
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biodegradable porous FeMn(–xAg) alloys: assessment of cytocompatibility, mechanical, magnetic and antibiofilm properties†
Aleksandra
Bartkowska
 *a,
Oriol
Careta
b,
Adam Benedict
Turner
c,
Andreu
Blanquer
b,
Elena
Ibañez
*b,
Margarita
Trobos
c,
Carme
Nogués
b,
Eva
Pellicer
*a and
Jordi
Sort
ad
*a,
Oriol
Careta
b,
Adam Benedict
Turner
c,
Andreu
Blanquer
b,
Elena
Ibañez
*b,
Margarita
Trobos
c,
Carme
Nogués
b,
Eva
Pellicer
*a and
Jordi
Sort
ad
aDepartament de Física, Universitat Autònoma de Barcelona, E-08193, Bellaterra (Cerdanyola del Vallès), Spain. E-mail: aleksandra.bartkowska@uab.cat; eva.pellicer@uab.cat
bDepartament de Biologia Cel·lular, Fisiologia i Immunologia, Universitat Autònoma de Barcelona, E-08193, Bellaterra (Cerdanyola del Vallès), Spain. E-mail: elena.ibanez@uab.cat
cDepartment of Biomaterials, Institute of Clinical Sciences, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
dInstitució Catalana de Recerca i Estudis Avançats (ICREA), Pg. Lluís Companys 23, E-08010, Barcelona, Spain
First published on 6th December 2022
Abstract
In this work, porous FeMn(–xAg) alloys are fabricated through powder metallurgy methods. The effects of porosity and Ag addition on the microstructure, biodegradability, magnetic and mechanical properties of the alloys are investigated. Studies on the cytocompatibility, inflammatory cytokine response and antibacterial effect are also conducted. The fabricated alloys exhibit a macro- and nanoporous structure, with uniformly distributed silver particles. The biodegradability tests reveal that the release of Mn to the Hank's solution is higher than that of Fe, without significant differences between the alloys. The degradation products consist mainly of Fe, Mn, O and compounds enriched in Ca, P and Cl. As-sintered alloys show a low saturation magnetization value (below 1 emu g−1), which does not increase significantly with immersion time. The results on biocompatibility indicate that all tested alloys are non-cytotoxic, but the addition of Ag might interfere with cell proliferation. However, the ions released by the FeMn(–xAg) alloys do not induce an inflammatory response in macrophages. The obtained results on microbiological interactions reveal that although no significant bactericidal effect is observed at 4 h between FeMn control and FeMn–5Ag, a significant reduction in the total biofilm biomass of both live and dead bacteria is observed after 24 h in Ag containing FeMn–5Ag surfaces.
1 Introduction
In recent years, the application of biodegradable metals (BMs) and polymers as potential candidates for temporary medical implants has been of growing interest.1–6 This class of materials is expected to corrode gradually in vivo once they have fulfilled their role.2,7 Their main advantage is the possibility of avoiding the implant removal surgery, hence limiting the cost of medical treatment and decreasing the risk of infections, which is increased during the revision surgery.8 It has been shown that biodegradable materials are in general less prone to biomaterial-associated infections than non-degradable materials, most probably due to the degradation processes that reduce the area available for bacterial colonization and, therefore, the formation of biofilm.9 Among various tested alloys, Mg-based and Fe-based alloys are especially attractive due to their very good biocompatibility.2,7 Mg-based alloys show very low toxicity and their Young's modulus is close to that of the human bone. However, their degradation rate is too rapid for some applications, resulting in H2 gas evolution and bubbling upon immersion. In addition, they are hard to process, and their strength and ductility are not satisfactory.Over the past few years, increasing attention has been paid to Fe-based alloys as they show good processability, mechanical strength and low toxicity,10–13 however, the degradation rate is too slow to be applied as biodegradable implants.7,14 Moreover, Fe-based alloys tend to be ferromagnetic, which is undesirable for magnetic resonance imaging (MRI) purposes. Thus, several approaches to improve the degradation rate of Fe-based alloys are being explored, such as alloying with different elements (e.g., Mn),11,13,15 using new fabrication methods to tune the microstructure such as selective laser melting (SLM),16,17 introducing porosity,17–21 adding small amounts of noble elements,22–25 or adding C to promote twinning-induced plasticity (TWIP),7,26–28 amongst others. The addition of elements such as Cu and Ag, which are more noble than Fe or Mn, is being investigated as a way of not only improving corrosion rates (through galvanic pairs) but also because they bring antimicrobial properties to the resulting alloys.22,25 Powder metallurgy is especially appealing to introduce insoluble elements like Ag to the Fe–Mn matrix in a homogenous manner. However, the effect of adding Ag in terms of its role in the degradation rate remains still controversial: while most authors agree that Ag causes increased degradation rates of Fe-based alloys,22–24,29 some studies report that Ag additions promote galvanic coupling only in the first stages of degradation and, after formation of the degradation layer, the Ag role is not being fulfilled anymore.27 Similarly, one can find dissimilar conclusions on the effect of silver on antibacterial activity. In the report by Liu30 no antimicrobial properties were confirmed in Fe70.3–Mn28.9–Ag0.8 alloy, while in the research published by Sotoudehbagha22 antibacterial activity in the alloys containing 1 and 3 wt% of Ag was observed.
The main aim of this study is to fabricate and characterize porous Fe–Mn alloys with different additions of Ag in terms of their microstructure, mechanical and magnetic properties, degradation, biocompatibility and antibacterial activity. For this purpose, four different compositions have been investigated: equiatomic FeMn, and FeMn with 1, 3 and 5 wt% of silver. The relatively high content of Mn is proposed to ensure the non-magnetic character of the investigated alloys. Up to date, neither a long-term biodegradability test nor a study of the evolution of magnetic properties upon immersion has been performed in terms of ion release and degradation surface analysis for porous, Ag-containing equiatomic Fe–Mn alloys.
2 Materials and methods
2.1 Fabrication of alloys
Commercial powders of iron (97% purity, Merck, Germany), manganese (≥99% purity, Merck, Germany) and silver (≥99.9% purity, Merck, Germany) were used to prepare four types of samples: FeMn, FeMn–1Ag, FeMn–3Ag and FeMn–5Ag, where the ratio of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn was adjusted to 50:50 (at%) and the additions of silver are given in wt% (1, 3 and 5 wt%, respectively). Mixtures of powders were alloyed using a planetary ball milling device (Pulverisette 5, Fritsch, Germany) under an Ar atmosphere at a rotation speed of 300 rpm, for 10 h and with a 10:1 ball-to-powder weight ratio. The procedure of weighing powders and sealing the container was conducted in a glove box under Ar atmosphere (H2O < 0.1 ppm, O2 < 0.1 ppm) to prevent oxidation and atmospheric contamination. The particle size of as-milled powders was measured using the ImageJ software (National Institutes of Health, USA) from scanning electron microscopy (SEM) images. After ball-milling, the powders were further refined in an agate mortar, to avoid leaving large agglomerates. Subsequently, as-prepared powders were uniaxially cold-pressed at 100 MPa to form cylindrical green pellets (discs) of 10 mm diameter and 2–3 mm in thickness. The green pellets were then loaded in a vacuum furnace and sintered at 900 °C at 5 × 10−5 mbar for 4 h at a heating rate of 1 °C min−1. As Mn has a high vapor pressure, higher sintering temperatures and longer dwelling times might lead to significant losses of Mn content due to sublimation,18,31 therefore the temperature was kept at 900 °C. Moreover, it has been shown that the losses of Mn in ball-milled powders are lower than in powders prepared using other methods.31
:Mn was adjusted to 50:50 (at%) and the additions of silver are given in wt% (1, 3 and 5 wt%, respectively). Mixtures of powders were alloyed using a planetary ball milling device (Pulverisette 5, Fritsch, Germany) under an Ar atmosphere at a rotation speed of 300 rpm, for 10 h and with a 10:1 ball-to-powder weight ratio. The procedure of weighing powders and sealing the container was conducted in a glove box under Ar atmosphere (H2O < 0.1 ppm, O2 < 0.1 ppm) to prevent oxidation and atmospheric contamination. The particle size of as-milled powders was measured using the ImageJ software (National Institutes of Health, USA) from scanning electron microscopy (SEM) images. After ball-milling, the powders were further refined in an agate mortar, to avoid leaving large agglomerates. Subsequently, as-prepared powders were uniaxially cold-pressed at 100 MPa to form cylindrical green pellets (discs) of 10 mm diameter and 2–3 mm in thickness. The green pellets were then loaded in a vacuum furnace and sintered at 900 °C at 5 × 10−5 mbar for 4 h at a heating rate of 1 °C min−1. As Mn has a high vapor pressure, higher sintering temperatures and longer dwelling times might lead to significant losses of Mn content due to sublimation,18,31 therefore the temperature was kept at 900 °C. Moreover, it has been shown that the losses of Mn in ball-milled powders are lower than in powders prepared using other methods.31
2.2 Structural characterization
The structural and compositional characterization of the fabricated materials was conducted using an SEM microscope (Zeiss Merlin, Zeiss, Germany) operated at 15 kV equipped with an energy-dispersive X-ray spectroscopy (EDS) detector. The phase compositions of ball-milled powders, sintered alloys and alloys after immersion in Hank's balanced salt solution (HBSS) were examined by X-ray diffraction (XRD, Panalytical X’Pert powder diffractometer, Malvern Panalytical, UK) using Cu Kα radiation. The measurements were conducted in an angular range from 20° to 100° with a step size of 0.026° in the Bragg-Brentano geometry. The XRD results were analyzed using Rietveld refinement (MAUD software),32 from which crystallite size and lattice parameters were determined. ImageJ software was used to assess the macroporosity of sintered alloys by analyzing the SEM images. Archimedes’ method was used to determine the density of the specimens. Then, the porosity values were estimated by comparing the theoretical (i.e., for the fully dense materials) and experimental densities, according to the formula (eqn (1)):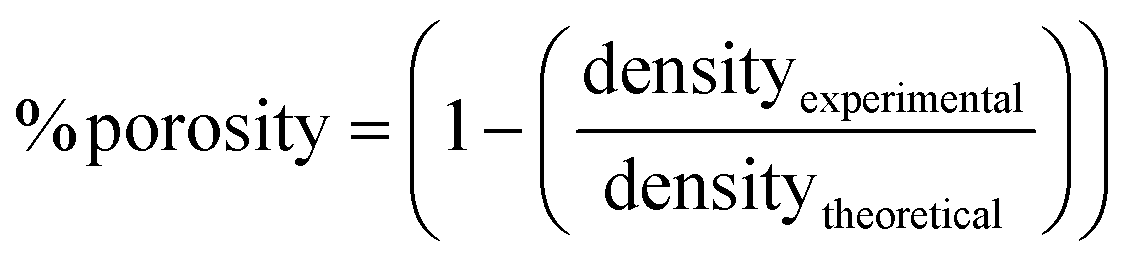 | (1) |
2.3 Biodegradability tests
Biodegradability tests were conducted by immersing samples of as-fabricated alloy pellets for various periods (7, 14, 28, 56, and 84 days) in HBSS at 37.5 ± 1 °C. Each sample, with a surface area of approximately 100 mm2, was immersed in a container containing approximately 30 mL HBSS. The volume of the solution adhered to the ASTM G31-72(2004) standard, using a volume/surface area ratio of 0.3 mL mm−2. At least three replicas were prepared at each time point. The HBSS (H8264, Merck, Germany) is a popular physiological solution used to reproduce the conditions in the human body, which contains CaCl2, MgSO4, KCl, KH2PO4, NaHCO3, NaCl, Na2HPO4 and glucose. After each period, 2 mL of solution from at least three replicas was taken for the ion release concentration analysis, which was conducted using inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7900, Agilent, USA). Before the ICP-MS analysis, the aliquots were diluted with HNO3. Samples immersed for 14, 28, 56, and 84 days were withdrawn from the containers for structural analysis of corrosion products formed both on the surface and the cross-section of the pellets. XRD analysis was performed to identify the corrosion products formed on the surface of FeMn and FeMn–5Ag alloys after 7 and 28 days of immersion.2.4 Characterization of mechanical and magnetic properties
The mechanical properties were studied using nanoindentation (Anton Paar NHT3 nanoindenter, Anton Paar, Austria). Before indentation, the discs were polished to a mirror-like surface, with the final polishing step using 1 μm diamond particles paste. Maximum applied loads of 100 and 500 mN were used to assess the behaviour of the material during deformation. A series of at least 50 measurements were performed on each sample to obtain statistically significant data. The load-displacement curves were used to calculate the hardness (HB) and reduced Young's modulus (Er), using the method of Oliver and Pharr.33 Apart from that, the elastic (Welast) and plastic (Wplast) energies of deformation were obtained from the load-displacement curves by assessing the areas between the unloading curve and displacement axis (Welast) and between the loading and unloading curves (Wplast). Finally, the elastic recovery was calculated as the ratio between elastic (Welast) and total (Welast + Wplast) energies during nanoindentation.A vibrating sample magnetometer (VSM, MicroSense, USA) was used to analyze the magnetic properties of the samples before (as-fabricated) and after incubation in HBSS, at room temperature. The maximum applied magnetic field was 20 kOe. The results were normalized over sample weight to obtain the value of saturation magnetization (MS).
2.5 Cell proliferation assays and cytotoxicity tests
For cell proliferation and cytotoxicity experiments, human osteosarcoma Saos-2 cells (ATCC HTB-85) were used. Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), under standard conditions (37 °C, 5% CO2).Experiments were conducted in a medium conditioned in the presence of the different FeMn(–xAg) alloys. To prepare the conditioned media, the samples were sterilized with UV light for 3 min and incubated in 15 mL of supplemented DMEM under standard conditions for 7, 14, 28, and 56 days. Then, the alloys were removed, and the conditioned media were kept at 4 °C protected from light until used. In parallel, control media were also prepared following the same incubation times and conditions but without the presence of any alloy (aged media). Osteoblasts (Saos-2 cells) proliferation was determined using Alamar Blue cell viability Reagent (Thermo Fisher Scientific) before (day 0) and after (day 3) cells’ exposure to the conditioned media. Briefly, 5 × 104 cells were seeded into each well of a 24-well plate with fresh medium. Cells were cultured for 24 h to allow cell attachment, and then the medium was replaced with fresh medium with 10% Alamar Blue, and cells were incubated for 4 h in standard conditions in the dark. Then, the supernatant was collected (day 0), and the fluorescence was measured at 585 nm wavelength after excitation at 560 nm on a Spark multimode microplate reader (Tecan, Männedorf, Switzerland). After supernatant collection, FeMn(–xAg) conditioned media for different incubation times were added to each well. Cells were cultured for 3 more days in a conditioned medium and the Alamar Blue assay was repeated according to the previously described protocol (day 3). Aged and fresh media were used as controls. Experiments were performed in triplicate.
Immediately after proliferation assays, cytotoxicity was assessed in cells incubated with media conditioned for 7 and 56 days using the live/dead viability/cytotoxicity kit for mammalian cells (Invitrogen), according to the manufacturer's protocol. Images from different regions of the well were captured using an Olympus IX71 inverted microscope equipped with epifluorescence.
2.6 Quantification of inflammatory cytokines secretion
THP-1 monocyte cells were used to analyze the potential immunological response induced by the FeMn(–xAg) alloys. Monocytes were grown in RPMI 1640 medium (Gibco) supplemented with 25% FBS (Gibco) under standard conditions. To differentiate monocytes into macrophages, 4 × 105 THP-1 cells were seeded into 24-well plates and treated with 0.16 μM phorbol-12-myristate-13-acetate (Sigma-Aldrich, Saint Louis, MO, USA) for 72 h. Then, cells were washed and incubated in a fresh medium for 24 h before carrying out the experiments. The fresh medium was replaced by a conditioned RPMI medium (prepared as explained above) and macrophages were incubated for 5 h and 24 h to allow the secretion of inflammatory cytokines. As a positive control, 1 μg mL−1 of lipopolysaccharide (LPS) (Sigma-Aldrich) was added to the macrophages culture. As a negative control, macrophages were cultured in a fresh medium. After 5 h and 24 h, supernatants were collected and used to quantify cytokine secretion. Inflammatory cytokines TNF-α, IL-1β and IL-6 were evaluated by flow cytometry using cytometric bead array (CBA) (Becton–Dickinson, East Rutherford, NJ, USA). Cytokine concentrations in the supernatant were analyzed according to the manufacturer's protocol. Negative control was considered as basal secretion level and used for statistical comparisons. Experiments were performed in triplicate.2.7 Assessment of S. aureus adhesion and viability
From a frozen stock, S. aureus ATCC 25923 was streaked onto a 5% horse blood agar (HBA) plate (Media Department, Clinical Microbiology Laboratory, Sahlgrenska University Hospital, Sweden) and incubated at 37 °C overnight. After incubation, colonies were taken and inoculated into 4 mL tryptic soy broth (TSB) (Scharlau, Barcelona, Spain) to achieve an OD546 of 0.13, equivalent to 108 CFU mL−1, which was diluted 1:1000 into 40 mL of TSB to achieve a suspension of 105 CFU mL−1. One mL of this suspension was then added to each well of a 48-well plate (Nunclon Delta-surface; Thermo Fisher Scientific, Waltham, USA) containing 9 mm diameter discs with the test surface (FeMn–5Ag) and controls of FeMn or Ti (Christers Finmekaniska AB, Skövde, Sweden). The discs were then incubated statically at 37 °C for 4 or 24 h to assess bacterial adhesion and biofilm formation as well as bacterial viability.
After each incubation period, each disc was rinsed three times in sterile 0.9% saline to remove non-adhered bacteria. Discs were then stained using LIVE/DEAD™ BacLight™ Bacterial Viability Kit, for microscopy & quantitative assays (Invitrogen, Waltham, USA) following the manufacturer's instructions. The discs were rinsed three times to remove any unbound stain and fluorescence was assessed in a plate reader (FluostarOmega, BMG LABTECH, Ortenberg, Germany) and confocal laser-scanning microscope (Nikon C2+, Nikon, Tokyo, Japan) with a 100× water dipping objective (CFI Plan 100XC W). Five z-stacks were taken at randomly chosen fields of view on each sample and images were taken at 3 μM intervals through the biofilm layers. Biofilm thickness and biomass were analyzed in BiofilmQ.34 From the plate reader, the average fluorescence intensity of each well scan was taken as the representative live/dead distribution for each disc. Experiments were performed in triplicate with duplicate material samples.
2.8 Statistical analysis
All quantitative data were analyzed with GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, USA) and presented as the mean ± standard error of the mean. Statistical differences were assessed by one-way analysis of variance (ANOVA) with a Bonferroni correction for cytocompatibility and inflammatory response analyses and with Fisher's least significant difference post-test for microbiological analyses. A value of p < 0.05 was considered significant.3 Results and discussion
3.1 Microstructure and compositional analysis
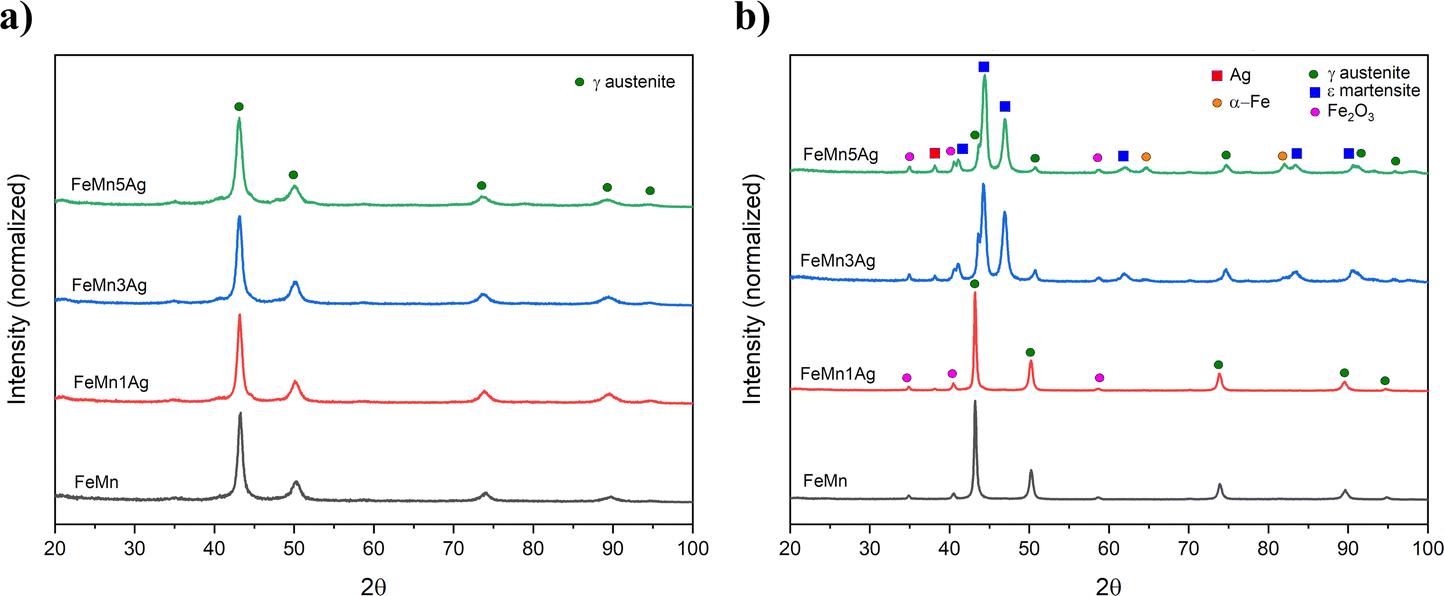 | ||
| Fig. 1 XRD patterns of (a) ball-milled powders and (b) sintered FeMn(–xAg) alloys. | ||
The FeMn and FeMn–1Ag sintered alloys maintain the austenitic structure, while FeMn–3Ag and FeMn–5Ag alloys consist of a duplex structure, comprising γ-austenite (fcc structure) and ε-martensite phase (hcp structure). The occurrence of the martensite phase was not expected as alloys containing above 27.3 at% of Mn are known to be mainly austenitic.13 This phase transition could be caused by the occurrence of discrete inhomogeneities in the element distribution, forming regions of lower Mn content and thus promoting the martensitic transformation. This phenomenon was already observed before by Zhang,21 but the explanation of martensitic transformation under furnace cooling conditions was not given. In this case, the martensitic transformation could be further enhanced by the presence of Ag. Nevertheless, both γ-austenite and ε-martensite phases are antiferromagnetic, therefore the occurrence of the martensitic phase should not interfere with MRI. A shift in the diffraction peaks towards higher 2θ angles is observed in the sintered alloys with higher Ag contents. The crystallite size of the alloys subjected to sintering increases when compared to as-milled powders, and values of 38, 39, 18 and 13 nm for FeMn, FeMn–1Ag, FeMn–3Ag and FeMn–5Ag, respectively, are obtained. Therefore, as in the case of powders, the addition of silver is leading to a decrease in the crystallite size. A peak of low intensity can be observed at 2θ = 35°, which was attributed to Fe2O3. The occurrence of the Fe2O3 phase was observed in other studies35 and it originates from the sintering method, which is always connected with some level of oxidation. Nevertheless, Fe2O3 is also biocompatible,37,38 therefore its presence should not compromise the biocompatibility of the tested alloys.
Fig. 2 shows SEM micrographs of the morphology of sintered (a–c) FeMn, (d–f) FeMn–1Ag, (g–i) FeMn–3Ag and (j–l) FeMn–5Ag alloys. The SEM images under various magnifications were analyzed using ImageJ software and the % area of macroporosity and the size of pores were calculated. Manganese is foreseen to diffuse into the iron matrix during sintering and create a solid solution, while silver may form non-soluble precipitates in the FeMn matrix, as the solubility of Ag in Fe is very low.39,40 The morphologies of the green pellets, i.e. pressed powders before sintering, are shown in Fig. S2 (ESI†). All tested alloys show a similar initial porosity in the range of 19–26%, as calculated using ImageJ based on SEM images. The process of sintering typically creates some additional level of porosity resulting from the rearranging of Fe and Mn, i.e., voids are left behind by the metals upon diffusion. Another factor causing porosity is the change in the crystalline structure from bcc-Fe and bcc-Mn to fcc-γ (FeMn) and hcp-ε (FeMn). This porosity is expected to have a beneficial impact on the proliferation of cells within the pores and, what is more, it can also improve the degradation rate due to an increased surface area exposed to the bodily environment.18,41 In the tested materials, two different levels of porosity can be distinguished – namely macroporosity with an average pore size of around 10 μm – and nanoporosity with a pore size of around 100 nm, as presented in Fig. 2(c, f, i and l). The calculated levels of total porosity are presented in Table 1, which shows that porosity increases with Ag content. While the addition of only 1 wt% of Ag does not cause a significant decrease in the density, the addition of 3 and 5 wt% of Ag leads to a higher total porosity (above 30%). This phenomenon is attributed mainly to the decreased compressibility due to the presence of non-alloyed silver particles in the ball-milled powder.42
 | ||
| Fig. 2 SEM micrographs of sintered and surface-polished samples of FeMn (a–c), FeMn–1Ag (d–f), FeMn–3Ag (g–i) and FeMn–5Ag (j–l) samples. The % area macroporosity was determined using ImageJ by analyzing SEM images of the regions of macroporosity. The unlabeled red arrows provide reference for the Ag particles. | ||
| Sample | wt% Fe | wt% Mn | wt% Ag | Macro-porosity (% area) | Density (g cm−3) | Total porosity (%) |
|---|---|---|---|---|---|---|
| FeMn | 54.3 | 45.7 | 0 | 18.39 ± 3.00 | 5.50 ± 0.16 | 28.2 ± 2.1 |
| FeMn–1Ag | 56.7 | 42.0 | 1.3 | 17.80 ± 1.54 | 5.38 ± 0.11 | 30.1 ± 1.4 |
| FeMn–3Ag | 54.9 | 42.5 | 2.6 | 20.61 ± 1.34 | 4.94 ± 0.04 | 36.3 ± 0.5 |
| FeMn–5Ag | 54.5 | 41.1 | 4.4 | 22.02 ± 0.30 | 4.87 ± 0.12 | 37.7 ± 0.6 |
Silver particles are uniformly distributed within the Fe–Mn matrix, as indicated by red arrows in Fig. 2. To further analyze the distribution of Ag precipitates in the Fe–Mn matrix, EDS compositional maps, presented in Fig. S3 and S4 (ESI†), show that Ag precipitates exhibit a polydisperse distribution within a size range of 0.5–3 μm. Larger agglomerates of Ag were observed, particularly in the FeMn–5Ag sintered alloy, as shown in Fig. S4b (ESI†). The presence of Ag-rich precipitates could in principle enhance the micro galvanic corrosion, hence inducing faster degradation rate of tested alloys.22–24
EDS compositional analysis of the samples furnished the weight percentage of individual elements, as listed in Table 1. The amounts of Ag are close to the nominal values (1, 3 and 5 wt%), while the weight percentages of Fe and Mn are slightly different from the nominal equiatomic composition (50.5 wt% Fe and 49.6 wt% Mn), with a lower amount of Mn than the initial value. Manganese is an element very prone to sublimation under high temperatures;39 therefore it may be assumed that partial sublimation of Mn occurred, lowering its final amount.
 | ||
| Fig. 3 Top-view SEM images of the degraded surface of FeMn (a–d) and FeMn–5Ag (e–h) incubated for 14 (a and e), 28 (b and f), 56 (c and g) and 84 (d and h) days in HBSS. (EDS analysis on the highlighted areas and locations is presented in Table 2). | ||
| Area/point | Elements (wt%) | ||||||
|---|---|---|---|---|---|---|---|
| Fe | Mn | O | Cl | P | Ca | ||
| FeMn | 1 | 34.6 | 22.5 | 38.3 | 0.5 | 3.1 | 1.0 |
| 2 | 19.3 | 20.1 | 30.9 | 12.9 | 8.4 | 8.4 | |
| FeMn–5Ag | 3 | 23.4 | — | 68.9 | 0.5 | 3.8 | 3.4 |
| 4 | 12.5 | 22.3 | 38.3 | — | 8.8 | 18.2 | |
| 5 | 40.5 | 27.1 | 31.1 | 1.4 | — | — | |
| 6 | 27.9 | 21.1 | 42.2 | 0.8 | 5.8 | 2.2 | |
| 7 | 6.3 | 42.9 | 25.1 | 15.4 | — | 10.4 | |
The globular precipitates, shown in Fig. 3f and h mainly consist of O, Mn, Ca and P. Increasing immersion period leads to the formation of a relatively thick oxidation layer, as shown in Fig. 3c and h. Even though the layer has the appearance of compact and homogenous, more detailed analysis (Fig. 3c) reveals a cracked structure caused most probably by dehydration due to air-drying after removal from HBSS.44,45 The cracked oxide layer is prone to detachment, as shown in Fig. 3h. After detachment, the exposed area allowed HBSS to penetrate further into the pores. As shown in Table 2, the degradation layer of samples incubated for 84 days (Fig. 3d and h) contains high levels of Cl−, namely 12.89 and 15.4 wt% for FeMn and FeMn–5Ag, respectively. The Cl− ions are aggressive to the surface of the alloy and they can break the passive oxidation layer, therefore leading to the formation of the pits.2,23,46 As can be seen in Fig. 3d, Cl-rich precipitates have formed on the exposed surface of FeMn alloys, most probably after Cl− ions have broken the oxide layer.
To gain further insight into the degradation products formed on the surfaces upon immersion, XRD analysis was performed on the FeMn and FeMn–5Ag samples after 7 and 28 days of immersion, as presented in Fig. S5 (ESI†). The XRD patterns obtained after 7 days did not demonstrate any additional features compared with the as-prepared samples (Fig. 1b). This can be explained by the thin nature of the oxide layer as well as the less abundant presence of other corrosion products. However, after 28 days of immersion, an increase in the intensity of some of the peaks was observed. Corrosion products such as MnPO4, MnCO3, Fe2O3 and MnO were identified. It is worth noting that some of the oxide peaks can overlap, therefore we might expect more corrosion products on the surface.
To unveil the degradation mechanism, cross-sections of all tested alloys immersed for 14, 28, 56 and 84 days in HBSS were examined (Fig. 4). One can observe the formation of a degradation layer at the utmost sample surface, which is particularly visible in the samples immersed for 14 and 28 days, with a thickness of around 30 μm. This layer was presumably cracked due to dehydration upon air drying or during the curing of the mounting resin, as mentioned before.11,27 Remarkably, this layer becomes thinner or even disappear for longer immersion periods.
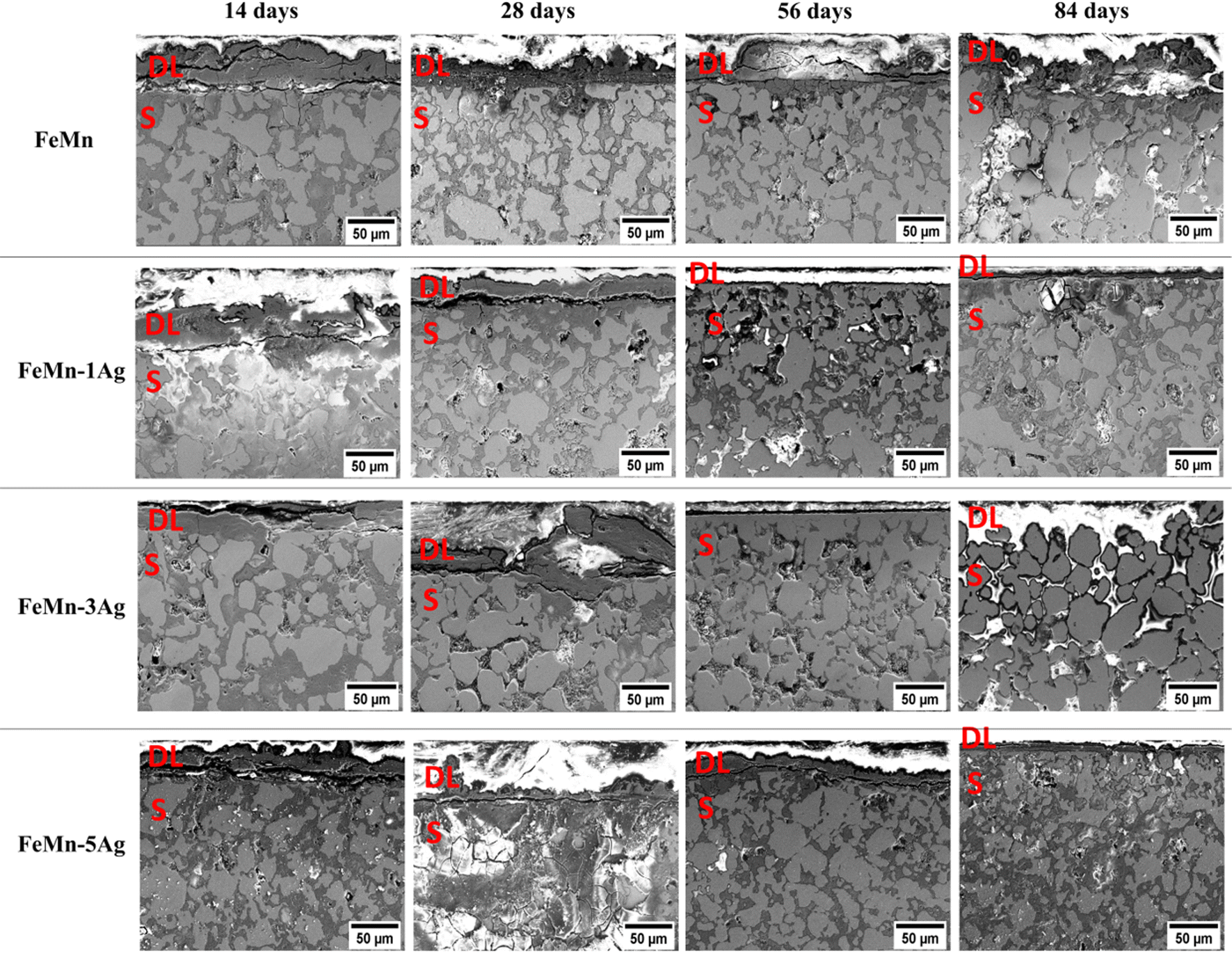 | ||
| Fig. 4 Cross-section SEM images of FeMn, FeMn–1Ag, FeMn–3Ag and FeMn–5Ag specimens after 14, 28, 56 and 84 days of immersion in HBSS. ‘S’ stands for sample and ‘DL’ to the degradation layer formed on top of the sample. | ||
At the same time, an increase of the incubation period favors higher penetration of the HBSS towards the bulk of the material since the HBSS is able to permeate through the open-pore structure of the pellets. As a result, corrosion products formed inside the pores, mainly consisting of iron and manganese oxides, as shown in Fig. S6 (ESI†). Brighter contrast (due to charging) is associated with low-conducting corrosion products made of O, Ca, P and Cl. When comparing individual alloys, the degradation layer of Ag-containing alloys seems to be thinner after 56 and 84 days of immersion than that of FeMn. Nevertheless, at the same time, the corrosion products consist mainly of metal hydroxides and carbonates, accompanied by calcium, phosphorus and chlorine elements, as mentioned before.11,20,47 The amount of Cl is notably higher in the Ag-containing alloys which can be attributed to localized pitting. Since porosity increases with Ag content, the Cl-rich corrosion products are accumulated inside the pores to a larger extent in samples with higher Ag wt%.
The EDS compositional maps of the cross-sections of the alloys immersed for 84 days, presented in Fig. S6 (ESI†), show the distribution of Fe and Mn alloying elements as well as of foreign elements that have accumulated inside the pores. Fe and Mn are uniformly distributed in the matrix, but the upper degradation layer shows a lower concentration of those elements than the bulk. The abundant presence of oxygen was observed, both in the degradation layer and the sample, mostly inside the pores. Other elements, such as Ca, Cl and P, were also present, mostly in the degradation layer. The EDS analyses performed in the degradation layer show that its Mn content is lower than that of Fe. For example, in the case of FeMn–5Ag, the content of Fe was around 4–5 times higher than the content of Mn (33.3 wt% of Fe and 8.5 wt% of Mn after 14 days of immersion, and 51.7 wt% of Fe and 11.1 wt% of Mn after 84 days of immersion). This suggests that the release of Mn ions to the HBSS occurs easier than the release of Fe ions.
The concentration of Fe and Mn ions released from the materials into the HBSS after 7, 14, 28, 56 and 84 days of immersion is presented in Fig. 5a and b. It can be observed that Mn release is higher than Fe release for all tested alloys and that the ion concentration increases with the immersion time, which is in agreement with the previously shown EDS results. Mn is more prone to be released due to a lower standard corrosion potential when compared with Fe (i.e., Mn2+ ions are more susceptible to be released to the HBSS than Fe2+ or Fe3+). In the first stage, up to 28 days of immersion, a sharp increase in the release is observed, especially in the case of Mn for all tested specimens and it tends to slow down upon further immersion. Nevertheless, an increase in both Mn and Fe release after 84 days of immersion is observed in the case of FeMn and FeMn–5Ag, as well as an increase of Fe release of the FeMn–1Ag sample.
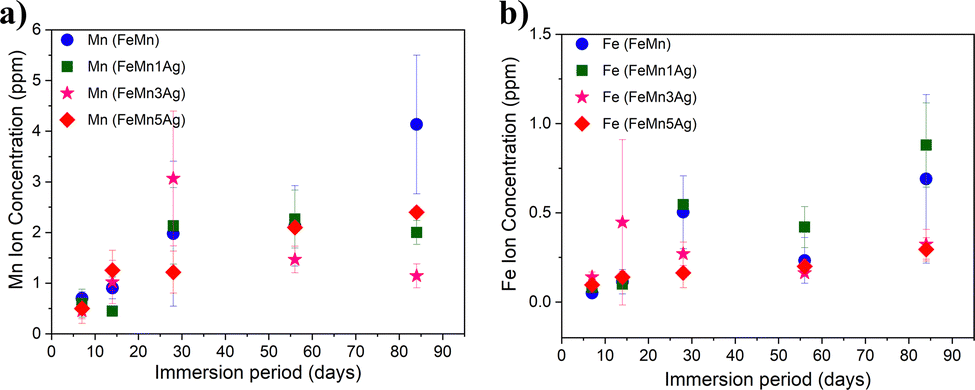 | ||
| Fig. 5 Concentration of (a) Mn and (b) Fe ions released into the HBSS at different immersion periods as measured with ICP-MS. Please note the different Y-axis scale in panels of Mn (a) and Fe (b) release. | ||
However, no significant differences in the amounts of released ions were observed between the tested alloys. The silver-containing alloys did not show an increase in the ion release of either Fe or Mn when compared to the alloy without Ag. Similar behavior has already been observed in literature with Fe–Mn–C–xAg alloy.26,27 Apparently, Ag had a negligible influence on the biodegradability of the material. Although Ag could in principle induce microgalvanic corrosion in contact with the less noble FeMn environment, the amounts of leached Fe and Mn ions in FeMn and FeMn–5Ag, as determined by ICP, were similar. This suggests that other effects, likely the porosity, play a major role.
The trend in the curve's shape of the ion concentration indicated a linear dependence up to 28 days and then a more stable increase during a longer immersion period. It can be mainly attributed to the degradation products formed on the surface that are partially blocking the dissolution of Fe and Mn at longer immersion periods. Due to the same reason, the corrosion rate does not increase with the immersion time for the Ag-containing alloys, as the release of silver is being blocked by corrosion products formed on the surface and inside the pores. Nevertheless, the release of silver was also measured, and it was 3 orders-of-magnitude lower than the release of Fe and Mn. Namely, the highest Ag release was observed for the FeMn–5Ag and was 1.23 ± 0.21 μg L−1 after 14 days of immersion, then it decreased to 0.88 ± 0.22 μg L−1 after 56 days of immersion and again increased up to 6.35 ± 3.65 μg L−1 after 84 days in HBSS. In the FeMn–1Ag and FeMn–3Ag, the highest concentration of Ag was also observed after 84 days of immersion and was 1.65 ± 0.55 μg L−1 and 1.3 ± 0.34 μg L−1, respectively. Thus, as could be suspected, the release of Ag ions increases with the initial Ag content. The increased amount of Ag released after 84 days of immersion, when compared with the amount released after 56 days, can be connected with the increased amounts of Cl-rich precipitates on the degradation surface that can contribute to breaking the degradation layer and, hence, enabling further degradation.
A previous study reported that Fe–Mn alloys (Fe35Mn and Fe25Mn), obtained through powder sintering and cold rolling and resintering cycles, released around 1.5 ppm and 2 ppm for Mn and Fe ions after 84 days of immersion, respectively.11 In another report, FeMnSiPd alloys obtained through arc melting,15 released around 1.5 ppm for both elements up to 120 days of immersion. The release of ions in our tested alloys was higher, presumably due to the higher level of porosity, which enhances the degradation.
3.2 Magnetic properties
The magnetic behavior of as-sintered and immersed alloys is presented in Fig. 6a–d. The as-sintered alloys (Fig. 6a) show very low saturation magnetization, with values of 0.019, 0.199, 0.626 and 1.077 emu g−1 for FeMn, FeMn–1Ag, FeMn–3Ag and FeMn–5Ag, respectively. For comparison, the value of saturation magnetization (MS) of the 304 austenitic stainless steel SS304, commonly used as implant material is 1.25 emu g−1.36,49 On the other hand, the MS value of pure iron is as high as 221.7 emu g−1.50 This means that the FeMn–5Ag alloy, which has the highest value of MS among the tested alloys, has only 0.49% the magnetization of Fe. In fact, the Fe-based alloys with manganese content above 30 wt% have been reported to be antiferromagnetic as they consist mainly of antiferromagnetic γ-austenite and ε-martensite phases.51,52 therefore enhancing the materials’ compatibility with MRI imaging. In our case, the MS value increased with higher Ag content, which might be attributed to slightly higher porosity of those alloys (which in turn could lead to higher amount of Fe2O3 after sintering).53,54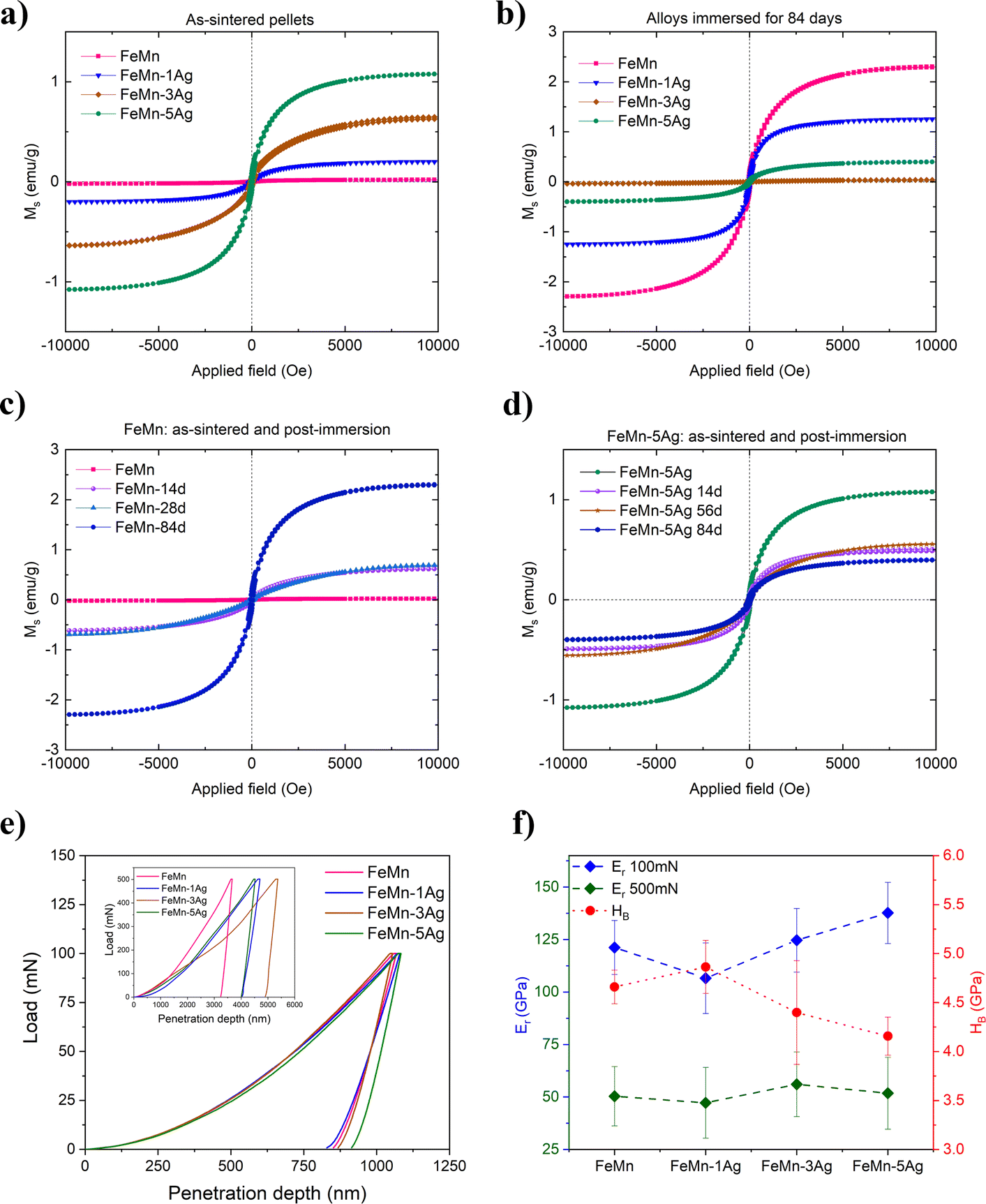 | ||
| Fig. 6 Magnetization curves (a–d) for (a) as-sintered samples, (b) alloys after immersion in HBSS for 84 days, and detailed changes of magnetization upon immersion for different periods for (c) FeMn and (d) FeMn–5Ag. Load-displacement curves (e) of the FeMn(–xAg) alloys under 100 and 500 mN (insert) maximum applied loads, and (f) corresponding reduced Young's modulus (Er) under 100 mN (blue) and 500 mN (green) of maximum applied loads, together with hardness (HB) (red). Please note the different Y-axis scales between the panels. | ||
Following immersion in HBSS, the magnetization of FeMn progressively increased with the immersion time, as shown in Fig. 6c. The alloys incubated for 84 days (Fig. 6b) showed a MS value of 2.301, 1.249, 0.0367 and 0.396 emu g−1 for FeMn, FeMn–1Ag, FeMn–3Ag and FeMn–5Ag, respectively. When comparing the gradual differences in the magnetic properties upon increasing immersion time, a similar trend was noticed in the FeMn alloy – the lowest value of magnetization is shown by the as-sintered material and it increased gradually – after 14 and 28 days the value was similar, around 0.690 emu g−1, with an increase up to 2.290 emu g−1 after 3 months of immersion. This observation can be ascribed to a higher release of Mn than Fe ions, which leads to a higher level of Fe-enriched regions in the material than before immersion. On the contrary, in the case of the FeMn–5Ag, an increase of the incubation time led to a small decrease in the MS values, starting from 1.077 emu g−1 for as-sintered alloy and then decreasing, firstly to around 0.550 emu g−1 after 14 days of immersion and then to a minimum value of 0.393 emu g−1 after 3 months of immersion. In any case, even though the MS values change upon degradation, the level of MS is sufficiently low to assure good compatibility of the alloys with MRI.
3.3 Mechanical properties
Nanoindentation was used to analyze the micro/nano-mechanical properties of the tested alloys. The tests were conducted under two different maximum loads, namely 100 and 500 mN. The lower load was used to extract information from the nanoporous regions of the material, while the higher loads may be able to assess the influence of both levels of porosity (macro and nano) on the reduced Young's modulus (Er) and hardness (HB). Representative load-displacement curves of tested alloys, together with calculated values of Er and HB are presented in Fig. 6e, f and Table 3, respectively. The load-displacement curves (Fig. 6e) show higher variability under higher load, which is mainly due to different levels of macroporosity among the tested alloys. On the other hand, the curves obtained under 100 mN of maximum load almost overlap and the resulting values of Er are more similar among the various samples, as shown in Fig. 6f. The nanoindentation tests conducted under a maximum applied load of 500 mN show that the alloys have a comparable value of Er, around 50 GPa, which is relatively close to that of a human bone (cortical bone has Young's modulus value of around 20 GPa).55 The average value of hardness performed under 500 mN was 1.48 GPa for FeMn, 1.2 GPa for FeMn–3Ag and 1.14 GPa for FeMn–5Ag.| Sample | E r (500 mN) GPa | E r (100 mN) GPa | H B (GPa) | H B/Er (wear resistance) | W elast/Wtotal | W plast/Wtotal |
|---|---|---|---|---|---|---|
| FeMn | 50.39 ± 14.16 | 121.24 ± 12.84 | 4.66 ± 0.17 | 0.038 ± 0.003 | 0.22 ± 0.03 | 0.78 ± 0.03 |
| FeMn–1Ag | 47.24 ± 16.86 | 106.60 ± 16.82 | 4.86 ± 0.27 | 0.046 ± 0.008 | 0.26 ± 0.04 | 0.74 ± 0.03 |
| FeMn–3Ag | 56.09 ± 15.36 | 124.65 ± 15.13 | 4.40 ± 0.53 | 0.035 ± 0.006 | 0.20 ± 0.03 | 0.80 ± 0.10 |
| FeMn–5Ag | 51.81 ± 17.13 | 137.67 ± 14.58 | 4.16 ± 0.19 | 0.030 ± 0.003 | 0.18 ± 0.02 | 0.82 ± 0.04 |
To understand the mechanical properties of the alloys, the results obtained under lower load (100 mN) were used to further calculate HB, Er, HB/Er ratio and energies of deformation, as presented in Table 3. The FeMn–1Ag alloy had the highest hardness and the lowest Young's modulus among all tested alloys, while the FeMn–5Ag had the lowest HB and the highest Er. The hardness values of all tested alloys are above 4 GPa, which is similar to that of fully dense Fe30Mn6Si1Pd.15 The values of hardness increase upon small additions of Ag and decrease at higher Ag amounts. As silver does not form an alloy with the Fe–Mn system and stays in a form of precipitates whose hardness is lower than the Fe–Mn matrix, the overall hardness of the material might be lower. The other factor influencing hardness values is the level of porosity (which increases with the Ag content), tending to reduce HB.
The HB/Er ratio is an indirect estimation of wear resistance. It has been shown that wear resistance is dependent on the hardness (HB) and the reciprocal value of elastic modulus (Er),56,57 thus the highest HB/Er ratio of FeMn–1Ag alloys indicates the highest wear resistance for this composition. In Table 3, the ratios of the elastic (Welast) and plastic (Wplast) energies versus the total (Wtotal) energy of deformation during nanoindentation are presented. All tested alloys demonstrate a higher contribution of plastic energy in the total energy of deformation. It is shown that the contribution of elastic energy is higher for the FeMn–1Ag alloy than for the other alloys, meaning that it possesses a slightly higher elastic recovery, which indicates the ability of the material to recover the initial shape after deformation.
Contrarily, the FeMn–5Ag alloy shows higher contribution of Wplast to the total energy of deformation. The so-called ‘plasticity index’ indicates higher plasticity of this alloy when compared to FeMn, FeMn–1Ag and FeMn–3Ag counterparts.
3.4 Cytocompatibility of FeMn(–xAg) alloys
To assess the alloys’ cytocompatibility, two different analyses were performed in vitro: cell proliferation and cytotoxicity. Whilst cell proliferation analysis allows to assess whether cells incubated with conditioned media can proliferate (i.e., increase their number over time), cytotoxicity analysis can determine whether the ions released by the alloys produce a toxic effect (i.e., a decrease in the number of live cells over time).First, we tried to perform direct cytocompatibility analyses, by growing the cells directly on the surface of the alloys. We observed that the cells did not adhere properly to the alloys (data not shown), probably due to morphological changes that the surface of the alloys experienced upon immersion (see Fig. 3 and 4). It has been described that Fe-based alloys show a relatively high corrosion and degradation rate that preclude cell adhesion.18 Hence, in order to analyze the effect of the alloys and of the ions released into the medium, we proceeded with the indirect cytocompatibility analyses, by culturing the cells in alloy-conditioned media.
In these experiments, two different control media were used: fresh media prepared just prior the experiment and media that were maintained in the incubator for the same time periods as the conditioned ones (aged medium; see Section 2.5).
We decided to include this aged medium as a control because we reasoned that the medium and serum components could degrade over time during the conditioning of the media with the alloys. Indeed, when comparing cell proliferation in both types of control media, we observed a significant decrease in Saos-2 cells proliferation when the cells were cultured in the 28 and 56 days aged media, the difference being higher in the last timepoint (Fig. 7). It is known that growth factors and other proteins degrade when stored at room temperature or 37 °C. Each protein has a different degradation rate but, to our knowledge, no research studies on serum degradation over time and its effect on cell proliferation have been published. However, the FBS information for users recommends to store the product at −10 to −40 °C, and not to use FBS stored at 2 to 8 °C for more than 4 weeks (Freezing and Thawing FBS and Other Sera). Because of the differences found in cell proliferation when using the two types of control media, the effect of the different conditioned media on cell proliferation was only compared with their corresponding aged medium.
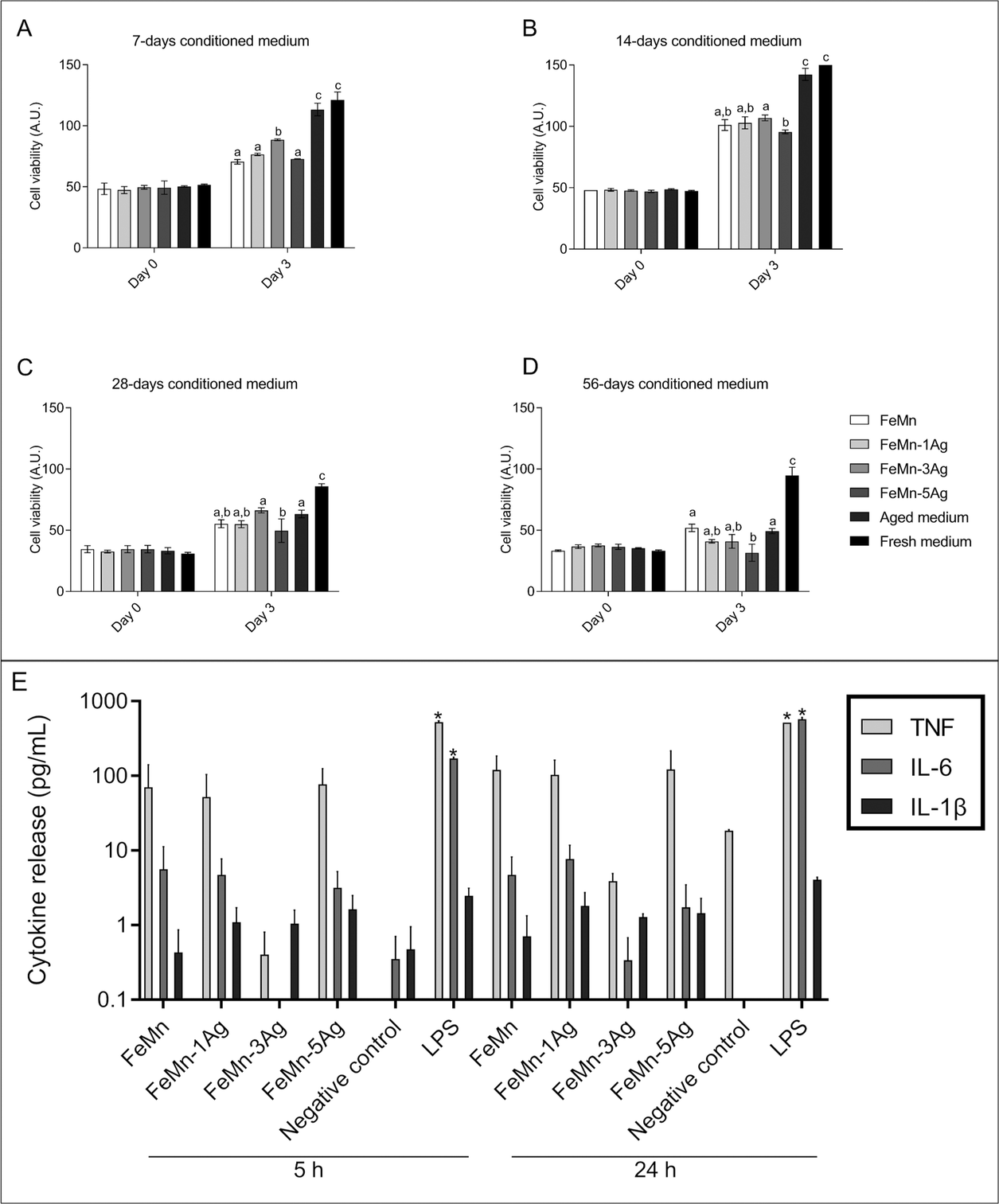 | ||
| Fig. 7 Cell proliferation of Saos-2 cells before (day 0) and after (day 3) culture with media conditioned for 7 (A), 14 (B), 28 (C) and 56 (D) days with FeMn, FeMn–1Ag, FeMn–3Ag and FeMn–5Ag alloys, measured by Alamar Blue fluorescence. Aged medium, consisting of DMEM medium incubated for the same incubation times and conditions but without any alloy, and fresh medium were used as controls. Bars with different alphabetical superscripts are significantly different from each other (p < 0.05), whereas bars with the same alphabetical superscripts are not significantly different from each other. (E) Logarithmic representation of cytokine release by macrophages exposed to 56 days FeMn(–Ag) conditioned media. Secretion was analyzed by the CBA test at 5 and 24 h of culture. LPS: Lipopolysaccharide, positive control. Asterisks above the columns indicate significant differences compared with the negative control. | ||
At day 0 (after one day in culture in fresh medium), the intensity of the signal obtained was similar among all samples of the same conditioned time group, with no significant differences (Fig. 7). After 3 days in culture in the conditioned media, cell viability significantly increased in 7 and 14 days conditioned media, regardless of the alloy composition (Fig. 7a and b), meaning that cells were able to proliferate. Nevertheless, cells cultured in the aged control medium showed a higher proliferation rate than in any of the alloys conditioned media tested. Some significant differences were found when comparing all conditioned media: cells cultured in FeMn–3Ag conditioned medium showed the highest proliferation rate among the 7 days conditioned media, and cells cultured in FeMn–5Ag conditioned medium showed the lowest proliferation rate among the 14 days conditioned media (Fig. 7a and b).
After 28 and 56 days of medium conditioning or aging, cell proliferation was similar between conditioned and aged media and among conditioned media, except for a significant decrease in the FeMn–5Ag conditioned medium compared with FeMn–3Ag at 28 days and with FeMn at 56 days (Fig. 7c and d).
Next, the live/dead assay was used to determine the cytotoxicity of the alloy-conditioned media. As shown in Fig. S7 (ESI†), the number of cells grown on wells cultured with media conditioned for 7 days with Fe–Mn, FeMn–1Ag, FeMn–3Ag and FeMn–5Ag was similar (A–D), but lower than the number of cells on wells cultured with the aged and the fresh control media (E and F). However, the number of cells grown on wells cultured with medium conditioned for 56 days was much lower. Specifically, cell numbers seemed to decrease as the silver percentage in the alloy increased (G–J). The number of cells growing in culture with the 56 days aged medium was also low (K), whereas cells cultured with fresh media appeared to be in monolayer and in a higher number (L). The number of live cells was higher than 90% in all cases. These results are in agreement with Alamar Blue results, and suggest that, while FeMn(–xAg) materials might interfere with cell proliferation, they are not cytotoxic.
The cytotoxicity of biodegradable metallic alloys is mainly attributed to the released degradation particles58 and metallic ions13 which can promote or inhibit both cell proliferation and metabolic activities. In addition, the corrosion of porous alloys can produce a general change in pH and osmolarity that can contribute to a decrease in the cell viability. The results obtained showed that media conditioned with the FeMn–5Ag alloy reduced cell proliferation compared with the other alloys containing less amount of Ag, indicating that certain amount of silver can alter cell proliferation. In fact, the FeMn–5Ag alloy was the one with the highest Ag release after 84 days in Hank's solution (6.35 μg L−1).
The effect of Ag on cell viability is controversial and depends on the state of the Ag, i.e. whether Ag is present in the culture as a salt, as nanoparticles, or as part of the alloy. In our case, Ag was incorporated within the FeMn matrix in the form of particles during the fabrication step.
It has been shown that the metal salt AgNO3 produces a high cytotoxic effect in mammalian cell cultures,59,60 with an IC50 of AgNO3 in the range of 2–11 μM (300–1900 μg L−1) depending on the cell type.61 On the other hand, studies using nanoparticles concluded that 6 μM silver nanoparticles do not have a significant impact on cytotoxicity of epithelial cells.62 Similar results were observed specifically using osteoblasts, as the presence of silver nanoparticles coating-alloys did not induce any cytotoxic effect on MG63 osteoblasts.63,64
Our results showed values at the 0.88–6.35 μg L−1 range of Ag released from the alloys. The highest values observed for FeMn–5Ag after 84 days were far from the concentrations considered cytotoxic. In this regard, our cytotoxic results are in agreement with those of previous studies (viability higher than 90%). However, the cell proliferation rate was reduced when osteoblasts were cultured in the presence of the alloy-conditioned media. After culture with 7 and 14 days conditioned medium, a decrease in cell proliferation was observed regardless of the alloy, which indicates that not only Ag can interfere with the cell cycle. The presence of Fe and Mn ions, or the relatively high corrosion rate could be responsible for the reduced proliferation rate. The high corrosion rate could increase the pH and osmolality, as previously observed.65 The release of Ag from the FeMn–5Ag after 28 and 56 of media conditioning could also contribute to the lowest cell proliferation observed.
3.5 Inflammatory cytokine response
To find out whether the ions released by alloys activate the secretion of inflammatory cytokines, 56 days conditioned medium was added to macrophage cultures. We analyzed the pro-inflammatory cytokines IL-1β, IL-6 and TNF, which are commonly produced by macrophages in response to bacterial endotoxin or LPS.66 According to Chanput et al., stimulating THP-1 macrophages with LPS results in a dramatic increase in the secretion of IL-1β, IL-6, and TNF, which reaches a plateau after 18 h of stimulation.67Results of the CBA showed that ions released by FeMn(–xAg) alloys did not activate the secretion of IL-1β, IL-6 or TNF in macrophages when compared with a non-stimulated control. However, when macrophages were exposed to LPS (positive control), an increase in the concentration of the three cytokines was detected in the culture medium, even though IL-1β increase was not statistically significant (Fig. 7e). Ubanako et al. also obtained the same results when using low amounts of LPS to stimulate THP-1 derived macrophages, that is, little or no secretion of IL-1β, IL-6 and TNF.68 It has been reported that the presence of metal debris and even low concentrations of metal ions can induce an inflammatory response.69 In this regard, it has been observed that the release of Fe, Mn and Ag could induce the secretion of inflammatory cytokines; Fe and Mn are considered no allergenic elements, although Ag is.70 However, the results obtained for cytokine release indicated that concentrations lower than 0.5 mg L−1 of Fe, 2.3 mg L−1 of Mn and 0.9 μg L−1 of Ag did not induce the inflammatory response in macrophages.
3.6 Microbial interactions with FeMn(–xAg)
In this investigation, the effect of FeMn and FeMn–5Ag alloys on S. aureus viability and biofilm formation was tested. The addition of silver was tested due to its widely known antimicrobial capacity.71,72 Biofilm biomass and thickness were assessed using confocal laser-scanning microscopy (CLSM) (Fig. 8A–D) and fluorescence plate reading (Fig. 8E–H) after 4 and 24 h of growth. Moreover, representative images of the biofilm formed on FeMn and FeMn–5Ag, obtained using CLSM are shown (Fig. 8I and J).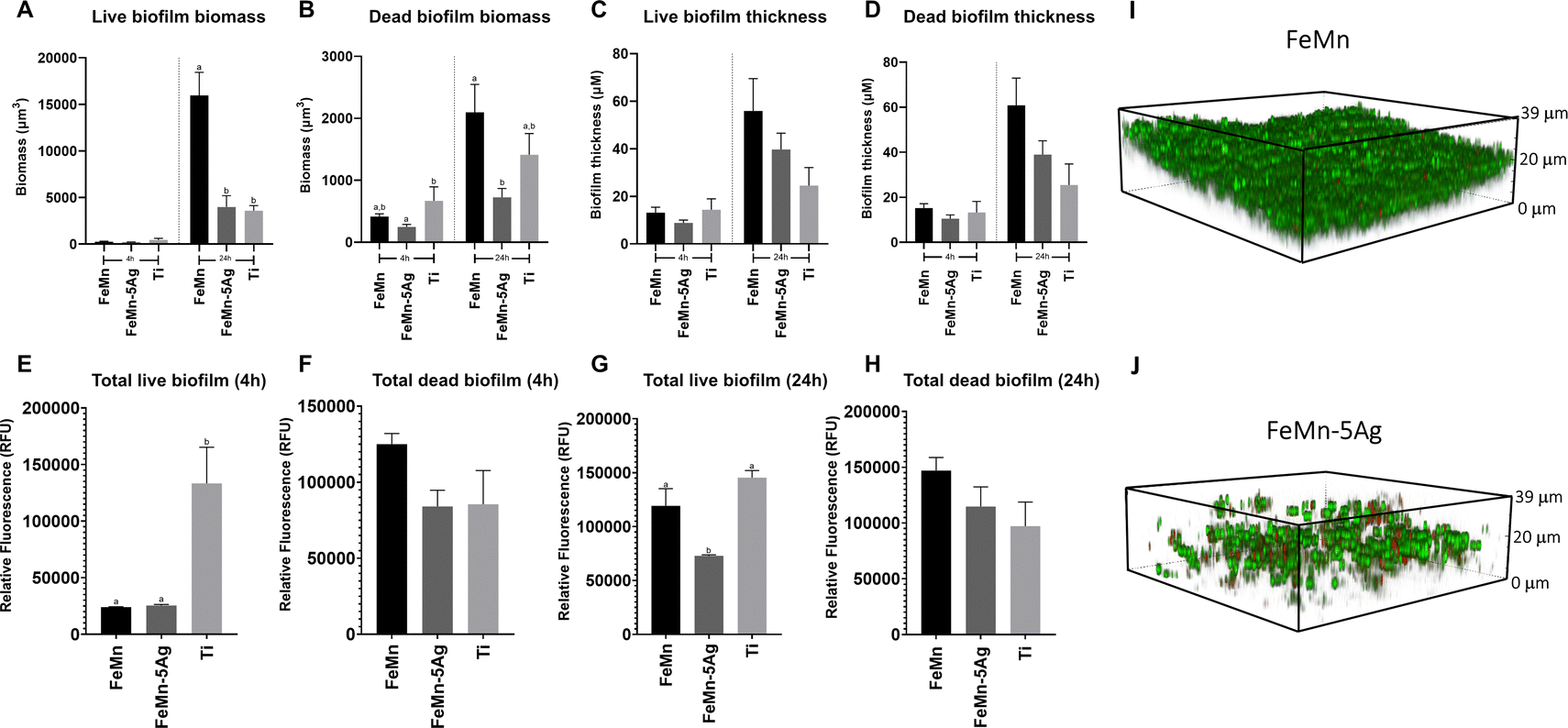 | ||
| Fig. 8 Staphylococcus aureus ATCC 25923 biofilm morphology on FeMn–5Ag compared to FeMn and machined titanium (Ti) controls after 4 and 24 h incubation, analysed using quantitative confocal laser-scanning microscopy (CLSM) to assess biofilm biomass (A and B) and thickness (C and D). Fluorescence intensity of total live and dead cells in biofilms grown for 4 h (E and F) and 24 h (G and H) on FeMn–5Ag, FeMn and Ti using fluorescence plate reading. Bars with different alphabetical superscripts are significantly different from each other (p < 0.05), whereas values with the same alphabetical superscripts are not significantly different from each other. CLSM images of the biofilm formed on the surface of FeMn (I) and FeMn–5Ag (J) alloys (NIS-Elements Viewer, Nikon). Green: Live S. aureus cells; Red: Dead S. aureus cells. | ||
When comparing the FeMn–5Ag to the FeMn material, whilst no significant difference in biofilm viability was observed at 4 h, a significant reduction in the total live and dead biomass was observed at the 24 h time point (Fig. 8A and B). Importantly, the total biofilm thickness on FeMn–5Ag was directly comparable to that of the titanium control (Fig. 8C and D), known for its biocompatibility in bone-implant applications, showing analogous performance of the FeMn–5Ag materials.
According to the fluorescence plate reading data, at 4 h the total biofilm viability was unaffected by the alloying of 5% silver compared to FeMn (Fig. 8E and F). A reduction in biofilm biomass on FeMn–5Ag compared to FeMn was also observed through fluorescent plate reading at 24 h for live (Fig. 8G) but not for dead (Fig. 8H) bacteria.
The current state of the literature regarding the addition of silver into FeMn alloys as a potential antimicrobial agent is disputed and dissimilar conclusions have been reported.21,29 The unchanged bacterial viability observed here by the addition of silver to FeMn is in agreement with certain literature,29 in which no reduction in S. aureus viability was observed after incubation with similar silver containing materials. With the concentrations of silver observed in the release assays undertaken in this study, it is likely that the total level of released silver into the culture medium was below the threshold for achieving a significant bactericidal effect.
Therefore, rather than a direct bactericidal activity of Ag, these results show that the FeMn–5Ag material may influence biofilm formation in S. aureus. The observed reduction in total biofilm biomass adhered to the FeMn–5Ag surface after 24 h is, to our knowledge, a novel finding (Fig. 8A and B). The potential mechanism for this is unknown but it could be related to the degradability of the bulk material, as seen in Fig. 3 and 4. By limiting the solid surface to which bacteria can adhere to, the ability for biofilm to form may be reduced, a concept also proposed by Daghighi et al.9
4 Conclusions
In this study, porous FeMn alloys with additions of Ag were fabricated through a powder metallurgy route. An in-depth study of the influence of porosity and Ag additions on the microstructure, biodegradability, degradation surface, magnetic and mechanical properties, has been undertaken. Furthermore, studies on cytocompatibility, inflammatory cell response and bacterial interactions have been conducted. The results revealed that:(a) FeMn and FeMn–1Ag sintered alloys consist mainly of a γ-austenite phase, while FeMn–3Ag and FeMn–5Ag alloys consist of a duplex structure, comprising γ-austenite (fcc structure) and ε-martensite phase (hcp structure).
(b) The alloys have a porous structure, consisting both of macro- and nanoporosity, with the total level of porosity varying from 28% for FeMn alloy up to 37% for FeMn–5Ag alloy.
(c) The biodegradability experiments, performed in HBSS up to 84 days of immersion, reveal that the release of Mn is higher than the release of Fe, which was attributed to a lower standard corrosion potential of Mn when compared to Fe. The degradation surface changes over time, with corrosion products consisting of Fe, Mn, O and enriched in Ca, P and Cl are present.
(d) The saturation magnetization value of FeMn alloys slightly increases with immersion time, with the highest Ms value after 84 days of immersion. Nevertheless, the Ms value even after 84 days is sufficiently low not to interfere with MRI imaging.
(e) No significant differences in the mechanical properties of the different alloys are observed, although the addition of 3% and 5% Ag tends to increase the Young's modulus and the plasticity index.
(f) The cytocompatibility experiments performed using conditioned media revealed that the investigated alloys are non-cytotoxic. Although Ag-containing alloys have shown to be cytocompatible, their cell proliferation is lower than that of FeMn.
(g) The inflammatory cell response experiment, conducted using media conditioned for 56 days, shows that the ions released by FeMn(–xAg) alloys do not induce an inflammatory response in macrophages in vitro.
(h) The viability and biofilm formation of Staphylococcus aureus with FeMn(–xAg) alloys were tested. The obtained results reveal that whilst no significant increase in bacterial killing is observed at 4 h between the FeMn and the FeMn–5Ag samples, a significant reduction in biofilm biomass of both live and dead cell populations is observed at 24 h.
(i) Overall, these results indicate a potential for a novel material that achieves identical performance compared to the current state of the art, whilst reducing early bacterial adhesion and biofilm formation.
Author contributions
A. Bartkowska: conceptualization; investigation; data curation; formal analysis; validation, visualization; writing – original draft. O. Careta: investigation; data curation; formal analysis; validation; visualization; writing – original draft. A. Turner: investigation; data curation; formal analysis; validation, visualization; writing – original draft. A. Blanquer: conceptualization; investigation; writing – review & editing; E. Ibáñez: conceptualization; methodology resources; supervision; writing – review & editing. M. Trobos: conceptualization; methodology; resources; supervision; writing – review & editing; funding acquisition C. Nogués: conceptualization; methodology; resources; supervision; writing – review & editing; project administration. E. Pellicer: conceptualization; methodology; resources; supervision; writing – review & editing; project administration. J. Sort: conceptualization; methodology; resources; supervision; writing – review & editing; project administration; funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 861046 (‘Bioremia’ European Training Network), the Spanish Ministry of Economy, Industry and Competitivity (Grant PID2020-116844RB-C21), the Generalitat de Catalunya (2017-SGR-0292 and 2017-SGR-503), the Swedish Research Council (2022-00853), the Swedish state under the agreement between the Swedish Government and the county councils, the ALF-agreement (ALFGBG-978896), the IngaBritt and Arne Lundberg Foundation (LU2021-0048); the Hjalmar Svensson Foundation, the Doctor Felix Neuberghs Foundation, and the Adlerbertska Foundation. The authors would like to thank the staff from the Servei de Microscòpia of Universitat Autònoma de Barcelona.Notes and references
- Y. Liu, Y. Zheng, X. H. Chen, J. A. Yang, H. Pan, D. Chen, L. Wang, J. Zhang, D. Zhu, S. Wu, K. W. K. Yeung, R. C. Zeng, Y. Han and S. Guan, Adv. Funct. Mater., 2019, 29, 1–21 Search PubMed.
- Y. F. Zheng, X. N. Gu and F. Witte, Mater. Sci. Eng., R, 2014, 77, 1–34 Search PubMed.
- H. Li, Y. Zheng and L. Qin, Prog. Nat. Sci.: Mater. Int., 2014, 24, 414–422 CrossRef CAS.
- J. Cheng, B. Liu, Y. H. Wu and Y. F. Zheng, J. Mater. Sci. Technol., 2013, 29, 619–627 CrossRef CAS.
- M. Moravej and D. Mantovani, Int. J. Mol. Sci., 2011, 12, 4250–4270 CrossRef CAS PubMed.
- S. Ganguly, S. Mondal, P. Das, P. Bhawal, P. Prasanna Maity, S. Ghosh, S. Dhara and N. C. Das, Int. J. Biol. Macromol., 2018, 111, 983–998 CrossRef CAS PubMed.
- J. Venezuela and M. S. Dargusch, Curr. Opin. Solid State Mater. Sci., 2020, 24, 100822 CrossRef CAS.
- H. J. Busscher, H. C. van der Mei, G. Subbiahdoss, P. C. Jutte, J. J. A. M. van den Dungen, S. A. J. Zaat, M. J. Schultz and D. W. Grainger, Sci. Transl. Med., 2012, 4, 153rv10 Search PubMed.
- S. Daghighi, J. Sjollema, H. C. van der Mei, H. J. Busscher and E. T. J. Rochford, Biomaterials, 2013, 34, 8013–8017 CrossRef CAS PubMed.
- J. He, F. L. He, D. W. Li, Y. L. Liu, Y. Y. Liu, Y. J. Ye and D. C. Yin, RSC Adv., 2016, 6, 112819–112838 RSC.
- H. Hermawan, A. Purnama, D. Dube, J. Couet and D. Mantovani, Acta Biomater., 2010, 6, 1852–1860 CrossRef CAS PubMed.
- R. Gorejová, L. Haverová, R. Oriňaková, A. Oriňak and M. Oriňak, J. Mater. Sci., 2019, 54, 1913–1947 CrossRef.
- H. Hermawan, D. Dubé and D. Mantovani, J. Biomed. Mater. Res., Part A, 2010, 93, 1–11 Search PubMed.
- M. Peuster, C. Hesse, T. Schloo, C. Fink, P. Beerbaum and C. von Schnakenburg, Biomaterials, 2006, 27, 4955–4962 CrossRef CAS PubMed.
- Y. P. Feng, A. Blanquer, J. Fornell, H. Zhang, P. Solsona, M. D. Baró, S. Suriñach, E. Ibáñez, E. García-Lecina, X. Wei, R. Li, L. Barrios, E. Pellicer, C. Nogués and J. Sort, J. Mater. Chem. B, 2016, 4, 6402–6412 RSC.
- D. T. Chou, D. Wells, D. Hong, B. Lee, H. Kuhn and P. N. Kumta, Acta Biomater., 2013, 9, 8593–8603 CrossRef CAS PubMed.
- P. Liu, D. Zhang, Y. Dai, J. Lin, Y. Li and C. Wen, Acta Biomater., 2020, 114, 485–496 CrossRef CAS PubMed.
- Y. P. Feng, N. Gaztelumendi, J. Fornell, H. Y. Zhang, P. Solsona, M. D. Baró, S. Suriñach, E. Ibáñez, L. Barrios, E. Pellicer, C. Nogués and J. Sort, J. Alloys Compd., 2017, 724, 1046–1056 CrossRef CAS.
- M. Heiden, E. Nauman and L. Stanciu, Adv. Healthcare Mater., 2017, 6, 1700120 CrossRef PubMed.
- S. M. Huang, E. A. Nauman and L. A. Stanciu, Mater. Sci. Eng., C, 2019, 99, 1048–1057 CrossRef CAS PubMed.
- Q. Zhang and P. Cao, Mater. Chem. Phys., 2015, 163, 394–401 CrossRef CAS.
- P. Sotoudehbagha, S. Sheibani, M. Khakbiz, S. Ebrahimi-Barough and H. Hermawan, Mater. Sci. Eng., C, 2018, 88, 88–94 CrossRef CAS PubMed.
- P. S. Bagha, M. Khakbiz, S. Sheibani, S. Ebrahimi-Barough, H. Hermawan, P. Sotoudeh Bagha, M. Khakbiz, S. Sheibani, S. Ebrahimi-Barough and H. Hermawan, ACS Biomater. Sci. Eng., 2020, 6, 2094–2106 CrossRef CAS PubMed.
- P. Sotoudeh Bagha, M. Khakbiz, S. Sheibani and H. Hermawan, J. Alloys Compd., 2018, 767, 955–965 CrossRef CAS.
- Z. Ma, M. Gao, D. Na, Y. Li, L. Tan and K. Yang, Mater. Sci. Eng., C, 2019, 103, 109718 CrossRef CAS PubMed.
- S. Loffredo, C. Paternoster, N. Giguère, M. Vedani and D. Mantovani, JOM, 2020, 72, 1892–1901 CrossRef CAS.
- S. Loffredo, S. Gambaro, L. Marin De Andrade, C. Paternoster, R. Casati, N. Giguère, M. Vedani and D. Mantovani, ACS Biomater. Sci. Eng., 2021, 7, 3669–3682 CrossRef CAS PubMed.
- M. Schinhammer, P. Steiger, F. Moszner, J. F. Löffler and P. J. Uggowitzer, Mater. Sci. Eng., C, 2013, 33, 1882–1893 CrossRef CAS PubMed.
- M. S. Dargusch, J. Venezuela, A. Dehghan-Manshadi, S. Johnston, N. Yang, K. Mardon, C. Lau and R. Allavena, Adv. Healthcare Mater., 2021, 10, 2000667 CrossRef CAS PubMed.
- R. Y. Liu, R. G. He, L. Q. Xu and S. F. Guo, Acta Metall. Sin., 2018, 31, 584–590 CrossRef CAS.
- Z. Xu, M. A. Hodgson and P. Cao, J. Mater. Res., 2017, 32, 644–655 CrossRef CAS.
- L. Lutterotti, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 334–340 CrossRef CAS.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564–1583 CrossRef CAS.
- R. Hartmann, H. Jeckel, E. Jelli, P. K. Singh, S. Vaidya, M. Bayer, D. K. H. Rode, L. Vidakovic, F. Díaz-Pascual, J. C. N. Fong, A. Dragoš, O. Lamprecht, J. G. Thöming, N. Netter, S. Häussler, C. D. Nadell, V. Sourjik, Á. T. Kovács, F. H. Yildiz and K. Drescher, Nat. Microbiol., 2021, 6, 151–156 CrossRef CAS PubMed.
- W. Huang, Calphad, 1989, 13, 243–252 CrossRef CAS.
- M. S. Dargusch, A. Dehghan-Manshadi, M. Shahbazi, J. Venezuela, X. Tran, J. Song, N. Liu, C. Xu, Q. Ye and C. Wen, ACS Biomater. Sci. Eng., 2019, 5, 1686–1702 CrossRef CAS PubMed.
- C. R. Ou, C. I. Shen and C. M. Ou, J. Supercond. Novel Magn., 2010, 23, 1197–1199 CrossRef CAS.
- M. Bhushan, D. Mohapatra, Y. Kumar and A. Kasi Viswanath, Mater. Sci. Eng. B, 2021, 268, 115119 CrossRef CAS.
- L. J. Swartzendruber, Bull. Alloy Phase Diagrams, 1984, 5, 560–564 CrossRef.
- C. G. Fink and V. S. de Marchi, Trans. Electrochem. Soc., 1938, 74, 271 CrossRef.
- A. H. Yusop, A. A. Bakir, N. A. Shaharom, M. R. Abdul Kadir and H. Hermawan, Int. J. Biomater., 2012, 2012, 641430 CAS.
- C. Tonna, C. Wang, D. Mei, S. V. Lamaka, M. L. Zheludkevich and J. Buhagiar, Bioact. Mater., 2022, 7, 426–440 CrossRef CAS PubMed.
- A. Šalak and M. Selecká, Alloying and sintering of manganese steels in terms of high manganese vapour pressure, in: Manganese in Powder Metallurgy Steels, Cambridge International Science Publishing Ltd; 2012, pp. 22–38 Search PubMed.
- E. Mouzou, C. Paternoster, R. Tolouei, A. Purnama, P. Chevallier, D. Dubé, F. Prima and D. Mantovani, Mater. Sci. Eng., C, 2016, 61, 564–573 CrossRef CAS PubMed.
- M. Moravej, A. Purnama, M. Fiset, J. Couet and D. Mantovani, Acta Biomater., 2010, 6, 1843–1851 CrossRef CAS PubMed.
- Y. Xin, K. Huo, H. Tao, G. Tang and P. K. Chu, Acta Biomater., 2008, 4, 2008–2015 CrossRef CAS PubMed.
- J. Čapek, J. Kubásek, D. Vojtěch, E. Jablonská, J. Lipov and T. Ruml, Mater. Sci. Eng., C, 2016, 58, 900–908 CrossRef PubMed.
- J. Čapek, K. Stehlíková, A. Michalcová, Š. Msallamová and D. Vojtěch, Mater. Chem. Phys., 2016, 181, 501–511 CrossRef.
- J. Childress, S. H. Liou and C. L. Chien, J. Appl. Phys., 1998, 64, 6059 CrossRef.
- H. Danan, A. Herr and A. J. P. Meyer, J. Appl. Phys., 2008, 39, 669 CrossRef.
- Y. Ishikawa and Y. Endoh, J. Appl. Phys., 2003, 39, 1318 CrossRef.
- S. Cotes, M. Sade and A. F. Guillermet, Metall. Mater. Trans. A, 1995, 26, 1957–1969 CrossRef.
- M. Tadic, M. Panjan, V. Damnjanovic and I. Milosevic, Appl. Surf. Sci., 2014, 320, 183–187 CrossRef CAS.
- D. Cao, H. Li, L. Pan, J. Li, X. Wang, P. Jing and X. Cheng, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- J. Y. Rho, R. B. Ashman and C. H. Turner, J. Biomech., 1993, 26, 111–119 CrossRef CAS PubMed.
- A. Leyland and A. Matthews, Wear, 2000, 246, 1–11 CrossRef CAS.
- E. Pellicer, S. Pané, K. M. Sivaraman, O. Ergeneman, S. Suriñach, M. D. Baró, B. J. Nelson and J. Sort, Mater. Chem. Phys., 2011, 130, 1380–1386 CrossRef CAS.
- X. Gu, Y. Zheng, Y. Cheng, S. Zhong and T. Xi, Biomaterials, 2009, 30, 484–498 CrossRef CAS PubMed.
- G. Schmalz, D. Arenholt-Bindslev, S. Pfüller and H. Schweikl, Altern. Lab. Anim., 1997, 25, 323–330 CrossRef.
- A. Yamamoto, R. Honma and M. Sumita, J. Biomed. Mater. Res., 1998, 39, 331–340 CrossRef CAS PubMed.
- D. Tie, F. Feyerabend, W. D. Müller, R. Schade, K. Liefeith, K. U. Kainer and R. Willumeit, Eur. Cells Mater., 2012, 25, 284–298 CrossRef PubMed.
- C. M. Santoro, N. L. Duchsherer and D. W. Grainger, Nanobiotechnology, 2007, 3, 55–65 CrossRef CAS PubMed.
- P. Jabłoński, M. Hebda, P. Pytlak, A. Kyzioł, H. Krawiec, Z. Grzesik and K. Kyzioł, Mater. Chem. Phys., 2020, 248, 122931 CrossRef.
- K. Kleszcz, M. Hebda, A. Kyzioł, H. Krawiec and K. Kyzioł, Appl. Surf. Sci., 2021, 557, 149795 CrossRef CAS.
- J. Fischer, D. Pröfrock, N. Hort, R. Willumeit and F. Feyerabend, Mater. Sci. Eng., B, 2011, 176, 830–834 CrossRef CAS.
- L. D. Hazlett, Prog. Retinal Eye Res., 2004, 23, 1–30 CrossRef CAS PubMed.
- W. Chanput, J. Mes, R. A. M. Vreeburg, H. F. J. Savelkoul and H. J. Wichers, Food Funct., 2010, 1, 254–261 RSC.
- P. Ubanako, N. Xelwa and M. Ntwasa, PLoS One, 2019, 14, e0222614 CrossRef CAS PubMed.
- J. C. Wataha, J. B. Lewis, K. R. Volkmann, P. E. Lockwood, F. L. W. Messer and S. Bouillaguet, J. Biomed. Mater. Res., Part B, 2004, 69, 11–17 CrossRef PubMed.
- A. Biesiekierski, J. Wang, M. Abdel-Hady Gepreel and C. Wen, Acta Biomater., 2012, 8, 1661–1669 CrossRef CAS PubMed.
- A. B. Lansdown, J. Wound Care, 2013, 11, 125–130 CrossRef PubMed.
- S. Subhadarshini, R. Singh, A. Mandal, S. Roy, S. Mandal, S. Mallik, D. K. Goswami, A. K. Das and N. C. Das, Langmuir, 2021, 37, 9356–9370 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of powders and cold-pressed pellets, EDS mappings taken before and after incubation in HBSS solution, XRD patterns after incubation in HBSS, and number of live/dead cells grown on wells cultured with media conditioned. See DOI: https://doi.org/10.1039/d2ma00867j |
| This journal is © The Royal Society of Chemistry 2023 |