, StatPearls, StatPearls Publishing, Treasure Island (FL), 2023 Search PubMed.
- A. Solokhina, D. Brückner, G. Bonkat and O. Braissant, Sci. Rep., 2017, 7, 9225 CrossRef PubMed.
- L. K. Vestby, T. Grønseth, R. Simm and L. L. Nesse, Antibiotics, 2020, 9, 59 CrossRef CAS PubMed.
- E. Roilides, M. Simitsopoulou, A. Katragkou and T. J. Walsh, Microbiol. Spectrum, 2015, 3, 3.3.22 CrossRef PubMed.
- J. P. Richards and A. K. Ojha, Microbiol. Spectrum, 2014, 2, 2.5.16 CrossRef PubMed.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34–40 CrossRef CAS PubMed.
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482–501 CAS.
- O. Ciofu, E. Rojo-Molinero, M. D. Macià and A. Oliver, APMIS, 2017, 125, 304–319 CrossRef PubMed.
- H. Wu, C. Moser, H.-Z. Wang, N. Høiby and Z.-J. Song, Int. J. Oral. Sci., 2015, 7, 1–7 CrossRef CAS PubMed.
- S. Adepu and S. Ramakrishna, Molecules, 2021, 26, 5905 CrossRef CAS PubMed.
- Z. Dong, Y. Sun, J. Chu, X. Zhang and H. Deng, J. Am. Chem. Soc., 2017, 139, 14209–14216 CrossRef CAS PubMed.
- R. Langer, Acc. Chem. Res., 2000, 33, 94–101 CrossRef CAS PubMed.
- J. Jacob, J. T. Haponiuk, S. Thomas and S. Gopi, Mater. Today Chem., 2018, 9, 43–55 CrossRef CAS.
- L. Pasqua, I. E. D. Napoli, M. D. Santo, M. Greco, E. Catizzone, D. Lombardo, G. Montera, A. Comandè, A. Nigro, C. Morelli and A. Leggio, Nanoscale Adv., 2019, 1, 3269–3278 RSC.
- E. Bagheri, L. Ansari, K. Abnous, S. M. Taghdisi, F. Charbgoo, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 277, 57–76 CrossRef CAS PubMed.
- J.-J. Zou, G. Wei, C. Xiong, Y. Yu, S. Li, L. Hu, S. Ma and J. Tian, Sci. Adv., 2022, 8, eabm4677 CrossRef CAS PubMed.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS PubMed.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- L. Robison, X. Gong, A. M. Evans, F. A. Son, X. Wang, L. R. Redfern, M. C. Wasson, Z. H. Syed, Z. Chen, K. B. Idrees, T. Islamoglu, M. Delferro, W. R. Dichtel, F.-X. Coudert, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 1503–1512 CrossRef CAS PubMed.
- Z. Chen, M. R. Mian, S.-J. Lee, H. Chen, X. Zhang, K. O. Kirlikovali, S. Shulda, P. Melix, A. S. Rosen, P. A. Parilla, T. Gennett, R. Q. Snurr, T. Islamoglu, T. Yildirim and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 18838–18843 CrossRef CAS PubMed.
- J. Tian, Z.-Y. Xu, D.-W. Zhang, H. Wang, S.-H. Xie, D.-W. Xu, Y.-H. Ren, H. Wang, Y. Liu and Z.-T. Li, Nat. Commun., 2016, 7, 11580 CrossRef CAS PubMed.
- L. Zhang, Z. Liu, Q. Deng, Y. Sang, K. Dong, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2021, 60, 3469–3474 CrossRef CAS PubMed.
- Q. Xia, W. Li, X. Zou, S. Zheng, Z. Liu, L. Li and F. Yan, Mater. Horiz., 2022, 9, 2881–2892 RSC.
- M. Mirzaei, H. Eshtiagh-Hosseini, Z. Karrabi, K. Molčanov, E. Eydizadeh, J. T. Mague, A. Bauzá and A. Frontera, CrystEngComm, 2014, 16, 5352–5363 RSC.
- M. Bazargan, M. Mirzaei, A. Franconetti and A. Frontera, Dalton Trans., 2019, 48, 5476–5490 RSC.
- Z. Rahmati, R. Khajavian and M. Mirzaei, Inorg. Chem. Front., 2021, 8, 3581–3586 RSC.
