
Singlet fission and triplet pair recombination in bipentacenes with a twist†‡
Lauren M.
Yablon§
a,
Samuel N.
Sanders§
a,
Ken
Miyazaki
 b,
Elango
Kumarasamy
a,
Guiying
He
cd,
Bonnie
Choi
a,
Nandini
Ananth
*b,
Matthew Y.
Sfeir
*cd and
Luis M.
Campos
*a
b,
Elango
Kumarasamy
a,
Guiying
He
cd,
Bonnie
Choi
a,
Nandini
Ananth
*b,
Matthew Y.
Sfeir
*cd and
Luis M.
Campos
*a
aDepartment of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: lcampos@columbia.edu
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA. E-mail: ananth@cornell.edu
cDepartment of Physics, Graduate Center, City University of New York, New York, New York 10016, USA. E-mail: msfeir@gc.cuny.edu
dPhotonics Initiative, Advanced Science Research Center, City University of New York, New York, New York 10031, USA
First published on 22nd October 2021
Abstract
We investigate triplet pair dynamics in pentacene dimers that have varying degrees of coplanarity (pentacene–pentacene twist angle). The fine-tuning of the twist angle was achieved by alternating connectivity at the 1-position or 2-positions of pentacene. This mix-and-match connectivity leads to tunable twist angles between the two covalently linked pentacenes. These twisted dimers allow us to investigate the subtle effects that the dihedral angle between the covalently linked pentacenes imparts on singlet fission and triplet pair recombination dynamics. We observe that as the dihedral angle between the two bonded pentacenes is increased, the rates of singlet fission decrease, while the accompanying decrease in triplet recombination rates is stark. Temperature-dependent transient optical studies combined with theoretical calculations show that the triplet pair recombination proceeds primarily through a direct multiexciton internal conversion process. Calculations further show that the significant decrease in recombination rates can be directly attributed to a corresponding decrease in the magnitude of the nonadiabatic coupling between the singlet multiexcitonic state and the ground state. These results highlight the importance of the twist angle in designing systems that exhibit rapid singlet fission, while maintaining long triplet pair lifetimes in pentacene dimers.
New conceptsThe fundamental understanding of multiexciton processes has generated a lot of interest across various disciplines. In organic chemistry, singlet fission (SF) is the mechanism by which a singlet exciton is converted into two triplet excitons. A great challenge is understanding how chemical structure impacts multiexciton dynamics to develop families of materials for optoelectronic applications. Here, we investigate how chemical connectivity impacts the rate of formation of triplet pairs and their recombination dynamics. The SF chromophores are pentacene dimers that are covalently coupled at different positions relative to each other. This leads to different dihedral twist angles between the planes of the two chromophores. Through experimental and theoretical calculations, we find that the twist angle drastically impacts the rates of intramolecular SF and the triplet pair recombination dynamics. This model system provides insight into the key factors governing SF as a function of chemical connectivity in bipentacenes. The structure–property relationship study of bipentacenes presented here is an effective fundamental model to addresses the rich multiexciton dynamics that have yet to be fully understood in intramolecular singlet fission processes. Moreover, the design is generalizable and can easily be extended to a variety of emerging chromophores that are not based on oligoacenes. |
Introduction
There has been an increased interest in materials that undergo singlet fission (SF) because of their potential to overcome the thermodynamic limit of efficiency in single-junction photovoltaics.1–3 In SF materials, the benefit for light harvesting results from the net reaction in which a photoexcited singlet exciton rapidly decays into two triplet excitons that can be independently harvested. The overall efficiency of this process is dependent on satisfying stringent energy conservation criteria while maintaining sufficient interchromophore coupling.4 Historically, satisfying these requirements was most readily accomplished in molecular crystals and films, where singlet fission occurs via an intermolecular process.5–10 More recently, intramolecular systems, such pentacene dimers, have emerged as ideal systems to investigate singlet fission (SF) because the number, connectivity, geometry, and energetics of interchromophore interactions can be precisely tuned through synthetic methods.11–19 Since isolated molecules can be studied in dilute solution, complications associated with excited-state dynamics in the solid-state can be avoided. In condensed media, photophysics vary widely due to a number of factors, such as packing interactions, morphology, defects, and grain boundaries.20–25The broad tunability of iSF chromophores has shown that a large diversity of triplet pair formation and decay dynamics can exist. Comparing the properties of various reported pentacene dimers11–14,16,18,26–31 and polymers32,33 reveals that variations in the molecular design34 can lead to drastic changes in the rates of iSF and triplet pair recombination. However, many studies have focused on establishing structure–property relationships of the triplet pair generation process. For example, it has been recently reported that a “direct coupling” mechanism can dominate in symmetric iSF chromophores with high energy charge-transfer (CT) states if the singlet and triplet pair are nearly resonant.26 Tuning the co-planarity of 2,2′-bipentacene (22BP) derivatives by introducing steric bulk at the 1- and 3-positions leads to slower iSF that scales as the square of the matrix element connecting the singlet and triplet pair: kSF ∼ |〈TT|Ĥel|S1〉|2, where Ĥel is proportional to the magnitude of the interchromophore Coulomb interaction.11,26 In other iSF chromophores that have an increased contribution from CT states, the dependence of the iSF rate on interchromophore coupling is even stronger.35 Furthermore, it has been found that introducing different bridging units between the two pentacenes can change the rates of singlet fission by orders of magnitude.27,29,30 Not only does the linker affect the singlet fission dynamics, but also the position at which the pentacenes are connected can affect the observed excited state dynamics.11,13
In all these iSF systems, it was noted that the triplet pair recombination rate was similarly affected by the interchromophore coupling, such that (in general) a slower rate of generation resulted in a longer triplet pair lifetime. Only recent multi-chromophore designs in which the chromophore interactions governing generation and recombination are spatially distinct have succeeded in overcoming this restriction.36,37 Still, an important outstanding question is the nature of interchromophore interactions that influence the triplet recombination dynamics. Unlike the triplet pair generation process in which only the net singlet triplet pair need be considered, triplet recombination involves a manifold of triplet pair states m(TT), with distinct total spin angular momenta (m = 1, 3, or 5) that evolve over time.4,38,39 While the decay of triplet pairs of finite magnetization can be studied directly using electron spin resonance techniques (m = 5)40,41 or indirectly by their population dynamics (m = 3),32,36 understanding the decay of 1(TT) (triplet pair in a net singlet configuration) is more elusive. Except in cases where the singlet exciton energy is nearly resonant (e.g., tetracene) permitting triplet pair fusion,42 the decay of 1(TT) is expected to be slow. This expectation stems from the consideration of a slow radiative process (since dipole coupled transitions to/from a doubly excited state are formally forbidden)43 and a slow non-radiative rate due to the large energy gap (∼1.7 eV in pentacene) between the triplet pair and ground state.44
Contrary to expectations, non-radiative recombination in a subset of iSF chromophores is extremely fast, with rapid triplet pair annihilation on time scales that are much shorter than the natural radiative lifetime of the monomer, implying an efficient relaxation mechanism involving 1(TT). For example, the excited state of monomeric TIPS–pentacene has a lifetime of ∼13 ns and a fluorescence high quantum yield (>70%).45 In 22BP, rapid formation of triplet pairs occurs in ∼0.8 ps, followed by rapid relaxation back to the group state on time scales of ∼500 ps,11 more that 20× faster than the monomer! Similar effects have been seen in other systems with strong chromophore–chromophore interactions, including dimers with different connectivity13,26 and in iSF polymers.46,47 For example, a soluble derivative of 6,6′-bipentacene (66BP) exhibits similarly fast recombination to 22BP, despite much larger twist angle.12,13,29 While decreasing interchromophore coupling strength has been shown to lessen this effect,14,27,48 a rigorous explanation for the cause of this rapid decay has not been definitively presented. We note that the fluorescence quantum yields remain low in these compounds. While weak 1(TT) emission in pentacene molecular crystals has been observed,49 no similar emission has been reported in iSF dimers. Unlike other dimers where the energetics favor rapid interconversion between singlets and triplet pairs, radiative emission processes are not strong enough to explain the observed lifetimes.42,50,51 As such, a rigorous description the origin of the enhanced radiationless transition rates for triplet pairs in iSF molecules represents a major outstanding problem in this field.
In this study, we sought to investigate how modifying interchromophore coupling, achieved by varying the interplanar twist angle using simple modifications to the connectivity, would affect triplet pair recombination. We focus on end-connected pentacene dimers, including 22BP, 1,2′-bipentacene (12BP) and 1,1′-bipentacene (11BP) derivatives (Fig. 1), which exhibit fast singlet fission and relatively long triplet pair lifetimes compared to center-ring linked 6,6′ dimers.13 Using temperature dependent transient optical measurements and multireference electronic calculations, we find that indeed the dominant recombination process in this series is a direct and fast radiationless transition from 1(TT) to the ground state. Furthermore, the decay rate is shown to be temperature independent using both theory and spectroscopic measurements, ruling out triplet fusion followed by delayed fluorescence as a viable decay process. Connectivities that exhibit weaker interchromophore coupling show a corresponding decrease in the non-radiative rate due to a reduction in the non-adiabatic (vibronic) coupling. These results provide a framework to understand the strong relationship between the generation and decay rates.
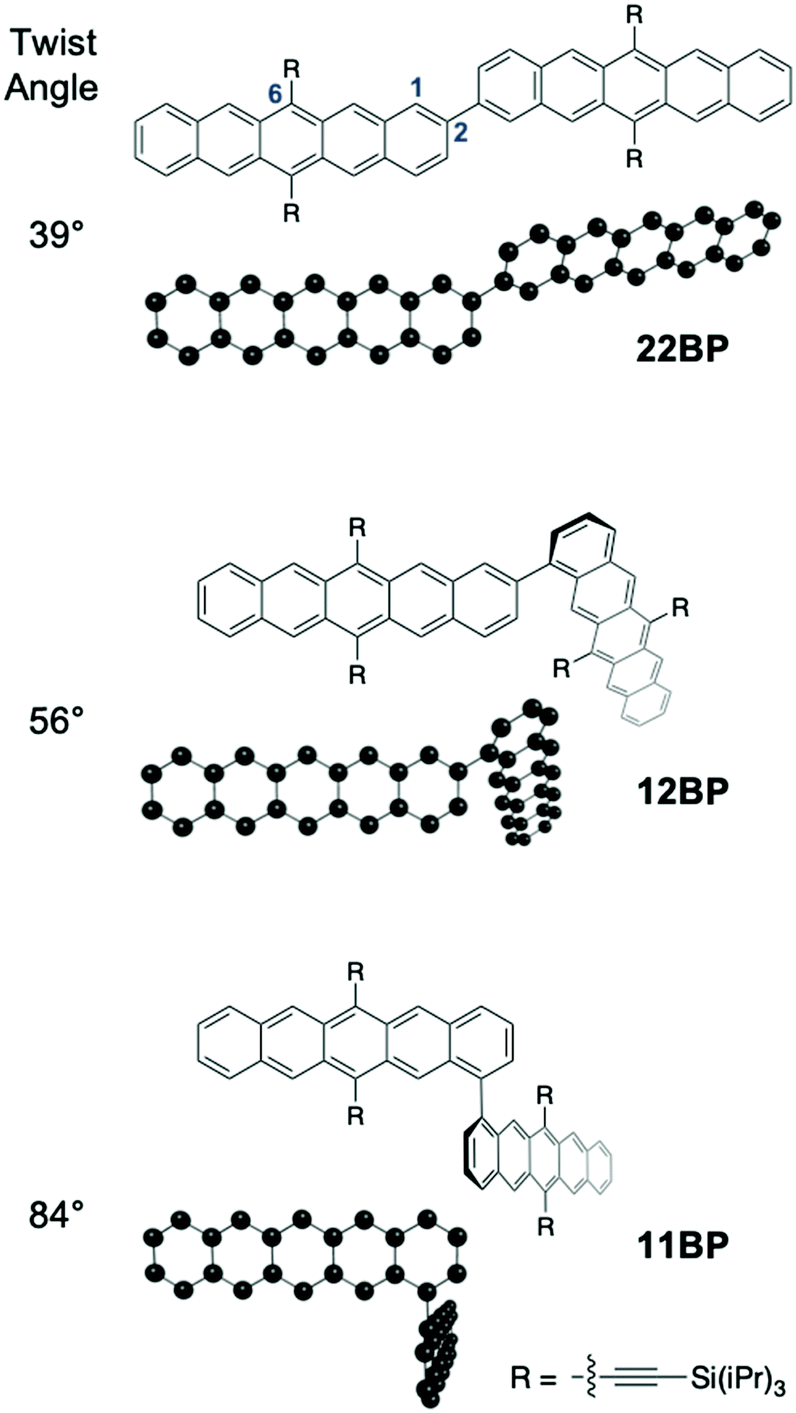 | ||
| Fig. 1 Minimized geometry revealing increasing interpentacene dihedral (twist) angle going from 22BP (39°), 12BP (56°) to 11BP (84°) (see ESI‡ for details). Only the pentacene monomers and their connectivities are shown in the optimized geometries for clarity. The ethynyl groups are used in determining the optimized geometries since they make a significant difference to the electronic structure. The 1, 2, and 6 positions on the pentacene are labeled on one of the pentacenes in 22BP. | ||
Results and discussion
We synthesized 12BP and 11BPvia the procedure discussed in the ESI‡ and 22BP according to a previous report.11 We used density functional theory to optimize the ground state geometry of these molecules and found that the dimers decrease in planarity from 22BP to 12BP to 11BP. Additional details on the calculations and coordinates for the geometry optimized structures are given in the ESI,‡ Appendix A. Overall, these three molecules exhibit a wide range of twist angles (Fig. 1)26 defined by the relative position of the planes between the pentacene units. For example, in 22BP this corresponds to the dihedral angle between atoms 1 and 2 of the first pentacene and atoms 2 and 3 of the second pentacene, using the standard numbering convention.52 In its optimized geometry, 22BP features an interplanar twist angle of about 39°, owing to the steric interactions between the hydrogens at the 1 and 3′ and 1′ and 3 positions. Linking the pentacenes at the 1 and 2′ positions (12BP) increases the steric hindrance between aryl hydrogens and increases the interplanar twist angle to 56°. Finally, when the pentacenes were connected at the 1 and 1′ positions (11BP), steric repulsion greatly increases the interplanar twist angle to 84°. It is also worth noting that free rotation of the pentacenes is also affected by changes in connectivity. The rotation in 12BP and 11BP is relatively hindered by the steric interaction between the pentacenes and the neighboring triisopropylsilyl (TIPS) group. Unlike previously reported approaches that use functionalization to modifying the dihedral twist angle,26 changing the connectivity has the advantage of a constant chemical structure, facilitating direct comparison between different dimers.In order to investigate how differing connectivity and geometry affect the triplet pair generation and recombination, we employed transient absorption spectroscopy (TAS) to probe the excited state dynamics of the dimers in dilute solution (Fig. 2a). The detailed singlet fission dynamics of 22BP have been previously detailed11 and the dynamics of 12BP and 11BP were quantified here using a similar procedure. Briefly, the transient features of the singlet exciton (S1) are quantified by comparison to time-resolved photoluminescence measurements and previously published spectra of similar pentacene compounds. The transient features corresponding to the triplet pair state (TT) are identified by comparison to triplet sensitization measurements (ESI‡). All three dimers featured qualitatively similar behavior, but with vastly different rate constants. Global analysis using a sequential decay model (S0 → S1 → TT → S0) was used to extract the photoinduced singlet and triplet pair spectra and to determine accurate time constants for singlet fission and the resulting triplet pair recombination processes (Fig. 2b).
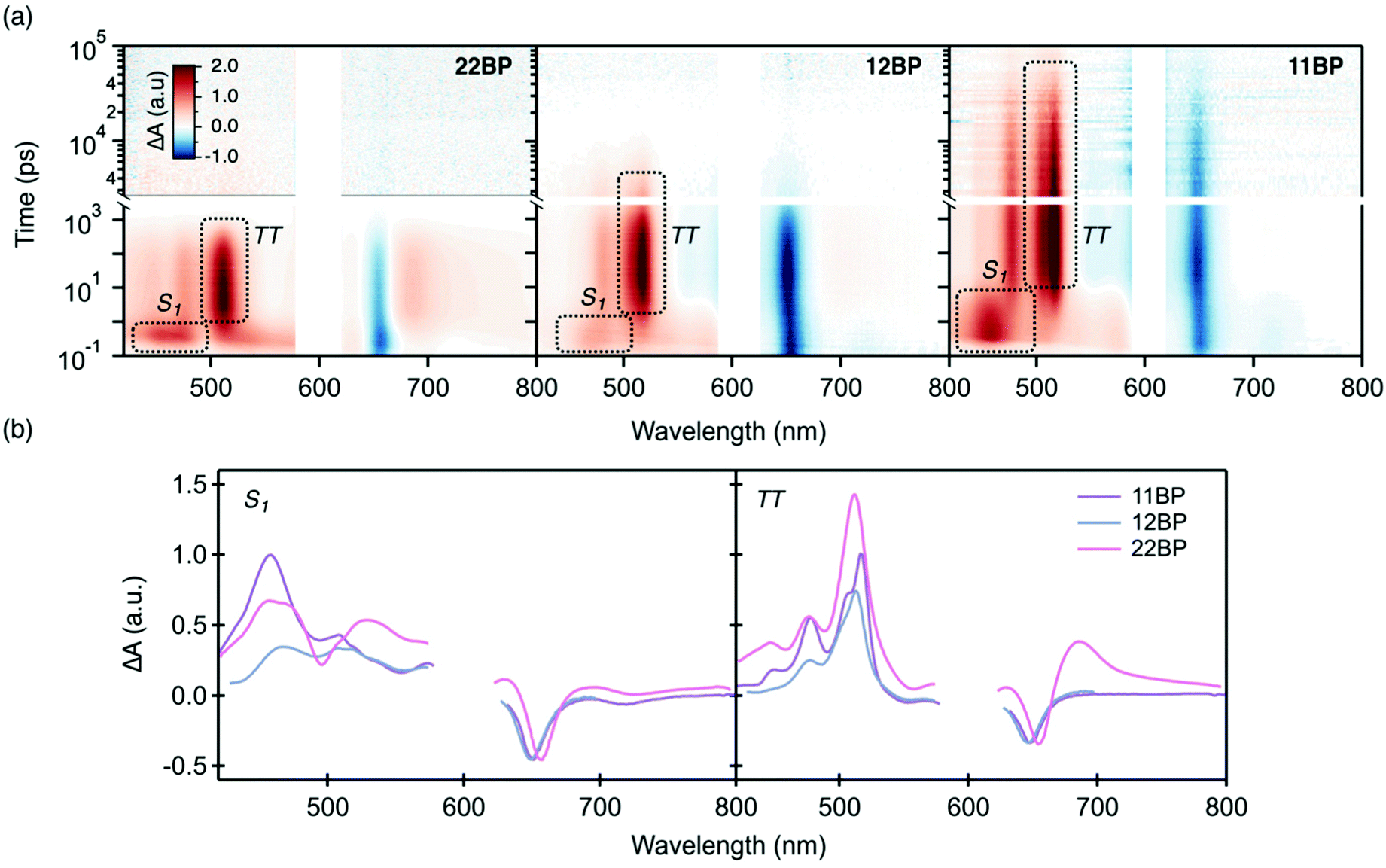 | ||
| Fig. 2 (a) Transient absorption spectra of 22BP, 12BP, and 11BP excited at 600 nm in chloroform. The most prominent singlet and triplet pair photoinduced absorptions are annotated. (b) Singlet (left) and triplet (right) signals of 11BP, 12BP, and 22BP as determined by global analysis. | ||
At early times the transient absorption spectra for optical pumping at 600 nm are dominated by the singlet photoinduced absorption, with a broad peak near ∼450 nm. The peak decays concurrently with the rise of the triplet photoinduced absorbance signal, dominated by a narrow photoinduced triplet–triplet absorption feature at ∼520 nm. The singlet and triplet spectra extracted from global analysis (Fig. 2b) are nearly identical all three compounds. The primary difference in the singlet spectra is the narrow bleach feature corresponding to the linear absorption near 500 nm that is superimposed on the broad positive background in 22BP and weakly visible in 12BP. The origin of this peak and its dependence on connectivity has been discussed previously.53 As excitation occurs into the lowest energy singlet state near 600 nm, this feature has no effect on the dynamics. In the triplet spectra, the primary differences between the different compounds occur in the NIR in the form of a photoinduced absorption feature (assigned to the normally forbidden T1 → T2 transition) that shows up as shoulder near the main ground state bleach (∼700 nm) and an additional excited state absorption feature near 1200 nm that has previous been assigned to a direct triplet to singlet transition (T1 → Sn, data in ESI‡).54,55 These two peaks have been previously shown to indicate a triplet pair with strong electronic interactions that mix the singlet and triplet potential energy surfaces.11,26,55,56 The lack of these features in 12BP and 11BP is consistent with a reduction in the interchromophore coupling strength. Throughout the iSF process, the total integrated intensity of the bleach feature near 660 nm is conserved (ESI‡), suggesting a quantitative singlet fission yield. Furthermore, at least one isosbestic point between the singlet and triplet transient spectra can be identified for each compound (ESI‡). These data along can be combined with kinetic arguments (singlet fission is orders of magnitude faster than radiative decay) to support a quantitative yield with high quality fits obtained from a sequential decay process (singlet to the triplet pair to the ground state) without inclusion of a parasitic loss process (Fig. S1, ESI‡).
While the three compounds exhibit a notable trend in the rate of iSF, the differences in the rates of triplet pair decay are more striking (Table 1). Although these rate constants are determined from global analysis of the full data set, the triplet pair dynamics can be visualized using kinetic traces through the maximum of the triplet photoinduced absorption (PIA) near 520 nm (Fig. 3). Though the singlet and triplet spectra are not completely separable at this wavelength (yielding a nonzero signal at early time), the dominant contribution is the rise and decay of the triplet photoinduced absorption signal, giving us a reasonable proxy for the triplet population. Our data set reveals that the more twisted dimers feature slower rates of singlet fission and slower triplet pair recombination. Additionally, we find that an increased interplanar twist angle slows triplet pair recombination more dramatically than their rate of formation by iSF. For example, the most planar molecule, 22BP, undergoes singlet fission the fastest, with a time constant of 0.76 ps, ∼10 times faster than the most twisted dimer, 11BP (∼7.1 ps). However, triplet pair recombination in 22BP (∼0.45 ns) is about 60 times shorter than the 11BP triplet pair lifetime (29 ns). Interestingly, the triplet pair lifetime in 22BP is ∼20 times shorter than the excited state (singlet) lifetime of the TIPS–pentacene monomer (13 ns). The triplet pair lifetime in 12BP is also shorter (3.2 ns) than the monomer lifetime, by a factor of ∼4. Importantly, the triplet population decays following a single time constant (τ(TT) in Table 1), suggesting that the dominant recombination process involves the loss of both triplets simultaneously from the net singlet triplet pair. This behavior is in stark contrast to other iSF compounds, in which a distinct biexponential decay behavior of the triplet pair indicates a sequential loss of single triplet excitons involving a triplet pair with a net multiplicity m different than one.36 Since only the net singlet m=1(TT) is formed in these strongly coupled dimers, we omit the m = 1 factor from our notation.
| Compound | τ iSF (ps) | τ (TT) (ns) | Dihedral angle (°) |
|---|---|---|---|
| 11BP | 7.1 | 29 | 84 |
| 12BP | 2.5 | 3.2 | 56 |
| 22BP | 0.76 | 0.45 | 39 |
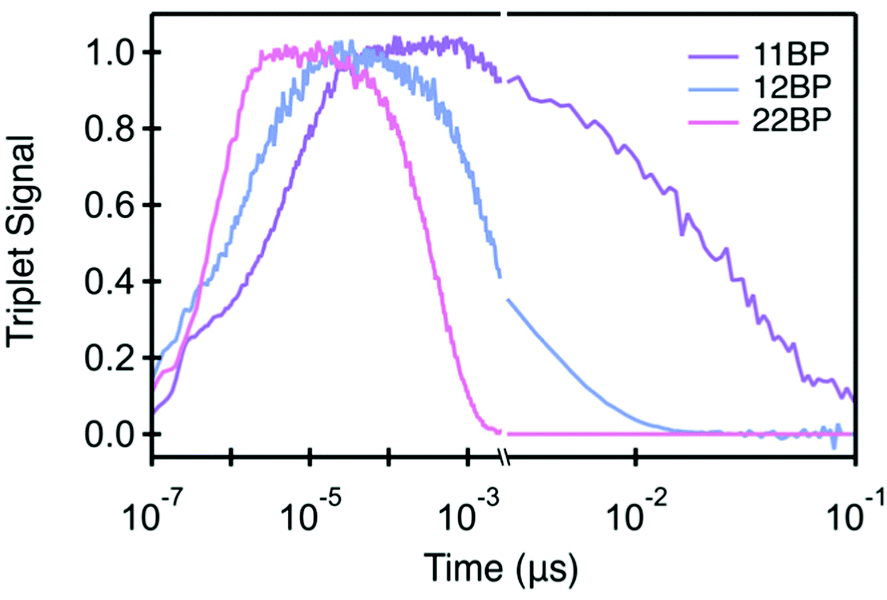 | ||
| Fig. 3 Triplet rise and decay for 11BP, 12BP, and 22BP. The triplet signal is normalized as shown. | ||
The only spin conserving process that can result in the loss of both triplets involves a direct decay of the net singlet triplet pair state (TT) to either the ground state (TT → S0) via a direct multiexciton internal conversion process or to the singlet excited state (TT → S1) via a thermally assisted triplet–triplet annihilation process. Regeneration of the excited state singlet exciton is typically only considered in singlet fission compounds with (nearly) degenerate triplet pair and singlet states, which exhibit a characteristic delayed fluorescence signal from the regenerated singlet following triplet fusion. This process has been observed in tetracene films and in tetracene–pentacene dimers,39,50,57 with obvious spectroscopic signatures, including reasonably high photoluminescence (PL) quantum yields and long-lived emission beyond the singlet lifetime. We note that neither of these signatures are present for these compounds (ESI‡), which have PL quantum yields below 1% and extremely rapid PL decay, nor for any other pentacene based chromophores.11 Though direct triplet pair emission has been observed in pentacene films on microsecond time scales,49 the overall yield is too low to explain the dramatic excited state lifetime shortening observed here.
To completely rule out repopulation of the singlet exciton via a thermally assisted triplet–triplet annihilation processes, we performed temperature dependent transient absorption measurements of 22BP embedded in an inert polystyrene (PS) matrix (Fig. 4). Similar measurements were performed in the solution phase over a smaller temperature range show identical behavior (ESI‡). As the temperature is reduced, we observe a small red-shift (<1.5 nm total) in the triplet excited state absorption feature near 520 nm and a small narrowing of the ground state bleach (Fig. 4a). More importantly, we observe that the triplet pair lifetime is completely temperature independent over the range of temperatures measured (90–300 K), ruling out regeneration of S1 through a thermally activated process. This result is consistent with expectations based on the widely reported energetics in pentacene based singlet fission materials, in which the energy of the singlet is expected to be more than 150–200 meV greater than the triplet pair energy.58,59 This corresponds to an energy barrier that is more than 20× the amount of thermal energy available at 90 K, rendering thermally assisted population of S1 extremely unlikely, consistent with our data.
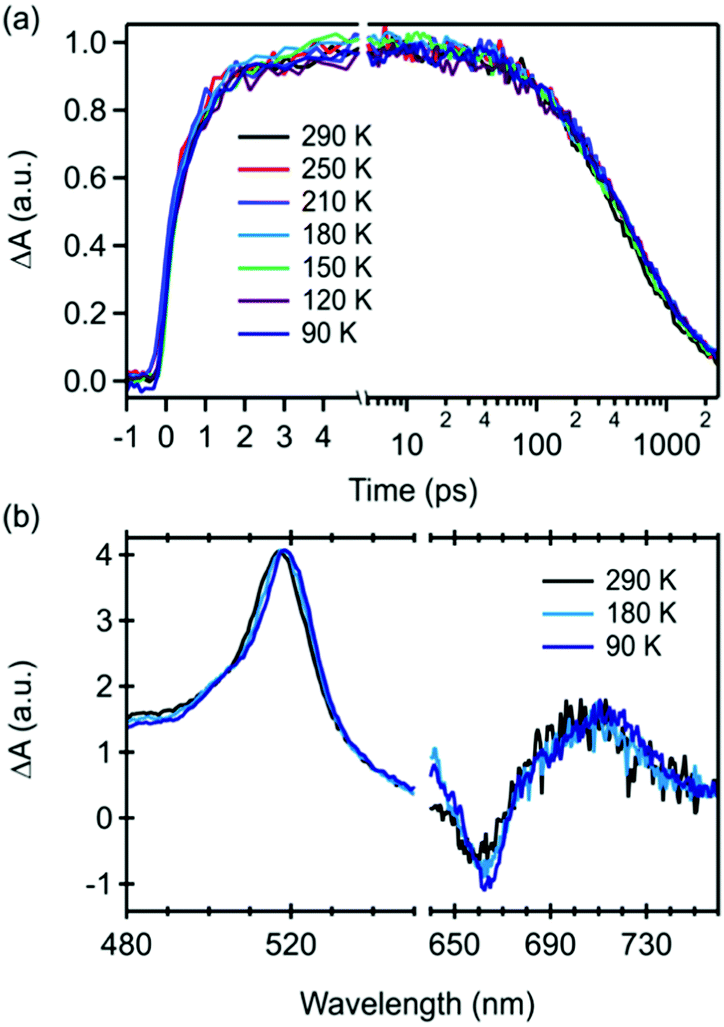 | ||
| Fig. 4 Transient absorption of 22BP in an inert polystyrene matrix over a range of temperatures 90–290 K. There is no effect of temperature on (a) triplet pair formation and decay and (b) triplet excited state absorption profile. | ||
To understand the radiationless multiexciton internal conversion process, we turn to theoretical studies. We calculate triplet pair recombination rates (krec) at 300 K for 11BP, 12BP, and 22BP using a new singularity-free formulation of the internal conversion (IC) rate60 that is based on an existing analytic approach, the vibrational correlation function formalism.61 The IC rate is formulated in the Fermi's golden rule limit, with interactions between the vibrational modes of different electronic states explicitly treated via the Duschinsky relation and the coupling between the two vibronic states obtained by calculating the nonadiabatic coupling vector. Details of the electronic structure methods used to obtain the optimized geometry of the S0 and TT states, the normal modes for each electronic state, the adiabatic energy gap between the two states, and the nonadiabatic coupling vector are provided in the ESI.‡
The recombination time constant (τTT = 1/krec) for three bipentacenes is labeled either S0 or TT to indicate the electronic state geometry at which the nonadiabatic coupling vector is evaluated. The results are summarized in Table 2. We find that the recombination time constant for 11BP is 10 times longer than 12BP and 60 times longer than 22BP. We note that the calculated timescales are within an order of magnitude of the experimental results and in keeping with experimental findings, recombination rates decrease as the dihedral angle increases. Our calculations also confirm the observed temperature independence of the recombination rate; we find the rate at 77 K for 22BP is nearly identical to the rate at 300 K as shown in Table 2. We find that the decrease in recombination rates with increasing twist angle can be directly attributed to a corresponding decrease in the magnitude of the nonadiabatic coupling vector. Our calculations confirm that despite the large energy gap between the triplet pair and the ground state, non-radiative recombination is an allowed process that can proceed rapidly in bipentacenes. We note iSF likely proceeds through a conical intersection (CI) in bipentacenes, so the IC rate formalism used here is not applicable.62 Given that the rate of a reaction through a CI is not simply proportional to the magnitude of the nonadiabatic coupling vector but strongly dependent on the topography of the potential energy surface in the vicinity of the CI, a direct dynamic simulation will be necessary to establish the origin of the much smaller observed slowdown in iSF rates with twist angle.
| Molecule | Geometry of evaluation | Calculated τTT (ns) | Experimental τTT (ns) |
|---|---|---|---|
| 11BP (300 K) | S0 | 300 | 29 |
| TT | 190 | ||
| 12BP (300 K) | S0 | 30 | 32 |
| TT | 13 | ||
| 22BP (300 K) | S0 | 4.8 | 0.45 |
| TT | 3.1 | ||
| 22BP (77 K) | S0 | 6.4 | |
| TT | 4.3 |
Conclusion
We have studied the correlation between interplanar twist angles in pentacene dimers and the triplet pair recombination dynamics, finding that a radiationless multiexciton internal conversion process dominates in strongly coupled molecular SF dimers. We found that with more twist, molecules showed a systematic reduction in rates of singlet fission dynamics while recombination rates decreased more dramatically. Because triplet pair recombination is slowed much more dramatically with twist angle than singlet fission is slowed, we can greatly increase triplet lifetimes by increasing the interplanar twist angle without sacrificing rapid singlet fission.36,63 This extension in the triplet lifetimes makes these materials very promising for future design principles of iSF chromophores, and demonstrates the important role structure–property relationships play in singlet fission.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0022036. S. N. S. and B. C. thank the NSF GRFP (grant number DGE 11-44155). This research used resources at the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility at Brookhaven National Laboratory under contract DE-SC0012704.References
- W. Shockley and H. J. Queisser, Detailed Balance Limit of Efficiency of P-n Junction Solar Cells, J. Appl. Phys., 1961, 32(3), 510–519, DOI:10.1063/1.1736034.
- M. A. Green, Third Generation Photovoltaics: Ultra-High Conversion Efficiency at Low Cost, Prog. Photovoltaics, 2001, 9(2), 123–135, DOI:10.1002/pip.360.
- M. C. Hanna and A. J. Nozik, Solar Conversion Efficiency of Photovoltaic and Photoelectrolysis Cells with Carrier Multiplication Absorbers, J. Appl. Phys., 2006, 100(7), 074510, DOI:10.1063/1.2356795.
- M. B. Smith and J. Michl, Singlet Fission, Chem. Rev., 2010, 110(11), 6891–6936, DOI:10.1021/cr1002613.
- S. Singh, W. J. Jones, W. Siebrand, B. P. Stoicheff and W. G. Schneider, Laser Generation of Excitons and Fluorescence in Anthracene Crystals, J. Chem. Phys., 1965, 42(1), 330–342, DOI:10.1063/1.1695695.
- R. E. Merrifield, P. Avakian and R. P. Groff, Fission of Singlet Excitons into Pairs of Triplet Excitons in Tetracene Crystals, Chem. Phys. Lett., 1969, 3(6), 386–388, DOI:10.1016/0009-2614(69)80144-2.
- C. E. Swenberg and W. T. Stacy, Bimolecular Radiationless Transitions in Crystalline Tetracene, Chem. Phys. Lett., 1968, 2(5), 327–328, DOI:10.1016/0009-2614(68)80087-9.
- C. Jundt, G. Klein, B. Sipp, J. Le Moigne, M. Joucla and A. A. Villaeys, Exciton Dynamics in Pentacene Thin Films Studied by Pump-Probe Spectroscopy, Chem. Phys. Lett., 1995, 241(1), 84–88, DOI:10.1016/0009-2614(95)00603-2.
- H. Marciniak, M. Fiebig, M. Huth, S. Schiefer, B. Nickel, F. Selmaier and S. Lochbrunner, Ultrafast Exciton Relaxation in Microcrystalline Pentacene Films, Phys. Rev. Lett., 2007, 99(17), 176402, DOI:10.1103/PhysRevLett.99.176402.
- M. W. B. Wilson, A. Rao, B. Ehrler and R. H. Friend, Singlet Exciton Fission in Polycrystalline Pentacene: From Photophysics toward Devices, Acc. Chem. Res., 2013, 46(6), 1330–1338, DOI:10.1021/ar300345h.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. T. Trinh, B. Choi, J. Xia, E. J. Taffet, J. Z. Low, J. R. Miller, X. Roy, X.-Y. Zhu, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Quantitative Intramolecular Singlet Fission in Bipentacenes, J. Am. Chem. Soc., 2015, 137(28), 8965–8972, DOI:10.1021/jacs.5b04986.
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss, J. Wu, R. H. Friend and N. C. Greenham, Tuneable Singlet Exciton Fission and Triplet–Triplet Annihilation in an Orthogonal Pentacene Dimer, Adv. Funct. Mater., 2015, 25(34), 5452–5461, DOI:10.1002/adfm.201501537.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Singlet Fission in Pentacene Dimers, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(17), 5325–5330, DOI:10.1073/pnas.1422436112.
- T. Sakuma, H. Sakai, Y. Araki, T. Mori, T. Wada, N. V. Tkachenko and T. Hasobe, Long-Lived Triplet Excited States of Bent-Shaped Pentacene Dimers by Intramolecular Singlet Fission, J. Phys. Chem. A, 2016, 120(11), 1867–1875, DOI:10.1021/acs.jpca.6b00988.
- A. Akdag, A. Wahab, P. Beran, L. Rulíšek, P. I. Dron, J. Ludvík and J. Michl, Covalent Dimers of 1,3-Diphenylisobenzofuran for Singlet Fission: Synthesis and Electrochemistry, J. Org. Chem., 2015, 80(1), 80–89, DOI:10.1021/jo502004r.
- C. Hetzer, D. M. Guldi and R. R. Tykwinski, Pentacene Dimers as a Critical Tool for the Investigation of Intramolecular Singlet Fission, Chem. – Eur. J., 2018, 24(33), 8245–8257, DOI:10.1002/chem.201705355.
- N. V. Korovina, J. Joy, X. Feng, C. Feltenberger, A. I. Krylov, S. E. Bradforth and M. E. Thompson, Linker-Dependent Singlet Fission in Tetracene Dimers, J. Am. Chem. Soc., 2018, 140(32), 10179–10190, DOI:10.1021/jacs.8b04401.
- H. Sakai, R. Inaya, H. Nagashima, S. Nakamura, Y. Kobori, N. V. Tkachenko and T. Hasobe, Multiexciton Dynamics Depending on Intramolecular Orientations in Pentacene Dimers: Recombination and Dissociation of Correlated Triplet Pairs, J. Phys. Chem. Lett., 2018, 9(12), 3354–3360, DOI:10.1021/acs.jpclett.8b01184.
- M. Chen, M. D. Krzyaniak, J. N. Nelson, Y. J. Bae, S. M. Harvey, R. D. Schaller, R. M. Young and M. R. Wasielewski, Quintet-Triplet Mixing Determines the Fate of the Multiexciton State Produced by Singlet Fission in a Terrylenediimide Dimer at Room Temperature, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(17), 8178–8183, DOI:10.1073/pnas.1820932116.
- N. R. Monahan, D. Sun, H. Tamura, K. W. Williams, B. Xu, Y. Zhong, B. Kumar, C. Nuckolls, A. R. Harutyunyan, G. Chen, H. L. Dai, D. Beljonne, Y. Rao and X. Y. Zhu, Dynamics of the Triplet-Pair State Reveals the Likely Coexistence of Coherent and Incoherent Singlet Fission in Crystalline Hexacene, Nat. Chem., 2017, 9(4), 341–346, DOI:10.1038/nchem.2665.
- G. B. Piland and C. J. Bardeen, How Morphology Affects Singlet Fission in Crystalline Tetracene, J. Phys. Chem. Lett., 2015, 6(10), 1841–1846, DOI:10.1021/acs.jpclett.5b00569.
- R. D. Pensack, A. J. Tilley, S. R. Parkin, T. S. Lee, M. M. Payne, D. Gao, A. A. Jahnke, D. G. Oblinsky, P.-F. Li, J. E. Anthony, D. S. Seferos and G. D. Scholes, Exciton Delocalization Drives Rapid Singlet Fission in Nanoparticles of Acene Derivatives, J. Am. Chem. Soc., 2015, 137(21), 6790–6803, DOI:10.1021/ja512668r.
- S. T. Roberts, R. E. Mcanally, J. N. Mastron, D. H. Webber, M. T. Whited, R. L. Brutchey, M. E. Thompson and S. E. Bradforth, Efficient Singlet Fission Discovered in a Disordered Acene Film, J. Am. Chem. Soc., 2012, 134(14), 6388–6400, DOI:10.1021/ja300504t.
- J. N. Mastron, S. T. Roberts, R. E. McAnally, M. E. Thompson and S. E. Bradforth, Aqueous Colloidal Acene Nanoparticles: A New Platform for Studying Singlet Fission, J. Phys. Chem. B, 2013, 117, 15519–15526, DOI:10.1021/jp4057972.
- D. Lubert-Perquel, E. Salvadori, M. Dyson, P. N. Stavrinou, R. Montis, H. Nagashima, Y. Kobori, S. Heutz and C. W. M. Kay, Identifying Triplet Pathways in Dilute Pentacene Films, Nat. Commun., 2018, 9(1), 4222, DOI:10.1038/s41467-018-06330-x.
- E. G. Fuemmeler, S. N. Sanders, A. B. Pun, E. Kumarasamy, T. Zeng, K. Miyata, M. L. Steigerwald, X.-Y. Zhu, M. Y. Sfeir, L. M. Campos and N. Ananth, A Direct Mechanism of Ultrafast Intramolecular Singlet Fission in Pentacene Dimers, ACS Cent. Sci., 2016, 2(5), 316–324, DOI:10.1021/acscentsci.6b00063.
- E. Kumarasamy, S. N. Sanders, M. J. Y. Tayebjee, A. Asadpoordarvish, T. J. H. Hele, E. G. Fuemmeler, A. B. Pun, L. M. Yablon, J. Z. Low, D. W. Paley, J. C. Dean, B. Choi, G. D. Scholes, M. L. Steigerwald, N. Ananth, D. R. McCamey, M. Y. Sfeir and L. M. Campos, Tuning Singlet Fission in π-Bridge-π Chromophores, J. Am. Chem. Soc., 2017, 139(36), 12488–12494, DOI:10.1021/jacs.7b05204.
- K. R. Parenti, G. He, S. N. Sanders, A. B. Pun, E. Kumarasamy, M. Y. Sfeir and L. M. Campos, Bridge Resonance Effects in Singlet Fission, J. Phys. Chem. A, 2020, 124(45), 9392–9399, DOI:10.1021/acs.jpca.0c08427.
- J. Zirzlmeier, R. Casillas, S. R. Reddy, P. B. Coto, D. Lehnherr, E. T. Chernick, I. Papadopoulos, M. Thoss, R. R. Tykwinski and D. M. Guldi, Solution-Based Intramolecular Singlet Fission in Cross-Conjugated Pentacene Dimers, Nanoscale, 2016, 8(19), 10113–10123, 10.1039/C6NR02493A.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, B. T. Phelan, M. D. Krzyaniak, S. R. Reddy, P. B. Coto, N. E. Horwitz, R. M. Young, F. J. White, F. Hampel, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Unified Model for Singlet Fission within a Non-Conjugated Covalent Pentacene Dimer, Nat. Commun., 2017, 8, 15171, DOI:10.1038/ncomms15171.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, S. R. Reddy, B. T. Phelan, M. D. Krzyaniak, M. K. Volland, P. B. Coto, R. M. Young, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Evidence for Charge-Transfer Mediation in the Primary Events of Singlet Fission in a Weakly Coupled Pentacene Dimer, Chem, 2018, 4(5), 1092–1111, DOI:10.1016/j.chempr.2018.04.006.
- L. M. Yablon, S. N. Sanders, H. Li, K. R. Parenti, E. Kumarasamy, K. J. Fallon, M. J. A. Hore, A. Cacciuto, M. Y. Sfeir and L. M. Campos, Persistent Multiexcitons from Polymers with Pendent Pentacenes, J. Am. Chem. Soc., 2019, 141(24), 9564–9569, DOI:10.1021/jacs.9b02241.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Singlet Fission in Polypentacene, Chem, 2016, 1(3), 505–511, DOI:10.1016/j.chempr.2016.08.016.
- J. Z. Low, S. N. Sanders and L. M. Campos, Correlating Structure and Function in Organic Electronics: From Single Molecule Transport to Singlet Fission, Chem. Mater., 2015, 27(16), 5453–5463, DOI:10.1021/cm502366x.
- A. M. Alvertis, S. Lukman, T. J. H. Hele, E. G. Fuemmeler, J. Feng, J. Wu, N. C. Greenham, A. W. Chin and A. J. Musser, Switching between Coherent and Incoherent Singlet Fission via Solvent-Induced Symmetry Breaking, J. Am. Chem. Soc., 2019, 141(44), 17558–17570, DOI:10.1021/jacs.9b05561.
- A. B. Pun, A. Asadpoordarvish, E. Kumarasamy, M. J. Y. Tayebjee, D. Niesner, D. R. McCamey, S. N. Sanders, L. M. Campos and M. Y. Sfeir, Ultra-Fast Intramolecular Singlet Fission to Persistent Multiexcitons by Molecular Design, Nat. Chem., 2019, 11(9), 821–828, DOI:10.1038/s41557-019-0297-7.
- N. V. Korovina, C. H. Chang and J. C. Johnson, Spatial Separation of Triplet Excitons Drives Endothermic Singlet Fission, Nat. Chem., 2020, 12(4), 391–398, DOI:10.1038/s41557-020-0422-7.
- R. E. Merrifield, Magnetic Effects on Triplet Exciton Interactions, Pure Appl. Chem., 1971, 481, DOI:10.1351/pac197127030481.
- J. J. Burdett and C. J. Bardeen, Quantum Beats in Crystalline Tetracene Delayed Fluorescence Due to Triplet Pair Coherences Produced by Direct Singlet Fission, J. Am. Chem. Soc., 2012, 134(20), 8597–8607, DOI:10.1021/ja301683w.
- L. R. Weiss, S. L. Bayliss, F. Kraffert, K. J. Thorley, J. E. Anthony, R. Bittl, R. H. Friend, A. Rao, N. C. Greenham and J. Behrends, Strongly Exchange-Coupled Triplet Pairs in an Organic Semiconductor, Nat. Phys., 2017, 13, 176–181, DOI:10.1038/nphys3908.
- M. J. Y. Tayebjee, S. N. Sanders, E. Kumarasamy, L. M. Campos, M. Y. Sfeir and D. R. McCamey, Quintet Multiexciton Dynamics in Singlet Fission, Nat. Phys., 2016, 13(2), 182–188, DOI:10.1038/nphys3909.
- A. B. Pun, S. N. Sanders, M. Y. Sfeir, L. M. Campos and D. N. Congreve, Annihilator Dimers Enhance Triplet Fusion Upconversion, Chem. Sci., 2019, 10(14), 3969–3975, 10.1039/C8SC03725F.
- I. A. Mikhailov, S. Tafur and A. E. Masunov, Double Excitations and State-to-State Transition Dipoles in π – π* Excited Singlet States of Linear Polyenes: Time-Dependent Density-Functional Theory versus Multiconfigurational Methods, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 77(1), 012510, DOI:10.1103/PhysRevA.77.012510.
- R. Englman and J. Jortner, The Energy Gap Law for Non-Radiative Decay in Large Molecules, J. Lumin., 1970, 1–2, 134–142, DOI:10.1016/0022-2313(70)90029-3.
- H. L. Stern, A. J. Musser, S. Gelinas, P. Parkinson, L. M. Herz, M. J. Bruzek, J. Anthony, R. H. Friend and B. J. Walker, Identification of a Triplet Pair Intermediate in Singlet Exciton Fission in Solution, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(25), 7656–7661, DOI:10.1073/pnas.1503471112.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X.-Y. Zhu, L. M. Campos and M. Y. Sfeir, A Design Strategy for Intramolecular Singlet Fission Mediated by Charge-Transfer States in Donor–Acceptor Organic Materials, Nat. Mater., 2015, 14(4), 426–433, DOI:10.1038/nmat4175.
- J. Hu, K. Xu, L. Shen, Q. Wu, G. He, J.-Y. Wang, J. Pei, J. Xia and M. Y. Sfeir, New Insights into the Design of Conjugated Polymers for Intramolecular Singlet Fission, Nat. Commun., 2018, 9(1), 2999, DOI:10.1038/s41467-018-05389-w.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, B. T. Phelan, M. D. Krzyaniak, S. R. Reddy, P. B. Coto, N. E. Horwitz, R. M. Young, F. J. White, F. Hampel, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Unified Model for Singlet Fission within a Non-Conjugated Covalent Pentacene Dimer, Nat. Commun., 2017, 8, 15171 CrossRef CAS PubMed.
- A. J. Musser and J. Clark, Triplet-Pair States in Organic Semiconductors, Annu. Rev. Phys. Chem., 2019, 70(1), 323–351, DOI:10.1146/annurev-physchem-042018-052435.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, K. Appavoo, M. L. Steigerwald, L. M. Campos and M. Y. Sfeir, Exciton Correlations in Intramolecular Singlet Fission, J. Am. Chem. Soc., 2016, 138(23), 7289–7297, DOI:10.1021/jacs.6b00657.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Intramolecular Singlet Fission in Oligoacene Heterodimers, Angew. Chem., Int. Ed., 2016, 55(10), 3373–3377, DOI:10.1002/anie.201510632.
- T. J. Chow, C.-C. Wu, T.-H. Chuangh, H.-H. Hsieh and H.-H. Huang, Synthesis and Applications of Soluble Pentacene Precursors and Related Compounds, US 2013/0017497A1, 2013 Search PubMed.
- T. J. H. Hele, E. G. Fuemmeler, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, L. M. Campos and N. Ananth, Anticipating Acene-Based Chromophore Spectra with Molecular Orbital Arguments, J. Phys. Chem. A, 2019, 123(13), 2527–2536, DOI:10.1021/acs.jpca.8b12222.
- S. Khan and S. Mazumdar, Diagrammatic Exciton Basis Theory of the Photophysics of Pentacene Dimers, J. Phys. Chem. Lett., 2017, 8(18), 4468–4478, DOI:10.1021/acs.jpclett.7b01829.
- M. T. Trinh, A. Pinkard, A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, L. M. Campos, X. Roy and X.-Y. Zhu, Distinct Properties of the Triplet Pair State from Singlet Fission, Sci. Adv., 2017, 3(7), e1700241, DOI:10.1126/sciadv.1700241.
- S. Khan and S. Mazumdar, Theory of Transient Excited State Absorptions in Solid Pentacene with Implications for Singlet Fission, 2017 Search PubMed.
- Y. Wan, Z. Guo, T. Zhu, S. Yan, J. Johnson and L. Huang, Cooperative Singlet and Triplet Exciton Transport in Tetracene Crystals Visualized by Ultrafast Microscopy, Nat. Chem., 2015, 7(10), 785–792, DOI:10.1038/nchem.2348.
- E. Busby, T. C. Berkelbach, B. Kumar, A. Chernikov, Y. Zhong, H. Hlaing, X.-Y. Zhu, T. F. Heinz, M. S. Hybertsen, M. Y. Sfeir, D. R. Reichman, C. Nuckolls and O. Yaffe, Multiphonon Relaxation Slows Singlet Fission in Crystalline Hexacene, J. Am. Chem. Soc., 2014, 136(30), 10654–10660, DOI:10.1021/ja503980c.
- S. Sanders, E. Kumarasamy, K. J. Fallon, M. Sfeir and L. Campos, Singlet Fission in a Hexacene Dimer: Energetics Dictate Dynamics, Chem. Sci., 2020, 11, 1079–1084, 10.1039/C9SC05066C.
- K. Miyazaki and N. Ananth, A Singularity-Free Internal Conversion Golden Rule Rate Formulation for Polyatomic Molecules, 2021, submitted.
- Q. Peng, Y. Niu, C. Deng and Z. Shuai, Vibration Correlation Function Formalism of Radiative and Non-Radiative Rates for Complex Molecules, Chem. Phys., 2010, 370, 215–222, DOI:10.1016/j.chemphys.2010.03.004.
- M. S. Schuurman and A. Stolow, Dynamics at Conical Intersections, Annu. Rev. Phys. Chem., 2018, 69(1), 427–450, DOI:10.1146/annurev-physchem-052516-050721.
- A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, D. N. Congreve and L. M. Campos, Triplet Harvesting from Intramolecular Singlet Fission in Polytetracene, Adv. Mater., 2017, 29(41), 1701416, DOI:10.1002/adma.201701416.
Footnotes |
| † In dedication to Seth Marder on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Includes synthetic and computational methods, characterization, spectroscopic data and analysis, supplementary figures, and supplementary tables. See DOI: 10.1039/d1mh01201k |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |