
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel α-mannose-functionalized poly(β-amino ester) nanoparticles as mRNA vaccines with increased antigen presenting cell selectivity in the spleen†
Nil
González-Ríos
 ab,
Margalida
Artigues
c,
Marta
Guerra-Rebollo
a,
Antoni
Planas
b,
Salvador
Borrós
a,
Magda
Faijes
*b and
Cristina
Fornaguera
*a
ab,
Margalida
Artigues
c,
Marta
Guerra-Rebollo
a,
Antoni
Planas
b,
Salvador
Borrós
a,
Magda
Faijes
*b and
Cristina
Fornaguera
*a
aGrup d’Enginyeria de Materials (Gemat), Institut Químic de Sarrià (IQS), Universitat Ramon Llull (URL), 08017 Barcelona, Spain. E-mail: cristina.fornaguera@iqs.url.edu
bLaboratoy of Biochemistry, Institut Químic de Sarrià (IQS), Universitat Ramon Llull (URL), 08017 Barcelona, Spain. E-mail: magda.faijes@iqs.url.edu
cAnalytical and Applied Chemistry Department, Institut Químic de Sarrià (IQS), Universitat Ramon Llull (URL), 08017 Barcelona, Spain
First published on 14th June 2023
Abstract
mRNA vaccination has emerged as a prominent therapy for the future of medicine. Despite the colossal advance in this technology and worldwide efficacy proof (ca. COVID vaccines), mRNA carriers still lack cell/tissue specificity, leading to possible side effects, and reduced efficacy among others. Herein we make use of the ubiquitous affinity of antigen-presenting cells (APC)s for glycosides to achieve specific targeting. To achieve this goal, we designed a new generation of α-mannosyl functionalized oligopeptide-terminated poly(β-aminoester). Fine formulation of these polymers with mRNA resulted in nanoparticles decorated with surface-exposed α-mannoses with sizes around 180 nm and positive surface charge. Notably, these particles maintained their properties after freeze-drying and subsequent redispersion. Finally, our mRNA carriers preferentially targeted and transfected APCs in vitro and in vivo. In conclusion, we demonstrated, at a preclinical level, that the mannose functionalization enables more selective targeting of APCs and, thus, these polymer and nanoparticles are candidates for a new generation of mRNA immunotherapy vaccines.
Introduction
Through evolution, the immune system has developed different mechanisms to avoid cancer development as well as to detect and eradicate pathogen infected cells. Despite its efficacy, malignant cancer cells can escape from immune recognition by developing resistance mechanisms, such as overexpression of immune-checkpoints, and therefore, mutated cells develop malignant tumors, which are still the second leading cause of worldwide mortality.1 Consequently, immunotherapies, which are defined as therapeutic approaches aimed to reactivate antitumor immune responses and overcome the different mechanisms that malignant cells present, should be a promising solution. As an example, immunotherapies based on antibodies targeting immune-checkpoint inhibitors (ICIs) have become the clinical standard of care for many tumors types, such as for melanoma,2,3 provided they achieve complete response rates to half of the treated patients. However, ICIs’ acute clinical toxicities have been reported, including those systemically affecting persistent sequelae which could be related to a lack of biodistribution control. Additionally, long-term implications have not yet been disclosed.4 In this scenario, it is required to find alternative immune therapeutic options, such as cancer vaccines.Beyond the use for infectious disease prophylaxis, vaccines have been reported recently as an efficient option to (re)activate the immune system against tumor antigens. Traditional vaccine formulations, however, are not adequate for this purpose, as there are so many possible antigens that tumors may present. Thus, mRNA vaccines that can be easily customized are becoming the core pillar of research. Many studies developing mRNA vaccines have already arrived at clinical stages, all of them using lipid carriers, which include, in addition to cationic lipids, poly(ethylene glycol) (PEG) modified surfaces, but none includes active targeting moieties.5 Although for SARS-CoV-2, these vaccines have demonstrated superior performance, constraints regarding the potential development of anti-PEG antibodies as well as poor arrival to target cells are appearing.6 In this context, the use of PEG-free, actively targeted alternative nanocarriers is an urgent need.
Poly β-(amino esters) (pBAE) are a group of polymers representing a promising choice for different reasons: their easy, versatile and tailored production; their ability to efficiently encapsulate mRNA generating small polymeric nanoparticles; the protection and controlled release of the genetic material; their biocompatibility and biodegradability; and their ability to promote cellular internalization and transfection.6–8 Additionally, in our group, we also demonstrated that the addition of a certain combination of oligopeptide end-modified cationic pBAEs enables in vivo administration and accumulation in the spleen,9 the target organ for vaccination. However, their selectivity is not fully controlled due to the lack of an active targeting moiety. For this reason, the main goal of this work is to set up a strategy to design a modified polymer, based on oligopeptide end modified-pBAEs, containing α-mannose functionalization on the lateral chains. Thus, these moieties will be exposed in the nanoparticle surface to selectively target them toward APCs, and even more specifically, to dendritic cells (DCs), where saccharide receptors are overexpressed. Among the different glycans with described affinity to the Carbohydrate Recognition Domain 4 (CRD4), one of the key receptors of DCs, α-mannose was selected as the targeting moiety thanks to: (1) previous bibliography using it as gold nanoparticle surface functionalization;10 (2) its good interaction with this receptor and its simple and, consequently, non-expensive production of targeted nanoparticles.11–13 Thus, in the present article, we present the methodology for the synthesis of PBAEs with different α-mannose-functionalization (Man-OM-pBAE), which is novel compared to previous polymers on the use of covalently attached mannoses to the lateral chains of the polymers, providing functionality ramification that positively contributes to the selective targeting to α-mannose receptors, their ability to form nanoparticles and their capacity to trigger an in vivo immune response.
Experimental
Materials
Reagents and solvents used for polymer synthesis were purchased from Sigma-Aldrich and Panreac. 2-(2-Chloroethoxy) ethanol and 2-[2-(2-Chloroethoxy) ethoxy] ethanol were acquired from Alfa Aesar and Acros Organics. Oligopeptides used for polymer modification (H-Cys-Lys-Lys-NH2, H-His-His-His-Cys-NH2) were obtained from Ontores (Shangai) with a purity higher than 97%. Antibodies for flow cytometry were purchased from Thermo-Fisher, Luciferase Polyclonal antibody-FITC (#200-102-150-0100) and PE anti-mouse CD209a (DC-SIGN) antibody 833003 from BioLegend. MRNA was purchased from Cellerna BiosciencePureBoostTM EGFP mRNA (2001M-1MG) and PureBoostTM Fluc mRNA (1001-1MG). Plasmid DNA (PmaxGFP) was purchased from Lonza.Cell culture
JAWSII (ATCC CRL-11904) cells were cultured in Eagle's minimum essential medium supplemented with 20% (v/v) heat-inactivated fetal bovine serum (FBS); 1 mmol L−1 sodium pyruvate, 4 mmol L−1 glutamine, and 5 ng mL−1 of granulocyte-macrophage colony stimulatory factor (GM-CSF) and 100 U mL−1 of penicillin–streptomycin. BEAS-2B (ATCC CRL-9609) cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v); 2 mmol L−1 of glutamine and 100 U mL−1 of penicillin–streptomycin. Both were cultured at 37 °C under a 5% CO2/95% air atmosphere. Cells were grown until 80–90% confluence before performing assays.Animals
Adult 6–8 week old Balb/c mice were purchased (Envigo) and kept under pathogen-free conditions in laminar flow boxes. Animal maintenance and experiments were performed under established guidelines of the Catalan Government and following protocol number 1121, approved by the Direcció General del Medi Natural.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (2:1), toluene:ethyl acetate, (4:1) and petroleum ether:ethyl acetate, (1:0.9) as eluents for each compound, respectively.
:ethyl acetate (2:1), toluene:ethyl acetate, (4:1) and petroleum ether:ethyl acetate, (1:0.9) as eluents for each compound, respectively.
The bromo/chloro-substituted mannosides (2.13, 1.86 and 1.95 mmol of each compound respectively) were mixed with 8 eq. of sodium azide in dimethylformamide (DMF) and stirred for 15 h at 60 °C. After removing the solvent under reduced pressure, the mixture was redissolved in ethyl acetate and filtered. Finally, the filtrate was evaporated and purified by flash chromatography using cyclohexane:ethyl acetate (2:1), petroleum ether:ethyl acetate (2:1), and petroleum ether:ethyl acetate (1:1), obtaining the products containing azide.
Next, the protected mannosides (0.7, 1.37 and 1.32 mmol of each compound respectively) were dissolved with dry MeOH and sodium methoxide (0.9 M in methanol). The reaction was stirred overnight at room temperature. Then, the mixture was neutralized with Amberlite IR 120 resin (H+), filtered, and freeze-dried.
Finally, the deprotected mannosides (0.6, 1.43 and 0.88 mmol of each compound respectively) were dissolved in a mixture of THF:H2O (1:1) with 2 eq. of Ph3P for 20 h at room temperature. After several cleaning steps with CHCl3, the aqueous phases were treated with Dowex® 50WX8. The final products were obtained after freeze-drying. Structures were characterized by 13C-NMR (see the spectra in Fig. S1–S3, ESI†).
Synthesis of C6
The synthesis of C6 polymer was performed according to Dosta et al., 2018.15 In brief C6 polymer was synthesized by the addition of hexylamine and 5-amino-1-pentanol to 1,4-butanediol diacrylate (1:1.1 ratio of amine: diacrylate). Polymerizations were done at 90 °C for 24 h and characterized by 1H-NMR using CDCl3 as the solvent.
Synthesis of Man1, Man2 and Man3 polymers
Acrylate-terminated polymers were synthesized by an addition reaction of primary amines to 1,4-butanediol diacrylate (at a 1:1.2 molar ratio of amine: diacrylate).
Mannose-containing polymers M(1–3) were synthesized as follows: Man1) 2-aminoethyl α-D-mannopyranoside, (38.8 mg, 0.174 mmol); Man2) 2-(2-aminoethoxy) ethyl α-D-mannopyranoside (52.5 mg, 0.196 mmol); or Man3) 2-[2-(2-amino ethoxy) ethoxy] ethyl α-D-mannopyranoside (32.6 mg, 0.105 mmol) and 1,4-butanediol diacrylate (according to molar ratio) mixture were stirred in the presence of 2 μL of trimethylamine and 400 μL of DMSO at 90 °C for 72–96 h. After cooling down, the resulting polymer was precipitated in water and freeze-dried. Characterization was performed by 13C-NMR using DMSO-d as the solvent. Structures were characterized by 13C-NMR using DMSO-d as the solvent (see the spectra in Fig. S4–S6, ESI†).
Synthesis of C6-CK3, C6-CH3, Man1-CK3, Man2-CK3, and Man3-CK3
Mannose oligopeptide-modified pBAE (Man-OM-pBAE) or without mannoses (OM-pBAE) as polymer references (C6-CK and C6-CH), were obtained by end-modification of an acrylate-terminated polymer with thiol-terminated oligopeptides at a 1:2.5 molar ratio in dimethyl sulfoxide as previously described in Dosta et al., 2018.15 In some cases, TCEP was added at a final concentration of 0.5 mM and 2 μL of triethylamine. The mixture was stirred overnight at room temperature, and the resulting polymer was obtained by precipitation in a mixture of diethyl ether and acetone (7:3; v:v). The different oligopeptide modifications were confirmed by H1-NMR (see spectra on Fig. S7–S9, ESI†) and UHPLC-Q-TOF. Polymers were dissolved in DMSO at 100 mg mL−1.
Synthesis of polymers labeled with Cy5
To label the polymers with cyanine 5 (Cy5), the following protocol was used.16 In brief, solutions of Cy5 NHS ester in DMSO and polymers C6-CK3 and Man2-CK3 were prepared in screw cap tubes. An excess of triethylamine was added to the polymer solution and then the Cy5 solution was poured inside. The solution was stirred for 20 h at 25 °C in a dark environment and purified in a mixture of 7:3 diethyl ether: acetone by dropwise addition. The sample was centrifuged at 4000 rpm for 10 min to remove the solvent. This step was repeated twice. Finally, the product was dried under vacuum and dissolved at 100 mg mL−1 of DMSO.
Synthesis of DNA labeled with Cy3
The labeling of pGFP was performed following the protocol Label IT® TrackerRM Intracellular Nucleic Acid Localization Kit with some modifications. For our imaging experiments, the ratio of DNA was increased to 0.5:10 (v/w) regarding the DNA labeling agent, and the reaction time was increased 2 h to increase the ratio of molecules with label.16 Finally, the DNA-Cy3 sample was quantified through fluorescence and absorbance with an Infinite M Plex Microplate reader from TECAN.
Structural characterization
1H and 13C NMR spectra were recorded in a Varian 500 MHz spectrometer operated at 298 K. For 1H NMR, the central peak of the DMSO-d6 multiplet (2.49 ppm) was used as a reference. For 13C NMR, the central peak of the DMSO-d6 multiplet (39.5 ppm) was used as a reference.UHPLC-QTOF samples were diluted 1/1000 in H2O and filtered with PVDF 0.22 μm filters. UHPLC-QTOF was performed using a UHPLC ExionLC™ AD System (AB Sciex, USA) coupled with an X500B QTOF System (MS/MS) (AB Sciex, USA) with electrospray ionization (ESI) in positive mode. Chromatographic separation was carried out using a bioZenTM 1.6 μm Peptide PS-C18, 150 × 2.1 mm, from Phenomenex. Mobile phase A was 0.1% trifluoro acetic acid in Milli-Q water and B was acetonitrile. A linear gradient program at a flow rate of 0.4 mL min−1 was used as follows: 0–10 min, from 5 to 35% (B); 10–11 min, from 35 to 100% (B); then 1.5 min to 100% (B) and back to 5% (B) for 2 min. The injection volume was set at 50 μL.
ESI source parameters: spray voltage, +5500 V; source temperature, 500 °C; nebulizer and auxiliary gas flow (N2), 60 psi and 60 psi; curtain gas flow (N2), 35 psi. Mass spectrometry analyses were performed applying a data-independent scan experiment (SWATH®) which uses a combination of a full-scan TOF spectrum (declustering potential (DP), 80 V; collision energy (CE), 10 V) with TOF MS/MS fragmentation using fixed Q1 transmission windows. Data were processed using SCIEX OS 2.2 acquisition software.
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2CH2NH2), 60.85 (C-6), 39.03 (OCH2- H2NH2) (Fig. S1, ESI†).
H2CH2NH2), 66.32 (OCH2CH2O), 60.87 (C-6), 39.06 (OCH2H2NH2). (Fig. S2, ESI†)
H2CH2NH2), 60.85 (C-6), 39.03 (OCH2- H2NH2) (Fig. S1, ESI†).
H2CH2NH2), 66.32 (OCH2CH2O), 60.87 (C-6), 39.06 (OCH2H2NH2). (Fig. S2, ESI†)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–C), 165.92 (CH2CH–(O)–O), 131.86 (H2CH–(CO)–O), 128.75 (CH2H–(CO)–O), 100.45 (C-1), 74.34 (C-2), 71.42 (C-3), 70.74 (C-4), 67.79 (C-5), 65.29 (O–H2–CH2–CH2–H2–O), 63.74 (OH2CH2NH2), 61.62 (C-6), 52.64 (OCH2H2NH2), 49.68 ((CO)–CH2–H2–N), 32.57 ((CO)–H2–CH2–N), 25.22 (O–CH2–H2–H2–CH2–O). (Fig. S4, ESI†) UHPLC-QTOF: n = 1 retention time: 10.02 min; m/z 620.2912 [M + H]+ (Fig. S10, ESI†). n = 2 retention time: 9.72 min; m/z 1041.4861 [M + H]+. n = 3 retention time: 8.54 min; m/z 1462.6809 [M + H]+. n = 4 retention time: 8.25 min; m/z 1883.8757 [M + H]+.
O)–C), 165.89 (CH2CH–(O)–O), 131.79 (H2CH–(CO)–O), 128.70 (CH2H–(CO)–O), 100.36 (C-1), 74.16 (C-2), 71.38 (C-3), 70.67 (C-4), 69.82(C–OCH2H2O), 69.26 (C-5), 67.35 (O–H2–CH2–N), 66.12(C–OH2CH2O), 64.11 & 63,72(O–H2–CH2–CH2–H2–O) 61.61 (C-6), 52.82 (OCH2H2NH2), 49.74 ((CO)–CH2–H2–N), 32.57 ((CO)–H2–CH2–N), 25.22 (O–CH2–H2–H2–CH2–O). (Fig. S5, ESI†). UHPLC-QTOF: n = 1 retention time: 10.34 min; m/z 664.3160 [M + H]+(Fig. S13, ESI†). n = 2 retention time: 9.25 min; m/z 1129.5301 [M + H]+. n = 3 retention time: 8.45 min; m/z 1594.7599 [M + H]+. n = 4 retention time: 8.67 min; m/z 2059.9795 [M + H]+, 1029.95 [M + H]2+ (Fig. S15, ESI†).
O)–C), 165.92 (CH2CH–(O)–O), 131.88 (H2CH–(CO)–O), 128.74 (CH2H–(CO)–O), 100.38 (C-1), 74.29 (C-2), 71.37 (C-3), 70.68 (C-4), 70.20(O–H2–CH2–O), 70.13 (O–H2–CH2–O), 69.94 (C–OCH2H2O), 69.36 & 66.14 (O–H2–CH2–CH2–H2–O), 67.37(C-5), 66.14 (O–H2–CH2–N), 64.13 (O–H2CH2N), 63.73 (O–CH2H2–OC–O) 61.65 (C-6), 52.86 (OCH2H2NH2), 49.75 ((CO)–CH2–H2–N), 32.65 ((CO)–H2–CH2–N), 25.22 (O–CH2–H2–H2–CH2–O). (Fig. S6, ESI†). UHPLC-QTOF: n = 1 retention time: 10.62 min; m/z 708.3432 [M + H]+(Fig. S16, ESI†). n = 2 retention time: 7.83 min; m/z 1217.5902 [M + H]+. n = 3 retention time: 9.33 min; m/z 1726.8397 [M + H]+. n = 4 retention time: 9.08 min; m/z 2236.0861 [M + H]+, 1029.95 [M + H]2+. (Fig. S17, ESI†).
O)–C), 165.92 (CH2CH–(O)–O), 131.86 (H2CH–(CO)–O), 128.75 (CH2H–(CO)–O), 100.45 (C-1), 74.34 (C-2), 71.42 (C-3), 70.74 (C-4), 67.79 (C-5), 65.29 (O–H2–CH2–CH2–H2–O), 63.74 (OH2CH2NH2), 61.62 (C-6), 52.64 (OCH2H2NH2), 49.68 ((CO)–CH2–H2–N), 32.57 ((CO)–H2–CH2–N), 25.22 (O–CH2–H2–H2–CH2–O). (Fig. S4, ESI†) UHPLC-QTOF: n = 1 retention time: 10.02 min; m/z 620.2912 [M + H]+ (Fig. S10, ESI†). n = 2 retention time: 9.72 min; m/z 1041.4861 [M + H]+. n = 3 retention time: 8.54 min; m/z 1462.6809 [M + H]+. n = 4 retention time: 8.25 min; m/z 1883.8757 [M + H]+.
O)–C), 165.89 (CH2CH–(O)–O), 131.79 (H2CH–(CO)–O), 128.70 (CH2H–(CO)–O), 100.36 (C-1), 74.16 (C-2), 71.38 (C-3), 70.67 (C-4), 69.82(C–OCH2H2O), 69.26 (C-5), 67.35 (O–H2–CH2–N), 66.12(C–OH2CH2O), 64.11 & 63,72(O–H2–CH2–CH2–H2–O) 61.61 (C-6), 52.82 (OCH2H2NH2), 49.74 ((CO)–CH2–H2–N), 32.57 ((CO)–H2–CH2–N), 25.22 (O–CH2–H2–H2–CH2–O). (Fig. S5, ESI†). UHPLC-QTOF: n = 1 retention time: 10.34 min; m/z 664.3160 [M + H]+(Fig. S13, ESI†). n = 2 retention time: 9.25 min; m/z 1129.5301 [M + H]+. n = 3 retention time: 8.45 min; m/z 1594.7599 [M + H]+. n = 4 retention time: 8.67 min; m/z 2059.9795 [M + H]+, 1029.95 [M + H]2+ (Fig. S15, ESI†).
O)–C), 165.92 (CH2CH–(O)–O), 131.88 (H2CH–(CO)–O), 128.74 (CH2H–(CO)–O), 100.38 (C-1), 74.29 (C-2), 71.37 (C-3), 70.68 (C-4), 70.20(O–H2–CH2–O), 70.13 (O–H2–CH2–O), 69.94 (C–OCH2H2O), 69.36 & 66.14 (O–H2–CH2–CH2–H2–O), 67.37(C-5), 66.14 (O–H2–CH2–N), 64.13 (O–H2CH2N), 63.73 (O–CH2H2–OC–O) 61.65 (C-6), 52.86 (OCH2H2NH2), 49.75 ((CO)–CH2–H2–N), 32.65 ((CO)–H2–CH2–N), 25.22 (O–CH2–H2–H2–CH2–O). (Fig. S6, ESI†). UHPLC-QTOF: n = 1 retention time: 10.62 min; m/z 708.3432 [M + H]+(Fig. S16, ESI†). n = 2 retention time: 7.83 min; m/z 1217.5902 [M + H]+. n = 3 retention time: 9.33 min; m/z 1726.8397 [M + H]+. n = 4 retention time: 9.08 min; m/z 2236.0861 [M + H]+, 1029.95 [M + H]2+. (Fig. S17, ESI†).
Determination of the buffering capacity of polymers
Buffering capacities of the polymers were checked by acid-base titration, following our previous studies.17 In brief, 20 mg of the polymers were dissolved in 10 mL of 0.1 M KCl solution. The pH of the solution was brought to pH 3 using 0.5 M HCl and titrated until reaching pH 11 using 0.1 M NaOH. The pH of the solution was measured after each addition using a Mettler Toledo S20 pH meter. Titration of 0.1 M KCl without polymer was used as a control.Synthesis and lyophilization of nanoparticles
α-Mannose-oligopeptide-pBAE (Man-OM-pBAE) nanoparticles were prepared following slight modifications of the Fornaguera et al., protocol.9 In brief, they were prepared at a 25:1 polymer:mRNA ratio, by mixing equal volumes of mRNA at 0.5 mg mL−1 with the polymer (with the polymer in a mixture of 45% C6-CK, 40% C6-CH and 15% of Man1-CK3, Man2-CK3 or Man3-CK3) at 12.5 mg mL−1, in 12.5 mM AcONa buffer solution. The mRNA was added over the polymer solution and mixed by pipetting, followed by 15 min incubation at 25 °C. For the formation of discrete structures, this mixture (volume = Vi) was nanoprecipitated in the same volume (Vi) of DEPC water. After that, the same volume (Vi) of a HEPES 20 mM + (2–4) wt% sucrose solution was also added for cryopreserving (depending on the formulation). Then, the nanoparticles were freeze-dried. The lyophilized powder was kept at −20 °C until use when it was reconstituted with DEPC water (Vi).
Biophysical characterization of polyplexes
Hydrodynamic diameter (nm), polydispersity index (PDI), and surface charge of nanoparticles were measured at 25 °C, 633 nm laser wavelength and 173° signal detector, using a ZetaSizer Nano ZS (Malvern Instruments Ltd, Malvern, UK). For size measurements, nanoparticles prepared at 0.25 mg mL−1 of mRNA were used, instead, a 1/10 dilution was used to measure surface charge. Three measurements of each nanoparticle batch were performed with 20 runs per measurement, considering the intensity approximation. Results correspond to the mean ± standard deviation of at least three independent nanoparticle batches. The hydrodynamic size was also measured by nanoparticle tracking analysis (NTA), using a NanoSight LM10 from Malvern, equipped with a fluorescent laser (λ = 488 nm). Results correspond to the mean ± standard deviation of at least three independent nanoparticle batches. Encapsulation efficiency was measured by the Quanti-It RiboGreen RNA assay kit (ThermoFisher Scientific) following the manufacturer's instructions and agarose gel retardation assay, as previously described.9 The morphology of the nanoparticles was determined using negatively stained samples through transmission electron microscopy. In brief, 10 μL of the nanoparticle dispersion were incubated with a carbon-coated copper grid, for 30 seconds. Then, the excess sample was removed and grids were stained for 10 seconds with uranyl acetate. Excess staining was removed again and samples were visualized with a JEOL 1400 TEM microscope.Labelling of nanoparticles using Concanavalin A-FITC
Detection of mannose on functionalized NPs was done according to Silva et al.18 In brief, the constitutive polymers of the nanoparticle were labeled with Cy5 and the genetic material was labeled with Cy3. Afterward, polyplexes were incubated with Concanavalin A (Con A)-FITC (0,7 mg mL−1) labeled for 1 h in PBS with BSA (3% w/v).Fluorescence resonance energy transfer (FRET) analysis
FRET measurements were carried out using an Infinite M Plex Microplate reader from TECAN and following Navalón-López, et al. 2023.19 In brief, nanoparticles labeled with Cy5 and Cy3 were prepared at the same label concentration with a Mirus labeling kit. Cy5 and Cy3 were used for the polymers (C6-CK3 and Man2-CK3) and the plasmid (pGFP) respectively. Three different negative controls were used, nanoparticles with only one fluorophore. The excitation wavelength was 535 nm to excite Cy3 and the emission wavelength was 675 nm to detect the emission of Cy5. The measurements were done at 25 °C at different times.In vitro cellular transfection and uptake
Cells were seeded in 96 well plates at 150000 cells mL−1 24 h before starting the experiment. Cells were incubated with 0.4 μg of mRNA coding for PureBoostTM EGFP mRNA/10000 cells encapsulated within the different formulations, using KH as a positive control. Non-treated cells were used as negative controls (results correspond to 100% viability, used to normalize the absorbance of the tested samples). After some time (described in each experiment) incubation in the presence of 10% FBS, uptake and transfection efficiency were measured by flow cytometry (NovoCyte, ACEA Biosciences).
Competition assay with free α-D-mannose
Jaws II cell line was harvested in 96 well plates at a concentration of 150000 cells mL−1 with the corresponding media (see above) 24 h before starting the experiment. Before introducing the nanoparticles with 0.4 μg of coding for PureBoostTM EGFP mRNA/10000 cells encapsulated within and labeled at 0.5% of Cy5, the 96 well plate was divided into 4 concentrations to be tested and the media were replaced with a fresh one containing different concentrations of α-D-mannose (0, 10, 1000 and 100000 nM20) and incubated for 1 h at 37 °C. After the polyplexes of OM-pBAE containing α-mannose were introduced, cells were incubated for 1 h at 37 °C. When the time was over, cells were centrifugated following ATCC conditions, trypsinized and fixed with 1% paraformaldehyde. Uptake was measured by flow cytometry (NovoCyte Flow Cytometer, ACEA Biosciences, Inc.) and fluorescence gating was determined using a negative control.
In vivo and ex vivo uptake and transfection efficiency evaluation
Mice (n = 4) were injected with 50 μg (200 μL) of mRNA encoding for PureBoostTM Fluc mRNA complexed with OM-pBAE polymers; 6 h post-injection animals were euthanized, the spleen was removed and disaggregated following the protocol from Pujol-Autonell et al.21 In brief, they were removed and placed in 2 wt% FBS in PBS media, at 4 °C. They were mechanically digested by manual tissue mashing in around 5 mL of media using a syringe plunger. The liquid was filtered through 70 μm nylon cell strainers to remove big fragments (conjunctival and stromal tissue). The filtered cell suspension was centrifuged for 10 min at 400 g, at 4 °C. The supernatant was removed, and the pellet was incubated with 5 mL hemolysis buffer (1.029 g Tris–HCl and 3.753 g ammonium chloride in 500 mL of milliQ water) for a maximum time of 5 min. 5 mL of the media was added, to stop the hemolytic activity, and the cells were recentrifuged twice for cleaning purposes. Finally, the cell density was counted, and cells were frozen with FBS/DMSO 90/10 for their further use. Spleen cellular dispersions were thawed and redispersed in media. The cells were counted using a Neubauer Chamber. Then, cells were permeabilized with PBS solution containing Tween20 (0.05%) and 3% BSA and finally incubated for 1 h at 25 °C with anti-DCSIGN-PE and anti-Luciferase-FITC. Finally, cells were washed with PBS and analyzed using a NovoCyte Flow Cytometer, ACEA Biosciences, Inc.). Results from the FC experiments correspond to means ± standard deviation of at least three independent cell suspensions, coming from 4 independent animals.Statistical analyses
Results correspond to the mean ± standard deviation of a sample size of n = 3. GraphPad Prism v8.0 software was used for the statistical analysis. A paired sample t-test has been applied to determine statistical differences. P values <0.05 (*), >0.01 (**), and >0.001 (***) were considered statistically significant.Results and discussion
To obtain an efficient mRNA antitumor vaccine, the transport of the genetic material to the target cells and consequently, the subcellular compartment, is a vital point, but also the interactions of the nanoparticles with the cell environment and how they deliver the genetic material, are fundamental steps. Furthermore, the side-effects of present vaccination options (muscle and joint pain, fever, fatigue…) due to the requirements of intramuscular administration, the unspecific generation of an innate immune response in terms of inflammation triggering caused by carriers or adjuvants22 and the poor biodistribution due to the puzzling targeting of current vaccines are well known. Thus, focusing on a strategy using selective targeting can overcome these shortcomings, and therefore, enhance the safety and performance of the vaccine. APCs are responsible for immune system activation and accordingly, the cell lineage to be targeted for efficient vaccination. As a differential characteristic from other immune cell subsets, they present on their surface a high amount of C-type lectin receptors (CLR) with the CRD-4. Taking advantage of this property, the objective of the present work was to selectively target our previously described pBAE nanoparticles to the Dendritic Cell-Specific ICAM-3 Grabbing Nonintegrin (DC-SIGN, CD209) and the macrophage mannose receptor (MMR, CD206), two CLRs exclusively overexpressed in DCs and macrophages, as a differential receptor from other cells in our body11–13,23,24). To do so, we designed α-mannosyl-functionalized pBAE, since it was previously described10 that both CD206 and CD209 receptors have a strong affinity for α-mannosyl glycoside residues. In the subsequent section, we describe the results obtained.Synthesis and characterization of oligopeptide end-modified pBAEs containing α-mannose in their lateral chains
In this study, the combination of the so-called C6-CK3 and C6-CH3 polymers was selected based on our previous publication demonstrating their preferred accumulation in the spleen and, specifically, in APCs.9 For the selective targeting of CRD4 of APCs, pBAE polymers with different moieties of α-mannose were synthesized by combining one of three different monomers (m0, m1, m2) with 1,4-butanediol diacrylate. The main difference between these mannose monomers is the length of the linker binding the carbohydrate to a primary amine.Acrylate-terminated polymers were obtained as a result of the conjugation of 1,4-butanediol diacrylate with the different mannosyl derivatives (m = 0, m = 1, m = 2) (Fig. 1i) with a slight excess of the acryl group. The chemical structures of Man1, Man2 and Man3 were confirmed by 13C-NMR and UHPLC-QTOF, and values between n = 1 to n = 4 were observed for all the synthesized polymers. The signals related to primary amines, as previously described by our group,9,25 disappeared at 40 ppm, confirming the synthesis of polymers containing α-mannose. Furthermore, α-mannose presence was confirmed by the signal of 100 ppm of anomeric carbon and the presence of signals regarding tertiary amines at 52, 49 and 32 ppm (see Fig. S3–S6, ESI†). In all cases, two or more molecular ions were observed corresponding to several adducts of a specific m/z (see the ESI†). Thus, it is possible to identify the real molecular ion ([M + H]+) with extra confidence for each compound. Moreover, the observed monoisotopic mass matches the expected monoisotopic mass with a mass error lower than 2 ppm in all the cases for n = 1 to n = 4 (Man1 (n = 1) 619.2834 Da, Man2 (n = 1) 663.3081 Da and Man3 (n = 1) 707.3353 Da). Subsequently, the capping was performed using a lysine-based oligopeptide (CKKK, known as CK3), through a Michael addition reaction between polymer end acrylates and cysteine thiol groups. The chemical structures of compounds Man1-CK3, Man2-CK3 and Man3-CK3 was confirmed by the reduction of acrylate signals in 1H-NMR (between 6.4–5.8 ppm) and the presence of typical displacements for oligopeptide structures, between 8.5–8 ppm, where the protons regarding the amine of the peptide bond appear, and also between 2–1 ppm where the signals regarding the aliphatic hydrogens of the lysine side chain appear (Fig. S7–S9, ESI†).
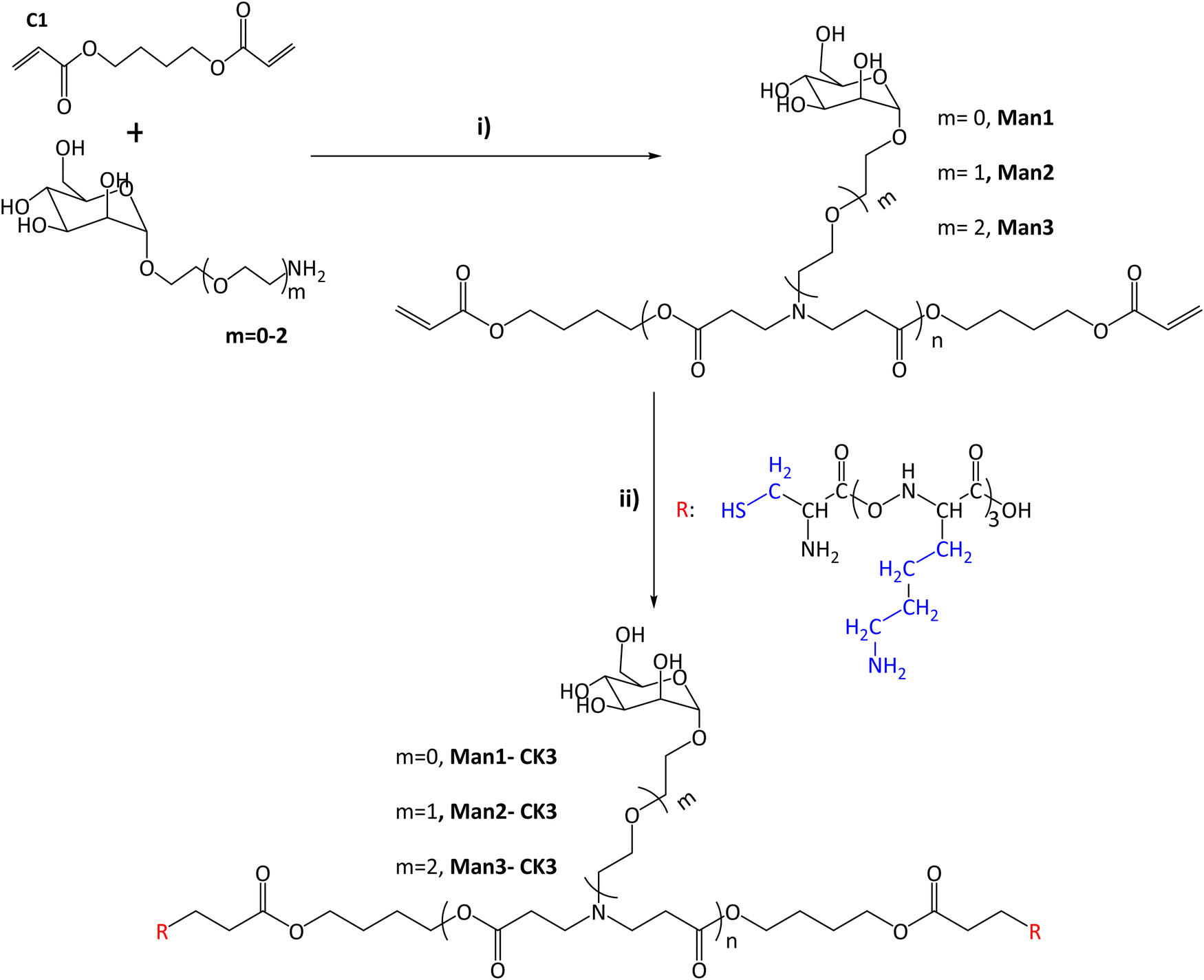 | ||
| Fig. 1 Synthesis of α-mannose containing polymers. (I) Michael addition of 1,4-butanediol diacrylate with monomers containing α-mannose. Different conditions and times were tested after reaching the final product 1:1.2 molar ratio (acrylate: m = 0–2). (II) End capping modification was carried out using oligopeptides based on Cys-Lys-Lys-Lys (CK3), in blue the side chains of the different peptides are indicated. The chemical structures were characterized with NMR 13C, 1H and UHPLC-QTOF (see Fig. S1–S20, ESI†). | ||
Finally, the expected molecular mass of the compounds was found using UHPLC-QTOF, obtaining the expected molecular weight for each polymer plus the addition of the peptides, and therefore confirming the synthesis. As an example, monoisotopic mass for n = 1 compounds is presented here: Man1-CK3 1629.8873 (Fig. S12, ESI†) Da, Man2-CK3 1673.9182 (Fig. S14, ESI†) Da and Man3-CK3 1717.9451 Da (Fig. S16, ESI†).
Complexation of oligopeptide-modified poly(B-amino ester)s containing α-mannose with mRNA forming nanometric polyplexes
Our group has different publications explaining how nanoparticles were formulated9,19,25–27 basically by the use of a mixture of different cationic polymers (C6-CK3 and C6-CH3) that electrostatically interact with the negatives charges of genetic materials, resulting in nanoparticles, named in the following as KH, that can encapsulate either plasmid DNA or mRNA. Here, we evaluated the ratios of the new mannose polymers that can be mixed with the standard KH nanoparticles, replacing part of the C6-CK3 polymer with a fraction of Man1-CK3, Man2-CK3 or Man3-CK3 polymer and therefore, obtaining new formulations. Thus, we evaluated the ratios of mannose polymers that could be used for a new formulation (Table 1) (based on 60:40 C6-CK3:C6-CH3 polymers we described before–25) without hampering their physicochemical attributes in terms of encapsulation efficiency, size and polydispersity index (PDI) of the nanoparticles, and Z-potential. (Fig. 2). The contribution of the different mannose-containing polymers is reduced from the C6-CK polymer amount, starting from 15% up to 60%, and maintaining the 40% of histidine-based polymers, previously described necessary not only for endosomal scaping but also to enable lyophilization.
| Formulations | Man1-CK3 (%) | Man2-CK3 (%) | Man3-CK3 (%) | C6-CK3 (%) | C6-CH3 (%) |
|---|---|---|---|---|---|
| KH | — | — | — | 60 | 40 |
| KHM1 15% | 15 | — | — | 45 | 40 |
| KHM1 30% | 30 | — | — | 30 | 40 |
| KHM1 45% | 45 | 15 | 40 | ||
| KHM1 60% | 60 | — | — | 0 | 40 |
| KHM2 15% | — | 15 | — | 45 | 40 |
| KHM2 30% | — | 30 | — | 30 | 40 |
| KHM2 60% | — | 60 | — | 0 | 40 |
| KHM3 15% | — | — | 15 | 45 | 40 |
| KHM3 30% | — | — | 30 | 30 | 40 |
| KHM3 60% | — | — | 60 | 0 | 40 |
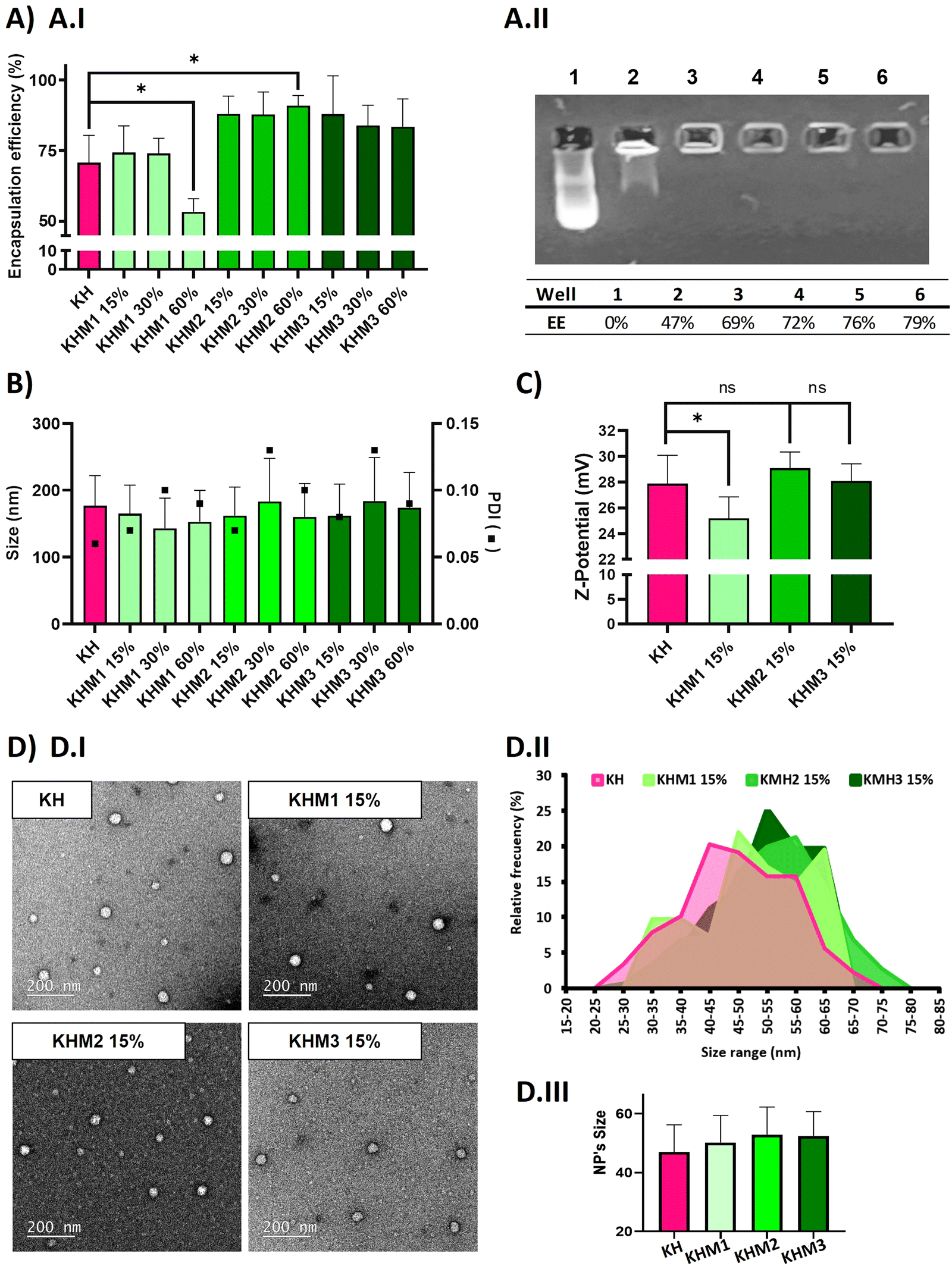 | ||
| Fig. 2 Physic-chemical analysis of different nanoparticle formulations containing α-mannose. In pink, the control KH, in soft green the formulations containing Man1-CK3, in medium green the formulations containing Man2-CK3 and in dark green the ones containing Man3-CK3. Polyplexes were prepared using mRNA eGFP with polymer at 25:1 ratio (w:w), diluted with RNAse-free water. Results are shown as mean and standard deviation of triplicates. (A) Average encapsulation efficiency of the different tested formulations with (A.I) Ribogreen® and (A.II) Gel retardation assay (quantified by ImageJ) of Man1-CK3 formulations, using DNA as the genetic material. (1-Free pDNA 2-KHM1 60% 3-KHM1 45% 4-KHM1 30% 5-KHM1 15% 6-KH) (B) size (bars) and Polydispersity Index (PDI) (square) of the analyzed populations by DLS. (C) Z-potential by DLS. p-Value: 0.05 (*), 0.01 (**), <0.001 (***). (D and D.I) TEM images of NP formulations KH, KHM1 15%, KHM2 15 and KHM3 15% with 12000X amplifications. (D.II) Analysis of the nanoparticles size distribution by ImageJ. (D.III) Mean and standard deviation of the nanoparticles. | ||
As expected, complexes without mannose-modified polymers formed polyplexes with a particle average size of 177 nm and 70% of encapsulation efficiency. The addition of Man1-CK3 at low ratios resulted in quite small NPs (around 150 nm), but upon increasing the ratio of Man1-CK3 from 15% to 60%, a decrease in encapsulation efficiency from 74% to 50% was appreciated. Differently, Man2-CK3 and Man3-CK3 addition resulted in NPs around 170 nm on average without a tendency to increase when higher ratios of Man2-CK3 or Man3-CK3 were used, upon checking the encapsulation efficiency (88, 87, 91%), it increases to almost 90% in all formulations, which is an advantageous parameter when compared to KH nanoparticles. These data allowed us to select formulations containing up to 15% of mannose polymers, a concentration of compromise between a percentage low enough to maintain the quality controls of our polyplexes while, at the same time, high enough to allow the selective targeting, as will be shown afterwards. Furthermore, quantitative values of encapsulation efficiency were also confirmed by agarose gel retardation assay (Fig. 2A.II) and quantified by ImageJ, only with Man1-CK3 formulations, obtaining results similar to Ribogreen® and therefore, demonstrating nanoparticles and technique robustness. Agarose gel electrophoresis is a semi-quantitative/qualitative method of assessing encapsulation efficiency and, consequently, and taking into account the same results as those obtained by RiboGreen, it was only applied to one formulation to demonstrate both techniques overlap. NPs were further analyzed in terms of Z-potential (Fig. 2C). Man1 NPs showed a significant decrease in surface charge, from around 28 mV of non-modified NPs to around 25 mV.
Additionally, the morphology of the particles was confirmed by electron microscopy. As shown in Fig. 2D, all formulations resulted in spherical nanoparticles with a normal distribution of diameter, showing mean sizes around 50–55 nm. Although no significant differences were found by the addition of mannose derivatizations, a slight tendency to increase size was observed when this functionalization was present. To note, as expected and according to previous results with polymeric nanoparticles(34), hard sphere diameters found by electron microscopy are markedly reduced as compared to hydrodynamic diameters found by light scattering due to the intrinsic nature of each technique and the use of hygroscopic polymers.
Confirmation of the exposure of mannose residues on the surface of the NPs
To target the DC-SIGN and/or MMR receptors present in APCs, mannose residues need to be expressed on the surface of the NPs. To confirm this, an array of different techniques was used. First, flow cytometry was performed, using Cyanine 5 as polymer labelling for tracking nanoparticles inside the cells, and localizing it with FITC-labelled ConcanavalinA (ConA), a CTLR with specific recognition of α-mannose. Nanoparticles were prepared with pmaxGFP and C6-CR3 as the labelled polymer, also containing the mannose polymers, followed by incubation with ConA-FITC. The results were compared with the KH in terms of Cy5 and FITC presence. The results suggest that nanoparticles containing polymers with α-mannose present a higher population of nanoparticles bound to ConA (Fig. 3A.IV–A.VI) 77%, 77% and 83% respectively for KHM1 15%, KHM2 15% and KHM3 15%. On the contrary, nanoparticles without the mannose functionalization (Fig. 3A.III, including gatings in Fig. 4A.I and A.II) almost did not present ConA, a significantly different result between the control samples and the tested ones, therefore, confirming the presence of α-mannose in the surface of the complex, and thus allowing the targeting.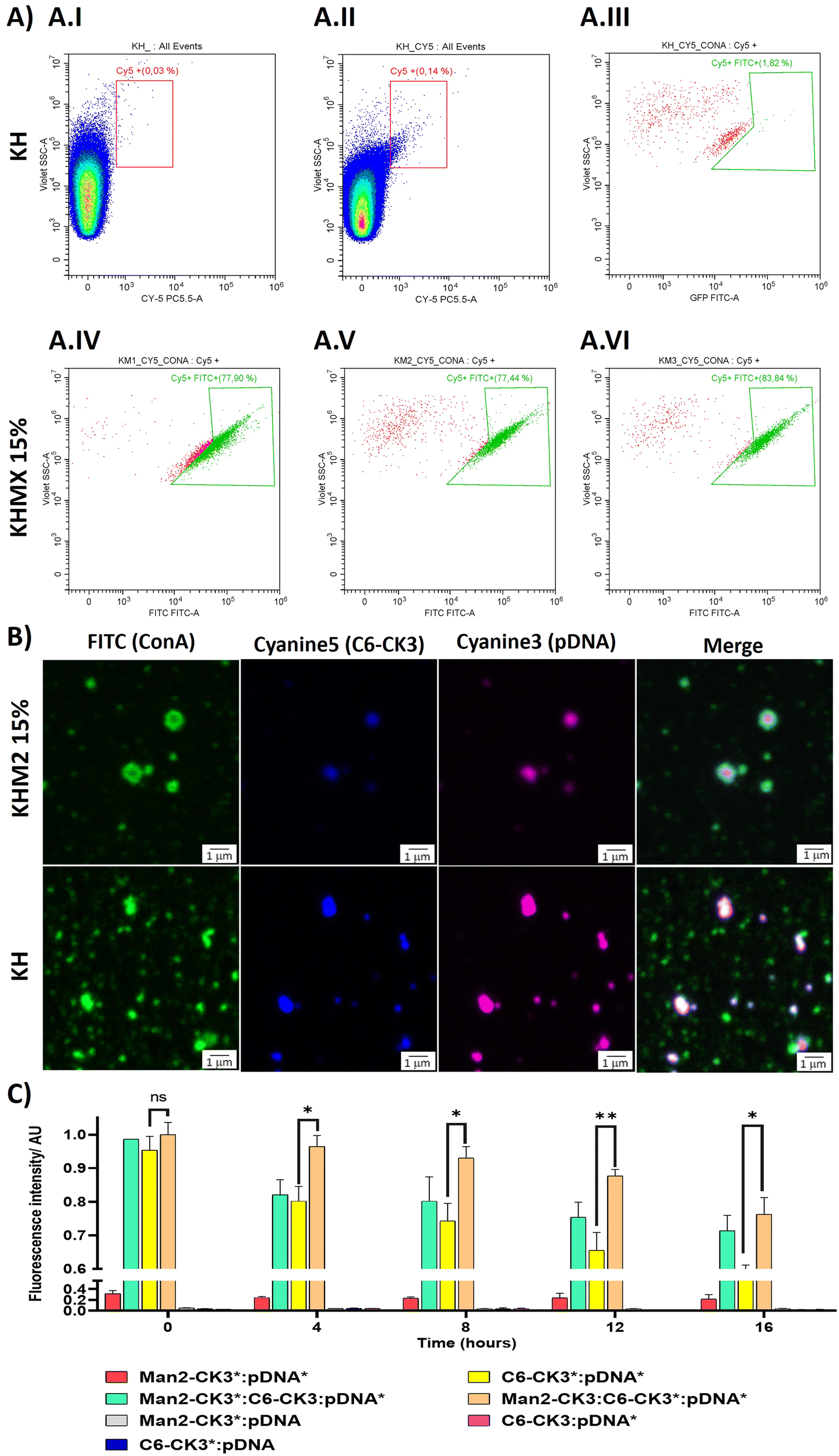 | ||
| Fig. 3 Surface characterization of the different formulations. (A) Nanoparticle analysis by flow cytometry (FC) labelling with Cy5 at 0.5% in polymer C6-CK3 and conjugating with Concanavalin A protein labelled with FITC. (A.I–A.III) KH sample used as the control for setting up Cy5 positive nanoparticles and FITC negative population, using Concanavalin A-FITC. (A.IV) Nanoparticles ensembled with KHM1 15% and ConA. (A.V) Nanoparticles ensembled with KHM2 15% and ConA. (A.VI) Nanoparticles ensembled with KHM3 15% and ConA. (B) Confocal microscopy of nanoparticles containing fluorophores, upon incubating with Concanavalin A labelled with FITC. (C) Fluorescence Resonance Energy Transfer between Cy3 (pDNA) and Cy5 (C6-CK3 polymer). Formulations tested were KHM2 15%, KHM2 60% and KH. The signal (*) indicates polymer or DNA labelled with a fluorophore. p-Value: 0.05 (*), 0.005 (**), >0.001 (***). | ||
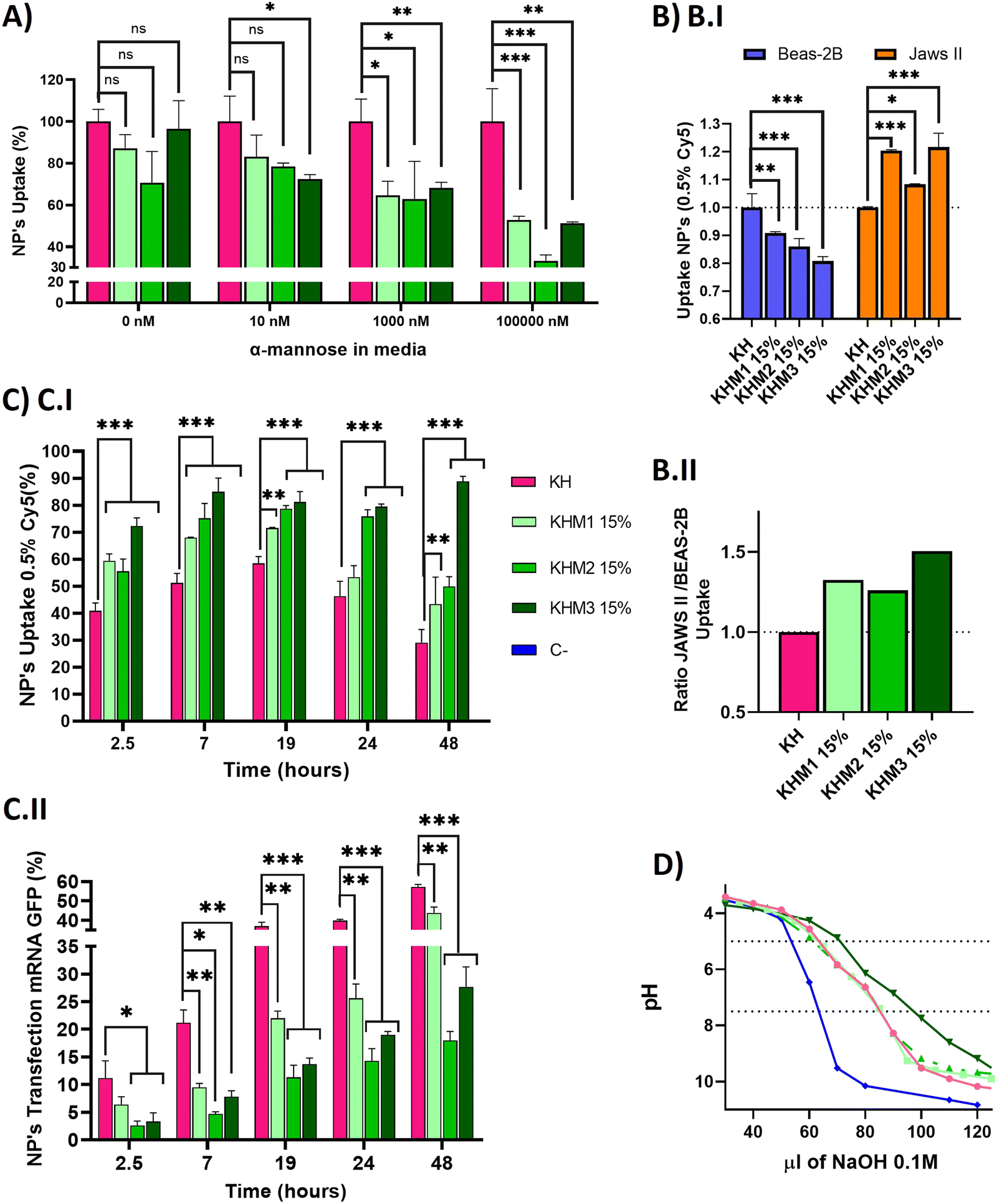 | ||
| Fig. 4 In vitro experiments carried out with different formulations containing Man1-CK3, Man2-CK3 and Man3-CK3 polymers at 15% with 0.5% of Cy5 in C6-CK3 been used as a reporter for FC. Nanoparticles were made with eGFP mRNA. (A) Competition assay with JAWS II at different concentrations of α-mannose in media to saturate the receptors CD206 and CD209. (B and B.I) Nanoparticles uptaken by different cell lines to compare the targeting effect of α-mannose polymers. (B.II) Ratio of mean values of nanoparticles uptake between JAWSII and BEAS-2B cell lines (C) In vitro kinetic (C.I) uptake and (C.II) transfection of selected formulations in JAWS II for 48 hours. (D) Buffering capacity of synthesized polymers. Titration curves of poly(β-amino ester)s at 0.2 mg mL−1. Polymer solutions were adjusted to pH 3 with HCl and consequently titrated with NaOH 0.1M until pH 10. C-refers to titration of the solution that does not contain polymer. C6-CK3 was used as a positive control. The dotted lines indicated the range between pH 5 and 7.5. p-Value: 0.05 (*), 0.005 (**), >0.001 (***). | ||
Subsequently, these results were confirmed by confocal microscopy. To remark the recent use of this technique for the analysis of nanometric vesicles with different immunostaining,28,29 although it must be taken into account that sizes observed for the complexes cannot be attributed to nanoparticle sizes due to the nanometric range below the limit of resolution of the technique. KHM2 15% and KH formulations were tested with the following labelling, and the labelled component is indicated with an asterisk: Man1-CK3-C6CK3*-C6CH3/pGFP* and C6-CK3*H/pGFP*, pmaxGFP (pGFP or pDNA) was used as the genetic material labelled with Cy3, incubated with concanavalin A-FITC for almost 1 h (Fig. 3B). The nanoparticles surface is homogenously enveloped by ConA-FITC in the sample of KHM2 15%, instead, in sample KH, ConA does not colocalize with the fluorescence of the nanoparticle components and is spread in discrete points in the media, clearly not related with the nanoparticle structure. Therefore, all this information points to the idea that the mannose-containing polymer is present in the nanoparticles and, advantageously, located on the surface of the nanoparticle.
Finally, fluorescence resonance energy transfer (FRET) experiments were performed to fully confirm the presence of the mannose-modified polymers in the nanoparticles, as we described before Navalón-López, et al. 2023.19 In brief, a couple of fluorophores (Cy3:Cy5) were used in order to detect FRET on the sample where higher FRET signals indicate a closer location of the polymer* (Cy5 labelled) to the pDNA* (Cy3 labelled). Thus, the disposition of the polymers inside the nanoparticle can be determined. As for confocal microscopy, KHM2 15% formulation was chosen as representative of the new polymers and compared with KH nanoparticles, using pmaxGFP as the genetic material (pDNA). In Fig. 3C the formulations with labelling on one polymer + labelling on the nucleic acid ([Man2-CK3:C6-CK3*:pDNA*], [Man2-CK3*:C6-CK3:pDNA*] and [C6-CK3*:pDNA*], as control) were the ones with the highest FRET values. At 0 hours, there were no significant differences between the control and the samples. At 4 hours, Man2-CK3:C6-CK3*:pDNA* presents the highest FRET value (90%), meaning that C6-CK3* polymer was closer to pDNA and Man2-CK3 was further away. Regarding the stability results, control nanoparticles presented a more abrupt decrease in the FRET signal on time, thus, they were not as stable as those including mannose. These results emphasize the stabilizing effect produced by the addition of α-mannose polymers in almost 20% regarding the Man2-CK3:C6-CK3*:pDNA* sample at 12 h. Furthermore, when Man2-CK3*:C6-CK3:pDNA* and Man2-CK3:C6-CK3*:pDNA* samples were compared, which are the same formulation but changing the polymer that was labelled (Man2-CK3 and C6-CK3 respectively), a different kinetic was appreciated: Man2-CK3*:C6-CK3:pDNA* FRET values decrease much faster, meaning that this polymer is located on a more external layer, and thus, hypothesized on the nanoparticle surface, at 12 h, obtaining a difference of 10% between both formulations.
With this array of experiments, then, we can conclude that the Man2-CK3 polymer was farther from DNA than the C6-CK3 polymer, exposed on the nanoparticles’ surface and therefore, adequate for contributing to the targeting effect.
Setting up a lyophilization process for mannose-functionalized nanoparticles that enables further clinical use
In order to transfer any nanotechnology to clinical practice, NP storage under mild conditions will be a clear advantage in terms of transport, logistics, batch processing and clinical use. However, nanomedicine stability in liquid dispersions is usually the bottleneck step.30–32 A solution for this issue is a dry-freeze process, allowing the preservation of nanoparticles in a ready-to-use mode, after the resuspension step. In our previous studies, we already set up a freeze-drying process, by the adjustment of the formulation by the addition of 4 wt% sucrose in Hepes 20 mM solution, as a lyoprotectant that also accompanies the pH and osmolality conditions of the bloodstream.27 Here, since polymers themselves contain sugar moieties, before starting in vitro and in vivo experiments, a readjustment of the formulation for its lyophilization was required by changing the sucrose percentage.Examining the data obtained in Table 2, we observed a trend between nanoparticle size and osmolality with sucrose concentration (w/w) after the freeze-drying procedure, as expected. According to size, for the current application, the nanoparticle size must be lower than 200–250 nm and according to osmolality, it should be near 300 mOsm kg−1,33 therefore, the ratio of sucrose at 2% (w/w) accomplishes both objectives, being the selected one for the lyophilization procedure used for further in vitro and in vivo studies.
| Sucrose (%) | Size (nm) | SD (nm) | PDI | Osmolality (mOsm kg−1) |
|---|---|---|---|---|
| Fresh NPs | 162.0 | 42.70 | 0.07 | — |
| 0 | 151.6 | 36.4 | 0.06 | 88 |
| 2 | 155.5 | 51.6 | 0.11 | 340 |
| 4 | 221.8 | 103.9 | 0.22 | 693 |
| 6 | 236.5 | 69.9 | 0.09 | 828 |
| 8 | 251.2 | 91.2 | 0.13 | 1032 |
| 10 | 230.9 | 82.2 | 0.13 | 1260 |
In vitro study of cell transfection efficacy of mannose-functionalized pBAE NPs
Once we confirmed adequate physicochemical properties for our aim, we studied the uptake and transfection efficiency of our nanoparticles. To determine the targeting contribution of Man1-CK3, Man2-CK3 and Man3-CK3 in different pBAE formulations, mRNA encoding for PureBoostTM EGFP mRNA was used to transfect JAWS II monocytic cells with Cy5-labelling on C6-CK3, to check transfection through GFP expression and uptake by Cy5 presence inside the cells by FC (Fig. 4). Preliminarily, the biocompatibility of both the polymers and the polyplexes, was confirmed in vitro in models of immune cells (see the results in Fig. S27 and S28, ESI†).A competition assay with free α-mannose was performed under different concentrations of α-mannose, by quantifying the uptake of the different nanoparticles after 1 hour of incubation and using KH nanoparticles as the negative control (without targeting; (Fig. 4A)). Regarding the uptake of mannose-containing polymers, and as expected, there were no significant differences at 0 nM of α-mannose between the tested polymers and KH control, since mannose-modified NPs remain cationic and thus, can penetrate cells through a non-receptor mediated mechanism. Nevertheless, when the α-mannose concentration in media was increased, the behavior between the samples and the control varied. As expected, and according to previous studies,20 uptake values decreased as competing mannose concentration increased. Previous studies already showed that at 10 nM mannose, the inhibition of DC-SIGN and MMR was 40% and 80%, respectively, for a mannose-protein conjugate.20 In our hands, an increase in the competitor dose was required, since we used the mannose without the protein conjugation for competition. 1000 nM was necessary to show significant differences between glyconanoparticles and KH nanoparticles, where KHM1 15%, KHM2 15%, and KHM3 15% had around 65% of uptake, despite the KH control being 100%. Finally, at 100000 nM, KHM1 15% and KHM3 15% were taken up at a rate of almost 50% while KHM2 15% decreased around 35% of uptake. It is important to note that, since no significant changes were observed in the physicochemical properties of the nanoparticles when adding this 15% of mannose modification, the effect of the mannose modification in the competition studies required these high competitor concentrations to be observed.
To summarize, as the mannose media concentration is increased, the uptake drastically decreases to 50–35% of the initial uptake. This information confirms that KHM1 15%, KHM2 15%, and KHM3 15%, were supposedly triggering the receptor-mediated endocytosis uptake as a major method of cell internalization due to CRD4 interaction. The expected complete inhibition of all the uptake at the highest α-mannose concentration in media was not observed due to the high cationic Z-potential of the nanoparticles (+20 mV) that promotes their non-specific interaction with anionic phospholipid cell membranes, and therefore, despite having a targeting effect, there is a part of cell internalization that comes from positively charged nanoparticles (between 35–50%).
Subsequently, selectivity studies included the quantification of the uptake in JAWSII, as a model for targeted immature dendritic cells from mice, which present MMR (CD206) and DC-SIGN (CD209),34 in contraposition with that of BEAS-2B cells, a model of non-target cells that do not present these receptors35 (Fig. 4B.I). For all mannose-functionalized particles, there is a clear effect of the polymer, being able to relatively decrease the uptake in BEAS-2B while increasing it in JAWS II. This behavior was found for the three polymer candidates, with only a slightly better ratio for Man1-CK3 and Man3-CK3 functionalization. In conclusion, we can confirm that polymers Man1-CK3, Man2-CK3 and Man3-CK3 significantly turn the balance to a preferential accumulation in targeted dendritic cells, as compared to a permissive epithelial cell line (Fig. 4B.II). Thus, the expected targeting to CD206 and/or CD209 receptors can be confirmed at an in vitro level.
Finally, a kinetic experiment was performed to compare the differences between polymers in terms of uptake and transfection, obtaining parameters about mRNA release from the different formulations into JAWS II cells (Fig. 4C). Longer times were tested to study uptake and transfection simultaneously. Starting at 2.5 h, we observed that the mannose-nanoparticles uptake is higher than that of the control KH. This difference could be attributed, as a difference to the Fig. 4A result of 0 nM of mannose, due to the interaction between glycopolymers and receptors, an effect that requires a lag phase that retards the uptake. Thus, at short times (<1.5 h), the mannose polymers need to adapt to the correct conformation to bind to the receptors CD206 and CD209,36 instead, KH can enter inside cells by pinocytosis or clathrin-independent/dynamin-dependent endocytosis, a non-specific and faster penetration route.37,38 Consequently, once glycopolymers were bound, the receptor prompts the nanoparticle internalization, facilitating nanoparticle entry and therefore, explaining the increase in the uptake comparing KHM1 15%, KHM2 15%, and KHM3 15% with KH (59%, 55%, 72%, and 41%). Thus, receptor-mediated endocytosis by using CD206 and/or CD209 receptors is corroborated.
At longer times (7 hours results), for KH nanoparticles, the uptake still increased up to 19 h. Instead, the maximum uptake was achieved for mannose-functionalized nanoparticles at 7 h, and maintained through 19 h, highlighting the uptake mediated by DC-SIGN receptors, which has the above-mentioned lag phase followed by fast endocytosis, and therefore, reaching the maximum uptake earlier than the control KH nanoparticles. After 19 h, the signal starts to decrease, indicating an expulsion/metabolization of the polymer, desired to avoid accumulation toxicity problems. At 48 h, only KHM3 15% presents a high signal of Cy5. We correlate the accumulation of Cy5 inside the cells in KHM3 15% nanoparticles with the increase of buffering capacity, detailed below, making the polymer much more difficult to eliminate from the cells due to a lower sensitivity to pH shifts.39 The decrease of the polymer signal is an indirect indication of the degradation of the polymer. As expected, our pBAE polymers, being biodegradable, start to be either degraded or expulsed from the cells, a positive point in terms of avoiding eventual toxicity that may be caused by nanoparticle accumulation inside cells.