- M. Bazargan, M. Mirzaei, A. Amiri and C. Ritchie, Microchim. Acta, 2021, 188, 108 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- G. Li and Y. Han, ACS Cent. Sci., 2022, 8, 150–152 CrossRef CAS PubMed.
- N. Singh, S. Qutub and N. M. Khashab, J. Mater. Chem. B, 2021, 9, 5925–5934 RSC.
- Z. Chao, J. Li, W. Jiang, C. Zhang, J. Ji, X. Hua, L. Xu, L. Han and L. Jia, Mater. Chem. Front., 2021, 5, 7617–7627 RSC.
- W. Zhu, J. Guo, J. O. Agola, J. G. Croissant, Z. Wang, J. Shang, E. Coker, B. Motevalli, A. Zimpel, S. Wuttke and C. J. Brinker, J. Am. Chem. Soc., 2019, 141, 7789–7796 CrossRef CAS PubMed.
- K. Wang, Y. Li, L.-H. Xie, X. Li and J.-R. Li, Chem. Soc. Rev., 2022, 51, 6417–6441 RSC.
- A. Hassanpoor, M. Mirzaei, M. N. Shahrak and A. M. Majcher, Dalton Trans., 2018, 47, 13849–13860 RSC.
- A. Abdar, A. Amiri and M. Mirzaei, J. Chromatogr. A, 2023, 1707, 464295 CrossRef CAS PubMed.
- A. Abdar, A. Amiri and M. Mirzaei, Microchem. J., 2022, 177, 107269 CrossRef CAS.
- S. Ghanei-Zare, M. Moghadasi, R. Khajavian, N. Akbarzadeh-T and M. Mirzaei, J. Mol. Struct., 2023, 1286, 135563 CrossRef CAS.
- R. Khajavian, M. Mirzaei and H. Alizadeh, Dalton Trans., 2020, 49, 13936–13947 RSC.
- M. Bazargan, F. Ghaemi, A. Amiri and M. Mirzaei, Coord. Chem. Rev., 2021, 445, 214107 CrossRef CAS.
- A. Amiri and M. Mirzaei, RSC 2023 ebook collection 10.1039/9781839167485.
- J. Yang and Y.-W. Yang, Small, 2020, 16, 1906846 CrossRef CAS PubMed.
- H. Sohrabi, S. Ghasemzadeh, S. Shakib, M. R. Majidi, A. Razmjou, Y. Yoon and A. Khataee, Ind. Eng. Chem. Res., 2023, 62, 4611–4627 CrossRef CAS.
- K. S. Butler, C. J. Pearce, E. A. Nail, G. A. Vincent and D. F. Sava Gallis, ACS Appl. Mater. Interfaces, 2020, 12, 31217–31224 CrossRef CAS PubMed.
- A. López-Olvera, J. G. Flores, J. Aguilar-Pliego, C. K. Brozek, A. Gutiérrez-Alejandre and I. A. Ibarra, Chem. Mater., 2021, 33, 6269–6276 CrossRef.
- Y. Yu, G. Chen, J. Guo, Y. Liu, J. Ren, T. Kong and Y. Zhao, Mater. Horiz., 2018, 5, 1137–1142 RSC.
- M. Nazari, A. S. Saljooghi, M. Ramezani, M. Alibolandi and M. Mirzaei, J. Mater. Chem. B, 2022, 10, 8824–8851 RSC.
- Z. Zhang, H. Zhang, D. Tian, A. Phan, M. Seididamyeh, M. Alanazi, Z. Ping Xu, Y. Sultanbawa and R. Zhang, Coord. Chem. Rev., 2024, 498, 215455 CrossRef CAS.
- Z. Sun, T. Li, T. Mei, Y. Liu, K. Wu, W. Le and Y. Hu, J. Mater. Chem. B, 2023, 11, 3273–3294 RSC.
- M. Moharramnejad, A. Ehsani, M. Shahi, S. Gharanli, H. Saremi, R. E. Malekshah, Z. S. Basmenj, S. Salmani and M. Mohammadi, J. Drug Delivery Sci. Technol., 2023, 81, 104285 CrossRef CAS.
- M. A. Simon, E. Anggraeni, F. E. Soetaredjo, S. P. Santoso, W. Irawaty, T. C. Thanh, S. B. Hartono, M. Yuliana and S. Ismadji, Sci. Rep., 2019, 9, 16907 CrossRef PubMed.
