
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBicyclobutanes: from curiosities to versatile reagents and covalent warheads
Christopher B.
Kelly
 *a,
John A.
Milligan
*b,
Leon J.
Tilley
c and
Taylor M.
Sodano
d
*a,
John A.
Milligan
*b,
Leon J.
Tilley
c and
Taylor M.
Sodano
d
aDiscovery Process Research, Janssen Research & Development LLC, 1400 McKean Road, Spring House, PA 19477, USA. E-mail: ckelly5@its.jnj.com
bDepartment of Biological and Chemical Sciences, College of Life Sciences, Thomas Jefferson University, 4201 Henry Avenue, Philadelphia, PA 19144, USA. E-mail: john.milligan@jefferson.edu
cDepartment of Chemistry, Stonehill College, 320 Washington Street, Easton, MA 02357, USA
dTherapeutics Discovery, Janssen Research & Development LLC, 1400 McKean Road, Spring House, PA 19477, USA
First published on 25th August 2022
Abstract
The unique chemistry of small, strained carbocyclic systems has long captivated organic chemists from a theoretical and fundamental standpoint. A resurgence of interest in strained carbocyclic species has been prompted by their potential as bioisosteres, high fraction of sp3 carbons, and limited appearance in the patent literature. Among strained ring systems, bicyclo[1.1.0]butane (BCB) stands apart as the smallest bicyclic carbocycle and is amongst the most strained carbocycles known. Despite the fact that BCBs have been synthesized and studied for well over 50 years, they have long been regarded as laboratory curiosities. However, new approaches for preparing, functionalizing, and using BCBs in “strain-release” transformations have positioned BCBs to be powerful synthetic workhorses. Further, the olefinic character of the bridgehead bond enables BCBs to be elaborated into various other ring systems and function as covalent warheads for bioconjugation. This review will discuss the recent developments in the synthesis and functionalization of BCBs as well as the applications of these strained rings in synthesis and drug discovery. An overview of the properties and the historical context of this interesting structure will be provided.
1. Introduction
The concept of molecular strain has fascinated chemists for decades.1 Studies on the properties and reactivity of strained carbocyclic systems have yielded numerous synthetically useful transformations. Indeed, the concept of “strain-release” reactions has become embedded into the modern lexicon of synthetic chemistry.2 Bicyclo[1.1.0]butanes (BCBs) are among the most structurally compact cyclic molecules known, yet despite their small size they have rich synthetic value because of their high strain energy. As such, they have been studied extensively from both a theoretical and preparative point of view, as featured in some recent reviews.2,3 The efforts of numerous laboratories in recent years have enabled BCBs to transition from curiosities of physical organic chemistry to modular building blocks for synthesis. This Minireview seeks to capture the evolutionary story of BCB in a concise manner from its discovery in the mid-20th century to its entrance as a synthetic “staple” in the 21st century, emphasizing the modern synthetic applications of BCB and its derivatives with special attention given to potential pharmaceutical applications. In this first section, fundamental aspects of BCB chemistry, ranging from properties to classic synthetic approaches, will be discussed.1.1 Properties and reactivity
A distinguishing feature of the BCB scaffold is its “butterfly” geometry, wherein the two “wings” adopt a 123° interflap angle (Scheme 1A). Consequently, the exo (1.194 Å), endo (1.167 Å), and bridgehead (1.142 Å) C–H bonds all differ in length.4 Interestingly, notwithstanding the significant amount of strain in BCB, the compound contains two essentially undistorted cyclopropane rings5 in which the bridgehead and side C–C bonds are almost identical in length at approximately 1.50 Å.6 These C–C bond lengths have been determined experimentally by vibrational7 and microwave spectroscopy,8 as well as electron diffraction studies.4a Earlier molecular orbital descriptions contrasted with these experimental findings by calculating more significant differences between the lengths of the two types of bonds (bridgehead shorter than side “bridging” bonds)6a,9 as well as other discrepancies leading to shorter calculated bond lengths for both types of bonds.10 However, more recent and increasingly powerful computational methods are in good agreement with experiment, predicting an ever decreasing difference between the two bond lengths. B3LYP/6-311G** level calculations11 still yield results pointing to the bridgehead bond being very slightly shorter than the side bonds, while MP2/cc-pVTZ and CCSD(T)/cc-pVTZ calculations show (to a slight degree) the opposite, with the latter method predicting equivalent bond lengths to within 0.0001 Å.6b | ||
| Scheme 1 Summary of fundamental properties and reactivity of BCB. | ||
Evidence for this unique structure can also be established by NMR spectroscopy.4a The proton NMR spectrum is very characteristic in terms of the significant chemical shift differences between the endo, exo, and bridgehead protons (0.489, 1.500, and 1.358 ppm in CDCl3), respectively (Scheme 1B).12 The enhanced s character of the shorter bridgehead methine C–H bonds is experimentally verifiable by observing the 13C–1H coupling constant (202 Hz).13 For context, this value lies between the 13C–1H coupling values observed for ethylene (156 Hz) and acetylene (249 Hz) (Scheme 1B). These observations, along with other spectroscopic evidence mentioned above, point to substantial π character of the strained central C–C bond.13,14
The quantification of strain energy in the BCB structure has been a topic of longstanding theoretical interest. Meier has recently reported a group contribution approach that has provided excellent agreement between theoretical (217.5 kJ mol−1) and experimental (217 kJ mol−1) formation enthalpies, and predicts a strain energy of 267 kJ mol−1 (∼64 kcal mol−1).15 Although the dominant contribution to the strain energy is most often cited to be Baeyer strain,16 bridgehead substituent effects also contribute significantly to the energy of the system. Dill has calculated the effects of a variety of substituents on the strain energy of BCB.17 Most notably, CF3, CN, and Li were seen to be stabilizing, although calculations for CN predicted less decrease in strain energy than suggested by experimental data. Hoz has shown the bridgehead substituent effects to be predominantly electronic, with little effects due to changes in Baeyer strain from potential deformation of the ring.18 Indeed, it is well-known that some electron withdrawing groups exhibit a stabilizing effect, and many of the early (and current) examples of BCBs contain such groups.3,13 Inagaki has also shown that bridgehead substituents can be a determining factor as to whether the bridgehead carbons exhibit “inverted” tetrahedral configurations (all four atoms bonded to carbon are on the same side of the plane containing the carbon) as a result of the extent to which they influence geminal delocalization between the bridgehead and side bonds.19 Based on a mean value for the angles of the six “substituents” on both carbon atoms of the bridgehead bond of 82°, Chaquin20 has recently classified the bridgehead bond in BCB as an “inverted bond” in which the smaller back lobes of the hybrid orbitals overlap to form the C–C σ-bond, while the larger lobes point outward.
Concomitant with the significant extent of s character of the bridgehead carbons, the strained central C–C bond in BCB can be described as being composed of hybrid orbitals which are nearly entirely p in nature (Scheme 1A). This hybridization gives rise to a bent σ-bond with a significant amount of π character (26.1% as determined by Newton).9a More recently, as part of a systematic correlation of σ-bond strength to bond angle for a series of hydrocarbons, Chaquin20 has indicated that the smaller bond angles in BCB correlate to decreased σ but increased compensatory (39.7%) π contribution (including some three-center, two electron bonds involving the methylenes). As expected, the overall bond strength does decrease as a result of strain. Dias calculated NICS (Nucleus Independent Chemical Shift) values and compared them to BRE (bond resonance energy) values based on a system modified for saturated hydrocarbons.21 A negative NICS value indicating that the 3-membered rings in BCBs are diatropic, in conjunction with positive BRE values, provide good evidence for significant σ-aromaticity characteristics (Scheme 1A).
The unusual bonding in BCB translates into fundamental differences in its acidic and basic behavior relative to unstrained hydrocarbons. The strain reduction in bridgehead-lithiated BCB noted by Dill17 correlates well to the relative acidity of the bridgehead hydrogens, rendering it a readily accessible and useful reactive intermediate in numerous syntheses.3,22 Whitman determined that these bridgehead C–H bonds have more s character (28%) than traditional Csp3 C–H bonds and are significantly more polarized than those of bicyclopentane, with correspondingly higher acidity (Scheme 1A).23 Kass has measured a gas phase acidity of BCB as 398 ± 2 kcal mol−1.24a Alkorta and Elguero have predicted a pKa of 37.9.24b The BCB system is also predicted to have some moderate gas phase hydrogen-bond acidity (H–B donating ability).25a In terms of basicity, Lee-Ruff has recently computed a proton affinity for BCB of 870 kJ mol−1, suggesting it is one of the most basic saturated hydrocarbons. Protonation is predicted to occur at the bridgehead carbon, leading to a cyclopropylcarbinyl cation.25b
Much of the chemistry of BCB is, in fact, due to the π character of the bridgehead bond.3,26 This C–C bond is known to react with a wide variety of electrophiles and nucleophiles, in processes ranging from solvolysis-type reactions to the addition of “hard” organometallic nucleophiles3 and a variety of thermal and photochemical cycloadditions.3,27 This bond has also been shown to react in tandem with bridgehead aryl groups as part of a conjugated system.28
The reactivity of the central C–C bond in BCB has also been leveraged in the development of cyclobutane-containing polymers formed by free-radical or anionic ring-opening of the corresponding BCB monomers (Scheme 1C).29 In fact, Wiberg's report on the first synthesis of a BCB structure (ethyl bicyclobutane-1-carboxylate) noted that this compound would polymerize spontaneously at room temperature.30 Hall subsequently reported the synthesis and polymerization of substituted BCB derivatives.31 These systems were found to co-polymerize with each other and with other vinyl monomers, and to have reactivities similar to corresponding vinyl compounds.32 Further work in this area showed the successful free radical polymerization and co-polymerization of a variety of BCBs with electron-withdrawing (nitrile, ester, ketone) substituents capable of stabilizing radical intermediates. Anionic and cationic conditions were not successful in this case.33 Indeed, poly(1-cyanobicyclobutane) (Scheme 1C) was studied for potential applications as a replacement for polyacrylonitrile and found to be a superior textile material, but was not utilized commercially due to high cost of monomer production (though some interest remains in the properties of the corresponding polyester material).31
The stereochemistry of these polymerizations has also been investigated. In general, polymerization using more highly substituted monomers or anionic polymerization seems to offer higher degrees of stereospecificity.34 Barfield reported approximately a 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 ratio of trans- to cis-linkages in anionic poly(bicyclobutane-1-carbonitrile) (PBBC).35 A subsequent study of PBBC prepared by radical polymerization found a 68:32 isomeric ratio, comparable to radical poly(1-methoxycarbonyl)bicyclobutane (68:32) and poly(bicyclobutane-1-carboxamide) (73:27).36 Hall found similar results for polymerization of a series of bicyclobutanecarboxylate esters; neither variation of the polymerization conditions nor the alkyl group on the ester caused appreciable variation from the roughly 66:33 ratio.31 The free radical polymerization of the more-substituted dimethyl bicyclobutane-1,3-dicarboxylate, however, gave a polymer with 95% trans stereochemistry.29 Anionic polymerization of methyl bicyclobutane-1-carboxylate itself gives trans-ratios of >90%, when initiated with tert-butyllithium/bis(2,6-di-tert-butylphenoxy)ethylaluminum in toluene at −78 °C, albeit with low initiator efficiency and broad molecular weight distribution.34 A mechanistic study concluded that attack of tert-butyllithium on the ester to incorporate less-reactive ketone unit anions was occurring as a side-reaction but was mostly suppressed by coordination of the ester to the aluminium Lewis acid.34
:25 ratio of trans- to cis-linkages in anionic poly(bicyclobutane-1-carbonitrile) (PBBC).35 A subsequent study of PBBC prepared by radical polymerization found a 68:32 isomeric ratio, comparable to radical poly(1-methoxycarbonyl)bicyclobutane (68:32) and poly(bicyclobutane-1-carboxamide) (73:27).36 Hall found similar results for polymerization of a series of bicyclobutanecarboxylate esters; neither variation of the polymerization conditions nor the alkyl group on the ester caused appreciable variation from the roughly 66:33 ratio.31 The free radical polymerization of the more-substituted dimethyl bicyclobutane-1,3-dicarboxylate, however, gave a polymer with 95% trans stereochemistry.29 Anionic polymerization of methyl bicyclobutane-1-carboxylate itself gives trans-ratios of >90%, when initiated with tert-butyllithium/bis(2,6-di-tert-butylphenoxy)ethylaluminum in toluene at −78 °C, albeit with low initiator efficiency and broad molecular weight distribution.34 A mechanistic study concluded that attack of tert-butyllithium on the ester to incorporate less-reactive ketone unit anions was occurring as a side-reaction but was mostly suppressed by coordination of the ester to the aluminium Lewis acid.34
Photochemical methods have been employed for synthesis (and degradation) of BCB, and the mechanistic pathways of these reactions have been studied.37 The 185 nm photolysis of BCB leads to the formation of butadiene and cyclobutene (Scheme 1D). Two possible pathways, one involving a concerted cleavage of two opposite non-bridgehead bonds, and the other involving cleavage of the bridgehead bond to a diradical followed by additional photolysis to an allylcarbene, were postulated. Isotopic labelling studies using both deuterium and 13C labelled substrates seem to suggest both mechanisms could be in operation.38
1.2 Classical synthetic approaches
The first known synthesis of a BCB was in 1959 when Wiberg and Ciula reported that treatment of ethyl 3-bromocyclobutane-1-carboxylate with sodium triphenylmethylide afforded an ester-substituted BCB (Scheme 2A).30 Wiberg and co-workers later generalized this approach for the synthesis of the parent, unsubstituted bicyclo[1.1.0]butane,39 including as an Organic Syntheses protocol (Scheme 2B).40 Soon after, Srinivasan demonstrated in 1963 that the unsubstituted BCB hydrocarbon could be formed as the minor product (along with cyclobutene) during the photolysis of 1,3-butadiene.41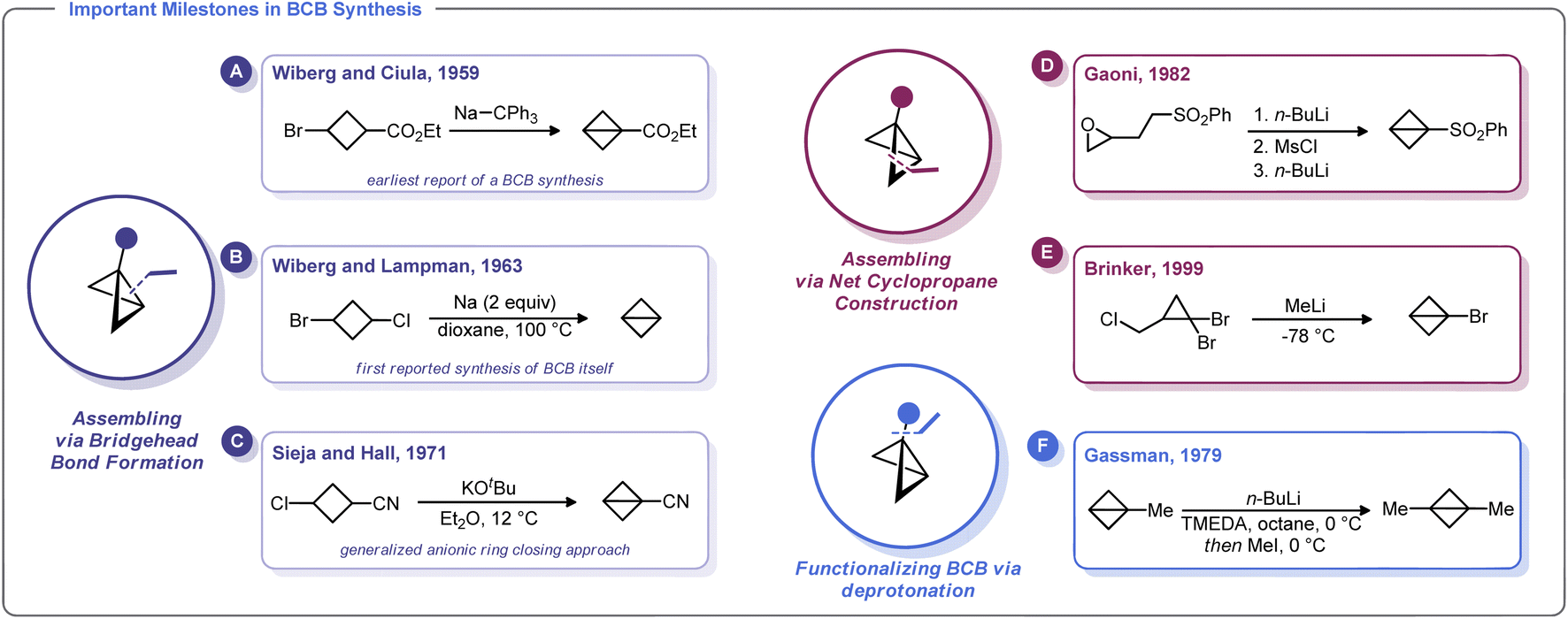 | ||
| Scheme 2 Synthetic approaches to BCB from landmark studies. | ||
In 1965, Vellturo eported the first electrochemical synthesis of a BCB, noting that the electrolysis of trans, trans, trans-1,3-dicarboxy-2,4-dicarbomethoxycyclobutane yielded 2,4-dicarbomethoxybicyclobutane.42 Subsequently, Rifi demonstrated in 1967 that BCBs can be prepared by anodic reduction of 1,3-dibrominated cyclobutanes, suggesting that the Wiberg-type trans-annular coupling reactions proceed through homolytic union of a biradical intermediate.43
Though clearly groundbreaking, these initial methods for BCB synthesis had a limited capacity to generate substituted derivatives. An advancement along these lines came with Sieja's 1971 report on the use of cyclobutanones (which are readily obtained by ketene/vinyl ether [2 + 2] cycloaddition) as an entry to BCB scaffolds with bridgehead substituents.44 Reaction of these 3-alkoxy cyclobutanones with PhMgBr gave the corresponding alcohols which were readily converted to tertiary chlorides. Treatment of these halides with magnesium metal in refluxing THF induces the formation of BCBs. Sieja and Hall also showed that this approach can be used to prepare BCBs with nitrile substituents, which can be used in polymerization studies and other ring-opening nucleophilic reactions (Scheme 2C). Indeed, treatment of 3-chlorocyclobutanecarbonitrile with potassium tert-butoxide gave facile access to a cyano-substituted BCB needed for polymerization studies.32a
An alternative approach to the strained BCB carbocycle was advanced by Gaoni in 1982 (Scheme 2D).45 Rather than using 1,3-disubstituted cyclobutane derivatives as starting materials, this approach used sequential intramolecular cyclopropanation reactions to access BCB sulfones. Starting from easily accessible epoxy-sulfones, treatment with n-BuLi triggers a cyclopropane formation with concomitant epoxide opening. The resulting alcohol can be converted into a sulfonate ester and subjected to a second lithiation/cyclopropanation sequence. Although somewhat circuitous, this approach is adaptable for a variety of substituents and for many years was the state-of-the-art method for the preparation of substituted BCBs. In fact, this strategy was relied upon by Baran and others in recent reports on strain-release functionalization (described later in this Minireview).46 Interestingly, Lindsay and co-workers recently developed a concise syntheis of sulfonyl-substituted BCBs from methyl sulfones and epichlorohydrin (and related epoxides) that proceeds via ring contraction of an activated cyclobutanol intermediate.47
One of the most versatile methods for the preparation of BCBs was developed by Brinker and co-workers in 1999 (Scheme 2E).22 This approach uses 1,1-dibromocyclopropanes as starting materials, which are easily accessed by dibromocarbene cyclopropanation of allyl chlorides. Treatment of this trihalide with two equivalents of an alkylithium results in a bicyclobutyllithium species, which can be trapped with a variety of electrophiles. Likewise, Gassman showed that lithiated BCBs could be accessed via direct deprotonation of the bridgehead C–H bond using an organolithium species. The resulting lithiated BCB could then be alkylated (Scheme 2F).48
2. Recent approaches to BCB construction
Recent efforts have sought to bolster the ability to access more complex BCB architectures. To do so, an array of technologies have been leveraged, ranging from transition metal catalysis to enzymatic processes. These efforts to derivatize the BCB core from simple building blocks using modern methods have greatly expanded the diversity of BCBs available to chemists. This section will highlight some of the recent advances along these lines.2.1 Transition metal-mediated catalysis
The ability of transition metal catalysts to execute precise complex transformations is rivalled only by nature. These catalysts enable chemists to forge traditionally challenging disconnections, with the added benefit of dictating the stereochemical outcome by ligand choice. However, despite their impressive capabilities, metal-mediated routes to access the BCB carbocycle have been sparse until recently.The conceptually most straightforward approach to BCB synthesis is a [2 + 1] reaction between a carbene equivalent and a cyclopropene, or double cyclopropanation of an alkyne. Although Wipf and co-workers demonstrated that this approach can be realized through a zirconium-mediated multicomponent transformation,49 the generality of accessing BCBs from alkynes and/or cyclopropenes using traditional Simmons-Smith chemistry is limited.50 However, metal-mediated decomposition of diazo species offers a convenient means to affect [2 + 1] addition with the opportunity for enantioselectivity. Early reports of this approach were advanced by both Ganem51 and Hussain52 (Scheme 3). Both studies used an intramolecular cyclopropanation of a homoallylic diazo species to build both rings of BCB at once rather than necessitating isolation of unstable cyclopropene intermediates. Only a few examples were documented in these reports, leaving open questions of generality and the potential for asymmetric BCB construction. In nearly concurrent reports in 2013, Davies53 and Fox54 were able to address these unknowns (Scheme 3). Using similar conditions to Ganem, Davies showed that the reaction proceeds with excellent diastereoselectivity when using homoallylic diazo species bearing a disubstituted olefin. Davies and co-workers later found that Rh2(R-BTPCP)4 could be used to affect enantioselective BCB synthesis. Highly enantioenriched 2-arylbicyclo[1.1.0]butane carboxylates could be assembled from 2-diazo-5-arylpent-4-enoates under remarkably mild conditions. Fox and co-workers were able to develop conditions that provided more substrate generality and generally higher enantioselectivity by using Rh2(S-NTTL)4 at cryogenic temperatures. In the case of the Fox report, the authors demonstrated that their enantioenriched BCB products could be used with organocuprate nucleophiles for homoconjugative addition to yield enantioenriched, densely functionalized cyclobutanes. Fox later used this stereoselective BCB formation-ring opening sequence in a synthesis of the cyclobutane-containing natural product piperarborenine B.55
 | ||
| Scheme 3 BCB synthesis via metal-mediated intramolecular [2 + 1] bicyclobutanation. | ||
2.2 Enzymatic catalysis
The importance of enzymatic catalysis cannot be overstated, and advances in the engineering of enzymes have allowed the power of biocatalysis to be unlocked in the context of organic synthesis. These processes benefit from remarkable selectivity, be it stereochemical or regiochemical in nature. Further, precise control of the enzymatic binding site allows for remarkable reaction efficiency. In a landmark paper for BCB synthesis, Arnold and co-workers built on the aforementioned Rh-catalysed BCB synthesis by designing a hemeprotein variant (P411-E10V78FS438A).56a Arnold's engineered enzyme facilitated intermolecular BCB construction using alkynes and ethyl diazoacetate with excellent total turnover number (TTNs) (Scheme 4). Indeed, naturally occurring BCBs are made by enzymatic catalysis.56b By tuning the enzyme towards engagement of cyclopropenes and alkynes, the authors were able to effect two sequential [2 + 1] reactions to produce bridge-symmetrical BCBs. One limitation of this work is specificity of the substrate. The reaction appears limited to ethyl diazoacetate and phenylacetylene derivatives. That said, this report demonstrated not only a scalable approach to trisubstituted BCB construction (producing nearly gram quantities of certain examples), but the concept that enzymes can be used to prepare strained carbocyclic molecules under remarkably mild conditions. The authors were able to demonstrate that their prepared BCBs could be readily derivatized via known approaches such as reduction and azide–alkyne “click” reactions.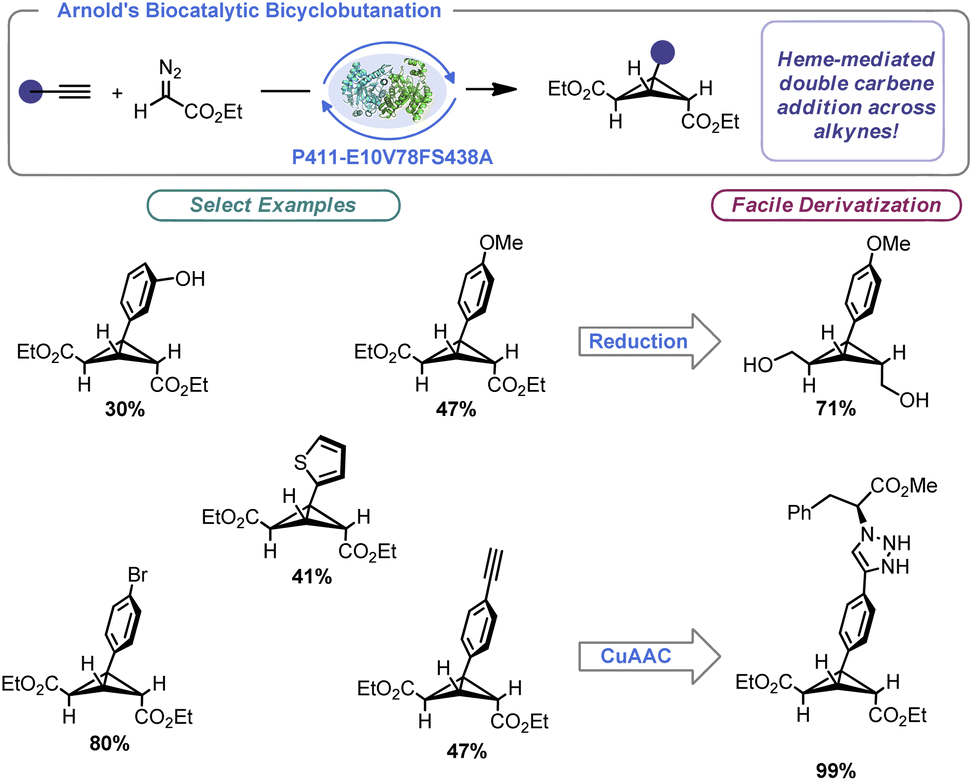 | ||
| Scheme 4 Enzymatic catalysis enables BCB synthesis. | ||
2.3 Cationic ring closure (CRC)
In 2011, Tilley and co-workers reported an unusual strategy to assemble perfluoroalkyl-substituted BCBs.57 Rather than proceed via the classic anionic approach, the authors reimagined ring closure as a cationic process wherein cation formation triggers ring contraction (Scheme 5A). To do so, the authors relied on the homohyperconjugative stabilization of carbocations by γ-silyl groups known as the γ-silyl effect,58 augmented by strong induction provided by an α-perfluoroalkyl group. The increased positive charge at the cationic center creates an “electron-deficient” carbocation59 and magnifies the precaudal (“through the tail”) participation by silicon, thus leading to 1,3 γ-silyl elimination. To determine the feasibility of this approach for BCB synthesis, the authors assembled a 3-(trimethylsilyl)cyclobutyl tosylate both with and without a perfluoroalkyl group at the putative cationic center (Scheme 5B). A W-type conformation is necessary to maximize precaudal participation and, as such, the (1s, 3s) diastereomer (with a cis relationship between the silyl and leaving groups) was specifically targeted. Although little to no BCB formation was observed with the des-CF3 system (only pyrolysis gave BCB formation, Scheme 5C), the authors observed exclusive ring closure under solvolytic conditions with the CF3 system, thus validating their approach. The authors invoke the so-called “perfluoroalkyl effect” which is known to kinetically stabilize strained systems by electron density delocalization, thereby relieving some strain energy.60 BCBs bearing CF3 and CF2CF3 groups were assembled in this manner in excellent yield despite their near room temperature boiling points. In a later report, Tilley and Leadbeater demonstrated that this same tactic could be used outside the constraints of the cyclobutyl system as a means to assemble perfluoroalkyl cyclopropanes (Scheme 5D).61 Tilley has continued investigating the efficacy of various electron-withdrawing groups on promoting silyl elimination of the cyclobutyl systems and has observed the BCBs to be the major products when the electron-withdrawing group is CN62 and CF2H.63 However, only small amounts of BCBs form in the case of the 2-fluoropyridin-3-yl cyclobutyl substrate, likely because of decreased necessity for silyl stabilization due to conjugation from the aromatic substituent.64 Creary has recently shown that cyclopropylcarbinyl cations initially formed during solvolysis of CF3, CN, and CO2CH3-containing sulfonate derivatives of cis-1-hydroxymethyl-2-trimethylsilylcyclopropane rearrange to the corresponding 3-trimethylsilylcyclobutyl cations and likewise undergo γ-silyl elimination to generate appreciable amounts of BCBs ranging from 71% (CF3) to 47% (CN) of the product mixture.65 Creary has also used the influence of γ-silyl groups in conjunction with cyclopropyl carbenes in an analogous fashion to furnish silyl-substituted BCBs.65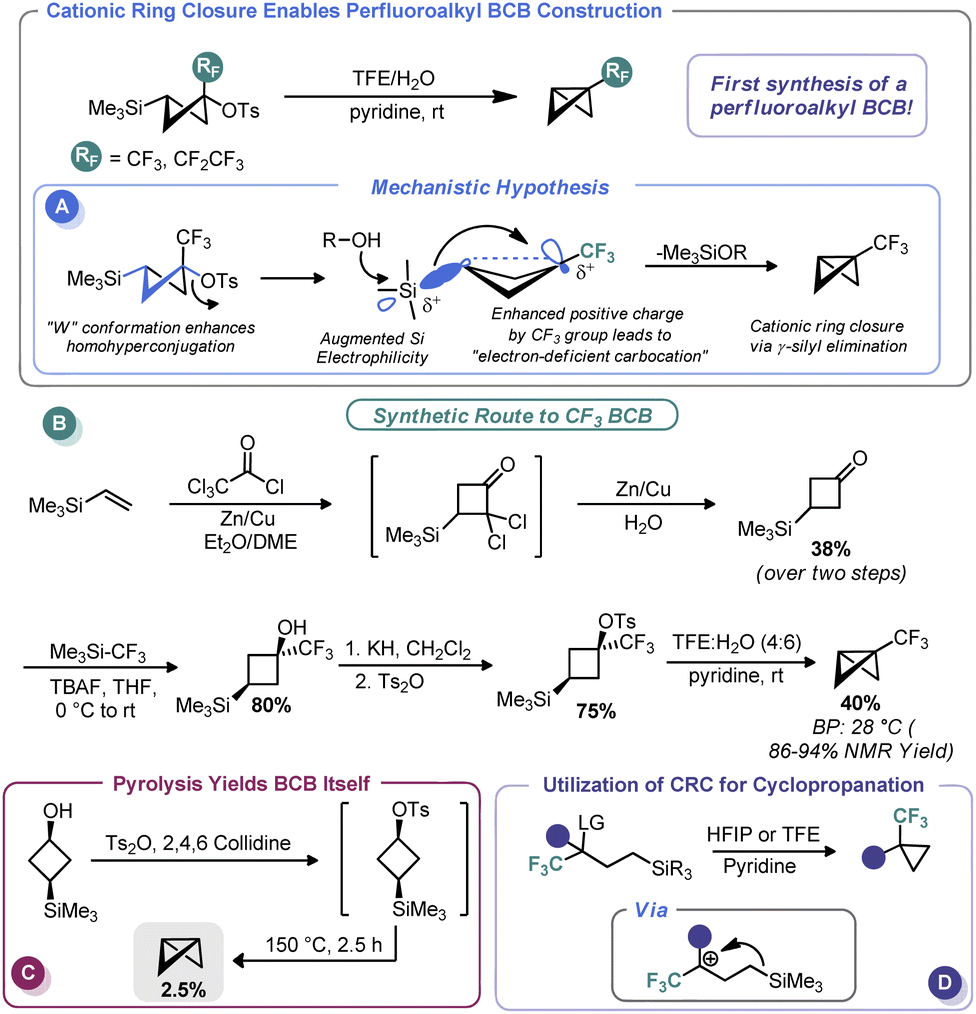 | ||
| Scheme 5 Synthesis of perfluoroalkyl-substituted BCBs via CRC. | ||
2.4 Metalation/functionalization sequences
Several groups have recently developed methods for the introduction of different functional groups at both the BCB bridgehead and methylene positions, which has been a longstanding challenge in the field (Scheme 6). In 2020, Malins and co-workers demonstrated that a bridgehead lithiated BCB (prepared by lithium halogen exchange with the corresponding bromide) could effectively engage Weinreb amides in an acylation process (Scheme 6A). Alternatively, a BCB Weinreb amide could be prepared from a lithiated BCB and N-methoxy-N-methylcyanoformamide, which can subsequently be used as an electrophilic handle to assemble BCB ketones.66 However, the need to exploit t-BuLi to prepare an anionic BCB equivalent yielded room for further development. To address this shortcoming, Anderson and co-workers reported that sulfonyl BCBs could be deprotonated using PhLi and converted to organozinc species in situ by treatment with ZnCl2.67 The resulting Negishi-type reagents could be engaged in cross-coupling with aryl iodides under mild conditions (Scheme 6B). A Pd0 source (Pd(dba)2) and 2-trifurylphosphine (tfp) were ideal for catalysis. The authors were able to show good compatibility with an array of aryl and heteroaryl iodides as well as other unsaturated species such as alkenes. An example of a non-sulfonyl BCB was also shown. The authors showcased their approach by using this non-sulfonyl system, an amide BCB, to prepare a bicyclopentyl species which yielded a building block of BCP-darapladib.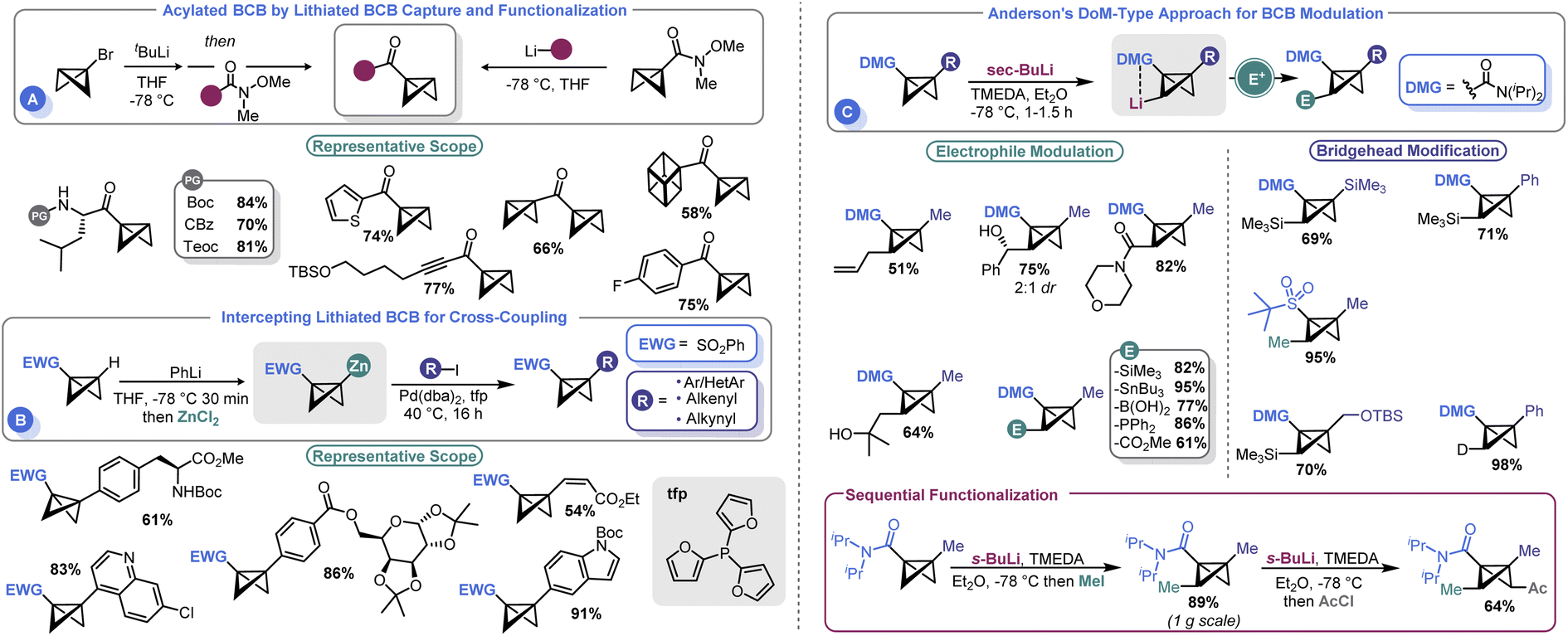 | ||
| Scheme 6 Functionalization of the BCB core via anionic BCB intermediates. | ||
A longstanding challenge in the field of BCB synthesis has been the lack of a facile means to introduce substituents on the methylene “wings” of the BCB structure. Anderson and co-workers recently advanced a solution to this problem by demonstrating that BCBs can be used in directed ortho metalation (DoM)-type reactions (Scheme 6C).68 This innovative approach uses a BCB with an amide at the bridgehead position to direct the deprotonation to the non-bridgehead methylene. sec-BuLi and TMEDA at temperatures as high as −40 °C were identified as ideal for executing this so-called “bridge-metalation”. Subsequent electrophilic quench furnished an array of BCBs bearing various functional groups. Further, the authors also demonstrated that sequential functionalization was possible, allowing both non-bridgehead sides of the BCB to be appended with electrophilic fragments. Desymmetrization strategies were pursued; the authors obtained a 2.7:1 diasteromeric ratio by using a C2-symmetric ligand.
3. Recent applications of BCB as a synthon
The innate strain energy of the BCB carbocycle lends itself to a broad array of synthetic applications. Great strides have been made in the development of heterolytic and homolytic ring-opening reactions of BCBs in recent years, thus allowing access to structurally complex cyclobutanes. Further, the reactivity of carbenes has been harnessed to convert BCBs into other highly sought small ring compounds. This section will provide an overview of some of these advances.3.1 Heterolytic strain-release functionalization
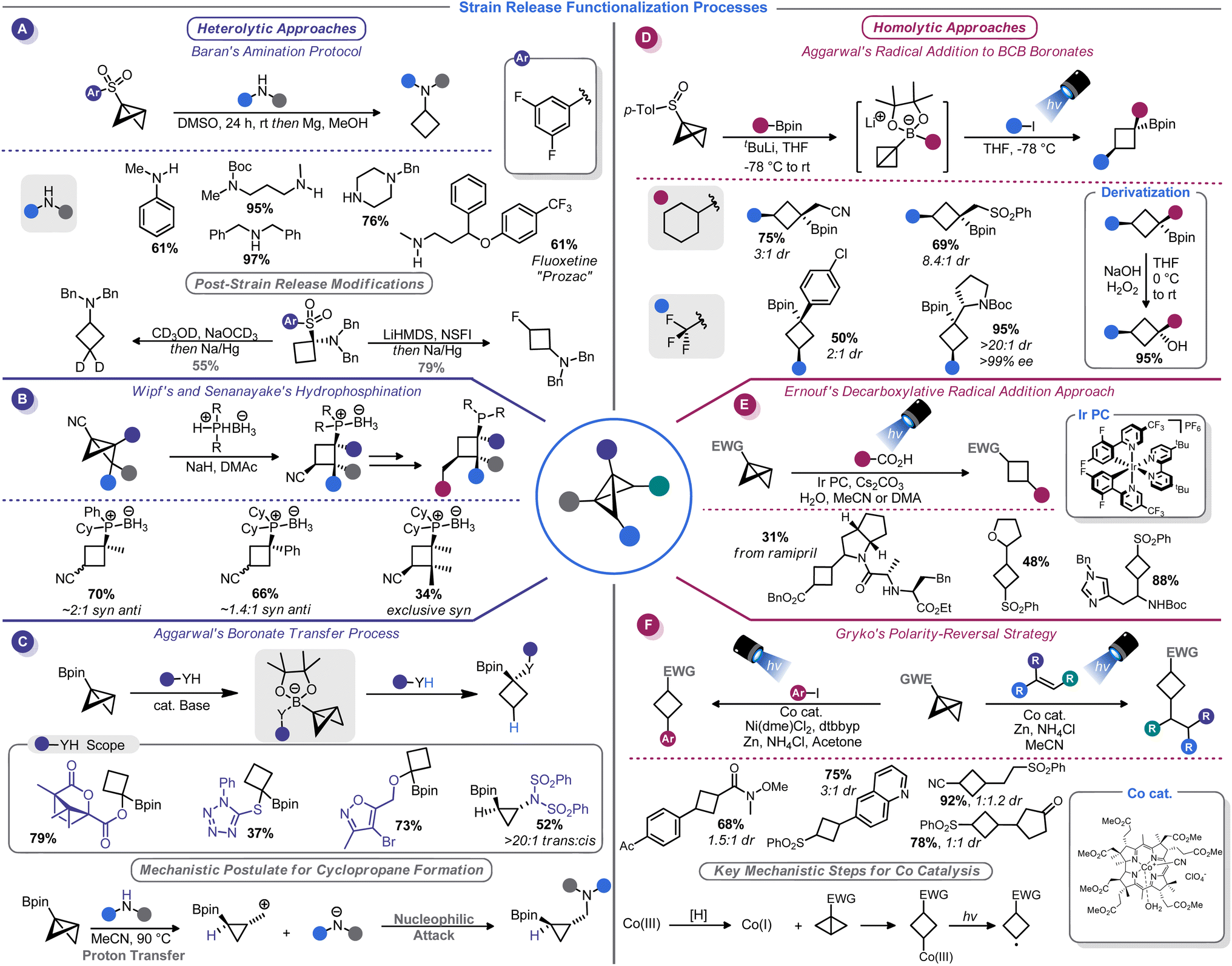
The substantial π character of the central C–C bond in BCBs renders them primed for addition reactions to form 1,3-disubstituted cyclobutanes. In fact, hydrolysis and alcoholysis reactions of BCBs were some of the first reactions to be studied as part of BCB structural confirmation studies.13 Hoz, Gaoni, and others thoroughly investigated the mechanistic and stereochemical aspects of various acid-mediated addition reactions to BCBs throughout the 1980s.69–71 Although these studies primarily had a mechanistic focus, several had rather useful synthetic applications. For example, the addition of hydrazoic acid to a BCB-carboxylate provides access to cyclobutane-containing amino acids.70Gaoni and co-workers carried out additional studies along these lines that focused on C–C bond formations enabled by organocuprate-mediated additions to BCB-sulfones.71 This concept was used for the synthesis of various substituted cyclobutanes, including a cyclobutane-containing pheromone natural product.72 Mechanistic studies suggest that the cuprate reagent approaches the central BCB bond selectively from the endo position, but the protonation of the resultant α-sulfonyl anion ultimately determines whether the cis or trans diastereomer predominates.73 The use of butylated hydroxytoluene (BHT) as a bulky proton source in a more recent application of this reaction validated this hypothesis, enabling highly diastereoselective cuprate addition to BCB-esters.54
A watershed moment for reinvigorating interest in BCBs came in 2016 when Baran and co-workers investigated the reaction of BCBs with amide nucleophiles. This approach, dubbed “strain-release amination”, is useful for the introduction of the cyclobutylamino motif on a variety of medicinally-relevant structures (Scheme 7A).46 Aryl sulfones were used as bridgehead activator groups, but they can be reductively removed. This strain-release approach was shown to be rather general: azabicyclobutanes and [1.1.1]propellane react in a similar manner with a variety of amine nucleophiles.46b Similarly, Milligan, Wipf, Busacca, and Senanayake showed that phosphine–borane anions can be used as nucleophiles to attack BCB-nitriles (Scheme 7B).74 This method provides convenient access to borane-protected tertiary phosphines with an appendage that can be used in the preparation of bidentate ligands.
 | ||
| Scheme 7 Strain-release heterolysis and homolysis enables rapid conversion of BCB into various synthons. | ||
Recently, Kerner and Wipf reported an alternative BCB activation strategy that involves a semi-pinacol rearrangement.75 In their approach, rearrangement of the BCB bridgehead group is triggered by a Brønsted or Lewis acid. This allowed the authors to generate ketones from BCB-alcohols. Aggarwal and co-workers have developed a series of conceptually similar reactions by using 1,2-boronate transfers (Scheme 7C).76 These reactions function by in situ formation of a BCB-pinacol boranate, 1,2-transfer, and functionalization by capture of an electrophile. Interestingly, subsequent studies found that alcohol nucleophiles can participate in these reactions to afford 1,1-disubstituted cyclobutanes.77
3.2 Homolytic strain-release functionalization
Just as there is a duality between Michael and Giese acceptors,78 so too can BCBs accept both anionic and odd-electron nucleophiles. Although evidence of the π character of the strained C–C bond was demonstrated in the earliest era of BCB studies,13,14 few homolytic functionalization reactions have been reported until rather recently (with the exception of a report of a photoinitiated radical chain process in the mid-1980s79). The increased development of radical-based ring opening reactions of BCBs is correlated to the broader renaissance of radical chemistry.80 Radical reactions unlock reactivity patterns that are simply not observed in two-electron processes, and photoredox technology serves as a vehicle to harness radicals for synthesis in a controlled, predicable manner. As such, interfacing BCBs with photocatalysis comprises the vast majority of advances in this arena. The bulk of reports on this topic focus on installation of carbon fragments rather than the heteroatom-centric reactivity seen with two-electron processes, thus making these two modes complimentary.Aggarwal and co-workers, building on their related work with in situ generated BCB boronate complexes,76,77 demonstrated that these boronates can be efficiently attacked by electrophilic radicals. Indeed, BCB-containing boronate complexes undergo trifluoromethylation via bridgehead bond homolysis upon irradiation with blue LED light (Scheme 7D).81 Others have shown that more typical BCBs with electron-withdrawing bridgehead groups, such as esters or sulfones, can be attacked by stabilized α-amino radicals generated from decarboxylation of amino acids82 (Scheme 7E) or oxidative activation of methyl anilines.83 As part of their studies on the titanium-mediated reductive generation of radicals, Lin and co-workers demonstrated that tertiary alkyl radicals are capable of adding to sulfonyl-substituted BCBs.84 A mechanistically distinct homolytic addition reaction of BCBs was demonstrated by Gryko and co-workers (Scheme 7F).85 Their approach used a vitamin B12-derived cobalt catalyst to generate a carbon-centered radical from the 3-position of a sulfonyl BCB. This radical could be added to a Giese acceptor such as an acrylate, or captured by a nickel catalyst to be used in cross-coupling.
3.3 Reactions with carbenes
Spurred by its bioisosteric value, high fraction of sp3 carbons, and broader intellectual property space, significant advances have been made with respect to incorporation of the bicyclo[1.1.1]pentyl (BCP) motif as part of potential therapeutic candidates.86 First recognized by Pfizer in 2012,87 BCPs have been identified as suitable bioisosteres of aromatic rings (Scheme 8). Early work in BCP synthesis was limited to simple monosubstituted BCPs. However, naked arenes are uncommon in drug molecules; rather, arenes are commonly decorated with fluorine atoms, which may be deployed during API development to offset undesired metabolism or modify bioavailability/permeability (although other atoms may be used).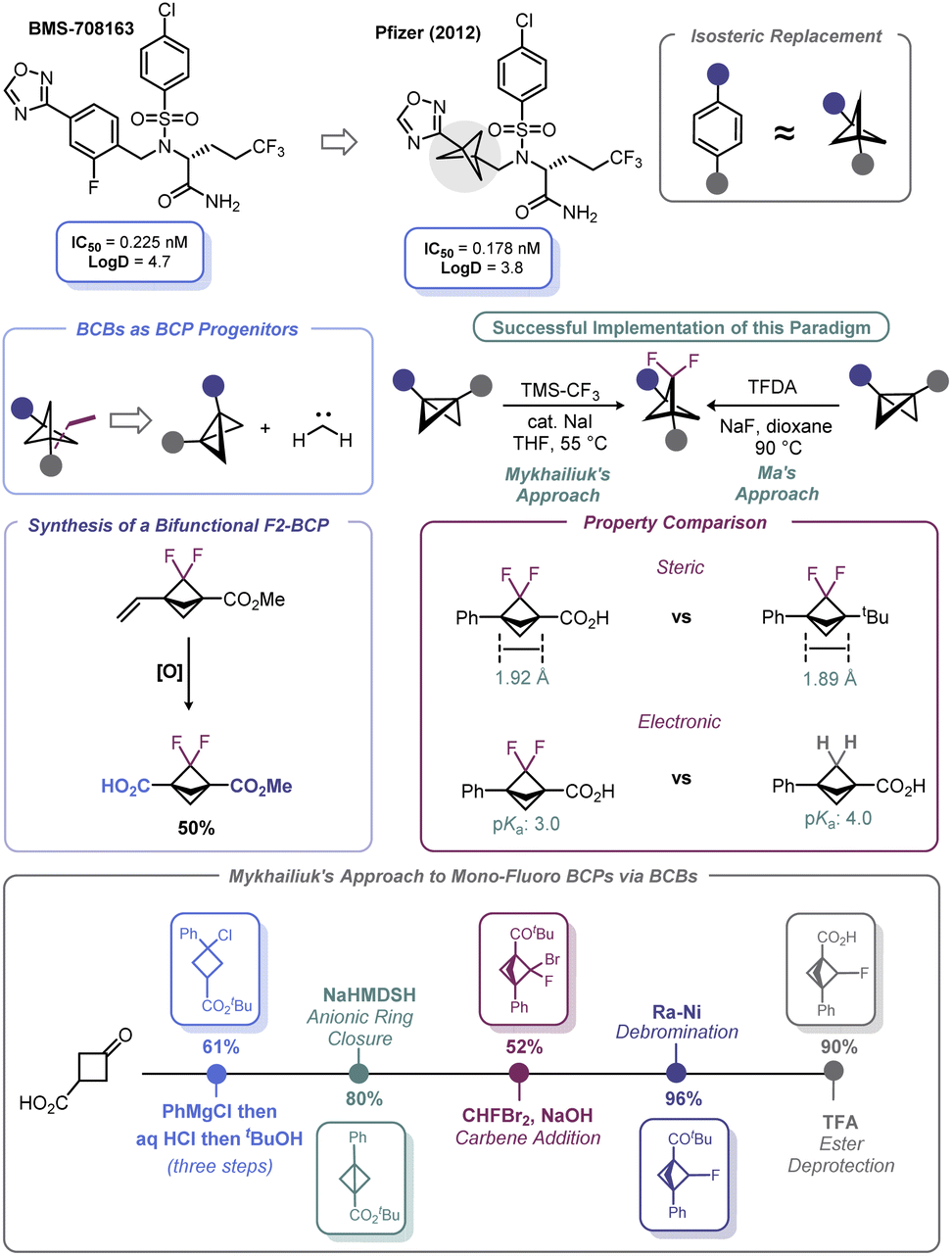 | ||
| Scheme 8 Accessing BCP species via carbene addition to BCB. | ||
Recognizing this limitation, reports by Ma88 and Mykhailiuk89 expounded (Scheme 8) upon the known propensity of BCBs to engage in [2 + 1] reactions with carbenes, which was first demonstrated by Applequist in the 1970s and early 1980s.90 These recent studies demonstrated that difluorobicyclopentyl (F2-BCP) building blocks could be assembled from BCBs by way of a net [2 + 1] addition between difluorocarbene and the BCB bridgehead bond. Ma elected to use trimethylsilyl 2-fluorosulfonyl-2,2-difluoroacetate (TFDA) as the difluorocarbene precursor, whereas Mykhailiuk used the practical approach reported by Prakash and Hu (from TMS–CF3 and NaI).91 Both approaches suffered from the limitation of requiring an aryl group as one of the substituents on the BCB. However, Mykhailiuk showed that a vinylic BCB can be used to overcome this limitation and produce a bifunctional BCP reagent. Analysis of the structural facets of these F2-BCPs revealed that the distance between the quaternary centers was unaffected despite minor deviations in bond lengths and angles. In addition, inductive effects were noted that lead to enhanced acidification of the bridgehead carboxylic acid groups on the BCP (Scheme 8). Recently, Mykhailiuk has used a conceptually similar approach to prepare mono-fluoro BCPs via addition of bromofluorocarbene across BCBs followed by dehalogenation with RANEY® Ni.92 The concept of using BCBs to access other bioisosteric motifs is discussed more at length in Section 4.
3.4 Reaction with transition-metal complexes
Numerous fundamental studies on the interaction between strained BCB hydrocarbons and transition metals such as Ag(I), Rh(I), and Pd(0) were conducted throughout the 1960's and 70's.26b These studies suggest that metals tend to facilitate strain-relieving ring opening reactions of BCBs, which typically result in vinyl carbene formation and terminate by isomerization to a cyclobutene or diene structure. While of limited utility from a synthetic perspective, these studies were instrumental in the advancement of modern organometallic catalysis.In 2008, Walczak and Wipf expanded upon these precedents by investigating cycloisomerization reactions of BCBs with a pendant allyl group.93 These substrates underwent isomerizations to yield cyclopropane-fused pyrrolidines and azepines in good yield with high levels of stereo- and regiocontrol. The fate of the BCB was dictated by catalyst choice, with a [Rh(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )2Cl2]2/PPh3 system yielding pyrrolidinyl products whereas [Rh(CO)2Cl2]2/dppe gave azepanyl compounds. Wipf's report laid the groundwork for further developments, and also highlighted some of the opportunities and pitfalls of interfacing BCBs with other transition metals (i.e. the propensity for polymerization, ring expansion, and rearrangements).
)2Cl2]2/PPh3 system yielding pyrrolidinyl products whereas [Rh(CO)2Cl2]2/dppe gave azepanyl compounds. Wipf's report laid the groundwork for further developments, and also highlighted some of the opportunities and pitfalls of interfacing BCBs with other transition metals (i.e. the propensity for polymerization, ring expansion, and rearrangements).
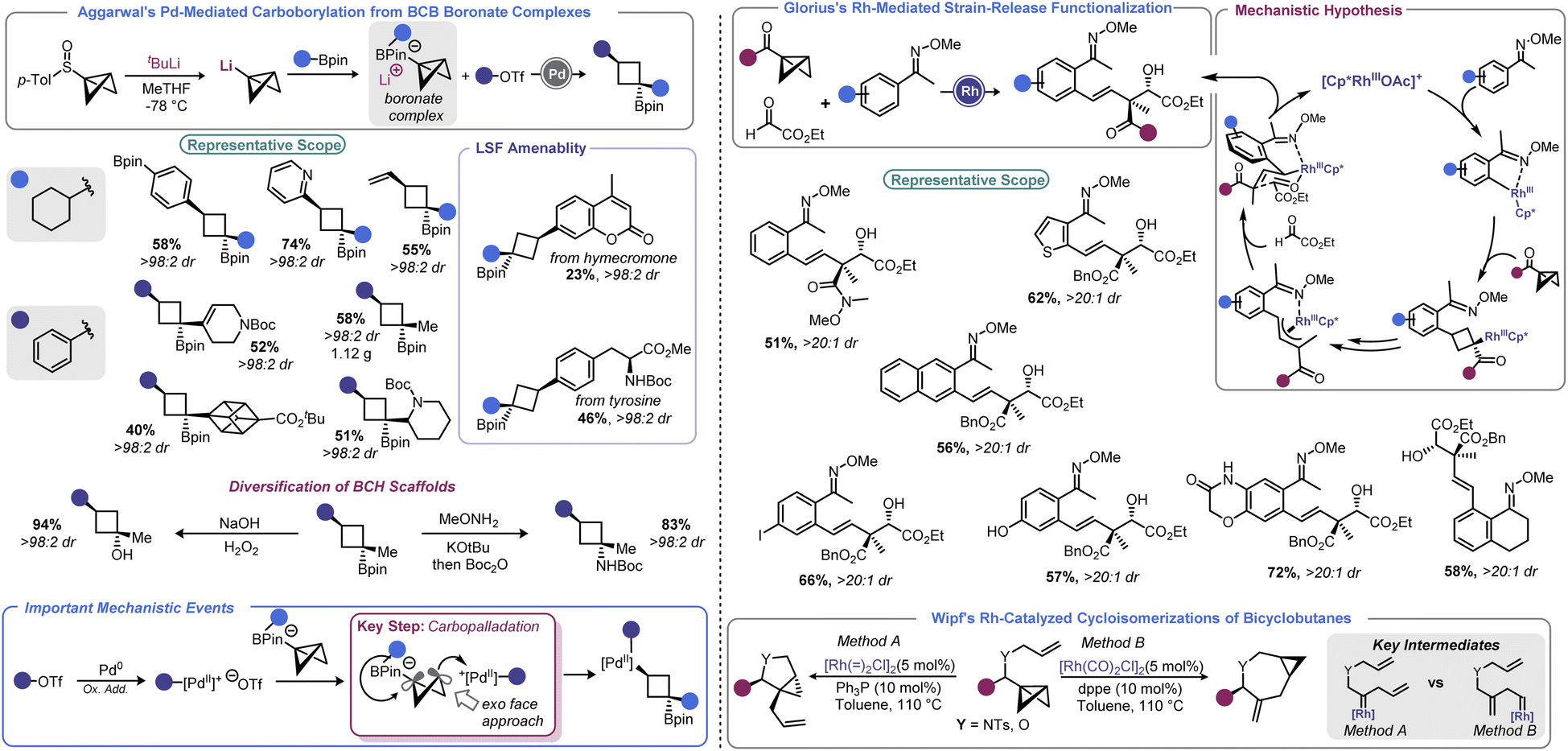
In 2019, Aggarwal devised a clever approach to overcome these challenges to effect a net carboborylation of the BCB scaffold (Scheme 9, left).94 Formation of a BCB boronate complex from lithiated BCB and a pinacol boronate ester allowed the authors to engage the central bond in catalysis without risk of transmetallation. Using aryl triflates and a Pd(0) catalyst with 1,1′-bis(diisopropylphosphino)ferrocene (dippf) as a ligand enabled facile arylation of the central bond triggered by a 1,2-metallate rearrangement event. Reductive elimination yields an arylated cyclobutane product and regenerates the Pd(0) complex. Because the oxidative addition complex must approach from the BCB's exo face (owing to steric constraints from the pinacol group), excellent dr is observed, with the Bpin group residing trans to the aryl group. Aryl and heteroaryl triflates were utilized as electrophiles in this process, and the authors noted that it was critical to generate lithiated BCB from a BCB sulfoxide rather than from the more traditional Brinker approach (from 1,1-dibromo-2-(chloromethyl)cyclopropane).22
 | ||
| Scheme 9 Pd and Rh catalysis enables synthesis of complex architectures promoted by strain-release. LSF = late-stage functionalization. | ||
In 2021, Glorius and co-workers revisited how Rh can be used to facilitate strain-release functionalizations with BCB (Scheme 9, right).95 Rather than use a Rh(I) system, the authors explored how a Rh(III) catalyst would interact with the strained central bond of a BCB. The authors posited that a rhodacycle (formed by C–H activation) should be prone to ligation and subsequent insertion by the bridgehead bond of BCB to give a Rh-cyclobutyl species. This new complex should decompose via β-carbon elimination to give a homoallylic Rh species. The latter undergoes π-allyl complex formation via β-hydride elimination/reinsertion at the benzylic position. This species isomerizes to the corresponding σ-allyl intermediate, then engages ethyl glyoxylate in alkylation. This last step sets a quaternary center and, after protodemetalation, resets the catalytic cycle. Because of the chair-like transition state in the final C–C bond forming step, the process proceeds with excellent diastereoselectivity. A range of homoallylic alcohols could be prepared in this fashion.
4. BCB in drug discovery & development
In addition to being important reagents for organic synthesis, BCBs have moved beyond the flask and into the realm of chemical biology. BCBs have been used as covalent warheads that react with specific residues in enzymatic pockets and building blocks to access traditionally challenging bioisosteric space, as well as serving as potential building blocks for therapeutic agents. Further, BCBs have themselves been targeted as potential small molecule inhibitors. This section will detail some of the strides made in this area.4.1 BCB-based therapeutics and chemical probes
In the pursuit of developing strain-release amination reactions useful in expanding the medicinal chemistry toolbox, the Baran lab has developed aryl sulfone bridgehead substituted BCBs as bench-stable “cyclobutylating” reagents (Scheme 7A).46 The reactivity of the central bond of these BCB reagents is tunable by varying the substituents off the sulfonyl arene; electron-donating groups (EDGs) attenuated the ring-opening reactivity, whereas electron-withdrawing groups (EWGs) increased the rate at which a nucleophile would undergo the substitution reaction. Of note, these investigations found that the reactivity could be tuned such that the central bond of 3,5-difluorophenyl sulfone BCB underwent selective strain-release opening with amines in the presence of alcohols.In addition to developing a robust method for the synthesis of diverse aminocyclobutanes, Baran and co-workers explored the application of the BCB reagents in a biologically relevant setting.46 When probing the feasibility of peptide functionalization with the aryl sulfone BCBs, it was found that these reagents were not only tolerant of aqueous media and phosphate buffer, but also showed exquisite chemoselectivity by reacting with the cysteine thiol over other nucleophilic amino acids (Scheme 10A). This selectivity translated to more complex peptides, with exclusive Cys-labeling observed with the BCB reagent, even when in direct competition with other nucleophilic residues (e.g. Tyr, Lys).46 A similar approach to this was used by Zhang and co-workers for the radioiodination of peptides.96 This demonstration of modular reactivity, stability in aqueous buffer, and chemoselectivity of aryl sulfone BCBs showed promise for the utility of these motifs for use in biological settings, such as in the covalent modification of proteins.
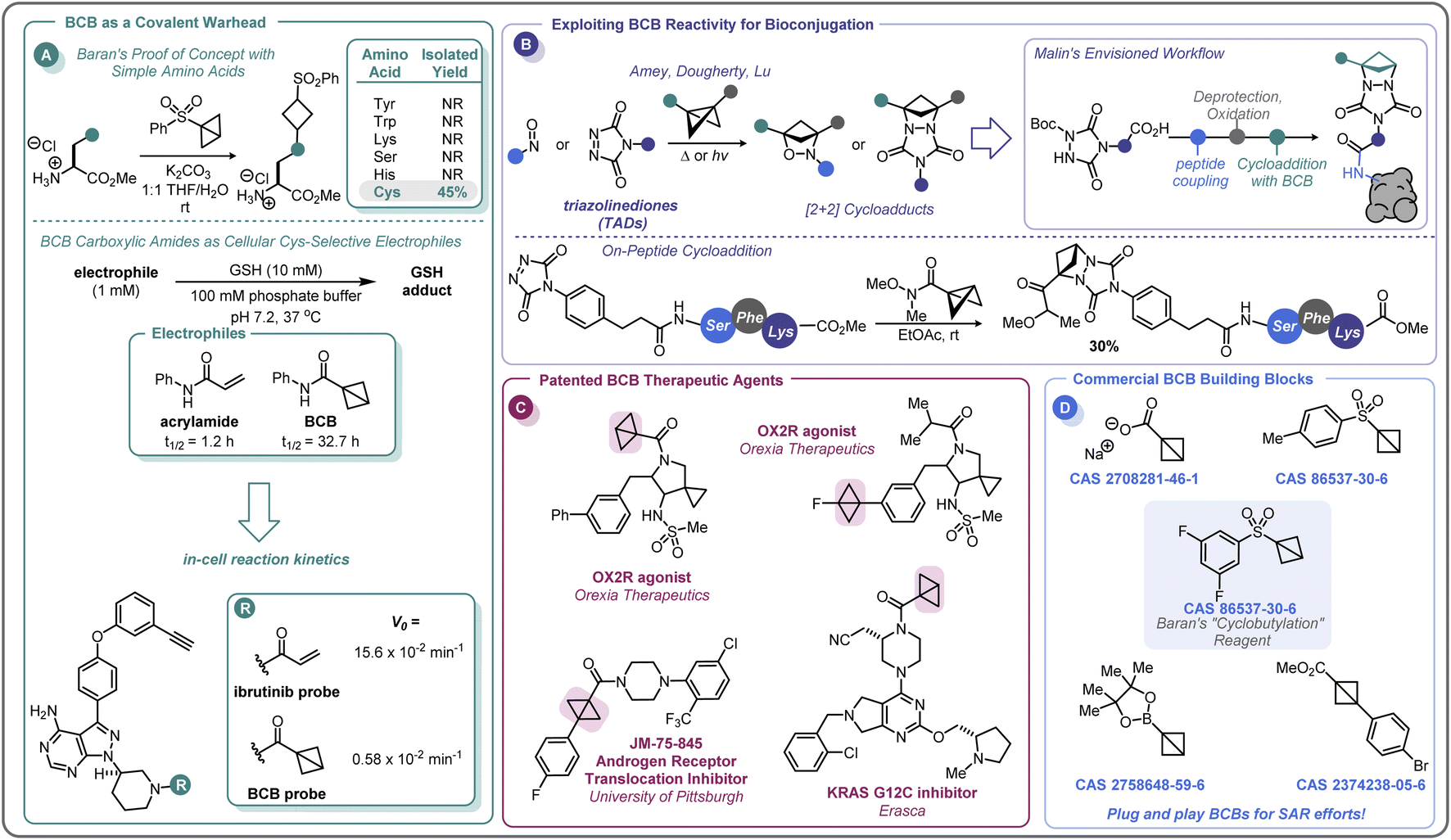 | ||
| Scheme 10 Landmark studies in the application of BCBs in medicinal chemistry and chemical biology. | ||
There has been keen interest within the pharmaceutical industry for developing covalent inhibitors of proteins. From 2011 to 2019, the FDA approved 14 new small-molecule drugs whose mechanism of action utilizes covalent inhibition.97 The majority of these compounds employ a Michael acceptor (e.g. acrylamide) as the covalent warhead and, while there are some strategies to control the electrophilicity of this motif,98 medicinal chemists are continuously seeking the right balance of reactivity with the target protein and selectivity against other biomolecules.99
In 2020, Ojida and co-workers were the first to demonstrate that BCB carboxylic amides could be used as Cys-selective electrophiles for the covalent inhibition of proteins in live cells (Scheme 10A).100 To gain an initial understanding of the reactivity of strain-release reagents as a new class of covalent warheads, N-phenylacrylamide was benchmarked against the BCB matched pair by measuring the half-life (t1/2) of each species in the presence of glutathione (GSH) under physiological conditions (Scheme 10A). Under these conditions, the acrylamide had a t1/2 of 1.2 hours, while the BCB was substantially more stable with a t1/2 over 30 hours. The Ojida laboratory then synthesized an analog of ibrutinib, an approved inhibitor of Bruton's tyrosine kinase (BTK), wherein the typical acrylamide warhead was replaced with a BCB carboxylic amide (Scheme 10A). The in-cell reaction kinetics showed a >25× difference in initial reaction rate (V0) between the ibrutinib probe (15.6 × 10−2 min−1) and the BCB probe (0.58 × 10−2 min−1), results that correlated with the observed GSH reactivity. Additionally, the authors were able to further optimize the linker from the BCB carboxylic amide warhead to the ibrutinib pyrazolopyrimidine core, increasing the V0 to a more moderate rate of 5.07 × 10−2 min−1 and achieving a cellular IC50 of 180 nM against wild-type BTK (not shown). Using the BCB probes developed during the course of this work, the authors also performed a chemical proteomics study to understand the selectivity profile of the acrylamide and BCB warheads in the context of ibrutinib. When Ramos cells were treated with 1 μM of either warhead for 4 hours, the two warheads were found to have distinct proteome reactivity profiles. Overall, 113 proteins were enriched for the acrylamide probe whereas only 50 proteins were enriched for the BCB probe, suggesting that BCBs have less off-target reactivity in this example.
It is worth mentioning that while BCBs offer a complementary approach to current technologies for targeted covalent inhibition based on their tunable reactivity, they are relatively unique in that a new stereocenter is formed in the 1,3-substituted cyclobutane Cys-adduct (Scheme 11). In general, the stereochemical outcome of nucleophilic addition to a BCB with a bridgehead EWG through a polar mechanism is dependent on the identity of the EWG and the rate of protonation.69 In certain situations where the mechanism of the substitution proceeds through a radical pathway, neither the cis- or trans-isomer is favored, leading to an equal distribution of the diastereomers (e.g.Scheme 7F).85 However, the environment within a protein binding pocket may override any thermodynamic, kinetic, or mechanistic stereochemical preference of the covalent adduct. Instead, the final geometry of the covalent linkage should largely depend on the conformation of the Cys-residue and the bound configuration of the inhibitor, although other factors may play a role as well, such as sterics within the binding pocket and interactions (e.g. hydrogen bonding) with the bridgehead EWG. To the best of our knowledge, at the time of publication there have been no crystal structures of a protein covalently bound to a BCB reported in the literature. More work is required to fully understand how these strain-release reagents behave as covalent warheads.
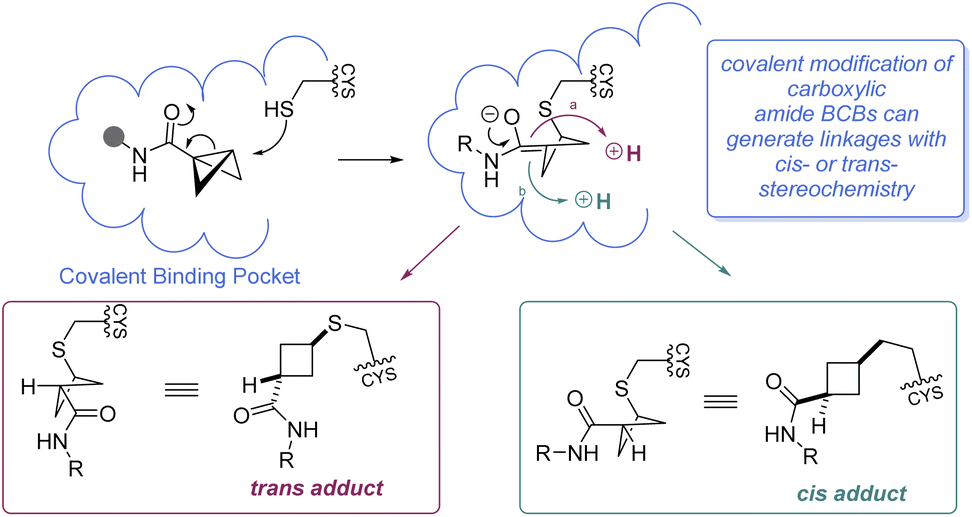 | ||
| Scheme 11 Mechanistic considerations for BCB covalent warheads. | ||
In addition to the direct covalent functionalization of peptides and proteins, BCBs are being investigated for their potential applications in bioconjugation chemistry as novel strain-release reagents for cycloaddition reactions. Recently, Malins has developed a cycloaddition reaction between acyl BCBs and triazolinediones (TADs) conjugated to peptides (Scheme 10B).101 This approach builds upon related prior work by Amey102 and Dougherty.103 The reaction proved to be mild and robust, proceeding at room temperature and tolerating a variety of solvents (e.g. THF, DCM, PhMe) and residual moisture. Additionally, amide, ester, and ketone substituted BCBs performed similarly well in the reaction. A tripeptide-conjugated TAD also successfully underwent the cycloaddition reaction (30% yield over two steps), and the authors noted that the TAD building blocks should be amenable to on-resin peptide synthesis, allowing for the potential to further expand the complexity of the peptide motif. In addition to modular peptide synthesis, the acyl functional handle from the BCB provides an opportunity for further diversification and installation of probes (e.g., alkynes for “click” chemistry, fluorophores, etc.), making this cycloaddition reaction an exciting area for further development as a bioconjugation platform.
The recent efforts to expand the use of BCBs in medicinal chemistry and pharmaceutical development have only begun to appear in the patent literature, with only a handful of compounds disclosed (Scheme 10C). In 2017, Wipf and co-workers disclosed a di-bridgehead substituted BCB in their work towards identifying small molecules that inhibit the nuclear translocation of the androgen receptor.104 Orexia Therapeutics filed a patent for orexin-2 receptor (OX2R) agonists for the treatment of narcolepsy that contained several structures with BCBs.105 These included a BCB bearing a bridgehead fluorine and a BCB carboxylic amide with a human OX2R pEC50 between 8 and 9, although the latter motif was not specified to be acting in a covalent manner. Erasca has patented a BCB carboxylic amide as the covalent warhead for a KRAS G12C inhibitor.106 As the KRAS G12C inhibitor patent space is highly congested, this last example highlights the importance of increasing the diversity of the building blocks used in drug development. BCBs and other small, strained carbocycles offer promise for not only generating new intellectual property, but also their increased Fsp3 and spatial geometry may impart benefits such as improved physicochemical properties and the ability to access novel chemical space.107 While initial intuition may suggest that the high strain energy of BCBs (vide supra) makes them incompatible with physiological conditions, this review has outlined numerous examples of BCBs that react with high chemoselectivity, and that do not require stringent air- and moisture-free conditions to store and handle. There is currently a small selection of commercially available bridgehead substituted BCB building blocks, including a sulfonyl arene, carboxylate, and boronic ester (Scheme 10D), that could easily integrate into routinely performed medicinal chemistry reactions or a parallel library workflow. With the complexity of functional groups and substitution patterns accessible on the BCB scaffold continually growing based on new methodology development, BCBs have the potential to move beyond a chemical curiosity to a versatile building block.
4.2 BCBs as entry points to new bioisosteric space
There has been an increasing interest in the utility of small carbocyclic species in medicinal chemistry.108 As mentioned earlier, BCB can be used as a viable starting point for BCP construction. With the success of BCPs as isosteres for arenes109 and the arsenal of methods that have now been realized for their construction,110 academic and industrial groups alike are exploring additional scaffolds as potential replacements for aromatic rings.Recently, the bicyclo[2.1.1]hexyl (BCH) group has been identified as an isosteric replacement for ortho- and meta-substituted benzenes.111 However, due to its unusual architecture, limited approaches are available to construct substituted BCHs. In principle, the fastest route to BCHs would be a [2 + 2] cycloaddition reaction between an olefin and the bridgehead bond of a BCB, but this transformation is not facile under traditional thermally-driven conditions. Very recently, near simultaneous reports by Procter112 and Glorius113 demonstrated two distinct approaches to addressing this synthetic impasse (Scheme 12, left). In Procter's report, a SmI2-catalysed radical relay process is employed to access a wide array of BCHs from the union of BCBs and olefins.112 The crux of this process is single electron reduction of a BCB ketone to generate a ketyl radical, which undergoes rapid ring opening. The resulting cyclobutyl radical can engage in Giese-type addition with an olefin. The resulting Giese adduct radical is well-positioned for a 5-exo-trig cyclization, reforming a ketyl radical. Regeneration of the carbonyl via back-electron transfer yields a carbonyl substituted BCH and the Sm(II) catalyst. Both the carbonyl group and the olefinic partner can be varied and the process tolerates a high degree of complexity. The authors later demonstrate a direct isosteric replacement of an ortho-substituted arene in phthalylsulfathiazole, a broad-spectrum antimicrobial agent.
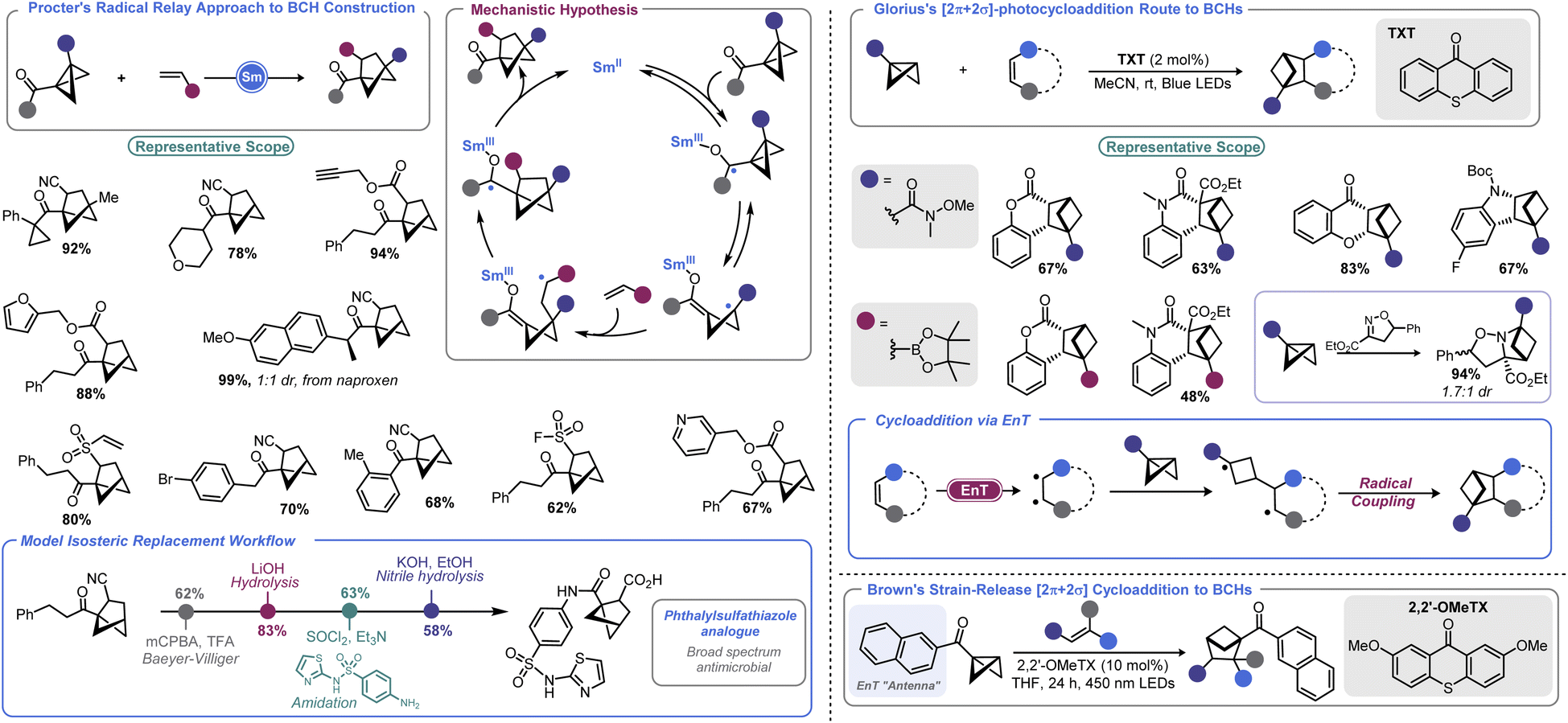 | ||
| Scheme 12 Accessing BCHs from BCBs. | ||
Glorius's approach relies on visible light to enable BCH construction by way of a [2π + 2σ]-photocycloaddition between olefins and BCBs.113 Photochemical cyclobutane synthesis via [2 + 2] cycloaddition is well-documented and proceeds with excellent stereochemical control both intra- and intermolecularly.114 However, use of a saturated system as one of the cycloaddition partners (i.e., using a σ bond) is more challenging. This may be in part due to the necessity to employ harsh UV irradiation when using non-polarized systems to facilitate cycloaddition. By using a triplet sensitizer (a thioxanthone, TXT) as a means to execute an energy transfer (EnT) process, Glorius and co-workers could effectively engage olefins with monosubstituted BCBs to produce BCH products under mild visible light irradiation. BCBs bearing functional handles such as Weinreb amides and boryl groups are well-tolerated in the described photochemical process, enabling BCH building blocks to be accessed. An isoxazoline partner can also be used to access polycyclic saturated heterocycles. DFT calculations conducted by the authors explained the regiochemical outcome, showing that a stepwise process with radical–radical recombination in the final mechanistic step was occurring.
Very recently, Brown and co-workers disclosed a related report where a naphthyl ketone is used as an “antenna” on the BCB itself.115 This allowed for visible light-induced formation of a triplet cyclobutyl diradical. Brown's approach allows a much wider array of olefin coupling partners, as this triplet cyclobutyl diradical can be captured by various alkenes. A final radical–radical recombination, as in Glorius's report, yields the BCH target.
To move beyond strictly carbocyclic isosteres (whose benefits of higher Csp3 may be countered by their high clogP), Leitch and co-workers explored if BCBs could be progenitors to 2-azabicyclo[2.1.1]hexanes (aza-BCHs) through a reaction with imines (Scheme 13).116 This hypothesis expands on the reports in the early 1980s by Amey and Dougherty,102,103 as well as the more recent work of Lu,27 who found that BCBs undergo strain-relieving cycloadditions upon irradiation with 1,2,4-triazoline-3,5-diones. The latter have been recently been popularized by Sarlah for dearomative cycloaddition reactions and utilized for the aforementioned bioconjugation reported by Malins.101,117 In contrast, this recent approach by Leitch uses a formal [3 + 2] cycloaddition using a BCB in which the central bond is polarized to give zwitterionic character, mimicking the well-known reactivity of donor–acceptor cyclopropanes.118 Lewis acids were used to induce enolate formation (and carbocation generation). From there, enolate attack (with potential aid by Lewis acid activation of the imine) yields an intermediate that undergoes ring closure. One challenge associated with any process proceeding through carbocations is the potential for E1 type elimination, and indeed, cyclobutene formation was observed by Leitch. The authors capitalized on this alternative pathway by devising conditions to exclusively form aza-BCHs or substituted cyclobutenes. Reaction temperature, the substitution pattern of the nitrogen on the imine, and the nature of the EWG all impacted the product distribution. As a means to overcome some of the architectural restrictions of their process, the authors demonstrated that cyclobutenes could be converted to aza-BCHs by way of an iodine-mediated intramolecular cyclization.
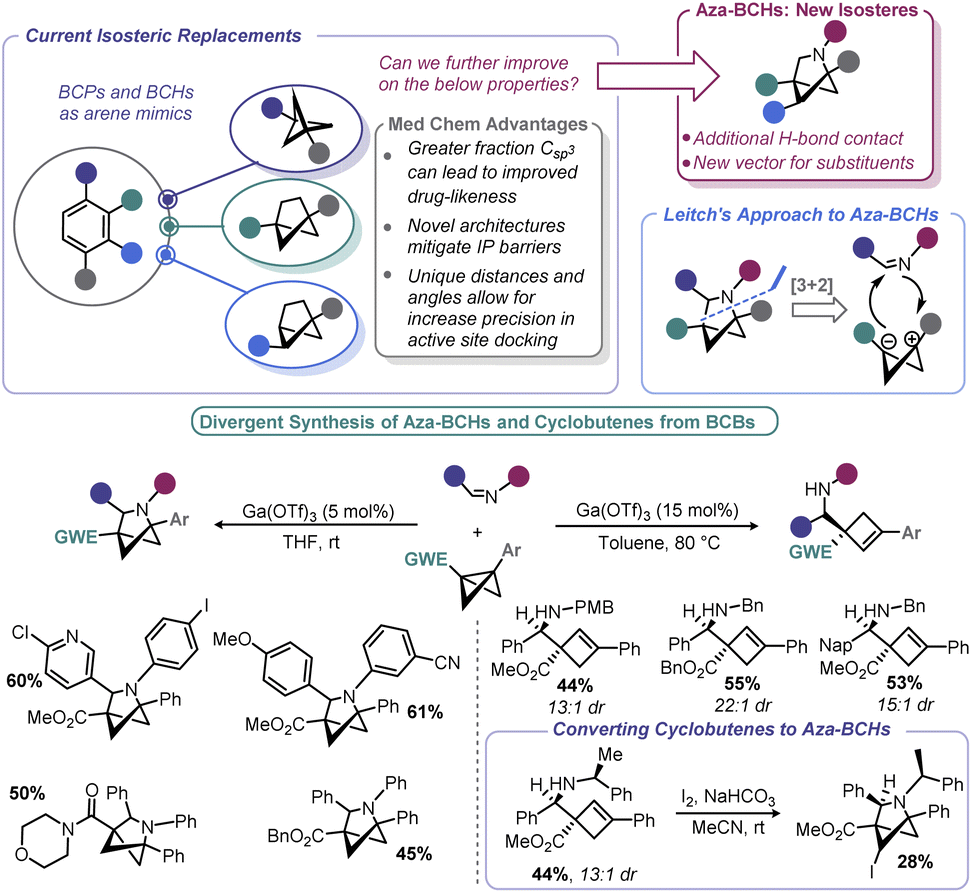 | ||
| Scheme 13 Leitch's approach to aza-BCHs from BCBs. | ||
4.3 As a means to access key building blocks for process-scale synthesis
As demonstrated in Sections 2 and 3 of this review, the BCB scaffold is a versatile handle for assembling an array of cyclobutyl species among other synthons. By tapping into the olefin-like, highly strained nature of the bridgehead C–C bond, nucleophiles can be added with relative ease. Very recently, researchers at Merck invoked a BCB as part of their route scouting efforts to prepare a small cyclobutyl building block, 3-(trifluoromethyl)cyclobutane-1-carboxylic acid (Scheme 14).119 The authors hoped to first prepare benzyl 3-(trifluoromethyl)bicyclo[1.1.0]butane-1-carboxylate before deprotecting the benzyl ester via hydrogenolysis. Synthesis of this BCB variant was accomplished in short order using a route analogous to Tilley's report57 (Scheme 5) but proceeding through anionic ring closure. Although the desired cyclobutane could be produced from this route, a combination of poor yield in the ring closing step coupled with a mixture of products produced during the final deprotection led to de-prioritization of this route over a related route featuring a cyclobutene.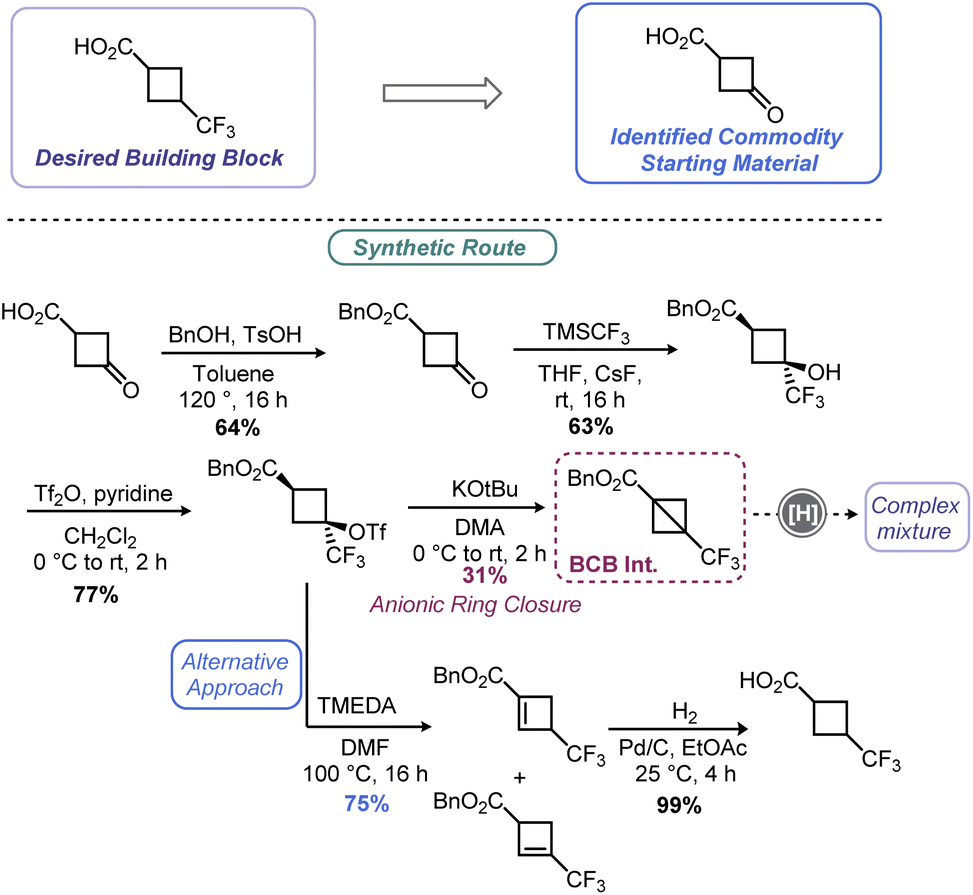 | ||
| Scheme 14 Merck's approach to a cyclobutyl building block via a BCB intermediate. | ||
5. Conclusions and outlook
Despite being a mere synthetic curiosity in the mid-20th century, the bicyclo[1.1.0]butyl motif has emerged in the 21st century as a powerful structure for organic synthesis. Although classical approaches have proven effective for assembling a range of BCB structures, several recent advances have further bolstered the arsenal available to synthetic chemists for the construction of more complex BCBs. Advances in our understanding of BCB chemistry have ultimately led to a broader profile of accessible BCB architectures and more widespread adoption by practitioners. Indeed, these innovations in BCB chemistry have unlocked approaches to synthetically challenging isosteric species. BCB's evolution from being viewed as “too reactive” to be synthetically tractable, to serving as a mainstay reagent with medicinal chemistry applications, is an important reminder of the opportunities that lie in the curiosities of the past.Author contributions
CBK and JAM co-led the organization of the review with input from LJT and TMS. JAM and LJT were responsible for the content Sections 1 and 2. CBK was responsible for preparing the figures for the manuscript as well as the Abstract, Section 3, and Section 5. TMS was responsible for Section 4. All authors provided feedback and edits for all sections.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Kerry Betz (Janssen) for helpful comments in preparing this manuscript.Notes and references
- K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312 CrossRef.
- (a) J. Turkowska, J. Durkaab and D. Gryko, Chem. Commun., 2020, 56, 5718 RSC; (b) J. A. Milligan and P. Wipf, Nat. Chem., 2016, 8, 296 CrossRef CAS PubMed.
- (a) A. Fawcett, Pure Appl. Chem., 2020, 92, 751 CrossRef CAS; (b) M. A. A. Walczak, T. Krainz and P. Wipf, Acc. Chem. Res., 2015, 48, 1149 CrossRef CAS PubMed.
- (a) S. Meiboom and L. C. Snyder, Acc. Chem. Res., 1971, 4, 81 CrossRef CAS; (b) P. G. Gassman, M. L. Greenlee, D. A. Dixon, S. Richsmeier and J. Z. Gougoutas, J. Am. Chem. Soc., 1983, 105, 5865 CrossRef CAS.
- A. de Meijere, S. I. Kozhushkov and H. Schill, Chem. Rev., 2006, 106, 4926 CrossRef CAS PubMed.
- (a) P. H. M. Budzelaar, E. Kraka, D. Cremer and P. R. Schleyer, J. Am. Chem. Soc., 1986, 108, 561 CrossRef CAS; (b) K. V. Berezin, V. V. Nechaev, M. K. Berezin, N. F. Stepanov and S. V. Krasnoshchekov, Opt. Spectrosc., 2014, 117, 366 CrossRef CAS.
- I. Haller and R. Srinivasan, J. Chem. Phys., 1964, 41, 2745 CrossRef CAS.
- (a) M. D. Harmony and K. Cox, J. Am. Chem. Soc., 1966, 88, 5049 CrossRef CAS; (b) M. D. Harmony, K. W. Cox, G. Nelson and K. B. Wiberg, J. Chem. Phys., 1969, 50, 1976 CrossRef.
- (a) M. D. Newton and J. M. Schulman, J. Am. Chem. Soc., 1972, 94, 767 CrossRef CAS; (b) J. F. Liebman and A. Greenberg, Chem. Rev., 1976, 76, 311 CrossRef CAS.
- (a) K. B. Wiberg, S. T. Waddell and R. E. Rosenberg, J. Am. Chem. Soc., 1990, 112, 2184 CrossRef CAS; (b) K. B. Wiberg, J. Am. Chem. Soc., 1983, 105, 1227 CrossRef CAS.
- (a) J. O. Jensen, J. Mol. Struct., 2003, 631, 157 CrossRef CAS; (b) R. Jamal and S. F. Al-Saidi, Phys. Chem.: Indian J., 2015, 10, 149 CAS; (c) D. M. Lemal, J. Org. Chem., 2009, 74, 2413 CrossRef CAS PubMed.
- K. Wuthrich, S. Meiboom and L. C. Snyder, J. Chem. Phys., 1970, 52, 230 CrossRef.
- K. B. Wiberg, G. M. Lampman, R. P. Ciula, D. S. Connor, P. Schertler and J. Lavanish, Tetrahedron, 1965, 21, 2749 CrossRef CAS.
- W. R. Moore and C. R. Costin, J. Am. Chem. Soc., 1971, 93, 4910 CrossRef CAS.
- R. J. Meier, ChemEngineering, 2021, 5, 24 CrossRef CAS.
- D. Barić and Z. B. Maksić, Theor. Chem. Acc., 2005, 114, 222 Search PubMed.
- J. D. Dill, A. Greenberg and J. F. Liebman, J. Am. Chem. Soc., 1979, 101, 6814 CrossRef CAS.
- E. Rozental, C. Azran, H. Basch and S. Hoz, Can. J. Chem., 1999, 77, 537 CrossRef CAS.
- S. Inagaki, T. Kakefu, T. Yamamoto and H. Wasada, J. Phys. Chem., 1996, 100, 9615 CrossRef CAS.
- R. Laplaza, J. Contreras-Garcia, F. Fuster, F. Volatron and P. Chaquin, Comput. Theor. Chem., 2022, 1207, 113505 CrossRef CAS.
- M. Makino, J. R. Dias and J. Aihara, J. Phys. Chem. A, 2020, 124, 4549 CrossRef CAS PubMed.
- J. Weber, U. Haslinger and U. H. Brinker, J. Org. Chem., 1999, 64, 6085 CrossRef CAS.
- R. Whitman and J. F. Chiang, J. Am. Chem. Soc., 1972, 94, 1126 CrossRef.
- (a) S. R. Kass and P. K. Chou, J. Am. Chem. Soc., 1988, 110, 7899 CrossRef CAS; (b) I. Alkorta and J. Elguero, Tetrahedron, 1997, 53, 9741 CrossRef CAS.
- (a) I. Alkorta, N. Campillo, I. Rozas and J. Elguero, J. Org. Chem., 1998, 63, 7759 CrossRef CAS; (b) A. Ragyanszki, B. Fiser, E. Lee-Ruff and J. F. Liebman, Photochem. Photobiol., 2021, 97, 1353 CrossRef CAS PubMed.
- (a) M. Pomerantz and E. W. Abrahamson, J. Am. Chem. Soc., 1966, 88, 3970 CrossRef CAS; (b) K. C. Bishop III, Chem. Rev., 1976, 76, 461 CrossRef.
- M. Wang, Y. Huang, C. Li and P. Lu, Org. Chem. Front., 2022, 9, 2149 RSC.
- M. Horner and S. Hünig, J. Am. Chem. Soc., 1977, 99, 6122 CrossRef CAS.
- H. K. Hall Jr and A. B. Padias, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 625 CrossRef.
- K. B. Wiberg and R. P. Ciula, J. Am. Chem. Soc., 1959, 81, 5261 CrossRef CAS.
- X. Drujon, G. Riess, H. K. Hall Jr and A. B. Padias, Macromolecules, 1993, 26, 1199 CrossRef CAS.
- (a) H. K. Hall Jr, E. P. Blanchard Jr, S. C. Cherkofsky, J. B. Sieja and W. A. Sheppard, J. Am. Chem. Soc., 1971, 93, 110 CrossRef; (b) T. D. Swartz and H. K. Hall Jr, J. Am. Chem. Soc., 1971, 93, 137 CrossRef CAS.
- H. K. Hall Jr, C. D. Smith, E. P. Blanchard Jr, S. C. Cherkofsky and J. B. Sieja, J. Am. Chem. Soc., 1971, 93, 121 CrossRef.
- T. Kawauchi, M. Nakamura, T. Kitayama, A. B. Padias and H. K. Hall Jr, Polym. J., 2005, 37, 439 CrossRef CAS.
- M. Barfield, L. Kao, H. K. Hall Jr and L. G. Snow, Macromolecules, 1984, 17, 240 CrossRef CAS.
- M. Barfield, R. J. H. Chan, H. K. Hall Jr and Y. Mou, Macromolecules, 1986, 19, 1343 CrossRef CAS.
- (a) J. Li, R. Stein and S. A. Lopez, J. Org. Chem., 2021, 86, 4061 CrossRef CAS PubMed; (b) W. J. Leigh, Chem. Rev., 1993, 93, 487 CrossRef CAS; (c) A. Waldemar, T. Oppenländer and G. Zang, J. Am. Chem. Soc., 1985, 107, 3921 CrossRef.
- (a) A. R. Rossi, Y. Wang and K. B. Wiberg, J. Phys. Chem. A, 2009, 113, 1686 CrossRef CAS PubMed; (b) A. F. Becknell, J. A. Berson and R. Srinivasan, J. Am. Chem. Soc., 1985, 107, 1076 CrossRef CAS.
- K. B. Wiberg and G. M. Lampman, Tetrahedron Lett., 1963, 4, 2173 CrossRef.
- G. M. Lampman, J. C. Aumiller, R. A. Fenoglio and K. B. Wiberg, Org. Synth., 1988, 51, 55 Search PubMed.
- R. Srinivasan, J. Am. Chem. Soc., 1963, 85, 4045 CrossRef CAS.
- A. F. Vellturo and G. W. Griffin, J. Am. Chem. Soc., 1965, 87, 3021 CrossRef CAS.
- M. R. Rifi, J. Am. Chem. Soc., 1967, 89, 4442 CrossRef CAS.
- J. B. Sieja, J. Am. Chem. Soc., 1971, 93, 130 CrossRef CAS.
- Y. Gaoni, J. Org. Chem., 1982, 47, 2564 CrossRef CAS.
- (a) R. Gianatassio, J. M. Lopchuk, J. Wang, C. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241 CrossRef CAS PubMed; (b) J. M. Lopchuck, K. Fjelbye, Y. Kawamata, L. R. Malins, C. Pan, R. Gianatassio, J. Wang, L. Prieto, J. Bradow, T. A. Brandt, M. R. Collins, J. Elleraas, J. Ewanicki, W. Farrell, O. O. Fadeyi, G. M. Gallego, J. J. Mosseau, R. Oliver, N. W. Sach, J. K. Smith, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 3209 CrossRef PubMed.
- M. Jung and V. N. G. Lindsay, J. Am. Chem. Soc., 2022, 144, 4764 CrossRef CAS PubMed.
- P. G. Gassman, M. J. Mullins, S. Richtsmeier and D. A. Dixon, J. Am. Chem. Soc., 1979, 101, 5793 CrossRef CAS.
- P. Wipf, C. R. J. Stephenson and K. Okumura, J. Am. Chem. Soc., 2003, 125, 14694 CrossRef CAS PubMed.
- (a) W. Mahler, J. Am. Chem. Soc., 1962, 84, 4600 CrossRef CAS; (b) S. Masamune, J. Am. Chem. Soc., 1964, 86, 735 CrossRef CAS.
- N. Ikota, N. Takamura, S. D. Young and B. Ganem, Tetrahedron Lett., 1981, 22, 4163 CrossRef CAS.
- M. S. Baird and H. H. Hussain, Tetrahedron, 1987, 43, 215 CrossRef CAS.
- C. Qin and H. M. L. Davies, Org. Lett., 2013, 15, 310 CrossRef CAS PubMed.
- R. Panish, S. R. Chintala, D. T. Boruta, Y. Fang, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2013, 135, 9283 CrossRef CAS PubMed.
- R. A. Panish, S. R. Chintala and J. M. Fox, Angew. Chem., Int. Ed., 2016, 55, 4983 CrossRef CAS PubMed.
- (a) K. Chen, X. Huang, S. B. J. Kan, R. K. Zhang and F. H. Arnold, Science, 2018, 360, 71 CrossRef CAS PubMed; (b) C. Schneider, K. Niisuke, W. E. Boeglin, M. Voehler, D. F. Stec, N. A. Porter and A. R. Brash, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18941 CrossRef CAS PubMed.
- C. B. Kelly, A. M. Colthart, B. D. Constant, S. R. Corning, L. N. E. Dubois, J. T. Genovese, J. L. Radziewicz, E. M. Sletten, K. R. Whitaker and L. J. Tilley, Org. Lett., 2011, 13, 1646 CrossRef CAS PubMed.
- (a) J. B. Lambert and R. B. Finzel, J. Am. Chem. Soc., 1982, 104, 2020 CrossRef CAS; (b) V. J. Shiner Jr and E. R. Davidson, J. Am. Chem. Soc., 1986, 108, 3135 CrossRef; (c) J. Coope, V. J. Shiner Jr and M. W. Ensinger, J. Am. Chem. Soc., 1990, 112, 2834 CrossRef CAS; (d) V. J. Shiner Jr, M. W. Ensinger and R. D. Rutkowske, J. Am. Chem. Soc., 1987, 109, 804 CrossRef; (e) L. J. Tilley and V. J. Shiner Jr, J. Phys. Org. Chem., 1999, 12, 564 CrossRef CAS; (f) J. Yoshida and M. J. Sugawara, J. Org. Chem., 2000, 65, 3135 CrossRef PubMed.
- (a) P. G. Gassman and J. B. Hall, J. Am. Chem. Soc., 1984, 106, 4267 CrossRef CAS; (b) P. G. Gassman and M. M. Doherty, J. Am. Chem. Soc., 1982, 104, 3742 CrossRef CAS; (c) D. W. Nelson, N. J. O'Reilly, J. Speier and P. G. Gassman, J. Org. Chem., 1994, 59, 8157 CrossRef CAS.
- (a) D. M. Lemal and L. H. Dunlap Jr, J. Am. Chem. Soc., 1972, 94, 6562 CrossRef CAS; (b) M. W. Grayston and D. M. Lemal, J. Am. Chem. Soc., 1976, 98, 1278 CrossRef CAS; (c) D. Wirth and D. M. Lemal, J. Am. Chem. Soc., 1982, 104, 847 CrossRef CAS; (d) D. M. Lemal, J. V. Staros and V. Austel, J. Am. Chem. Soc., 1969, 91, 3373 CrossRef CAS.
- M. A. Mercadnte, C. B. Kelly, T. A. Hamlin, K. R. D. Chiaie, M. D. Drago, K. K. Duffy, M. T. Dumas, D. C. Fager, B. L. C. Glod, K. E. Hansen, C. R. Hill, R. M. Leising, C. L. Lynes, A. E. MacInnis, M. R. McGohey, S. A. Murray, M. C. Piquette, S. L. Roy, R. M. Smith, K. R. Sullivan, B. H. Truong, K. M. Vailonis, V. Gorbatyuk, N. E. Leadbeater and L. J. Tilley, Chem. Sci., 2014, 5, 3983 RSC.
- A. Moriondo, Synthesis of 1-cyanobicyclobutane through γ-silyl elimination, Undergraduate thesis, Stonehill College, Easton, MA, 2018 Search PubMed.
- N. Thomas, Investigation of the Difluoromethylation of Enolizable Ketones and the Synthesis of Difluoromethylbicyclobutane, Undergraduate thesis, Stonehill College, Easton, MA, 2022 Search PubMed.
- G. Giardino, Synthesis of Acetylinic and Pyridyl Bicyclobutanes via γ−silyl Elimination, Undergraduate thesis, Stonehill College, Easton, MA, 2019 Search PubMed.
- (a) X. Creary, Beilstein J. Org. Chem., 2019, 15, 1769 CrossRef CAS PubMed; (b) X. Creary, J. Am. Chem. Soc., 2013, 135, 6570 CrossRef CAS PubMed.
- B. D. Schwartz, M. Y. Zhang, R. H. Attard, M. G. Gardiner and L. R. Malins, Chem.–Eur. J., 2020, 26, 2808 CrossRef CAS PubMed.
- R. E. McNamee, M. M. Haugland, J. Nugent, R. Chan, K. E. Christensen and E. A. Anderson, Chem. Sci., 2021, 12, 7480 RSC.
- R. E. McNamee, A. L. Thompson and E. A. Anderson, J. Am. Chem. Soc., 2021, 143, 21246 CrossRef CAS PubMed.
- (a) S. Hoz and D. Aurbach, J. Org. Chem., 1984, 49, 4144 CrossRef CAS; (b) S. Hoz and D. Aurbach, J. Org. Chem., 1984, 49, 3285 CrossRef CAS; (c) S. Hoz, M. Livneh and D. Cohen, J. Org. Chem., 1986, 51, 4537 CrossRef CAS; (d) S. Hoz, C. Azran and A. Sella, J. Am. Chem. Soc., 1996, 118, 5456 CrossRef CAS.
- Y. Gaoni, Tetrahedron Lett., 1988, 29, 1591 CrossRef CAS.
- (a) Y. Gaoni and A. Tomažič, J. Org. Chem., 1985, 50, 2948 CrossRef CAS; (b) Y. Gaoni, Tetrahedron, 1989, 45, 2819 CrossRef CAS.
- Y. Gaoni, Tetrahedron Lett., 1982, 23, 5215 CrossRef CAS.
- Y. Gaoni, A. Tomažič and E. Potgieter, J. Org. Chem., 1985, 50, 2943 CrossRef CAS.
- J. A. Milligan, C. A. Busacca, C. H. Senanayake and P. Wipf, Org. Lett., 2016, 18, 4300 CrossRef CAS PubMed.
- M. J. Kerner and P. Wipf, Org. Lett., 2021, 23, 3615 CrossRef CAS PubMed.
- S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 16766 CrossRef CAS PubMed.
- L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2021, 60, 212 CrossRef CAS PubMed.
- A. L. G. Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116 CrossRef PubMed.
- P. G. Gassman and G. T. Carroll, J. Org. Chem., 1984, 49, 2074 CrossRef CAS.
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS PubMed.
- M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511 CrossRef CAS PubMed.
- G. Ernouf, E. Chirkin, L. Rhyman, P. Ramasami and J. Cintrat, Angew. Chem., Int. Ed., 2020, 59, 2618 CrossRef CAS PubMed.
- C. J. Pratt, R. A. Aycock, M. D. King and N. T. Jui, Synlett, 2020, 31, 51 CrossRef CAS PubMed.
- X. Wu, W. Hao, K. Ye, B. Jiang, G. Pombar, Z. Song and S. Lin, J. Am. Chem. Soc., 2018, 140, 14836 CrossRef CAS PubMed.
- M. Ociepa, A. J. Wierzba, J. Turkowska and D. Gryko, J. Am. Chem. Soc., 2020, 142, 5355 CrossRef CAS PubMed.
- (a) M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046 CrossRef CAS PubMed; (b) J. M. Anderson, N. D. Measom, J. A. Murphy and D. L. Poole, Angew. Chem., Int. Ed., 2021, 60, 24754 CrossRef CAS PubMed.
- A. F. Stepan, C. Subramanyam, I. V. Efemov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffman, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, J. Med. Chem., 2012, 55, 3414 CrossRef CAS PubMed.
- X. Ma, D. L. Sloman, Y. Han and D. J. Bennett, Org. Lett., 2019, 21, 7199 CrossRef CAS PubMed.
- R. M. Bycheck, V. Hutskalova, Y. P. Bas, O. A. Zaporozhets, S. Zozulya, V. V. Levterov and P. K. Mykhailiuk, J. Org. Chem., 2019, 84, 15106 CrossRef PubMed.
- (a) D. E. Applequist and J. W. Wheeler, Tetrahedron Lett., 1977, 18, 3411 CrossRef; (b) D. E. Applequist, T. L. Renken and J. W. Wheeler, J. Org. Chem., 1982, 47, 4985 CrossRef CAS.
- F. Wang, T. Luo, J. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153–7157 CrossRef CAS PubMed.
- R. Bychek and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2022, 61, e202205103 CrossRef CAS PubMed.
- M. A. A. Walczak and P. Wipf, J. Am. Chem. Soc., 2008, 130, 6924 CrossRef CAS PubMed.
- A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117 CrossRef CAS PubMed.
- T. Pinkert, M. Das, M. L. Schrader and F. Glorius, J. Am. Chem. Soc., 2021, 143, 7648 CrossRef CAS PubMed.
- P. Zhang, R. Zhuang, X. Wang, H. Liu, J. Li, X. Su, X. Chen and X. Zhang, Bioconjugate Chem., 2018, 29, 467 CrossRef CAS PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876 RSC.
- M. E. Flanagan, J. A. Abramite, D. P. Anderson, A. Aulabaugh, U. P. Dahal, A. M. Gilbert, C. Li, J. Montgomery, S. R. Oppenheimer, T. Ryder, B. P. Schuff, D. P. Uccello, G. S. Walker, Y. Wu, M. F. Brown, J. M. Chen, M. M. Hayward, M. C. Noe, R. S. Obach, L. Philippe, V. Shanmugasundaram, M. J. Shapiro, J. Starr, J. Stroh and Y. Che, J. Med. Chem., 2014, 57, 10072 CrossRef CAS PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673 CrossRef CAS PubMed.
- K. Tokunaga, M. Sato, K. Kuwata, C. Miura, H. Fuchida, N. Matsunaga, S. Koyanagi, S. Ohdo, N. Shindo and A. Ojida, J. Am. Chem. Soc., 2020, 142, 18522 CrossRef CAS PubMed.
- B. D. Schwartz, A. P. Smyth, P. E. Nashar, M. G. Gardiner and L. R. Malins, Org. Lett., 2022, 24, 1268 CrossRef CAS PubMed.
- R. L. Amey and B. E. Smart, J. Org. Chem., 1981, 46, 4090 CrossRef CAS.
- M. H. Chang and D. A. Dougherty, J. Org. Chem., 1981, 46, 4092 CrossRef CAS.
- S. Tai, K. Takubo, J. A. Milligan, P. Wipf, J. K. Johnson, E. M. Skoda, J. B. Nelson and Z. Wang, International Patent, WO 2017165822 A8, 2017.
- B. Lefker, K. Gibson, M. Spendiff, P. Humphries, S. Bucknell, W. Zawodny and R. Porter, International Patent, WO 2022051596 A1, 2022.
- T. Mukaiyama, P. C. Chua and J.-M. Vernier, International Patent, WO 2021173923 A1, 2021.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (b) F. Lovering, MedChemComm, 2013, 4, 515 RSC.
- M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448 RSC.
- O. O. Grygorenko, D. M. Volochnyuk and B. V. Vashchenko, Eur. J. Org. Chem., 2021, 6478 CrossRef CAS.
- J. Kanazawa and M. Uchiyama, Synlett, 2019, 30, 1 CrossRef CAS.
- (a) P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839 RSC; (b) A. Denisenko, P. Garbuz, S. V. Shishkina, N. M. Voloshchuk and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 20515 CrossRef CAS PubMed.
- S. Agasti, F. Beltran, E. Pye, N. Kaltsoyannis, G. Crisenza and D. Procter, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-v93kv.
- R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477 CrossRef CAS PubMed.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed.
- R. Guo, Y. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988 CrossRef CAS PubMed.
- K. Dhake, K. J. Woelk, J. Becia, A. Un, S. E. Jenny and D. C. Leitch, Angew. Chem., Int. Ed., 2022, 61, e202204719 CrossRef CAS PubMed.
- C. J. Huck and D. Sarlah, Chem, 2020, 6, 1589 CAS.
- N. R. O'Connor, J. L. Wood and B. M. Stoltz, Isr. J. Chem., 2016, 56, 431 CrossRef PubMed.
- Z. J. Song, J. Qi, M. H. Emmert, J. Wang, X. Yang and D. Xiao, Org. Process Res. Dev., 2021, 25, 82 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |