
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA free boratriptycene-type Lewis superacid†
Marcel
Henkelmann
a,
Andreas
Omlor
b,
Michael
Bolte
 a,
Volker
Schünemann
b,
Hans-Wolfram
Lerner
a,
Jozef
Noga
c,
Peter
Hrobárik
*c and
Matthias
Wagner
*a
a,
Volker
Schünemann
b,
Hans-Wolfram
Lerner
a,
Jozef
Noga
c,
Peter
Hrobárik
*c and
Matthias
Wagner
*a
aInstitute of Inorganic Chemistry, Goethe University Frankfurt, Max-von-Laue-Straße 7, 60438 Frankfurt am Main, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
bPhysics Department, University Kaiserslautern, Erwin-Schrödinger-Straße 56, 67663 Kaiserslautern, Germany
cDepartment of Inorganic Chemistry, Faculty of Natural Sciences, Comenius University, Ilkovicova 6, 84215 Bratislava, Slovakia. E-mail: peter.hrobarik@uniba.sk
First published on 7th December 2021
Abstract
Bicyclic pyrazabole-bridged ferrocenes with BH groups at their bridgehead positions were prepared from [Li(thf)]2[1,1′-fc(BH3)2] and pyrazole or 3,5-dimethylpyrazole in the presence of Me3SiCl (1 or 1Me, respectively; 1,1′-fc = 1,1′-ferrocenylene); Me3SiH and H2 are released as byproducts. Treatment of 1 or 1Me with 1 eq. of the hydride scavenger [Ph3C][B(C6F5)4] afforded the borenium salts [2][B(C6F5)4] (72%) and [2Me][B(C6F5)4] (77%). According to X-ray crystallography, [2Me]+ contains one trigonal-planar borenium cation, the cyclopentadienyl (Cp) rings of the 1,1′-fc fragment remain parallel to each other, but the Cp–B bond vector is bent out of the Cp plane by an unprecedentedly large dip angle α* of 40.6°. The Fe⋯B(sp2) distance is very short (2.365(4) Å) and the 11B NMR signal of the cationic B(sp2) center is remarkably upfield shifted (23.4 ppm), suggesting a direct Fe(3d) → B(2p) donor–acceptor interaction. Although this interpretation is confirmed by quantum-chemical calculations, the coupling between the associated orbitals corresponds to an energy of only 12 kJ mol−1. Accordingly, both the experimental (e.g., Gutmann–Beckett acceptor number AN = 111) and theoretical assessment (e.g., Et3PO and F−-ion affinities) of the Lewis acidity proves that [2]+ is among the strongest boron-based Lewis acids available to date.
Introduction
The rise to prominence of boron-based Lewis acids began when their application potential as activators for metal-catalyzed homogeneous olefin polymerization was recognized.1 The advent of the ‘Frustrated Lewis Pairs’ led to a further surge of interest, which has remained at a high level ever since.1,2 Among all the remarkable developments in the field, we focus here particularly on Lewis acidic organoboranes (‘Lewis superacids’).3One way to increase the affinity of a triorganylborane (BR3) for even weak Lewis bases is to introduce electronegative halogen substituents at the periphery.4 Classic examples include the compounds B(C6F5)3 of Massey and Park5 or B(C6Cl5)3 of Ashley and O'Hare.6 As an alternative, mesomerically electron-withdrawing substituents have also been employed.7 The affinity of a boron center (in particular) for anionic ligands can also be increased by the Coulomb attraction of an adjacent positively charged group.8 Gabbaï et al. took advantage of this effect when they used the phosphonium borane ([A]+; Fig. 1) to bind F− ions in water despite their high hydration enthalpy.9,10 Our group combined a high fluorine load with the positive charge of a phosphonium center to create a Lewis acid [B]+, stronger than B(C6F5)3 and suitable as a catalyst for Diels–Alder reactions.11 By shifting the positive charge even closer to the boron center, Corey et al. obtained chiral oxazaborolidinium cations [C]+ that serve as broadly applicable catalysts in (enantioselective) [4 + 2]- and [3 + 2]-cycloaddition reactions.12 Most trivalent compounds BR3 with an empty boron 2p orbital adopt trigonal planar configurations. Only after coordination of a Lewis basic donor (Do) is the BC3 moiety pyramidalized to allow for a (distorted) tetrahedral geometry of the BR3·Do adduct. However, when a rigid organic framework is designed to impose a structural constraint on the B atom that forces the BC3 core out of planarity, the free Lewis acid becomes destabilized but also better preorganized for ligand coordination. As a consequence, the organoborane becomes a better electron-pair acceptor.3a–c,13 Mikhailov realized such a structural motif by the synthesis of 1-boraadamantane,14,15 while Berionni et al. released a non-planar 9-boratriptycene (D) by in situ protonolysis of corresponding borate-complexes [R4P][tBuC6H4–D] with HNTf2 in CH2Cl2 (Tf = O2SCF3).16 In the resulting mixture, D is in an association–dissociation equilibrium with the weakly coordinating [NTf2]− ion and was isolated in the form of its trapping products with various N-, P-, or O-centered Lewis bases.17 Structural data from X-ray crystallography are not available for either free 1-boraadamantane or D. Gas-phase electron diffraction on 3-methyl-1-boraadamantane at 100 °C revealed that the B atom in the equilibrium structure has a (slightly) pyramidal configuration with ∑(C–B–C) = 349.4(4)° and av. d(B–C) = 1.556(5) Å.18 The experimentally determined structural parameters were confirmed by quantum-chemical calculations, which further indicate significant hyperconjugation between the vacant boron 2p orbital and its adjacent C–C σ bonds (average stabilization energy: 64.4 kJ mol−1; electron occupancy q(B(2p)): 0.17 e−).18,19 Compared to 1-boraadamantane, the computed molecular structure of D shows a more pyramidalized BC3 core (∑(C–B–C) = 339.3°) while q(B(2p)) is lower (0.08 e−).16
 | ||
| Fig. 1 Organoboranes of particularly high Lewis acidity due to introduced positive charges and fluorine substituents ([A]+, [B]+, [C]+) or structurally enforced pyramidalization (D). Mes = 2,4,6-Me3C6H2, Tf = O2SCF3. | ||
Taking into account the above developments over the last two decades, we have now designed a novel ferrocenylborane-based10h,20,21 Lewis superacid [2]+ guided by the following criteria (Scheme 1): (i) facile synthesis inspired by the self-assembly of pyrazaboles or scorpionate ligands;22 (ii) a positively charged B center with electronegative N(sp2) atoms in its first coordination sphere, which cannot donate π-electron density to the vacant p orbital on the B atom due to the orthogonal positions of the filled p orbitals on the N atoms;23–25 (iii) a strained scaffold analogous to boratriptycene. The 1,1′-ferrocenylene bridge in [2]+ has two functions to prevent the molecule from exceeding its stability limits: (i) a weak and easily displaced Fe → B+ through-space interaction should help to buffer the electron deficiency of the free borenium cation in its resting state.10h,20 (ii) Because of the facile distortion of the normally parallel cyclopentadienyl rings toward a tilted orientation, the 1,1′-ferrocenylene unit can act as a hinge that mitigates an overly strained pyramidal configuration of the B atom. The synthesis and key properties of [2]+-type compounds will be described herein.
 | ||
| Scheme 1 Synthesis of pyrazabole-bridged ansa-ferrocenes 1 and 1Me; hydride abstraction to afford the borenium-ion salts [2][B(C6F5)4] and [2Me][B(C6F5)4]. Reagents and conditions: (i) THF, −78 °C (30 min) to 70 °C (6 h). (ii) THF, room temperature (overnight) to 70 °C (3 h). (iii) CH2Cl2, room temperature, 1 min. R = C6F5. | ||
Results and discussion
Synthesis of [2][B(C6F5)4] and [2Me][B(C6F5)4]
Treatment of the readily available hydridoborate salt [Li(thf)]2[1,1′-fc(BH3)2]26 with pyrazole or 3,5-dimethylpyrazole and Me3SiCl afforded the pyrazabole-bridged ansa-ferrocenes 1 and 1Me in yields of 39% and 55%, respectively (Scheme 1; 1,1′-fc = 1,1′-ferrocenylene); Me3SiH and H2 are formed as the byproducts. Clean and quantitative hydride abstraction was achieved through the reaction of 1 or 1Me with the hydride scavenger [Ph3C][B(C6F5)4] in CH2Cl2. After precipitation with n-pentane, [2][B(C6F5)4] (72%) and [2Me][B(C6F5)4] (77%) were isolated in good yields. Crystals of the borenium salts suitable for X-ray analysis were grown by layering their concentrated solutions in CH2Cl2 with n-hexane.27X-ray crystallography
Both precursor/borenium-ion pairs, 1/[2][B(C6F5)4] and 1Me/[2Me][B(C6F5)4], were studied by X-ray diffraction. Since the crystal lattice of [2][B(C6F5)4] contains three crystallographically independent entities in the asymmetric unit, only the molecular structures of 1Me/[2Me][B(C6F5)4] are compared here (Fig. 2). We verified that all the conclusions drawn are also valid for the pair 1/[2][B(C6F5)4] (see the ESI†). | ||
| Fig. 2 Molecular structures of 1Me (left) and [2Me][B(C6F5)4] (right) in the solid state. C-bonded H atoms and the [B(C6F5)4]− anion are omitted for clarity. Selected structural parameters are compiled in the table. | ||
The 1,1′-ferrocenylene moieties in 1Me/[2Me]+ adopt almost perfectly staggered conformations with torsion angles  close to zero (Ci = B-bonded C atom; COG = cyclopentadienyl-ring centroid). No significant differences are also observed between the B–C (av. 1.596 Å) or B–N (av. 1.567 Å) bond lengths in 1Me and the B(2)–C (1.613(6) Å) or B(2)–N (av. 1.568 Å) bond lengths in [2Me]+. The configuration of the second B atom, however, changes fundamentally from tetrahedral to trigonal-planar upon going from 1Me to [2Me]+ (sum of angles around B(1) = 359.7°). Accordingly, the B(1)–C (1.497(6) Å) and B(1)–N (av. 1.506 Å) bonds of [2Me]+ are contracted relative to the corresponding bonds at B(2). This contraction is about twice as pronounced for B(1)–C (Δ = 0.116 Å) as for B(1)–N (Δ = 0.062 Å). The B(1)–C bond of [2Me]+ is also considerably shorter than the exocyclic B–C bond between the orthogonally positioned planes in 9-Mes-9-borafluorene (1.562 Å), which we take as an archetypal B(sp2)–C(sp2) single bond.28 The B(1)–N bonds of [2Me]+ are still longer than the average B–N bond in propeller-shaped B(pyrMe2)3 (1.443 Å; pyrMe2 = 2,5-dimethylpyrrolyl), for which B
close to zero (Ci = B-bonded C atom; COG = cyclopentadienyl-ring centroid). No significant differences are also observed between the B–C (av. 1.596 Å) or B–N (av. 1.567 Å) bond lengths in 1Me and the B(2)–C (1.613(6) Å) or B(2)–N (av. 1.568 Å) bond lengths in [2Me]+. The configuration of the second B atom, however, changes fundamentally from tetrahedral to trigonal-planar upon going from 1Me to [2Me]+ (sum of angles around B(1) = 359.7°). Accordingly, the B(1)–C (1.497(6) Å) and B(1)–N (av. 1.506 Å) bonds of [2Me]+ are contracted relative to the corresponding bonds at B(2). This contraction is about twice as pronounced for B(1)–C (Δ = 0.116 Å) as for B(1)–N (Δ = 0.062 Å). The B(1)–C bond of [2Me]+ is also considerably shorter than the exocyclic B–C bond between the orthogonally positioned planes in 9-Mes-9-borafluorene (1.562 Å), which we take as an archetypal B(sp2)–C(sp2) single bond.28 The B(1)–N bonds of [2Me]+ are still longer than the average B–N bond in propeller-shaped B(pyrMe2)3 (1.443 Å; pyrMe2 = 2,5-dimethylpyrrolyl), for which B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N π bonding is negligible.29 The most important structural features of [2Me]+ are the pronounced dip angle α* = 40.6° and the short Fe⋯B(sp2) contact of 2.365(4) Å (α* = 180°–α; α = COG–Ci–B(1)). Corresponding distortions have already been observed for other ferrocenylboranes, but the dip angles are much smaller even in the most extreme cases, i.e., FcBBr2: α* = 18.9°,20a 9-Fc-9-borafluorene: α* = 25.5°,20d FcBC4Ph4: α* = 29.4° (Fc = ferrocenyl).20f The planarization of one bridgehead B atom does not result in a substantial tilting of the ferrocenylene unit: 1Me/[2Me]+ show largely coplanar cyclopentadienyl rings with Cp//Cp′ angles of 5.2(2)/5.4(2)°. The ‘hinge-effect’ of the 1,1′-fc bridge, which was assumed in the design of the molecular framework, obviously does not play a major role. Considerable differences are observed for the pyrazabole moieties of 1Mevs. [2Me]+: the gap between the two pyrazol-1-yl (pz) rings is significantly wider in 1Me than in [2Me]+ (pz//pz′ = 146.8(1)° vs. 118.7(2)°) and the dihedral angle between the best plane through the four N atoms and the B(2)-bonded Cp ring amounts to a value of 88.3° in 1Me, but only to 71.6° in [2Me]+.
N π bonding is negligible.29 The most important structural features of [2Me]+ are the pronounced dip angle α* = 40.6° and the short Fe⋯B(sp2) contact of 2.365(4) Å (α* = 180°–α; α = COG–Ci–B(1)). Corresponding distortions have already been observed for other ferrocenylboranes, but the dip angles are much smaller even in the most extreme cases, i.e., FcBBr2: α* = 18.9°,20a 9-Fc-9-borafluorene: α* = 25.5°,20d FcBC4Ph4: α* = 29.4° (Fc = ferrocenyl).20f The planarization of one bridgehead B atom does not result in a substantial tilting of the ferrocenylene unit: 1Me/[2Me]+ show largely coplanar cyclopentadienyl rings with Cp//Cp′ angles of 5.2(2)/5.4(2)°. The ‘hinge-effect’ of the 1,1′-fc bridge, which was assumed in the design of the molecular framework, obviously does not play a major role. Considerable differences are observed for the pyrazabole moieties of 1Mevs. [2Me]+: the gap between the two pyrazol-1-yl (pz) rings is significantly wider in 1Me than in [2Me]+ (pz//pz′ = 146.8(1)° vs. 118.7(2)°) and the dihedral angle between the best plane through the four N atoms and the B(2)-bonded Cp ring amounts to a value of 88.3° in 1Me, but only to 71.6° in [2Me]+.
NMR spectroscopy
NMR spectra were recorded in CDCl3 or CD2Cl2. The changes in the 1H, 11B, and 13C{1H} NMR spectra upon going from 1 to [2]+ or 1Me to [2Me]+ are very similar and will be discussed here exemplarily for the 1Me/[2Me]+ pair.The 11B NMR spectrum of the precursor molecule 1Me shows a resonance in the typical region of tetracoordinated boron nuclei (−7.6 ppm).30 After H− abstraction, the spectral parameters of the residual BH moiety of [2Me]+ remain largely the same, whereas the newly generated cationic B center gives rise to a signal at 23.4 ppm. Its chemical shift is highly unusual for tricoordinated B sites, which should be much less shielded.30 Since the formation of adduct equilibria with the weakly coordinating solvent molecules CD2Cl2 is unlikely (as also supported by our DFT simulations), we attribute the peculiar NMR feature either to a true Fe⋯B bonding interaction or to a magnetic anisotropy effect of the Fe d-electrons in close proximity to the B cation. We note in this context that upfield shifts of the 11B resonances have also been observed for FcBBr2vs. PhBBr2 (46.7 vs. 56.6 ppm),31 9-Fc-9-borafluorene vs. 9-Ph-9-borafluorene (53.0 vs. 64.5 ppm),20d and FcBC4Ph4vs. PhBC4Ph4 (47.4 vs. 65.4 ppm);20f along this series, the shift difference increases as the dip angle α* increases.
H−-ion abstraction leads to a symmetry breaking that renders the two Cp rings of [2Me]+ chemically inequivalent. The assignment of resonances to Cp′–B+ or Cp–B(H) was achieved by means of a NOESY spectrum (cf.Scheme 1 for the atom labeling): an intense cross peak was observed between a pzCH3 signal at δ(1H) = 2.56 and a CpH resonance at δ(1H) = 4.11 ppm; a second, significantly weaker, cross peak was detected between the second pzCH3 signal (2.48 ppm) and a CpH resonance at 3.31 ppm (pz = pyrazol-1-yl). Through-space interactions of two nuclear spins that are in closer proximity to each other tend to result in a more pronounced NOE. Since we know from the solid-state structure of [2Me][B(C6F5)4] that the average distance between CpH-2′ and pzCH3-3′ is 2.94 Å and thus approximately 30% shorter than the distance between CpH-2 and pzCH3-3 (4.19 Å), we assign the proton signals at 4.11/3.31 ppm to CpH-2′/CpH-2 and those at 2.56/2.48 to pzCH3-3′/pzCH3-3. The proton and carbon nuclei in the Cp′ ring of [2Me]+ are considerably deshielded relative to those of 1Me due to the electron-withdrawing effect of the attached borenium cation (Δδ: CpH-3′ = 1.40, CpH-2′ = 0.78 ppm; CpC-3′ = 15.5, CpC-2′ = 9.3 ppm). A qualitatively similar, albeit less marked, effect is also visible for the Cp ring of [2Me]+vs.1Me. Also all 1H and 13C NMR resonances of the pyrazolyl rings of [2Me]+ experience downfield shifts compared to the corresponding signals of 1Me.
Dynamic hydride-exchange equilibrium between 1 and [2]+
On one occasion, in the attempt to synthesize the borenium-cation salt [2][B(C6F5)4], we inadvertently added only about 0.8 eq. of [Ph3C][B(C6F5)4] to 1 (NMR-scale experiment; CD2Cl2). A subsequent 1H NMR spectroscopic investigation of the sample revealed well-resolved resonances of the byproduct Ph3CH, but extremely broad signals of the ferrocene-containing species. We attributed this effect to a dynamic exchange equilibrium of hydride ions between tetracoordinated BH and tricoordinated B+ sites. To test this assumption, we intentionally prepared a mixture of 1 (1.0 eq.) and [Ph3C][B(C6F5)4] (0.9 eq.) in CD2Cl2 and were able to reproduce the previous observation. After addition of [nBu4N][BH4] (1.0 eq. of the [BH4]− ion) to the sample, sharp proton resonances of 1 reappeared. The hydride abstraction/addition sequence was accompanied by only little decomposition (mainly producing ferrocene), and the proton-integral ratios of the re-generated 1 and Ph3CH conformed to an almost equimolar distribution (see the ESI† for plots of the spectra). In a second experiment, we prepared a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1 and [2][B(C6F5)4] (CD2Cl2); the observed 1H and 11B NMR resonances were broadened almost beyond detection (except for the [B(C6F5)4]−-anion signal). Upon cooling from room temperature to 243 K, the resonances became slightly sharper and the number of distinguishable signals increased as the exchange equilibrium was slowed down. We finally repeated the latter experiment with the 3,5-dimethylpyrazolyl derivatives 1Me and [2Me][B(C6F5)4]. Here, the line-broadening was much less pronounced and we were still able to identify all proton resonances of the two species in the NMR spectrum of the mixture. This result clearly shows that the introduced methyl groups provide considerable steric shielding of the boron centers, further confirming our hypothesis of a ‘hydride jump’ as the origin of the line-broadening effect.
:1 mixture of 1 and [2][B(C6F5)4] (CD2Cl2); the observed 1H and 11B NMR resonances were broadened almost beyond detection (except for the [B(C6F5)4]−-anion signal). Upon cooling from room temperature to 243 K, the resonances became slightly sharper and the number of distinguishable signals increased as the exchange equilibrium was slowed down. We finally repeated the latter experiment with the 3,5-dimethylpyrazolyl derivatives 1Me and [2Me][B(C6F5)4]. Here, the line-broadening was much less pronounced and we were still able to identify all proton resonances of the two species in the NMR spectrum of the mixture. This result clearly shows that the introduced methyl groups provide considerable steric shielding of the boron centers, further confirming our hypothesis of a ‘hydride jump’ as the origin of the line-broadening effect.
Electronic structures of [2]+/[2Me]+ and quantum-chemical calculations
Our experimental results consistently point toward the existence of a donor–acceptor Fe(3d) → B(2p) interaction in the borenium ions [2]+/[2Me]+, similar to the Fe → Si+ interaction in ferrocene-stabilized silylium ions.32 Comparable interactions have been also found for complexes of so-called Z-type ligands, in which Lewis-basic metal atoms donate charge density to Lewis-acidic boron sites located in the ligand backbones.33 Closely related examples to our system are certain metallaboratranes, i.e., complexes containing κ4-tris(2-mercaptoimidazolyl)borane ligands that interact with the coordinated metal atom via three dative S → M σ bonds and one retrodative M → B σ bond.34The experimentally determined very short Fe⋯B+ contacts in [2]+ and [2Me]+ (Fig. 2) are well reproduced at the DFT level (PBE-D3(BJ)/def2-TZVPP), which provides optimized Fe⋯B+ distances of 2.34 Å and 2.38 Å for [2]+ and [2Me]+, respectively; these values are practically unaffected by considering/neglecting corrections for dispersion forces. It was also theoretically confirmed that the dip angles α* are the largest among all ferrocene-stabilized boranes or borenium ions and that they are comparable to the corresponding dip angles in [Fc–SiR2]+ cations32 (α* = 41.9° for [2]+ and 40.0° for [2Me]+; Table S4†). Moreover, we found an excellent agreement between the computed and experimental 11B NMR chemical shift values (Table S5 and Fig. S66†), demonstrating that the optimized/solid-state structures prevail in solution (elongated Fe⋯B+ distances would result in downfield-shifted 11B(sp2) resonances).
The question now arises, to what extent the short Fe⋯B+ contacts in [2]+ and [2Me]+ can be attributed to a ‘through-space’ Fe(3d) → B(2p) donor–acceptor interaction. Structural strain imparted by the bridging rigid pyrazabole moieties and the avoidance of a pyramidalized tricoordinated B(sp2) center probably also play a role. According to an NBO second-order perturbation energy analysis, the stabilization energy E(2) for the coupling between the occupied ‘lone-pair’ orbital on Fe and the vacant B(2p) orbital corresponds to only 12 kJ mol−1, which is about eight times less than the stabilization energy found for the Fe → Si+ interaction in [Fc–SiR2]+ ions.32 The relative weakness of the Fe(3d) → B(2p) interaction in [2]+/[2Me]+ is further indicated by the following facts: (i) the B(sp2) center is trigonal planar rather than inwardly pyramidalized. (ii) An optimization of the model system Fc–Bpz2, formally obtained from [2]+ by removing the B(sp3)H tether to the second Cp ring, leads to a notably longer Fe⋯B contact of 2.70 Å, even after protonation of the pyrazole moieties or N,N′-chelation of a [BH2]+ fragment to introduce the overall positive charge (Fig. S63†). The impact of structural strain on the Fe⋯B+ distance is also evident from a comparison of the energies of [2]+ in its optimized geometry and in a hypothetical geometry with outwardly pyramidalized borenium center and diminished Fe(3d) → B(2p) interaction ([2pyram]+; the structure was obtained from that of 1 by hydride abstraction while keeping the dihedral angle θ(N–B–N–Ci) fixed during the optimization; Fig. S64†). Relative to [2]+, [2pyram]+ is energetically disfavored by 117 kJ mol−1, which is much more than the stabilization energy of the Fe(3d) → B(2p) interaction. Mayer bond orders (MBO = 0.43 for [2]+, 0.40 for [2Me]+) and QTAIM delocalization indices (DI = 0.21 for [2]+, 0.18 for [2Me]+) suggest a small covalency for the ‘through-space’ Fe(3d) → B(2p) interaction in [2]+ and [2Me]+, where these parameters are about twice as large as those found for previously reported Fc–B analogues (Table S4†). The partial covalency of the Fe(3d) → B(2p) interaction in [2]+ and [2Me]+ is also seen in ELI-D and NLMO analyses (Fig. 3), both revealing a three-center two-electron (3c2e) bond between the Fe(II) center, the B(sp2) cation, and the attached Ci atom of the Cp ring. In the simplified Lewis structure notation, the 3c2e bond in [2]+ and [2Me]+ results from a sharing of the respective Fe–Ci electron bonding pair, which has a large Fe(3d) character, with the vacant B(2p) orbital. The 3c2e bond thus can be viewed as a donor–acceptor interaction (see Fig. 3b for the composition of corresponding NLMO orbitals). In contrast, the corresponding 3c2e bond in structurally related [Fc–SiR2]+ ions is formed with the Ci atom of the other Cp ring. This difference might be attributed to the larger covalent/ionic radii of tricoordinated Si+ compared to tricoordinated B+ ions.
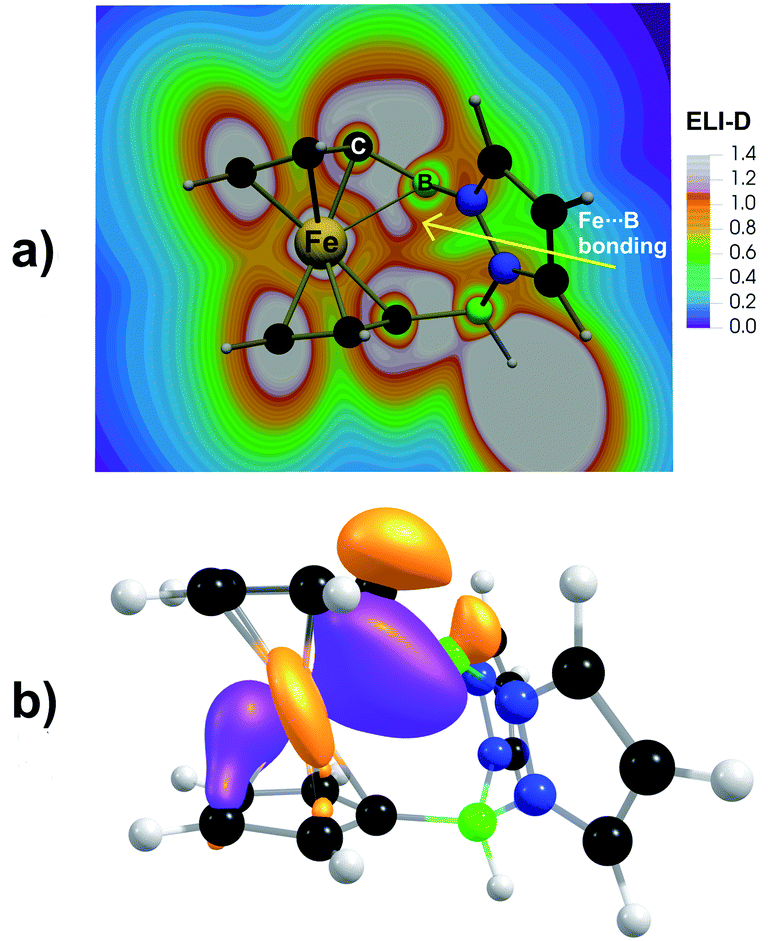 | ||
| Fig. 3 (a) Electron localizability indicator (ELI-D) analysis of bonding in [2]+. The selected plane includes the Fe and two B atoms. The gray-white regions represent ELF maxima (bonding attractors), showing a donor–acceptor Fe⋯B interaction. (b) Natural localized molecular orbital (NLMO) corresponding to a 3c2e bond between Fe(II), B(sp2), and the adjacent Ci atom in [2]+ (isosurface plot: ±0.05 au). NLMO composition is as follows: 51% Fe (99% d), 15% B (10% s, 90% p), 26% C (99% p). A similar NLMO picture was obtained for [2Me]+ with the following composition: 50% Fe (99% d), 14% B (10% s, 90% p), 28% C (99% p). | ||
The metal → B interaction found for [2]+ is computed to become significantly stronger when the Fe(II) center in the metallocene scaffold is replaced by heavier Ru(II) or Os(II) ions with more diffuse 4d or 5d valence orbitals, respectively. This is concluded mainly from the shorter M⋯B+ contacts (M = Ru, Os) despite the larger ionic radii of these M(II) centers, and also from the larger M⋯B+ covalency revealed by the larger QTAIM delocalization indices and Mayer bond orders (Table S4†).
Despite the existing Fe(3d) → B(2p) interaction and a hence conceivable charge transfer leading to the generation of Fe(III)/B˙ radical pairs, the electronic ground states of [2]+ and [2Me]+ remain diamagnetic with Fe(II) and B+ centers, which is confirmed by computed and experimental NMR shifts as well as 57Fe Mössbauer spectral parameters (Fig. S51, S52 and Tables S1, S6†). The electron-spin density in the lowest-lying triplet state of [2]+ (147 kJ mol−1 above the singlet ground state) resides almost exclusively on the Fe(II) center, with just small positive/negative fractions at the C atoms of both Cp rings (Fig. S65†). The same holds for higher-lying triplet excited states and thus, even upon singlet-triplet excitation, the Fe(3d) → B(2p) interaction would not be expected to promote the formation of an Fe(III)/B˙ radical pair. A qualitatively similar picture, but with notably larger energy gap between singlet and triplet states, is obtained for the ruthenocene analogue of [2]+ (ΔE = 280 kJ mol−1). In striking contrast, the osmocene congener (which is also predicted to feature a singlet ground state with Os(II) and B+ centers) is computed to display a distinct Os(III)/B˙ character in its relaxed triplet excited state (ΔE = 238 kJ mol−1).
Finally, we propose that the Fe(3d) → B(2p) interaction in [2]+/[2Me]+, while not significantly reducing the Lewis acidity of the ferrocenyl borenium ions (see below), may nevertheless provide the very extra stability needed to isolate the free Lewis acids (compare the case of the [D–NTf2]− ↔ D + [NTf2]− equilibrium shown in Fig. 1).
Assessment of the Lewis acidity of the borenium cation [2]+ by the Gutmann–Beckett NMR method, competition experiments with B(C6F5)3 and computational studies
The sterically less encumbered borenium-ion derivative [2]+ was selected for a comparative assessment of Lewis acidities. We first determined the acceptor number (AN) of [2]+ on the scale developed by Gutmann and Beckett.35 To this end, mixtures of [2][B(C6F5)4] and Et3PO were prepared in CD2Cl2 and 31P{1H} NMR spectra were recorded (see the ESI† for full details). We consistently observed a resonance at δ(31P) = 91.0 ppm for the adduct [2·OPEt3][B(C6F5)4]. The signal is shifted to lower field by 40.7 ppm and 50.0 ppm, compared to the resonance of pure Et3PO in CD2Cl2 (50.3 ppm) or n-hexane (41.0 ppm), respectively. From these data, an acceptor number AN([2]+) = 111 was calculated, which is significantly higher than the acceptor number of B(C6F5)3 (AN = 77–82; CD2Cl2), higher than the acceptor numbers of so far reported Fc-borenium ions (cf. Table S7 in ESI†), and close to that of neat BBr3 (AN = 109) or BI3 (AN = 115).36The pyridine adduct [2·py][B(C6F5)4] was synthesized on a preparative scale and characterized by NMR-spectroscopy and X-ray crystallography (Scheme 2). Five 1H and five 13C resonances are observed for the pyridine ligand in the corresponding room-temperature NMR spectra, demonstrating a sterically restricted rotation about the B–N(py) bond on the NMR time scale (CD2Cl2). The p-CH group of the coordinated pyridine molecule should be least affected by magnetic anisotropy effects originating from the surrounding π-electron clouds. Its continuous deshielding upon going from free pyridine (δ(1H)/δ(13C) = 7.67/136.1 ppm)11b to B(C6F5)3·py (8.21/143.2 ppm)11b and [2·py][B(C6F5)4] (8.56/147.8 ppm) therefore indicates increasing charge polarization induced by increasingly strong N–B donor–acceptor binding (cf. [Hpy]Cl: 8.51/146.1 ppm (ref. 11b)). Consistent with this, the N(py)–B bond in [2·py][B(C6F5)4] (1.603(3) Å) is shorter than that in B(C6F5)3·py (1.614(2) Å,37 1.628(2) Å (ref. 38)), but identical to the N–B bond in the adducts [B·py][O3SCF3] (1.599(4) Å (ref. 11b)) and B(C6F5)3·DMAP, which contains the stronger donor p-dimethylaminopyridine (1.602(6) Å (ref. 39)).
 | ||
| Scheme 2 Synthesis and solid-state structure of the pyridine adduct [2·py][B(C6F5)4]; C-bonded H atoms and the [B(C6F5)4]− anion are omitted for clarity. Reagents and conditions: (i) CHCl3, room temperature. R = C6F5. | ||
We also performed competition experiments using the combinations [2·Do][B(C6F5)4]/B(C6F5)3 and [2][B(C6F5)4]/B(C6F5)3·Do in CD2Cl2 (Do = Et3PO or pyridine). Irrespective of the ligand employed, we found that the transfer of Do from B(C6F5)3 to [2]+ was faster and occurred to a much higher extent than vice versa, which clearly points to a higher Lewis acidity of [2]+ compared to B(C6F5)3 (see the ESI† for plots of corresponding NMR spectra). A quantitative determination of exchange-equilibrium constants was not possible, because [2][B(C6F5)4] is not stable over the long term toward B(C6F5)3. An observed slow increase in the amount of B(C6F5)3·Do over the course of several days may therefore well be preceded by a slow B(C6F5)3-induced degradation of [2]+/[2·Do]+ and subsequent trapping of the released ligand by B(C6F5)3.
To confirm the exceptionally strong Lewis acidity of [2]+ indicated by the above experiments, we compared the Et3PO affinities and Gutmann–Beckett acceptor numbers for [2]+/[2Me]+, the Ru/Os congeners of [2]+, a series of structurally related neutral/ionic Fc–B Lewis acids, and variously substituted boranes also on the basis of quantum-chemical calculations (Table S7 and Fig. S67, S68†). When taking into account the solvation and counterion effects (where appropriate), we observe a very good correlation between experimental and computed 31P NMR shifts for a range of 28 Lewis acid adducts with Et3PO (Table S7 and Fig. S67†); the linear regression is improved almost to perfection (R2 = 0.991) when the data points for the boron trihalides BX3 are omitted (X = F, Cl, Br, I). In accordance with the experimental NMR studies, the DFT calculations indicate that [2]+ is one of the strongest Lewis acids among hitherto reported boron-containing species.
Given that the assessment of Lewis acidity based on the Gutmann–Beckett method is sometimes under criticism, in particular when comparing the Lewis acidity of ionic and neutral borane species,40 we also calculated the F−-ion affinities (FIAs) for the above series of Lewis acids (Table S9†). The calculations were performed along the lines of Christe's method using F2CO as the standard, which avoids the theoretical treatment of free F− in the gas phase.41,42 Fig. S69 and S70† show that there is a reasonably good correlation between the computed FIAs and the 31P NMR shifts, acceptor numbers, and Et3PO affinities for all compounds studied. Thus, also according to the FIAs, [2]+ is one of the strongest Lewis acids among the boron-containing systems: Apart from Ingleson's transient [CatB][CbBr6] (Cat = catecholato, [CbBr6]− = [closo-1-H-CB11H5Br6]−),43 [2]+ outperforms all neutral and ionic Fc–B species as well as variously substituted boranes and borenium ions, including Corey's oxazaborolidinium cations [C]+,12 and Jäkle's borenium Lewis acids.40 A borderline case is the hypothetical free 9-phosphonium-10-boratriptycene cation, which could not yet be isolated (cf. the structural motif of D featuring a [PH]+ rather than a CH group at the bridgehead position; Fig. 1):17a while the computed Gutmann–Beckett acceptor number of [2]+ is higher than that of 9-phosphonium-10-boratriptycene (ANcalcd = 110 vs. 83; Table S7†), the FIA of [2]+ is lower (FIA = 396 vs. 474 kJ mol−1; Table S9†).
We finally note that replacing the Fe(II) center in [2]+ with Ru(II) or Os(II) leads to more shielded 31P nuclei in corresponding Et3PO adducts, which indicates a reduced Lewis acidity of the B+ centers, consistent with stronger M → B donation (see above).
Optoelectronic properties of 1/[2][B(C6F5)4] and 1Me/[2Me][B(C6F5)4]
Cyclic voltammograms and UV/vis spectra were recorded in CH2Cl2 at room temperature. The Fe(II)/Fe(III) transitions of 1 and 1Me occur at halve-wave potentials of E1/2 = −0.23 V and −0.32 V, respectively (vs. FcH/FcH+). Their 1,1′-fc moieties are therefore easier to oxidize than the reference compound ferrocene, indicating a net electron-donating effect of the two tetracoordinated B substituents. Jäkle et al. reported on a pronounced Lewis acidity enhancement of neutral dibora-diferrocenes upon oxidation of their 1,2-ferrocenylene moieties.44 In a similar vein, we have also attempted the Fe-centered oxidation of [2][B(C6F5)4] and [2Me][B(C6F5)4] with the goal of generating dicationic borenium ions, but have not been successful so far. The longest-wavelength absorptions of 1 and 1Me in CH2Cl2 are broad and appear at λmax = 451 nm, near the most bathochromic band in the UV-vis spectrum of parent ferrocene (441 nm).45 The experimentally determined absorption maximum is well reproduced by TD-DFT calculations on 1, which predict a transition at λmax(calc) = 447 nm (fOSC = 0.001), attributable to a charge transfer from the 1,1′-fc unit to the pyrazole rings (HOMO−1 → LUMO). [2][B(C6F5)4] and [2Me][B(C6F5)4] show their longest-wavelength absorptions at λmax = 378 and 391 nm, respectively (CH2Cl2; Fig. 4). According to TD-DFT calculations, the first absorption peak of [2]+ at λmax = 378 nm (λmax(calc) = 383 nm; fOSC = 0.004) is mainly associated with a charge transfer from Fe(II) d-orbitals (HOMO−2) to the Cp rings (LUMO+1). The second intense peak at 309 nm (λmax(calc) = 317 nm; fOSC = 0.082) can be attributed to an Fe(II) → B2pz2 (HOMO−2 → LUMO) transfer (Fig. 4). However, the transferred electron density accumulates predominantly at the pz moieties, and the charge at the B+ center does not change much upon excitation (Table S12†) due to the delocalized nature of the corresponding MOs (ground-state [2]+: q(B(2p)) = 0.90 e−, excited-state [2]+: q(B(2p)) = 0.91 e−). | ||
| Fig. 4 Top: experimentally determined UV/vis absorption spectrum of [2][B(C6F5)4] in CH2Cl2. Bottom: computed key molecular orbitals of [2]+ and lowest energy electronic transitions (TD-DFT PBE0-10HF/def2-TZVP/CH2Cl2). | ||
Conclusions
By exploiting the facile self-assembly of the pyrazabole scaffold, we have devised a straightforward route to boratriptycene-type precursors that have two pyrazole bridges and one 1,1′-ferrocenylene linker between two bridgehead BH moieties (1, 1Me). H− abstraction with the [Ph3C]+ cation generates bridgehead borenium cations of exceptional Lewis acidity that can nevertheless be isolated in free form ([2][B(C6F5)4], [2Me][B(C6F5)4]). A weak Fe–B(sp2) through-space interaction likely assists in stabilizing the systems without appreciably diminishing their electron-pair affinities. Although the borenium centers of [2]+ and [2Me]+ adopt trigonal-planar configurations (X-ray crystallography, DFT calculations), huge dips of the B atoms with respect to their attached Cp rings indicate pretensioned ‘entatic’ states of the molecules,46 which should significantly promote Lewis acid–base pairing. Since a plethora of pyrazole derivatives are available, the degree of steric protection of the electrophilic borenium center can be varied easily (cf. [2]+ with pyrazole bridges vs. [2Me]+ with bulky 3,5-dimethylpyrazole bridges). By using chiral substituents in the 3,5-positions of the pyrazole rings, even chiral Lewis acids should be within reach. As an outlook, we note that an exchange of Fe for Os would lead to a related Lewis acid that is computed to exhibit a distinct Os(III)/B˙ character in its relaxed triplet excited state and thus could open new avenues for exploring the evolving chemistry of in situ-generated boron radicals.47Data availability
Experimental and computational data associated with this article have been provided in the ESI.†Author contributions
M. H. synthesized and characterized all compounds. J. N. and P. H. performed all quantum-chemical calculations. A. O. and V. S. recorded and interpreted the Mössbauer spectra. M. B. performed the X-ray crystal structure analyses. H.-W. L. and M. W. supervised the project. The manuscript was written by M. W. and P. H. and edited by all the co-authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. W. and M. H. thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for financial support (WA 864/6-1). V. S. acknowledges the support of the DFG through TRR 88 142808194 3MET. J. N. and P. H. acknowledge the support by the Slovak Research and Development Agency (grant no. APVV-17-0324), the Grant Agency of the Ministry of Education of the Slovak Republic (VEGA project no. 1/0712/18), and by the European Union's Horizon 2020 research and innovation programme under grant no. 810701 (LAMatCU).Notes and references
- (a) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC; (b) E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed; (c) M. Bochmann, S. J. Lancaster, M. D. Hannant, A. Rodriguez, M. Schormann, D. A. Walker and T. J. Woodman, Pure Appl. Chem., 2003, 75, 1183–1195 CAS; (d) G. Erker, Chem. Commun., 2003, 1469–1476 RSC; (e) W. E. Piers, Adv. Organomet. Chem., 2005, 52, 1–76 CAS; (f) F. Jäkle, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, R. H. Crabtree, C. M. Lukehart, D. A. Atwood and R. A. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed; (b) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (c) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed; (d) A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef CAS.
- (a) L. A. Mück, A. Y. Timoshkin and G. Frenking, Inorg. Chem., 2012, 51, 640–646 CrossRef PubMed; (b) A. Y. Timoshkin and K. Morokuma, Phys. Chem. Chem. Phys., 2012, 14, 14911–14916 RSC; (c) T. M. Gilbert, Dalton Trans., 2012, 41, 9046–9055 RSC; (d) E. I. Davydova, T. N. Sevastianova and A. Y. Timoshkin, Coord. Chem. Rev., 2015, 297–298, 91–126 CrossRef CAS.
- (a) P. A. Chase, W. E. Piers and B. O. Patrick, J. Am. Chem. Soc., 2000, 122, 12911–12912 CrossRef CAS; (b) C. Fan, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2009, 48, 2955–2958 CrossRef CAS PubMed; (c) A. Y. Houghton, V. A. Karttunen, W. E. Piers and H. M. Tuononen, Chem. Commun., 2014, 50, 1295–1298 RSC; (d) A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed; (e) J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC.
- (a) A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS; (b) A. G. Massey and A. J. Park, J. Organomet. Chem., 1966, 5, 218–225 CrossRef CAS.
- A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- (a) J. Landmann, F. Keppner, D. B. Hofmann, J. A. P. Sprenger, M. Häring, S. H. Zottnick, K. Müller-Buschbaum, N. V. Ignat'ev and M. Finze, Angew. Chem., Int. Ed., 2017, 56, 2795–2799 CrossRef CAS PubMed; (b) E. L. Bennett, E. J. Lawrence, R. J. Blagg, A. S. Mullen, F. MacMillan, A. W. Ehlers, D. J. Scott, J. S. Sapsford, A. E. Ashley, G. G. Wildgoose and J. C. Slootweg, Angew. Chem., Int. Ed., 2019, 58, 8362–8366 CrossRef CAS PubMed.
- (a) T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed; (b) H. Zhao, L. A. Leamer and F. P. Gabbaï, Dalton Trans., 2013, 42, 8164–8178 RSC.
- Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 3363–3369 CrossRef CAS PubMed.
- For selected examples of conceptually related positively charged boron Lewis acids, see: (a) C. Dusemund, K. R. A. Samankumara Sandanayake and S. Shinkai, J. Chem. Soc., Chem. Commun., 1995, 333–334 RSC; (b) C.-W. Chiu and F. P. Gabbaï, J. Am. Chem. Soc., 2006, 128, 14248–14249 CrossRef CAS PubMed; (c) T. Agou, J. Kobayashi and T. Kawashima, Inorg. Chem., 2006, 45, 9137–9144 CrossRef CAS PubMed; (d) T. W. Hudnall and F. P. Gabbaï, J. Am. Chem. Soc., 2007, 129, 11978–11986 CrossRef CAS PubMed; (e) M. H. Lee, T. Agou, J. Kobayashi, T. Kawashima and F. P. Gabbaï, Chem. Commun., 2007, 1133–1135 RSC; (f) K. Venkatasubbaiah, I. Nowik, R. H. Herber and F. Jäkle, Chem. Commun., 2007, 2154–2156 RSC; (g) G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407–3414 RSC; (h) J. K. Day, C. Bresner, N. D. Coombs, I. A. Fallis, L.-L. Ooi and S. Aldridge, Inorg. Chem., 2008, 47, 793–804 CrossRef CAS PubMed; (i) T. W. Hudnall, Y. M. Kim, M. W. Bebbington, D. Bourissou and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 10890–10891 CrossRef CAS PubMed; (j) Y. Kim, H. Zhao and F. P. Gabbaï, Angew. Chem., Int. Ed., 2009, 48, 4957–4960 CrossRef CAS PubMed; (k) D. Cao, H. Zhao and F. P. Gabbaï, New J. Chem., 2011, 35, 2299–2305 RSC.
- (a) A. Schnurr, H. Vitze, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 6012–6019 CrossRef CAS; (b) A. Schnurr, M. Bolte, H.-W. Lerner and M. Wagner, Eur. J. Inorg. Chem., 2012, 2012, 112–120 CrossRef CAS.
- (a) E. J. Corey, T. Shibata and T. W. Lee, J. Am. Chem. Soc., 2002, 124, 3808–3809 CrossRef CAS PubMed; (b) D. H. Ryu, T. W. Lee and E. J. Corey, J. Am. Chem. Soc., 2002, 124, 9992–9993 CrossRef CAS PubMed; (c) G. Zhou and E. J. Corey, J. Am. Chem. Soc., 2005, 127, 11958–11959 CrossRef CAS PubMed.
- (a) A. Chardon, A. Osi, D. Mahaut, A. Ben Saida and G. Berionni, Synlett, 2020, 31, 1639–1648 CrossRef CAS; (b) K. T. Giju, A. K. Phukan and E. D. Jemmis, Angew. Chem., Int. Ed., 2003, 42, 539–542 CrossRef CAS PubMed.
- B. M. Mikhailov, Pure Appl. Chem., 1983, 55, 1439–1452 CAS.
- (a) S. Y. Erdyakov, A. V. Ignatenko, T. V. Potapova, K. A. Lyssenko, M. E. Gurskii and Y. N. Bubnov, Org. Lett., 2009, 11, 2872–2875 CrossRef CAS PubMed; (b) M. E. Gurskii, G. D. Kolomnikova, S. V. Baranin and Y. N. Bubnov, Mendeleev Commun., 2018, 28, 366–368 CrossRef CAS.
- A. Chardon, A. Osi, D. Mahaut, T.-H. Doan, N. Tumanov, J. Wouters, L. Fusaro, B. Champagne and G. Berionni, Angew. Chem., Int. Ed., 2020, 59, 12402–12406 CrossRef CAS PubMed.
- For D-type compounds containing (a) a P atom or (b) an S+ cation instead of the CH group at the second bridgehead position, see: (a) A. Ben Saida, A. Chardon, A. Osi, N. Tumanov, J. Wouters, A. I. Adjieufack, B. Champagne and G. Berionni, Angew. Chem., Int. Ed., 2019, 58, 16889–16893 CrossRef CAS PubMed; (b) A. Osi, D. Mahaut, N. Tumanov, L. Fusaro, J. Wouters, B. Champagne, A. Chardon and G. Berionni, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202112342 . For related Lewis base adducts of 1-borabarrelenes, see:; (c) T. K. Wood, W. E. Piers, B. A. Keay and M. Parvez, Org. Lett., 2006, 8, 2875–2878 CrossRef CAS PubMed.
- Y. V. Vishnevskiy, M. A. Abaev, A. N. Rykov, M. E. Gurskii, P. A. Belyakov, S. Y. Erdyakov, Y. N. Bubnov and N. W. Mitzel, Chem.–Eur. J., 2012, 18, 10585–10594 CrossRef CAS PubMed.
- (a) B. Wrackmeyer, W. Milius, O. L. Tok and Y. N. Bubnov, Chem.–Eur. J., 2002, 8, 1537–1543 CrossRef CAS PubMed; (b) C. E. Wagner, J.-S. Kim and K. J. Shea, J. Am. Chem. Soc., 2003, 125, 12179–12195 CrossRef CAS PubMed.
- (a) A. Appel, F. Jäkle, T. Priermeier, R. Schmid and M. Wagner, Organometallics, 1996, 15, 1188–1194 CrossRef CAS; (b) K. Ma, M. Scheibitz, S. Scholz and M. Wagner, J. Organomet. Chem., 2002, 652, 11–19 CrossRef CAS; (c) M. Scheibitz, M. Bolte, J. W. Bats, H.-W. Lerner, I. Nowik, R. H. Herber, A. Krapp, M. Lein, M. C. Holthausen and M. Wagner, Chem.–Eur. J., 2005, 11, 584–603 CrossRef CAS PubMed; (d) L. Kaufmann, H. Vitze, M. Bolte, H. W. Lerner and M. Wagner, Organometallics, 2008, 27, 6215–6221 CrossRef CAS; (e) U. D. Eckensberger, K. Kunz, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2008, 27, 764–768 CrossRef CAS; (f) H. Braunschweig, I. Fernández, G. Frenking and T. Kupfer, Angew. Chem., Int. Ed., 2008, 47, 1951–1954 CrossRef CAS PubMed; (g) H. Braunschweig, C.-W. Chiu, D. Gamon, M. Kaupp, I. Krummenacher, T. Kupfer, R. Müller and K. Radacki, Chem.–Eur. J., 2012, 18, 11732–11746 CrossRef CAS PubMed.
- (a) M. Fontani, F. Peters, W. Scherer, W. Wachter, M. Wagner and P. Zanello, Eur. J. Inorg. Chem., 1998, 1998, 1453–1465 CrossRef; (b) M. Grosche, E. Herdtweck, F. Peters and M. Wagner, Organometallics, 1999, 18, 4669–4672 CrossRef CAS; (c) M. Scheibitz, J. W. Bats, M. Bolte and M. Wagner, Eur. J. Inorg. Chem., 2003, 2003, 2049–2053 CrossRef.
- (a) F. Jäkle, T. Priermeier and M. Wagner, J. Chem. Soc., Chem. Commun., 1995, 1765–1766 RSC; (b) F. Jäkle, M. Mattner, T. Priermeier and M. Wagner, J. Organomet. Chem., 1995, 502, 123–130 CrossRef; (c) E. Herdtweck, F. Jäkle, G. Opromolla, M. Spiegler, M. Wagner and P. Zanello, Organometallics, 1996, 15, 5524–5535 CrossRef CAS; (d) F. Jäkle, T. Priermeier and M. Wagner, Organometallics, 1996, 15, 2033–2040 CrossRef; (e) E. Herdtweck, F. Jäkle and M. Wagner, Organometallics, 1997, 16, 4737–4745 CrossRef CAS.
- For review articles on borocations, see: (a) D. Franz and S. Inoue, Chem.–Eur. J., 2019, 25, 2898–2926 CrossRef CAS PubMed; (b) M. J. Ingleson, in Topics in Organomet. Chem., Springer, 2015, vol. 49, pp. 39–71 Search PubMed; (c) T. S. de Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef CAS PubMed; (d) W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed.
- For selected adducts of cage-like boron Lewis acids that have been prepared from tripodal triamido or triaryloxy ligands to establish BN3 or BO3 environments, see: (a) M. Yasuda, S. Yoshioka, S. Yamasaki, T. Somyo, K. Chiba and A. Baba, Org. Lett., 2006, 8, 761–764 CrossRef CAS PubMed; (b) H. Zhu and E. Y. Chen, Inorg. Chem., 2007, 46, 1481–1487 CrossRef CAS PubMed; (c) H. Nakajima, M. Yasuda, R. Takeda and A. Baba, Angew. Chem., Int. Ed., 2012, 51, 3867–3870 CrossRef CAS PubMed; (d) A. Konishi, K. Nakaoka, H. Nakajima, K. Chiba, A. Baba and M. Yasuda, Chem.–Eur. J., 2017, 23, 5219–5223 CrossRef CAS PubMed.
- Boron compounds of the forms (C6F5)nB(OC6F5)3−n (n = 0–2) are also known. Their Lewis acidities with respect to Et3PO increase as n decreases, which is due to the negative inductive effect of the electronegative O atoms. In these particular cases, this −I effect is not compensated by strong OB π-backbonding, since the oxygen-bonded C6F5 substituents themselves have a strong π-electron withdrawing character:
(a) D. Naumann, H. Butler and R. Gnann, Z. Anorg. Allg. Chem., 1992, 618, 74–76 CrossRef CAS;
(b) G. J. P. Britovsek, J. Ugolotti and A. J. P. White, Organometallics, 2005, 24, 1685–1691 CrossRef CAS.
- M. Scheibitz, H. Li, J. Schnorr, A. Sánchez Perucha, M. Bolte, H.-W. Lerner, F. Jäkle and M. Wagner, J. Am. Chem. Soc., 2009, 131, 16319–16329 CrossRef CAS PubMed.
- Comparable hydride-abstraction reactions from pentamethylazaferrocene–borane adducts have been described: B. Bentivegna, C. I. Mariani, J. R. Smith, S. Ma, A. L. Rheingold and T. J. Brunker, Organometallics, 2014, 33, 2820–2830 CrossRef CAS.
- A. Iida, A. Sekioka and S. Yamaguchi, Chem. Sci., 2012, 3, 1461–1466 RSC.
-
(a) B. Wrackmeyer, Z. Anorg. Allg. Chem., 2015, 641, 2525–2529 CrossRef CAS;
(b) B. Wrackmeyer, B. Schwarze and W. Milius, Inorg. Chim. Acta, 1996, 241, 87–93 CrossRef CAS . The conclusion that the B–N bonds in this compound have essentially single-bond character is based on the following considerations (i) the electron lone pairs on the N atoms contribute mainly to the heteroaromatic systems, which would be perturbed by BN π bonding, (ii) the Me2pyr rings are strongly twisted against the BN3 plane (dihedral angles = 36.7°, 48.5°, 50.8°), (iii) three nitrogen lone pairs would compete for one vacant boron 2p orbital, which necessarily reduces any double-bond character of the individual B–N bonds..
- H. Nöth and B. Wrackmeyer, Nuclear Magnetic Resonance Spectroscopy of Boron Compounds, in NMR – Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer, Berlin, Heidelberg, New York, 1978 Search PubMed.
- T. Renk, W. Ruf and W. Siebert, J. Organomet. Chem., 1976, 120, 1–25 CrossRef CAS.
- K. Müther, P. Hrobárik, V. Hrobáriková, M. Kaupp and M. Oestreich, Chem.–Eur. J., 2013, 19, 16579–16594 CrossRef PubMed.
- (a) D. F. Shriver, Acc. Chem. Res., 1970, 3, 231–238 CrossRef CAS; (b) H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., Int. Ed., 2006, 45, 5254–5274 CrossRef CAS PubMed; (c) A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859–871 RSC; (d) H. Braunschweig and R. D. Dewhurst, Dalton Trans., 2011, 40, 549–558 RSC; (e) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed.
- (a) J. S. Figueroa, J. G. Melnick and G. Parkin, Inorg. Chem., 2006, 45, 7056–7058 CrossRef CAS PubMed; (b) M. Sircoglou, S. Bontemps, G. Bouhadir, N. Saffon, K. Miqueu, W. Gu, M. Mercy, C.-H. Chen, B. M. Foxman, L. Maron, O. V. Ozerov and D. Bourissou, J. Am. Chem. Soc., 2008, 130, 16729–16738 CrossRef CAS PubMed.
- (a) V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS; (b) M. A. Beckett, D. S. Brassington, S. J. Coles and M. B. Hursthouse, Inorg. Chem. Commun., 2000, 3, 530–533 CrossRef CAS.
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- K. Tanifuji, S. Tajima, Y. Ohki and K. Tatsumi, Inorg. Chem., 2016, 55, 4512–4518 CrossRef CAS PubMed.
- F. Focante, P. Mercandelli, A. Sironi and L. Resconi, Coord. Chem. Rev., 2006, 250, 170–188 CrossRef CAS.
- M. J. G. Lesley, A. Woodward, N. J. Taylor, T. B. Marder, I. Cazenobe, I. Ledoux, J. Zyss, A. Thornton, D. W. Bruce and A. K. Kakkar, Chem. Mater., 1998, 10, 1355–1365 CrossRef CAS.
- (a) J. Chen, R. A. Lalancette and F. Jäkle, Chem. Commun., 2013, 49, 4893–4895 RSC; (b) P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202114550.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151–153 CrossRef CAS.
- The inclusion of an implicit solvation model is necessary to obtain reliable FIAs when comparing neutral and ionic boron-based Lewis acids in condensed (solution) phase to reflect the experimentally observed trends (the gas-phase results overestimate the Lewis acidities of cationic species due to unshielded Coulomb attraction and charge-effect contributions; Tables S8 vs. S9†)..
- A. Del Grosso, R. G. Pritchard, C. A. Muryn and M. J. Ingleson, Organometallics, 2010, 29, 241–249 CrossRef CAS.
- K. Venkatasubbaiah, L. N. Zakharov, W. S. Kassel, A. L. Rheingold and F. Jäkle, Angew. Chem., Int. Ed., 2005, 44, 5428–5433 CrossRef CAS PubMed.
- H. B. Gray, Y. S. Sohn and N. Hendrickson, J. Am. Chem. Soc., 1971, 93, 3603–3612 CrossRef.
- (a) B. L. Vallee and R. J. P. Williams, Proc. Natl. Acad. Sci. U. S. A., 1968, 59, 498–505 CrossRef CAS PubMed; (b) R. J. P. Williams, Eur. J. Biochem., 1995, 234, 363–381 CrossRef CAS PubMed.
- (a) J. T. Henthorn and T. Agapie, Angew. Chem., Int. Ed., 2014, 53, 12893–12896 CrossRef CAS PubMed; (b) L. L. Liu, L. L. Cao, Y. Shao and D. W. Stephan, J. Am. Chem. Soc., 2017, 139, 10062–10071 CrossRef CAS PubMed; (c) X. Tao, C. G. Daniliuc, R. Knitsch, M. R. Hansen, H. Eckert, M. Lübbesmeyer, A. Studer, G. Kehr and G. Erker, Chem. Sci., 2018, 9, 8011–8018 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, computational, and crystallographic data. CCDC 2120063–2120067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06404e |
| This journal is © The Royal Society of Chemistry 2022 |