- G. Wyszogrodzka-Gaweł, P. Dorożyński, S. Giovagnoli, W. Strzempek, E. Pesta, W. P. Węglarz, B. Gil, E. Menaszek and P. Kulinowski, Pharmaceutics, 2019, 11, 687 CrossRef PubMed.
- A. P. Acharya, K. B. Sezginel, H. P. Gideon, A. C. Greene, H. D. Lawson, S. Inamdar, Y. Tang, A. J. Fraser, K. V. Patel, C. Liu, N. L. Rosi, S. Y. Chan, J. L. Flynn, C. E. Wilmer and S. R. Little, J. Controlled Release, 2022, 352, 242–255 CrossRef CAS PubMed.
- K. Sun, L. Li, X. Yu, L. Liu, Q. Meng, F. Wang and R. Zhang, J. Colloid Interface Sci., 2017, 486, 128–135 CrossRef CAS PubMed.
- C. Gajadeera, M. J. Willby, K. D. Green, P. Shaul, M. Fridman, S. Garneau-Tsodikova, J. E. Posey and O. V. Tsodikov, J. Antibiot., 2015, 68, 153–157 CrossRef CAS PubMed.
- P. Kumar, N. Kaur, P. Tiwari, A. K. Gupta and S. M. Mobin, ACS Mater. Lett., 2023, 5, 1100–1108 CrossRef CAS.
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- J. E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, CrystEngComm, 2013, 15, 9258–9264 RSC.
- Y. Zhou, L. Liu, Y. Cao, S. Yu, C. He and X. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 22581–22592 CrossRef CAS PubMed.
- R. Pati, R. Sahu, J. Panda and A. Sonawane, Sci. Rep., 2016, 6, 24184 CrossRef CAS PubMed.
- A. Padhi, S. K. Naik, S. Sengupta, G. Ganguli and A. Sonawane, Microbes Infect., 2016, 18, 224–236 CrossRef CAS PubMed.
- S. N. Ansari, P. Kumar, A. K. Gupta, P. Mathur and S. M. Mobin, Inorg. Chem., 2019, 58, 9723–9732 CrossRef CAS PubMed.
- A. Kawamura, A. R. Greenwood, A. S. Filatov, A. T. Gallagher, G. Galli and J. S. Anderson, Inorg. Chem., 2017, 56, 3349–3356 CrossRef CAS PubMed.
- Y. Zeng, T. Lan, M. Li, G. Yuan, F. Li, J. Liao, J. Yang, Y. Yang and N. Liu, J. Chem. Eng. Data, 2021, 66, 749–760 CrossRef CAS.
- D. Liu, Y.-J. Chang and J.-P. Lang, CrystEngComm, 2011, 13, 1851–1857 RSC.
- H. V. Doan, H. Amer Hamzah, P. Karikkethu Prabhakaran, C. Petrillo and V. P. Ting, Nano-Micro Lett., 2019, 11, 54 CrossRef CAS PubMed.
- L. Peng, S. Yang, S. Jawahery, S. M. Moosavi, A. J. Huckaba, M. Asgari, E. Oveisi, M. K. Nazeeruddin, B. Smit and W. L. Queen, J. Am. Chem. Soc., 2019, 141, 12397–12405 CrossRef CAS PubMed.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- G. Férey, Chem. Soc. Rev., 2007, 37, 191–214 RSC.
- S. Rojas, I. Colinet, D. Cunha, T. Hidalgo, F. Salles, C. Serre, N. Guillou and P. Horcajada, ACS Omega, 2018, 3, 2994–3003 CrossRef CAS PubMed.
- J. A. Kellum, Crit. Care, 2000, 4, 6 CrossRef CAS PubMed.
- Z. Li, Y. Peng, X. Xia, Z. Cao, Y. Deng and B. Tang, Sci. Rep., 2019, 9, 17570 CrossRef PubMed.
- B. E. Souza, L. Donà, K. Titov, P. Bruzzese, Z. Zeng, Y. Zhang, A. S. Babal, A. F. Möslein, M. D. Frogley, M. Wolna, G. Cinque, B. Civalleri and J.-C. Tan, ACS Appl. Mater. Interfaces, 2020, 12, 5147–5156 CrossRef CAS PubMed.
- M. Chakraborty, S. Dasgupta, C. Soundrapandian, J. Chakraborty, S. Ghosh, M. K. Mitra and D. Basu, J. Solid State Chem., 2011, 184, 2439–2445 CrossRef CAS.
- C. X. (Cynthia) Lin, S. Z. Qiao, C. Z. Yu, S. Ismadji and G. Q. (Max) Lu, Microporous Mesoporous Mater., 2009, 117, 213–219 CrossRef.
- P. Costa and J. M. S. Lobo, Eur. J. Pharm. Sci., 2001, 13(2), 123–133 CrossRef CAS PubMed.
- Strategies to Modify the Drug Release from Pharmaceutical Systems, ed. M. L. Bruschi, Woodhead Publishing, 2015, pp. 63–86 Search PubMed.
- R. Yoshida, K. Sakai, T. Okano and Y. Sakurai, Polym. J., 1991, 23, 1111–1121 CrossRef CAS.
- N. V. Mulye and S. J. Turco, Drug Dev. Ind. Pharm., 1995, 21, 943–953 CrossRef CAS.
- J. V. Natarajan, C. Nugraha, X. W. Ng and S. Venkatraman, J. Controlled Release, 2014, 193, 122–138 CrossRef CAS PubMed.
- D. D. Mal, S. Khilari and D. Pradhan, Green Chem., 2018, 20, 2279–2289 RSC.
- K. Jiang, L. Zhang, Q. Hu, X. Zhang, J. Zhang, Y. Cui, Y. Yang, B. Li and G. Qian, Microporous Mesoporous Mater., 2019, 275, 229–234 CrossRef CAS.
- H. Wu, Q. Geng, Y. Li, Y. Song, J. Chu, R. Zhou, X. Ning, S. Dong and D. Yuan, J. Ind. Eng. Chem., 2022, 114, 475–482 CrossRef CAS.
- D. Han, X. Liu and S. Wu, Chem. Soc. Rev., 2022, 51, 7138–7169 RSC.
- M. Yang, J. Zhang, Y. Wei, J. Zhang and C. Tao, Nano Res., 2022, 15, 6220–6242 CrossRef CAS PubMed.
- J. H. Jo, H.-C. Kim, S. Huh, Y. Kim and D. N. Lee, Dalton Trans., 2019, 48, 8084–8093 RSC.
- Frontiers|Beyond Risk: Bacterial Biofilms and Their Regulating Approaches, https://www.frontiersin.org/articles/10.3389/fmicb.2020.00928/full, (accessed April 24, 2023).
- J. Esteban and M. García-Coca, Front. Microbiol., 2018, 8, 2651 CrossRef PubMed.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- C. Michaelis and E. Grohmann, Antibiotics, 2023, 12, 328 CrossRef CAS PubMed.
- L. Friedman and R. Kolter, Mol. Microbiol., 2003, 51, 675–690 CrossRef PubMed.
- C. Solano, B. García, J. Valle, C. Berasain, J.-M. Ghigo, C. Gamazo and I. Lasa, Mol. Microbiol., 2002, 43, 793–808 CrossRef CAS PubMed.
- J. M. Chen, G. J. German, D. C. Alexander, H. Ren, T. Tan and J. Liu, J. Bacteriol., 2006, 188, 633–641 CrossRef CAS PubMed.
- P. Maan, A. Kumar, J. Kaur and J. Kaur, Front. Cell. Infect. Microbiol., 2018, 8, 421 CrossRef CAS PubMed.
- O. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef PubMed.
- E. R. Erwin, A. P. Addison, S. F. John, O. A. Olaleye and R. C. Rosell, Tuberculosis, 2019, 116, S66–S70 CrossRef CAS PubMed.
- M. Combrink, D. T. Loots and I. du Preez, Toxicol. Lett., 2020, 322, 104–110 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental section includes the synthesis of the H4L linker, its characterization studies (mass spectrometry, NMR spectroscopy), activation of IITI-3 (PXRD), linearity plot, DLS hydrodynamic average particle size, kinetic models, and zeta potential. See DOI: https://doi.org/10.1039/d3tb02292g |
| ‡ Pawan Kumar (Design and synthesis of Cu-MOF, characterization and drug delivery) and Ananyaashree Behera (Involved in Biological studies) contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |