
The journey of iron-based electrocatalytic materials for nitrogen reduction reaction: from current status to future prospects
Yi-Han
Wang
b,
Ji-Hong
Dong
a,
Zhenquan
Tan
 a,
Xiao-Feng
Wang
c and
Xue-Zhi
Song
*a
a,
Xiao-Feng
Wang
c and
Xue-Zhi
Song
*a
aState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: songxz@dlut.edu.cn
bLeicester International Institute, Dalian University of Technology, Panjin 124221, China
cKey Laboratory of Materials Modification by Laser, Ion and Electron Beams, Ministry of Education, School of Physics, Dalian University of Technology, Dalian 116024, China
First published on 1st May 2023
Abstract
Ammonia (NH3) is the most important chemical and carbon-free energy carrier, which is currently mainly produced by the energy-intensive Haber–Bosch process at high temperatures and pressures. In the spirit of global carbon neutrality and sustainable development, electrochemical nitrogen reduction reaction (NRR) is expected as a promising alternative to the traditional Haber–Bosch process for industrial ammonia production due to both low energy consumption and zero emission of carbon dioxide. However, further breakthroughs in electrochemical NRR strongly depend on the efficiency and selectivity of electrocatalysts. The element Fe has been found to be an important component in nitrogenase enzymes, generating extensive research interest. As one of the most cost-effective and abundant transition metals, iron-based materials have presented great potential for electrochemical NRR. In this review, we summarize recent advances in iron-based materials (including its oxides, chalcogenides, borides, single Fe-atom catalysts, and bimetallic catalysts especially for Fe–Mo-based compounds) towards highly efficient N2-to-NH3 conversion under ambient conditions. Based on recent theoretical and experimental studies, we focus on the structure–property relationship. Strategies to boost NRR performances and perspectives for future developments are comprehensively presented in synthetic protocols, structure modification, activity/selectivity enhancement, and reaction mechanisms to provide insightful guidance for the field of NRR studies.
 Yi-Han Wang | Yi-Han Wang is pursuing his B.S. degree at Dalian University of Technology. His research focuses on the design and preparation of electrocatalysts for NRR under the supervision of Dr Xue-Zhi Song. |
 Ji-Hong Dong | Ji-Hong Dong received her B.S. degree in chemical engineering from Dalian University of Technology. Now, she is pursuing the M.S. degree under the supervision of Associate Prof. Xue-Zhi Song. Her research focuses on the design and preparation of electrocatalysts for NRR. |
 Zhenquan Tan | Zhenquan Tan is a Full Professor at School of Chemical Engineering in Dalian University of Technology (China). He received his B.S. degree in chemistry at Peking University (China) in 2002, and received his PhD degree in chemistry from Tohoku University (Japan) in 2008. His current research interests are focused on the inorganic nanomaterials and their applications in photo-/electrocatalysis, energy storage and conversion, and anti-tumor nanodrug delivery systems. |
 Xiao-Feng Wang | Xiao-Feng Wang is an associate professor in Dalian University of Technology, China. She received her PhD degree in Material Physics and Chemistry from Shandong University, China in 2014. She was a visiting scholar at A. J. Drexel Nanomaterials Institute, Drexel University, during 2018. Her research interests focus on nanomaterials and their applications in gas sensors and catalysis. |
 Xue-Zhi Song | Xue-Zhi Song received his B.S. degree from Lanzhou University (China) in 2009. He completed his PhD degree in Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (China) in 2015. He is currently an Associate Professor in Dalian University of Technology (China). His research interest focuses on functional inorganic nanomaterials for electrocatalysis in HER, OER, and NRR. |
1. Introduction
Ammonia is one of the most principal chemicals and carbon-free energy carriers and also plays an indispensable role in sustaining life.1 Its worldwide annual production is estimated to be 150–200 million tons, providing a hint of insufficiency due to the booming human demands.2 Despite the fact that N2 makes up more than 78% of the atmosphere, it cannot be directly utilized by industry or living organisms.3 Modern industrial large-scale ammonia production still relies on the Haber–Bosch process, which was invented in the 20th century and has contributed to human society for hundreds of years.4 However, the Haber–Bosch technology entails thermocatalytic conversion of N2 and H2 at high temperature (400–500 °C) and intense pressure (200–300 atm).5–7 These conditions determine that Haber–Bosch process is extremely energy-intensive, accounting for 2% of the global energy consumption, accompanied by large greenhouse gas emissions (300–400 million tons of CO2 annually).8,9 Therefore, a greener and more efficient NH3 synthesis method under mild conditions is desired to support the world's sustainable development and carbon neutrality goals.10,11Currently, it has been proven that biocatalysis, photocatalysis, and electrocatalysis are clean pathways for ammonia synthesis from nitrogen reduction.12–15 Electrochemical synthesis of ammonia is considered the most promising alternative to the Haber–Bosch process, due to its mild and stable reaction conditions, abundant hydrogen source (direct extraction from electrolyte instead of fossil fuels), zero CO2 emissions, and flexible and easily-controlled production process by adjusting external parameters.16 In the electrochemical ammonia synthesis, NH3 is produced from N2 and H2O under ambient conditions by inputting electrical energy, with the catalyst playing a decisive role. In fact, the breakthrough development of the electrochemical ammonia synthesis is hampered by some inherent limitations: (1) the ultra-low solubility of N2 in water (6.8 × 10−4 M, 1.01 × 105 Pa, 298 K) is not conducive to maintaining high reactant concentrations, resulting in the unsatisfying ammonia production rate. (2) Low Faraday efficiency (FE). The improvement of FE is hindered by competing hydrogen evolution reactions (HER).17 The electrochemical NRR and HER are both thermodynamically available under reaction conditions. Not only that but HER is preferred from a kinetic point of view because it is a feasible two-electron process. By comparison, electrocatalytic NRR is a multi-step kinetic retardation process, involving six protons and six electrons as well as the activation of inert nitrogen bonds.18 (3) The stability and ammonia yield rate of the catalyst is challenging to fulfill the requirements of industrial production.19
From the molecular mechanistic level, the primary challenge of NRR results from the inertness of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond, which is the strongest among the diatomic molecules and lacks a permanent dipole.20 A high energy input of 945 kJ mol−1 is required to cleave the NN triple bond, which well explains the harsh conditions of the Haber–Bosch process.21 The kinetically difficult electron transfer reaction and the thermodynamically forbidden protonation of N2 to N2H+ also make the electrochemical NRR more difficult.22 Researchers have developed numerous catalysts with good performances to address the above challenges of electrochemical NRR, including noble metals, transition metal (TM) compounds, and metal-free materials.23–33 Among these materials, iron-based catalysts stand out as a non-noble metal protagonist with the advantages of low cost, large-scale industrial synthesis, and high catalytic efficiency. Iron (Fe) is the only element that coexists among the three biological nitrogen-fixing enzymes, suggesting that iron plays an important role in NRR.21 Moreover, Fe is an active metal easy to be compounded with other elements, such as modifying it into nitrides, hydroxides, oxides, sulfides, and phosphides, which may exhibit distinguished catalytic activation and stability. More importantly, the electronic configuration of Fe provides good solutions to activate the N2 molecule. As a transition metal, the Fe atom has an electronic configuration of 3d64s2, indicating four unpaired d-spin electrons. These abundant d-orbital electrons and unoccupied orbitals activate the inert NN triple bond by driving the well-known donation-acceptance process.34–40 To be more precise, the empty or half-occupied orbitals in Fe allow σ donation to accommodate electrons from N2 bonding orbitals (σ orbitals). Meanwhile, the affluent d-electrons in Fe atoms could be injected into the antibonding orbitals (π* orbitals) in N2 by back-donation. As a result, the loss of electrons from the bonding orbitals and the injection of electrons into antibonding orbitals greatly destabilize N2 and expedite the break of the NN bond. It is acknowledged that the NRR activity of the transition metals is usually closely related to the occupancy of eg and t2g orbitals in the catalyst. Fe atom has a manipulable local spin state of t2g4eg2. Both experimental and theoretical analysis indicate that the heightened eg electrons penetration to the antibonding π-orbital of N2 and regulation of the adsorption strength of nitrogen species can be realized by customized spin-polarization configuration.41 Moreover, the electron jump between high-spin iron and ferrous in the appropriate Fe lattice provides good electronic conductivity and poor adsorption of protons, which may lead to more ideal efficiency and selectivity towards NRR.42,43 From a macroscopic view in the design of catalysts, Fe is easily modified for various construction strategies, including vacancy engineering, heteroatom doping, facet control, single atom dispersion, and even metal–organic frameworks (MOFs).21,44–46 These strategies usually enhance N2 adsorption by redistribution of charge density in the catalyst and provide more active sites to improve catalytic efficiency of NRR.47,48
N triple bond, which is the strongest among the diatomic molecules and lacks a permanent dipole.20 A high energy input of 945 kJ mol−1 is required to cleave the NN triple bond, which well explains the harsh conditions of the Haber–Bosch process.21 The kinetically difficult electron transfer reaction and the thermodynamically forbidden protonation of N2 to N2H+ also make the electrochemical NRR more difficult.22 Researchers have developed numerous catalysts with good performances to address the above challenges of electrochemical NRR, including noble metals, transition metal (TM) compounds, and metal-free materials.23–33 Among these materials, iron-based catalysts stand out as a non-noble metal protagonist with the advantages of low cost, large-scale industrial synthesis, and high catalytic efficiency. Iron (Fe) is the only element that coexists among the three biological nitrogen-fixing enzymes, suggesting that iron plays an important role in NRR.21 Moreover, Fe is an active metal easy to be compounded with other elements, such as modifying it into nitrides, hydroxides, oxides, sulfides, and phosphides, which may exhibit distinguished catalytic activation and stability. More importantly, the electronic configuration of Fe provides good solutions to activate the N2 molecule. As a transition metal, the Fe atom has an electronic configuration of 3d64s2, indicating four unpaired d-spin electrons. These abundant d-orbital electrons and unoccupied orbitals activate the inert NN triple bond by driving the well-known donation-acceptance process.34–40 To be more precise, the empty or half-occupied orbitals in Fe allow σ donation to accommodate electrons from N2 bonding orbitals (σ orbitals). Meanwhile, the affluent d-electrons in Fe atoms could be injected into the antibonding orbitals (π* orbitals) in N2 by back-donation. As a result, the loss of electrons from the bonding orbitals and the injection of electrons into antibonding orbitals greatly destabilize N2 and expedite the break of the NN bond. It is acknowledged that the NRR activity of the transition metals is usually closely related to the occupancy of eg and t2g orbitals in the catalyst. Fe atom has a manipulable local spin state of t2g4eg2. Both experimental and theoretical analysis indicate that the heightened eg electrons penetration to the antibonding π-orbital of N2 and regulation of the adsorption strength of nitrogen species can be realized by customized spin-polarization configuration.41 Moreover, the electron jump between high-spin iron and ferrous in the appropriate Fe lattice provides good electronic conductivity and poor adsorption of protons, which may lead to more ideal efficiency and selectivity towards NRR.42,43 From a macroscopic view in the design of catalysts, Fe is easily modified for various construction strategies, including vacancy engineering, heteroatom doping, facet control, single atom dispersion, and even metal–organic frameworks (MOFs).21,44–46 These strategies usually enhance N2 adsorption by redistribution of charge density in the catalyst and provide more active sites to improve catalytic efficiency of NRR.47,48
Based on these facts, this review introduces the recent progress of iron-based catalysts for electrocatalytic NRR under ambient conditions. Firstly, we systematically present the recent advances on iron catalysts for NRR according to the types of iron compounds, which are iron oxides, iron sulfides, Fe-containing bimetallic materials, especially iron-molybdenum compounds, iron borides, and single iron atoms catalysts (Fig. 1). Furthermore, we analyze the available approaches to design advanced electronic and interface structures for different types of iron-based catalysts and relationships between structures and properties from experimental and theoretical perspectives. Finally, we prospect the challenges and potential strategies for further enhancement of NRR performance in terms of FE, ammonia yield, and selectivity.
 | ||
Fig. 1 Scheme organization of the main contents of this review. Iron oxides: reproduced with permission: copyright 2021, Elsevier.49 Copyright 2022, Wiley-VCH.50 Copyright 2022, Royal Society of Chemistry.51 Iron chalcogenides: reproduced with permission: copyright 2021, Elsevier.52 Copyright 2021, Wiley-VCH.53 Fe–Mo-based catalysts: reproduced with permission: copyright 2019, Elsevier.54 Copyright 2020, Royal Society of Chemistry.55 Copyright 2020, American Chemical Society.56 Iron borides: reproduced with permission: copyright 2020, Elsevier.57 Copyright 2019, Royal Society of Chemistry.58 Fe-based SACs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :reproduced with permission: copyright 2022, Wiley-VCH.59,60 :reproduced with permission: copyright 2022, Wiley-VCH.59,60 | ||
2. Fundamental principles of the electrocatalytic NRR
Understanding the characteristics of nitrogen molecules and the reaction mechanism of NRR is the basis of superior NRR electrocatalyst design.2.1 Properties of N2
The very stable nitrogen molecule is formed by the linear combination of two N atoms, each of them has five valence electrons, arranged in the configuration of 2s22p3. According to molecular orbital theory, the valent orbitals of N2 molecules are composed of four bonding orbitals (σ2s, σ2p, and two π) and four antibonding orbitals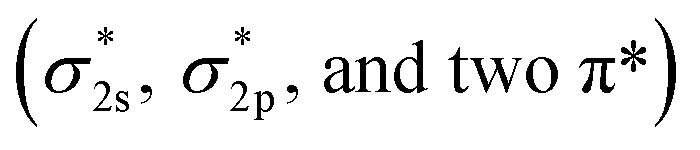 , in which two π* orbitals and one
, in which two π* orbitals and one 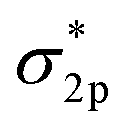 orbital display an empty state, while other orbitals are fully occupied by the coupled spin electrons (Fig. 2A).61 The three occupied σ2p and two π orbital bonds also contribute to the high-energy triple bond.62 The large energy gap (10.82 eV) between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) of N2, the high ionization potential (15.58 eV), and the negative electron affinity (−1.9 eV) all account for the resistance of N2 to Lewis acid/base and electron-transfer processes.63–68 In the view of thermodynamics, NRR is practicable with the overall negative Gibbs free energy. In fact, as the first step of the overall reaction, the adsorption and activation of N2 are intractable since their non-polarity and high stability. The initial endothermic protonation of N2 to form N2H+ (ΔH0 = +37.6 kJ mol−1) is thermodynamically forbidden. Another challenge is the kinetically complex nitrogen hydrogenation process, which involves multiple electron transfers and complex intermediates.44 These intrinsic features render the N2 fixation via electro-reduction of N2 to NH3 of great challenge and difficulty. Nevertheless, the NN bond can be weakened by σ donation and π back-donation between N2 and Fe atoms (Fig. 2B).
orbital display an empty state, while other orbitals are fully occupied by the coupled spin electrons (Fig. 2A).61 The three occupied σ2p and two π orbital bonds also contribute to the high-energy triple bond.62 The large energy gap (10.82 eV) between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) of N2, the high ionization potential (15.58 eV), and the negative electron affinity (−1.9 eV) all account for the resistance of N2 to Lewis acid/base and electron-transfer processes.63–68 In the view of thermodynamics, NRR is practicable with the overall negative Gibbs free energy. In fact, as the first step of the overall reaction, the adsorption and activation of N2 are intractable since their non-polarity and high stability. The initial endothermic protonation of N2 to form N2H+ (ΔH0 = +37.6 kJ mol−1) is thermodynamically forbidden. Another challenge is the kinetically complex nitrogen hydrogenation process, which involves multiple electron transfers and complex intermediates.44 These intrinsic features render the N2 fixation via electro-reduction of N2 to NH3 of great challenge and difficulty. Nevertheless, the NN bond can be weakened by σ donation and π back-donation between N2 and Fe atoms (Fig. 2B).
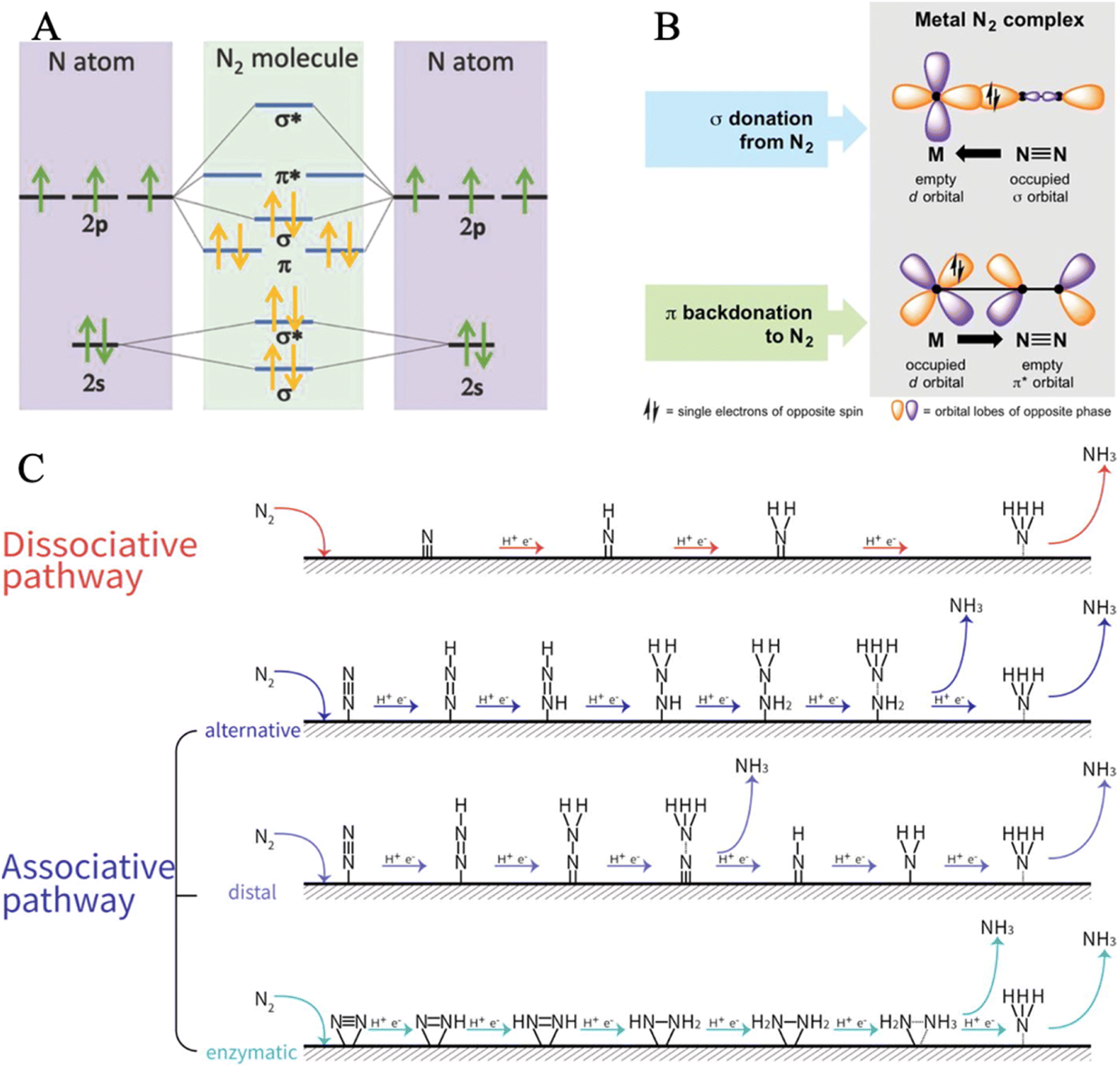 | ||
| Fig. 2 (A) Diagram of N atomic orbitals and N2 molecular orbital diagram. Reproduced with permission: copyright 2018, Wiley-VCH.66 (B) Binding modes of N2 molecules with transition metals. Reproduced with permission: copyright 2018, American Association for the Advancement of Science.79 (C) Reaction pathways for the electrochemical nitrogen reduction to ammonia. Reproduced with permission: copyright 2021, Elsevier.12 | ||
This will provide guidance for the rational design of catalytic active sites starting from addressing the intrinsic inertness of N2.
2.2 NRR mechanisms
The net NRR can be expressed as the equations below:In acidic media
| N2 + 6H+ + 6e− → 2NH3 |
In basic media
| N2 + 6H2O + 6e− → 2NH3 + 6OH− |
The detailed NRR process mainly involves the following three steps: (i) the chemisorption of N2 molecules on the active sites; (ii) the dissociation and hydrogenation of nitrogen; and (iii) the desorption of the ammonia molecules from the catalyst surface. According to the adsorption configurations and the hydrogenation reaction sequences of nitrogen, two basic pathways have been proposed (i) the dissociative mechanism and (ii) the associative mechanism (Fig. 2C).69–73
N bond has a large energy barrier and usually occurs only at high temperatures (such as in the Haber–Bosch process), which is unlikely to be a favorable pathway for electrochemical NRR under ambient conditions.
N bond provides thermodynamically favorable pathways, namely, associative pathways, where the hydrogenation of N2 occurs prior to the breaking of the NN bond. The two different adsorption modes (end-on and side-on) initiate varying associative pathways. It can be classified into three possible reaction pathways depending on the binding pattern of N2 and the hydrogenation sequence. As for the end-on adsorbed N2, it accepts an electron and proton to generate *N2H, and then both the alternative pathway and the distal pathway can proceed.
Associative-alternative pathway. In the alternative pathway, both N atoms are reduced successively with alternative hydrogenation. The N
N bond remains unbroken until the last hydrogenation and two NH3 molecules are almost concurrently desorbed.
Associative-distal pathway. In the distal pathway, the distal N atom accomplishes hydrogenation to form and release an NH3 molecule before any hydrogenation of the proximal N atom.
Associative-enzymatic pathway. In this particular case, N2 is absorbed in a side-on adsorption mode and each nitrogen atom is hydrogenated enzymatically, which is similar to the alternating pathway. Notably, this pathway is favored in the presence of biological nitrogenases and some catalysts that have a strong bind with N2. The hydrazine (N2H4), formed as an intermediate of the enzymatic pathway, is mostly a minor product thanks to the high energy barrier of its desorption from the catalyst surface.12 The enzymatic routes that occur on iron-based catalyst surfaces were reported in several works.74–76
In addition to the classical NRR pathways mentioned above, novel pathways have been discovered in practice by in situ spectroscopies.77,78 Shao's group proposed a new two-step reaction pathway for NRR, based on surface-enhanced infrared-absorption spectroscopy and differential electrochemical mass spectrometry data.78 In such a pathway, the absorbed nitrogen molecule reduces to 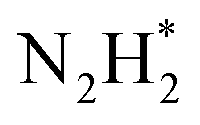 by a two-electron transfer process, followed by its desorption and decomposition to form NH3 (3N2H2 → 2NH3 + 2N2). These characterization techniques and findings provide important tools and new insights into the reaction mechanism of NRR on metal surfaces, which can inspire further understanding of the NRR mechanism and rational design of the more advanced electrocatalysts.
by a two-electron transfer process, followed by its desorption and decomposition to form NH3 (3N2H2 → 2NH3 + 2N2). These characterization techniques and findings provide important tools and new insights into the reaction mechanism of NRR on metal surfaces, which can inspire further understanding of the NRR mechanism and rational design of the more advanced electrocatalysts.
3. Iron-based electrocatalytic materials
Electrocatalysis is a surface-sensitive process, and surface and subsurface configurations directly affect catalytic performance.80,81 Various material modification strategies, such as vacancies, defects, high-index facets (HIFs), and strains have been developed to design NRR catalysts, including iron-based catalysts.44,82–86 These strategies will be introduced, respectively in the category/section to which they belong. Amongst them, defect and vacancy engineering (e.g., oxygen vacancies (OVs), sulfur vacancies (SVs), and tellurium vacancies (TeVs)) are more common and effective to generate active sites and enhance intrinsic NRR activity for the following reasons. Firstly, by creating defects and vacancies in the catalyst, the electronic structure of the electrocatalyst can be manipulated to improve conductivity and promote N2 adsorption and activation, thereby reducing the activation energy barrier.87,88 Besides, the coordination environment of the metal centers can be adjusted to produce low-coordinated and unsaturated metal sites, thus increasing the extrinsic catalytic activity of the electrocatalyst by increasing the number of active sites.893.1 Iron oxide
Metal oxides are widely used in the field of chemical catalysis studies, due to their low cost, wide source, tunable activity, and environment benignity.90 Interestingly, plenty of works about iron oxides and their derivatives for electrochemical ammonia synthesis have been reported, providing a wealth of experience for the further development and application of iron oxides as efficient NRR catalysts.91–93 Iron oxides are suggested to offer good long-term durability and cycling stability in a neutral electrochemical environment.49 Also, some works have pointed to the beneficial effects of pairing iron oxides with highly conductive catalyst substrates to accelerate electron transfer efficiency and achieve good electrochemical performance.94,95 Iron oxides have the natural defect of OVs, tailoring their electronic structures and charge distribution for increasing the quantity of active sites with a modulated energy barrier. On the one hand, surface OVs could accommodate the electron-accumulating Lewis base as well as catalytically reactive unsaturated sites to guide NRR to proceed through pathways with low energy barriers. On the other hand, theoretical studies revealed that the electrons tend to accumulate around the valence band maximum of metal oxides with OVs, and thus facilitate N2 adsorption and N2 activation by stretching the NN triple bond.96–99
Stoichiometric Fe3O4 and Fe2O3 and their derived structures have been extensively studied as iron oxides for NRR electrocatalysis. Hu and coworkers reported a Fe/Fe3O4 hybrid catalyst via thermal oxidization of Fe foil and subsequent in situ electrochemical reduction (Fig. 3B).100 X-ray photoelectron spectroscopy (XPS) (Fig. 3C) revealed the composition and chemical state changes of the surface layer in the three samples. The Fe0 on the Fe foil surface was completely oxidized and then reduced to both Fe0 and oxidic Fe components by the subsequent electrochemical reduction. Furthermore, the presence of α-Fe and Fe3O4 crystallites in the as-prepared catalyst was confirmed by X-ray diffraction (XRD) (Fig. 3D). Such Fe/Fe3O4 catalyst demonstrated significantly better NRR selectivity than several commercial iron materials (Fig. 3A), reaching an NH3 yield of 0.19 μg cm−2 h−1 and a FE of 8.29% in the phosphate buffer solution. Similarly, Xie et al. developed a one-step green synthesis of Fe–Fe3O4 catalyst via the reduction of FeSO4 in an aqueous solution using KBH4.50 High-resolution transmission electron microscope (HRTEM) image (Fig. 3E) depicted the presence of atom-vacancy defects in Fe–Fe3O4 nanocrystals, which resulted in the excellent NRR performance by enhancing the nitrogen adhesive ability. Electrochemical impedance spectroscopy (EIS) suggested that Fe–Fe3O4 had a significantly enhanced charge transfer capacity. During five cycle tests, no obvious changes in the current density curves (Fig. 3F), NH3 yield rate, and FE (Fig. 3G) were observed, indicating the excellent stability of Fe–Fe3O4 for NRR. Porous structures with high specific surface area have been built to enhance the catalytic performance by increasing active surface area. In a recent study, Ying et al. reported porous Fe3O4 nanosheets via regeneration from deep eutectic solvent (DES) and subsequent annealing (Fig. 3H).49 A series of annealing temperatures was applied, sample Fe3O4-300, which was sintered at 300 °C had the largest specific surface area (88.30 m2 g−1), exhibiting an NH3 yield rate of 12.09 μg h−1 mgcat−1 and a remarkable FE of 34.38% (Fig. 3I).
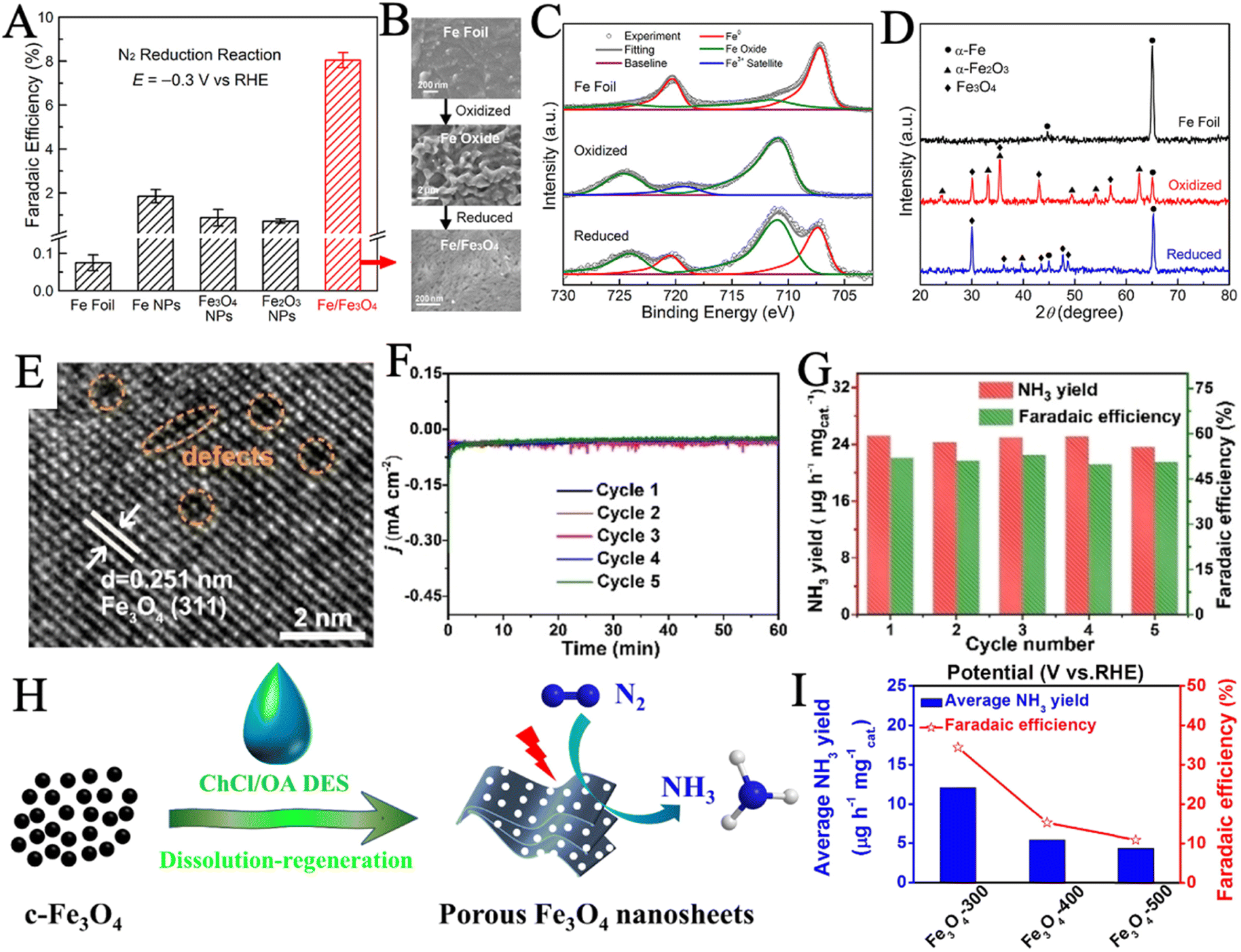 | ||
| Fig. 3 (A) FE of Fe/Fe3O4 and several commercial iron materials. (B) SEM images of the Fe foil, the 300 °C oxidized sample, and the sample after the reduction. XPS spectra of the Fe 2p peaks (C) and grazing-incidence XRD patterns (D) of the three samples. Reproduced with permission: copyright 2018, American Chemical Society.100 (E) HRTEM images of Fe–Fe3O4 catalyst. Chronoamperometry curves (F), NH3 production rate and FE (G) of Fe–Fe3O4 catalyst during the 5 cycles tests. Reproduced with permission: copyright 2022, Wiley-VCH.50 (H) Synthetic route schematic of porous Fe3O4 nanosheets. (I) Average NH3 production rate and FEs (at −0.1 V) of samples were annealed at different temperatures. Reproduced with permission: copyright 2021, Elsevier.49 | ||
Except for the widely studied aqueous electrolyte systems, advances of iron-based catalysts have also been made in non-aqueous electrolyte systems to address some inherent difficulties.18,101,102 Ammonia electro-synthesis in aqueous systems suffer from low N2 solubility and competitive HER.103 Typically, the reduction of water to hydrogen occurs in the same region of potential with NRR, resulting in low FEs.104 Calculations and experiments showed that nitrogen is much more soluble in some ionic liquids (ILs) than in water.105,106 The improved N2 solubility in IL electrolytes is an effective way to increase N2 adsorption at the active sites.11 Besides, ILs are hydrophobic and can serve as excellent non-aqueous substances because they can lower the water content at an optimal level to effectively restrain the evolution of H2.104 MacFarlane and coworkers practically explored the NRR performance of Fe deposited working electrode substrates (including fluorine-doped tin oxide glass, nickel foam, and stainless steel cloth) in a hydrophobic, high N2-solubility IL electrolyte ([C4mpyr][eFAP] and [P6,6,6,14][eFAP], respectively).104 The strong interaction between N2 and [eFAP]− was proposed by DFT calculations, which explained the high solubility of N2. This work also suggests the influence of the viscosity of ILs on electrochemical performance, that lower viscosity can support higher mass transport to produce higher current densities and yields. Although this NRR system achieved an incredibly high FE of 60%, the reduction currents obtained on low surface area substrates are too small and the yield needs to be improved for practical applications. In another work, MacFarlane's group further focused on the rational electrode–electrolyte design and demonstrated the effectiveness of using highly fluorinated solvents to realize efficient electrochemical ammonia synthesis.107 The mixture of 1H,1H,5H-octafluoropentyl-1,1,2,2-tetrafluoroethyl ether and [C4mpyr][eFAP] were chosen and characterized to determine the optimum solvent–IL ratio to achieve a maximum conductivity of 1.95 mS cm−1 and electrochemical window of 3.40 V, which was ideal for NRR. The core–shell α-Fe@Fe3O4 catalyst was applied in such an IL mixture and accomplished an ammonia yield rate of ∼2.35 × 10−11 mol s−1 cmECSA−2 and NRR selectivity of ∼32%.
Thus far, most reported studies of iron oxide catalysts also focused on Fe2O3.100 High-index facets are usually represented by a set of Miller indices {hkl} with at least one index greater than 1. These facets have a high density of exposed low-coordination atoms and are therefore more open and active than the tightly packed {111} and {100} low-index facets.108 The energy barrier of HER can be elevated at some specific HIFs, while a more feasible NRR pathway is opened to improve NRR selectivity.109 Xie et al. combined the facet modulation and nanoarray structure design to improve the NRR performance of α-Fe2O3.110 The α-Fe2O3 nanorod arrays (NAs) with exposed (104) HIF offered abundant unsaturated Fe atomic dangling bonds as the NRR active sites. Moreover, the nanoarray structure achieved an enhanced electrochemical surface area (ECSA) to fully display the active sites and maintain sufficient mass diffusion. Thus, the selectively exposed (104) surface coupled with the high ECSA of the nanoarray structure achieves the superior electrocatalytic performance of α-Fe2O3 NAs. Unfortunately, current studies on HIFs to optimize the electrocatalytic selectivity and activity are popular in noble metal-based materials and their alloys, such as Pt and Au.87,111,112 The lattice facets control engineering on iron materials still needs to be explored.
Given the fact that adjusting the OV concentration is an effective method for improving the oxide catalyst properties for NRR, various strategies have been applied for this target, such as annealing and heteroatom doping. High-temperature annealing is a feasible way to increase the concentration of OVs. Cui et al. reported an o-Fe2O3–Ar/CNT catalyst that was annealed under argon (Ar) utilizing commercial hematite nanopowder as raw material.44 It was found that the NRR performance of o-Fe2O3–Ar with the highest OV concentration (12.26%) was better than that of o-Fe2O3-air and Fe2O3-untreated. After 16 hours of the chronoamperometry test, the NH3 production rate and FE decreased significantly, suggesting that the stability of the structure needs to be improved. However, high-temperature annealing is an energy-intensive process, which is typically carried out in a reducing atmosphere. Thus, the introduction of OVs by doping with low valence metals is highly desirable to be developed as a relatively environmentally friendly synthesis method. Yu et al. developed zinc-doped Fe2O3 nanoparticles via a wet chemical route at only 160 °C.113 Zinc has a similar ion radius to that of Fe3+, which is able to induce OVs in Fe2O3 nanoparticles. In neutral media, it offers a maximum FE of 10.4% and an NH3 formation rate of 15.1 μg h−1 mgcat−1 at −0.5 V vs. RHE. Furthermore, Zhang et al. were the first to propose a task-specific ionic liquid strategy for OV-rich α-Fe2O3 nanocubes (denoted as Fe2O3-IL), achieving an NH3 production rate of 32.13 μg h−1 mgcat−1 with a FE of 6.63% at 0.3 V vs. RHE.114 The ionic liquid n-octylammonium formate (OAF) simultaneously worked as the reaction medium and structure-directing template for growing α-Fe2O3 nanocubes while the reductant formic anion of ionic liquid drove rich OVs in situ in α-Fe2O3. This innovative approach allows the OV concentration to be adjusted under mild conditions. Additionally, the low surface tensions of OAF compared with water resulted in a higher nucleation rate of Fe2O3, than the growth rate. Consequently, Fe2O3-IL had a much smaller particle size of 23.2 ± 1.8 nm than that of Fe2O3 synthesized in a water medium. As a result, a larger specific surface area and more active sites were delivered in the catalytic process.
Most of the catalysts we mentioned above were prepared with the additive of surfactants through solution methods. However, the surfactants had a passivation effect on the catalyst surface, which decreased the activity of the catalyst. Therefore, developing a strategy to construct a clean surface could be critical for achieving high activity.115 Herein, Wang et al. reported porous Fe2O3 nanorods grown on carbon cloth (p-Fe2O3/CC).116 The EDX element mapping image suggested that Fe and O elements with a ratio of about 2/3 are uniformly distributed throughout the nanorods. With the assistance of Na2SO4 as a structure-directing agent, the self-supported structure of p-Fe2O3/CC with a large surface area provided abundant active sites and accessible channels for promoting mass/charge transfer during the NRR process. Furthermore, Wang et al. successfully synthesized clean-surface Fe2O3 nanoparticles without any surfactants or structure-directing agents.115 Fe(NO3)3 was dispersed in glycerine for prior hydrothermal treatment, followed by high-temperature calcination. This clean-surface catalyst exhibited better performance with a high yield of NH3 22 μg h−1 mgcat−1 and a FE of 3.5% at −0.5 V vs. RHE. The as-synthesized catalyst showed good catalytic and structural stability even without a binder.
The conventional preparation of iron oxide materials mostly requires high-temperature annealing, which is not convenient and highly energy intensive. Preparation strategies at room temperature need more exploration. In addition, iron oxide suffers from poor electrical conductivity, which is against the high catalytic efficiency. Good electron donors can be introduced to modulate the electronic structure of the catalyst. Many of the current iron oxide electrocatalysts for NRR struggle with low ammonia yields or FEs. Further theoretical studies are recommended to explore their catalytic intrinsic principles and thus guide the catalyst improvement.
3.2 Iron chalcogenides
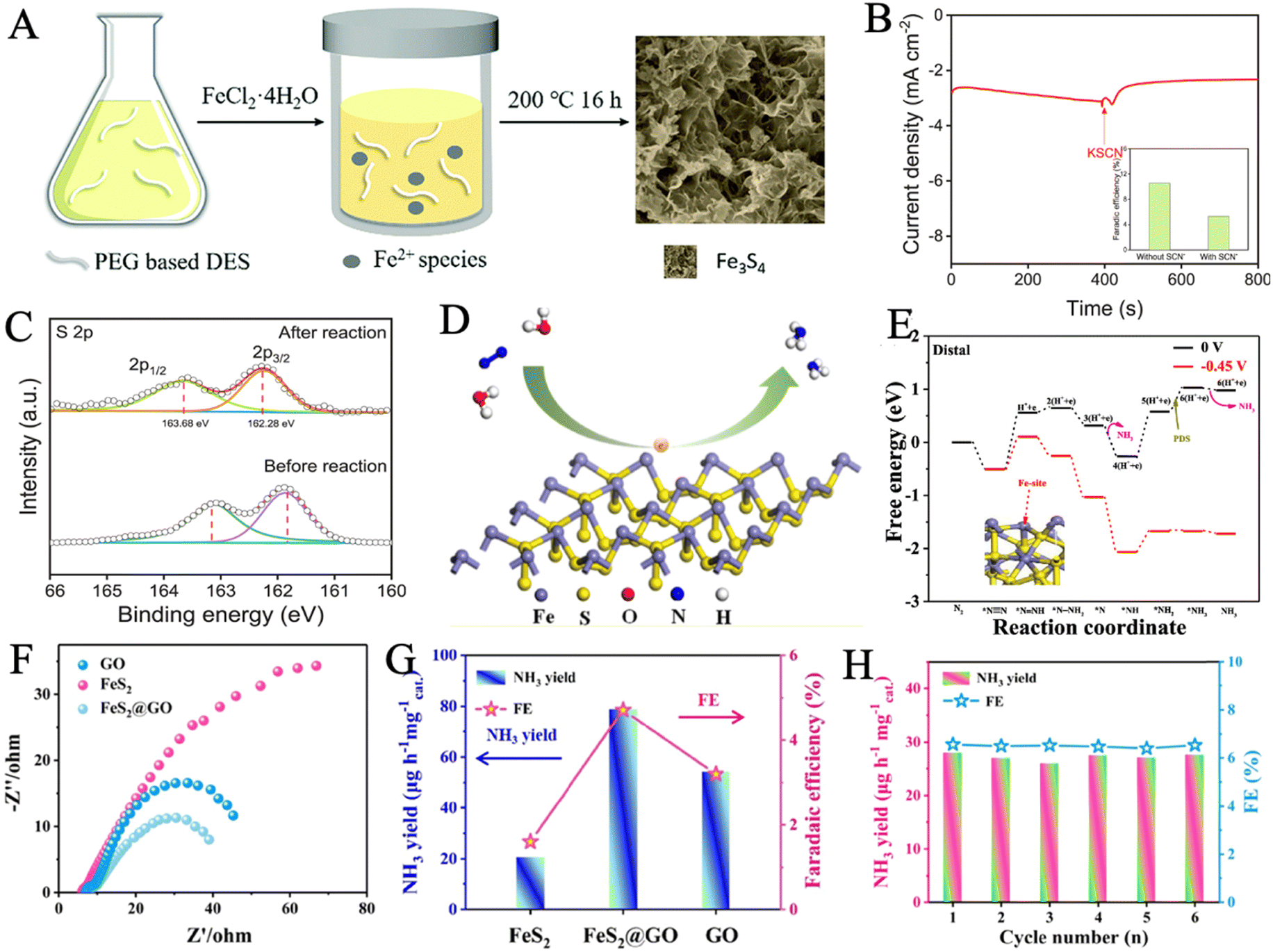 | ||
| Fig. 4 (A) Schematic of the formation of Fe3S4 in the PEG based DES. Reproduced with permission: copyright 2018, Royal Society of Chemistry123 (B) chronoamperometry curve of Vs-FePS3 in N2-saturated electrolyte with the addition of SCN− ion. Inset: FEs with and without the SCN− ion. (C) XPS spectra of S 2p of Vs-FePS3 before and after NRR. Reproduced with permission: copyright 2021, Elsevier.52 (D) Schematic diagram of coordination configuration in FeS2. (E) Free energy profile for NRR in Fe atoms as the active sites. Reproduced with permission: copyright 2020, American Chemical Society.126 (F) Nyquist plots and (G) ammonia yield and FE of FeS2@GO, GO and FeS2 at −0.3 V in N2-saturated 0.1 M HCl solution. (H) Six-time recycling test of FeS2@GO. Reproduced with permission: copyright 2021, American Chemical Society.128 | ||
Inducing surface defects is an available strategy to increase the active sites of NRR via heteroatom doping. In a recent work, a surface-defective FeS2 catalyst was obtained by sulfurization of stainless-steel foil at 500 °C.125 The fitting of the XPS spectrum sulfur S 2p peaks and the HRTEM image proved the existence of abundant SVs. Meanwhile, such defects were considered to be the active sites for the stable and efficient adsorption of dinitrogen molecules. It was strongly advised that the generation of an active hydrogen atom was primarily caused by the exposed (210) facet of iron pyrite. The stainless-steel-based catalyst possessed a considerably higher FE of 14.6% in contrast to the pure iron-based catalyst in 0.1 M Li2SO4 at −0.2 V. Because of the existence of Cr species, the SS-based electrode with anti-corrosion properties presented a superior catalytic stability under acidic conditions. Hou et al. reported a porous exfoliated FePS3 nanosheet with rich SVs (Vs-FePS3 NSs), which was formed through electrochemical exfoliation and hydrogenation.52 Vs-FePS3 NSs showed significant activity in the electrocatalytic NRR with a high NH3 yield rate of 3.88 μg h−1 cm−2 at −0.25 V and a FE of 12.36% at −0.20 V versus RHE due to their rich SV and unique 2D morphology. To identify the actual active sites of Vs-FePS3 NSs for NRR, SCN− ion was used to act as a poisoning reagent for metal-centered active sites. A significant decrease appeared in the current density and FE (from 12.36% to 5.31%), indicating that the active Fe site was effectively shielded (Fig. 4B). The shift of S 2p main peaks to higher binding energies in high-resolution XPS spectra suggested that the SVs boosted the hydrogenation of N2 (Fig. 4C).
The suitable ligand configuration of the active sites plays a significant role in enhancing the catalytic performance. It is found that a metal sulfur cluster with four permanent ligands in a twisted tetrahedral ligation shell is an effective active center of NRR. This coordination structure is well demonstrated in FeS2, where the NRR process proceeds preferably on Fe atoms (Fig. 4D). In a recent study, a FeS2 NRR catalyst was synthesized via the one-step hydrothermal method.126 The catalyst exhibited an outstanding NRR performance, achieving a high NH3 yield rate of 37.2 μg h−1 mgcat−1 and a FE of 11.2% at −0.5 V versus RHE. The current density and original structure of FeS2 had no significant change after 12 h of electrolysis, indicating excellent durability and stability. Fe atoms were proven to be the active sites instead of S atoms by poisoning experiments and free energy profile for NRR in Fe sites and S sites. The reaction mechanism was investigated by DFT calculations that showed that the associative distal pathway was preferred and the conversion from *NH2 to *NH3 was regarded as the potential-determining step (PDS) (Fig. 4E).
The heterostructure of iron sulfide-based catalysts has also been developed to enhance their catalytic performance. The interface within heterostructures can usually regulate the electronic structure and expose extra active sites simultaneously. Chen and coworkers designed a hybrid catalyst of crystalline Fe2O3 with amorphous FeS (Fe2O3/FeS) via a DES-regulated fabrication, followed by an annealing process.127 The hybrid catalyst exhibited abundant channels and a high specific surface area of up to 103.94 m2 g−1, providing more exposed electrochemical active sites for N2 fixation and accelerating mass convection. Interface engineering between crystalline Fe2O3 and amorphous FeS optimized electron distribution and surface structure, being conducive to the absorption and activation of the N2 molecules. In another work, Gao et al. created a graphene oxide-wrapped pyrite nanoparticle (FeS2@GO) via Fe(NO)3, NH3·H2O, and graphene oxide as a precursor.128 The coupling effect between the catalyst and the substrate accelerated the electron transport property, FeS2@GO exhibited the smallest charge transfer resistance, highest NH3 production, and FE, much better than those of pure FeS2 and GO (Fig. 4F and G). Meanwhile, the NH3 yield rate and FE remained almost constant during the six-time recycling tests (Fig. 4H). Importantly, the low-coordinated Fe atoms are activated as highly active sites, where the free energy change ΔG (*N2) of N2 binding was reduced to −0.57 V, leading to stronger binding of N2 and longer NN bond of absorbed *N2 with respect to pristine FeS2 (100).
Further progress in iron sulfide can be made by modulating the electronic structures of electrocatalysts by doping, tailoring surface vacancies, defect engineering, and/or heterointerface engineering. Meanwhile, refining and creating more active sites are also proposed to optimize the adsorption and activation of the nitrogen species.
N triple bond to 1.136 Å (Fig. 5B). According to the Gibbs free energy diagrams, the mixed pathway is the most favored reaction route. In addition, the FeTe2 effectively suppressed HER, via a highly positive adsorption-free energy of *H of 0.71 eV compared to the negative ΔG*N2 of −0.27 eV (Fig. 5C). 2D ferromagnetic materials with large spin moments are newly discovered as high-performance NRR electrocatalysts. In theory, the Fe3GeTe2 monolayer was explored as an NRR electrocatalyst through DFT calculations.130 The highly centralized spin-polarization on the exposed Fe4 center in defective Fe3GeTe2 with TeVs contributed to strong 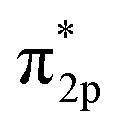 back-donation and effective activation of the NN triple bond (Fig. 5D). Importantly, an enzymatic pathway is preferred, and all six hydrogenation steps are exothermic and thus spontaneous, during the NRR process (N2 + 6H+ + 6e− = 2NH3) (Fig. 5E). HER can also be suppressed by the rapid termination of N2 molecules on TeVs.
back-donation and effective activation of the NN triple bond (Fig. 5D). Importantly, an enzymatic pathway is preferred, and all six hydrogenation steps are exothermic and thus spontaneous, during the NRR process (N2 + 6H+ + 6e− = 2NH3) (Fig. 5E). HER can also be suppressed by the rapid termination of N2 molecules on TeVs.
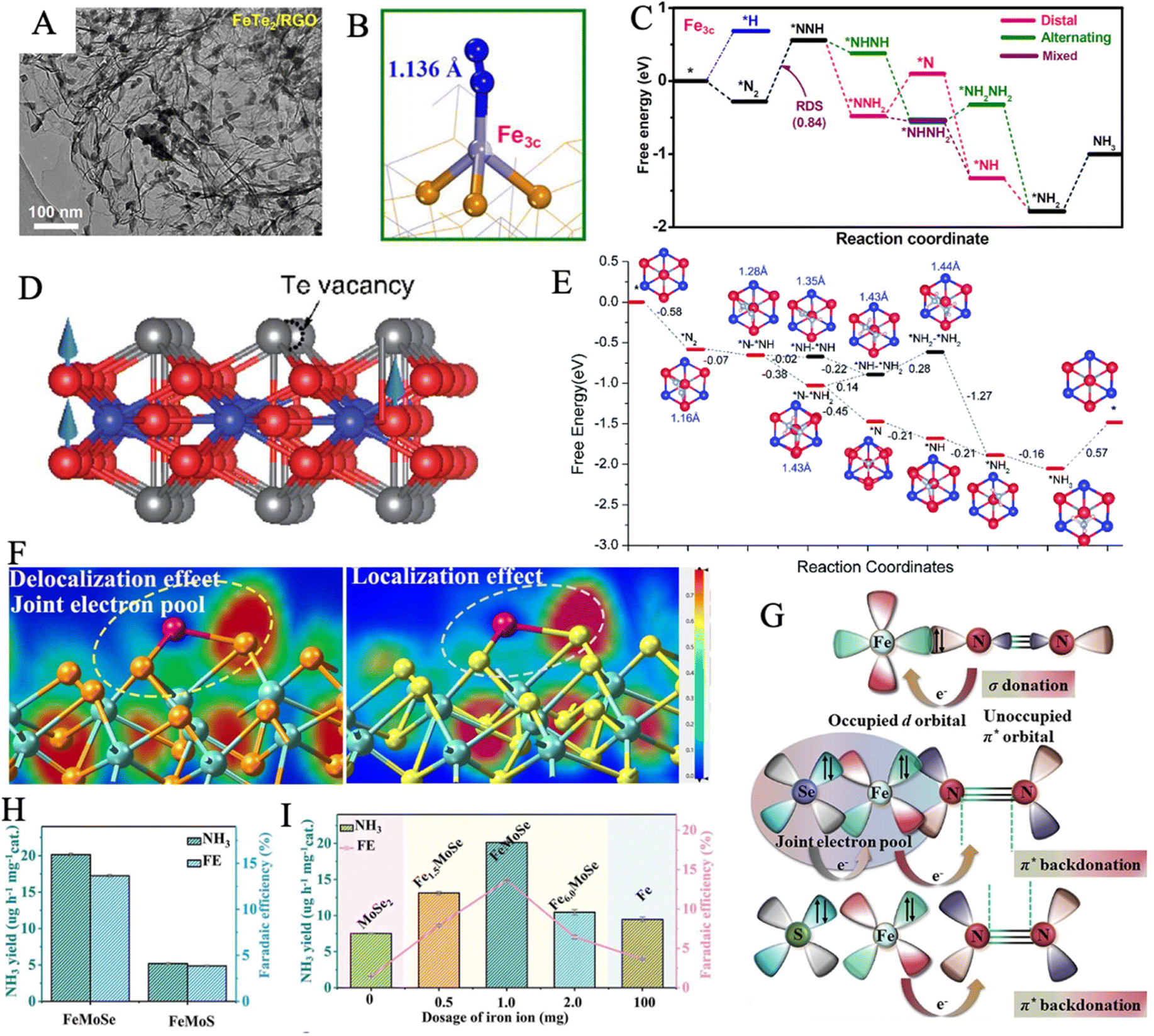 | ||
| Fig. 5 (A) TEM image of FeTe2/RGO. (B) Optimized structures of *N2 over Fe3c sites. (C) Free energy profiles of three NRR pathways on Fe3c site at U = 0. Reproduced with permission: copyright 2021, Elsevier.129 (D) Side views of the defective Fe3GeTe2 monolayer. (color code: red, Fe; bule, Ge; gray, Te) The blue arrows stand for local magnetic moments of Fe atoms up aligned. (E) Possible reaction pathways for NRR. Reproduced with permission: copyright 2021, Royal Society of Chemistry.130 (F) Electron location function of FeMoSe (left), FeMoS (right), in which the Fe, Mo, Se and S atoms are denoted by red, green, yellow and cyan balls, respectively. (G) Schematic illustration for N2 binding to FeMoSe and FeMoS. NH3 yields and FEs of FeMoSe, FeMoS (H) and other comparative samples (I).132 | ||
2D layered double hydroxide (LDH) with evenly distributed ferric ions exhibited N2 fixation activity, but the intrinsic resistivity and poor charge transfer properties astricted its further application. A NiFe-LDH-derived NiFe-selenide (Ni0.75Fe0.25Se2) was designed to solve this weakness, the introduction of selenium conspicuously accelerated the p–d coupled electron transfer during nitrogen reduction.131 Through theoretical analysis, the selenization of iron caused lattice distortion, which may arise from the uneven occupation of t42g and e2g orbitals in Fe2+. More N2 molecule adsorption sites were introduced by lattice distortion regulation. A bio-inspired joint electron pool was realized in FeMo(Se, Te) by replacing sulfur with selenium or tellurium.132 Notably, Se and Te had a lower electron-negativity as compared with S, allowing a more expansive electron cloud to form the joint electron pool, which originated from the delocalized electronic structure (Fig. 5F). The density of state (DOS) profiles of the top valence band near the Fermi level in FeMoSe is mainly composed of Fe d, Mo d, and Se p valence orbitals, illustrating a higher electron density and charge transport ability than those of FeMoS and MoSe2. Theoretical models for the joint electron pool displayed stronger nitrogen adsorption, which is critical for the subsequent hydrogenation steps. As these, FeMoSe had a greater ability to inject electrons into N2 (Fig. 5G), exhibiting an NH3 yield rate of 20.14 μg h−1 mgcat−1 and a FE of 13.65%, which were far better than those of FeMoS and other comparative samples (Fig. 5H and I).
Briefly, the novel metal chalcogenides may be promising electrocatalysts with high efficiency for ammonia synthesis from gaseous dinitrogen. There is a large space for scientists to apply multi-element doping and heterostructure engineering similar to that used in iron sulfides to further promote NRR electrocatalysis. More in-depth research about iron selenide/iron telluride is expected to promote this electrocatalytic NRR field.
3.3 Bimetallic Fe-containing catalysts
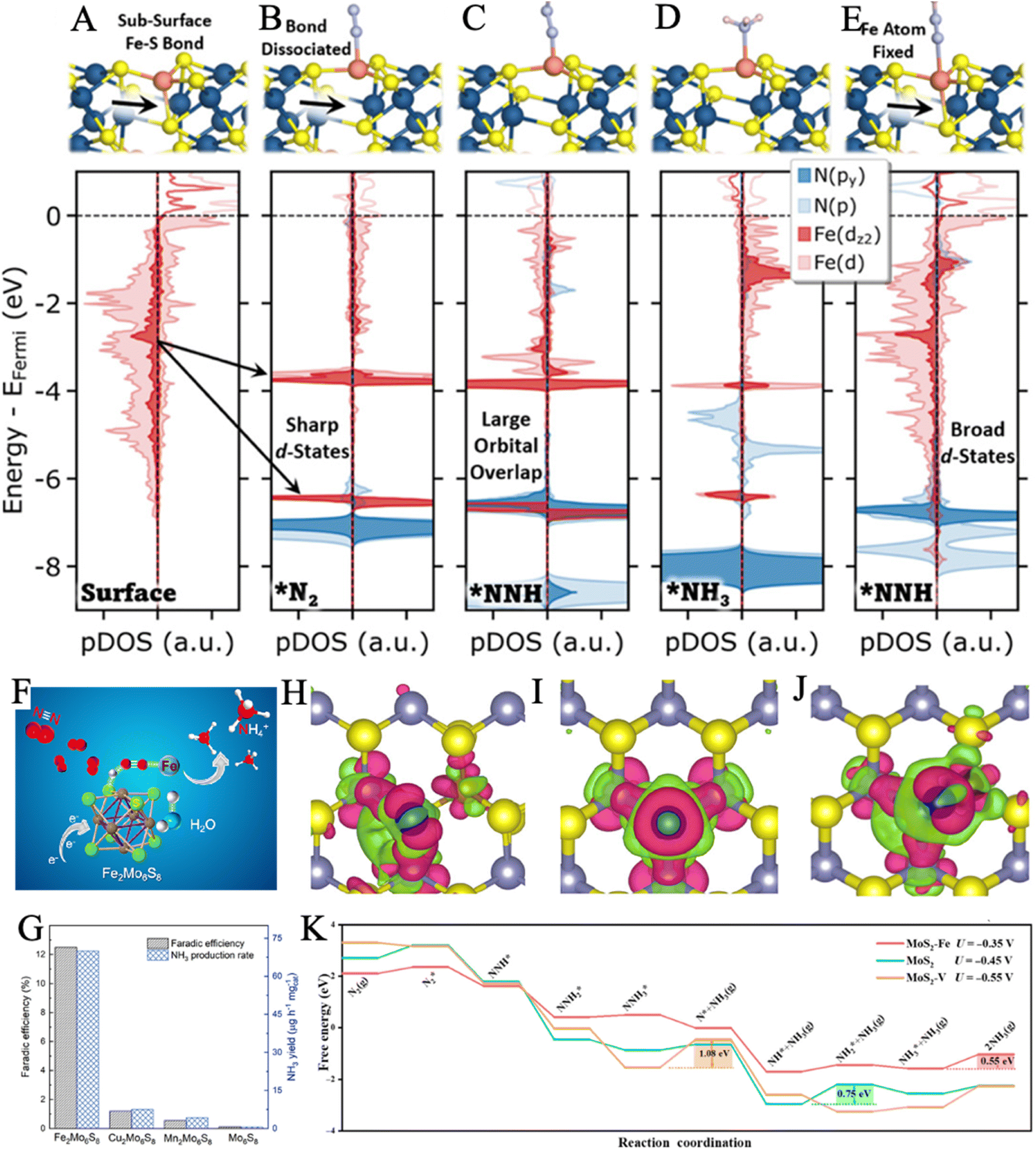 | ||
| Fig. 6 Structure and projected density of states (pDOS) for the clean Fe2Mo6S8 surface (A), the Fe2Mo6S8 surface with *N2 (B), *NNH (C), *NH3 (D), and *NNH (E) with the Fe surface atom fixed at its position on the clean surface. Reproduced with permission: copyright 2022, American Chemical Society.136 (F) Illustration of the proposed binding patterns for N2 absorption and subsequential conversion to NH3. (G) Comparison of FE and NH3 yield for Mo6S8 and M2Mo6S8 (M = Fe, Mn, and Cu) electrocatalysts. Reproduced with permission: copyright 2021, American Chemical Society.137 (H–J) Charge density difference of MoS2–Fe, MoS2, MoS2–V (from left to right). Electron excess and deficiency are denoted by red and green isosurfaces, respectively. Isosurface was set as 0.003 Å−3. (K) Free-energy profiles of the NRR of MoS2–Fe, MoS2, and MoS2–V. Reproduced with permission: copyright 2022, reproduced with permission: copyright 2021, Springer.140 | ||
3.3.1.1. Heteroatom doping. Single Fe-based or Mo-based catalysts can be mutually doped to achieve better N2 fixation performance by modifying their intrinsic nature. MoS2 was recently indicated to be a potential material for NRR, as its sufficient Mo (at IV valence states) active sites are able to accept the electrons from N2.6,56,79 Nevertheless, its further application is limited by the strong adsorption capacity of Mo to N2, which impedes the desorption of it and release of NH3, leading to strong HER competition.138,139 The doping of Fe was proved to efficaciously modulate the electron density state of the catalyst. The roles of Fe doping for N2 adsorption, N2 activation, and NH3 desorption were further revealed by Sun and coworkers, through a combination of experiments and theoretical calculations.140 The Fe-doped MoS2 electrocatalyst demonstrated an NH3 yield of 20.11 μg h−1 mgcat−1 at −0.35 V vs. RHE, which was significantly higher than that of its MoS2 and MoS2–V counterparts. When N2 molecules were adsorbed on the catalyst, the charge of MoS2–V was localized between V and N2 molecules, leading to the strong adsorption ability of N2, while the electrons of MoS2–Fe were more concentrated on Mo, which may favor the ammonia release (Fig. 6H–J). According to the free energy profiles, MoS2–Fe had the lowest rate-determining step (RDS) energy barrier, indicating a more thermodynamically favorable pathway on MoS2–Fe (Fig. 6K). Their work even provided enlightenment on how to achieve the optimal balance between the three key stages (nitrogen adsorption, hydrogenation, and ammonia desorption) of NRR for superior electrocatalysis by introducing proper doping materials to tune the surface electronic structure.
Progress has also been made in bionic design from FeMoS-nitrogenases.141 In recent work, Du and coworkers designed a Mo(IV)-doped FeS2 (FeS2–Mo) via the hydrothermal reaction of MoCl5, Fe(NO3)2, and thioacetamide.56 Energy Dispersive Spectrometer (EDS) mapping indicated that Mo, Fe, and S elements were distributed uniformly throughout the nanosheets. Electrochemical measurements suggested that pure FeS2 presented HER negative and a relatively low yield rate for NH3. However, the NRR activity was significantly promoted after Mo doping and increased gradually with the Mo content, proving that Mo could act as active sites. The Mo-doped-modified catalyst exhibited a high FE of 14.41% and an NH3 yield rate of 26.15 μg h−1 mgcat−1. Afterwards, the optimal pathways for NRR in FeS2 and FeS2–Mo were proposed by DFT calculations. Moreover, the doping of Mo opened up an associative distal pathway in FeS2–Mo, instead of the associative alternating pathway in FeS2. The reaction energy barrier of the critical dinitrogen hydrogenation step (NN* + H → NNH*) was reduced from 1.24 eV to 0.62 eV. Therefore, the FeS2 substrate was regarded to suppress the competitive HER, while the doping of Mo favored the adsorption and activation of nitrogen. Similarly, Su et al. achieved the suppression of HER by designing atomically dispersed Fe-decorated MoS2 (Fe–MoS2).142 The edge S atoms in MoS2 were generally considered active sites for HER, while the Mo atoms acted as active centers for NRR.135 Therefore, better selectivity for NRR could be accomplished by Fe modification of the S sites without shielding the Mo sites to achieve high FE (18.8% at −0.3 V versus RHE). DFT calculations proved that the energy barriers of HER in Fe–MoS2 (0.15 eV) were obviously magnified with respect to pure MoS2 (0.03 eV). It is worth mentioning that our group predicted and achieved Fe–O–Mo subunits for highly active NRR in Mo-doped Fe2O3 (Fig. 7A).143 The introduction of Mo destroyed the ordered crystal structure and enriched lattice defects, creating more active sites. It also tuned the electronic structures of Fe2O3 to enhance the electron injection into N2, resulting in an elongated and weakened nitrogen bond (Fig. 7C and D). The as-synthesized material showed much higher ammonia yields (21.3 μg h−1 mgcat−1) and FE (11.2%) than pure Fe2O3, proving the significant role of Mo doping (Fig. 7B). An optimized reaction pathway with a lower energy barrier was confirmed on Mo-doped Fe2O3(110) in comparison with pristine α-Fe2O3(110) (Fig. 7E). Unfortunately, this work could not achieve the doping amount control. It needs further effort to reveal the phase transition mechanism of the catalytic material during the long-term electrocatalytic process.
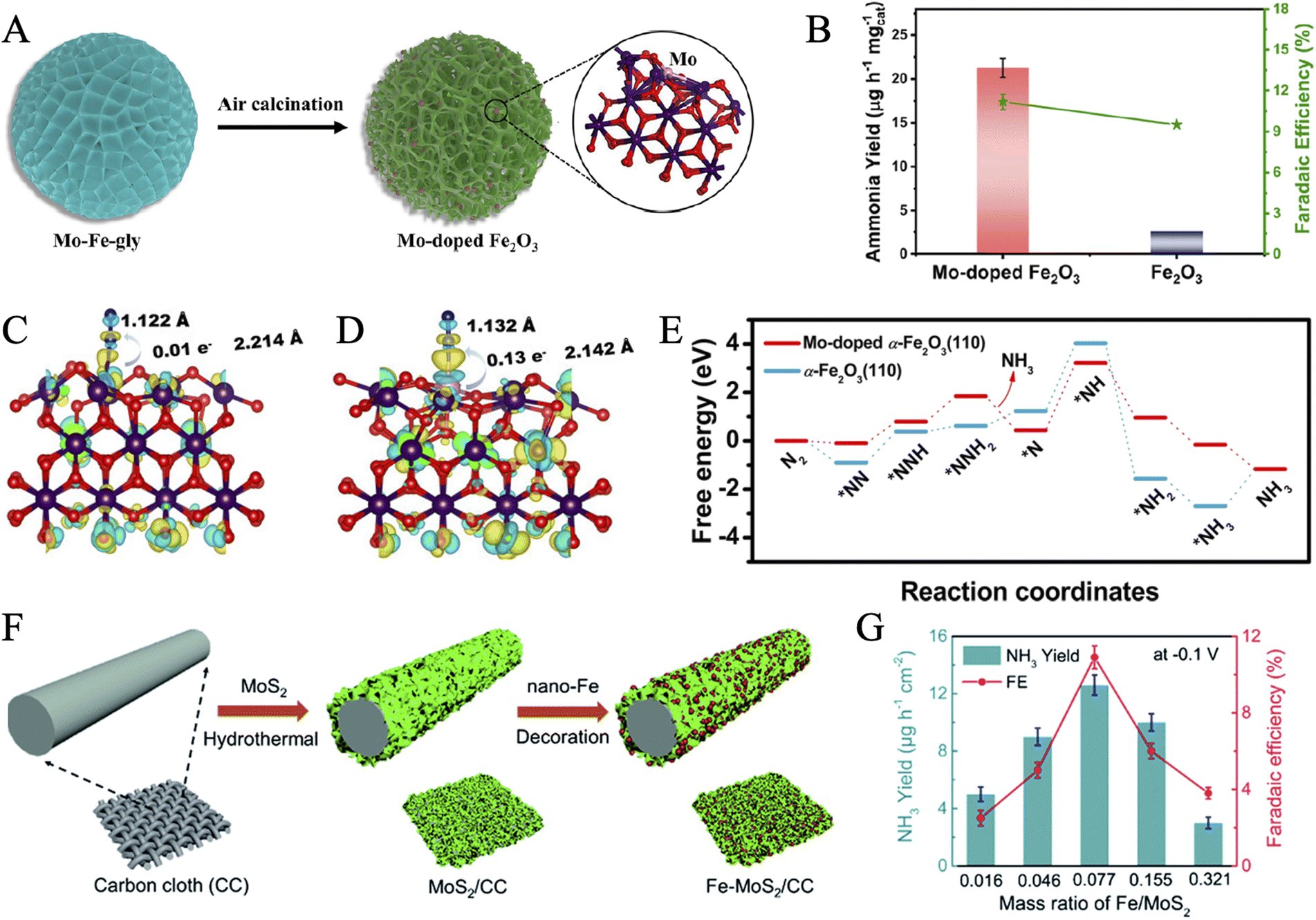 | ||
| Fig. 7 (A) Diagrammatic representation of the formation of Mo-doped Fe2O3 porous nanospheres. (B) Comparison of NH3 production rates and FEs at −0.6 V for Mo-doped Fe2O3 and Fe2O3. The charge density difference of N2-adsorbed α-Fe2O3(110) (C) and Mo-doped α-Fe2O3(110) (D) with an isosurface value of 0.006 e Å−3. (E) Free energy diagram for the NRR process on α-Fe2O3(110) and Mo-doped α-Fe2O3(110), respectively. Reproduced with permission: copyright 2022, American Chemical Society.143 (F) Schematic illustration for the formation routes of Fe–MoS2/CC. (G) NH3 yield and FE with different mass ratios of Fe–MoS2. Loading mass: 2.15 mg cm−2. Reproduced with permission: copyright 2019, Royal Society of Chemistry.152 | ||
Doping heteroatoms is effective to promote the electrocatalytic activity of basic materials. Further endeavors should be devoted to disclosing the position of the doping exogenous atom, the coordination and valent state, and the corresponding function during the whole catalytic process.
3.3.1.2. Interface engineering. Interface engineering also provides an effective scheme for conjugate iron and molybdenum compounds. A single material may have too strong or too weak adsorption capacity for nitrogen species, while the hybrid structure can effectively modulate the energy barrier in the reaction stages.144,145 Abundant active sites and high specific surface area can also be obtained by rational interface engineering.146,147 Moreover, two or more components could induce electronic coupling and generate a fast electron transport channel to speed up charge transfer.148–151
A FeS2 and MoO2 nanocomposite anchored on a three-dimensional (3D) graphene aerogel (MoO2/FeS2/GA) was developed by Liu and coworkers.146 FeS2 and MoO2 nanoparticles were embedded as active sites to absorb and activate N2 in an interconnected 3D graphene-based mesoporous aerogel, which provided good conductivity and large accessible specific surface areas. Synergistic effects of such a ternary microstructure exhibited superior electrochemical performance over either single structure, endowed the hybrid catalyst with an impressively high FE of 37.44% and NH3 yield of 40.18 μg h−1 mgcat−1 at −0.25 V in 0.1 M HCl. Zhao et al. reported Fe nanodot-decorated MoS2 nanosheets on carbon cloth (Fe–MoS2/CC) via a chemical reduction strategy (Fig. 7F).152 The strong electronic interaction of Fe and MoS2 was confirmed by the shift of XPS binding energies of Mo and S and the red shifts in Raman spectra. Furthermore, the effect of Fe loading on the NRR performance was also explored via electrochemical measurements. The best NH3 yield reached 12.5 μg h−1 cm−2 with a FE of 10.8% at a Fe/MoS2 mass ratio of 0.077 (Fig. 7G). When the Fe content got too large, the recession in NRR performance was speculated to be attributed to the degraded structures. In another work, FeS-dotted MoS2 nanosheets covered carbon fiber cloth (CFC) (FeS@MoS2/CFC) was prepared through a facile single-step hydrothermal preparation.54 FeS and MoS2 uniformly supported the highly conductive CFC and successfully simulated the composition of the active site in FeMoS-cofactor. Notably, it exhibited considerable NRR catalytic activity over a wide pH range.
3.3.1.3. MOF precursors design. Metal–organic frameworks (MOFs) are widely used to design composite electrocatalysts due to their porous structure with high specific surface area for accommodating active sites and facilitating charge/mass transfer.153–158 Moreover, the desirable nanostructure of the catalysts could be controlled by the morphological structure inheritance of MOFs during thermal annealing processes. The organic ligands in MOFs can be pyrolyzed into porous carbons to improve the conductivity and stability of the derivatives and serve as a support for the active metal sites.159–162 A handful of excellent reviews have focused on the derived nanomaterials from typical MOFs precursors, including zeolite imidazolate frameworks (ZIFs), Prussian blue analogs (PBAs), as electrocatalysts in energy-related fields, such as hydrogen evolution reaction, oxygen evolution reaction, and oxygen reduction reaction.157,163–169 These will further provide guidance for advanced materials exploration for NRR electrocatalysis.
Chen et al. reported a one-step pyrolysis–phosphating process from MoFe-MOF precursors to fabricate phosphorus-doped carbon microspheres (MoFe-PC), as a cost-effective and highly active catalyst for N2 fixation under ambient conditions.170 Electrochemical measurements confirmed that P-doped MoFe-PC (34.23 μg h−1 mgcat−1, FE: 16.83%) showed more excellent activity than MoFe–C (24.73 μg h−1 mgcat−1, FE: 12.47%). On the one hand, the introduction of P to form a P–C connection increased the conductivity of the hybrids by optimizing the electronic structure. On the other hand, the synergistic interaction between bimetallic oxides and P-doped carbon ensured abundant active sites for the adsorption and activation of nitrogen. Chen and coworkers applied a nanostructure tailoring strategy to successfully synthesize unique hollow NiαFe1−α–MoS2 nanocubes through vulcanization treatment of Ni–Fe PBA by ammonium thiomolybdate.171 The as-prepared material showed high stability during the two-hour NRR process at various applied potentials (Fig. 8A). The charge redistribution of the catalyst in an optimal atomic ratio of the NiFe (α = 0.3) as compared to the pure MoS2 was presented by the calculated electron density difference (EDD) mappings (Fig. 8B–C). Based on the electron transfer pathways of NiαFe1−α–MoS2 catalysts, the spin-polarized density of states, and the corresponding partial density of states, it was suggested that the coupling of S 2p and Mo 3d orbitals generated the Ni0.3Fe0.7 active sites, which could easily donate electrons to the absorbed N2 molecules. Furthermore, a possible pathway of the NRR on the NiFe–MoS2 NCs catalyst was proposed by electrochemical-FTIR spectroscopy analysis and DFT calculations (Fig. 8D and E). Another Fe-containing MOF precursor, famous as MIL, has also been demonstrated to synthesize Fe2O3@MoS2 composites for NRR electrocatalysis and achieved a remarkable NH3 yield rate of 112.15 μg h−1 mgcat−1 and a FE of 8.62%.150
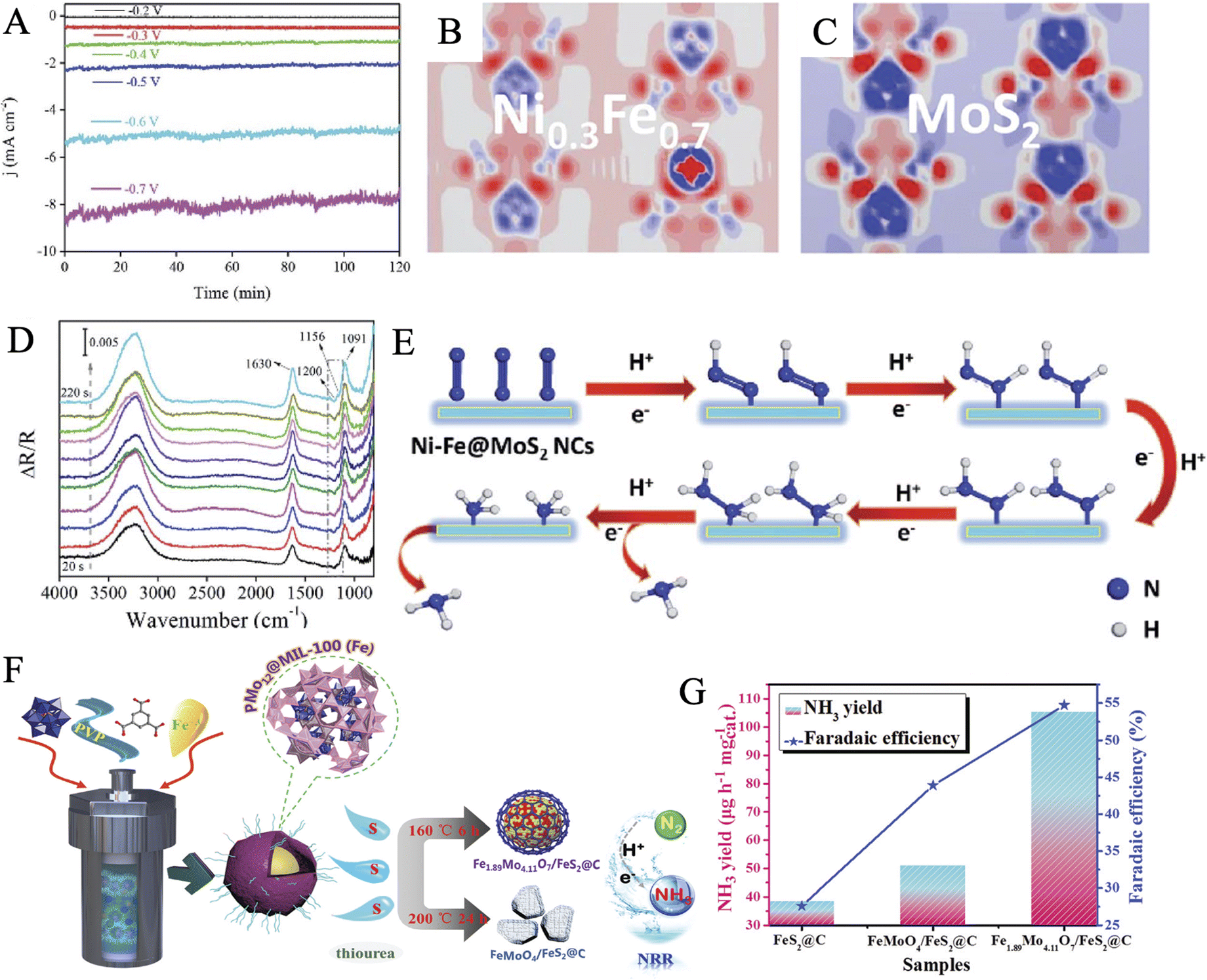 | ||
| Fig. 8 (A) Time-dependent current density (j) of Ni–Fe–MoS2 at various potentials. Electron density difference of the optimized structures of MoS2 (B) and Ni0.3–Fe0.7@MoS2 (C) (electron excess and deficiency are denoted by red and blue, respectively). (D) In situ electrochemical-FTIR spectra of the NRR process on the Ni–Fe–MoS2 NCs catalyst. (E) Proposed electrocatalytic mechanism on Ni–Fe@MoS2. Reproduced with permission: copyright 2020, Royal Society of Chemistry.171 (F) Schematic diagram of the preparation routes of Fe1.89Mo4.11O7/FeS2@C and FeMoO4/FeS2@C composites. (G) Comparison of NH3 production rate and FE of FeS2@C, FeMoO4@FeS2@C, and Fe1.89Mo4.11O7@FeS2@C (1.6 wt%) at the optimum voltage, respectively. Reproduced with permission: copyright 2020, Royal Society of Chemistry.55 | ||
Polyoxometalate-based metal–organic frameworks (POMOFs) combine the advantages of both MOFs and POMs to provide specific target structures derived from organic ligands and serve as a source of metals such as Co, Fe, Ni, Mo.169,172,173 Lately, Ma et al. made an exploration using PMo12@MOF-100(Fe)@PVP as a precursor to synthesize Fe1.89Mo4.11O7/FeS2@C for efficient electrocatalysis of nitrogen reduction (Fig. 8F).55 Experiments and characterization analysis concluded that the ratio of Fe and Mo could be controlled by accurately regulating the sulfuration time and temperature. Besides, a certain amount of PVP was convinced to be converted into a defected graphitization configuration, according to the intensity ratio of the D band and G band in Raman spectroscopy. Most of the FeS2 was successfully encapsulated in the graphitic carbon layers leading to obtaining good conductivity. Fe1.89Mo4.11O7/FeS2@C exhibited an extremely high NH3 yield of 105.3 μg h−1 mgcat−1 and a FE of 54.7% at −0.4 V (Fig. 8G), as well as long-period electrolytic stability. The above works open new schemes for the rational design and fabrication of bimetallic MoFe-composites derived from MOFs for NRR. It can be further envisioned that the MOFs and even COFs (COF = covalent organic framework) containing Fe and Mo simultaneously may be used as electrocatalysts and through coordination modulation to regulate the electrocatalytic NRR performance. Moreover, MOFs and COFs may also be utilized as templates or precursors to structurally direct functional nanomaterials with special nano-morphologies, multi-component, and even heterostructures for high-performance NRR electrocatalysis. Thus, Fe,Mo-MOF/COF should be paid much attention to for the materials synthesis and structure design.
3.3.1.4. Ferric molybdate materials. Ferric molybdate materials are a kind of NRR catalysts that are widely available, easy to prepare, and cost-efficient. Wu et al. reported a one-step solvothermal method to synthesize FeMoO4 nanorods (NRs) from FeCl2 and Na2MoO4.174 Thanks to synergy interaction between Mo and Fe, the as-prepared FeMoO4 NRs exhibited superior NH3 yield (17.51 μg h−1 mgcat−1) and FE (10.53%) than that of Fe2O3 or MoO3, in 0.1 M Na2SO4. The current density remained almost unchanged in the long-term stability test for 12 hours under different applied potentials, indicating superb electrochemical stability. Notably, no N element was included in the raw materials, which helped to prove the source of the product ammonia. The catalytic performance and potential mechanism of FeMoO4 nanorods were also investigated by Chu and workers.175 The bimetallic nature of FeMoO4 could bring about accelerated electron transport and improved reaction kinetics, and thus it demonstrated a smaller charge transfer resistance than Fe2O3 and MoO3. Besides, the considerable elongation of N–N bond (1.128 Å) and decrease of Mo–N distance (2.014 Å) from Mullikan charge analysis suggested effective *N2 polarization and activation on 3-fold coordinated Mo (Mo3c) (Fig. 9A). Xian et al. synthesized Fe2(MoO4)3 nanoparticles (FMO-NPs) through a wet chemical process (Fig. 9B).176 No by-product N2H4 was detected during electrolysis, and an impressive NH3 yield (18.16 μg h−1 mgcat−1) and FE (9.1%) were obtained. Ulteriorly, distal and alternating pathways at the Fe2(MoO4)3 (110) surface were studied through material calculations (Fig. 9C). The catalyst surface showed strong adsorption of nitrogen molecules, as a deep downhill of −1.22 eV at this step. Then, the first hydrogenation step (*N2 → *NNH) was confirmed to be the PDS. At the following hydrogenation of the N atom, the bond lengths of N
N and N–Fe showed regular elongation and curtailment, respectively. Importantly, the suitable desorption energy barriers of the distal pathway ensured the smooth release of NH3 and reaction completion.
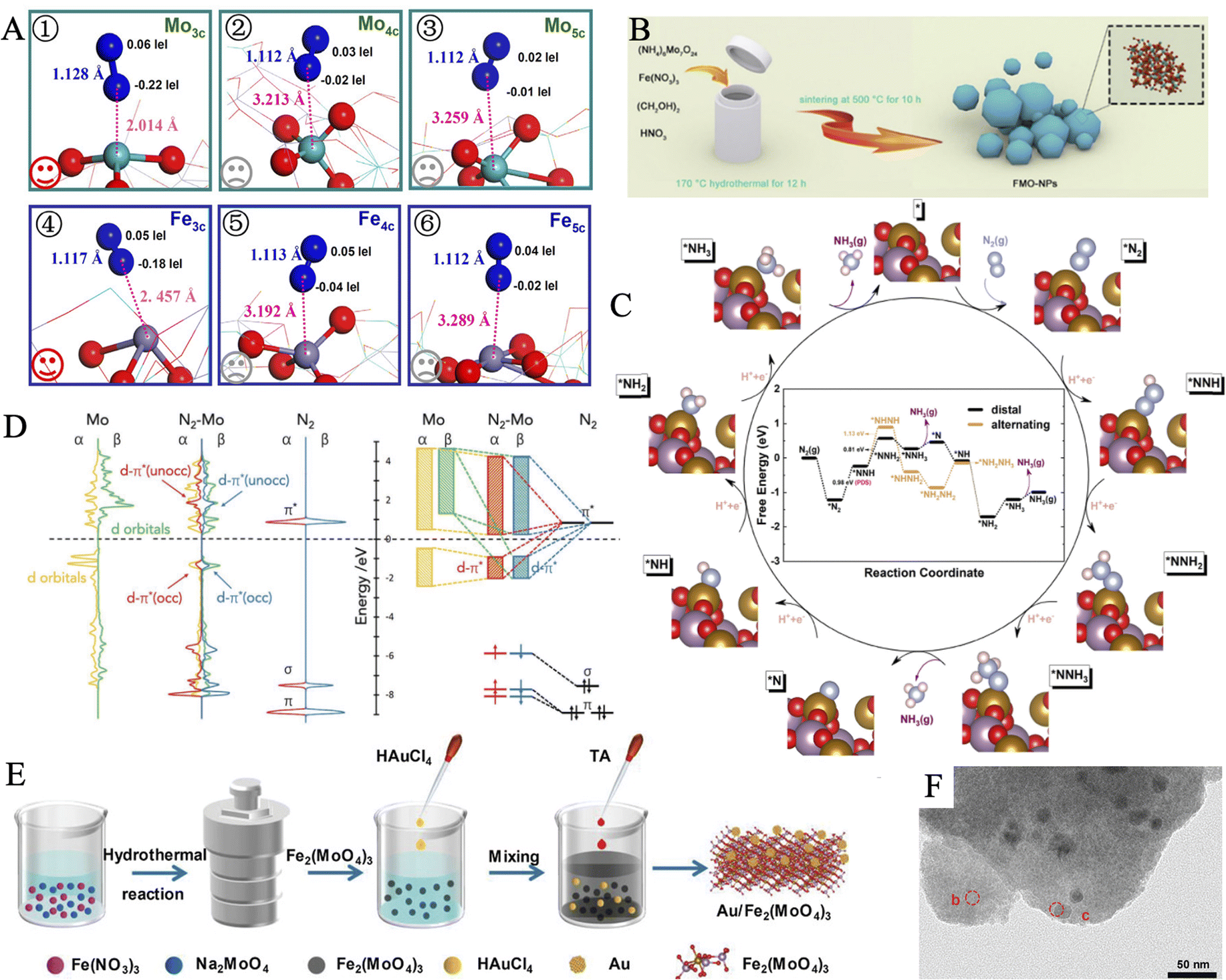 | ||
Fig. 9 (A) Optimized structures for N2 adsorption on various types of Fe and Mo atoms. Reproduced with permission: copyright 2020, American Chemical Society.175 (B) Schematic illustration of the synthesis for FMO-NPs. (C) Variation of free energy for the distal and alternating pathways at the Fe2(MoO4)3 (110) surface. Reproduced with permission: copyright 2020, American Chemical Society.176 (D) The p-DOS and schematic illustrations of 4d orbitals of Mo atoms on Au/Fe2(MoO4)3, 2p-orbitals of the N2 gas molecule, and their interaction within 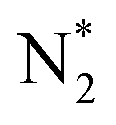 -Au/Fe2(MoO4)3. (E) Synthetic route of Au/Fe2(MoO4)3. (F) TEM image of Au/Fe2(MoO4)3. Reproduced with permission: copyright 2021, Wiley-VCH.178 -Au/Fe2(MoO4)3. (E) Synthetic route of Au/Fe2(MoO4)3. (F) TEM image of Au/Fe2(MoO4)3. Reproduced with permission: copyright 2021, Wiley-VCH.178 | ||
The deposition of noble metal nanoparticles is universal to improve the electrocatalytic activity. The above-mentioned iron molybdate materials demonstrate satisfying NRR activities, meanwhile, Au has generally low activity of HER.177 Therefore, it is a reasonable scheme to introduce Au to optimize the atomic structure of Fe2(MoO4)3 and enhance the performances of NRR. Yao et al. supported Au on the Fe2(MoO4)3 substrate to construct an Au/Fe2(MoO4)3 hybrid via a facile room-temperature reduction approach, achieving an excellent FE of 18.79% at −0.4 V (Fig. 9E and F).178 Compared with Fe2(MoO4)3 and Au/Fe2(MoO4)3via the analysis of temperature-programmed desorption of N2, the later one showed stronger binding strength of N2, which originated from the Au regulating promotion. To get a profound understanding of the activation of NN, the projected density of states plots of Mo atom and N2 were calculated. The d-σ and d-π* orbitals hybridization facilitated the electron transfer from Mo 4d orbitals to N2 2p orbitals (Fig. 9D), thus effectively weakening the inert NN triple bond. As a result, the reaction energy barrier of RDS of the distal pathway at Mo active centers was reduced from the former 0.98 to 0.6 eV after Au loading. Unfortunately, these catalytic ferric molybdate materials with large particle sizes suffer from insufficient active sites. In the future, material scientists may develop novel synthetic routes to decrease the size to expose more sites. The electrocatalytic performance can be further enhanced by simultaneous heteroatom doping and heterostructure engineering.
Due to the coupling effect, the multicomponent nanostructures generally demonstrate more advanced NRR performance than their individual counterparts.32,184 Introducing the second metal compound to a Fe-based material will be a compelling strategy to attain further improved performance. For example, doping Co elements into Fe-based materials can be an effective way to rationally regulate the electronic structure of catalysts due to the small atomic size and electronegativity of Co atoms, and the d–d coupling reduces the free energy barrier for NN triple bond dissociation. This idea was confirmed experimentally by Chen and coworkers by designing the Co-doped Fe3S4 nanoflowers (Fig. 10A). The introduced Co behaved as a new active center to promote N2 absorption and reduce the energy barrier, achieving a good catalytic performance (Fig. 10B).185
 | ||
| Fig. 10 (A) Schematic of Co-doped Fe3S4 nanoflowers. (B) NH3 yields and FEs of Co-doped Fe3S4 nanoflowers at a series of applied potentials. Reproduced with permission: copyright 2020, American Chemical Society.185 (C) TEM image of Au–Fe3O4 NPs. (D) N2 temperature-programmed desorption curves of different catalysts. (E) The geometric structure of various intermediate states during NRR, in which the hydrogen, oxygen, nitrogen, iron, and gold atoms are denoted by white, red, blue, slate blue, and yellow balls, respectively. (F) Free energy diagram of the NRR pathways on Au–Fe3O4 (101), Fe3O4 (101), and Au (111). Reproduced with permission: copyright 2020, Wiley-VCH184 (G) Illustration of synthesis and electronic interactions of dendritic Fe2O3/Cu electrocatalyst. (H) XPS valence band spectra of Fe2O3/Cu, Cu, and Fe2O3. Reproduced with permission: copyright 2019, Elsevier.186 | ||
Zhang et al. prepared heterogeneous Au–Fe3O4 nanoparticles (NPs) (Fig. 10C) via a one-pot wet-chemical reaction of HAuCl4 and diironnonacarbonyl (Fe2(CO)9).184 Such Au–Fe3O4 NPs exhibited good performance with a high ammonia yield of 21.42 μg h−1 mgcat−1 and a favorable FE of 10.54%. The N2 temperature-programmed desorption (TPD) (Fig. 10D), and surface valence band spectra together confirmed that Au–Fe3O4 NPs had a strong synergistic effect for adsorbing reaction species and suitable interface for charge and proton transfer. The theoretical calculations showed that Fe performed as an active site to fix N2 into *N2H while the introduction of Au boosted the adsorption of NRR intermediates and provided an energetic-favorable pathway for NRR (Fig. 10E and F). Huang et al. developed a Fe2O3/Cu composite catalyst by electro-deposition in the presence of FeCl2 and CuCl2 as electrolytes (Fig. 10G), which reached its highest yield of 15.66 μg h−1 mgcat−1 with a FE of 24.4% at −0.1 V vs. RHE.186 The composite improved the electron transfer on Fe2O3, as copper is a good conductor of electrons. As d-band theory suggests, the closer the d-band center approaches the Fermi level, the stronger the adsorption is. The electronic interactions between Fe2O3 and Cu elevated the d-state center (Fig. 10H), enabling strong adsorption and back-bonding of N2 molecules, leading to high catalytic activity.
3.4 Iron boride
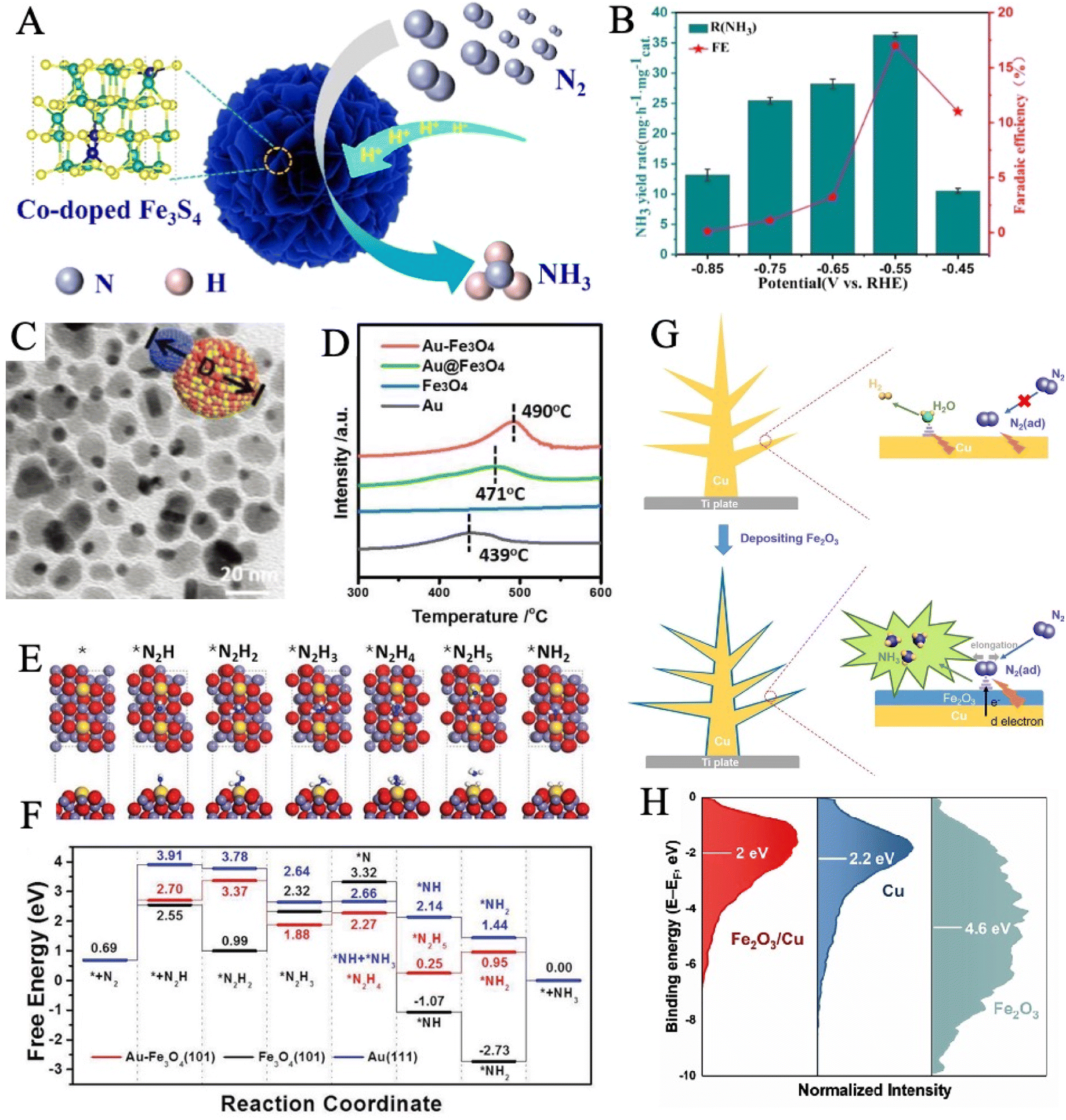
Transition metal borides, also known as MBenes, are an emergent class of catalysts that are still in their infancy in the electrochemical NRR field.187–189 MBenes as NRR electrocatalysts have the advantage of dual active edges from both the exposed metal end and the exposed boride end.190 In the previous section, we have elaborated on how the electrons and orbitals of iron activate nitrogen bonds. Similarly, boron has three valence electrons in the 2s22p1 configuration with an electron-deficient feature and an empty orbital to accept lone pair electrons from N2. B-to-N π-back bonding could be formed between the sp3 hybrid orbital of the boron atom and 2p orbital of N2, which is conducive to N2 adsorption on the B-active ends (Fig. 11A).79,191–195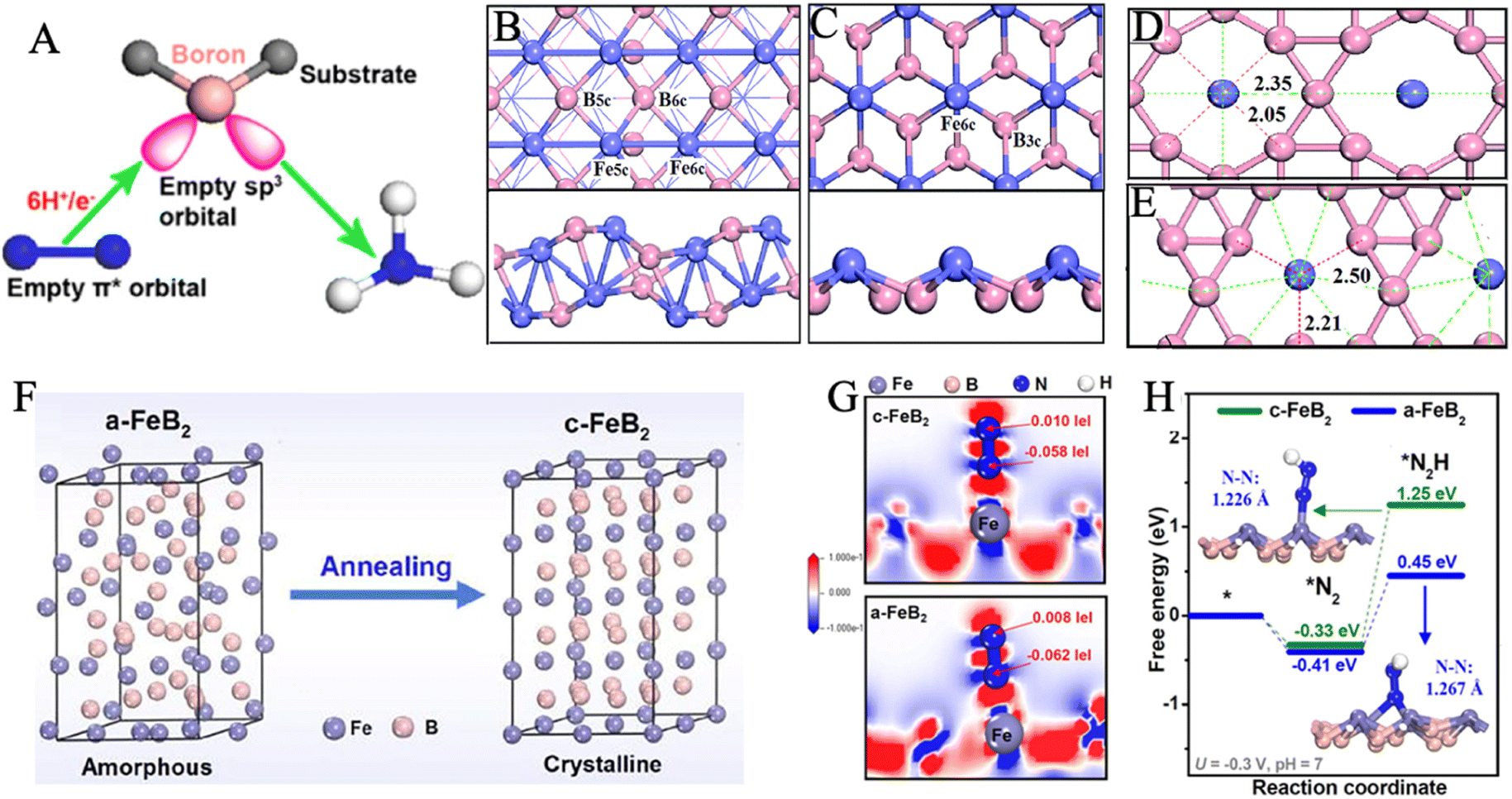 | ||
| Fig. 11 (A) Schematic of π-backdonation between boron and N2. Reproduced with permission: copyright 2019, American Chemical Society.192 Top and side views of FeB (B) and FeB2 (C). Fe and B are represented as purple and pink balls, respectively. Models and bonding network for (D) FeB6 (α) and (E) FeB6 (β). The length of Fe–B5c (short) and Fe–B6c (long) bonds are labeled in units of Å. Reproduced with permission: copyright 2019, Royal Society of Chemistry.58 (F) Illustration of the phase transformation from a-FeB2 to c-FeB2. (G) Charge density difference: the electron accumulation and depletion are represented as red and blue regions, respectively. (H) Free energy profiles of *N2 and *N2H adsorption on c-FeB2 and a-FeB2. Reproduced with permission: copyright 2021, Elsevier.57 | ||
In a theoretical study, four iron-borides were constructed and explored as potential NRR catalysts, including FeB, FeB2, FeB6(α), and FeB6(β) under the framework of DFT (Fig. 11B–E).58 The authors proposed that Fe–B bonding could remarkably affect Fe–N interaction by examining the free energy differences in reaction pathways on the Fe-sites of FeB2 and FeB, respectively. Additionally, the NH3 release issue had been solved by establishing the hypercoordinated FeB6(α) and FeB6(β). Among these, FeB6(β) offered the lowest ΔGmax of 0.68 eV, owing to the low oxidized Fe in the β-phase. Thus, N2 could acquire electrons as a newly oxidizing reagent and be activated on the lightly oxidized Fe sites. In the evaluation of selectivity, the calculated ΔG(H*) for the four catalysts is less than the ΔGmax of NRR, but H* adsorption is weak in contrast to N2 adsorption. The heterostructure construction was also applied to design high-performance iron boride-based electrocatalysts. In the current work, the process of NRR was studied on iron atoms supported by Bn-doped graphyne (GY).196 A distal pathway was successfully identified with a minimum limiting potential of −0.53 V using DFT calculations. Additionally, the FeB@GY surface could effectively promote electron transfer efficiency and significantly improve the stability of intermediate adsorption during NRR. The substitution of B and N atoms for the C atoms in the doped GY structure is suitable for improving surface activity. From the results of the Bader charge analysis and charge density difference, it was detected that the reaction intermediates could not only accept electrons but also extract electrons from FeB@GY. The analysis of the partial density of states indicated a strong interaction between Fe-3d and N-2p in addition to B-2p, which was caused by the existence of the overlapped orbitals.
Chu's group pioneered the experimental synthesis of amorphous FeB2 porous nanosheets (α-FeB2 PNSs) through a facile reflux method.57 Amorphization engineering produced vast unsaturated coordination sites and surface-exposed defects, which served as active sites to promote the electrocatalytic reaction energetics and kinetics. In 0.5 M LiClO4 electrolyte, α-FeB2 PNSs showed a maximum NH3 yield rate of 39.8 μg h−1 mgcat−1 at −0.3 V and a FE of 16.7% at −0.2 V. Furthermore, the chrono-potentiometric response showed no obvious decays for 20 h of consecutive electrolysis, with a 92.8% retention of the initial NH3 yield, indicating excellent long-term stability. In this work, crystalline FeB2 PNSs (c-FeB2 PNSs) were also prepared by annealing at 600 °C for 3 h (Fig. 11F). When comparing the NRR performance, c-FeB2 showed smaller ECSA and weaker N2 absorption than a-FeB2. Meanwhile, a-FeB2 had strong charge transfer between *N2 and active substrates and more injected electrons to activate the N2 molecule to a greater extent, as observed in Fig. 11G. A lower G*N2H (0.45 eV) on a-FeB2 was provided to stabilize *N2H and reduce the initial reduction barrier (Fig. 11H). Currently, most research on TMBs (TM = transition metal) especially iron borides as NRR electrocatalysts is focused on the theoretical calculations.58,197–200 There is a deficiency of reports about experimental fruits on iron borides for NRR electrocatalysis, probably due to the harsh preparation conditions.
3.5 Fe-based single-atom catalysts
Up to now, single-atom catalysts (SACs), for which isolated atoms are anchored on various substrates to generate stable configuration, have been extensively investigated for nitrogen reduction reactions.34,201–204 Compared with traditional catalyst materials, decreasing metal nanoparticles into single atoms can significantly improve the catalytic activity and selectivity towards NRR.205 The SACs may maximize atom utilization and effectively reduce economic costs in large-scale industrial production.206–208 Meanwhile, the high dispersibility of SACs ensures their larger specific surface area and more exposed active sites, leading to stronger catalytic activity.202,209 Moreover, the selection of substrate plays an important role in improving anchoring stability and modulating the electronic structure of SACs.207,210 The interactions between substrates and loaded metal atoms give rise to tunable electronic structures, which favors the adsorption and activation pathway of the reactants at the catalytic sites to lower the energy barrier of NRR, in conjunction with unsaturated coordination environments.211,212 However, some problems still exist when single atoms are applied as catalysts such as low stabilities for long cycle catalysis, low active metal atom loadings, difficulties in efficiently dispersing and isolating individual atoms, and the complex preparation process.213,214 As a result, developing suitable substrates to achieve stable and efficient loading of single atoms is an urgent need.Nitrogen-doped carbon is one of the most encouraging candidates due to its enhanced electronic conduction, optimized charge distribution, and electronic structure of active sites, which is caused by the interfacial interactions between iron atoms and the surrounding nitrogen/carbon atoms.210 As revealed by DFT calculations, atomically dispersed Fe–N3 site on nitrogen-doped graphene (Fe–N3/graphene) can substantially activate the inert NN bond of N2 with high spin-polarization and a localized magnetic moment.215 Yang et al. anchored Fe single-atoms on a nitrogen-doped carbon substrate (Fe SAC/N–C) to realize the coordination structure of Fe–N3.210 The HAADF-STEM images indicated that the majority of Fe objects were downsizing to single atoms with a range of 0.1 to 0.2 nm, and there were no obvious Fe nanoparticles or clusters. The catalyst achieved an enhanced NH3 yield rate of 53.12 μg h−1 mgcat−1 and a high FE of 39.6% in 0.1 M KOH with ignored decay in seven consecutive electrolysis cycles. Computational studies provided direct evidence of the robust binding capability of Fe–N3 catalytic active sites to N2 molecules to achieve the high selectivity of Fe SAC/N–C for NH3 formation. The calculated Gibbs free energy diagrams illustrated that the alternating pathway was a more convincing mechanism on highly dispersed Fe sites compared with the distal pathway, as its lower energy barrier of ΔG = 0.915 eV (Fig. 12A).
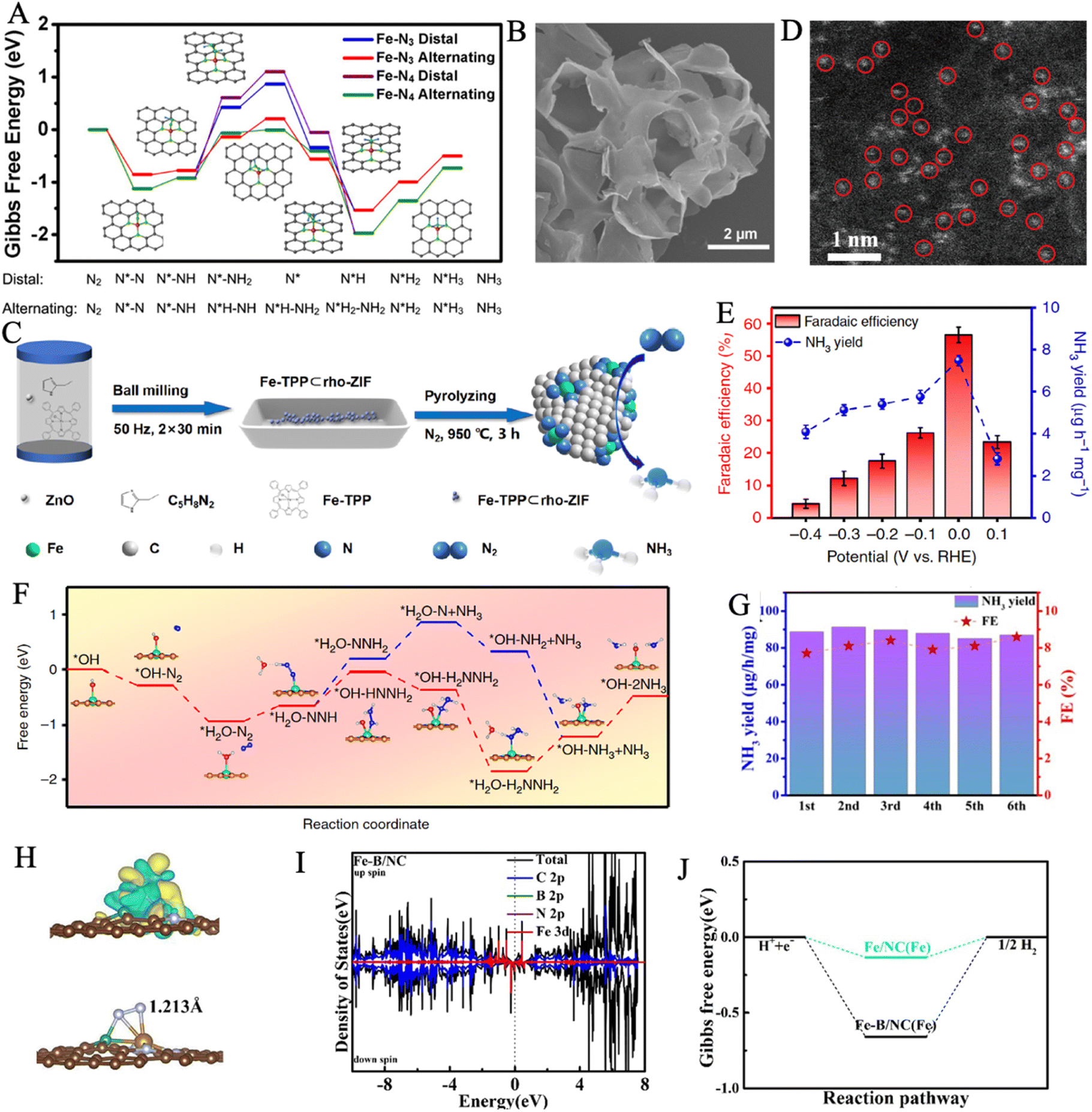 | ||
| Fig. 12 (A) Gibbs free energy profiles of NRR on Fe–N3 and Fe–N4 through alternating and distal pathways. The inset shows the optimized structure of Fe–N3 coordination and the corresponding adsorption of the NRR intermediates. Reproduced with permission: copyright 2020, American Chemical Society.210 (B) SEM images of carbon substrate. Reproduced with permission: copyright 2019, American Chemical Society.216 (C) Schematic illustration of the synthesis routes of Fe–N/C-CPs. (D) HAADF-STEM image and magnified image of Fe–N/C. The Fe-SA centers are marked by red circles. Reproduced with permission: copyright 2021, American Chemical Society.213 (E) NH3 yield rates and FEs at each given potential of FeSA–N–C. (F) Free energy profiles for NRR on the FeSA–N–C catalyst at U = 0 V. The inset represents the corresponding adsorption models (H, C, N, O, and Fe atoms are represented with gray, orange, blue, red and green spheres, respectively). Reproduced with permission: copyright 2019, Springer.218 (G) NH3 yields and FEs during six-recycling tests at −0.45 V vs. RHE. (H) Charge density difference and the N–N bond length after N2 adsorption (I) The density of states for C 2p, B 2p, N 2p and Fe 3d for Fe–B/NC. (J) Gibbs free energy profiles of the H2 evolution on the as-prepared sample. Reproduced with permission: copyright 2022, Elsevier.222 | ||
Fe–N4 configurations are also developed for electrocatalytic ammonia synthesis. He et al. reported an atomically dispersed iron phthalocyanine (FePc) molecules on porous carbon with Fe–N4 coordination structure, which achieved a dramatically high NH3 yield rate of 137.95 μg h−1 mgcat−1 at a low potential of −0.3 V (vs. RHE).216 The SEM image showed the comb-like structure of 3D carbon substrate with massive nanopores, which contributed to the high loading ability of 7.43 wt% (the loading of FePc) (Fig. 12B). The current density decreased significantly in poisoning treatment by SCN− suggests that the Fe center of FePc is the dominant active site for NH3 formation. Liu et al. explored the realization of high Fe–N4 loading capacity.213 Fe-TPP, C5H8N2, and N,N-diethylformylamide (DEF) etc. were milled in a zirconia-ball grinding mill to form the Fe-TPP⊂rho-ZIF precursor, and Fe–N/C was obtained by pyrolyzing in a tube heating furnace at 950 °C for 3 h under an N2 atmosphere (Fig. 12C). The HAADF-STEM image indicated that the Fe atoms were uniformly distributed on the carbon substrate, in which the Fe single-atom loading reached 3.5 wt% (Fig. 12D). The sublimation of Zn in the pyrolysis process of the precursor was favorable to expose the catalytic sites and increase the specific surface area,217 which reached up to 1088.96 m2 g−1. Wang et al. reported a FeSA–N–C catalyst with an extremely high FE of over 56.55% (Fig. 12E).218 Single Fe atoms were uniformly loaded throughout the N-doped carbon support, and no Fe nanoparticles or aggregations were obviously detected. The adsorption behavior of N2 toward Fe active sites was illustrated by molecular dynamics simulations, which suggested a lower energy barrier of 2.38 kJ mol−1 at approximately 0.54 nm for efficient N2 access. As depicted in Fig. 12F, the alternating pathway with lower energy uphill of 0.28 eV was more favorable than the distal pathway. Additionally, the high energy barriers (2.91 eV) for *H adsorption resulted, in particular, sluggish HER kinetics. Based on the above characteristics, the desired selectivity and FE were achieved by enhancing nitrogen reduction and suppressing hydrogen evolution.
Fe SACs that anchored on strain-engineered graphene substrates were modeled and systematically examined by first-principles calculations, suggesting that curvature strongly influences the catalytic activity and selectivity of SACs for NRR.219 Bu et al. studied the NRR pathways for a dual-atom Fe2 species on graphitic carbon nitride (g-C3N4) by changing external conditions.220 It was found that applying an appropriate range of tensile to the graphitic carbon nitride (g-C3N4) substrate could reduce the overpotential values, which arose from a break in the intrinsic scaling relationship of the adsorption energy between the *N2 and *HNN adsorbates. More influences of structural distortion on the NRR catalytic properties of Fe, N codoped carbon (Fe–N–C) were explored.221 The NRR intermediate species *NNH could be strongly bound at the FeN4 and FeN3 active sites embedded in strained graphene layers, contributing to the enhanced NRR activity. In contrast to *NNH, *H showed a greater adsorption energy barrier, which indicated the less favored competitive HER and higher selectivity of NRR. Their computational work provides evidence for improving the catalytic activity and selectivity of Fe–N–C materials towards NRR by tuning the degree of compressive strain in their graphene layers. Although the theoretical studies of strain engineering on Fe-SACs have achieved promising results, the predictions need to be further confirmed by experimental works.
The doping of multi-element atoms into the substrate is an effective and well-explored strategy for adjusting the Fe active site configuration to enhance NRR. Wang et al. developed boron-induced electron-rich SACs (Fe–B/NC) by dispersing single Fe sites on boron, nitrogen-co-doped carbon.222 The desired selectivity and stability were proved by no detectable N2H4 and six recycling tests (Fig. 12G). The introduction of B atoms with low electronegativity (χ = 2.0) broke the symmetric electron distribution of the Fe–N4 coordination, engendering electron donor (B)/acceptor (N) pairs along the B–Fe–N bridge to enhance electron transfer. After being adsorbed on B–Fe sites, the triple bond in N2 was elongated by 0.115 Å with a comparison of free N2, due to back-donation from B and Fe to N2, which was confirmed by the Bader charge after N2 adsorption analysis (Fig. 12H). Moreover, the coupling with B 2p could delocalize the electrons in the Fe 3d orbital (Fig. 12I), increasing the electron conductivity for N2 adsorption/activation, thereby enhancing the NRR performance. In addition, the catalyst exhibited a hindered effect on HER relative to the boron-free sample, owing to a large ΔGH* (Fig. 12J). Another coordination mode for anchoring Fe SA has also been explored, typically as Fe-(O–C2)4 single-atom sites on lignocellulose-derived carbon with impressive catalytic activity (32.1 μg h−1 mgcat−1 equal to 5350 μg h−1 mgFe−1), by Zhao and coworkers.45
In addition to nitrogen-doped carbon, other substrates also can be applied to support Fe single atoms as SACs for NRR. Porous materials with large surface areas or vacancies are ideal supports that can accommodate ample active sites, realizing the high metal loading. Chen et al. prepared S-coordinated Fe SACs on mesoporous TiO2 (Fe1Sx@TiO2), which were constructed by a lattice-confined strategy.60 FeS2O2 sites were confirmed as active centers by theoretical calculations. No peak was observed for a Fe–Fe bond in the Fe-K edge EXAFS spectra of Fe1Sx@TiO2, indicating that Fe atoms are distributed individually (Fig. 13A). Abundant OVs and Ti–S coordination bonds were detected by Ti K-edge EXAFS spectra, resulting in high dispersion stability. The finite element analysis showed that confinement of opened and ordered mesopores could promote mass transfer and provide a large active surface area. As a result, the catalyst showed a good NH3 production rate of 18.3 μg h−1 mgcat−1 with 17.3% FE, and the origin of NH3 was verified via an isotopic-labeling experiment (Fig. 13B). Moreover, the relationship between Fe loading capability and NRR performance was also investigated, when the ratio of Fe:Ti is 6:100, NRR activity reached maximum without Fe aggregation. First-principles calculations revealed the existence of an enzymatic mechanism with a low overpotential of 0.21 V and side-on adsorption of N2 in MoS2-supported Fe2 clusters (Fe2/MoS2), which is conducive to effective NN bond activation.223 Li et al. creatively designed a single Fe atom supported on atomically thin molybdenum disulfide (SACs-MoS2–Fe) to form high curvatures with interfacial protrusion-like shape (Fig. 13C).224 Triggering interfacial polarization by curvature-rich surface and interface demonstrates potential universality in wide-scope electrocatalysis, including the promising and energy-effective breaking of NN bond. Fe single atoms worked as external adatoms conjugating with MoS2 to create 3D protrusions, instead of interior dopants substituting in MoS2 skeletons, which was distinct from extensively reported metal–Nx and defect-trapped-SACs. The unique geometry stimulated electron injection into NN efficiently enabled N2 polarization and reduction. When the atomic ratio of Fe and MoS2 reached 2.0, SACs-MoS2–Fe2 demonstrated the best catalytic performance of ammonia yield (97.5–6 μg h−1 cm−2) and FE (31.6–2%) at −0.2 V.
 | ||
| Fig. 13 (A) k3-weighted Fourier-transform EXAFS spectra of the Fe R-space for FeS2 and Fe foil, Fe1Sx@TiO2 and Fe–TiO2, the inset is the first peak of Fe1Sx@TiO2 and Fe–TiO2. (B) 1H-NMR spectra of 14NH4+ and 15NH4+ produced in the NRR process. Reproduced with permission: copyright 2022, Wiley-VCH.60 (C) 3D topographic potential distribution images of MoS2 and pure MoS2. Reproduced with permission: copyright 2020, Elsevier.224 (D) In situ FTIR spectra obtained on the surfaces of FeMo/NC, Fe/NC, and Mo/NC SACs, respectively (from left to right). (E) Proposed NRR pathways on FeMo/NC, Fe/NC, and Mo/NC surfaces. Reproduced with permission: copyright 2022, Springer.231 (F) NH3 yields and FEs on various applied potentials. (G) Fe and Co K edge EXAFS fitting curves of Fe/Co–O–C-1.0 at R space. Inset is the predicted bimetallic Fe–Co coordination configuration in Fe/Co–O–C-1.0. (C, O, Fe, and Co are denoted by brown, red, orange, and blue, respectively). Reproduced with permission: copyright 2022, Springer.233 (H) Differential charge densities of absorbed N2 on Pd and PdFe1, where electron accumulation and depletion are denoted by yellow and cyan, respectively. (I) 3D topographic atom images of PdFe1. Reproduced with permission: copyright 2022, Wiley-VCH.59 | ||
Diatomic catalysts (DACs) derived from single-atom catalysts have been widely explored for highly efficient electrocatalysis.225–228 According to related investigations, the catalytic activity of graphene-based Mn, Ni, Co, and Fe DACs exceeds that of the corresponding SACs.229,230 Zhang and coworkers reported FeMo/NC DSACs as a “dual-site” material by anchoring isolated Fe and Mo atoms on hierarchical N-doped carbon nanotubes.231 This catalyst demonstrated an NH3 yield of 26.8 μg h−1 mgcat−1 and a FE of 11.8, which is 1.6 and 2.5 times larger than those of Mo/NC and Fe/NC. Time-dependent in situ Fourier transform infrared spectroscopy (in situ FTIR) measurements were conducted for dual-site FeMo/NC DSACs and single-site Fe/NC and Mo/NC to investigate the synergy mechanism of Mo and Fe single atoms (Fig. 13D). Three positive-going bands of N–N stretching, –NH2 wagging, and H–N–H bending were appeared at the in situ FTIR spectra of FeMo/NC, suggesting the formation of *N2Hy intermediates during the NRR process. No positive-going N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching was observed on the FeMo/NC surface, which implied that the *N2Hx intermediates (0 ≤ x ≤ 2) had no obstacle to separate into a single bond N–N of *N2Hy intermediates (1 ≤ y ≤ 4). The NRR over FeMo/NC may follow an associative mechanism (Fig. 13E), due to the existence of the three main intermediates, as well as no N2H4 was detected. Ma et al. revealed the catalytic principle of Fe/Mo–N–C from the point of view of the spin moment of the active centers through theoretical calculations.232 The neighboring Fe could modulate the spin state of the Mo center in MoN4 to switch from a high-spin state to a medium-spin state, effectively relieving the strong overlap between the Mo 4d orbital and the NxHy intermediates, facilitating the desorption of NH3, and ultimately leading to a lower overpotential. Recent work reported that an extraordinary NRR performance of 579.2 ± 27.8 μg h−1 mgcat−1 NH3 yield rate and 79.0 ± 3.8% FE could be achieved on an atomically dispersed, bimetallic Fe–Co active site catalyst (Fe/Co–O–C-r) (Fig. 13F).233 The observation and fitting of EXAFS spectra of Fe/Co–O–C-1.0 suggested that the atomically dispersed Fe, Co atoms are anchored on carbonized BC (CBC) by a bimetallic Fe–Co arrangement anchoring to graphitic carbon through six O-bridging bonds, for which, [(O–C2)3Fe–Co(O–C2)3] is the likely bimetallic unit (Fig. 13G). Calculations pointed out that an energy-favorable transformation from [(O–C2)3Fe–Co(O–C2)3] to [(O–C2)3Fe–Co(O–C)C2] can occur at the end of an NRR cycle on desorption of *NH3. [(O–C2)3Fe–Co(O–C)C2] was further confirmed to be the actual active site of Fe/Co–O–C-r and resulted in remarkable NRR performance.
N stretching was observed on the FeMo/NC surface, which implied that the *N2Hx intermediates (0 ≤ x ≤ 2) had no obstacle to separate into a single bond N–N of *N2Hy intermediates (1 ≤ y ≤ 4). The NRR over FeMo/NC may follow an associative mechanism (Fig. 13E), due to the existence of the three main intermediates, as well as no N2H4 was detected. Ma et al. revealed the catalytic principle of Fe/Mo–N–C from the point of view of the spin moment of the active centers through theoretical calculations.232 The neighboring Fe could modulate the spin state of the Mo center in MoN4 to switch from a high-spin state to a medium-spin state, effectively relieving the strong overlap between the Mo 4d orbital and the NxHy intermediates, facilitating the desorption of NH3, and ultimately leading to a lower overpotential. Recent work reported that an extraordinary NRR performance of 579.2 ± 27.8 μg h−1 mgcat−1 NH3 yield rate and 79.0 ± 3.8% FE could be achieved on an atomically dispersed, bimetallic Fe–Co active site catalyst (Fe/Co–O–C-r) (Fig. 13F).233 The observation and fitting of EXAFS spectra of Fe/Co–O–C-1.0 suggested that the atomically dispersed Fe, Co atoms are anchored on carbonized BC (CBC) by a bimetallic Fe–Co arrangement anchoring to graphitic carbon through six O-bridging bonds, for which, [(O–C2)3Fe–Co(O–C2)3] is the likely bimetallic unit (Fig. 13G). Calculations pointed out that an energy-favorable transformation from [(O–C2)3Fe–Co(O–C2)3] to [(O–C2)3Fe–Co(O–C)C2] can occur at the end of an NRR cycle on desorption of *NH3. [(O–C2)3Fe–Co(O–C)C2] was further confirmed to be the actual active site of Fe/Co–O–C-r and resulted in remarkable NRR performance.
Traditional SACs have the inherent disadvantage that single metal atoms tend to agglomerate due to their large surface free energy. Avoiding the aggregation of individual atoms at the expense of low metal loading limits the improvement of NRR performance and practical applications. A new variety of SACs, single-atom alloys (SAAs), in which, the isolated metal atoms are anchored on the substrate metal has been proposed with integrated merits.234–236 Specifically, the extremely strong metal–support interactions endow the high thermodynamic stability to break limitations in metal loading. Moreover, the alloying effect of SAAs tunes the electronic structure of atomic metal sites to enable an advantageous balance between the appropriate binding of intermediates and the effective dissociation of reactants. Li et al. first developed a PdFe1 single-atom alloy metallene, which demonstrated an NH3 yield of 111.9 μg h−1 mgcat−1 and a FE of 37.8% at −0.2 V vs. RHE.59 Preliminary theoretical computations indicated the enhancement of N2 affinity on PdFe1 and the Lewis acid–base interaction facilitated the following N2 polarization and cleavage (Fig. 13H). In the free energy diagrams, the energy barrier of the rate-limiting step was reduced to 0.72 eV, due to the efficient N2 activation on Fe sites. Encouraged by the above theoretical results, the preconceived PdFe1 (Fe: 3.91 at%) was prepared via a one-step wet chemistry strategy. The low-peak-intensity spots in 3D topographic atom images suggested that Fe atoms successfully substituted the partial Pd atoms on the PdFe1 substrate (Fig. 13I). Thus, this stably distributed the SAA catalyst and presented a steady current density during the 100 h of continuous electrolysis in chronoamperometry, and no apparent recession in NRR yields and FEs within ten cycling tests were observed.
4. Conclusions and outlook
Significant and encouraging advancements in ambient electrochemical NRR for gaseous nitrogen molecules to NH3 have been made in the past five years and even the decade. However, efficient nitrogen reduction under ambient conditions faces challenges for both fundamental research and practical applications. These substantial barriers include, for example, the inertness and low water solubility of N2 molecules, weak N2 adsorption on catalyst surfaces, high overpotentials, low catalytic lifetimes, and the involvement of multi-proton and multi-electron pathways leading to low selectivity. To negotiate these obstacles and achieve efficient NRR with a high production rate, high FE, and high energy utilization efficiency, functional Fe-based materials have been paid much attention to and explored for this promising field, possibly inspired by both the Fe-containing industrial catalyst and biological enzyme. In this review, a brief catalytic mechanism for the electrocatalytic NRR and a comprehensive overview of the research fruits of Fe-based electrocatalytic materials for NRR are presented. Table 1 summarized and compared the electrocatalytic performances of the representative Fe-based electrocatalysts, including iron oxide, sulfides, bimetallic Fe-containing materials, single-atom, and even dual-atomic materials. Unfortunately, the activity, selectivity, and longevity of the Fe-based materials and others are not satisfactory. Therefore, to optimize the electrocatalytic performance and further promote significant development, we propose the following perspectives and further development directions (Fig. 14).| Catalyst | Electrolyte | NH3 yield | Quantification methods | FE (%) | Isotope labeling | Ref. |
|---|---|---|---|---|---|---|
| o-Fe2O3–Ar/CNT | 0.1 M KOH | 0.46 μg h−1 cm−2 | Nessler colorimetry | 6.0 | No | 44 |
| Fe/Fe3O4-300 | 0.1 M phosphate buffer solution | 0.19 μg h−1 cm−2 | Indophenol blue colorimetry | 8.29 | No | 100 |
| Fe–Fe3O4 nanoparticles | 0.1 M Na2SO4 | 24.6 μg h−1 mgcat−1 | Indophenol blue colorimetry | 53.2 | No | 50 |
| Fe3O4-300 nanosheet | 0.1 M Na2SO4 | 12.09 μg h−1 mgcat−1 | Indophenol blue colorimetry | 34.38 | Yes | 49 |
| Fe–Fe3O4 nanoparticles | 0.1 M Na2SO4 | 22 μg h−1 mgcat−1 | Indophenol blue colorimetry | 3.5 | No | 115 |
| p-Fe2O3/CC | 0.1 M Na2SO4 | 6.78 μg h−1 cm−2 | Indophenol blue colorimetry | 7.69 | No | 116 |
| Zn doped Fe2O3 | 0.1 M Na2SO4 | 15.1 μg h−1 mgcat−1 | Indophenol blue colorimetry | 10.4 | Yes | 113 |
| α-Fe2O3 nanocubes | 0.1 M KOH | 32.13 μg h−1 mgcat−1 | Indophenol blue colorimetry | 6.63 | Yes | 114 |
| Au–Fe3O4 nanoparticles | 0.1 M KOH | 21.42 μg h−1 mgcat−1 | Nessler colorimetry | 10.54 | Yes | 184 |
| Fe2O3/Cu | 0.1 M KOH | 15.66 μg h−1 mgcat−1 | Indophenol blue colorimetry | 24.4 | No | 186 |
| Fe3S4 nanosheets | 0.1 M HCl | 75.4 μg h−1 mgcat−1 | Indophenol blue colorimetry | 6.45 | Yes | 123 |
| FeSx/Fe | 0.1 M KOH | 4.13 × 10−10 mol s−1 cm−2 | Ion chromatography, indophenol blue colorimetry, Nessler colorimetry | 17.6 | Yes | 124 |
| Vs-FePS3 NSs | 0.1 M HCl | 3.88 μg h−1 cm−2 | Ion chromatography, indophenol blue colorimetry | 12.36 | No | 52 |
| FeS2 | 0.1 M Na2SO4 | 37.2 μg h−1 mgcat−1 | Indophenol blue colorimetry | 11.2 | No | 126 |
| FeS2 | 0.1 M Li2SO4 | 11.5 μg h−1 mgFe−1 | Nessler colorimetry | 14.6 | No | 125 |
| Fe2O3/FeS | 0.1 M KOH | 34.31 μg h−1 mgcat−1 | Indophenol blue colorimetry | 18.06 | Yes | 127 |
| FeS2@GO | 0.1 M HCl & 0.1 M Na2SO4 | 78.6 μg h−1 mgcat−1 in 0.1 M HCl, 27.9 μg h−1 mgcat−1 in 0.1 M Na2SO4 | Indophenol blue colorimetry | 4.7 in 0.1 M HCl, 6.8 in 0.1 M Na2SO4 | No | 128 |
| FeTe2/RGO | 0.5 M LiClO4 | 39.2 μg h−1 mgcat−1 | Indophenol blue colorimetry | 18.1 | Yes | 129 |
| FeMoSe | 0.1 M Na2SO4 | 20.14 μg h−1 mgcat−1 | Indophenol blue colorimetry | 13.65 | Yes | 132 |
| FeS2–Mo | 0.1 M KOH | 26.15 μg h−1 mgcat−1 | Indophenol blue colorimetry, Nessler colorimetry, ion chromatography | 14.41 | Yes | 56 |
| Fe2Mo6S8 | 0.5 M Na2SO4 mixed with 0.1 M sodium citrate buffer | 70 μg h−1 mgcat−1 | Indophenol blue colorimetry | 12.5 | Yes | 137 |
| MoS2–Fe | 0.1 M Na2SO4 | 20.11 μg h−1 mgcat−1 | Indophenol blue colorimetry | 15.72 | No | 140 |
| Fe–MoS2 | 0.5 M K2SO4 | 8.63 μg h−1 mgcat−1 | Indophenol blue colorimetry | 18.8 | Yes | 142 |
| Mo-doped Fe2O3 | 0.1 M Na2SO4 | 21.3 μg h−1 mgcat−1 | Indophenol blue colorimetry | 11.2 | No | 143 |
| MoO2/FeS2/GA | 0.1 M HCl | 40.18 μg h−1 mgcat−1 | Indophenol blue colorimetry | 37.44 | Yes | 146 |
| FeS@MoS2/CFC | 0.1 M Na2SO4 | 8.45 μg h−1 cm−2 | Indophenol blue colorimetry | 2.96 | No | 54 |
| Fe–MoS2/CC | 0.1 M KOH | 12.5 μg h−1 cm−2 | Indophenol blue colorimetry | 10.8 | Yes | 152 |
| SACs-MoS2-Fe | 0.1 M KCl | 97.5 μg h−1 cm−2 | Indophenol blue colorimetry | 31.6 | Yes | 224 |
| MoFe-PC | 0.1 M HCl | 34.23 μg h−1 mgcat−1 | Indophenol blue colorimetry | 16.83 | No | 170 |
| Ni0.3–Fe0.7@MoS2 | 0.1 M Na2SO4 | 128.17 μg h−1 mgcat−1 | Indophenol blue colorimetry | 11.34 | No | 171 |
| Fe1.89Mo4.11O7/FeS2@C | 0.5 M K2SO4 | 105.3 μg h−1 mgcat−1 | Nessler colorimetry | 54.7 | Yes | 55 |
| FeMoO4 nanorods | 0.1 M Na2SO4 | 17.51 μg h−1 mgcat−1 | Indophenol blue colorimetry | 10.53 | No | 174 |
| FeMoO4 nanorods | 0.5 M LiClO4 | 45.8 μg h−1 mgcat−1 | Indophenol blue colorimetry | 13.2 | Yes | 175 |
| Fe2(MoO4)3 nanoparticles | 0.1 M Na2SO4 | 18.16 μg h−1 mgcat−1 | Indophenol blue colorimetry | 9.1 | No | 176 |
| Au/Fe2(MoO4)3 | 0.2 M Na2SO4 | 7.61 μg h−1 mgcat−1 | 1H-NMR method | Yes | 178 | |
| α-FeB2 PNS | 0.5 M LiClO4 | 39.8 μg h−1 mgcat−1 | Indophenol blue colorimetry | 16.7 | Yes | 57 |
| Fe SAC/N–C | 0.1 M KOH | 53.12 μg h−1 mgcat−1 | Indophenol blue colorimetry | 39.6 | Yes | 210 |
| Fe–N/C | 0.5 M K2SO4 | 2.27 μg h−1 mgcat−1 | Indophenol blue colorimetry | 7.67 | Yes | 213 |
| FePc/C | 0.1 M Na2SO4 | 137.95 μg h−1 mgFePc−1 | Indophenol blue colorimetry | 10.05 | No | 216 |
| 10.25 μg h−1 mgcat−1 | ||||||
| FeSA–N–C | 0.1 M KOH | 7.48 μg h−1 mgcat−1 | Indophenol blue colorimetry | 56.55 | Yes | 218 |
| Fe–B/NC | 0.1 M KOH | 88.7 μg h−1 mgcat−1 | Indophenol blue colorimetry | 9.2 | Yes | 222 |
| Fe1Sx@TiO2 | 0.1 M HCl | 18.3 μg h−1 mgcat−1 | Indophenol blue colorimetry | 17.3 | Yes | 60 |
| FeMo/NC DSACs | 0.1 M Na2SO4 | 26.8 μg h−1 mgcat−1 | Indophenol blue colorimetry | 11.8 | No | 231 |
| Fe/Co–O–C-r | 0.1 M Na2SO4 | 579 μg h−1 mgcat−1 | Indophenol blue colorimetry, 1H-NMR method | 79.0 | Yes | 233 |
| PdFe1 | 0.5 M LiClO4 | 111.9 μg h−1 mgcat−1 | Indophenol blue colorimetry | 37.8 | Yes | 59 |
 | ||
| Fig. 14 Strategies to boost NRR performances and perspectives for future developments of iron-based catalysts. Regulate in-depth mechanism: reproduced with permission: copyright 2022, Wiley-VCH.59 Coupling strategy: reproduced with permission: copyright 2020, Royal Society of Chemistry.237 Copyright 2022, Elsevier.150 Copyright 2020, Elsevier.224 Theoretical calculation: reproduced with permission: copyright 2022, American Chemical Society.136 | ||
4.1 From an in-depth mechanistic reaction pathway
According to the possible associative distal/alternating pathway, and enzymatic pathway, the nitrogen adsorption ability, activation ability, and even the N2 adsorption mode of the metal site are very crucial. The Fe element with variable valence will be endowed with different electronic configurations.238 Furthermore, the crystal field and the local coordination environments can be further elaborately designed to modulate the spin states, including high spin, low spin, and even intermedia spin, of the Fe center.71 It may provide a perspective solution to strengthen the activation ability. From another point of view, the microscopic reaction mechanism indicates that the hydrogenation process is another key factor to complete NRR. However, few researchers pay attention to hydrogen sources, especially for the hydrogen intermediate H* in the pathway and their transfer process. Wang and coworkers recognized that the directly significant suppression of HER may be counterproductive for electrocatalytic NRR because N2 hydrogenation also needs active H species.239 It is anticipated that scientific research should focus more on the solution of the intermedia H*-related issues, including its source, its production, its transfer, and subsequent hydrogenation for N2. Understanding these above two key points may provide meaningful guidance to explore catalytic materials with advanced structures and performances.4.2 Exploring coupling strategies for materials design
The strategies of enzyme mimicking, elemental doping, heterostructures engineering, and single-atom dispersion on functional supports, etc. presented in this review have all yielded exciting results. These tactics are usually adopted to optimize the binding energy of the reactants and key intermediates through the synergistic effect, thereby enhancing the intrinsic per-site activity. More exposed active sites will promote the electrochemical reaction, which is believed to maximize extrinsic activity. Advanced nanoarchitectures, such as 2D nanosheets, hierarchical structures, and porous structures are advantageous to facilitate charge/mass transportation. Moreover, the compositions and the local coordination environments are also needed to disclose their importance. These mentioned strategies are not single-functioned and not self-governed. A deeper understanding and sensible coupling of these strategies can further improve both the intrinsic and extrinsic catalytic activity of iron-based catalysts, simultaneously.240 According to the first point of perspective, the creation of multi-model active sites may be efficient to improve the catalytic performance. The real function of each site and their distance, interaction, or synergistic effect should be further investigated in depth. Therefore, it is of great significance to develop new synthetic methods to accomplish the above coupling strategies and analyze the design principles that would provide great opportunities for rational exploration of superior NRR electrocatalysts.4.3 Establishing viable and standard protocols for electrocatalytic NRR evaluation
Up to now, the researchers have used different methods to quantify produced ammonia, including commonly used colorimetry (indophenol blue and Nessler's reagent), ion chromatography, and NMR.241 Unfortunately, some of them are not completely reliable. For example, ion chromatography detection is susceptible to interference from other cations (e.g. Li+, K+, etc.). Therefore, it is recommended to integrate multiple methods under repeatable experiments to produce meaningful data with an error bar. In order to exclude exogenous nitrogen pollutants (NH3, nitrate/nitrite, and nitrous oxide), the N2 feed gas, electrolytes, and electrocatalysts need to be purified for NRR tests. It is necessary to conduct isotope (15N2) labelling experiments to identify the origin of the ammonia, a key experiment to verify the true electrocatalytic positive. Moreover, the configurations of the electrolytic cell may be further studied to guide its rational design. The flow cell within an enclosed system may be better to ensure a continuous supply of nitrogen with enough solubility to the catalyst surface. It will also realize the cycling of 15N2 to reduce both economic cost and contamination. It is also urgently required to develop more selective, precise, real-time on-line methods for NH3 determination. Therefore, it is a burdensome system engineering to standardize the evaluation protocol, yet, is of great significance for materials comparison, and this NRR field is rapidly growing.4.4 Combining theoretical calculations and in situ characterizations
The true reaction pathway in electrocatalytic NRR is expected to unravel from fundamental research that integrates experiments and theoretical calculations. In situ/operando measurements by various techniques, such as surface-enhanced infrared absorption spectroscopy, Raman, near ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS), X-ray absorption spectroscopy (XAS) by synchrotron radiation and others, can directly define *NxHy intermediate products, revealing the actual catalytic active sites and even understand the deactivation reasons.242 These will help researchers to clarify the elementary reactions during the NRR process and build up more valid structure models for theoretical investigations. Great endeavors will be devoted to investigating the adsorption strength of different intermediates, and the Gibbs free energy change during NRR, helping to reveal the mechanism from a theoretical view. Improving the resolution of these operando techniques and providing a programmable measurement database are needed. Different in situ methods with varying functions are recommended for in-depth investigations. The combination of theoretical calculations and in situ characterizations would bridge the gap between mechanistic thoughts and experimental electrocatalytic performances.243 It will further underpin the structure–property relationships and guide new directions for electrocatalytic materials design.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (No. 22271036), the Fundamental Research Funds for the Central Universities of China (DUT22LK15), and the Open Funds of the State Key Laboratory of Rare Earth Resource Utilization (RERU2022011).Notes and references
- J. Deng, J. A. Iñiguez and C. Liu, Joule, 2018, 2, 846–856 CrossRef CAS
.
- S. Chen, X. Liu, J. Xiong, L. Mi, X.-Z. Song and Y. Li, J. Mater. Chem. A, 2022, 10, 6927–6949 RSC
- A. Liu, Y. Yang, X. Ren, Q. Zhao, M. Gao, W. Guan, F. Meng, L. Gao, Q. Yang, X. Liang and T. Ma, ChemSusChem, 2020, 13, 3766–3788 CrossRef CAS PubMed
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS
- F. Rehman, M. Delowar Hossain, A. Tyagi, D. Lu, B. Yuan and Z. Luo, Mater. TodayMater. Today, 2021, 44, 136–167 CrossRef CAS
- M. Arif, M. Babar, U. Azhar, M. Sagir, M. Bilal Tahir, M. Asim Mushtaq, G. Yasin, M. Mubashir, J. Wei Roy Chong, K. Shiong Khoo and P. Loke Show, Chem. Eng. J., 2023, 451, 138320 CrossRef CAS
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC
- Q. Wang, J. Guo and P. Chen, J. Energy Chem., 2019, 36, 25–36 CrossRef
- K. Ithisuphalap, H. Zhang, L. Guo, Q. Yang, H. Yang and G. Wu, Small Methods, 2018, 3, 1800352 CrossRef
- T. Wu, W. Fan, Y. Zhang and F. Zhang, Mater. Today Phys., 2021, 16, 100310 CrossRef CAS
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, EnergyChem, 2019, 1, 100011 CrossRef
- L. Niu, L. An, X. Wang and Z. Sun, J. Energy Chem., 2021, 61, 304–318 CrossRef CAS
- S. Zhang, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, EnergyChem, 2019, 1, 100013 CrossRef
- V. Rosca, M. Duca, M. T. de Groot and M. T. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed
- C. Zhang, Z. Wang, J. Lei, L. Ma, B. I. Yakobson and J. M. Tour, Small, 2022, 18, e2106327 CrossRef PubMed
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS
- L. Du, L. Xing, G. Zhang, X. Liu, D. Rawach and S. Sun, SusMat, 2021, 1, 150–173 CrossRef CAS
- Y. Yao, J. Wang, U. B. Shahid, M. Gu, H. Wang, H. Li and M. Shao, Electrochem. Energy Rev., 2020, 3, 239–270 CrossRef CAS
- X. Zhu, S. Mou, Q. Peng, Q. Liu, Y. Luo, G. Chen, S. Gao and X. Sun, J. Mater. Chem. A, 2020, 8, 1545–1556 RSC
- X. W. Lv, C. C. Weng and Z. Y. Yuan, ChemSusChem, 2020, 13, 3061–3078 CrossRef CAS PubMed
- B. Ma, H. Zhao, T. Li, Q. Liu, Y. Luo, C. Li, S. Lu, A. M. Asiri, D. Ma and X. Sun, Nano Res., 2020, 14, 555–569 CrossRef
- Y. Pang, C. Su, G. Jia, L. Xu and Z. Shao, Chem. Soc. Rev., 2021, 50, 12744–12787 RSC
- Y. Fu, T. Li, G. Zhou, J. Guo, Y. Ao, Y. Hu, J. Shen, L. Liu and X. Wu, Nano Lett., 2020, 20, 4960–4967 CrossRef CAS PubMed
- X. Guo, X. Wan and J. Shui, Cell Rep. Phys. Sci.Cell Rep. Phy. Sci., 2021, 2, 100447 CrossRef CAS
- L. Zhang, X. Xue, M. Gao, J. Zhao, T. Yan, C. Yu, L. Zhao, X. Ren and Q. Wei, New J. Chem., 2021, 45, 11457–11460 RSC
- W. Zhang, K. Mao, J. Low, H. Liu, Y. Bo, J. Ma, Q. Liu, Y. Jiang, J. Yang, Y. Pan, Z. Qi, R. Long, L. Song and Y. Xiong, Nano Res., 2021, 14, 3234–3239 CrossRef CAS
- Y. Zhang, J. Hu, C. Zhang, Q. Qi, S. Luo, K. Chen, L. Liu and M. K. H. Leung, J. Mater. Chem. A, 2021, 9, 5060–5066 RSC
- H. Wang, J. Si, T. Zhang, Y. Li, B. Yang, Z. Li, J. Chen, Z. Wen, C. Yuan, L. Lei and Y. Hou, Appl. Catal., B, 2020, 270, 118892 CrossRef CAS
- R. Zhao, C. Liu, X. Zhang, X. Zhu, P. Wei, L. Ji, Y. Guo, S. Gao, Y. Luo, Z. Wang and X. Sun, J. Mater. Chem. A, 2020, 8, 77–81 RSC
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677 RSC
- H.-M. Liu, S.-H. Han, Y. Zhao, Y.-Y. Zhu, X.-L. Tian, J.-H. Zeng, J.-X. Jiang, B. Y. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC
- H. Bai, F. Wang, Y. Liu, C. Ma, J. Ding and W. Fan, Colloids Surf., A, 2022, 655, 130312 CrossRef CAS
- C. Lv, C. Yan, G. Chen, Y. Ding, J. Sun, Y. Zhou and G. Yu, Angew. Chem., Int. Ed., 2018, 130, 6181–6184 CrossRef
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lu, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS PubMed
- X. Zhao, G. Hu, G. F. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, e2007650 CrossRef PubMed
- Z. Wang, F. Gong, L. Zhang, R. Wang, L. Ji, Q. Liu, Y. Luo, H. Guo, Y. Li, P. Gao, X. Shi, B. Li, B. Tang and X. Sun, Adv. Sci., 2019, 6, 1801182 CrossRef PubMed
- P. Wei, H. Xie, X. Zhu, R. Zhao, L. Ji, X. Tong, Y. Luo, G. Cui, Z. Wang and X. Sun, ACS Sustainable Chem. Eng., 2019, 8, 29–33 CrossRef
- P. Zhao, Z. Lu and S. Liu, J. Nanosci. Nanotechnol., 2018, 18, 3348–3355 CrossRef CAS PubMed
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. Wang, Small Methods, 2018, 3, 1800376 CrossRef
- C. N. R. Rao and G. Ranga Rao, Surf. Sci. Rep., 1991, 13, 223–263 CrossRef
- Y. Li, Y. Ji, Y. Zhao, J. Chen, S. Zheng, X. Sang, B. Yang, Z. Li, L. Lei, Z. Wen, X. Feng and Y. Hou, Adv. Mater., 2022, 34, e2202240 CrossRef PubMed
- D. Zheng, N. Wang, M. Wang, S. Ding, C. Ma, M. Y. Darensbourg, M. B. Hall and L. Sun, J. Am. Chem. Soc., 2014, 136, 16817–16823 CrossRef CAS PubMed
- J. Luo, Y. Hu, L. Xiao, G. Zhang, H. Guo, G. Hao and W. Jiang, Nanotechnology, 2019, 31, 085708 CrossRef PubMed
- X. Cui, C. Tang, X. M. Liu, C. Wang, W. Ma and Q. Zhang, Chem.–Eur. J., 2018, 24, 18494–18501 CrossRef CAS PubMed
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed
- M. Yang, Z. Jin, C. Wang, X. Cao, X. Wang, H. Ma, H. Pang, L. Tan and G. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 55040–55050 CrossRef CAS PubMed
- Q. Li, Y. Guo, Y. Tian, W. Liu and K. Chu, J. Mater. Chem. A, 2020, 8, 16195–16202 RSC
- W. Tong, B. Huang, P. Wang, Q. Shao and X. Huang, Natl. Sci. Rev., 2021, 8, nwaa088 CrossRef CAS PubMed
- H. Ying, T. Chen, C. Zhang, J. Bi, Z. Li and J. Hao, J. Colloid Interface Sci., 2021, 602, 64–72 CrossRef CAS PubMed
- H. Q. Xie, X. Zheng, Q. Y. Feng, X. P. Chen, Z. H. Zou, Q. X. Wang, J. Tang, Y. Li and Y. Ling, ChemSusChem, 2022, 15, e202200919 CAS
- X. Wang, J. Zheng, P. Li, X. B. Yin, S. Wang, B. Zhang, J. Xu and M. Zhang, Dalton Trans., 2022, 51, 3170–3179 RSC
- H. Wang, Z. Li, Y. Li, B. Yang, J. Chen, L. Lei, S. Wang and Y. Hou, Nano Energy, 2021, 81, 105613 CrossRef CAS
- Y. Zhao, F. Li, W. Li, Y. Li, C. Liu, Z. Zhao, Y. Shan, Y. Ji and L. Sun, Angew. Chem., Int. Ed., 2021, 60, 20331–20341 CrossRef CAS PubMed
- Y. Guo, Z. Yao, B. J. J. Timmer, X. Sheng, L. Fan, Y. Li, F. Zhang and L. Sun, Nano Energy, 2019, 62, 282–288 CrossRef CAS
- X. Wang, Z. Feng, B. Xiao, J. Zhao, H. Ma, Y. Tian, H. Pang and L. Tan, Green Chem., 2020, 22, 6157–6169 RSC
- H.-B. Wang, J.-Q. Wang, R. Zhang, C.-Q. Cheng, K.-W. Qiu, Y.-j. Yang, J. Mao, H. Liu, M. Du, C.-K. Dong and X.-W. Du, ACS Catal., 2020, 10, 4914–4921 CrossRef CAS
- K. Chu, W. Gu, Q. Li, Y. Liu, Y. Tian and W. Liu, J. Energy Chem., 2021, 53, 82–89 CrossRef CAS
- Q. Li, C. Liu, S. Qiu, F. Zhou, L. He, X. Zhang and C. Sun, J. Mater. Chem. A, 2019, 7, 21507–21513 RSC
- X. Li, P. Shen, Y. Luo, Y. Li, Y. Guo, H. Zhang and K. Chu, Angew. Chem., Int. Ed., 2022, 61, e202205923 CAS
- J. Chen, Y. Kang, W. Zhang, Z. Zhang, Y. Chen, Y. Yang, L. Duan, Y. Li and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202203022 CAS
- L. Zhang, G. F. Chen, L. X. Ding and H. Wang, Chem.–Eur. J., 2019, 25, 12464–12485 CrossRef CAS PubMed
- K. C. Macleod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed
- C. J. van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC
- H. P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Norskov, Science, 2005, 307, 555–558 CrossRef CAS PubMed
- S. Wang, F. Ichihara, H. Pang, H. Chen and J. Ye, Adv. Funct. Mater., 2018, 28, 1803309 CrossRef
- X. L. Ma, J. C. Liu, H. Xiao and J. Li, J. Am. Chem. Soc., 2018, 140, 46–49 CrossRef CAS PubMed
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef
- J. G. Howalt, T. Bligaard, J. Rossmeisl and T. Vegge, Phys. Chem. Chem. Phys., 2013, 15, 7785–7795 RSC
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Norskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC
- X. F. Li, Q. K. Li, J. Cheng, L. Liu, Q. Yan, Y. Wu, X. H. Zhang, Z. Y. Wang, Q. Qiu and Y. Luo, J. Am. Chem. Soc., 2016, 138, 8706–8709 CrossRef CAS PubMed
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019, 27, 69–90 CrossRef CAS
- C. Ling, X. Bai, Y. Ouyang, A. Du and J. Wang, J. Phys. Chem. C, 2018, 122, 16842–16847 CrossRef CAS
- G. Liu, L. Niu, Z. Ma, L. An, D. Qu, D. Wang, X. Wang and Z. Sun, Nano Res., 2022, 15, 5940–5945 CrossRef CAS
- Z. Wei, J. He, Y. Yang, Z. Xia, Y. Feng and J. Ma, J. Energy Chem., 2021, 53, 303–308 CrossRef CAS
- A. Jain, M. Bar Sadan and A. Ramasubramaniam, J. Phys. Chem. C, 2021, 125, 19980–19990 CrossRef CAS
- D. Yao, C. Tang, L. Li, B. Xia, A. Vasileff, H. Jin, Y. Zhang and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 2001289 CrossRef CAS
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, Angew. Chem., Int. Ed., 2020, 59, 10479–10483 CrossRef CAS PubMed
- M. A. Legare, G. Belanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef CAS PubMed
- P. Chen, Y. Tong, C. Wu and Y. Xie, Acc. Chem. Res., 2018, 51, 2857–2866 CrossRef CAS PubMed
- C. Tang, H. F. Wang and Q. Zhang, Acc. Chem. Res., 2018, 51, 881–889 CrossRef CAS PubMed
- C. Yang, Y. Zhu, J. Liu, Y. Qin, H. Wang, H. Liu, Y. Chen, Z. Zhang and W. Hu, Nano Energy, 2020, 77, 105126 CrossRef CAS
- Z. He, Y. Jiang, X. Cui, Z. Liu, X. Meng, J. Wan and F. Ma, ACS Appl. Nano Mater., 2022, 5, 5470–5478 CrossRef CAS
- J. Wang, G. Li, T. Wei, S. Zhou, X. Ji and X. Liu, Nanoscale, 2021, 13, 3036–3041 RSC
- Y. Ling, F. M. D. Kazim, S. Ma, Q. Zhang, K. Qu, Y. Wang, S. Xiao, W. Cai and Z. Yang, J. Mater. Chem. A, 2020, 8, 12996–13003 RSC
- Y. J. Mao, L. Wei, X. S. Zhao, Y. S. Wei, J. W. Li, T. Sheng, F. C. Zhu, N. Tian, Z. Y. Zhou and S. G. Sun, Chem. Commun., 2019, 55, 9335–9338 RSC
- S. Dou, X. Wang and S. Wang, Small Methods, 2019, 3, 1800211 CrossRef
- H. Fei, T. Guo, Y. Xin, L. Wang, R. Liu, D. Wang, F. Liu and Z. Wu, Appl. Catal., B, 2022, 300, 120733 CrossRef CAS
- D. Guo, S. Wang, J. Xu, W. Zheng and D. Wang, J. Energy Chem., 2022, 65, 448–468 CrossRef CAS
- K. Chu, Y.-p. Liu, Y.-b. Li, H. Zhang and Y. Tian, J. Mater. Chem. A, 2019, 7, 4389–4394 RSC
- D. Bao, Q. Zhang, F. L. Meng, H. X. Zhong, M. M. Shi, Y. Zhang, J. M. Yan, Q. Jiang and X. B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed
- B. Cui, J. Zhang, S. Liu, X. Liu, W. Xiang, L. Liu, H. Xin, M. J. Lefler and S. Licht, Green Chem., 2017, 19, 298–304 RSC
- M. Zhao, C. Guo, L. Gao, H. Yang, C. Liu, X. Kuang, X. Sun and Q. Wei, ChemCatChem, 2021, 13, 4990–4997 CrossRef CAS
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed
- J. Li, X. Zhu, T. Wang, Y. Luo and X. Sun, Inorg. Chem. Front., 2019, 6, 2682–2685 RSC
- B. Xu, L. Xia, F. Zhou, R. Zhao, H. Chen, T. Wang, Q. Zhou, Q. Liu, G. Cui, X. Xiong, F. Gong and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 2889–2893 CrossRef CAS
- F. Lai, J. Feng, X. Ye, W. Zong, G. He, C. Yang, W. Wang, Y.-E. Miao, B. Pan, W. Yan, T. Liu and I. P. Parkin, J. Mater. Chem. A, 2020, 8, 1652–1659 RSC
- C. Wang, L.-L. Gu, S.-Y. Qiu, J. Gao, Y.-C. Zhang, K.-X. Wang, J.-J. Zou, P.-J. Zuo and X.-D. Zhu, Appl. Catal., B, 2021, 297, 120452 CrossRef CAS
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed
- L. Hu, A. Khaniya, J. Wang, G. Chen, W. E. Kaden and X. Feng, ACS Catal., 2018, 8, 9312–9319 CrossRef CAS
- J. Wang, S. Chen, Z. Li, G. Li and X. Liu, ChemElectroChem, 2020, 7, 1067–1079 CrossRef CAS
- K. Kim, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F1523–F1526 CrossRef CAS
- B. Wang, T. Li, F. Gong, M. H. D. Othman and R. Xiao, Fuel Process. Technol., 2022, 235, 107380 CrossRef CAS
- F. Zhou, L. M. Azofra, M. Ali, M. Kar, A. N. Simonov, C. McDonnell-Worth, C. Sun, X. Zhang and D. R. MacFarlane, Energy Environ. Sci., 2017, 10, 2516–2520 RSC
- S. Stevanovic and M. F. Costa Gomes, J. Chem. Thermodyn., 2013, 59, 65–71 CrossRef CAS
- C. S. M. Kang, X. Zhang and D. R. MacFarlane, J. Phys. Chem. C, 2018, 122, 24550–24558 CrossRef CAS
- B. H. R. Suryanto, C. S. M. Kang, D. Wang, C. Xiao, F. Zhou, L. M. Azofra, L. Cavallo, X. Zhang and D. R. MacFarlane, ACS Energy Lett., 2018, 3, 1219–1224 CrossRef CAS
- S. Sun, X. Zhang, J. Cui, Q. Yang and S. Liang, Nanoscale, 2019, 11, 15739–15762 RSC
- W. Zhang, Y. Shen, F. Pang, D. Quek, W. Niu, W. Wang and P. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 41613–41619 CrossRef CAS PubMed
- A. Xie, L. Xiao, Q. Qiao and J. Liu, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2022, 37, 807–814 CrossRef CAS
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed
- C. Xiao, B.-A. Lu, P. Xue, N. Tian, Z.-Y. Zhou, X. Lin, W.-F. Lin and S.-G. Sun, Joule, 2020, 4, 2562–2598 CrossRef CAS
- S. Yu, Q. Wang, J. Wang, Y. Xiang, X. Niu and T. Li, Int. J. Hydrogen Energy, 2021, 46, 14331–14337 CrossRef CAS
- C. Zhang, S. Liu, T. Chen, Z. Li and J. Hao, Chem. Commun., 2019, 55, 7370–7373 RSC
- M. Wang, F. Li and J. Liu, RSC Adv., 2020, 10, 29575–29579 RSC
- Z. Wang, K. Zheng, S. Liu, Z. Dai, Y. Xu, X. Li, H. Wang and L. Wang, ACS Sustainable Chem. Eng., 2019, 7, 11754–11759 CrossRef CAS
- K. Chu, H. Nan, Q. Li, Y. Guo, Y. Tian and W. Liu, J. Energy Chem., 2021, 53, 132–138 CrossRef CAS
- J. Wang, H. Nan, Y. Tian and K. Chu, ACS Sustainable Chem. Eng., 2020, 8, 12733–12740 CrossRef CAS
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, e1800191 CrossRef PubMed
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed
- X.-f. Zheng, J.-l. Xia, Z.-y. Nie, Y. Wang, Y. Yu, N. Ei Houda Bouroubi, J.-h. Chen and H.-c. Liu, Chem. Eng. J., 2022, 442, 136350 CrossRef CAS
- X. Zhao, X. Lan, D. Yu, H. Fu, Z. Liu and T. Mu, Chem. Commun., 2018, 54, 13010–13013 RSC
- W. Xiong, Z. Guo, S. Zhao, Q. Wang, Q. Xu and X. Wang, J. Mater. Chem. A, 2019, 7, 19977–19983 RSC
- D. Feng, X. Zhang, Y. Sun and T. Ma, Nano Mater. Sci., 2020, 2, 132–139 CrossRef
- H. Du, C. Yang, W. Pu, L. Zeng and J. Gong, ACS Sustainable Chem. Eng., 2020, 8, 10572–10580 CrossRef CAS
- T. Chen, H. Ying, C. Zhang, J. Bi, Z. Li and J. Hao, Chem. Commun., 2021, 57, 6688–6691 RSC
- L. Gao, C. Guo, M. Zhao, H. Yang, X. Ma, C. Liu, X. Liu, X. Sun and Q. Wei, ACS Appl. Mater. Interfaces, 2021, 13, 50027–50036 CrossRef CAS PubMed
- Y. Guo, Y. Cheng, Q. Li and K. Chu, J. Energy Chem., 2021, 56, 259–263 CrossRef CAS
- Z. Ma, C. Xiao, Z. Cui, W. Du, Q. Li, R. Sa and C. Sun, J. Mater. Chem. A, 2021, 9, 6945–6954 RSC
- S. Yang, W. Ye, D. Zhang, X. Fang and D. Yan, Inorg. Chem. Front., 2021, 8, 1762–1770 RSC
- Y. Sun, S. Ding, B. Xia, J. Duan, M. Antonietti and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e202115198 CAS
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef
- S. Chen, X. Liu, J. Xiong, L. Mi and Y. Li, Mater. Today Nano, 2022, 18, 100202 CrossRef CAS
- N. R. Singstock and C. B. Musgrave, J. Am. Chem. Soc., 2022, 144, 12800–12806 CrossRef CAS PubMed
- K. Lu, F. Xia, B. Li, Y. Liu, I. B. Abdul Razak, S. Gao, J. Kaelin, D. E. Brown and Y. Cheng, ACS Nano, 2021, 15, 16887–16895 CrossRef CAS PubMed
- L. Zeng, S. Chen, J. van der Zalm, X. Li and A. Chen, Chem. Commun., 2019, 55, 7386–7389 RSC
- J. Zhang, X. Tian, M. Liu, H. Guo, J. Zhou, Q. Fang, Z. Liu, Q. Wu and J. Lou, J. Am. Chem. Soc., 2019, 141, 19269–19275 CrossRef CAS PubMed
- L. Niu, D. Wang, K. Xu, W. Hao, L. An, Z. Kang and Z. Sun, Nano Res., 2021, 14, 4093–4099 CrossRef CAS
- J. Guo, T. Tadesse Tsega, I. Ul Islam, A. Iqbal, J. Zai and X. Qian, Chin. Chem. Lett., 2020, 31, 2487–2490 CrossRef CAS
- H. Y. Su, L. L. Chen, Y. Z. Chen, R. Si, Y. T. Wu, X. N. Wu, Z. G. Geng, W. H. Zhang and J. Zeng, Angew. Chem., Int. Ed., 2020, 59, 20411–20416 CrossRef CAS PubMed
- Z. Y. Niu, L. Jiao, T. Zhang, X. M. Zhao, X. F. Wang, Z. Tan, L. Z. Liu, S. Chen and X. Z. Song, ACS Appl. Mater. Interfaces, 2022, 14, 55559–55567 CrossRef CAS PubMed
- H. Wang, W. Fu, X. Yang, Z. Huang, J. Li, H. Zhang and Y. Wang, J. Mater. Chem. A, 2020, 8, 6926–6956 RSC
- M. Kim, M. A. R. Anjum, M. Choi, H. Y. Jeong, S. H. Choi, N. Park and J. S. Lee, Adv. Funct. Mater., 2020, 30, 2002536 CrossRef CAS
- J. Liu, W. Kong, Z. Jin, Y. Han, J. Sun, L. Ma, Y. Niu and Y. Xu, J. Mater. Chem. A, 2020, 8, 19278–19282 RSC
- X.-Z. Song, F.-F. Sun, Y.-L. Meng, Z.-W. Wang, Q.-F. Su and Z. Tan, New J. Chem., 2019, 43, 3601–3608 RSC
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed
- Y. Xu, X. Jiang, G. Shao, H. Xiang, S. Si, X. Li, T. S. Hu, G. Hong, S. Dong, H. Li, Y. Feng and S. Liu, Energy Environ. Mater., 2020, 4, 117–125 CrossRef
- C. Ma, D. Liu, Y. Zhang, J. Yong Lee, J. Tian, B. Liu and S. Yan, Chem. Eng. J., 2022, 430, 132694 CrossRef CAS
- X. Z. Song, W. Y. Zhu, J. C. Ni, Y. H. Zhao, T. Zhang, Z. Tan, L. Z. Liu and X. F. Wang, ACS Appl. Mater. Interfaces, 2022, 14(29), 33151–33160 CrossRef CAS PubMed
- X. Zhao, X. Zhang, Z. Xue, W. Chen, Z. Zhou and T. Mu, J. Mater. Chem. A, 2019, 7, 27417–27422 RSC
- E. Hu, Y. Yao, Y. Cui and G. Qian, Mater. Today Nano, 2021, 15, 100124 CrossRef CAS
- C. Li, X. Sun, Y. Yao and G. Hong, Mater. Today Nano, 2021, 13, 100105 CrossRef CAS
- W. Ma, F. Wu, P. Yu and L. Mao, Chem. Sci., 2021, 12, 7908–7917 RSC
- Z. Li, M. Song, W. Zhu, W. Zhuang, X. Du and L. Tian, Coord. Chem. Rev., 2021, 439, 213946 CrossRef CAS
- X. Z. Song, N. Zhang, X. F. Wang and Z. Tan, Mater. Today Energy, 2021, 19, 100597 CrossRef CAS
- Y. Yang, S. Feng, J. Su, Y. Gong and J. Wang, ACS Sustainable Chem. Eng., 2022, 10, 14064–14072 CrossRef CAS
- Y. Li, Z. Yin, M. Cui, S. Chen and T. Ma, Mater. Today Energy, 2020, 18, 100565 CrossRef CAS
- S. Chen, M. Cui, Z. Yin, J. Xiong, L. Mi and Y. Li, ChemSusChem, 2021, 14, 73–93 CrossRef CAS PubMed
- X.-Z. Song, Y.-H. Zhao, W.-B. Yang, Y.-L. Meng, X. Chen, Z.-Y. Niu, X.-F. Wang and Z. Tan, ACS Appl. Nano Mater., 2021, 4, 13450–13458 CrossRef CAS
- X. Z. Song, H. Wang, Z. Li, Y. L. Meng, Z. Tan and M. Zhu, Chem. Commun., 2021, 57, 3022–3025 RSC
- X. Song, S. Song, D. Wang and H. Zhang, Small Methods, 2021, 5, e2001000 CrossRef PubMed
- H. He, H.-M. Wen, H.-K. Li and H.-W. Zhang, Coord. Chem. Rev., 2022, 471, 214761 CrossRef CAS
- K. A. Adegoke, O. R. Adegoke, R. A. Adigun, N. W. Maxakato and O. S. Bello, Coord. Chem. Rev., 2022, 473, 214817 CrossRef CAS
- L. Xiao, Z. Wang and J. Guan, Coord. Chem. Rev., 2022, 472, 214777 CrossRef CAS
- L. Zou, Y. S. Wei, C. C. Hou, C. Li and Q. Xu, Small, 2021, 17, e2004809 CrossRef PubMed
- Y. Pan, R. Abazari, Y. Wu, J. Gao and Q. Zhang, Electrochem. Commun., 2021, 126, 107024 CrossRef CAS
- Z. H. Wang, X. F. Wang, Z. Tan and X. Z. Song, Mater. Today Energy, 2021, 19, 100618 CrossRef CAS
- S. Chen, H. Jang, J. Wang, Q. Qin, X. Liu and J. Cho, J. Mater. Chem. A, 2020, 8, 2099–2104 RSC
- L. Zeng, X. Li, S. Chen, J. Wen, W. Huang and A. Chen, J. Mater. Chem. A, 2020, 8, 7339–7349 RSC
- B. Li, X. Yu, H. Pang, Q. Shen, Y. Hou, H. Ma and J. Xin, Chem. Commun., 2020, 56, 7199–7202 RSC
- G. Wang, T. Chen, C. J. Gomez-Garcia, F. Zhang, M. Zhang, H. Ma, H. Pang, X. Wang and L. Tan, Small, 2020, 16, e2001626 CrossRef PubMed
- J. Wu, Z. Wang, S. Li, S. Niu, Y. Zhang, J. Hu, J. Zhao and P. Xu, Chem. Commun., 2020, 56, 6834–6837 RSC
- K. Chu, Q. Q. Li, Y. H. Cheng and Y. P. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 11789–11796 CrossRef CAS PubMed
- H. Xian, H. Guo, Z. Chen, G. Yu, A. A. Alshehri, K. A. Alzahrani, F. Hao, R. Song and T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 2445–2451 CrossRef CAS PubMed
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef
- J. Yao, Y. Zhou, J. M. Yan and Q. Jiang, Adv. Energy Mater., 2021, 11, 2003701 CrossRef CAS
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed
- J. D. Lee, J. B. Miller, A. V. Shneidman, L. Sun, J. F. Weaver, J. Aizenberg, J. Biener, J. A. Boscoboinik, A. C. Foucher, A. I. Frenkel, J. E. S. van der Hoeven, B. Kozinsky, N. Marcella, M. M. Montemore, H. T. Ngan, C. R. O'Connor, C. J. Owen, D. J. Stacchiola, E. A. Stach, R. J. Madix, P. Sautet and C. M. Friend, Chem. Rev., 2022, 122, 8758–8808 CrossRef CAS PubMed
- A. Das, A. S. Nair, S. C. Mandal and B. Pathak, ACS Appl. Nano Mater., 2021, 4, 7758–7770 CrossRef CAS
- R. Chen, D. Chen and Y. Xiao, Mater. Today Energy, 2021, 20, 100684 CrossRef CAS
- A. Das, S. C. Mandal, A. S. Nair and B. Pathak, ACS Phys. Chem. Au, 2022, 2, 125–135 CrossRef CAS PubMed
- J. Zhang, Y. Ji, P. Wang, Q. Shao, Y. Li and X. Huang, Adv. Funct. Mater., 2019, 30, 1906579 CrossRef
- X. Chen, H. Yin, X. Yang, W. Zhang, D. Xiao, Z. Lu, Y. Zhang and P. Zhang, Inorg. Chem., 2022, 61, 20123–20132 CrossRef CAS PubMed
- C. Huang, L. Shang, P. Han, Z. Gu, A. M. Al-Enizi, T. M. Almutairi, N. Cao and G. Zheng, J. Colloid Interface Sci., 2019, 552, 312–318 CrossRef CAS PubMed
- A. Biswas, S. Bhardwaj, T. Boruah and R. S. Dey, Mater. Adv., 2022, 3, 5207–5233 RSC
- S. Carenco, D. Portehault, C. Boissiere, N. Mezailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS PubMed
- X. Yang, C. Shang, S. Zhou and J. Zhao, Nanoscale Horiz., 2020, 5, 1106–1115 RSC
- S. Zhou, X. Yang, W. Pei, Z. Jiang and J. Zhao, JPhys Energy, 2020, 3, 012002 CrossRef
- F. Ma, Y. Jiao, G. Gao, Y. Gu, A. Bilic, Z. Chen and A. Du, Nano Lett., 2016, 16, 3022–3028 CrossRef CAS PubMed
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed
- X. Liu, Y. Jiao, Y. Zheng and S.-Z. Qiao, ACS Catal., 2020, 10, 1847–1854 CrossRef CAS
- Z. Chen, J. Zhao, L. Yin and Z. Chen, J. Mater. Chem. A, 2019, 7, 13284–13292 RSC
- X. Mao, S. Zhou, C. Yan, Z. Zhu and A. Du, Phys. Chem. Chem. Phys., 2019, 21, 1110–1116 RSC
- W. Song, Z. Fu, L. Fu and C. He, J. Electrochem. Soc., 2021, 168, 066503 CrossRef CAS
- G. Qin, Q. Cui, A. Du, W. Wang and Q. Sun, ChemCatChem, 2019, 11, 2624–2633 CrossRef CAS
- Z. Pu, T. Liu, G. Zhang, X. Liu, M. A. Gauthier, Z. Chen and S. Sun, Small Methods, 2021, 5, e2100699 CrossRef PubMed
- L. Ge, W. Xu, C. Chen, C. Tang, L. Xu and Z. Chen, J. Phys. Chem. Lett., 2020, 11, 5241–5247 CrossRef CAS
- S. Qi, Y. Fan, L. Zhao, W. Li and M. Zhao, Appl. Surf. Sci., 2021, 536, 147742 CrossRef CAS
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W. C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, e1800396 CrossRef
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS
- Z. Fu, M. Wu, Q. Li, C. Ling and J. Wang, Mater. Horiz., 2023, 10, 852–858 RSC
- L. Li, W. Yu, W. Gong, H. Wang, C.-L. Chiang, Y. Lin, J. Zhao, L. Zhang, J.-M. Lee and G. Zou, Appl. Catal., B, 2023, 321, 122038 CrossRef CAS
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, e1801995 CrossRef PubMed
- X. Li, P. Cui, W. Zhong, J. Li, X. Wang, Z. Wang and J. Jiang, Chem. Commun., 2016, 52, 13233–13236 RSC
- H. Jeong, S. Shin and H. Lee, ACS Nano, 2020, 14, 14355–14374 CrossRef CAS PubMed
- H. Yang, Y. Liu, Y. Luo, S. Lu, B. Su and J. Ma, ACS Sustainable Chem. Eng., 2020, 8, 12809–12816 CrossRef CAS
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef
- R. Qin, P. Liu, G. Fu and N. Zheng, Small Methods, 2018, 2, 1700286 CrossRef
- Y. Liu, Z. Zhao, W. Wei, X. Jin, G. Wang, K. Li and Y. Lin, ACS Appl. Nano Mater., 2021, 4, 13001–13009 CrossRef CAS
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS
- X. Guo and S. Huang, Electrochim. Acta, 2018, 284, 392–399 CrossRef CAS
- C. He, Z.-Y. Wu, L. Zhao, M. Ming, Y. Zhang, Y. Yi and J.-S. Hu, ACS Catal., 2019, 9, 7311–7317 CrossRef CAS
- W. Wei, X. Shi, P. Gao, S. Wang, W. Hu, X. Zhao, Y. Ni, X. Xu, Y. Xu, W. Yan, H. Ji and M. Cao, Nano Energy, 2018, 52, 29–37 CrossRef CAS
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 341 CrossRef CAS PubMed
- D. Liu, H. Ai, W. T. Lou, F. Li, K. H. Lo, S. Wang and H. Pan, Sustainable Energy Fuels, 2020, 4, 3773–3779 RSC
- B. Wang, S. Huang, L. Yang, Q. Fu and Y. Bu, J. Phys. Chem. C, 2021, 125, 14253–14262 CrossRef CAS
- W. Shan and G. Wang, J. Phys. Chem. C, 2021, 125, 16004–16012 CrossRef CAS
- Y. Wang, H. Li, F. Peng and F. Gao, Chem. Eng. J., 2022, 445, 136692 CrossRef CAS
- H. Zhang, C. Cui and Z. Luo, J. Phys. Chem. C, 2020, 124, 6260–6266 CrossRef CAS
- J. Li, S. Chen, F. Quan, G. Zhan, F. Jia, Z. Ai and L. Zhang, Chem, 2020, 6, 885–901 CAS
- X. Lv, W. Wei, B. Huang, Y. Dai and T. Frauenheim, Nano Lett., 2021, 21, 1871–1878 CrossRef CAS PubMed
- H. Li, Z. Zhao, Q. Cai, L. Yin and J. Zhao, J. Mater. Chem. A, 2020, 8, 4533–4543 RSC
- F. Li and Q. Tang, Nanoscale, 2019, 11, 18769–18778 RSC
- W. Ren, X. Tan, J. Qu, S. Li, J. Li, X. Liu, S. P. Ringer, J. M. Cairney, K. Wang, S. C. Smith and C. Zhao, Nat. Commun., 2021, 12, 1449 CrossRef CAS PubMed
- D. Ma, Z. Zeng, L. Liu and Y. Jia, J. Energy Chem., 2021, 54, 501–509 CrossRef CAS
- D. Ma, Z. Zeng, L. Liu, X. Huang and Y. Jia, J. Phys. Chem. C, 2019, 123, 19066–19076 CrossRef CAS
- W. Cui, B. Geng, X. Chu, J. He, L. Jia, X. Han, X. Wang, S. Song and H. Zhang, Nano Res., 2022 DOI:10.1007/s12274-022-5246-x
- S. Gao, X. Liu, Z. Wang, Y. Lu, R. Sa, Q. Li, C. Sun, X. Chen and Z. Ma, J. Colloid Interface Sci., 2023, 630, 215–223 CrossRef CAS PubMed
- S. Zhang, M. Han, T. Shi, H. Zhang, Y. Lin, X. Zheng, L. R. Zheng, H. Zhou, C. Chen, Y. Zhang, G. Wang, H. Yin and H. Zhao, Nat. Sustain., 2022, 6, 169–179 CrossRef
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Chem. Soc. Rev., 2021, 50, 569–588 RSC
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed
- Z. Xu, Z. Ao, M. Yang and S. Wang, J. Hazard. Mater., 2022, 424, 127427 CrossRef CAS PubMed
- K. Chu, Y.-h. Cheng, Q.-q. Li, Y.-p. Liu and Y. Tian, J. Mater. Chem. A, 2020, 8, 5865–5873 RSC
- Z. Chen, H. Niu, J. Ding, H. Liu, P. H. Chen, Y. H. Lu, Y. R. Lu, W. Zuo, L. Han, Y. Guo, S. F. Hung and Y. Zhai, Angew. Chem., Int. Ed., 2021, 133, 25608–25614 CrossRef
- J. Chen, H. Cheng, L.-X. Ding and H. Wang, Mater. Chem. Front., 2021, 5, 5954–5969 RSC
- Y. Yang, M. Luo, W. Zhang, Y. Sun, X. Chen and S. Guo, Chem, 2018, 4, 2054–2083 CAS
- I. E. Khalil, C. Xue, W. Liu, X. Li, Y. Shen, S. Li, W. Zhang and F. Huo, Adv. Funct. Mater., 2021, 31, 2010052 CrossRef CAS
- Z. Xu, Z. Liang, W. Guo and R. Zou, Coord. Chem. Rev., 2021, 436, 213824 CrossRef CAS
- Z. Chen, C. Liu, L. Sun and T. Wang, ACS Catal., 2022, 12, 8936–8975 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |