
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facilitating the acidic oxygen reduction of Fe–N–C catalysts by fluorine-doping†‡
Xiafang
Tao§
 ab,
Ruihu
Lu§
c,
Lingmei
Ni
d,
Vladislav
Gridin
d,
Samir H.
Al-Hilfi
b,
Zijie
Qiu
b,
Yan
Zhao
c,
Ulrike I.
Kramm
*d,
Yazhou
Zhou
*ab and
Klaus
Müllen
*b
ab,
Ruihu
Lu§
c,
Lingmei
Ni
d,
Vladislav
Gridin
d,
Samir H.
Al-Hilfi
b,
Zijie
Qiu
b,
Yan
Zhao
c,
Ulrike I.
Kramm
*d,
Yazhou
Zhou
*ab and
Klaus
Müllen
*b
aSchool of Materials Science and Engineering, Jiangsu University, Zhenjiang, Jiangsu 212013, China. E-mail: yazhou@ujs.edu.cn
bMax Planck Institute for Polymer Research, 55128 Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de
cState Key Laboratory of Silicate Materials for Architectures International School of Materials Science and Engineering, Wuhan University of Technology, Wuhan, Hubei 430070, China
dDepartment of Materials and Earth Science and Department of Chemistry, Technical University Darmstadt, Otto-Berndt-Straße 3, 64287 Darmstadt, Germany. E-mail: kramm@ese.tu-darmstadt.de
First published on 9th November 2021
Abstract
As the alternatives to expensive Pt-based materials for the oxygen reduction reaction (ORR), iron/nitrogen co-doped carbon catalysts (FeNC) with dense FeNx active sites are promising candidates to promote the commercialization of proton exchange membrane fuel cells. Herein, we report a synthetic approach using perfluorotetradecanoic acid (PFTA)-modified metal–organic frameworks as precursors for the synthesis of fluorine-doped FeNC (F-FeNC) with improved ORR performance. The utilization of PFTA surfactants causes profound changes of the catalyst structure including F-doping into graphitic carbon, increased micropore surface area and Brunauer–Emmett–Teller (BET) surface area (up to 1085 m2 g−1), as well as dense FeNx sites. The F-FeNC catalyst exhibits an improved ORR activity with a high E1/2 of 0.83 V (VS. RHE) compared to the pristine FeNC material (E1/2 = 0.80 V). A fast decay occurs in the first 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles for the F-FeNC catalyst, but high durability is still maintained up to another 50000 cycles. Density functional theory calculations reveal that the strongly withdrawing fluorine atoms doped on the graphitic carbon can optimize the electronic structure of the FeNx active center and decrease the adsorption energy of ORR intermediates.
000 potential cycles for the F-FeNC catalyst, but high durability is still maintained up to another 50000 cycles. Density functional theory calculations reveal that the strongly withdrawing fluorine atoms doped on the graphitic carbon can optimize the electronic structure of the FeNx active center and decrease the adsorption energy of ORR intermediates.
New conceptsIron and nitrogen co-doped carbon catalysts (FeNC) play an important role as potential alternatives to expensive Pt-based catalysts in proton exchange membrane fuel cells. The relatively strong adsorption of oxygen reduction reaction (ORR)-related species on FeNx sites largely limits the ORR kinetics. The electron-withdrawing heteroatoms in FeNC catalysts is regarded as a promising strategy to improve the kinetic activity. This effect should be even more pronounced in the case of fluorine (F) as F possesses the highest electronegativity. To date, studies about F-modified FeNC catalysts (F-FeNC) of high ORR activity have been little reported. Here, we developed a new synthetic approach by pyrolysis of perfluorotetradecanoic acid (PFTA)-modified metal–organic frameworks for improving the ORR performance of F-FeNC in acidic solution. The utilization of PFTA was proven to be beneficial to achieve the increased content of micropores, high Brunauer–Emmett–Teller surface area (1085 m2 g−1), dense FeN4 active sites, as well as fluorine-doping (F-doping). Density functional theory calculations revealed that the strongly electron-withdrawing F atoms doped into carbon can optimize the electronic structure of the FeN4 active center and decrease the adsorption energy of ORR intermediates. |
Introduction
Hydrogen-based sustainable energy technologies, particularly proton exchange membrane fuel cells (PEMFCs), hold promise for achieving carbon neutrality and reducing the threats of climate change.1–4 While the development of PEMFCs has been significantly advanced over the past decades, their commercialization is still hindered by the need for platinum-based catalysts, which are expensive and scarce.5–7 Therefore, searching for non-noble metals to catalyze the oxygen reduction reaction (ORR) in PEMFCs is highly rewarding.8–10Transition metal and nitrogen co-doped carbon materials (M–N–C, M = Fe, Co, Mn, etc.) have been developed as catalysts for ORR, in which atomically dispersed MNx moieties on carbon supports are generally considered as the main active sites to directly absorb O2 and catalyze the subsequent ORR kinetics.11–17 In particular, the Fe–N–C catalysts demonstrate an encouraging ORR performance in acidic solution and are promising alternatives to Pt-based catalysts in PEMFCs.18–21 Previous studies have revealed that heteroatom doping (e.g. N, O, S, and P) into Fe–N–C catalysts or binding with Fe atoms can determine the ORR performance by balancing the adsorption and desorption of intermediates at FeNx sites.20,22–25 Recently, in Mun's work, the ORR activity of the FeN4 catalytic center could be tailored by introducing S-functionalities into carbon black.26 Li et al. demonstrated a higher activity of the N–FeN4 than for the FeN4 moiety.27 Han et al. reported that due to its electronegativity, chlorine atoms vertically coordinated with the FeN4 center that can facilitate electron transfer from Fe atom to oxygen intermediates during the ORR.24 This effect should be even more pronounced in the case of fluorine (F) as F possesses the highest electronegativity.28,29 However, F-modified Fe–N–C catalysts are rarely studied in the literature.30 Surface modification of carbon black supports by grafting with trifluoromethylphenyl groups is reported to improve the stability, however, lower the activity of Fe–N–C catalysts.31 This is caused by the increased hydrophobicity of carbon black, which keeps the oxidants from reaching the triple-phase boundary.31 The fluorination of Fe–N–C samples with fluorine gas has led to the poisoning of the Fe active sites and dramatic ORR degradation.32 Attempts at F-doping into carbon supports without poisoning active sites can therefore affect ORR performance.
In this work, we explore an approach to disperse the FeNx active sites on N, F co-doped graphitic carbon materials (F-FeNC) for improved ORR performance. This F-FeNC catalyst was prepared by pyrolysis of perfluorotetradecanoic acid (PFTA)-modified Fe-doped zeolitic imidazolate frameworks (PFTA/Fe-ZIFs), wherein PFTA served as fluorine source. The PFTA is proven to be beneficial to maintain the microporous structure of ZIFs, achieving the high Brunauer–Emmett–Teller (BET) surface area (1085 m2 g−1). The surface and bulk characterizations reveal that the F-FeNC catalyst possesses densely embedded single-atom Fe sites and fluorine-doping into graphitic carbon. As a result, the prepared catalysts deliver a superior ORR activity with a half-wave potential of 0.83 V versus the reversible hydrogen electrode (vs. RHE) in acidic solution as compared to the pristine FeNC material.
Results and discussion
Synthesis and characterization of F-FeNC catalysts
As illustrated in Fig. 1a, the F-FeNC catalysts were prepared by high-temperature pyrolysis of PFTA/Fe-ZIFs. Details of the method are described in the (ESI‡). In brief, a methanol solution containing zinc nitrate hexahydrate, ferric nitrate nonahydrate, 2-dimethylimidazole, and PFTA surfactant in a molar ratio of 75:1:190:2 was hydrothermally treated at 60 °C overnight to synthesize the PFTA/Fe-ZIFs. The PFTA surfactant was in situ introduced into the frameworks of PFTA/Fe-ZIFs through the coordination between carboxyl groups of PFTA and the metal atom. The presence of carboxyl groups and C–F bonds in PFTA/Fe-ZIFs precursors is proven by X-ray photoelectron spectroscopy (XPS) (Fig. S1, ESI‡). Notably, Fe-ZIFs and PFTA/Fe-ZIFs showed similar diffraction patterns (Fig. S2a, ESI‡), uniform morphological features (Fig. S3, ESI‡), and similar pore size distributions (Fig. S4, ESI‡), suggesting that the introduction of a limited amount of PFTA does not change the crystalline structure of ZIFs. The PFTA/Fe-ZIFs precursors were then thermally annealed in an argon atmosphere at 1000 °C to obtain a black powder. During high-temperature pyrolysis, the hydrocarbons of PFTA/Fe-ZIFs were carbonized into partially graphitic carbon, as evidenced by powder X-ray diffraction (PXRD) patterns (Fig. S2b, ESI‡) and Raman spectra (Fig. 2a).33 The inclusion of nitrogen dopants into the graphitic carbon provided a large number of anchoring sites for bonding Fe atoms. Samples prepared by adding different amounts of PFTA surfactants were denoted as F-FeNC-1, F-FeNC-2, and F-FeNC-3 based on the molar ratio of PFTA to 2-dimethylimidazole (169:1, 85:1, and 68:1, respectively) (see Method, ESI‡). For comparison, the F–NC sample without Fe and the FeNC sample without fluorine were synthesized in an analogous fashion.
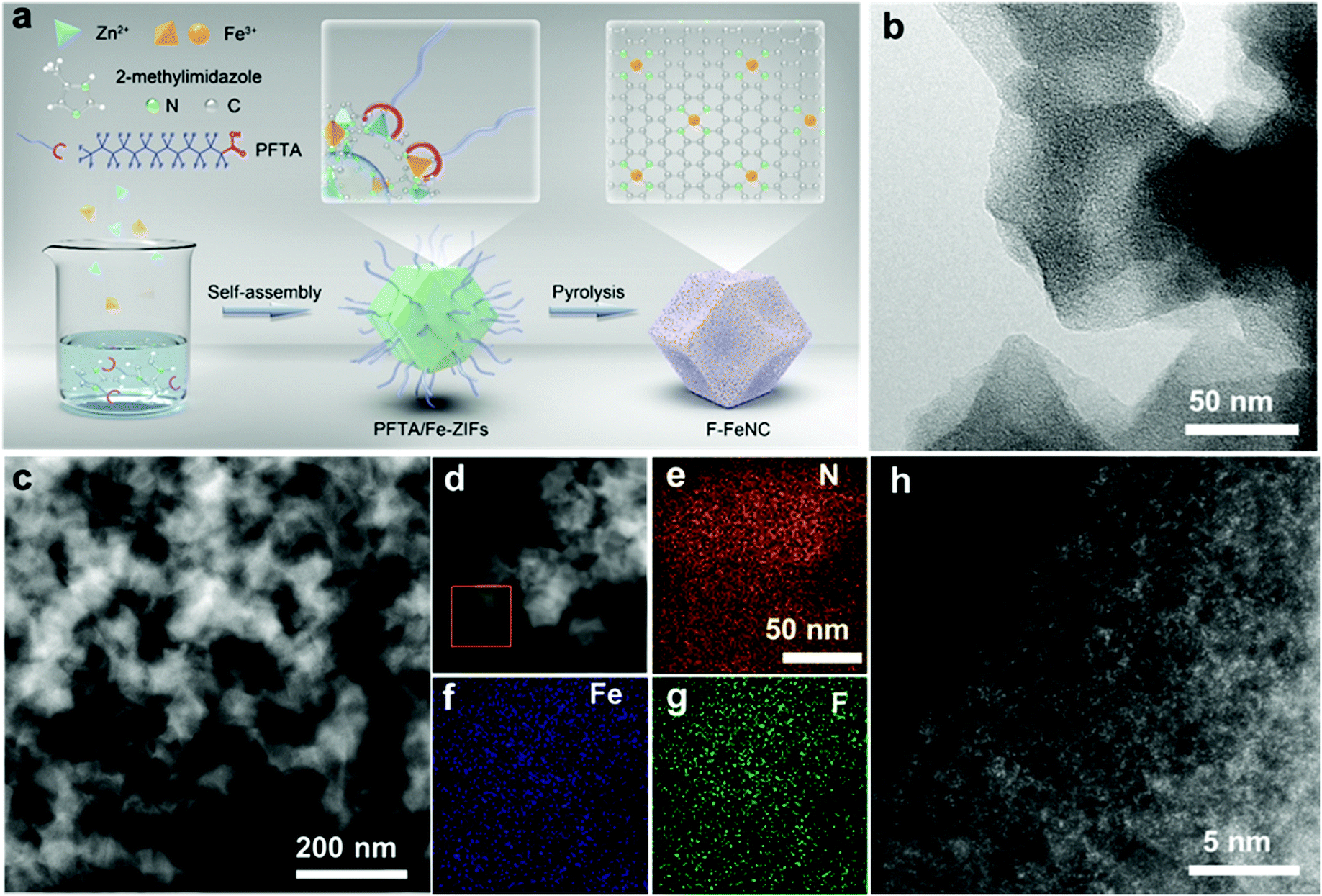 | ||
| Fig. 1 (a) Illustration of the synthesis of F-FeNC catalysts. (b) TEM and (c and d) STEM of the obtained F-FeNC-2 catalyst. Elemental mapping of (e) N, (f) Fe, and (g) F of the highlighted area in (d). (h) Aberration-corrected HAADF-STEM image of F-FeNC-2. | ||
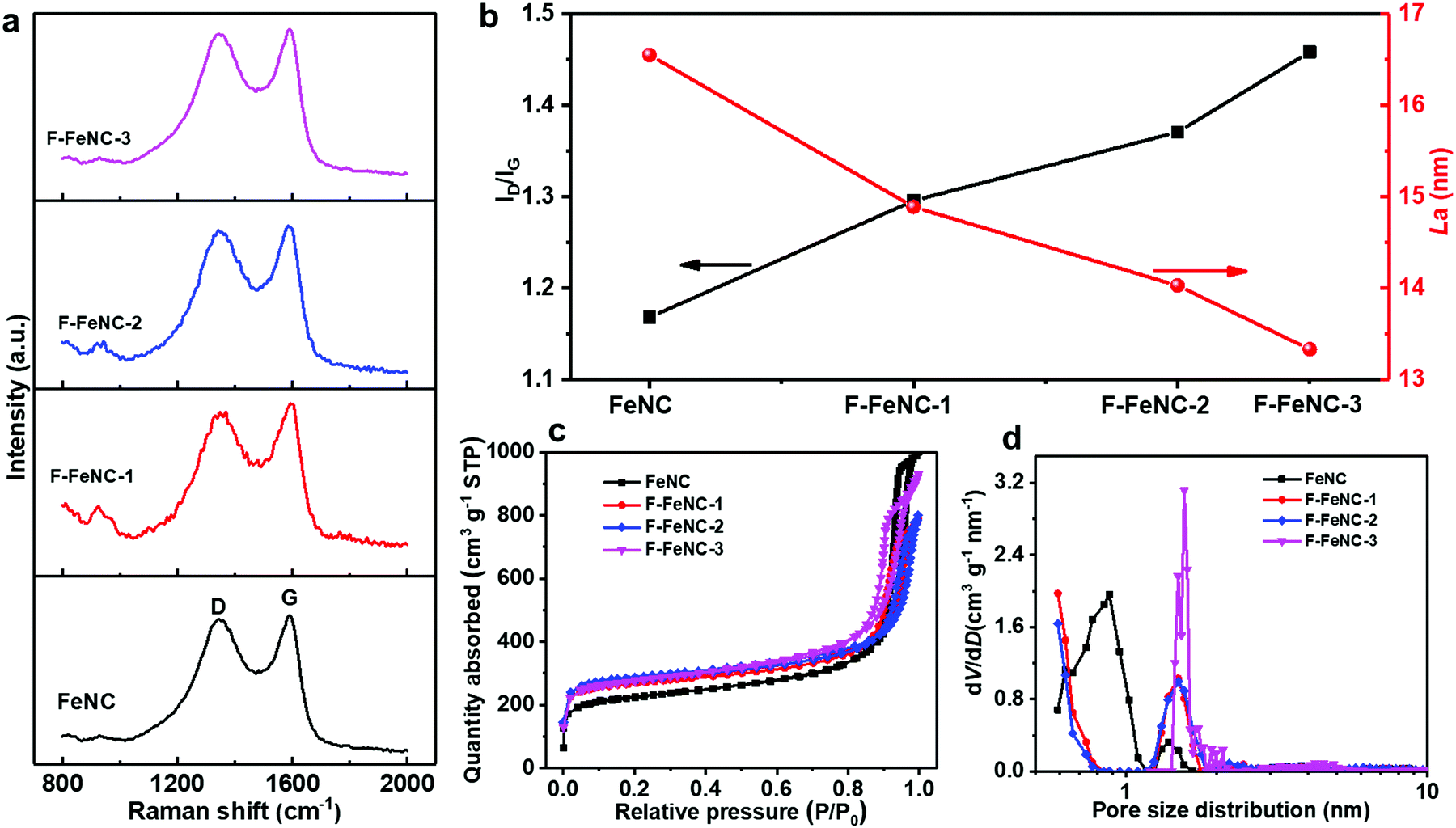 | ||
| Fig. 2 (a) Raman spectra, (b) La sizes and the ratio of the G and D bands (ID/IG) in the Raman spectra, (c) N2 adsorption/desorption isotherms and (d) pore size distributions processed from the non-local density functional theory (NLDFT) analysis FeNC, F-FeNC-1, F-FeNC-2, and F-FeNC-3. | ||
The morphologies of the new catalysts were characterized by transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) revealing carbon nanoparticles with 100 nm size in F-FeNC-2 (Fig. 1b and c). No obvious Fe-based particles with crystalline phases were observed in the high-resolution STEM (HR-STEM) image, which was also supported by selected area electron diffraction (SAED, Fig. S5f, ESI‡) analysis. Therefore, Fe species were highly dispersed as single atoms. Distinguishable signals for N, F, and Fe were detected in corresponding elemental mapping images and electron energy loss spectroscopy (EELS) across the carbon nanoparticles, indicating uniform distributions of these elements in the F-FeNC-2 sample (Fig. 1d–g). The bright spots in the aberration-corrected HAADF-STEM image corresponded to single heavy atoms. This gave evidence that the Fe atoms seem mainly atomically dispersed within the graphitic carbons (Fig. 1h). Similar morphologies were observed for the reference materials (Fig. S5 and S6, ESI‡).
The effect of PFTA surfactants on the characteristics of graphitic carbon in the catalysts was analyzed using Raman spectroscopy. Two prominent peaks located at around 1345 and 1585 cm−1 were attributed to the D band and the G band, respectively (Fig. 2a).34 The ratio of the intensity of D and G bands (ID/IG) and the crystallite size of the carbon plane (La) were correlated with the amount of PFTA surfactant in FeNC, F-FeNC-1, F-FeNC-2, and F-FeNC-3 (Fig. 2b and Table S4, ESI‡). The ID/IG values, which are commonly utilized to determine the density of defects increased significantly with higher amounts of PFTA surfactants, indicating that more defects were created in the F-FeNC catalysts by F-doping. In the meanwhile, the increased density of defects induced the formation of smaller carbon planes, as evidenced by the decreased La value.
The porosity of the catalysts was assessed by nitrogen-sorption analysis and a non-local density functional theory (NLDFT) model was used to determine the pore size distributions. Type IV isotherms with a steep increase in Vads at relatively low and high N2 pressures indicated the preferred existence of micropores and large outer surfaces in all the samples (Fig. 2c). The BET surface area of the F-FeNC-2 catalysts was 1085 m2 g−1, higher than that of the FeNC catalyst (809 m2 g−1) (Table S5, ESI‡). The larger surface area originated from the increased micropore surface area (Smicropore) of 665 m2 g−1 compared with that of FeNC (362 m2 g−1), which was further confirmed by the pore size distributions in which ultramicropore and micropore were centered at 0.6 and 1.5 nm, respectively (Fig. 2d). When using higher contents of PFTA, the SMicropore of F-FeNC-3 decreased (561 m2 g−1), which was attributed to the carbonization of excessive PFTA which may block the ultramicropores. Notably, all the F-FeNC samples showed the high SMicropore, suggesting that the addition of PFTA is beneficial for the formation of micropores.
The surface-near elemental composition of FeNC and F-FeNC were obtained from XPS. The high-resolution C 1s XPS spectra of all samples displayed similar C 1s peaks (Fig. S7a, ESI‡).35,36 The weak C–F peaks located at 685.5–688.0 eV in the high-resolution F 1s XPS spectra (Fig. S7b, ESI‡) suggested that fluorine atoms were doped into graphitic carbon.37,38 The percentage of Fe3+ species was increased from 27% for the FeNC sample to 48% for F-FeNC-3 based on fitting results of Fe 2p3/2 spectra (Table S1, ESI‡). The higher oxidation state of the Fe site was ascribed to the surrounding fluorine heteroatoms with high electronegativity. The high-resolution N 1s spectra in Fig. S8 (ESI‡) were deconvoluted into five types of N species with binding energies of 402.1, 401.2, 400.3, 399.4, and 398.4 eV which could be assigned to oxidized-, graphitic-, pyrrolic-N, FeNx, and pyridinic-N, respectively.34,39 All the F-FeNC catalysts displayed a higher percentage of FeNx (11–16%) than that in the FeNC (∼9.2%). This finding suggested the formation of more FeNx active sites by PFTA surfactant addition (Table S2, ESI‡). The fluorine contents measured by XPS were 0.1, 0.1, and 0.2 at% for F-FeNC-1, F-FeNC-2, and F-FeNC-3, respectively (Table S3, ESI‡). The Fe content was determined to be 1.68 wt% for F-FeNC-2 by inductively coupled plasma atomic emission spectroscopy (ICP-AES), higher than that of the FeNC reference material (1.49 wt%) and other F-FeNC catalysts.
To gain deeper insight into the nature of Fe-related species, 57Fe Mössbauer spectroscopy was employed for both F-FeNC-2 and FeNC catalysts. The Mössbauer spectrum of FeNC was fitted with two major doublets of D1 and D2 that correspond to FeN4 species (Fig. S9a and Table S6, ESI‡), whereas three doublets of D1, D2, and D3 were observed for the F-FeNC-2 sample (3a).14,15,17,21,40 Based on the fact that similar D3 sites were already found in other fluorine-free FeNC catalysts,14,15 it may conclude that a fluorine coordination to iron can be excluded. This is in agreement with the previous conclusion that F atoms in F-FeNC were present as F-doping into the graphitic carbon. In addition to the doublets, the FeNC sample exhibited a sextet and singlet, whereas no sextet was found in the F-FeNC-2 sample (3b), suggesting a possible formation of more Fe active sites in F–FeNC.
Further information on the binding situation of Fe atoms was obtained from X-ray absorption spectroscopy (XAS). The Fe K-edge X-ray absorption near-edge structure (XANES) displayed a shift towards higher energy from FeNC to F-FeNC-2, confirming the increased oxidation state of Fe due to the F-doping in the surrounding carbon (Fig. 3c),41,42 which is consistent with the XPS analysis. More detailed information on the mode of coordination of the Fe site was obtained from the Fe K-edge extended X-ray absorption fine structure (EXAFS) analysis. A prominent peak was observed at ∼1.48 Å in the radial distribution function obtained for the Fourier transformed (FT) EXAFS for the F-FeNC (Fig. 3d), which can be assigned to the Fe–N (O) coordination. The signal of Fe–Fe bonds with typical bond distances of around 2 Å were very weak in both FeNC and F-FeNC catalysts pointing toward more abundant single-atom Fe sites. Two backscattering paths of Fe–N and Fe–O were performed for fitting EXAFS spectra (Fig. 3e and f). For the Fe–N path, the coordination number of Fe atom was calculated to be 5.4 ± 0.4 with a Fe–N distance of 2.00 ± 0.02 Å (Table S7, ESI‡). The EXAFS fitting for the F-FeNC could also be obtained by a mixture of Fe–N and Fe–O paths, with coordination numbers of 3.9 ± 0.9 and 1.1 ± 0.7, respectively. Based on this fitting, we propose two structures for the Fe center, i.e. an in-plane FeN4 with either an axial nitrogen ligand (FeN4–N, Fig. 3e inset) or an axial oxygen-containing ligand (e.g. as FeN4–O), respectively. In contrast, the FeNC samples possess mainly FeN4 sites (Table S7, ESI‡).
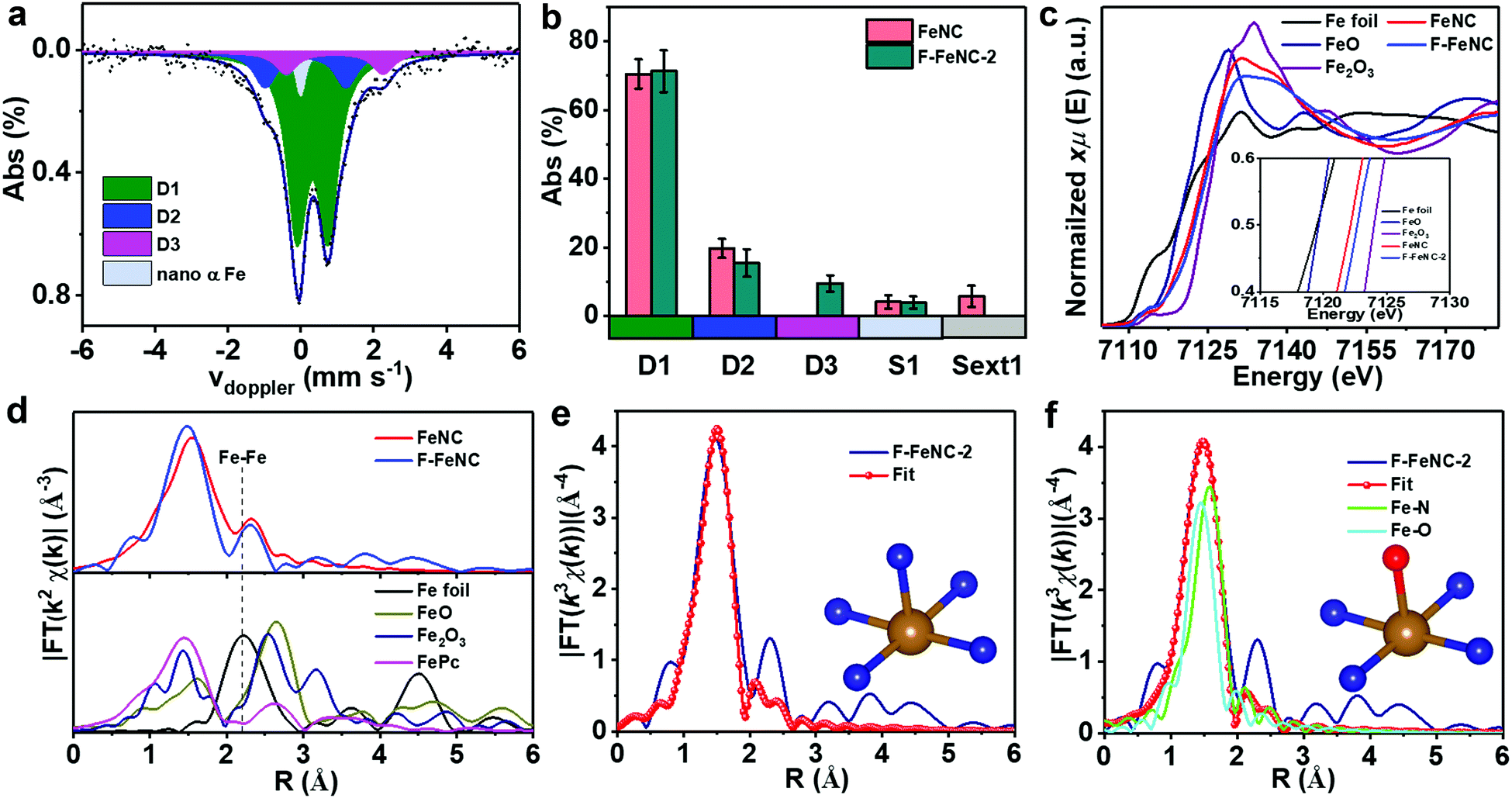 | ||
| Fig. 3 (a) RT 57Fe Mössbauer spectra of F-FeNC-2, (b) comparison of Mössbauer parameters of the doublets for the FeNC and F-FeNC-2 catalysts, (c) K-edge XANES spectra with a zoomed-in view of the pre-peak region in the inset, (d) Fourier transformed of k3-weighted EXAFS of the FeNC, F-FeNC-2, and Fe foil, iron phthalocyanine (FePc), FeO, Fe2O3 reference materials. EXAFS fitting curves of F-FeNC-2 using (e) Fe–N, (f) a mixture of Fe–N and Fe–O paths, inset is corresponding structure, where blue, red, and golden balls represent the N, O, and Fe atoms, respectively. | ||
ORR performance
The electrocatalytic ORR performance of F-FeNC and control FeNC samples was, first, investigated in an O2-saturated 0.5 M H2SO4 electrolyte using a rotating disk electrode (RDE) technique. A commercial Pt/C catalyst (20 wt% Pt, Fuelcell store) as a reference was evaluated in 0.1 M HClO4 solution. All potentials were calibrated against a hydrogen electrode (RHE). ORR polarization curves clearly revealed the effect of fluorine incorporation (Fig. 4a). The FeNC sample demonstrated an ORR onset potential (Eonset) of 0.93 V (Eonset: the potential at current density of 0.1 mA cm−2) and a half-wave potential (E1/2) of 0.80 V (vs. RHE). By using PFTA, the ORR activity of F-FeNC-1 was enhanced. When increasing the use of PFTA, the F-FeNC-2 achieved the best ORR activity with a high E1/2 of 0.83 V which was only 20 mV lower than that of the Pt/C reference catalyst (E1/2, 0.85 V). At 0.85 V, the F-FeNC-2 material gave a three times higher kinetic current density (Jk) than that of the bare FeNC sample (1.63 vs. 0.55 mA cm−2, Fig. 4b). Moreover, the Tafel slopes of all samples fell into the range from 66 to 70 mV dec−1 (Fig. S10 and Table S8, ESI‡). Further increasing the PFTA caused a decline in the activity of F-FeNC-3. Due to a high affinity of SCN- anion to Fe ion, the SCN- can poison FeNx active sites and lead to a significant suppression of the ORR activity.26,43 The poisoning test using an electrolyte containing SCN− ions and the comparison with the ORR polarization curve of the Fe-free F–NC revealed that the ORR activity was mainly stemming from the FeNx sites (Fig. 4f).26,43 Compared to reference samples, the F-FeNC-2 exhibited a higher density of FeNx active sites because of the larger surface area of micropores and the higher contents of FeNx, achieving the enhanced ORR activity.42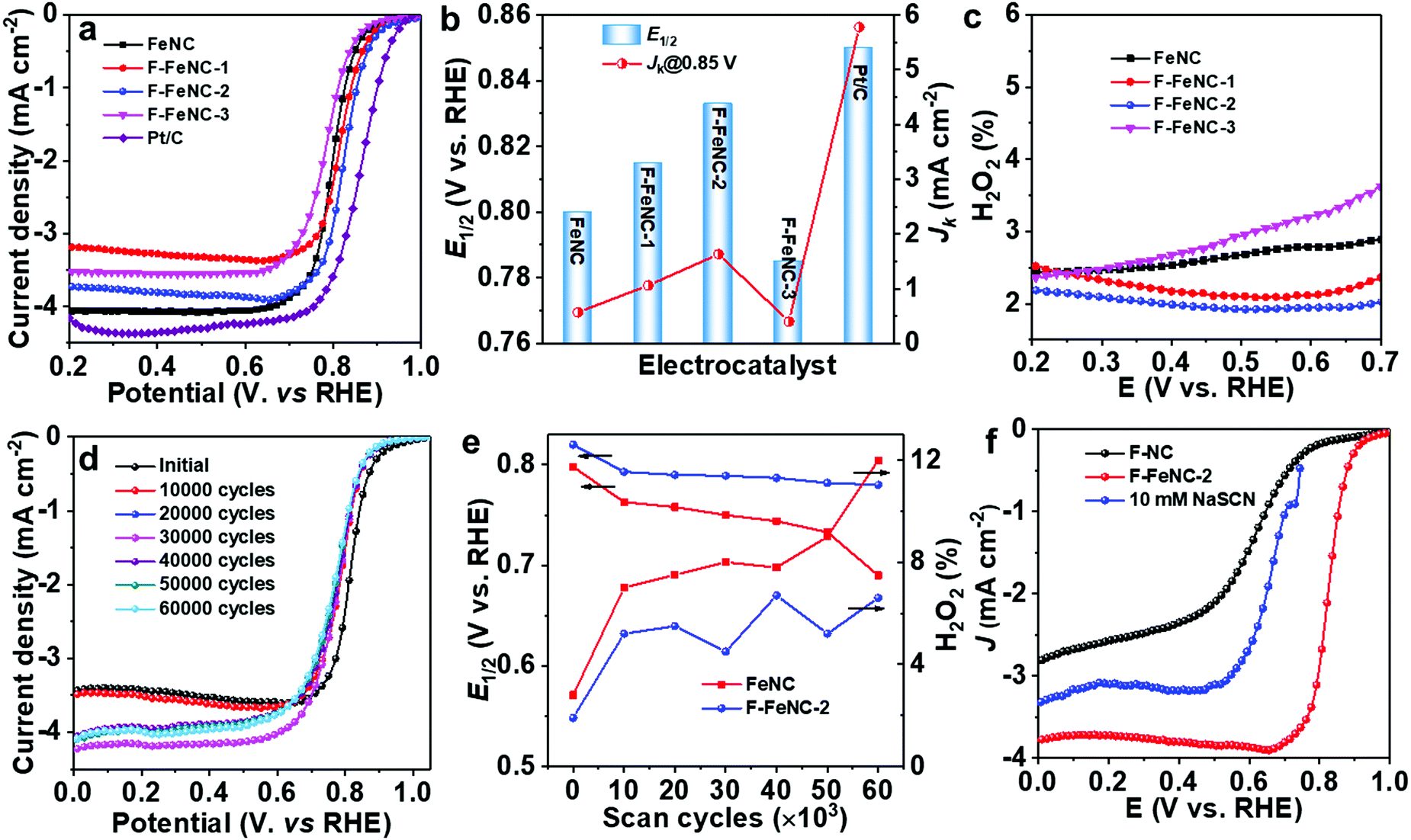 | ||
| Fig. 4 (a) Steady-state ORR polarization plots recorded with a rotation speed of 900 r.p.m. and a scan rate of 5 mV s−1, (b) Jk at 0.85 V and E1/2 and (c) calculated H2O2 yield for FeNC, F-FeNC-1, F-FeNC-2, F-FeNC-3, and commercial Pt/C catalysts, (d) ORR polarization plots of the F-FeNC-2 and (e) the changes of E1/2 and H2O2 yield for FeNC and F-FeNC catalysts during the durability tests. (f) ORR polarization curves of F–NC and F-FeNC and results of poisoning test with SCN− of F-FeNC. | ||
To examine the ORR mechanism of the catalysts, the rotating ring-disk electrode (RRDE) technique was applied for monitoring the formation of H2O2 during the tests. As shown in Fig. 4c, the H2O2 yield for F-FeNC-2 was below 1.7% in the potential range from 0.2 to 0.7 V, which was lower than that of the FeNC sample. The low H2O2 yield and the calculated electron-transfer number of 3.98 were firm evidence of a four-electron pathway for oxygen reduction.33 The stabilities of F-FeNC and FeNC catalysts were evaluated using accelerated stress tests (ASTs) by cycling the potential from 0.6 to 1.0 V (vs. RHE) in an O2-saturated 0.5 M H2SO4 electrolyte (Fig. 4d). The changes of E1/2 and H2O2 yield were calculated after every 10000 cycles (Fig. 4e). After the first 10000 cycles, both catalysts showed a steep activity degradation with E1/2 losses for FeNC and F-FeNC of 35 and 27 mV due to the removal of unstable sites in acidic solution. After continuing the potential cycling for another 50000 cycles, a significant activity decline with another 73 mV loss in E1/2 and increased H2O2 product were observed for FeNC (Fig. 4e and Fig. S11, ESI‡) due to the carbon corrosion and demetalation of FeNxvia Fenton (-like) reactions between the Fe centers and the intermediate product of H2O2.44,45 In contrast, the F-FeNC-2 presented improved durability with a small loss of only 13 mV in E1/2. The involved F-doing on graphitic carbon can enhance the durability of Fe–N–C catalyst by the mitigation of the oxidative corrosion of the carbon support, which is one of the major decay mechanisms of FeNC catalyst.21,31 The F-FeNC-2 catalyst was further studied as the cathode in a PEMFC in an H2-air system (1 bar back pressure for H2 and air), and compared with commercial Pt/C catalyst. The F-FeNC-2 catalyst demonstrated a peak power density of 122 mW cm−2 (Fig. S12a, ESI‡). A stability test at a constant voltage of 0.5 V with F-FeNC-2 catalyst as cathodic catalyst is shown in Fig. S12b (ESI‡), exhibiting much poorer stability performance than in the RRDE test. The reasons for this difference are still a matter of debate but observed for several Fe–N–C catalysts.32,46
Theoretical study of F-FeNC on ORR
To understand the effect of F-doping, we investigated the adsorption behavior and corresponding free-energy evolution on the Fe active sites by first-principles calculations (see Computational method, ESI‡). The appropriate structure, denoted as FeN4–aX–Fx, was constructed based on EXAFS fitting and XPS analyses and consisted of a porphyrin-like FeN4 with an axial ligand (X represents C-, N-, and O-containing axial ligands), and fluorine atoms doped in graphene layers (x = 2, 4, and 8) (Table S9, ESI‡). The control structures of FeN4, FeN4–Fx and FeN4–aX without F-doping, were considered for comparison. The optimized ORR intermediates (*OOH, *OH, and *O) adsorbed Fe sites were also included. The Gibbs free energy profiles for the ORR process on F-FeNC catalysts were investigated under zero potential (U = 0 V), highest potential (U = biggest output potential), and equilibrium potential (U = 1.23 V) (Fig. S13–S16, ESI‡). The calculated free energy changes suggest that the reduction of *OH is the rate-determining step (RDS) for the ORR process on most of FeN4–aX–Fx, catalysts, whereas the RDS of FeN4–aO–F4 and FeN4–aO–F8 catalysts is the formation of *OOH. The adsorption energies of *OH, *OOH and *O display linear correlations (Fig. 5a). This scaling relations lead to a volcano relationship between the ORR catalytic activity and the adsorption energy of *OH (ΔG*OH) (Fig. 5b). The principle for enhancement of intrinsic ORR activity is to balance the *OH adsorption on the Fe active site that is neither too strong nor too weak.47 The FeN4 exhibits a high ΔG*OH of 0.50 eV and an overpotential of 1.73 V. The activity of FeN4 site is mostly limited by the *OH reduction due to a strong *OH adsorption. The FeN4–Fx and FeN4–aX–Fx structures located at the right side of FeN4 point toward a reduced ΔG*OH on the FeN4, certifying that electron-withdrawing F-doping weakens the intermediates adsorption on active sites.26 The FeN4–aO–F2 with a low content of F-doping reaches the top of the volcano plot and is thus identified as the most active ORR catalyst.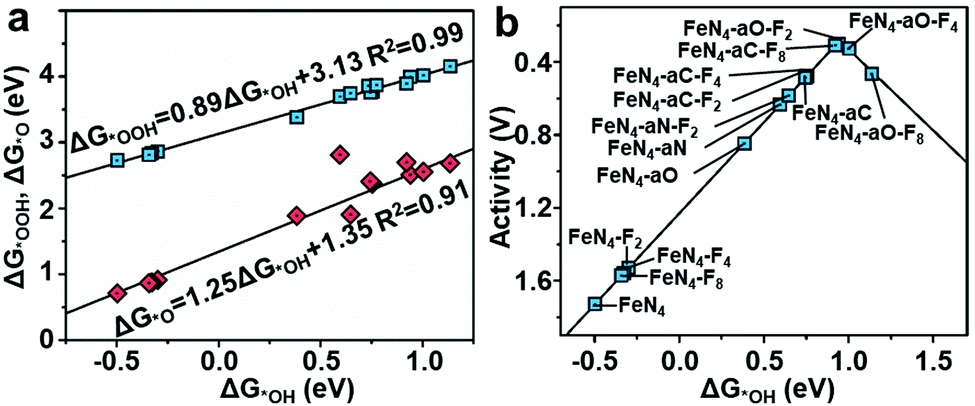 | ||
| Fig. 5 (a) The scaling relations of ΔG*OOHvs. ΔG*OH and ΔG*Ovs. ΔG*OH. (b) A volcano plot for the ORR overpotential against the adsorption energy ΔG*OH, wherein the letter “a” denotes the axial ligands and the subscripted number denotes the number of fluorine atoms that are embedded into graphene layers. | ||
Due to the low contents of fluorine in the catalyst, it can be questioned if the enhanced performance of F-FeNC catalysts is only related to the fluorine doping. Indeed, it might not be only governed by the fluorine content but maybe cooperatively by one or all of the following three aspects. Firstly, PFTA plays an important role in maintaining the microporous structure of ZIFs, achieving a higher surface area of micropores and BET surface area. More micropores improve the accessibility of FeNx sites for ORR. Secondly, the F-FeNC-2 sample possesses a high density of active sites due to the high loading of single Fe atoms. Thirdly, the fluorine atoms doped into graphitic carbon can balance the adsorption of oxygen intermediate, facilitating the intrinsic activity on FeN4 sites. Overall, different from the fluorinated FeNC catalysts in literature,31,32 our experimental and theoretical results indicate that the use of PFTA-modified ZIFs as precursors can influence both the Fe active sites and carbon structure of the F-FeNC catalyst, thus leading to very good ORR performance in acidic media.
Conclusions
In summary, a new synthetic approach using PFTA-modified ZIFs has been developed to prepare microporous Fe–N–C catalysts. Compared with the FeNC without fluorine incorporation, the prepared F-FeNC catalyst exhibited strongly enhanced ORR performance in terms of the onset and have-wave potentials, as well as durability. This enhancement is associated with the larger surface area of micropores (665 vs. 362 m2 g−1) and BET surface area (1085 vs. 809 m2 g−1), abundant single Fe atoms, and strong electron-withdrawing F-doping compared to F-free FeNC. We believe that the findings demonstrated here could be extended to fabricate other F-modified M–N–C catalysts with high performance for the hydrogen/oxygen evolution reaction, electrochemical carbon dioxide reduction reaction, nitrogen fixation reaction, and other (electro-) catalytic applications.Author contributions
Y. Z and K. M conceived and designed the experiments. X. T and Y. Z conducted the synthesis, characterization and RDE measurements of catalysts. U. I. K and L. N recorded 57Fe Mössbauer transmission spectra and did the data analysis. U. I. K and V. G performed the PEM fuel cell investigations. R. L and Y. Z carried out computational studies. X. T, Z. Q, U. I. K, Y. Z and K. M co-wrote the paper. The research was supervised by U. I. K, Y. Z and K. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Max Planck Society and the Natural Science Foundation of China (51702129 and 51972150). Y. Z acknowledges the support from the China Postdoctoral Science Foundation (2018M630527) and the China Scholarship Council for financial support (201708320150). X. T thanks the scholarship from Jiangsu University. U. I. K, L. N and V. G thank the German federal ministry of education and research (BMBF) for financial support via the NanoMatFutur young research group FeNC–StRedO (03XP0092). We thank Umicore for providing the Pt/C catalyst for FC testing. Open Access funding provided by the Max Planck Society.References
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- G. Karanfil, Int. J. Energy Res., 2020, 44, 4–25 CrossRef.
- L. L. Chen, X. L. Xu, W. X. Yang and J. B. Jia, Chin. Chem. Lett., 2020, 31, 626–634 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- Y. Z. Zhou, C. H. Yen, Y. H. Hu, C. M. Wang, X. N. Cheng, C. M. Wai, J. Yang and Y. H. Lin, J. Mater. Chem. A, 2016, 4, 18628–18638 RSC.
- A. A. Gewirth, J. A. Varnell and A. M. DiAscro, Chem. Rev., 2018, 118, 2313–2339 CrossRef CAS.
- B. P. Setzler, Z. B. Zhuang, J. A. Wittkopf and Y. S. Yan, Nat. Nanotechnol., 2016, 11, 1020–1025 CrossRef CAS PubMed.
- X. Wan, X. F. Liu, Y. C. Li, R. H. Yu, L. R. Zheng, W. S. Yan, H. Wang, M. Xu and J. L. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- M. Chen, Y. He, J. S. Spendelow and G. Wu, ACS Energy Lett., 2019, 4, 1619–1633 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–483 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed.
- U. I. Kramm, M. Lefevre, P. Bogdanoff, D. Schmeisser and J. P. Dodelet, J. Phys. Chem. Lett., 2014, 5, 3750–3756 CrossRef CAS.
- U. I. Kramm, M. Lefevre, N. Larouche, D. Schmeisser and J. P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978–985 CrossRef CAS.
- U. I. Kramm, L. M. Ni and S. Wagner, Adv. Mater., 2019, 31, 1805623 CrossRef.
- L. Ni, C. Gallenkamp, S. Paul, M. Kübler, P. Theis, S. Chabbra, K. Hofmann, E. Bill, A. Schnegg, B. Albert, V. Krewald and U. I. Kramm, Adv. Energy Sustainab. Res., 2021, 2, 2000064 CrossRef.
- S. T. Thompson and D. Papageorgopoulos, Nat. Catal., 2019, 2, 558–561 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M. X. Chen, E. Jin, J. Yang, H. W. Liang, Y. Zhao, X. Feng, A. Narita and K. Mullen, Nat. Commun., 2020, 11, 5892 CrossRef CAS.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2020, 4, 10–19 CrossRef.
- X. H. Yan, X. L. Li, C. H. Fu, C. Lin, H. M. Hu, S. Y. Shen, G. H. Wei and J. L. Zhang, Prog. Nat. Sci.: Mater. Int., 2020, 30, 896–904 CrossRef CAS.
- K. Hu, L. Tao, D. D. Liu, J. Huo and S. Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 19379–19385 CrossRef CAS.
- Y. H. Han, Y. G. Wang, R. R. Xu, W. X. Chen, L. R. Zheng, A. J. Han, Y. Q. Zhu, J. Zhang, H. B. Zhang, J. Luo, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- K. J. Chen, K. Liu, P. D. An, H. J. W. Li, Y. Y. Lin, J. H. Hu, C. K. Jia, J. W. Fu, H. M. Li, H. Liu, Z. Lin, W. Z. Li, J. H. Li, Y. R. Lu, T. S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS.
- X. N. Li, C. S. Cao, S. F. Hung, Y. R. Lu, W. Z. Cai, A. I. Rykov, S. Miao, S. B. Xi, H. B. Yang, Z. H. Hu, J. H. Wang, J. Y. Zhao, E. E. Alp, W. Xu, T. S. Chan, H. M. Chen, Q. H. Xiong, H. Xiao, Y. Q. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS.
- X. J. Sun, Y. W. Zhang, P. Song, J. Pan, L. Zhuang, W. L. Xu and W. Xing, ACS Catal., 2013, 3, 1726–1729 CrossRef CAS.
- S. G. Peera, A. Arunchander and A. K. Sahu, Nanoscale, 2016, 8, 14650–14664 RSC.
- Y. Chang, J. X. Chen, J. C. Jia, X. Hu, H. J. Yang, M. L. Jia and Z. H. Wen, Appl. Catal., B, 2021, 284 Search PubMed.
- Y. C. Wang, P. F. Zhu, H. Yang, L. Huang, Q. H. Wu, M. Rauf, J. Y. Zhang, J. Dong, K. Wang, Z. Y. Zhou and S. G. Sun, ChemElectroChem, 2018, 5, 1914–1921 CrossRef CAS.
- G. X. Zhang, X. H. Yang, M. Dubois, M. Herraiz, R. Chenitz, M. Lefevre, M. Cherif, F. Vidal, V. P. Glibin, S. H. Sun and J. P. Dodelet, Energy Environ. Sci., 2019, 12, 3015–3037 RSC.
- J. Z. Li, M. J. Chen, D. A. Cullen, S. Hwang, M. Y. Wang, B. Y. Li, K. X. Liu, S. Karakalos, M. Lucero, H. G. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. X. Feng, D. Su, K. L. More, G. F. Wang, Z. B. Wang and G. Wu, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- H. G. Zhang, S. Hwang, M. Y. Wang, Z. X. Feng, S. Karakalos, L. L. Luo, Z. Qiao, X. H. Xie, C. M. Wang, D. Su, Y. Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- Y. Z. Zhou, C. H. Yen, S. F. Fu, G. H. Yang, C. Z. Zhu, D. Du, P. C. Wo, X. N. Cheng, J. Yang, C. M. Wai and Y. H. Lin, Green Chem., 2015, 17, 3552–3560 RSC.
- Y. Z. Zhou, J. Yang, C. Z. Zhu, D. Du, X. N. Cheng, C. H. Yen, C. M. Wai and Y. H. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 25863–25874 CrossRef CAS.
- P. Z. Chen, T. P. Zhou, S. B. Wang, N. Zhang, Y. Tong, H. X. Ju, W. S. Chu, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 15471–15475 CrossRef CAS.
- G. Panomsuwan, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 3, 9972–9981 RSC.
- Y. Li, Y. Z. Zhou, H. J. Wen, J. Yang, C. Maouche, Q. Q. Liu, Y. Y. Wu, C. Cheng, J. Zhu and X. N. Cheng, Dalton Trans., 2018, 47, 14992–15001 RSC.
- S. Wagner, H. Auerbach, C. E. Tait, I. Martinaiou, S. C. N. Kumar, C. Kubel, I. Sergeev, H. C. Wille, J. Behrends, J. A. Wolny, V. Schunemann and U. I. Kramm, Angew. Chem., Int. Ed., 2019, 58, 10486–10492 CrossRef CAS PubMed.
- Q. H. Li, W. X. Chen, H. Xiao, Y. Gong, Z. Li, L. R. Zheng, X. S. Zheng, W. S. Yan, W. C. Cheong, R. A. Shen, N. H. Fu, L. Gu, Z. B. Zhuang, C. Chen, D. S. Wang, Q. Peng, J. Li and Y. D. Li, Adv. Mater., 2018, 30, 1800588 CrossRef.
- Q. Wang, Y. Q. Yang, F. F. Sun, G. B. Chen, J. Wang, L. S. Peng, W. T. Chen, L. Shang, J. Q. Zhao, D. Sun-Waterhouse, T. R. Zhang and G. I. N. Waterhouse, Adv. Energy Mater., 2021, 11 Search PubMed.
- Q. Wang, Z. Y. Zhou, Y. J. Lai, Y. You, J. G. Liu, X. L. Wu, E. Terefe, C. Chen, L. Song, M. Rauf, N. Tian and S. G. Sun, J. Am. Chem. Soc., 2014, 136, 10882–10885 CrossRef CAS PubMed.
- G. Bae, M. W. Chung, S. G. Ji, F. Jaouen and C. H. Choi, ACS Catal., 2020, 10, 8485–8495 CrossRef CAS.
- C. H. Choi, H. K. Lim, M. W. Chung, G. Chon, N. R. Sahraie, A. Altin, M. T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Drazic, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- D. Banham, S. Ye, K. Pei, J. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Seth Marder on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh01307f |
| § These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2022 |