
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic polymer nanomaterials for the production of high value compounds
Julian
Heuer
and
Calum T. J.
Ferguson
 *
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: ferguson@mpip-mainz.mpg.de
First published on 7th January 2022
Abstract
Nanotechnology has provided a platform for producing new photocatalytic materials, where the reduction in length scales has been used to amplify the efficiency of these light active materials. The progression to nano-based photocatalysts has been driven by the increase in surface area that is achieved. Furthermore, nanophotocatalysts based on porous polymers or gel materials are often more active as reagents can more easily partition across the whole photocatalyst. Here, reducing the diffusional path length for substrates across the porous/gel material increases the quantity of accessible active sites in the photocatalytic material. The formation of nanophotocatalytic materials has also enabled the formation of functional nanoparticles that can be used in different conditions traditionally inaccessible to bulk catalysts. Specifically, aqueous compatible nanophotocatalytic materials have been reported, enabling greener reaction conditions and new applications of photocatalysts.
 Julian Heuer | Julian Heuer received his B.Sc. in Chemistry in 2018 at the University of Münster, focusing on the insitu polymerization of electrolytes for lithium-ion batteries. He then undertook his M.Sc. in Münster, working on supramolecular, thermoresponsive nanocarriers in the Ravoo group. Since December 2020 he is pursuing his Ph.D. the department of Physical Chemistry of Polymers at the Max-Planck-Institute for Polymer Research working with Dr. Calum T. J. Ferguson and Prof. Katharina Landfester. His research interests are focused on selective photocatalysis, biomimetic materials and radical polymer synthesis. |
 Calum T. J. Ferguson | Calum Ferguson undertook an integrated Masters in Chemistry at The University of Edinburgh. He then attained his PhD from the University of Leeds, UK, in 2018. After the completion of his doctoral studies, he joined the department of Physical Chemistry of Polymers at the Max Planck Institute for Polymer Research (MPIP), working with Prof. Katharina Landfester. Early 2020, he started as a research group leader at MPIP. His research interests include controlled radical polymer synthesis, photocatalytic classical polymers, organic small molecule photocatalysts, and the formation of bio-mimicking polymers and colloids. |
Introduction
Sunlight is a clean, renewable energy source that can be harnessed to catalyse chemical reactions. Increasingly, the use of this energy source has been targeted as a cleaner, more environmentally friendly alternative to thermal energy. The emergence of photocatalytic materials has facilitated this shift. Mimicking natural processes such as photosynthesis, these materials utilise solar energy to enable chemical reactions to proceed. Initially, organo-metallic photocatalytic complexes, mainly based on iridium or ruthenium were investigated for mediation of photocatalytic organic reactions.1,2 Recently, a shift towards exclusively organic compounds has been observed. Furthermore, in order to enhance the recyclability of photocatalytic materials there has been a shift towards heterogeneous photocatalysts. These materials are often bulk polymeric materials that can be easily recycled, through centrifugation, cleaned and reused. Typically, conjugated donor–acceptor polymers are used as photocatalytic materials, with adjustable molecular orbitals and physical properties.3,4 Furthermore, the incorporation of small molecule photocatalysts into classical polymers has recently been reported as new heterogeneous photocatalytic systems.5Photocatalytic systems use visible light to enable a broad range of chemical reactions to proceed including water splitting,6–9 CO2 reduction,10–13 C–C coupling reactions,14,15 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage,16,17 pericyclic reactions,18,19 metal reduction,20,21 enzyme co-factor activation,22,23 trifluoromethylation of arenes,24 oxidation of sulfides,25,26 dehalogenation of haloketones,27 heterocycle formation28 and enantioselective alpha-alkylation.29 These reactions proceed through the formation of photogenerated radical species within the photocatalytic material. The lifetime of these active species varies between photocatalytic materials and in the reaction conditions, but the highest concentration of these active agents is found at the photocatalytic centre. Therefore, for efficient photocatalytic conversions the reagents must be in close proximity of the photocatalytic material.
C bond cleavage,16,17 pericyclic reactions,18,19 metal reduction,20,21 enzyme co-factor activation,22,23 trifluoromethylation of arenes,24 oxidation of sulfides,25,26 dehalogenation of haloketones,27 heterocycle formation28 and enantioselective alpha-alkylation.29 These reactions proceed through the formation of photogenerated radical species within the photocatalytic material. The lifetime of these active species varies between photocatalytic materials and in the reaction conditions, but the highest concentration of these active agents is found at the photocatalytic centre. Therefore, for efficient photocatalytic conversions the reagents must be in close proximity of the photocatalytic material.
