
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the interaction of ex situ biofilms with plasmonic metal nanoparticles using surface-enhanced Raman spectroscopy†
Wafaa
Aljuhani
a,
Yingrui
Zhang
a,
Matthew P.
Wylie
 b,
Yikai
Xu
a,
Colin P.
McCoy
b and
Steven E. J.
Bell
*a
b,
Yikai
Xu
a,
Colin P.
McCoy
b and
Steven E. J.
Bell
*a
aSchool of Chemistry and Chemical Engineering, David Keir Building, Queen's University Belfast, BT9 5AG Belfast, UK. E-mail: s.bell@qub.ac.uk
bSchool of Pharmacy, Queen's University Belfast, BT9 7BL Belfast, UK
First published on 5th April 2023
Abstract
Biofilms are complex environments where matrix effects from components such as extracellular polymeric substances and proteins can strongly affect SERS performance. Here the interactions between SERS-enhancing Ag and Au particles were studied using ex situ biofilms (es-biofilms), which were more homogenous than in situ biofilm samples. This allowed systematic quantitative studies, where samples could be accurately diluted and analysed, to be carried out. Strong signals from intrinsic marker compounds were found for the Pseudomonas aeruginosa and Staphylococcus aureus extracted es-biofilms, which the standard addition method showed were due to 2 × 10–3 mol dm−3 pyocyanin or the equivalent of 1 × 10–4 mol dm−3 adenine, respectively. The es-biofilms hindered aggregation of Ag colloids more than Au but for both Au and Ag nanospheres the presence of es-biofilm reduced SERS signals through a combination of poorer aggregation and blocking of surface sites. For Ag, the effect of lower aggregation was to reduce the signals by a factor of ca. 2×, while site blocking gave a further 10× reduction for adenine. Similar results were found for Au nanospheres with adenine, although these particles gave low enhancement with pyocyanin. Nanostars were found to be unaffected by reduced aggregation and also showed lower site blocking effects, giving more reproducible signals than those from aggregated particles, which compensated for their lower enhancement factor. These results provide a rational basis for selecting enhancing substrates for use in in situ studies, where the further complexity means that it is important to begin with well-understood and controllable enhancing media.
Introduction
Bacterial biofilms are the predominant form of microbial life on Earth. They occur in many settings, most notably in water distribution systems and as pathological infections in humans and animals. Indeed 80% of all human bacterial infections are associated with biofilm formation.1 These infections are particularly challenging to treat by conventional antibiotics because the microorganisms in the biofilm are protected by a strong sticky matrix formed by extracellular polymeric substances (EPS).1 The EPS matrix has a complex structure, which primarily consists of polysaccharides, proteins, lipids and DNA. Bacteria in biofilms are particularly difficult to eradicate because the matrix prevents ingress of some antibiotics.2It is important that methods which allow detailed information of the chemical composition and structure of the EPS matrix, preferably in situ, are developed, since this will underpin strategies for administering, and monitoring the effectiveness of, biocides and for developing antifouling strategies. This is a challenging problem since biofilms are extremely complex, so studies normally concentrate on detecting and analysing just some of the components, for example by focusing on the bacteria within the films or the molecular structure of the EPS.3–5 These studies are made more difficult by the fact that the biofilms are very heterogenous so the structure and chemical properties will differ in different regions of the biofilm.3 For example, the bacteria tend to be located near the lower surface, i.e. close to the substrate on which the film has grown.3 We are interested in detecting small molecules present in the biofilms because this is relevant to understanding quorum sensing, response of bacteria in the films to external stimuli and the transport of antibiotics from the external surface to the interior where the bacteria are located. Current techniques do not provide the required combination of sensitivity and chemical specificity that is required to fully address these areas.3
The most widely used methods for quantification and identification of biofilm matrix use optical microscopy, which is typically combined with staining of various components to provide a level of chemical specificity.4 In addition, confocal laser scanning microscopy (CLSM) offers the possibility of 3D mapping of the biofilm.4 Staining techniques can give good information on the gross physical structure of the biofilms, for example, crystal violet (CV) is extensively used as a biofilm stain and forms the basis for thickness measurements based on the amount of the positively charged CV adsorbed to the negatively charged EPS components, which is quantified by measuring the UV/Vis absorbance of the dye.3 Similarly, other staining techniques are available for live/dead analysis of bacteria and for visualization of EPS components including carbohydrates, proteins and extracellular DNA (eDNA).3,4 However, staining techniques require long sample preparation times and are not suitable for real time monitoring of changes within a single biofilm. In addition, spectral overlap means that typically only two components can be analysed simultaneously.3 Alternatively, scanning electron microscopy (SEM) can provide much higher spatial resolution than optical methods and it allows the distribution of cells and EPS to be monitored but it has no chemical specificity. In addition, conventional SEM does not allow monitoring of living biofilm structures since the imaging requires high vacuum conditions and therefore dehydrated samples, whose structure is likely to be very different from the natural structure.5 This problem may be alleviated to some extent using environmental SEM methods.6
A promising approach to obtaining molecularly specific information on biofilms is the use of vibrational spectroscopy and both infrared and Raman spectroscopy can provide rapid and direct information on both the chemical composition and, through mapping/imaging the structure of biofilm.7 The low Raman scattering cross section of water makes it more suitable than IR for the analysis of water-containing samples like biofilms.7 Raman spectroscopy has many other advantages that would seem to make it ideal for studying biofilms, it is a rapid non-invasive technique which provides detailed chemical information with no sample preparation. For example, Stimulated Raman Scattering (SRS) Microscopy is a promising technique for studying biofilms, especially in situ, as it can provide information on the chemical composition and structure of the biofilm reasonably quickly.8 However, a major drawback of Raman spectroscopy is the relative weakness of Raman scattering, especially when a low limit of detection is required.9 This limitation can be addressed by using surface-enhanced Raman scattering (SERS), where the signal may be enhanced by factor of up to ×107 by placing the sample of interest in close proximity of a nanostructured noble metal surface.10
Several studies using SERS for in situ analysis of biofilms have been published.11–16 Most of these studies involved detection of pyocyanin, which is of interest for quorum sensing but also in the current context has been important in development of the technique since it gives very large SERS signals even at the low concentrations found in biofilms. The enhancing substrates used in previous studies were often colloidal silver nanoparticles (AgNPs) synthesized with a reducing agent such as sodium citrate and hydroxylamine hydrochloride.11–13 These particles were normally added to previously grown biofilms, subsequent adsorption of the particles to the exterior surface of the biofilms then allowed biofilm components in the vicinity of the absorbed surface particles to be detected.
Conversely, an alternative approach is to grow the biofilm on top of the enhancing substrate, which allows the detection of the molecules on the bottom of the biofilm.14–16
Our long-term objective is to look at other small molecules, in addition to pyocyanin, since this compound is limited to Pseudomonas aeruginosa (P. aeruginosa) biofilms and also to carry out detection of molecules within the EPS of the biofilm, rather than at the upper or lower surfaces.16 This raises real challenges since the EPS is an unfavourable matrix for SERS detection. This is because the components in EPS may act to reduce the enhancement, either by preventing the aggregation, which is necessary to form plasmonic hotspots or by blocking access to the surface through formation of an adsorbed protein and/or a polysaccharide layer.17,18 In addition, the biofilms are physically and chemically heterogeneous, which adds an additional layer of complexity to interpretation of their spectra while the high viscosity of the matrix can hinder particle diffusion.18
In the current study, initial SERS studies on intact biofilm highlighted the difficulties outlined above. For that reason, further experiments were performed using ex situ biofilms (es-biofilms) where biofilms were diluted and removed from their culture plates to produce a controllable homogenous liquid, thus removing complications associated with the structure and gel-like properties of the in situ biofilm. This allowed the biofilm-nanoparticle interactions that affect the SERS performance to be studied systematically. These results are interesting in their own right but more importantly they provide the understanding needed to underpin well-controlled studies on intact biofilms.
Materials and methods
All reagents were purchased from Sigma-Aldrich. Milli-Q water with a resistivity of 18.2 MΩ cm was used for all experiments.Preparation of the nanoparticle colloids
Ag colloid was prepared using the method reported by Lee and Meisel.19 Au colloid was prepared using the method reported by Frens with slight modification.20 Briefly, 0.05 g of gold(III) chloride (HAuCl4) was dissolved in 50 mL of deionized water and stirred and heated under reflux until boiling. Once boiling was reached, 5.6 ml of 1% (w/w) aqueous trisodium citrate was added to the precursor solution all at once. This resulted in a colour change of the solution from pale yellow to wine red. This colloid was then left to react for another 15 min before being cooled to room temperature. The colloid mixture was stored below 4 °C until required. Nanostar (NS) colloid was synthesized using a modified seedless protocol.21 Briefly, 360 μL of 10 mM gold(III) chloride (HAuCl4) and 20 μL of 10 mM silver nitrate (AgNO3) were mixed in 10 mL of deionised water in a 50 mL centrifuge tube under vortex for 30 s. Then, 60 μL of 100 mM L-ascorbic acid (C6H8O6) was quickly added all at once and the sample vortexed for another 30 s. The solution turned from faint yellow to greenish blue almost immediately after adding the reducing agent. The freshly synthesized NS colloid was stabilized by adding 1 ml of 1% hydroxyethyl cellulose (250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MW) and vortexing for 1 min. It was then centrifuged at 300 rcf for 1 h, resuspended in 1 mL of H2O, and was stored at room temperature until required. All particle concentrations were measured with a Malvern NanoSight NS300 instrument and the particle concentrations were equalised to that of the Ag colloid by diluting the Au colloid and concentrating the NS by centrifugation/resuspension, as discussed above.
000 MW) and vortexing for 1 min. It was then centrifuged at 300 rcf for 1 h, resuspended in 1 mL of H2O, and was stored at room temperature until required. All particle concentrations were measured with a Malvern NanoSight NS300 instrument and the particle concentrations were equalised to that of the Ag colloid by diluting the Au colloid and concentrating the NS by centrifugation/resuspension, as discussed above.
Characterization
The size distribution of the colloidal nanoparticles was obtained by Nanoparticle Tracking Analysis (NTA) using a Malvern NanoSight NS300. UV/Vis extinction measurements were recorded using an Agilent 8453 single beam diode array spectrometer. For the aggregation experiments, samples were prepared by adding 10 μL of 1 M MgSO4 to 100 μL of the chosen colloid and then allowed to sit for 30 s to allow aggregation to take place. They were then diluted with deionized water by a factor of 100× for the NanoSight measurements or 10× for UV/Vis measurements. For experiments measuring the effect of added biofilm, 50 μL of biofilm was added to 100 μL colloid and allowed to react for 30 s before addition of 15 μL of 1 M MgSO4.For the NanoSight measurements, all samples were analysed for 30 s and run 3 times. The measurements shown are the average of the 3 runs. A Quanta FEG 250 scanning electron microscope was used for imaging of the nanoparticles interacting with the intact biofilm. This sample was prepared for imaging by spotting colloids onto the in situ biofilm and drying. Samples were imaged with a 10 kV accelerating voltage and a spot size of 4.0. TEM characterization of the nanostars were conducted by Joel JEM-1400 plus Transmission Electron Microscope and TALOS F200X G2: Scanning/transmission electron microscope (S/TEM). The NS samples were prepared for imaging by adding one drop of the colloid onto carbon films (S160, 200 mesh Cu (25)) and allowing them to dry at room temperature.
SERS analysis
Measurements were carried out using an Avalon RamanStation R2 benchtop Raman spectrometer, which is equipped with a 785 nm external cavity diode laser with a maximum power of 160 mW (10 × 10 s accumulation time). With this system, the 801 cm−1 band of a sample of pure cyclohexane gives a signal of 1100 cts per s. All spectra are shown with no pre-processing other than scaling the intensities for display purposes. In cases where samples were aggregated with salt, 20 μL of MgSO4 was added to 200 μL of the chosen colloid. In all the experiments, 20 μL of the analyte were added to the colloid before the addition of any salts, if needed. In the experiments where thiophenol signals were obtained in presence of es-biofilm, 20 μL of 10–4 mol dm−3 thiophenol was added to a mixture of 20 μL S. aureus es-biofilm plus 200 μL colloid, then 22 μL of MgSO4 was added if aggregation was needed. In cases where thiophenol was added first, 20 μL of the es-biofilm was added to a mixture of 20 μL thiophenol – 200 μL colloid, then 22 μL of MgSO4 was added if aggregation was needed.For the standard addition method experiments, the control sample with no added analyte contained 150 μL NS colloid, 50 μL biofilm and 20 μL water. For pyocyanin measurements, four samples with final concentrations of added analyte of 1.0, 2.0, 3.0, 4.0 × 10–4 M were prepared. For adenine, the standard additions were recorded with final added concentrations of 1.0, 2.0, 3.0, 4.0 × 10–5 M.
Biofilm preparation
For ex situ biofilm preparation, a previously reported protocol was followed with some modifications.22An overnight cell culture of P. aeruginosa (PAO1) and Staphylococcus aureus (S. aureus, ATCC 29213) were diluted in Tryptic Soy Broth (TSB) to obtain 106 cfu ml−1 inoculum. 150 μL of this inoculum was inoculated into a 96-well microplate (sterilized, Thermo Scientific) and incubated at 37 °C and 100 rpm for 24 h. After 24 h, the broth was replaced with 150 μL of fresh medium and incubated again for 24 h. At 48 h, the medium was removed, and each well was rinsed three times in water to remove any remaining medium and unattached cells. 5 μL of sterile water was then added to the microplate and it was sonicated for 15 min to transfer the biofilm attached to the walls of the wells into the water. This process has previously been shown to have no effect on the viability of S. aureus samples.23 Finally, the ex situ biofilm solutions in each well were aspirated into a container for further SERS analysis. For in situ experiments, quartz slides were added to each well of the 96-well microplate and cultured as described above. After biofilm growth was complete, the quartz slides were rinsed in situ with water and then removed for SERS analysis.
Results and discussion
In this work, three different enhancing substrates, citrate-reduced silver colloid, citrate-reduced gold colloid and Au/Ag nanostars (NS), were used to obtain SERS spectra of P. aeruginosa and S. aureus biofilms.Initial experiments were carried out using simple quasi-spherical Au and Ag particles added to intact biofilms of P. aeruginosa and S. aureus, to establish the feasibility of this approach. In these experiments the particles were aggregated with salt and a droplet was placed on top of the pre-grown biofilms. It was found that the aggregated particles adsorbed to the biofilms and formed a visible layer on the surface which did not detach on rinsing. However, the particle distribution was very inhomogeneous at all length scales. Under low power visible microscopy, darker and lighter areas were observed on the mm scale, while in the SEM images, even those areas which had the highest particle density consisted of random aggregates on the μm and nm scales (see Fig. 1b).
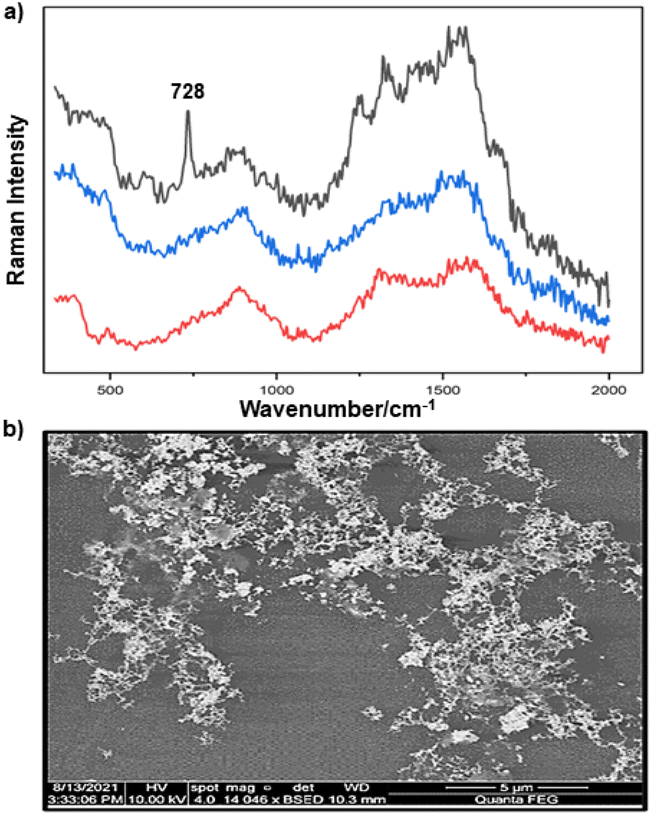 | ||
| Fig. 1 (a) Au colloid–enhanced spectra recorded at three different positions of an in situ S. aureus biofilm. (b) SEM image of AuNPs adsorbed onto the surface of S. aureus biofilm. Scale bar = 5 μm. | ||
For the S. aureus biofilm, it was found that using a Raman microscope with a relatively large spot diameter of ca. 60 μm, SERS spectra with weak bands at 728 cm−1 could be obtained from some of the samples. This band is characteristic of adenine-containing molecules and attributed to the ring breathing vibrational mode of adenine,24 but even in the best spectra these were superimposed on a broad background and in most cases no features other than the background were observed (Fig. 1a). Similar results were found for both Au and Ag enhancing particles. Similarly, for the P. aeruginosa samples, only a weak pyocyanin band was observed at 1354 cm−1 (Fig. S1†). The signal at 1354 cm−1 has been assigned to a combination of C–C stretching, C–N stretching and C–H in-plane bending modes of the pyocyanin aromatic ring.16 It is difficult to attribute the reasons for the low signal intensity and poor reproducibility since numerous different factors are important in measurements of this type. For example, it is well known that using SERS for direct detection of target analytes (i.e. microbial metabolites) in complex biological environments is complicated by the possibility of competition between the target analyte and the matrix components for binding to the metallic surface. In particular, biological environments are rich in proteins that can adsorb onto the surface of unmodified spherical nanoparticles such as the Ag and Au colloids used here. These can compete with other components of the biofilm or hinder access of other molecules, such as the analytical targets, to the enhancing surface.9 Moreover, protein adsorption is also known to prevent the aggregation of the particles, which is necessary for generating SERS “hot spots”.17 While the SEM image in Fig. 1 showed that some particle aggregation could be observed for dried colloid/biofilm samples, it was not clear if the deposits were aggregates which had formed above the films and subsequently deposited or had formed within the top surface layer of the biofilm. This distinction is important because it is generally accepted that pre-aggregation creates aggregates in which many of the hot spots are inaccessible, since they lie within the particle assembly. Of course, there is an additional uncertainty, in that the concentration of the target molecules and their distribution within the biofilm is also not known in advance, so that the signal may vary across the sample due to heterogeneity in the film, as well as due to differences in the enhancing aggregates. In order to remove as many variables as possible but retain the important parts of the measurements, the focus of the studies was shifted from in situ measurements to ex situ measurements, where the biofilms were removed from the surface on which they were cultured and mixed, so that they could be treated as a homogenous controllable liquid. The preparation of the ex situ biofilm (es-biofilm) was carried out in a way which was designed to minimise disruption of the sample. The biofilms were grown in 96 well plates and extracted by adding the minimum amount of water (5 μL) to each well and could then be removed by pipette. Larger volumes were prepared by combining the extracts from numerous wells. In subsequent experiments, the addition of colloidal suspensions of nanoparticles and/or aqueous salt solutions resulted in ex situ samples which had a significantly higher water content than the in situ biofilms, which reduced their viscosity and made them easier to handle. In addition, this also removed complications associated with the structure and gel-like properties of the in situ biofilm. Of course this means that this approach cannot be used to monitor the structure which was present in the original biofilm but this is an inevitable consequence of the method. Initial es-biofilm experiments were carried out with P. aeruginosa. In this case, the es-biofilm was added to the colloid, to allow any interactions to occur, before the salt was added to promote aggregation. As shown in Fig. 2, the SERS spectra recorded with the Ag colloid in the 400–1800 cm−1 range are dominated by signals due to pyocyanin, a major virulence factor produced by P. aeruginosa. This assignment was confirmed by comparison of the es-biofilm signals with that of a 1 × 10–4 mol dm−3 solution of pyocyanin in water (also shown in Fig. 2), which is the concentration in the biofilm after dilution for SERS (see below). Detection of pyocyanin is as expected from the literature since pyocyanin has a very large scattering cross-section at this excitation wavelength.16 A significant advantage of using es-biofilm is that it can be used almost like a normal aqueous sample. For example, it is useful to establish the concentration of the pyocyanin in the biofilms. One way to measure this would be to use UV/Vis absorption spectroscopy, which would, in principle, be possible, since the pyocyanin is strongly coloured but in practice is difficult due to the low optical density of the relatively thin biofilms and the fact that the in situ biofilms are also heterogeneous. More generally, it is important to have a method available which could be used for other targets where UV/Vis absorption measurements are not possible, due to low extinction coefficients, such as the adenine measurements discussed below. Therefore, for the quantitative SERS measurements, the standard addition method was used, since this removes the effect of the matrix on the sample, which is particularly important with the es-biofilms where strong interference from the matrix is expected. Since the es-biofilms are essentially homogenous liquids the experiments simply involved adding the enhancing Au/Ag nanostars (NS) to a series of es-biofilm samples with increasing concentrations of added pyocyanin. The plot of signal intensity versus added pyocyanin could then be extrapolated to give the concentration of pyocyanin in the es-biofilm samples at zero added pyocyanin (see Fig. S2†). This measurement gave a pyocyanin concentration of 6 × 10–4 mol dm−3 in the diluted SERS samples used for the standard addition, which contained the NS and es-biofilm, meaning that the concentration in the extracted es-biofilm is 2 × 10–3 mol dm−3. Once the concentration of the pyocyanin in the es-biofilm was known, the effect of the biofilm matrix on the spectra could be determined by comparing the es-biofilm data with those of the same concentration of pyocyanin in a simple aqueous medium. Without this data it was possible to observe that the in situ measurements showed only small signals, but it was not clear if this was due to matrix effects that reduced the intensity or which one of the many factors that could cause this effect were responsible. However, direct comparison of the SERS spectra from Ag colloid with 1 × 10–4 mol dm−3 pyocyanin in the es-biofilm matrix and aqueous solution showed that the intensity of the pyocyanin band at 1354 cm−1 reduced by 90% in presence of biofilm (Fig. 2). The question of whether this is caused by loss of aggregation in the es-biofilm sample or the components of the biofilm blocking access to the surface sites is discussed below, where additional data are available. However, these data show that the matrix effects are large, even in the diluted es-biofilm samples. One final observation was that these data also show a small band at 730 cm−1, which does not match with the pyocyanin spectra (expanded spectra comparing both are shown in the Fig. S3†). This peak is in the position expected for the strongest band in adenine or adenine-containing molecules, including ATP, NAD, RNA and DNA.24 Further studies on S. aureus (see below) show this band much more clearly. Surprisingly, with Au colloid, the spectra of the es-biofilm showed features that were identical to those observed for the blank sample, with no obvious pyocyanin bands (Fig. 2). This lack of signal might be due to the lower affinity of pyocyanin for the surface of the Au particles used here than for the Ag particles. To test if this was the case, the SERS spectrum of a 1 × 10–4 mol dm−3 aqueous sample of pyocyanin (the same concentration as used in the experiments with Ag colloid) was recorded. The spectrum, shown in Fig. 2, is very similar to that of the Au colloid alone and there are only very weak pyocyanin bands, demonstrating that pyocyanin has low affinity for the Au colloids used in these studies. The complete lack of pyocyanin bands in the biofilm is presumably due to the combination of this low affinity and the effect of the EPS matrix interfering with the binding or aggregation of the Au colloid. While P. aeruginosa is a good test organism because many P. aeruginosa strains produce pyocyanin, which is very easy to detect, it is clearly different from most bacteria because they do not produce this strong marker compound.22 It was therefore important to move to a more representative biofilm to be able to get a better understanding of SERS in biofilms. Here S. aureus was chosen because it is clinically relevant and readily forms biofilms. Although the es-biofilms of S. aureus did not contain any pyocyanin, it was still possible to observe strong bands in the SERS spectra obtained using both Ag and Au colloids (Fig. 3). In this case the signals closely matched those of adenine, whose spectra were also recorded using the same colloids and are shown for comparison in Fig. 3. Since there are so many potential sources of “adenine-like” signals, including ATP, NAD etc., considerably more work would be necessary to unambiguously assign the source of the “adenine” peak. However, an interesting possibility is that the adenine-like signals are due to external DNA (eDNA) present in the es-biofilm. eDNA is an important structural component of biofilms, and its Raman spectrum, like that of all DNA, is dominated by scattering from the adenine bases.24,25 Also notable in the spectra of the S. aureus es-biofilms is the lack of any bands associated with the EPS matrix, presumably because any weak bands are masked here by the adenine signal, in the same way as they would be masked by the pyocyanin for P. aeruginosa spectra. Again, since we are using ex situ samples it was straightforward to use standard addition method to demonstrate that the signal from the diluted es-biofilm was equivalent to that created by 4 × 10−5 mol dm−3 adenine (Fig. S4†), although of course this does carry the assumption that the scattering cross-section of the unidentified adenine-type compound in the biofilm is the same as that of simple adenine. This gives an adenine concentration in the undiluted extract of 1 × 10–4 mol dm−3. The standard addition method also allows us to compare the intensities of the intrinsic marker compounds in the biofilms with those in simple aqueous solution and therefore to measure the extent to which the presence of the es-biofilm affects the intensity of the SERS signals. The data from the P. aeruginosa biofilm showed that with the Ag colloid the ex situ matrix reduces the pyocyanin signal by 90%, although the same comparison could not be made for Au colloid due to the low signal intensities (Fig. 2). With the S. aureus biofilm, the 730 cm−1 adenine marker band was reduced by 70% in the es-biofilm for Ag colloid. In this case it was possible to also compare the effect with Au colloid where the intensity reduction was similar (80%) (Fig. 3). As discussed above, these large reductions in signal intensity for both Ag and Au colloids in presence of biofilm might be a result of the matrix components, possibly proteins, blocking access to the enhancing surface and/or interfering with the aggregation. These two effects can be decoupled because fortunately, it is relatively easy to detect aggregation in metal colloids, since the aggregates have very different UV/Vis spectra from individual particles.16 In this work, UV/Vis measurements provided a simple method to assess the ability of EPS to reduce the salt-induced nanoparticle aggregation. For this purpose, UV/Vis spectra were recorded for both Ag and Au colloid before and after addition of 1 M MgSO4 and in the presence and absence of es-biofilm. S. aureus biofilm was used for these experiments because the pyocyanin in P. aeruginosa biofilm can also cause colloidal aggregation, making it more difficult to assess the effect of biofilm proteins on the overall aggregation.26 The UV/Vis spectrum of unaggregated Ag colloid shows a single extinction band at 413 nm, this band is not perturbed by the addition of es-biofilm, showing that the biofilm did not induce aggregation of the colloid. For the simple aqueous colloid, the addition of 1 M of MgSO4 resulted in a second peak appearing at longer wavelengths, indicating the formation of aggregates, as expected. However, in the presence of es-biofilm, addition of the salt to the silver colloid gave a new band which was less shifted to the red than was the case for the samples without biofilm present (Fig. 4a). It is well known that the location of the second band strongly depends on the size of the aggregates, so the smaller shift of the second peak indicates formation of smaller aggregates in the presence of biofilm. This is a clear indication that the es-biofilm can indeed hinder aggregation for Ag colloid. These observations were further confirmed with Dynamic Light Scattering (DLS) which showed that while salt addition did significantly increase the average size of the particles/aggregates in both cases, the extent of aggregation was lower in the sample containing es-biofilm than that in a simple aqueous solution. This is particularly clear at lower end of the size range, where even after salt addition the biofilm-containing samples still retained a large number of particles in the 20–70 nm size range (Fig. 4b). The data showing that the es-biofilm reduces aggregation helps to explain why the biofilm addition did not create aggregates in the absence of added salt, even though aggregation might be expected since the biofilm was grown in a medium which contained NaCl. Presumably here the effect of the salt which was added as part of the es-biofilm was countered by the ability of the biofilm to also reduce aggregation through protein adsorption. While these results show that the biofilm affects aggregation, they do not allow the effect of aggregation and of blocking of the surface to be separated, since the Ag colloid does still aggregate to a significant extent, even in the presence of es-biofilm and so might be SERS active, although less enhancing than simple aqueous samples. In order to resolve this problem, experiments were carried out with a very strongly binding analyte, thiophenol, which we can be confident will adsorb to the surface and not then be displaced by other biofilm components.27Fig. 5 compares the spectra of 10–4 mol M−3 thiophenol with simple Ag colloid and Ag colloid with added es-biofilm. Two biofilm experiments were carried out. In the first of these, the thiophenol was added to the Ag colloid, to allow binding before the es-biofilm was added. In this case, a relatively small reduction of approximately 2× in the intensity of the 1570 cm−1 thiophenol signal (assigned to ring C–C stretching)27 was observed, compared to the control sample. This can be attributed to the reduced aggregation caused by the es-biofilm which was also observed in the UV/Vis experiments above. In the second experiment, the es-biofilm was added to the Ag colloid before the thiophenol, so in this case the components of the biofilm were free to interact with the colloid and block access to the active sites, as well as interfere with the aggregation. In this experiment the thiophenol signal was reduced much more, falling by 95% compared to simple Ag colloid. This suggests that the effect of the biofilm is to reduce the enhancement factor of the colloid due to poor aggregation by a factor of 2×, however the effect of site blocking is larger, since it adds an additional factor of 10× when the signal is measured 100 s after mixing. Although these values are for thiophenol, the overall reduction in signal is similar to the 70% reduction value observed for adenine in S. aureus and also the 90% value observed for pyocyanin in P. aeruginosa. This suggests that there is a similar pattern of behaviour across all these samples. For detection of pyocyanin, the results were different for Au and Ag colloids, so it is useful to also investigate the play-off between aggregation and surface site blocking for Au colloid. The initial experiments were similar to those for the Ag colloid, where the UV/Vis spectra were recorded when salt was added to samples containing es-biofilm and simple Au colloid controls. In this case, the unaggregated Au colloid shows the expected single extinction band at 520 nm, while a second peak appeared at longer wavelength when salt was added, indicating the formation of aggregates in the usual way. However, in contrast to the Ag colloid, where addition of es-biofilm did not change the extinction spectrum, here the addition led to the appearance of a new peak shifted to longer wavelength, although not as far as when salt was added to simple colloid (Fig. 6a). This different behaviour of the Ag and Au colloids in the presence of es-biofilm indicates that they interact differently with the biofilm's components. Specifically, the interaction between the Au colloid and the es-biofilm which hinders aggregation must be weaker than is the case for Ag (Fig. 6c). As a result, in the Au colloid spectrum, the colloid is sufficiently aggregated to give a weak adenine signal at 730 cm−1 even without added salt, while the unaggregated Ag colloid gives only a featureless broad scattering background. It has been previously reported that the binding affinity of specific proteins to the metal surface can differ between Ag and Au nanoparticles but of course in the current study it is not possible to attribute effects to specific components within the very complex mixture that is a biofilm.28
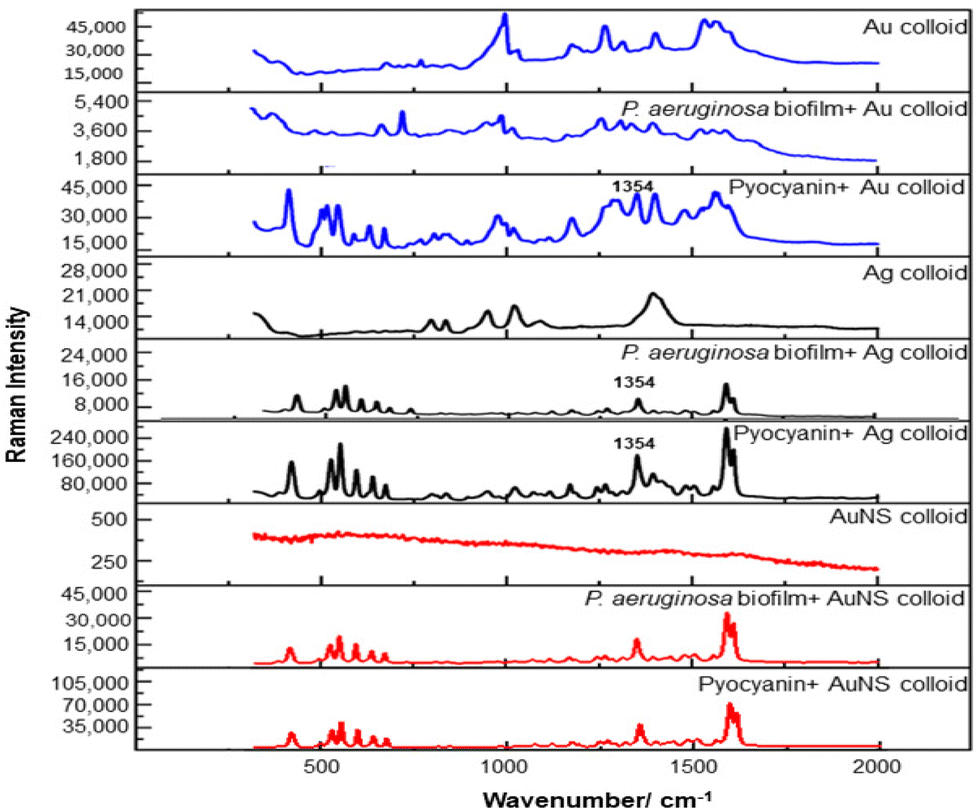 | ||
| Fig. 2 Comparison of SERS spectra of P. aeruginosa es-biofilm and 10−4 M pyocyanin using Ag, Au and NS colloids. | ||
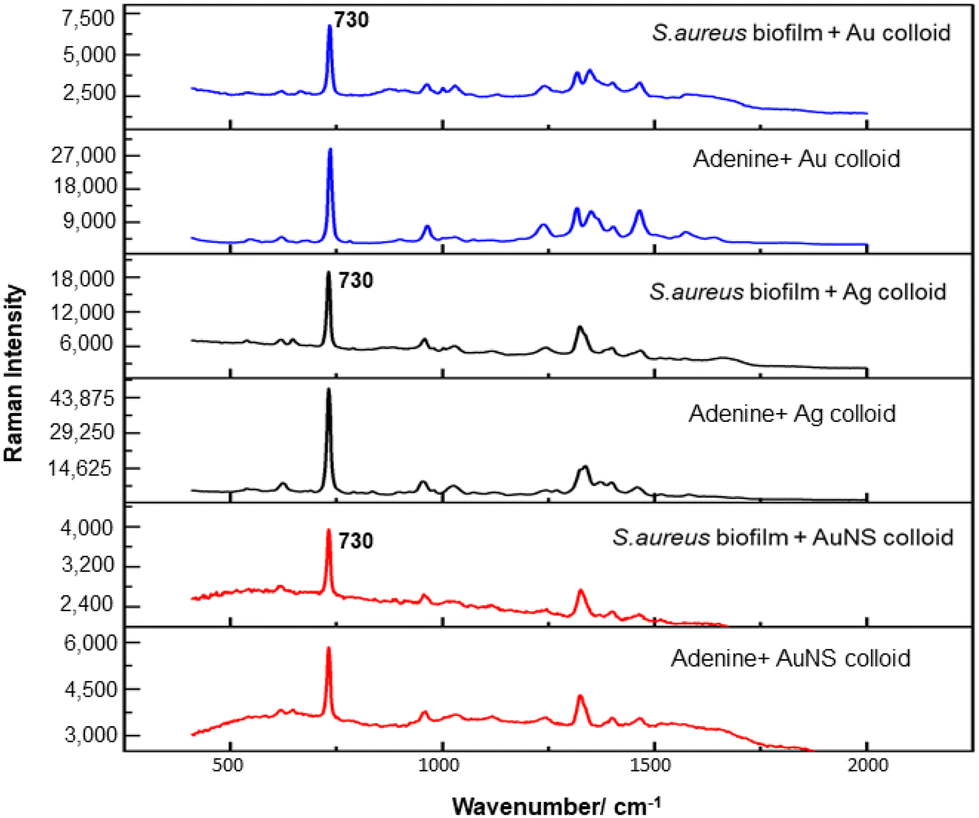 | ||
| Fig. 3 Comparison of SERS spectra of S. aureus es- biofilm and 10−5 M adenine using Ag, Au and NS colloids. | ||
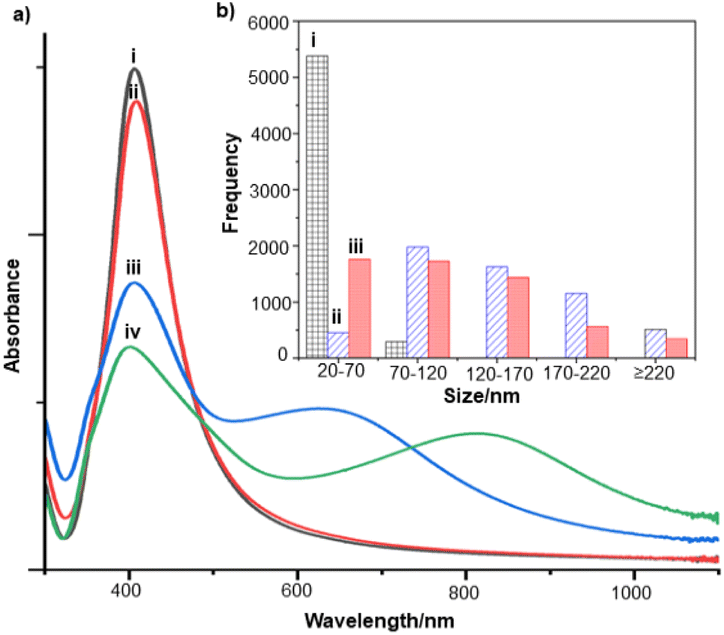 | ||
| Fig. 4 (a) UV-vis absorption spectra of Ag colloid, (i) Unaggregated (ii) with S. aureus es-biofilm, (iii) with S. aureus es-biofilm and after addition of 1 M MgSO4 and (iv) after addition of 1 M MgSO4 only. (b) Particle size distribution of Ag colloid (i) before aggregation, (ii) after addition of 1 M MgSO4 and (iii) with S. aureus es-biofilm after addition of 1 M MgSO4. | ||
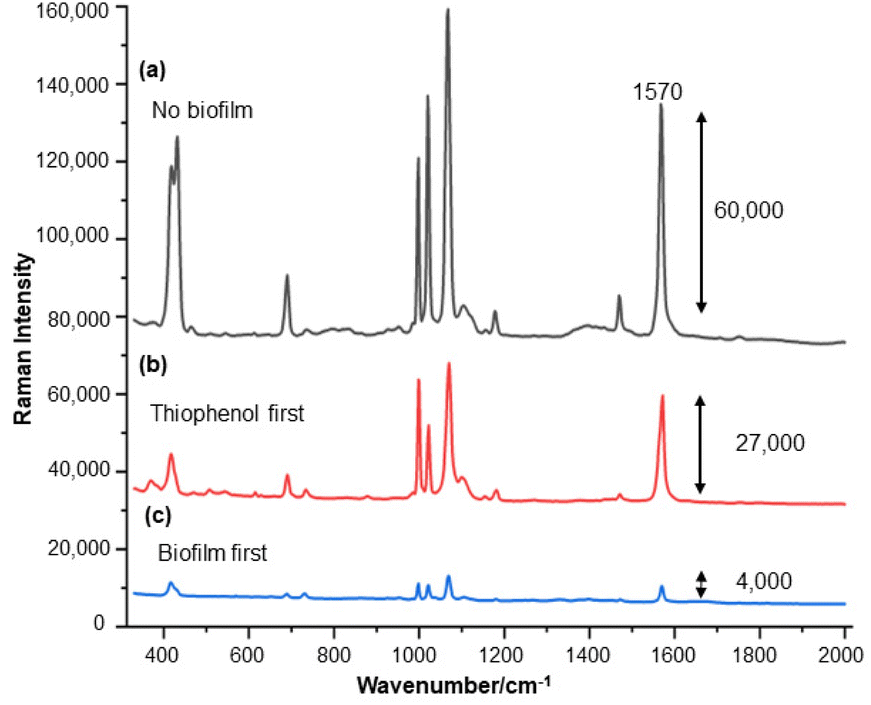 | ||
| Fig. 5 SERS spectra of 10−4 M thiophenol on Ag colloid aggregated with 1 M MgSO4. (a) Thiophenol only, (b) thiophenol-treated sample after addition of S. aureus es-biofilm (c) sample where S. aureus es-biofilm was added to colloid before addition of thiophenol. | ||
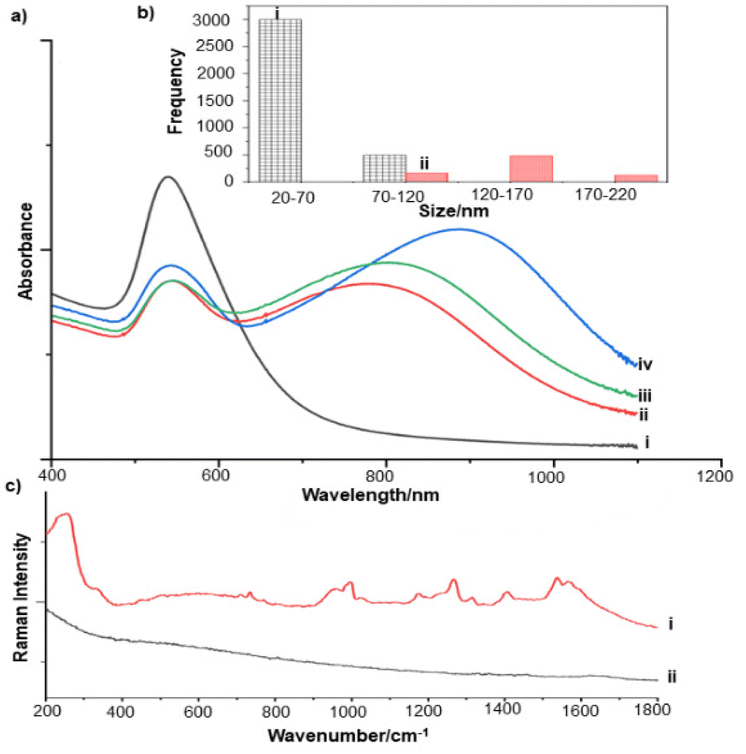 | ||
| Fig. 6 (a) UV-vis absorption spectra of Au colloid, (i) Unaggregated (ii) with S. aureus es-biofilm, (iii) with S. aureus es-biofilm and after addition of 1 M MgSO4 and (iv) after addition of 1 M MgSO4 only. (b) Particle size distribution of Au colloid (i) before aggregation, (ii) after addition of S. aureus es-biofilm. (c) SERS spectra of Au colloid (i) and Ag colloid (ii) after addition of S. aureus es-biofilm with no additional salt. | ||
Interestingly, addition of 15 μL of 1 M salt to the Au colloid which was pre-aggregated by es-biofilm did not induce significant additional aggregation, since the long wavelength peak in the UV/vis absorption spectrum barely shifted on salt addition. This indicates that the nanoparticles were already aggregated to almost the fullest extent possible in the presence of the biofilm (Fig. 6a), although this was still less than is possible with simple aqueous Au colloid, which is similar to what is observed with the Ag colloid (Fig. 6a and 4a). Again, the ability of the biofilm to aggregate Au colloid was further confirmed with DLS, which shows formation of larger aggregates upon biofilm addition (Fig. 6b). Similarly, when thiophenol was added after the es-biofilm and the sample was aggregated the signal dropped by 90% (see Fig. S5†), very similar to the value observed for the Ag colloid.
It is useful to note that all the experiments described above were carried out using es-biofilm which was initially diluted by the addition of water to allow it to be removed from the 96 well plate and this stock solution of extracted biofilm was then further diluted by a factor of >20× by addition of colloid and water. The observation of significant effects on particle aggregation and site blocking within relatively dilute es-biofilm solution suggests that even larger effects will be observed in in situ biofilm experiments, where a higher concentration of biofilm components will be present.
To avoid aggregation problems in the presence of biofilm, nanostars (NS) were also investigated because it is known that they give SERS enhancement without aggregation. The NS used in this work were prepared as previously reported and comprise a central metal core from which multiple sharp spikes protrude.21 The key element of these substrates is the spikes, which serve as efficient nanoantennae, so that the electromagnetic field is expected to be very high at the end of each tip, giving rise to multiple intrinsic SERS hot spots within a single nanoparticle.29
For these NS, aggregation is not desirable, indeed it is a problem since they are known to spontaneously aggregate, which makes them difficult to store and handle. It also reduces the plasmonic enhancement they provide. The propensity to aggregate is believed to be associated with the asymmetric electric double layers in the spikes.30 In the current work the NS were stabilised by adding a low concentration of hydroxyethyl cellulose (HEC). Importantly, this polymer does not change the viscosity of the colloid significantly or chemically adsorb onto the surface of the NS, so that it can prevent aggregation while leaving the surface of the particles free for binding.‡
The extent of aggregation of the NS in the presence of es-biofilm was first checked with UV/Vis spectroscopy, which shows a broad extinction peak centred around 780 nm, that is characteristic of the unaggregated NS (Fig. 7a). This peak does not shift noticeably in the presence of the S. aureus es-biofilm, although the large width of the peak would make it more difficult to observe peak shifts and broadening than was the case for the Ag and Au nanoparticles shown above. As a result, the stability of NS was further investigated by DLS. Fig. 7b compares the size distribution of NS in water and in the presence of biofilm. It is clear that there is some aggregation, since the size distribution shifts slightly to larger sizes, but the effect is very much smaller than is observed for Ag and Au colloids.
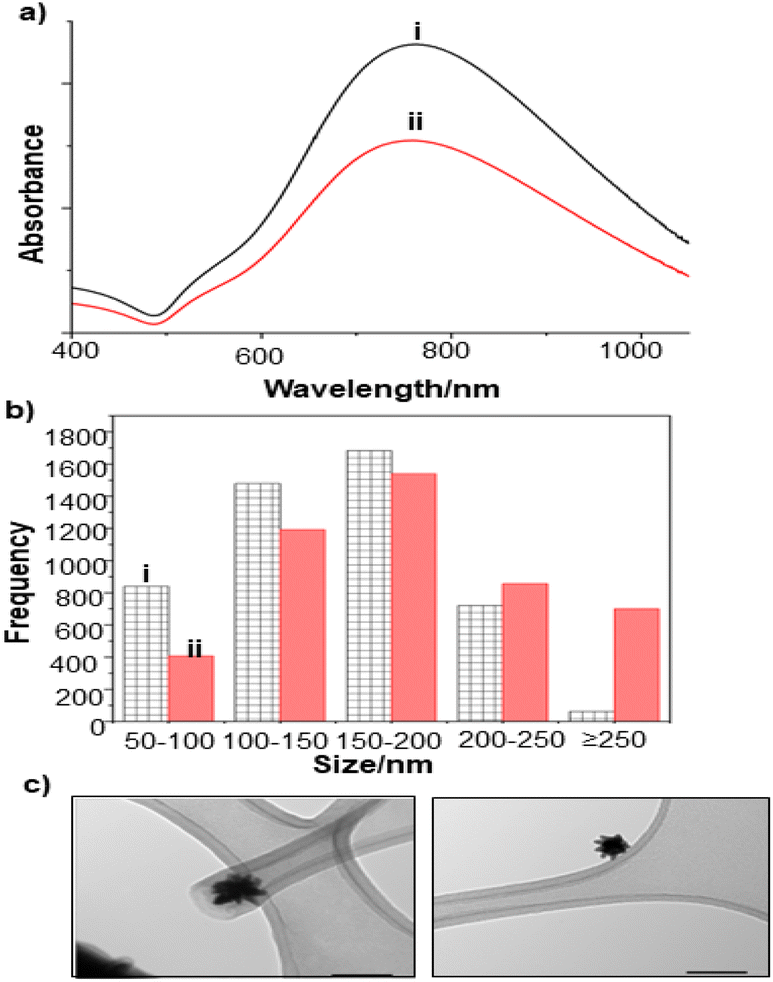 | ||
| Fig. 7 (a) UV-vis absorption spectra of NS colloid, (i) Unaggregated (ii) with S. aureus es-biofilm (b) Particle size distribution of NS colloid (i) before aggregation, (ii) after addition of S. aureus es-biofilm. (c) TEM image of the NS colloid. Scale bar = 100 nm. | ||
SERS studies of the HEC-stabilised NS were carried out in the presence of both P. aeruginosa and S. aureus es-biofilms. For P. aeruginosa, strong bands associated with pyocyanin were observed. This contrasts with the result for Au colloid where pyocyanin binding was weak. Presumably in this case the presence of Ag along with Au in the NS increases pyocyanin's affinity for the surface. Importantly, the intensity of the pyocyanin signals was only reduced to 50% of the value in the control sample when es-biofilm was added (Fig. 2). The observed decrease was significantly less than that observed with Au and Ag colloids 100 s after mixing. Furthermore, this smaller decrease can be attributed to blocking access to the active sites, rather than a difference in aggregation and would therefore be expected to remain around this value even in in situ biofilms while, as discussed above the reduction in signal for aggregated particles would be expected to even larger under in situ conditions. These results are consistent with previous study which reported that star-shaped gold nanoparticles showed lower fouling due to reduced protein corona formation.31
Studies using NS and S. aureus es-biofilm were found to be consistent with the P. aeruginosa results, in that the samples show strong adenine bands, and these adenine bands again show only a small reduction to 43% of the control value when es-biofilm is added (Fig. 3). This suggests that the degree to which each of the es-biofilms can block the binding sites is quite similar. Similarly, as shown in Fig. S6,† the signal intensity for thiophenol added to a simple NS colloid was only reduced by 50% when it was added to NS colloid which was pre-treated with S. aureus es-biofilm. This was expected on the basis of the pyocyanin and adenine data, assuming that the thiophenol is not able to displace the blocking groups from the active sites.
The overall results for the NS colloid are very good. The absolute SERS signals obtained for simple aqueous solution of the pyocyanin and adenine are significantly smaller than those for the Ag and Au colloids (with the exception of the pyocyanin/Au combination which is already limited by low affinity). However, the fact that the SERS signals from NS colloid are significantly less affected by addition of the es-biofilm than are those from simple Ag and Au colloids means that in the es-biofilm-containing samples the absolute intensities obtained with all three enhancing materials are similar. This is a meaningful comparison because the concentration of NS colloid used was similar to that in the Ag and Au colloids. Of course the NS are significantly larger than the individual Au or Ag nanoparticles used here and this would be expected to reduce their diffusion within intact biofilms.18 However, this is a misleading comparison since, for SERS experiments, the Au and Ag nanoparticles are used as aggregates which contain numerous particles and therefore would also be expected to diffuse more slowly than individual particles.
A significant advantage of the NS particles is that they sit as numerous randomly distributed individual particles within the es-biofilm and this homogenous particle distribution leads to a more uniform signal enhancement. In contrast, the Ag and Au colloids need to be aggregated with salt, which means that the samples are intrinsically more inhomogeneous, with local high concentration regions and areas where the number of particles is very low. This leads to much more variation in the absolute SERS intensity recorded at different points within the es-biofilm compared to the NS samples (Fig. 8 and S7†).
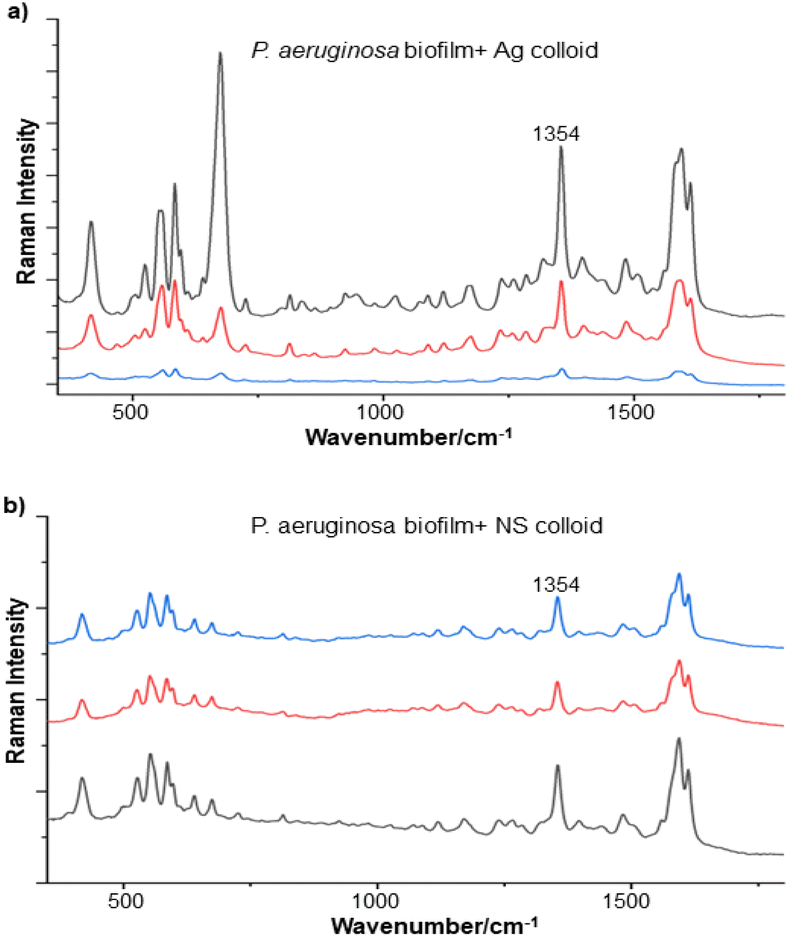 | ||
| Fig. 8 Surface enhanced Raman spectra of P. aeruginosa es-biofilm obtained with (a) Ag colloid and (b) NS colloid. Three technical replicates recorded on the same day using the same es-biofilm sample are shown for each colloid. | ||
Conclusion
The work presented above provides a new approach for using SERS to study biofilms. This ex situ method allowed for quantitative measurements of the intrinsic marker compounds present in biofilms in a way which is very difficult or impossible to carry out in very heterogeneous in situ biofilm samples. In particular, it allowed standard addition methods to be used to quantify the signals from the intrinsic biomarkers and therefore the various factors which control the overall signals which are detected in the complex biofilm matrix to be untangled. Here, the fact that strong signals could be detected from intrinsic marker compounds in two different species and that these could be quantified even in the presence of strong matrix interference demonstrates the power of this approach. Similarly, the observation and quantification of matrix effects on different enhancing media is central to further studies in this area. It is clear that both hindered aggregation and blocking of surface sites are important in determining overall performance. In the current study, the nanostars tested were found to be superior to standard aggregated Ag and Au colloidal nanospheres and this will be important for our future studies of in situ biofilm samples. In addition, this work also provides a general approach which can be adopted more widely as a basis for developing and validating SERS enhancing media for biofilm studies in a systematic way. This will allow them to be optimised before moving on to much more complex in situ measurements where their performance relative to other materials would otherwise be very difficult to determine.Author contributions
W. A. contributed to conceptualization, methodology, investigation, data analysis, writing – original draft, review and editing; M. W. contributed to methodology, review and editing; Y. X. contributed to methodology; Y. Z. contributed to investigation; C. P. M contributed to conceptualization, review and editing, and S. E. J. B. contributed to conceptualisation, methodology, data analysis, resources, supervision, writing – original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. A. acknowledges the Ministry of Education in Saudi Arabia for funding support. Y. Z. acknowledges the Chinese Scholarship Council (202008370188) for funding support. Y. X. acknowledges the Leverhulme Trust Early Career Fellowship (grant ECF2020703).References
- L. K. Vestby, T. Grønseth, R. Simm and L. L. Nesse, Antibiotics, 2020, 9, 1–29 CrossRef PubMed.
- H. C. Flemming, T. R. Neu and D. J. Wozniak, J. Bacteriol., 2007, 189, 7945–7947 CrossRef CAS PubMed.
- M. Magana, C. Sereti, A. Ioannidis, C. A. Mitchell, A. R. Ball, E. Magiorkinis, S. Chatzipanagiotou, M. R. Hamblin, M. Hadjifrangiskou and G. P. Tegos, Clin. Microbiol. Rev., 2018, 31, 1–49 CrossRef PubMed.
- M. Okshevsky and R. L. Meyer, J. Microbiol. Methods, 2014, 105, 102–104 CrossRef CAS PubMed.
- M. Alhede, K. Qvortrup, R. Liebrechts, N. Høiby, M. Givskov and T. Bjarnsholt, FEMS Immunol. Med. Microbiol., 2012, 65, 335–342 CrossRef CAS PubMed.
- A. M. Donald, Nat. Mater., 2003, 2, 511–516 CrossRef CAS PubMed.
- N. P. Ivleva, P. Kubryk and R. Niessner, Anal. Bioanal. Chem., 2017, 409, 4353–4375 CrossRef CAS PubMed.
- B. Kideog, W. Zheng, Y. Ma and Z. Huang, Theranostics, 2019, 9, 1348–1357 CrossRef PubMed.
- C. Zong, M. Xu, L. J. Xu, T. Wei, X. Ma, X. S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- S. Keleştemur, E. Avci and M. Çulha, Chemosensors, 2018, 6, 1–15 CrossRef.
- N. P. Ivleva, M. Wagner, H. Horn, R. Niessner and C. Haisch, Anal. Chem., 2008, 80, 8538–8544 CrossRef CAS PubMed.
- S. Keleştemur and M. Çulha, Appl. Spectrosc., 2017, 71, 1180–1188 CrossRef PubMed.
- E. Efeoglu and M. Culha, Appl. Spectrosc., 2013, 67, 498–505 CrossRef CAS PubMed.
- S. De Marchi, D. García-Lojo, G. Bodelon, J. Pérez-Juste and I. Pastoriza-Santos, ACS Appl. Mater. Interfaces, 2022, 13, 61587–61597 CrossRef PubMed.
- C. Q. Nguyen, W. J. Thrift, A. Bhattacharjee, S. Ranjbar, T. Gallagher, M. Darvishzadeh-Varcheie, R. N. Sanderson, F. Capolino, K. Whiteson, P. Baldi, A. I. Hochbaum and R. Ragan, ACS Appl. Mater. Interfaces, 2018, 10, 12364–12373 CrossRef CAS PubMed.
- G. Bodelón, V. Montes-García, V. López-Puente, E. H. Hill, C. Hamon, M. N. Sanz-Ortiz, S. Rodal-Cedeira, C. Costas, S. Celiksoy, I. Pérez-Juste, L. Scarabelli, A. La Porta, J. Pérez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzán, Nat. Mater., 2016, 15, 1203–1211 CrossRef PubMed.
- S. Dominguez-medina, J. Blankenburg, J. Olson, C. F. Landes and S. Link, ACS Sustainable Chem. Eng., 2013, 1, 833–842 CrossRef CAS PubMed.
- T. O. Peulen and K. J. Wilkinson, Environ. Sci. Technol., 2011, 45, 3367–3373 CrossRef CAS PubMed.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- G. Frens, Nat., Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- S. He, Y. M. E. Kyaw, E. K. M. Tan, L. Bekale, M. W. C. Kang, S. S. Y. Kim, I. Tan, K. P. Lam and J. C. Y. Kah, Anal. Chem., 2018, 90, 6071–6080 CrossRef CAS PubMed.
- G. Bodelón, V. Montes-garcía, V. López-puente and E. H. Hill, Nat. Mater., 2017, 15, 1203–1211 CrossRef PubMed.
- 1 P. B. Flynn, W. G. Graham and B. F. Gilmore, Lett. Appl. Microbiol., 2019, 68, 344–349 CrossRef PubMed.
- L. J. Xu, Z. C. Lei, J. Li, C. Zong, C. J. Yang and B. Ren, J. Am. Chem. Soc., 2015, 137, 5149–5154 CrossRef CAS PubMed.
- F. Monticolo, E. Palomba, P. Termolino, P. Chiaiese, E. de Alteriis, S. Mazzoleni and M. L. Chiusano, Front. Plant Sci., 2020, 11, 1–15 CrossRef PubMed.
- Y. Tanaka, E. H. Khoo, N. A. B. M. Salleh, S. L. Teo, S. Y. Ow, L. Sutarlie and X. Su, Analyst, 2021, 146, 6924–6934 RSC.
- F. Sun, D. D. Galvan, P. Jain and Q. Yu, Chem. Commun., 2017, 53, 4550–4561 RSC.
- W. Lai, Q. Wang, L. Li, Z. Hu, J. Chen and Q. Fang, Colloids Surf., B, 2017, 152, 317–325 CrossRef CAS PubMed.
- A. Shiohara, Y. Wang and L. M. Liz-Marzán, J. Photochem. Photobiol., C, 2014, 21, 2–25 CrossRef CAS.
- Z. Ye, C. Li, M. Celentano, M. Lindley, T. O'Reilly, A. J. Greer, Y. Huang, C. Hardacre, S. J. Haigh, Y. Xu and S. E. J. Bell, JACS Au, 2022, 2, 178–187 CrossRef CAS PubMed.
- A. Tukova, I. C. Kuschnerus, A. Garcia-Bennett, Y. Wang and A. Rodger, Nanomaterials, 2021, 11, 1–17 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: SERS spectra, standard addition plots. See DOI: https://doi.org/10.1039/d3an00301a |
| ‡ The protection against aggregation provided by HEC does have a limit, so that increasing the NS concentration above the value used here does lead to significant aggregation, even in the presence of HEC. In use, the polymer is expected to give minimal perturbation because any small effects which might be present in the undiluted colloid stock solution will be reduced even further when the colloid is diluted by adding to the biofilm. |
| This journal is © The Royal Society of Chemistry 2023 |