Regarding transfection results, they are reported in Fig. 4C.II, to directly study the functionality of the nanoparticles and indirectly study the release kinetics of the mRNA. KH nanoparticles resulted in the highest yields at any time (2.5–48 h), followed closely by KHM1 15%, and more distantly KHM2 15% and KHM3 15%. The highest transfection yield was at 48 h, in which we can appreciate 60% for KH, followed by KHM1 15% with 40% and finally KHM2 15% and KHM3 15% with 20% and 25% respectively.
Examining both data together, KH and KHM1 15% had a similar profile for genetic material release from nanoparticles, in which there is uptake, up to 6–7 h where 90% of nanoparticles are inside the cells, followed by a high increase in transfection due to the proton sponge effect and protein expression (between 7–19 h), continued with a slow decrease on the uptake at the same time due to polymer degradation. In comparison, KHM2 15% and KHM3 15% slowed the mRNA release and therefore, the transfection yield is lower, specifically for KHM3 15%, which did not overpass 10% transfection. This effect could be driven due the polymer KHM3 15% buffering or enveloping excessively.
With all these results, we can conclude that the presence of mannose-functionalized polymers was markedly changing the nanoparticles’ behavior compared with KH nanoparticles, significantly increasing the uptake and decreasing the transfection rates. Despite decreasing GFP expression levels, the obtained values for KHM2 15% and KHM3 15% were similar to those reported previously for Lipofectamine2000,9 since JAWSII is a highly restrictive cell line to transfection, thus, these formulations were maintained for further evaluation. In order to verify if the hypothesized mRNA release delay was occurring, the buffering capacity of the synthesized polymers was checked, with the proton sponge effect being the main trigger of polyplex disassembly and genetic material release from nanoparticles directly to cytoplasm thus enabling mRNA translation and consequent expression.40 The buffering capacity of the new polymers was determined by base titration between 5 and 7.5 pH (Fig. 4D). The highest buffering capacity was observed with Man3-CK3, but in contrast, Man1-CK3 and Man2-CK3 polymers had the same buffering profile as the control C6-CK3.
These results suggest that the presence of mannose on the polymers Man1-CK3 and Man2-CK3 does not hamper their buffering capacity, allowing the endosomal escape of the genetic material; but, for the Man3-CK polymer, on the contrary, we appreciate an increase on this feature that could make the endosomal escape difficult, and therefore, slowing down transfection yields and explaining the transfection kinetic of KHM3 15%; thus confirming the hypothesis above.
It is important to highlight the fact that, altogether, these data are a clear indication of the capacity of the particles to release the loading, the mRNA, inside the endosomes and then to disrupt the endosomes’ integrity allowing the release of the mRNA to the cytoplasm, their consequent translation to protein and further expression of the encoded protein. This mechanism is taking place for all the formulations but, as widely discussed in this section, the release profile depends on the mannose modification used.
Ex vivo confirmation of antigen-presenting cells’ selectivity thanks to the mannose-functionalization
Once it was confirmed that the new family of mannose-containing polymers drives targeting properties in vitro, an in vivo experiment was carried out. This experiment aimed to confirm that the different mannose-functionalized formulations can target CD209+ cell populations, commonly overexpressed by DC.41 Thus, mice were intravenously transfected with Cy5 labelled nanoparticles containing PureBoostTM Fluc mRNA, and after 6 h the spleens were removed and mechanically disaggregated. Finally, labelling with an antibody against CD209 was carried out for cell identification by FC (Fig. 5).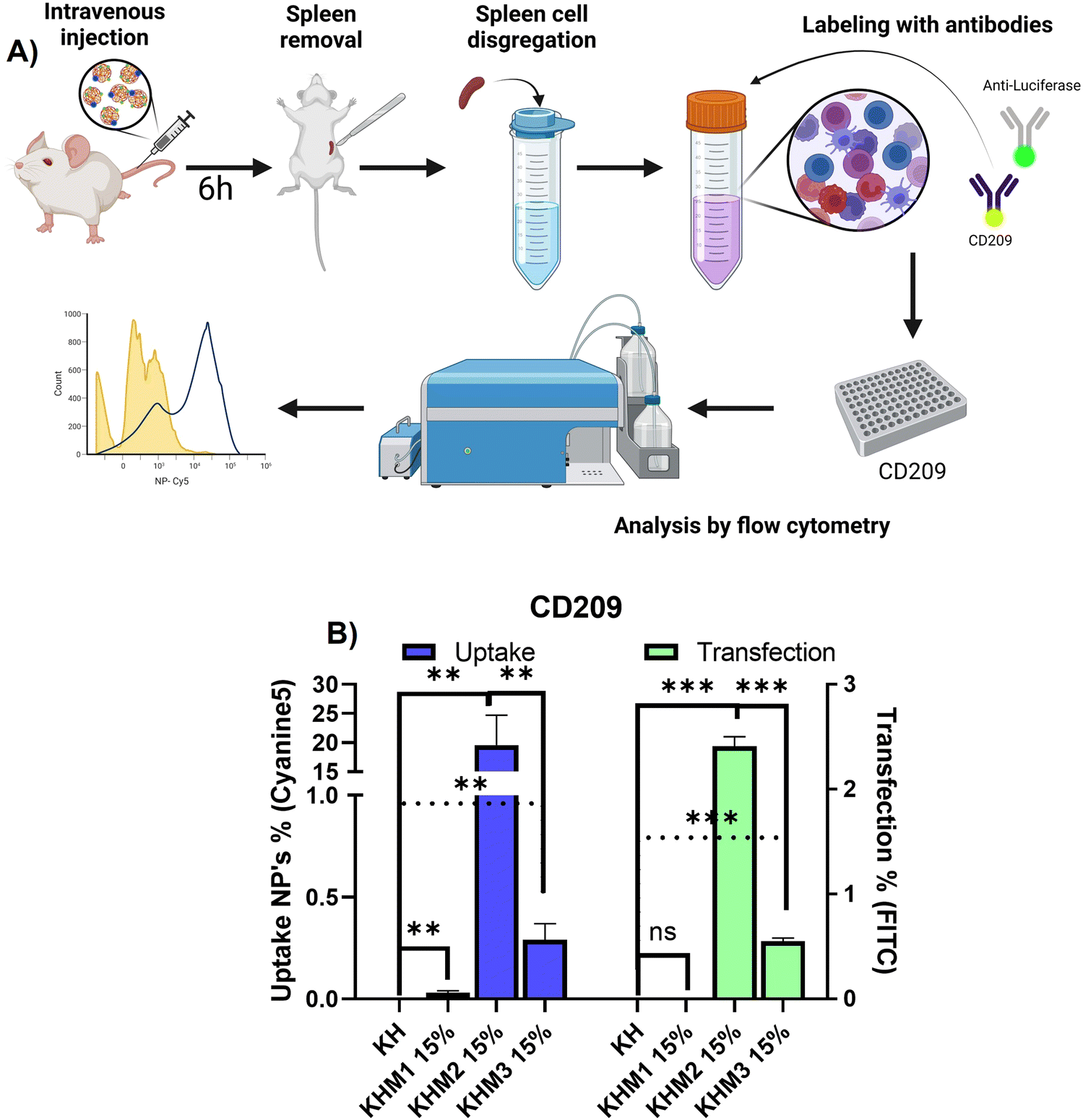 | ||
| Fig. 5 In vivo experiment with 3 highlighted formulations and KH control (n = 4). (A) Scheme of the procedure that was carried out to remove and analyze spleen cells. (B) Ex vivo results of uptake and transfection for cell population CD209+. | ||
From the different formulations, KHM2 15% standard as the most promising, since it resulted in the highest uptake values (around 20%) with cells that present DC-SIGN. Additionally, for those samples, a transfection of 2.5% was obtained. These results interestingly contrast with the control KH nanoparticles, where there is no presence of uptake in either transfection.
From the resulting data related to the uptake, we can infer that the type of cells that are being transfected could be mo-DC due two major approaches: it is reported that pBAE could drive into proinflammatory pathways related to interferon-regulatory factor, being this modulating DC subset and driving monocytes to mo-DC.42,43 Moreover, some reports suggest that mo-DC is overexpressing DC-SIGN inside the spleen.44 Finally, to conclude which subtype nanoparticles are going to, further studies should be done with more markers for cell identification by FC. Focusing on transfection rates, previous works reported that for some gene therapy applications transfection percentages lower than 10% can trigger a therapeutic effect or change gene expression,45,46 and therefore, the application of KHM2 15% is a promising candidate to follow up.
Conclusions
In this article, we reported, for the first time, the functionalization of poly(β aminoester) polymers with α-mannose lateral chains, for the selective vectorization of resulting nanoparticles to APCs and their use as mRNA vaccines. This is the first time that lateral chains of the pBAE polymers have been functionalized with mannoses, providing ramification of the functionalization that contributes to the specific and selective targeting of mannose receptors. As demonstrated through the manuscript, polymers were obtained, as well as nanoparticles with the α-mannose exposed on their surface. Furthermore, we also demonstrated in vitro and in vivo the selectivity of this new nano-delivery system to preferentially target and transfect APCs. Thus, the achievement of our aim opens the possibility to develop a new family of mRNA polymeric vaccines for immunotherapy approaches. Additionally, the presented synthetic strategy could be used to generate libraries of glycopolymers and resulting mRNA-loaded nanoparticles as promising candidates targeting cells with overexpressed glycan receptors.Author contributions
Conceptualization: CF; MF; data curation: CF; resources: CF, SB, AP; formal analysis: NGR, MA, MGR; funding acquisition: CF, SB, AP; investigation: CF, MF; methodology: NGR; MA; MGR; project administration: CF, MF; software: NGR, MA, MGR; supervision: CF, MF, SB, AP; validation: CF, MF; visualization: NGR; writing – original draft: NGR, CF, MA; writing – review and editing: CF.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Experimental support from Maria Stampa Lopez-Pinto and Laura Olmo in some polymer synthesis, from Anna Fotopoulou in nanoparticle characterization and from BD team, for the CytoFlex experiments, is acknowledged. Funding from AGAUR-Generalitat de Catalunya (2022 SGR 00537 and 2022 SGR 00535) from MICIN/AEI (PID2019-104350RB-I00; PID2021-125910OB-I00, MCIN/AEI/10.13039/501100011033/FEDER, UE), from the Institute of Health Carlos III (ISCIII) (AC22/00042), and FCAECC (TRNSC213882FORN) from the TRANSCAN framework and by the Joint Transnational Initiative ERA-NET TRANSCAN-3, European Commission is acknowledged. This article is also based upon work from COST Action CA21154 and COST CIG 17104, supported by COST (European Cooperation in Science and Technology).References
- A. Bhatia and Y. Kumar, Cellular and molecular mechanisms in cancer immune escape: A comprehensive review, Expert Rev. Clin. Immunol., 2014, 10(1), 41–62 CrossRef CAS PubMed.
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, A guide to cancer immunotherapy: from T cell basic science to clinical practice, Nat. Rev. Immunol., 2020, 20(11), 651–668 CrossRef CAS PubMed.
- K. Esfahani, L. Roudaia, N. Buhlaiga, S. V. Del Rincon, N. Papneja and W. H. Miller, A review of cancer immunotherapy: From the past, to the present, to the future, Current Oncol., 2020, 27(S2), 87–97 CrossRef PubMed.
- D. B. Johnson, C. A. Nebhan, J. J. Moslehi and J. M. Balko, Immune-checkpoint inhibitors: long-term implications of toxicity, Nat. Rev. Clin. Oncol., 2022, 19(4), 254–267 CrossRef PubMed.
- K. van der Maaden, J. Heuts, M. Camps, M. Pontier, A. Terwisscha van Scheltinga and W. Jiskoot, et al., Hollow microneedle-mediated micro-injections of a liposomal HPV E743–63 synthetic long peptide vaccine for efficient induction of cytotoxic and T-helper responses, J. Controlled Release, 2018, 269, 347–354 CrossRef CAS PubMed.
- D. Shi, D. Beasock, A. Fessler, J. Szebeni, J. Y. Ljubimova and K. A. Afonin, et al., To PEGylate or not to PEGylate: Immunological properties of nanomedicine's most popular component, polyethylene glycol and its alternatives, Adv. Drug Deliv. Rev., 2022, 180, 114079 CrossRef CAS PubMed.
- Y. Liu, Y. Li, D. Keskin and L. Shi, Poly(β-Amino Esters): Synthesis, Formulations, and Their Biomedical Applications, Adv. Healthcare Mater., 2019, 8(2), 1801359 Search PubMed.
- H. Guerrero-Cázares, S. Y. Tzeng, N. P. Young, A. O. Abutaleb, A. Quiñones-Hinojosa and J. J. Green, Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo, ACS Nano, 2014, 8(5), 5141–5153 CrossRef PubMed.
- C. Fornaguera, M. Guerra-Rebollo, M. Á. Lázaro, C. Castells-Sala, O. Meca-Cortés and V. Ramos-Pérez, et al., mRNA Delivery System for Targeting Antigen-Presenting Cells In Vivo, Adv. Healthcare Mater., 2018, 7(17), 1800335 CrossRef PubMed.
- B. Arnáiz, O. Martínez-Ávila, J. M. Falcon-Perez and S. Penadés, Cellular uptake of gold nanoparticles bearing HIV gp120 oligomannosides, Bioconjug. Chem., 2012, 23(4), 814–825 CrossRef PubMed.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez and O. Blixt, et al., Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR, Nat. Struct. Mol. Biol., 2004, 11(7), 591–598 CrossRef CAS PubMed.
- H. Feinberg, S. A. F. Jégouzo, Y. Lasanajak, D. F. Smith, K. Drickamer and W. I. Weis, et al., Structural analysis of carbohydrate binding by the macrophage mannose receptor CD206, J. Biol. Chem., 2021, 296(22), 1–18 Search PubMed.
- C. G. Figdor, Y. van Kooyk and G. J. Adema, C-type lectin receptors on dendritic cells and Langerhans cells, Nat. Rev. Immunol., 2002, 2(2), 77–84 CrossRef CAS PubMed.
- T. K. Lindhorst, S. Kötter, U. Krallmann-Wenzel and S. Ehlers, Trivalent α-D-mannoside clusters as inhibitors of type-1 fimbriae-mediated adhesion of Escherichia coli: Structural variation and biotinylation, J. Chem. Soc., 2001, 7, 823–831 Search PubMed.
- P. Dosta, V. Ramos and S. Borrós, Stable and efficient generation of poly(β-amino ester)s for RNAi delivery, Mol. Syst. Des. Eng., 2018, 3(4), 677–689 RSC.
- R. Riera, N. Feiner-Gràcia, C. Fornaguera, A. Cascante, S. Borrós and L. Albertazzi, Tracking DNA complexation state of pBAE polyplexes in cells with super resolution microscopy, Nanoscale, 2019, 11(38), 17869–17877 RSC.
- N. Segovia, P. Dosta, A. Cascante, V. Ramos and S. Borrós, Oligopeptide-terminated poly(β-amino ester)s for highly efficient gene delivery and intracellular localization, Acta Biomater., 2014, 10(5), 2147–2158 CrossRef CAS PubMed.
- J. M. Silva, G. Vandermeulen, V. G. Oliveira, S. N. Pinto, C. Rodrigues and A. Salgado, et al., Development of functionalized nanoparticles for vaccine delivery to dendritic cells: A mechanistic approach, Nanomedicine, 2014, 9(17), 2639–2656 CrossRef CAS PubMed.
- M. Navalón-López, A. Dols-Perez, S. Grijalvo, C. Fornaguera and S. Borrós, Unravelling the role of individual components in pBAE/polynucleotide polyplexes in the synthesis of tailored carriers for specific applications: on the road to rational formulations, Nanoscale Adv., 2023, 5, 1611–1623 RSC.
- R. T. Lee, T. L. Hsu, S. K. Huang, S. L. Hsieh, C. H. Wong and Y. C. Lee, Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities, Glycobiology, 2011, 21(4), 512–520 CrossRef CAS PubMed.
- I. Pujol-Autonell, M. J. Mansilla, S. Rodriguez-Fernandez, M. Cano-Sarabia, J. Navarro-Barriuso and R. M. Ampudia, et al., Liposome-based immunotherapy against autoimmune diseases: therapeutic effect on multiple sclerosis, Nanomedicine, 2017, 12(11), 1231–1242 CrossRef CAS PubMed.
- O. Abu-Hammad, H. Alduraidi, S. Abu-Hammad, A. Alnazzawi, H. Babkair and A. Abu-Hammad, et al., Side effects reported by jordanian healthcare workers who received covid-19 vaccines, Vaccines, 2021, 9(6), 1–10 Search PubMed.
- M. K. Hossain and K. A. Wall, Use of dendritic cell receptors as targets for enhancing anti-cancer immune responses, Cancers, 2019,(3), 11 Search PubMed.
- B. J. Appelmelk, I. van Die and S. J. van Vliet, Vandenbroucke-Grauls CMJE, Geijtenbeek TBH, van Kooyk Y. Cutting Edge: Carbohydrate Profiling Identifies New Pathogens That Interact with Dendritic Cell-Specific ICAM-3-Grabbing Nonintegrin on Dendritic Cells, J. Immunol., 2003, 170(4), 1635–1639 CrossRef CAS PubMed.
- P. Dosta, N. Segovia, A. Cascante, V. Ramos and S. Borrós, Surface charge tunability as a powerful strategy to control electrostatic interaction for high efficiency silencing, using tailored oligopeptide-modified poly(beta-amino ester)s (PBAEs), Acta Biomater., 2015, 20, 82–93 CrossRef CAS PubMed.
- R. Riera, N. Feiner-Gracia, C. Fornaguera, A. Cascante, S. Borrós and L. Albertazzi, Tracking the DNA complexation state of pBAE polyplexes in cells with super resolution microscopy, Nanoscale, 2019, 11(38), 17869–17877 RSC.
- C. Fornaguera, C. Castells-Sala, M. A. Lázaro, A. Cascante and S. Borrós, Development of an optimized freeze-drying protocol for OM-PBAE nucleic acid polyplexes, Int. J. Pharm., 2019, 569, 118612 CrossRef CAS PubMed.
- A. Mondal, K. A. Ashiq, P. Phulpagar, D. K. Singh and A. Shiras, Effective Visualization and Easy Tracking of Extracellular Vesicles in Glioma Cells, Biol. Proced. Online, 2019, 21, 4 CrossRef PubMed.
- C. Bagci, M. Sever-bahcekapili, N. Belder, A. P. S. Bennett, S. E. Erdener and T. Dalkara, Overview of extracellular vesicle characterization techniques and introduction to combined reflectance and fluorescence confocal microscopy to distinguish extracellular vesicle subpopulations, Neurophotonics, 2022, 9(2), 021903 CAS.
- S. J. Lim and C. K. Kim, Formulation parameters determining the physicochemical characteristics of solid lipid nanoparticles loaded with all-trans retinoic acid, Int. J. Pharm., 2002, 243, 135–146 CrossRef CAS PubMed.
- C. Chen, D. Han, C. Cai and X. Tang, An overview of liposome lyophilization and its future potential, J. Controlled Release, 2010, 142(3), 299–311 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Freeze-drying of nanoparticles: Formulation, process and storage considerations, Adv. Drug Deliv. Rev., 2006, 58(15), 1688–1713 CrossRef CAS PubMed.
- M. Rasouli, Basic concepts and practical equations on osmolality: Biochemical approach, Clin. Biochem., 2016, 49(12), 936–941 CrossRef CAS PubMed.
- P. R. Taylor, L. Martinez-Pomares, M. Stacey, H. H. Lin, G. D. Brown and S. Gordon, Macrophage receptors and immune recognition, Annu. Rev. Immunol., 2005, 23, 901–944 CrossRef CAS PubMed.
- X. Han, T. Na, T. Wu and B. Z. Yuan, Human lung epithelial BEAS-2B cells exhibit characteristics of mesenchymal stem cells, PLoS One, 2020, 15(1), 1–18 Search PubMed.
- U. Švajger, M. Anderluh, M. Jeras and N. Obermajer, C-type lectin DC-SIGN: An adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity, Cell Signal, 2010, 22(10), 1397–1405 CrossRef PubMed.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics, Nat. Nanotechnol., 2021, 16(3), 266–276 CrossRef CAS PubMed.
- B. Yameen, W. Il Choi, C. Vilos, A. Swami, J. Shi and O. C. Farokhzad, Insight into nanoparticle cellular uptake and intracellular targeting, J. Controlled Release, 2014, 190, 485–499 CrossRef CAS PubMed.
- H. Azevedo and R. Reis, Understanding the Enzymatic Degradation of Biodegradable Polymers and Strategies to Control Their Degradation Rate, Biodegrad. Syst. Tissue Eng. Regener. Med., 2004, 177–202 Search PubMed.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery, Bioconjug. Chem., 2019, 30(2), 263–272 CrossRef CAS PubMed.
- E. J. Soilleux, L. S. Morris, G. Leslie, J. Chehimi, Q. Luo and E. Levroney, et al., Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro, J. Leukoc Biol., 2002, 71(3), 445–457 CrossRef CAS PubMed.
- T. Tamura, P. Tailor, K. Yamaoka, H. J. Kong, H. Tsujimura and J. J. O’Shea, et al., IFN Regulatory Factor-4 and -8 Govern Dendritic Cell Subset Development and Their Functional Diversity, J. Immunol., 2005, 174(5), 2573–2581 CrossRef CAS PubMed.
- N. M. Dold, Q. Zeng, X. Zeng and C. M. Jewell, A poly(beta-amino ester) activates macrophages independent of NF-κB signaling, Acta Biomater., 2018, 68, 168–177 CrossRef CAS PubMed.
- Y. Y. Hey and H. C. O’Neill, Murine spleen contains a diversity of myeloid and dendritic cells distinct in antigen presenting function, J. Cell. Mol. Med., 2012, 16(11), 2611–2619 CrossRef CAS PubMed.
- M. A. Oberli, A. M. Reichmuth, J. Robert Dorkin, M. J. Mitchell, O. S. Fenton and A. Jaklenec, et al., Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy HHS Public Access Author manuscript, Nano Lett., 2017, 17(3), 1326–1335 CrossRef CAS PubMed.
- S. Hamdy, A. Haddadi, R. W. Hung and A. Lavasanifar, Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations, Adv. Drug Deliv. Rev., 2011, 63(10–11), 943–955 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00607g |
| This journal is © The Royal Society of Chemistry 2023 |