Multiple strategies have been implemented to maximise reagent and photocatalytic proximity, most notably by increasing the surface area of photocatalytic materials. The emergence of porous organic polymers (POP) and the subsequent utilisation of this material class for photocatalysis has yielded high surface area photocatalysts. Conjugated mesoporous polymers (CMP) and subsequently covalent triazine frameworks (CTF) have been widely reported for visible light mediated photocatalysis. These bulk systems are large conjugated networks with defined nanostructures, maximising the surface area, therefore, allowing diffusion into the network. However, as these materials are often in the 100's of μm to mm range, the mass transport across all of the photocatalytic material is often a challenge. Bulk gel photocatalytic materials have been reported that have photocatalytic sites incorporated into the gel structure.18 These gel networks allow for the diffusion of reagents into the networks, enabling their catalytic conversion. Similarly, to conjugated porous polymers, mass-transfer is once again a challenge limiting these materials. Recently, the production of photocatalytic nanomaterials has been investigated as a method to produce heterogeneous photocatalysts that require shorter diffusional path lengths.
With this review we wish to highlight an emerging class of photocatalytic nanomaterials. Despite its young age, several impressive examples have been published, underlining the demand for a summary. Discussing porous polymers, followed by polymeric gels and ending with self-assembled nanoparticles, a broad spectrum of different photocatalytic nanomaterials is covered in this review, visualising their synthesis, benefits and possible applications.
Porous photocatalytic nanomaterials
Nanomaterials have been used to maximise the surface area of photocatalytic materials. Further enhancement of total surface area has also been achieved by the utilisation of porous structures. Initially bulk porous photocatalysts were produced, comprised of a highly cross-linked aromatic 3D network.3,4 These porous polymers offer several advantages over their solid analogues including higher surface-to-volume ratio, easy synthesis, high tunability of the donor–acceptor structure, recyclability, and increased turnover rate for catalytic reactions.3,30–32A prominent class of porous nanomaterials are conjugated microporous polymers (CMPs). These CMPs comprise a π-conjugated backbone, featuring electron density donor- and electron density acceptor-type monomers.33 CMPs are synthesised in a one-pot procedure, where the composition of the CMP is controlled by the monomer-types and their molar ratios. This flexibility in synthesis has allowed generation of a wide-range of CMPs over the last decade, targeting different applications. CMPs represent well-established materials in the fields of organic synthesis,16 CO2 reduction and H2O splitting.34
Recently, nano-based CMP systems have been reported that further enhance the photocatalysts surface area.35 Moreover, due to the nanosize of these materials, the diffusion length of substrates into photocatalytic material is reduced, creating higher performing systems. Nano-CMPs are typically formed using a mini-emulsion strategy where the conjugated polymers are formed within droplets and are dispersed in water. This dispensability in water has enabled the shift to undertaking photocatalytic reaction in water, a greener solvent. Ma et al. produced a series of nanomaterials by either Suzuki–Miyaura or Sonogashira–Hagihara cross-coupling polycondensation reactions in an oil-in-water mini-emulsion, followed by solvent evaporation.36 Spherical, rod and ring shaped nanomaterials were produced depending on the monomers selected, where increasing the concentration of benzothiadiazole within the polymer led to non-spherical structures. Here, nano-CMPs could be used for rapid photocatalytic degradation of rhodamine B, with ring shaped nano-CMPs being the most efficient. It is unclear whether the morphology of the nano-CMP or the polymer composition plays a more important role in photocatalytic efficiency.
Ma et al. also investigated the use of CMP nanoparticles for the regeneration of the cofactor NAD+. Via oil-in-water mini-emulsion polymerisation, triethynylbenzene and oxadiazole components were polymerized, yielding 50–80 nm spherical, microporous CMP nanoparticles. With encapsulation of these nanoparticles into giant polymer vesicles, an effective nanoreactor was established, which could be used in combination with modified enzymes for photobiocatalysis.38
Finally, Ma et al. investigated the use of CMP nanoparticles for use in photodynamic therapy (PDT). Here, the aqueous compatibility of the nanomaterials enabled the eradication of bacteria using non-toxic and pure organic systems. Both Escherichia coli K-12 and Bacillus subtilis where inactivated by photogenerated singlet oxygen (Fig. 1).37 In addition to CMPs, covalent triazine frameworks (CTFs) are an interesting class of porous polymers. Similarly to CMPs, CTFs are porous networks formed from an aromatic backbone, but here, triazine donor units are used.39 Typically, cyanide containing monomers undergo a one-pot trimerization reaction to form triazine groups, allowing for tunability via monomer selection.40–42
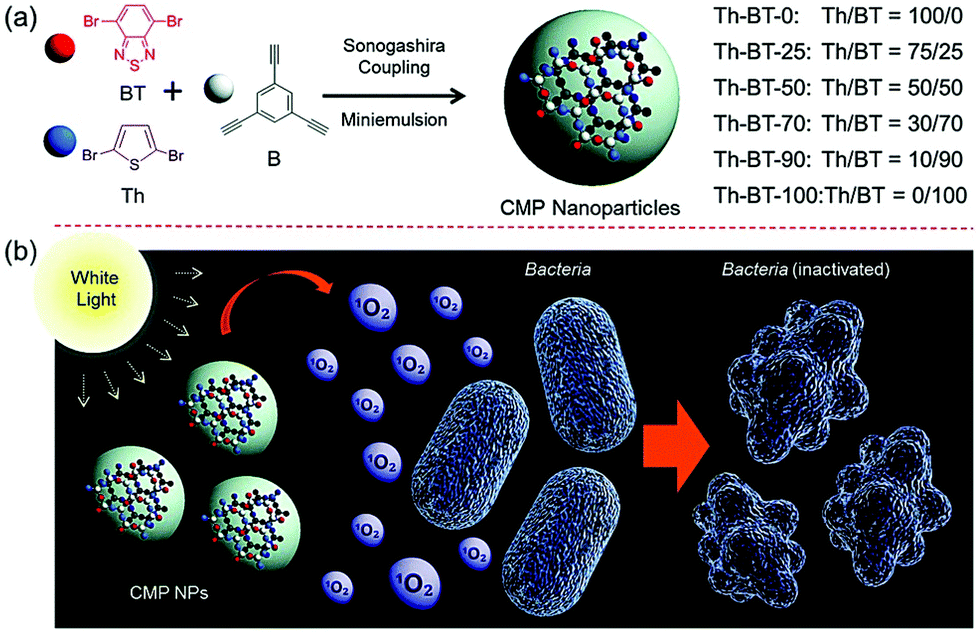 | ||
| Fig. 1 (a) Synthesis route for the production of photocatalytic CMP nanoparticles by Sonogashira–Hagihara cross-couplings. (b) Bacterial deactivation by active oxygen. Reproduced from ref. 37 with permission from Royal Society of Chemistry, copyright 2016. | ||
The production of CTF materials was initially undertaken using conventional liquid-phase approaches in molten ZnCl243,44 or trifluoromethanesulfonic acid (TfOH).45 Unfortunately, these methods do not produce materials with regular morphologies and defined optical properties. Huang et al. have recently reported a size-controllable confinement synthesis of CTF nanoparticles. Since the synthesis of CTFs usually occurs in an ionothermal procedure, with harsh reaction conditions, formation of controlled CTF nanoparticles with defined morphologies has not been possible. In a combined sol–gel emulsion, assisted by TfOH vapour, confined silica templates were used to encapsulate a thiophene-derived dinitrile monomer (Fig. 2). After polymerization of these monomers, the silica template was removed using an etching procedure, giving CTF-NP with a discrete particle size, confirmed by BET, TEM and EDX measurements. With an additional incorporation of an electron-withdrawing benzothiadiazole group into the polymer, an increased charge-transfer was induced, where higher BT content shifts the LUMO energy to more positive potentials, altering the energetic levels of the system. The photo generated charge separation was boosted by the incorporation of the strong electron acceptor 2,1,3-benzothiadiazole, increasing electron localisation and resulting in very high conversion rates and selectivity of a [3 + 2] cycloaddition. When compared to the bulk polymer, an 18-fold increase in conversion rate was observed.30
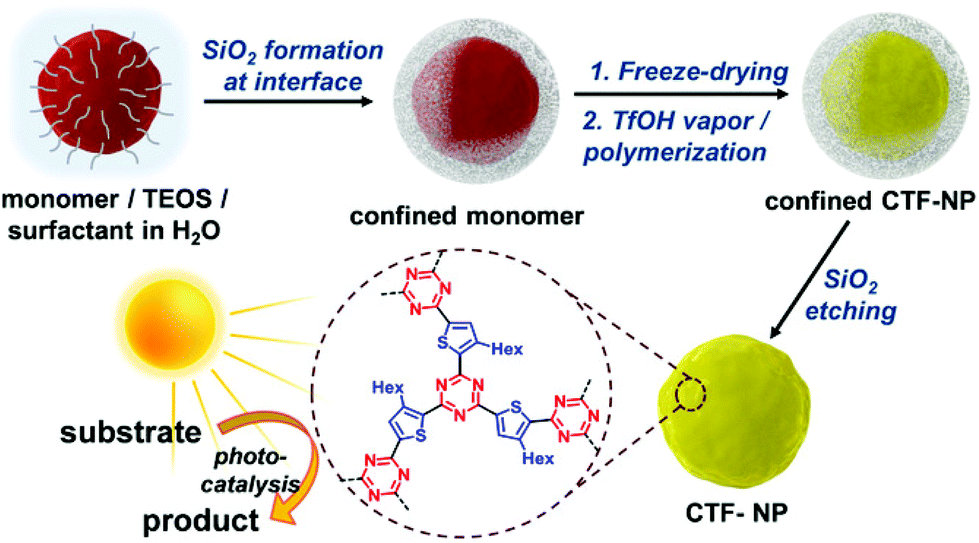 | ||
| Fig. 2 Synthesis route for nanoparticle CTFs using a silica shell to confine the monomers and allow polymerisation in TfOH vapour. Reproduced from ref. 30 with permission from John Wiley and Sons, copyright 2020. | ||
Lastly, a new type of porous polymer nanoparticles was presented by Ferguson et al.46 Here, hyper cross-linked porous nanoparticles were synthesised containing a benzothiadiazole based vinyl-monomer, using RAFT-PISA. PEG based polymeric stabilising units were used, allowing the polymer particles to be dispersed in a range of different continuous phases. This allows for widespread applications, since an optimal solvent selection for the photocatalytic reaction can be provided. Ferguson et al. demonstrated that this new porous material could be used in water, acetonitrile or toluene, whilst the hydrophobic small molecule photocatalyst shows no solubility in water.
Photocatalytic gels
Cross-linked polymer gel networks containing photoactive species have recently gained significant interest. Both organo and hydrogel based systems have been implemented for the production of high value compounds. Similarly to porous polymers, polymer gel networks enable enhanced reagent and catalyst contact. Here, photocatalytic gels have been produced to allow easy partitioning of reagents into the gel network, where they can be readily transformed. To date conjugated,47 polymer/inorganic,48 polymer/metal49 and classical polymer18 gel networks have been investigated.Similar to the previously discussed porous polymeric nanomaterials, photocatalytically modified gels exhibit significant advances over linear, bulk polymers. Besides an enlargement of the photocatalytically available surface, an enhancement of compatible solvents can be achieved. Moreover, the implementation of photocatalysts onto highly cross-linked gels, allow for the introduction of new functionality. Bulk gels have shown promising catalytic efficacy. However, most of the catalytic centres are often not available due to spatial hindrance inside the polymer gel. Similarly to CMPs and CTFs, recently nano-based gel systems have been reported to tackle spatial hindrance. Reagents in the nano/microgel systems are required to diffuse much shorter distances compared to bulk system. As the diffusion of reagents to the catalyst is often the rate limiting step in photocatalytic systems, significantly reducing the length scales required, vastly increases the photocatalytic performance of these materials.
Petrizza et al. prepared one of the first examples of photocatalytic polymer microgels. Here, a cross-linked block-copolymer was synthesized by free radical mini-emulsion polymerization of vinyl acetate and vinyl caprolactam was produced (Fig. 3). Into this hydrogel, a vinyl benzyl derivative of rose bengal (RB) was copolymerised into the gel network, resulting in covalently bound, swellable microgel particles. Polymer microgels around 200 nm in size were produced that under irradiation of light could create reactive oxygen species, which could be used to synthesise 6-hydroxy-(2H)-pyran-3-one and 1,2-dibenzoylbenzene.50
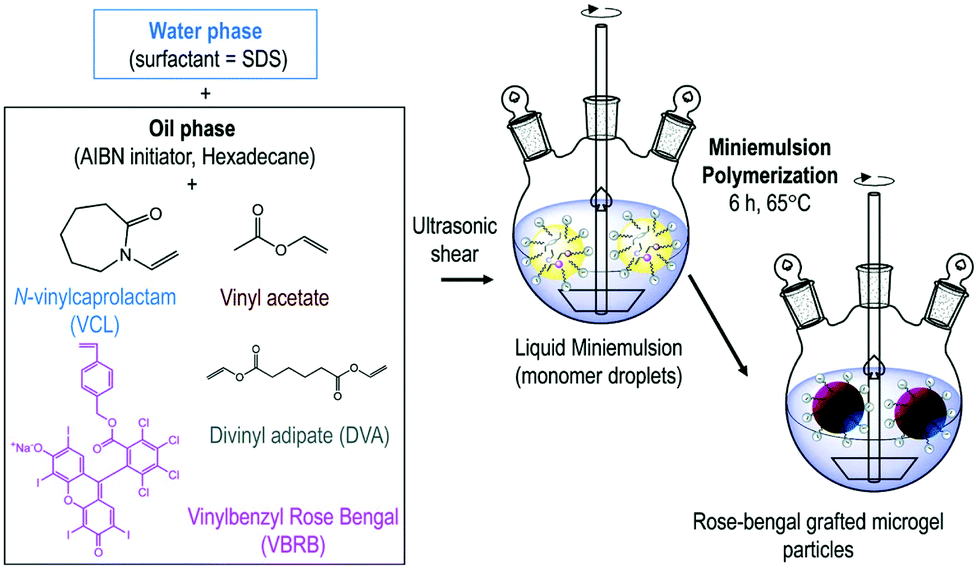 | ||
| Fig. 3 Synthesis of photoactive microgels by mini-emulsion polymerisation. A dispersed phase of N-vinylcaprolactam, vinyl acetate, divinyl adipate, vinyl benzene rose bengal, AIBN and hexadecane was polymerised to produce rose bengal grafted microgels. Reproduced from ref. 50 with permission from Royal Society of Chemistry, copyright 2019. | ||
Radjagobalou et al. further expanded the use of photocatalytic microgels, showing their use in flow chemistry. Poly(N-vinylcaprolactam-covinyl acetate-co-vinylbenzyl Rose Bengal) microgels was used for the photooxygenation of α-terpine. The microporous structural properties of the photoactive microgels allowed not only for the diffusion of the reagent to the catalytic centres, but also for the application into a spiral flow chemistry setup. A similar product conversion to free RB was observed, moreover advantages in a decrease of photobleaching as well as a fixed microgel for continuous usage was observed.51
Further to the synthesis of polymer nanogels that can be swollen to allow photocatalysis Ferguson et al. produced a responsive system that could be turned on or off (Fig. 4). Photocatalytic nano/microgels were synthesised by copolymerising diphenylbenzothiadiazoleacrylamide together with N-isopropylacrylamide (NIPAM) and poly(ethylene glycol) dimethacrylate as cross-linker. The gels produced had an average diameter of around 190 nm at ambient temperature. NIPAM groups display lower critical solution temperature (LCST) behaviour, therefore, a thermoresponsiveness was established into the polymer network. Upon heating to 40 °C, a shrinkage of the nanogel to 70 nm was observed, giving a compressed, and non photocatalytically active material. When cooled down to 25 °C, swelling of the particles back to 190 nm was observed, demonstrating the reversibility of the system. Reaction control by light and temperature was extensively investigated for NAD+ regeneration in water, and as well as an oxidation, reduction and redox catalytic conversion. Reaction control by temperature and light was achieved, with additional high conversion rates of at least 87% presented.52
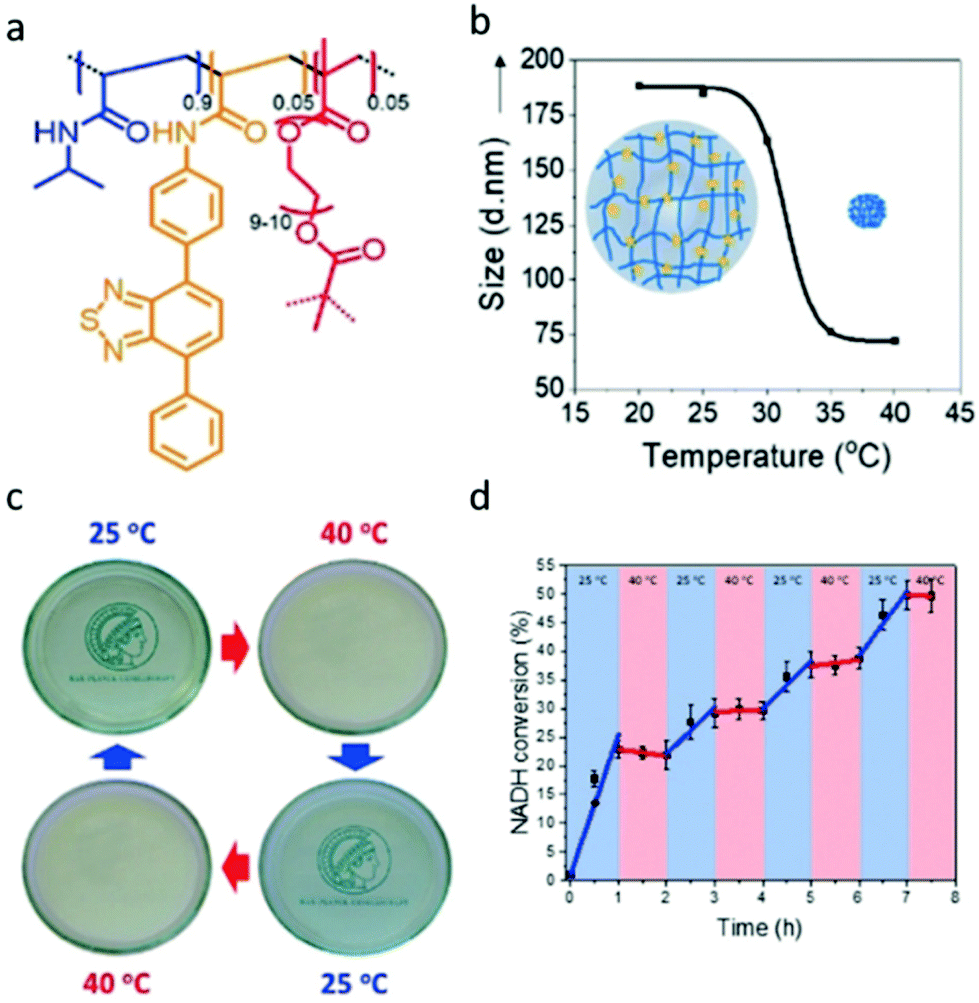 | ||
| Fig. 4 (a) Photocatalytic temperature-responsive nanogels structure. (b) Temperature-dependent size of nanogels. (c) Reversible phase changes by changing temperature. (d) Temperature-dependent photocatalytic conversion of the enzyme cofactor NADH to NAD+. Reproduced from ref. 52 with permission from John Wiley and Sons, copyright 2019. | ||
Self-assembled polymer photocatalysts
Classical polymer photocatalysts have recently emerged as an alternative to conjugated photocatalytic systems.5 Here, a small molecular photocatalyst is modified so that it can be incorporated into a classical polymer. This incorporation can be during the polymerisation of the polymer, or as a post-polymerisation modification. This methodology enables a broad range of photocatalytic materials to be synthesised, where the physical properties of the polymer can be easily varied whilst maintaining photocatalytic activity.Controlled radical polymerisation techniques, such as reversible addition–fragmentation chain-transfer (RAFT) and atom transfer radical polymerization (ATRP), allow more complex polymer architectures to be synthesised. Here, well-defined block copolymers can be synthesised, where the location of any given monomer on the polymer chain is known. This spatial control of monomers has been utilised when forming classical polymer photocatalysts. The photocatalytic moiety can easily be incorporated within a desired polymeric block. Furthermore, polymerisation induced self-assembly (PISA) enables superstructures (including nanoparticles) to be formed in solution. Typically, reactions are undertaken in aqueous or alcoholic conditions, where initially a hydrophilic macro chain transfer agent is chain extended by a more hydrophobic monomer. As the polymer chain extends the second block becomes insoluble in the continuous phase, driving self-assembly.
Recently, RAFT mediated PISA has been used by multiple research groups to form photocatalytic classical polymer nanoparticles. Here, organic dyes have been modified to include vinyl functionality enabling their copolymerisation within RAFT polymerisation. Interestingly, the inclusion of the photoactive monomer into both the hydrophobic particle core and hydrophilic corona has been investigated. Lessard et al. chain extended a PEG macroCTA with Eosin Y modified with a methacrylate functional group and benzyl methacrylate.53 The synthesis was undertaken in the absence of any initiator where the photocatalytic monomer was able to self-initiate the polymerisation through photo-induced electron/energy transfer-RAFT (PET-RAFT) (Fig. 5). Further to the self-synthesising polymer nanoparticles Lessard et al. used a PMMA Eosin Y for the successful photo-mediated oxidation of 4-methoxyphenyl boronic acid to 4-methoxyphenol.53
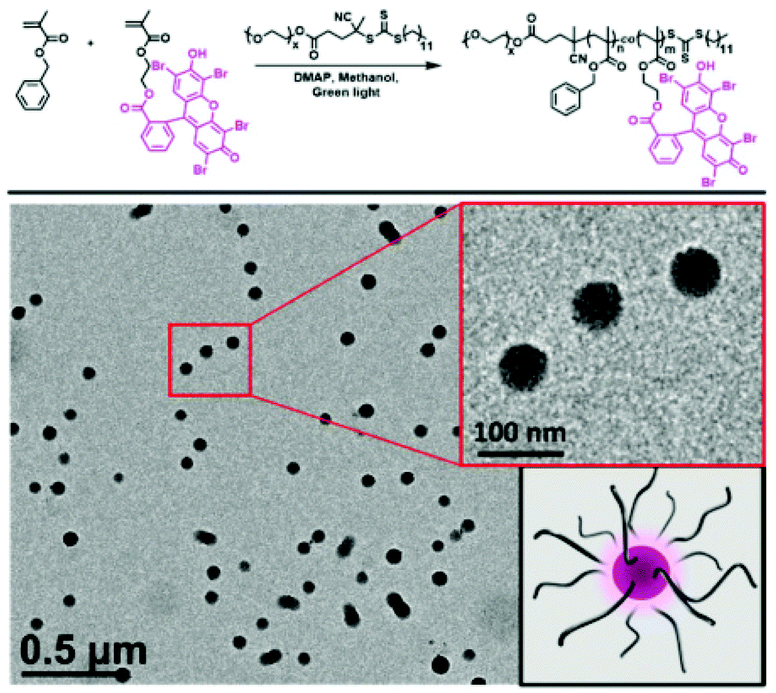 | ||
| Fig. 5 Eosin Y containing polymer nanoparticles produced by PET RAFT-PISA. Synthesised in the absence of initiator sub 100 nm photoactive polymer nanoparticles are formed. Reproduced from ref. 53 with permission from Royal Society of Chemistry, copyright 2021. | ||
Recently Korpusik et al. incorporated a methacrylate functionalized RB derivative into a diblock copolymer, using PISA-RAFT. An azide-modified macro chain transfer agent (mCTA) enabled modification of the polymer nanoparticles to be achieved by a subsequent HUISGEN-cyclization reaction of DNA aptamers. This conjugation of aptamers onto the surface resulted in enhanced cellular uptake of the nanoparticles. Decreased aggregation of photocatalyst (compared to free RB) was observed due to the equal distribution of the active molecule across the hydrophobic portion of the polymer chains. The enhanced distribution lead to increased singlet oxygen production, creating a promising material for use in photodynamic therapy.54
Boussiron et al. also utilised RAFT mediated PISA to form photocatalytic classical polymer nanoparticles.55 Here, RB was selected as the photocatalytic unit, which was again modified to contain a polymerizable group, an acrylate or styrene derivative. Early work by Boussiron et al. produced photoactive latex particles, where rose bengal was incorporated into an acrylic acid macroCTA and then chain extended with n-butyl acrylate.55 This self-assembled photoactive particles showed low polydispersities and sub 100 nm diameters, typical for the polymerisation technique used and suggesting the inclusion of the bulk photocatalyst did not significantly impede the polymerisation or self-assembly process. Furthermore, the photoactive nanoparticles produced readily produced reactive oxygen species upon irradiation with visible light. Boussiron et al. further investigated the inclusion of rose bengal into self-assembled nanoparticles to determine the optimum position of the photocatalytic material within the self-assembled nanoparticle.56 Boussiron et al. demonstrated the easy incorporation of the photoactive material into both environments, combining multiple different monomers.56 Independent on the location of the photoactive monomer sub 100 nm particles were again formed. No particular difference in the photooxidation reaction of furfuryl alcohol was observed when RB was found in the core, corona or as a free molecule. Conversely, Ibrahimova et al. found significant differences in performance of a BODIPY based photoactive monomer when it was incorporated into the hydrophilic or hydrophobic portion of a self-assembled polymersome.57 Here, significantly higher performance was observed for the photoperoxidation of anthracene 9,10-divinylsulfonate by singlet oxygen when the BODIPY was located in the corona compared to the core. It is currently unclear what causes the differences in performance between these systems, it may be due to the differences in monomers, photoactive molecules, reaction being catalysed or the polymeric structures.
Conclusions and perspectives
Nanotechnology has enabled the synthesis of a broad range of polymer structures. The application of nanomaterials in photocatalytic systems has yielded highly efficient materials that combat some of the main challenges in heterogeneous photocatalysis. Specifically, the use of nanomaterials in photocatalytic porous or gel materials has mitigated some of the challenges with mass transfer into the light active material.Here, diffusional path lengths are significantly reduced for nano-based systems compared to bulk material, where not all of the photocatalytically active surface area can be fully utilised.
Furthermore, photocatalytic nanomaterials can often be used in a broader range of solvents, including greener solvents such as water.
The use of photocatalytic systems in aqueous environments also enables combinations with biomaterials such as enzymes to be more readily realised. The application of nanotechnology has been reported here for three main classes of polymer photocatalysts: self-assembled, porous and gel photocatalytic polymers. For each of these classes the use of nanotechnology has allowed more control of the physical properties of the light active material compared to the analogous bulk systems. Future research will likely follow this trend where advanced nanomaterials could be synthesised for specialised applications.
It is likely that increasingly defined nanomaterials will be synthesised to increase the catalytic efficacy, solvent compatibility and even selectiveness of photocatalytic materials. Conceivable options include controllability of chemo- and regio-selectivity, controlled diastereomer conversion, responsive drug delivery systems, but even regeneration of value compounds in flow chemistry. There is a broad range of different photocatalytic nanostructures that could feasibly be produced, however, a balance must be made between the benefit of producing more complex systems and the difficulty in synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the MaxSynBio consortium that is jointly funded by the Federal Ministry of Education and Research of Germany (BMBF) and the Max Planck Society (MPG). Open Access funding provided by the Max Planck Society.References
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- J. Twilton, P. Zhang, M. H. Shaw, R. W. Evans and D. W. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- Z. Qian and K. A. Zhang, Sol. RRL, 2021, 5, 2000489 CrossRef CAS.
- J. Byun and K. A. Zhang, Mater. Horiz., 2020, 7, 15–31 RSC.
- C. T. Ferguson and K. A. Zhang, ACS Catal., 2021, 11, 9547–9560 CrossRef CAS.
- S. Bi, Z. A. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang, E. Brunner, X. Wang and F. Zhang, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang and X. Zhang, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2013, 2, 353–358 CrossRef.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176, 44–52 Search PubMed.
- C. Yang, W. Huang, L. C. da Silva, K. A. Zhang and X. Wang, Chem. – Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., 2019, 131, 3471–3475 CrossRef.
- X.-H. Li, M. Baar, S. Blechert and M. Antonietti, Sci. Rep., 2013, 3, 1743 CrossRef.
- A. Mills and C. O'Rourke, Catal. Today, 2014, 230, 256–264 CrossRef CAS.
- C. Ayed, L. Caire Da Silva, D. Wang and K. A. I. Zhang, J. Mater. Chem. A, 2018, 6, 22145–22151 RSC.
- A. K. Singh, R. Chawla and L. D. S. Yadav, Tetrahedron Lett., 2015, 56, 653–656 CrossRef CAS.
- N. Huber, R. Li, C. T. Ferguson, D. W. Gehrig, C. Ramanan, P. W. Blom, K. Landfester and K. A. Zhang, Catal. Sci. Technol., 2020, 10, 2092–2099 RSC.
- C. Yang, R. Li, K. A. Zhang, W. Lin, K. Landfester and X. Wang, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemCatChem, 2016, 8, 694–698 CrossRef CAS.
- C. Lu, P. Zhang, S. Jiang, X. Wu, S. Song, M. Zhu, Z. Lou, Z. Li, F. Liu, Y. Liu, Y. Wang and Z. Le, Appl. Catal., B, 2017, 200, 378–385 CrossRef CAS.
- S. Lin, S. Sun, K. Wang, K. Shen, B. Ma, Y. Ren and X. Fan, Nanomaterials, 2018, 8, 127 CrossRef PubMed.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486–1493 RSC.
- D. A. Nagib and D. W. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2012, 48, 5355–5357 RSC.
- Y. Huang, Z. Xin, W. Yao, Q. Hu, Z. Li, L. Xiao, B. Yang and J. Zhang, Chem. Commun., 2018, 54, 13587–13590 RSC.
- T. Maji, A. Karmakar and O. Reiser, J. Org. Chem., 2010, 76, 736–739 CrossRef PubMed.
- W. Wei, R. Li, N. Huber, G. Kizilsavas, C. T. J. Ferguson, K. Landfester and K. A. I. Zhang, ChemCatChem, 2021, 13, 3410 CrossRef CAS.
- Z. Li, W. Zhang, Q. Zhao, H. Gu, Y. Li, G. Zhang, F. Zhang and X. Fan, ACS Sustainable Chem. Eng., 2015, 3, 468–474 CrossRef CAS.
- W. Huang, N. Huber, S. Jiang, K. Landfester and K. A. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18368–18373 CrossRef CAS PubMed.
- L. Jiao, Y. Hu, H. Ju, C. Wang, M.-R. Gao, Q. Yang, J. Zhu, S.-H. Yu and H.-L. Jiang, J. Mater. Chem. A, 2017, 5, 23170–23178 RSC.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- S. Bhowmik, R. G. Jadhav and A. K. Das, J. Phys. Chem. C, 2018, 122, 274–284 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76–80 CrossRef PubMed.
- L. Li and Z. Cai, Polym. Chem., 2016, 7, 4937–4943 RSC.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- B. C. Ma, S. Ghasimi, K. Landfester and K. A. Zhang, J. Mater. Chem. B, 2016, 4, 5112–5118 RSC.
- B. C. Ma, L. Caire da Silva, S.-M. Jo, F. Wurm, M. Bannwarth, K. A. Zhang, K. Sundmacher and K. Landfester, ChemBioChem, 2019, 20, 2529–2682 CrossRef.
- H. S. Jena, C. Krishnaraj, G. Wang, K. Leus, J. Schmidt, N. Chaoui and P. Van Der Voort, Chem. Mater., 2018, 30, 4102–4111 CrossRef CAS.
- M. Liu, L. Guo, S. Jin and B. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant and A. I. Cooper, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- P. Katekomol, J. r. m. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319–326 CrossRef CAS.
- W. Huang, Z. J. Wang, B. C. Ma, S. Ghasimi, D. Gehrig, F. Laquai, K. Landfester and K. A. Zhang, J. Mater. Chem. A, 2016, 4, 7555–7559 RSC.
- C. T. Ferguson, N. Huber, T. Kuckhoff, K. A. Zhang and K. Landfester, J. Mater. Chem. A, 2020, 8, 1072–1076 RSC.
- J. Byun, K. Landfester and K. A. Zhang, Chem. Mater., 2019, 31, 3381–3387 CrossRef CAS.
- M. Liu, Y. Ishida, Y. Ebina, T. Sasaki and T. Aida, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- J. Yang, Z. Li and H. Zhu, Appl. Catal., B, 2017, 217, 603–614 CrossRef CAS.
- L. Petrizza, M. Le Bechec, E. Decompte, H. El Hadri, S. Lacombe and M. Save, Polym. Chem., 2019, 10, 3170–3179 RSC.
- R. Radjagobalou, J.-F. Blanco, L. Petrizza, M. Le Bechec, O. Dechy-Cabaret, S. Lacombe, M. Save and K. Loubiere, ACS Sustainable Chem. Eng., 2020, 8, 18568–18576 CrossRef CAS.
- C. T. Ferguson, N. Huber, K. Landfester and K. A. Zhang, Angew. Chem., 2019, 131, 10677–10681 CrossRef.
- J. J. Lessard, G. M. Scheutz, A. B. Korpusik, R. A. Olson, C. A. Figg and B. S. Sumerlin, Polym. Chem., 2021, 12, 2205–2209 RSC.
- A. B. Korpusik, Y. Tan, J. B. Garrison, W. Tan and B. S. Sumerlin, Macromolecules, 2021, 54, 7354–7363 CrossRef CAS.
- C. Boussiron, M. Le Bechec, L. Petrizza, J. Sabalot, S. Lacombe and M. Save, Macromol. Rapid Commun., 2019, 40, 1800329 CrossRef PubMed.
- C. Boussiron, M. Le Bechec, J. Sabalot, S. Lacombe and M. Save, Polym. Chem., 2021, 12, 134–147 RSC.
- V. Ibrahimova, S. A. Denisov, K. Vanvarenberg, P. Verwilst, V. Préat, J.-M. Guigner, N. D. McClenaghan, S. Lecommandoux and C.-A. Fustin, Nanoscale, 2017, 9, 11180–11186 RSC.
| This journal is © The Royal Society of Chemistry 2022 |