
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural alterations on the TEMPO scaffold and their impact on the performance as active materials for redox flow batteries†
Philip
Rohland
ab,
Oliver
Nolte
ab,
Kristin
Schreyer
ab,
Helmar
Görls
c,
Martin D.
Hager
 ab and
Ulrich S.
Schubert
*ab
ab and
Ulrich S.
Schubert
*ab
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstrasse 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de
bCenter for Energy and Environmental Chemistry Jena (CEEC Jena), Friedrich Schiller University Jena, Philosophenweg 7a, 07743 Jena, Germany
cInstitute for Inorganic and Analytical Chemistry (IAAC), Friedrich-Schiller University Jena, Humboldtstrasse 8, 07743 Jena, Germany
First published on 26th March 2022
Abstract
Trimethylammonium-2,2,6,6-tetramethylpiperidine-1-oxyl chloride (TMA-TEMPO) has been intensively studied for its usage in aqueous organic redox flow batteries. Straightforward synthesis, reliable electrochemistry, fast kinetics and high cycling stability are the advantages of this active material. Nevertheless, it has been shown that elevated temperatures and high states of charge accelerate the decomposition of this material. Hence, a comparative study was performed with five new and one known TEMPO derivatives, to elucidate the structure–stability relationship of the TEMPO scaffold and to investigate the influence on the battery performance. The results show that the introduction of linkers between the solubility-promoting group and the piperidyl core or enhanced shielding of the radical has a great impact on the stability during cycling.
Introduction
Conventional energy production based on fossil energy sources, such as coal or natural gas, contributes to increasing amounts of greenhouse gasses in the atmosphere. Therefore, there is an urgent need for decarbonization of the energy production around the world. Renewables offer a viable alternative, with solar and wind power being the most prominent representatives.3–6 However, unlike conventional power plants based on nuclear power or fossil fuels, these technologies suffer from a discontinuous power output, depending on weather conditions and daytime. To compensate supply shortages or overproduction in a smart grid, the implementation of large-scale energy storage capabilities is highly important. Nowadays, many different technologies have been evaluated for long-term storage of electrical energy.7 Since they enable decentralized and scalable applications, electrochemical storage technologies, in particular battery systems, are by far the most practical and promising solution. Among other batteries based on lead and lithium ions, redox flow batteries (RFB) stand out due to the possibility of tailoring their characteristics according to a broad range of applications with long lifetimes and low self-discharge.8 Until now, the all-vanadium RFB is the most technologically advanced RFB type. However, the technology failed to reach high market share in the large-scale battery sector.8–10 The main reason is the cost-intensive, toxic and highly corrosive vanadium electrolyte based on concentrated sulphuric acid.11 Organic-based RFB electrolytes may be able to overcome these drawbacks by providing inexpensive water-based electrolytes, making the technology environmentally benign and even more competitive to the widely used lithium and sodium batteries.Currently developed organic and all organic redox flow batteries (AORFB) use, e.g., alloxazine,12,13 flavine,14 quinones,15–17 ferrocene,18,19 stable organic radicals20–22 and viologen derivatives,21,23,24 showing the wide range of possible scaffolds and the possibility to tailor the characteristics of the storage materials. Nevertheless, many organic electrolytes do not meet the requirements for long cycle stability and additionally only a narrow number of molecules were tested under more realistic conditions, e.g., at higher temperatures or dwell times in vulnerable redox state like high state of charge (SOC).17,25
Scaffolds based on the 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) radicals are among the best-investigated active materials and considered as one of the most promising catholyte materials.26 However, there are a few studies reporting about the thermal decomposition of TEMPO radicals and their associated oxoammonium cations.1 Thus, we developed a new battery cycling protocol to investigate the influence of temperature and dwell time at high SOCs on the lifetime of the active material.27
In this work, we study six TEMPO derivatives with five of them being synthesised for the first time. The molecules are then compared with N,N,N-2,2,6,6-heptamethylpiperidinyloxy-4-ammonium chloride (TMA-TEMPO), the current forerunner, regarding their suitability as new catholyte materials for ORFBs and to elucidate a structure–stability relationship. We therefore focus on the influence of structural changes on the TEMPO scaffold and on the molecular stability of the compounds upon thermal stresses and dwell times at high SOCs. We were able to demonstrate that the introduction of a linker between the piperidyl core and the solubility promoting group or enhanced shielding of the radical has a great impact on the stability, leading to a TEMPO derivative which is stable at room temperature even if dwell times are applied. However, the compound was found to be less stable at an elevated temperature of 40 °C.
Scope
In a recent publication from our group, we investigated the decomposition of TMA-TEMPO (1) and the associated oxoammonium cation 1+ in detail. The results of the extensive gas chromatography–mass spectrometry experiments support the findings by Ma et al. and Takata et al.1,2 The proposed mechanism is summarized in Scheme 1. Furthermore, the findings of the study suggest three possible decomposition pathways (see Scheme 2).27 The first starts with a Hoffmann-like elimination of the trimethylammonium group and generation of a double bond. Liu et al. recently proposed that the introduction of a linker between the solubility-promoting group (SPG) and the piperidyl core may result in an increase of the stability of the active material.26 The authors suggested that the decoupling of the strong electron-withdrawing trimethyl ammonium group from the piperidine core reduces electronic effects, like inductive effects, that can promote ring opening reactions. Furthermore, the introduction of a linker may prevent the Hoffmann-like elimination that results in the formation of endocyclic double bonds. To investigate these effects, we decided to synthesise the amide molecule 2 as well as the sulfonamide derivate 3 and compare them to the ether system 4, which Liu26 had introduced previously. The electron-withdrawing effect expected for both the amide and the sulfonamide groups is lower compared to that of the ether 4 and, thus, the decomposition rate may be further reduced. Additionally, these linkers may be more stable towards possible pH changes, caused by water splitting or other side reactions during battery cycling.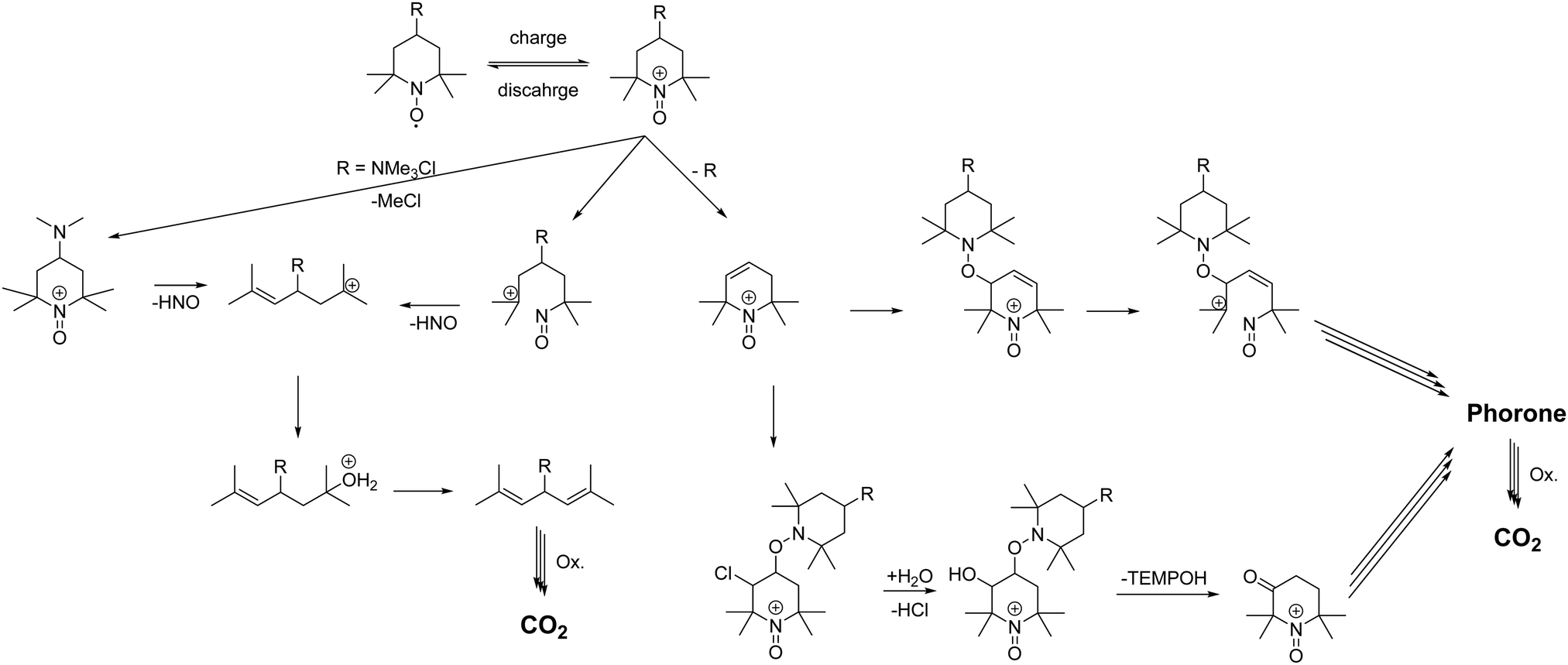 | ||
| Scheme 1 Schematic representation of the proposed decomposition mechanism of TEMPO derivatives according to Ma et al. and Takata et al.1,2 | ||
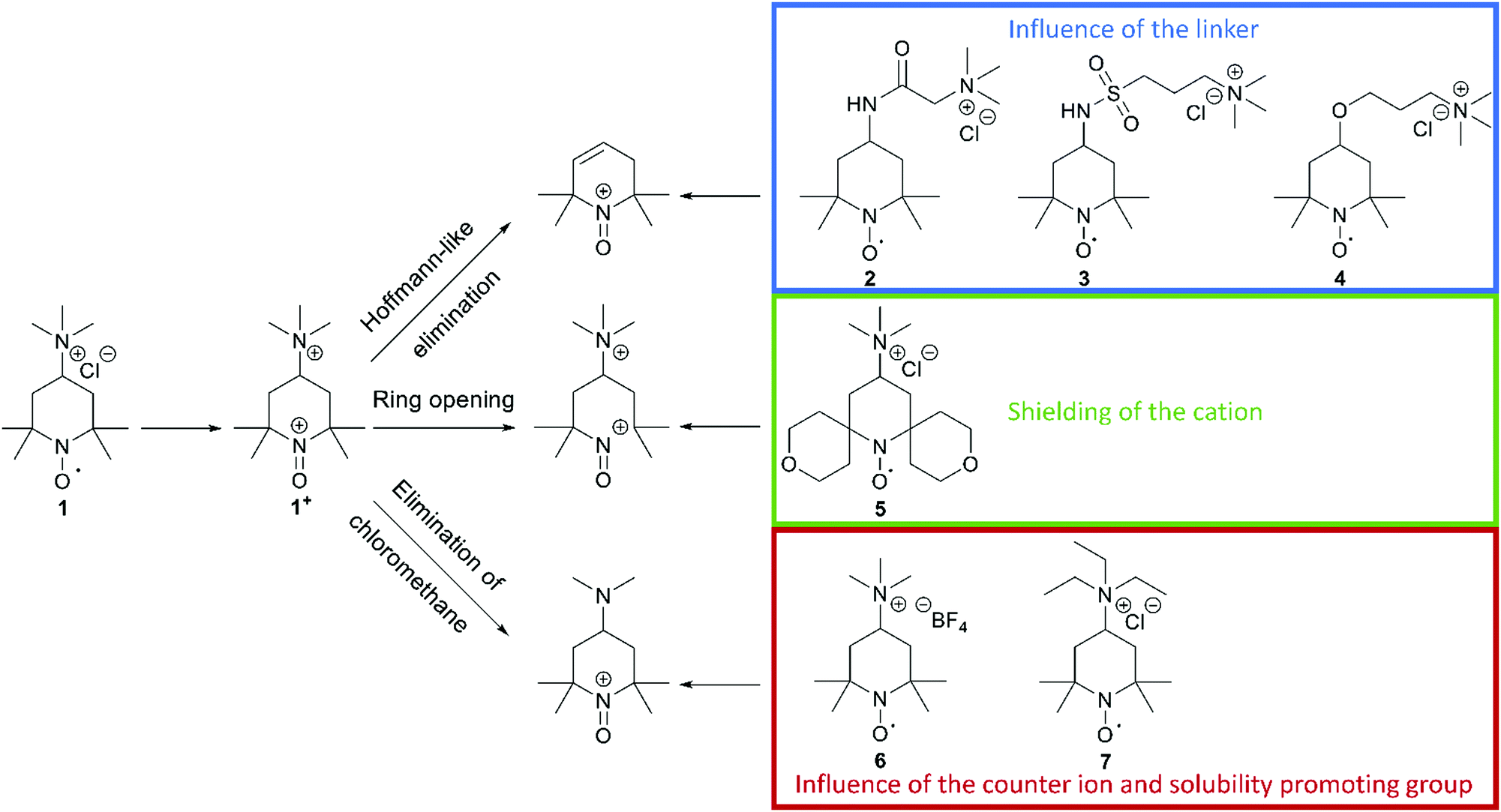 | ||
| Scheme 2 Schematic representation of the three suggested decomposition pathways of TMA-TEMPO 1 and the proposed TEMPO derivatives 2–7 to influence the respective decomposition pathway by the introduction of different linkers between the solubilizing group and the TEMPO moiety (top), a shielding of the formed cation utilizing a spiro derivate (middle) and influencing the elimination of chloromethane by different anions and the exchange of methyl to ethyl (bottom). | ||
The second decomposition pathway starts directly with a ring-opening reaction by breaking one of the C–N bonds next to the oxoammonium cation. As the TEMPO radical and its oxoammonium cation are stabilised by the shielding effect of the four neighbouring methyl groups, we intended to enhance this shielding effect by introducing tetrahydropyran rings. In the resulting spiro-TEMPO compound 5, the reactive site of the molecule should be better protected and, thus, possible ring-opening reactions with subsequent decomposition reactions may be hindered.
The last pathway includes the loss of chloromethane. Here, the nucleophilic character of the counterion may play an important role. Thus, we decided to substitute the chloride ion with the less nucleophilic tetrafluoroborate counterion to yield the TMA-TEMPO BF4 salt 6. Furthermore, we intended to investigate if the switch to a triethyl-ammonium-bearing derivative 7 might suppress the formation of halogenoalkanes or the elimination of pathway one.
Experimental section
Chemicals and materials
Chemicals for synthesis were purchased with a purity exceeding 95% and were used as received. Details can be obtained from the ESI.† Deionized water was used for all experiments. The ion-selective membrane (FAA-3-50, Fumasep GmbH, Germany) was cut into appropriate pieces and prewetted for at least 24 h in 1.5 M aq. sodium chloride or sodium tetrafluoroborate solution. GFA-6 graphite felts from SGL SE, Germany, were used as porous electrodes.Electrochemical analytics
Cyclic voltammetry (CV)
Cyclic voltammetry was performed using a VMP-3 (Bio-Logic, France) potentiostat/galvanostat using a standard three-electrode setup with an AgCl/Ag reference electrode (Ag wire in 3 M aq. potassium chloride solution), a platinum wire counter electrode and a glassy carbon working electrode (GCWE) with a diameter of 1.6 mm.The diffusion coefficients for the oxidized TEMPO derivatives were calculated according to the Randles–Sevcik approach.28
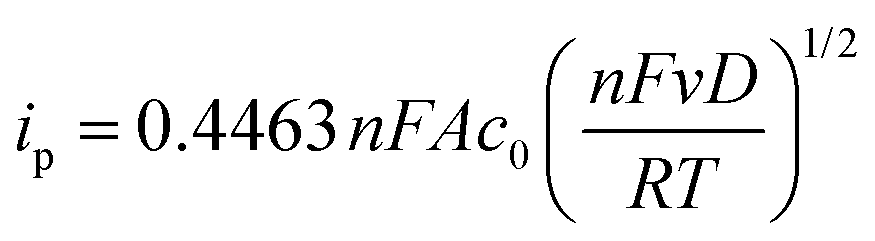 | (1) |
The rate constant was determined using the Nicholson approach.29,30
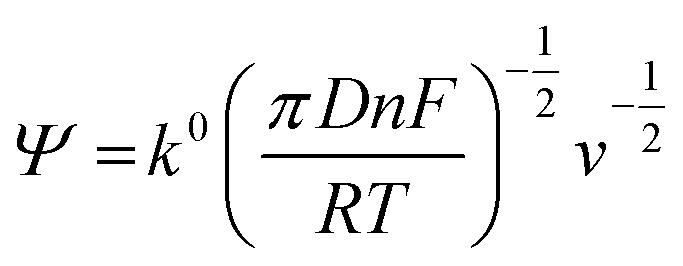 | (2) |
Rotating-disk electrode voltammetry (RDE)
Measurements were conducted with the same setup but with a GCWE diameter of 5 mm and an EDI 101 rotator controlled by a CTV101 (Radiometer analytical, France). The Data of the RDE measurements were analyzed using the Levich equation, | (3) |
The applied Koutecký–Levich equation,
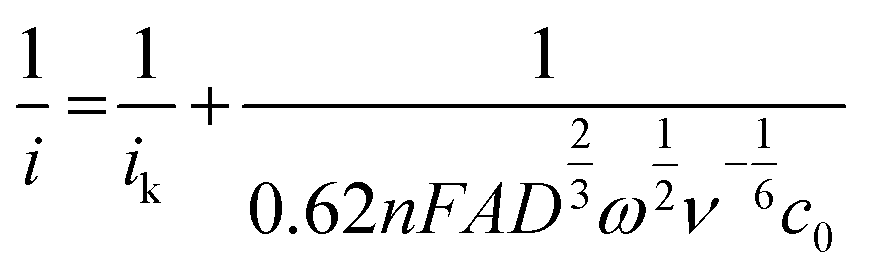 | (4) |
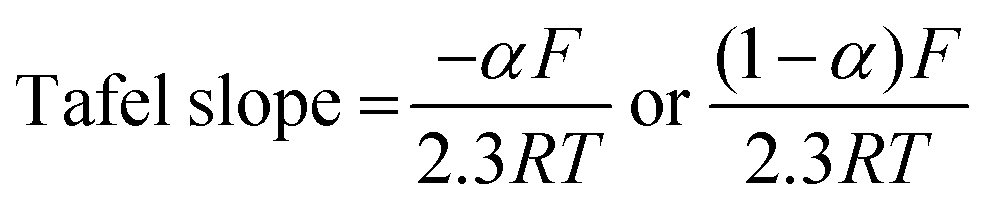 | (5) |
General optical titration procedure
Standard solution preparation
The purchased 0.1 M cerium(IV)-sulphate solution was diluted with water to obtain a 0.025 M solution.Titer determination
Approx. 0.125 mmol ammonium iron(II) sulphate was dissolved in 45 mL of water, and 5 mL of 10 vol% sulfuric acid as well as two drops of ferroin solution were added.Sample preparation
Approx. 0.125 mmol TEMPO derivative was dissolved in 45 mL of water, and 5 mL 10 vol% sulfuric acid as well as two drops of ferroin solution were added.Titration
The sample/titer solution was continuously stirred and the standard solution was added dropwise using a 10 mL burette (AS grade, Duran®, Germany) until the color of the sample solution turned from red to slightly yellow and the color change remained at least for 5 seconds.General potentiometric titration procedure
The same sample, titer and standard solution preparation procedures as described for optical titration were used but without the ferroin indicator. The dispensing voluminal interval was adjusted according to the expected equivalence point, derived from optical titration. To measure the redox potential, a GMH 3530 digital pH-/mV-/thermometer (Greisinger electronic GmbH, Germany) equipped with a GR 105 redox electrode (Greisinger electronic GmbH, Germany, reference electrode: Ag wire in 3 M aq. potassium chloride solution) was used.The added volume of cerium(IV)-sulphate solution was plotted over the measured potential and an adjusted eqn (6) according to Levie31 was fitted to the data set.
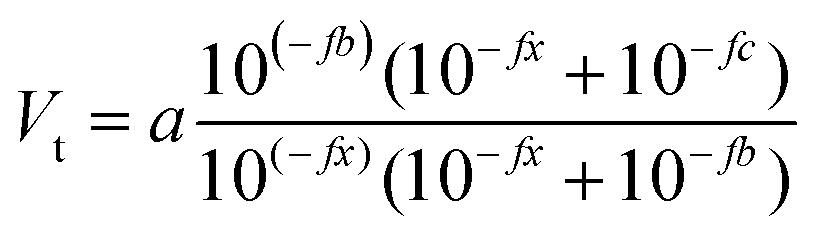 | (6) |
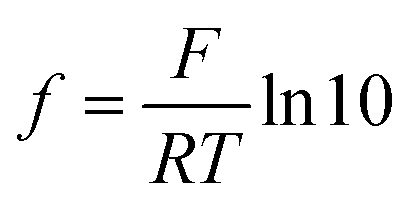 , which describes the pre-factor in the Nernst equation and was set to a value of 17.2, measured potential x)
, which describes the pre-factor in the Nernst equation and was set to a value of 17.2, measured potential x)
Battery experiments
All battery tests were performed under normal atmosphere. As a potentiostat/galvanostat a VMP3 (BioLogic, France) was used. The electrolyte was pumped by a Hei-Flow Value 01 equipped with a C8 multi-channel pumping head (Heidolph, Germany) at a flow rate of approx. 25 mL min−1 through Norprene® A-60-G (øinn 1.6 mm, øout 4.8 mm, Saint-Gobain, France) peristaltic tubes into the cell. For all battery tests, a flat-type redox flow cell according to the design of Jena Batteries GmbH, Germany, with an active membrane area of 5 cm2 was utilized. A detailed overview of the cell components is provided in the ESI.†The room temperature was 22 °C. For tempering, the samples were placed into an oil bath and heated to 40 °C using a magnetic stirrer/hot plate (Hei-Tec, Heidolph, Germany) equipped with an external temperature sensor (PT 1000, Heidolph, Germany).
General method for SOC 50% samples
2 mmol of the synthesized TEMPO derivative was mixed with 20 mL of water and 20 mmol of the appropriate supporting electrolyte (sodium chloride or tetrafluoroborate). The sample was charged against 25 mL of 0.1 M aq. methyl viologen solution mixed with 20 mmol sodium chloride or tetrafluoroborate. The applied current was 80 mA cm−2 until a cell voltage of 1.45 V was reached. Afterwards, the battery was charged potentiostatically at 1.45 V until the current decreased below 0.5 mA cm−2. This electrolyte was considered as fully charged and mixed with a solution of 2 mmol of the same TEMPO derivative and 20 mmol sodium chloride or tetrafluoroborate in 20 mL of water. The solution was pumped through the cell for approx. 10 min and the SOC of the obtained sample was considered as 50%.General cycling protocol
The received SOC-50% sample was split into 12 mL of 0.1 M aq. TEMPO derivative solution for the capacity-limiting side (CLS) and 18 mL of 0.1 M aq. TEMPO derivative solution for the capacity non-limiting side (CNLS), except for TMA-TEMPO, where 15 mL of 0.1 M aq. solution for CLS and 20 mL of 0.1 M aq. solution as CNLS were used. For all splitting processes, the same 1, 2, 5 and 10 mL volumetric pipettes (Duran®, Germany) were used.The unbalanced, compositionally symmetric cell was cycled 50 times between +0.45 V and −0.45 V with 76 mA cm−2. After the cell voltage limits were reached, the battery was charged/discharged potentiostatically until the current dropped below the threshold of 0.5 mA cm−2. After finishing the 50 cycles, the battery was charged to maximal SOC and a hold time of 24 h was applied. During these hold times the pumps of the battery were not paused or slowed down. This cycling protocol was repeated three times and 25 additional cycles were applied at the end. Afterwards, the reservoir of the capacity-limiting side was heated up to 40 °C and the cycling protocol described above was applied again (see ESI† Fig. S15).
Synthesis
In general, the syntheses of the desired derivatives can be classified in two different approaches (see Scheme 3): the first starts with a reductive amination of the triacetone amine precursors I with subsequent quaternization, leading to the trialkylammonium derivative III, which is oxidized to the radical IV in the final step. The second route starts with the respective aminoxyl radical V bearing an amino or hydroxyl group in the 4-position and the introduction of halogen-bearing linkers (VI). The desired product VII is subsequently synthesised via nucleophilic substitution with trimethyl amine. Detailed synthesis procedures of all compounds are available in the ESI.†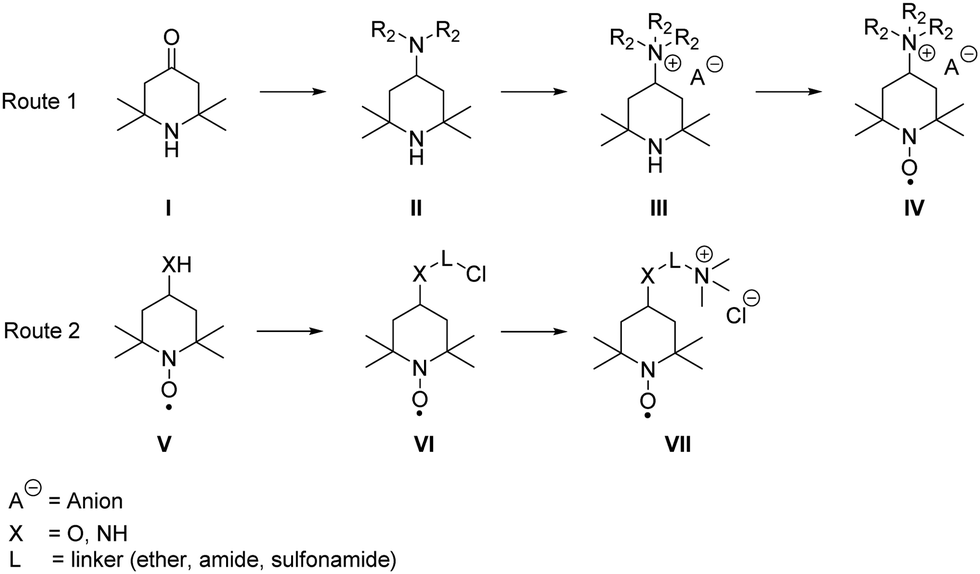 | ||
| Scheme 3 Schematic representation of the used general synthesis routes for the desired TEMPO derivatives. | ||
Results and discussion
Electrochemical characteristics
In order to elucidate the structure–stability relationship the degree of oxidation (DoO) of the TEMPO derivatives was first analysed, followed by electrochemical characterisation studies, before battery experiments were conducted.The DoOs of the TEMPO derivatives were determined using a cerimetric redox titration procedure with the indicator ferroin. To overcome the subjective titration endpoint determination based on color change, the change of the redox potential during the titration was measured using a potentiometer (see Fig. S2 and S3, ESI†).
As the curve for eqn (6) depends on the redox potential of the TEMPO derivative, we tried to validate our new fitting approach by comparing with the redox potential delivered by CV measurements. The differences between the redox potentials obtained by the CV method and from the fitting curve are mostly below 10 mV (see Table 1) with standard deviation of 7 mV or below. The shape of the CV curves (see ESI† chapter: CV-RDE-Measurements) is typical for TEMPO derivatives. The plot of the peak currents over the square root of the scan speed revealed linear behavior except for the spiro-Tempo 5 where the curves flattened at low scan speeds, indicating a non-reversible redox reaction upon oxidation. Furthermore, compound 5 could not be determined by titration. This may be caused by the hydrolization of the ether bonds of the attached rings and the subsequent oxidation of the resulting alcohols in the acidic environment. Using both titration methods, neither a stable color change during optical titration nor a significant change in the potential could be observed. Thus, we assumed that the oxoammonium cation is not stable and decomposes within a short time frame.
| Compound | E 1/2CV[V] | E 1/2tit[V] | ΔE1/2 [mV] | DoOopt. [%] | DOOpot [%] |
|---|---|---|---|---|---|
| 1 21 | 0.754+/−1 mV | 0.747+/−1 mV | 6 | 99+/−0.3 | 103+/−0.5 |
| 2 | 0.644+/−1 mV | 0.652+/−1 mV | 7 | 86+/−0.1 | 88+/−0.2 |
| 3 | 0.648+/−1 mV | 0.652+/−1 mV | 4 | 101+/−0.1 | 100+/−0.8 |
| 4 26 | 0.623+/−2 mV | 0.610+/−2 mV | 13 | 81+/−0.1 | 74+/−0.7 |
| 5 | 0.883+/−7 mV | — | — | — | — |
| 6 | 0.755+/−1 mV | 0.753+/−1 mV | 2 | 79+/−0.2 | 78+/−0.2 |
| 7 | 0.841+/−6 mV | 0.754+/−1 mV | 85 | 59+/−0.6 | 58+/−1 |
In contrast to the redox potentials of the other TEMPO derivatives determined by titration and by CV (Table 1), TEA-TEMPO 7 exhibited an extraordinarily high difference of 85 mV. Up to now, we do not have a plausible explanation for this observation and an in-depth investigation of this behavior is the topic of further research. Our first assumption is that derivatives with a DoO of below approx. 80% are not suitable for a simultaneous determination of the redox potential and the DoO.
The DoOs of the other TEMPO derivatives were mostly over 80% with two derivatives with approx. 100% degree of oxidation. Lower radical contents might be caused by the reduction or disproportionation reactions during the purification processes. However, a reduced DoO may only affect the overall electrolyte capacity but not the cyclability.20,22,32 Thus, we assumed that the stability of the TEMPO derivatives in the following experiments is not compromised by the residual amount of the respective nonoxidized precursors. All in all, we were able to develop a new titration protocol for water-soluble TEMPO derivatives, which is independent from subjective visual perception of the titrator and furthermore enables the direct measurement of the redox potential of the analyte within the same measurement.
Furthermore, the titration procedure can be automated using a titration machine. However, the main drawback of the method is its long measuring time caused by the equilibration time of the redox electrode. Additional electrochemical properties were determined by rotating disk electrode (RDE) voltammetry (see ESI† section: CV-RDE Measurements).
The electron-transfer rates k0, the diffusion coefficients D and the transfer coefficients α, determined via RDE, were found to be 1.6 to 3.0 × 10−3 cm s−1, 3.7 to 4.7 × 10−6 cm2 s−1 and 0.54 to 0.66, respectively (see Table 2). To overcome some potential errors due to the fast redox kinetics of the analyte material, the electron-transfer rates k0 and the diffusion coefficients D were also determined using both the Nicholson and the Randles–Sevcik approach.28–30 The findings of the different methods are comparable to each other and to the published data for TMA-TEMPO in aq. solutions at glassy carbon electrodes and other organic active materials, like quinones.21,33 It seems that structural deviations at the TEMPO scaffold do not affect the electrochemical properties or kinetic parameters significantly. This is not surprising since the molecular orbitals of the radical as well as the oxoammonium cation that are involved in the redox reaction are mainly located at the NO moiety of the TEMPO scaffold.34
As already indicated during the CV measurements, the spiro-TEMPO 5 cannot be analysed using RDE due to the absence of a plateau during the oxidation process (see ESI† section: CV-RDE measurements), likely because of stability problems of the attached cyclic ethers, as discussed above.
The TEA-TEMPO 7 likewise cannot be analyzed (see ESI† section: CV-RDE measurements), most likely due to the high content of non-oxidized educt and possible interactions between an educt and a product. A simple evaluation with the Levich equations was thus not possible.
While compound 5 is not stable enough for battery applications, triethyl ammonium TEMPO-derivative 7 was excluded due to its low DoO and insufficient yields in the synthesis, which make them both unsuitable candidates for further large-scale applications. Based on these findings, compounds 5 and 7 were not investigated further.
Battery experiments
In order to test the stability of the proposed active materials in a battery setup, the unbalanced, compositionally symmetric cell setup was employed. The main advantage is that the utilization of the same active material as a posolyte as well as a negolyte largely avoids crossover. This concept was firstly introduced by the group of Brushett35 in 2016, who used the same electrolyte with an initial SOC of 50% for both half-cells, creating a battery that cycles around a cell voltage of 0 V (Fig. 2). Goulet and Aziz further developed this idea and reported an elaborate cycling protocol including a capacity-limiting half-cell, hold times and potentiostatic charging.25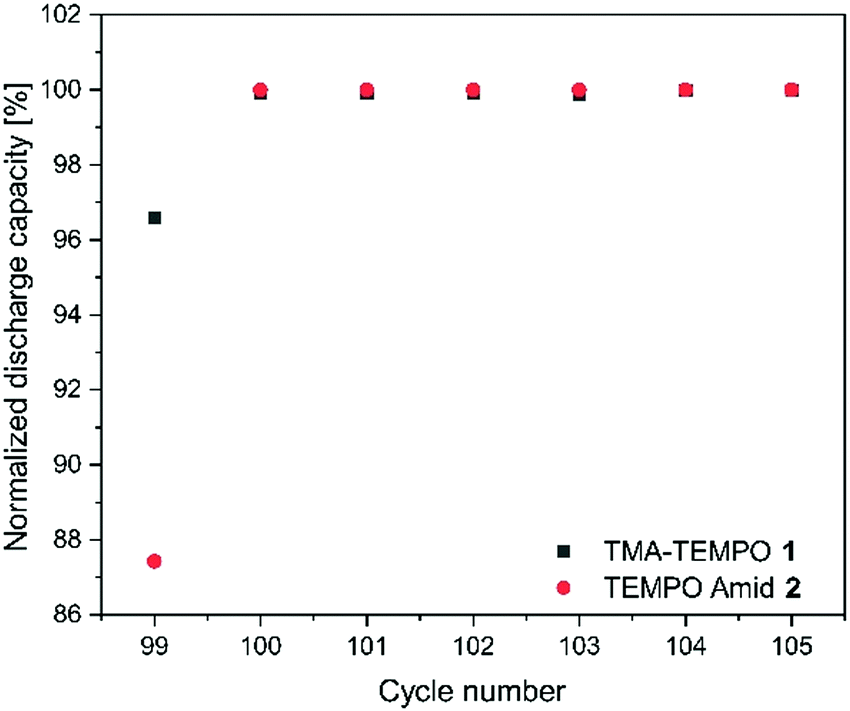 | ||
| Fig. 1 Illustration of the discharge capacity of the first seven cycles of the TMA-TEMPO 1 and the amide 2 after the second hold time of 24 h. The capacity was normalized to the highest capacity in the shown interval. The first cycle showed a massively reduced discharge capacity. | ||
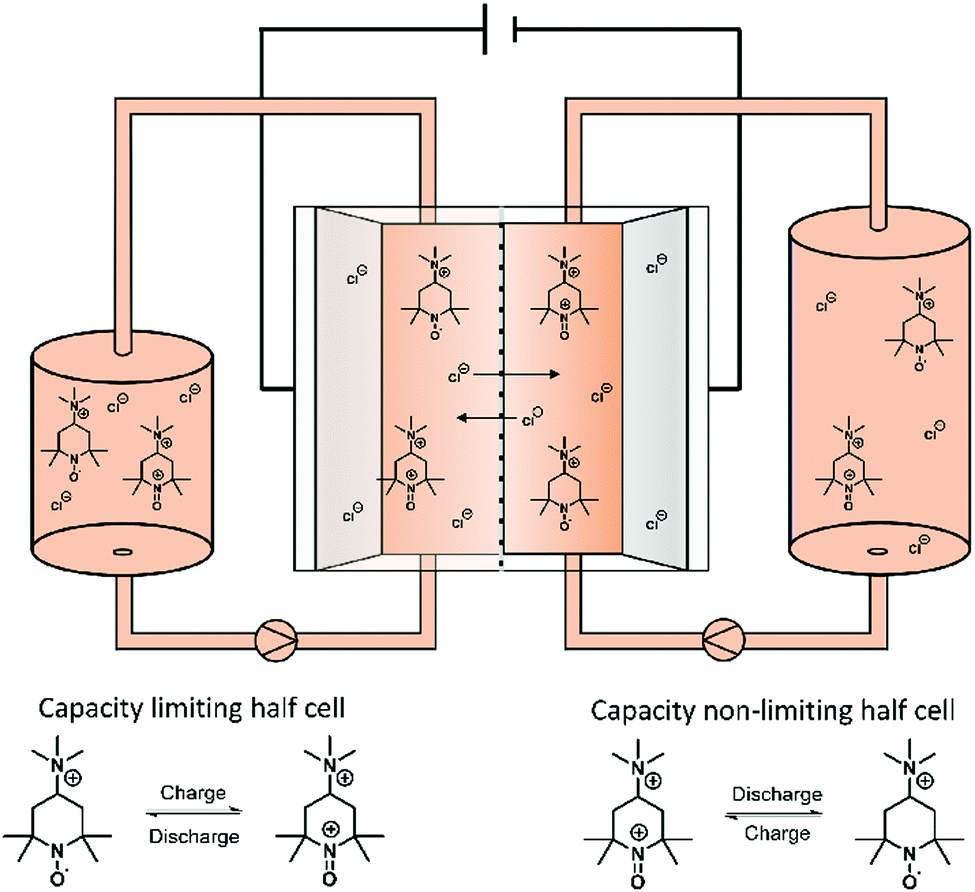 | ||
| Fig. 2 Illustration of a symmetric TMA-TEMPO redox flow battery set up as well as the corresponding half-cell reaction. | ||
In order to study the influence of temperature and rest times at high SOC values, the cycling protocol was adapted by our group. The adapted protocol featured potentiostatic cycling near the extreme SOC values of the capacity-limiting electrolyte with a limiting current of 0.5 mA cm−2. Every 50 cycles, a hold time of 24 h at maximum SOC was applied. After 175 cycles, the capacity-limiting reservoir was placed in an oil bath at 40 °C and the described protocol was repeated. This stress test should mimic unfavorable, but still realistic conditions.
The utilized low concentration of 0.1 M was used to keep the experiment in a manageable time frame. For comparability reasons, we normalized the highest achieved capacity during the test protocol for each derivative to 100% and calculated every other datapoint relative to this capacity.
At first, we investigated the TMA-TEMPO system 1 in order to create a benchmark to which we can then compare our new derivatives. TMA-TEMPO 1 showed a relatively stable charge and discharge capacity in the first 50 cycles; however, after an initial peak the electrolyte capacity started to decay with an increased dip after the first hold time of 24 h. While under room temperature conditions (22 °C) the capacity loss during the hold times is only moderately higher than that during cycling (1.5% d−1 and 0.9% d−1, respectively, see Table 3), increasing the temperature to 40 °C results in an increased decay during hold times (2.0% d−1). Interestingly, the capacity fade during cycling is comparable to that at room temperature (0.8% d−1). After the completion of the test protocol, 73.0% rest capacity could be recovered (see Table 3).
| Decay 22 °C [% d−1] | Decay 40 °C [% d−1] | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start rel. discharge capacity | 1. Cycling period | 1. Hold time | 2. Cycling period | 2. Hold time | 3. Cycling period | 3. Hold time | 4. Cycling period | Final rel. discharge capacity | 1. Cycling period | 1. Hold time | 2. Cycling period | 2. Hold time | 3. Cycling period | 3. Hold time | 4. Cycling period | Final rel. discharge capacity | |
| 1B | 89.07 | 0.28 | 1.25 | 1.24 | 1.63 | 1.36 | 1.57 | 0.55 | 81.80 | 1.00 | 2.06 | 0.51 | 1.90 | 0.92 | 2.14 | 0.60 | 72.95 |
| 2 | 94.74 | 6.79 | 12.80 | 2.10 | 10.36 | 1.01 | 10.18 | 0.84 | 76.84 | 0.45 | 11.32 | 0.92 | 11.35 | 0.97 | 11.44 | 0.97 | 71.33 |
| 3 | 99.24 | 20.45 | 13.75 | 2.57 | 10.02 | 0.92 | 8.90 | 0.35 | 32.47 | 0.76 | 8.98 | 0.35 | 8.47 | 0.15 | 8.10 | 0.43 | 27.09 |
| 4 | 99.95 | 28.31 | 16.35 | 3.30 | 9.77 | 1.52 | 8.15 | 1.11 | 36.59 | 1.16 | 10.60 | 0.87 | 9.00 | 0.60 | 8.80 | 0.97 | 28.47 |
| 6 | 80.43 | +1.98 | +1.12 | 0.16 | — | 73.60 | — | ||||||||||
As mentioned before, the nature of the linker to the solubilizing moiety may have a pronounced influence on the stability of the active material during battery experiments. We first investigated the TEMPO-amide system 2. While 6.6% of the capacity is lost during the first 50 cycles, the decay stabilizes during the next 100 cycles and declines linearly afterwards until the end of the experiment. All in all, it is comparable to TMA-TEMPO 1 (0.9% d−1 20 °C, 0.8.% d−1 40 °C). In contrast, the loss during the hold time is, with an average of 11.6% d−1, much higher (1.5% d−11, see Table 3).
This decay can mostly be reversed during the next consecutive charging (see Fig. 1), indicating a much more pronounced but also highly reversible capacity loss mechanism. A self-discharge mechanism whose kinetics are relatively slow, so it cannot be measured during cycling, is the most plausible explanation (a possible mechanism is explained below). Nevertheless, the TEMPO-amide compound 2, even with the initial comparably high loss, shows a similar stability as the known TMA-TEMPO system 1 (see Fig. 3). In comparison, the TEMPO-sulfamide molecule 3 is significantly more unstable. The experiment showed an exponential capacity decay (see Fig. 3), and only 50% of the initial capacity remained after the first 50 cycles, which corresponds to a decay of over 20% d−1. This trend continues over the next cycles, although not that pronounced. After the first 175 cycles, only 32% of the initial capacity can be recovered. At higher temperatures, the decay is 1.2% per day (Fig. 3), which is comparable to that of the other derivatives. All in all, the sulfamide system 3 revealed a strongly pronounced capacity decay during cycling, which shows its insufficiency as a battery active material. Originally intended to be a more stable linker for the SPG, the amide compound 2 and sulfamide molecule 3 failed to improve the stability of the TEMPO scaffold. Whereas the amide revealed a similar stability to TMA-TEMPO, the sulfamide is much more prone to capacity decay. We think that oxidative cleavage of the linker might be the most probable explanation. While amides are stable against the oxidative cleavage of the C–N bond up to a potential of approx. 1.25 V vs. Ag/AgCl,36 it was shown that the N–S bond of sulfamides can be cleaved at a redox potential of 0.75 to 0.95 V vs. Ag/AgCl,37–40 which is similar to the oxidation potential of the TEMPO derivatives.
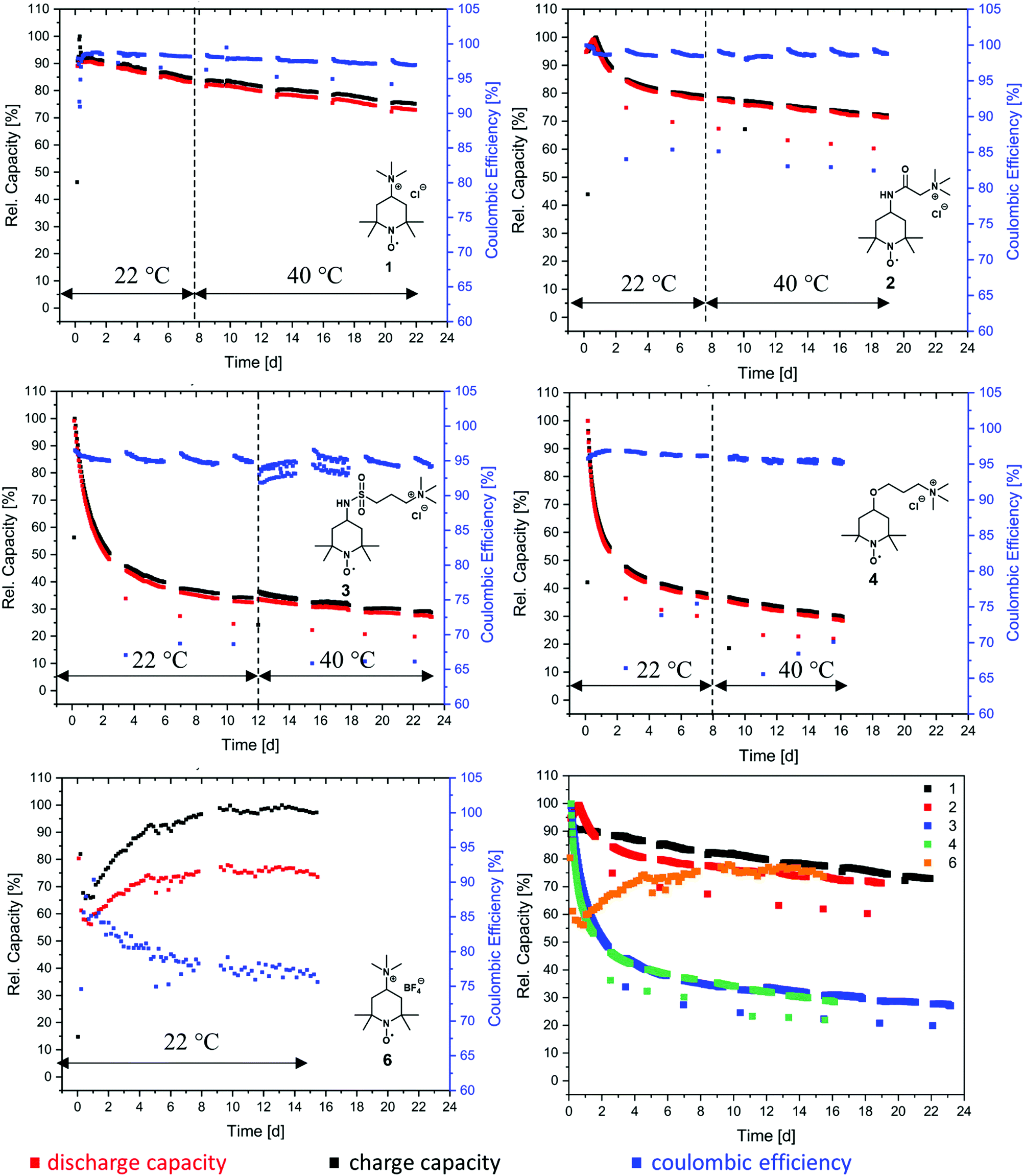 | ||
| Fig. 3 Overview of the battery performance of different TEMPO-derivatives in symmetrical redox flow cell tests cycled potentiostatically with 76 mA cm2 with a consecutively added galvanostatic part (current limitation of 0.5 mA cm2). The cells consist of 12 mL vs. 18 mL 0.1 M active material solution, except for TMA-TEMPO, where 15 mL vs. 20 mL solutions were used. All electrolytes consist of 1.5 M appropriate supporting salt in water. The charge capacity is represented in black, the discharge capacity in red and the coulombic efficiency in blue. (Lower right) Comparison of the discharge capacity of the TEMPO derivatives. | ||
Thus, an autoxidative cleavage of the N–S sulfamide bond by the oxoammonium cation itself is considered as a reason for the fast decay of the battery capacity. The resulting 4-amino-TEMPO is more likely to undergo degradation reactions than TMA-TEMPO 1, the amide 2 or the sulfamide 3.41 This trend was also demonstrated for the 4-hydroxy-TEMPO, which was shown to be significantly more unstable compared to TMA-TEMPO.42 During hold times, the capacity loss of 3 is much more pronounced than that of the TMA-TEMPO system 1 and is 10.2% d−1 (see Table 3), which is in the range of the amide compound 2. The loss can be largely recovered in the next cycle. Once again, a reversible self-discharge mechanism with slow kinetics is a good explanation for this behavior.
The TEMPO-ether molecule 4 shows a comparable behaviour of the battery capacity to the sulfamide system 3. After an initial exponential decay within the first 50 cycles, the fade stabilizes and becomes linear. At the end of the segment conducted at room temperature, only 36% of the initial capacity can be retained and 28% remaining capacity was accessible after the consecutive test at 40 °C (see Table 3). TEMPO-based oxoammonium cations are known to be capable of cleaving ether bonds oxidatively.43 The resulting 4-hydroxy TEMPO undergoes further irreversible degradation reactions, as discussed before. The finding of Liu that the introduction of an ether between the solubility-promoting trimethylammonium group and the TEMPO scaffold increases the stability26 could not be confirmed by our setup (under the used conditions, e.g., no hold times in vulnerable SOC, other voltage limits).
Ma et al. showed that TEMPO can undergo multiple bond breaking reactions in acidic aqueous media resulting in unsaturated phorones (see Scheme 1).1 Takata and co-workers demonstrated that oxoammonium cations undergo rapid 1,2 electrophilic additions with double bonds.2 Subsequent cleavage of the addition products leads to acid formation, reduction of the oxoammonium cation to hydroxylamine and oxidation of the double bond to an alcohol/ketone (see Scheme 1). Ma et al. stated that for every decomposed TEMPO molecule approx. four hydroxylamine molecules are formed, which is in good agreement with the mechanism formulated by Takata et al. and our findings in the GC-MS study.27
These findings could possibly explain why the coulombic efficiencies of the molecules 1 to 4 are comparably low. Fast cycling of the active material, avoidance of hold times in the oxidized state and high cut off currents would lower the oxoammonium cation concentration and, thus, bypass the described mechanism. Furthermore, the formed acids do not only catalyse the processes Ma et al. described, but they also influence the cleavage of the linker. The amide 2, sulfonamide 3 and the ether 4 linkers are thus under double stress; on the one hand the oxidative cleavage and on the other hand the acidic catalysed cleavage could take place. Nevertheless, the low coulombic efficiency and the huge healable capacity drop after hold times is most likely attributed to the formed hydroxylamine, which needs to be electrochemically reoxidized to the radical. However, we repeated the measurements of Liu et al. of the TEMPO-ether 4 with lower cut-off voltages and higher cut-off currents (see ESI† Fig. S22). The results show that the decay rate can be massively reduced by carefully adjusting the cycling parameter of the molecule. However, the decomposition rate is with 3.1% d−1, during pure cycling, reduced but the TEMPO-amide 2 or the TMA-TEMPO 1 is still more stable. Further post-mortem analyses like pH, CV or NMR (see ESI† Fig. S23 and S24) support these findings, but give no additional hinds on the ongoing processes.
Compared to halogenide anions, the BF4− anion is known to be less nucleophilic.44,45 Therefore, compound 6 was synthesised to investigate the influence of the counterion on the stability of TMA-TEMPO. As expected, the salt revealed only a low solubility in the used aqueous electrolyte, with a maximum concentration of around 0.1 M. During the cycling test, only 50% of the theoretical capacity (see ESI† section: battery experiments) can be addressed. Furthermore, the achieved charge and discharge current densities are relatively low, resulting in exceptionally long cycling times.
Although the membrane was prewetted for 24 h in a 1.5 M aq. sodium tetrafluoroborate solution, the anion exchange might be hindered, which would explain the long charging and discharging times.
However, over the first cycles the capacity slowly increases before remaining nearly stable for the rest of the experiment. This atypical behavior could be explained by the low solubility and possible dissolution-participation equilibria which would also explain the low accessible capacity of the electrolyte. The influence of the counter ion on the stability could thus not be derived. Furthermore, the test could not be completed as the solubility of the BF4-salt was insufficiently low and precipitation took place in the cell. After 79 cycles, the membrane was perforated either because of clogging and the associated pressure increase or because of conductive-salt crystals, which had grown through the membrane (see pictures in the ESI† section: photos of BF4-salt precipitate).
As discussed above, we think that the main reason for the low coulombic efficiencies is attributed to the formal oxidation of a double bond to an alcohol by an oxoammonium cation with its reduction to the hydroxylamine as discussed by Takata (see Scheme 1).2 The alcohol is then oxidized to the ketone and the corresponding keto–enol-tautomerism produces additional double bonds which are oxidized. This mechanism continues until CO2 is formed.1
The formation of these double bonds needs to be avoided for a more stable TEMPO derivative. The ether 4 and the sulfonamide 3 linked compound can be cleaved oxidatively and/or acid catalysed resulting in a lesser stable 4-hydroxy or 4-amino TEMPO. These compounds can be oxidized easely to the carbonylic or carbonyl-equivalent. The subsequent keto–enol-tautomerism produces double bonds which needs to be avoided. Since the amide 2 is (more) stable against the decay mechanism, linker cleavage as the decomposition starting point can be omitted. Furthermore, the amide 2 should also hinder the Hoffman-like elimination pathway and decomposition of the solubility promoting group via chloromethane evolution. Nevertheless, the amide 2 and the TMA-TEMPO system 1 are comparably stable. We thus hypothesize that the comparable capacity loss of the TMA-TEMPO system 1 and the TEMPO-amide compound 2 is attributed to the N–C bond break in the TEMPO scaffold and the subsequent formation of oxidizable double bonds. A cleavage of this bond must be prevented either by further shielding of the radical or stabilization via electronic effects caused by substituents vicinal to the N–O radical.
Conclusions
TMA-TEMPO is among the best investigated and most stable posolyte materials for redox batteries. Nevertheless, it does suffer from decomposition and reversible self-discharge reactions. We synthesised five new TEMPO derivatives as well as one known and compared their electrochemical parameters as well as their stability with those of TMA-TEMPO.At first, we developed a new evaluation method for redox titration with which, in one measurement, the DoO and the half-cell potential of the analyte can be obtained, which were validated by both CV and optical titrations. However, while the DoO is accurate even at lower values, the half-cell potentials are only precise when the degree of oxidation is sufficiently high. While the method needs a comparably long measuring time, it nevertheless reveals great potential due to its possible automatization.
In subsequent battery experiments, a selection of promising candidates were tested using a cycling protocol involving dwell times at high SOCs at room temperature and 40 °C. The introduction of a tetra fluoroborate counter ion to TMA-TEMPO lowered the solubility so much that no reliable statements about possible introduced stability changes could be made.
The usage of an amide linker revealed no significant stability changes in comparison to TMA-TEMPO. Nevertheless, a more pronounced but also highly reversible self-discharge mechanism was visible, whose details are the subject of ongoing studies.
However, linkers connected via a sulfonamide or an ether unit did show pronounced signs of capacity loss even at low temperatures; these were less accentuated for TMA-TEMPO. The main reason for this may be a possible oxidative cleavage of the linker by the corresponding oxoammonium cation, although Liu et al. did not observe such a degradation process during their investigation of the ether-linked compound 5.26 This may be caused by the differences in the cycling protocols. We are intending to emphasise concentration-dependent studies as well as dwell times in vulnerable SOCs for future investigations.
Author contributions
Philip Rohland: conceptualization, methodology, formal analysis, data curation, visualization, writing – original draft (lead) investigation, and synthesis (equal); Oliver Nolte: investigation (equal), formal analysis (supporting), writing – original draft (supporting), and writing – review and editing: critical review (equal); Kristin Schreyer: synthesis (equal), investigation (supporting), and writing – review and editing: critical review (equal); Helmar Görls: XRC (lead), investigation (supporting), and writing – review and editing: critical review (equal); Martin D. Hager: writing – review and editing: critical review (equal), supervision of P. R., O. N., and K. S. (equal), and funding acquisition (equal); Ulrich S. Schubert: writing – review and editing: critical review (equal), supervision of P. R., O. N., and K. S. (equal), and funding acquisition (equal).Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge the European Social Fund (ESF), the Thüringer Aufbaubank (TAB) and the Thuringian Ministry of Economic Affairs, Science and Digital Society (TMWWdG) for financial support. We further want to thank Christian Friebe and Christian Stolze for their help and suggestions.References
- Y. Ma, C. Loyns, P. Price and V. Chechik, Org. Biomol. Chem., 2011, 9, 5573–5578 RSC.
- T. Takata, Y. Tsujino, S. Nakanishi, K. Nakamura, E. Yoshida and T. Endo, Chem. Lett., 1999, 937–938 CrossRef CAS.
- C. R. Chamorro, M. E. Mondéjar, R. Ramos, J. J. Segovia, M. C. Martín and M. A. Villamañán, Energy, 2012, 42, 10–18 CrossRef.
- E. von Sperling, Energy Proc., 2012, 18, 110–118 CrossRef CAS.
- B. Obama, Science, 2017, 355, 126–129 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- I. Hadjipaschalis, A. Poullikkas and V. Efthimiou, Renewable Sustainable Energy Rev., 2009, 13, 1513–1522 CrossRef.
- P. Alotto, M. Guarnieri and F. Moro, Renewable Sustainable Energy Rev., 2014, 29, 325–335 CrossRef CAS.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 129, 702–729 CrossRef.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. Liu, J. Appl. Electrochem., 2011, 41, 1137 CrossRef CAS.
- J. Noack, N. Roznyatovskaya, T. Herr and P. Fischer, Angew. Chem., Int. Ed., 2015, 54, 9776–9809 CrossRef CAS PubMed.
- K. Lin, R. Gómez-Bombarelli, E. S. Beh, L. Tong, Q. Chen, A. Valle, A. Aspuru-Guzik, M. J. Aziz and R. G. Gordon, Nat. Energy, 2016, 1 Search PubMed.
- W. Lee, B. W. Kwon and Y. Kwon, ACS Appl. Mater. Interfaces, 2018, 10, 36882–36891 CrossRef CAS PubMed.
- A. Orita, M. G. Verde, M. Sakai and Y. S. Meng, Nat. Commun., 2016, 7, 13230 CrossRef CAS PubMed.
- B. Yang, L. Hoober-Burkhardt, S. Krishnamoorthy, A. Murali, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2016, 163, A1442–A1449 CrossRef CAS.
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed.
- D. G. Kwabi, K. Lin, Y. Ji, E. F. Kerr, M.-A. Goulet, D. De Porcellinis, D. P. Tabor, D. A. Pollack, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Joule, 2018, 2, 1907–1908 CrossRef.
- Y. Zhu, F. Yang, Z. Niu, H. Wu, Y. He, H. Zhu, J. Ye, Y. Zhao and X. Zhang, J. Power Sources, 2019, 417, 83–89 CrossRef CAS.
- E. S. Beh, D. De Porcellinis, R. L. Gracia, K. T. Xia, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2017, 2, 639–644 CrossRef CAS.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2016, 55, 14427–14430 CrossRef CAS PubMed.
- J. Winsberg, T. Janoschka, S. Morgenstern, T. Hagemann, S. Muench, G. Hauffman, J. F. Gohy, M. D. Hager and U. S. Schubert, Adv. Mater., 2016, 28, 2238–2243 CrossRef CAS PubMed.
- J. Luo, B. Hu, C. Debruler and T. L. Liu, Angew. Chem., Int. Ed., 2018, 57, 231–235 CrossRef CAS PubMed.
- C. DeBruler, B. Hu, J. Moss, J. Luo and T. L. Liu, ACS Energy Lett., 2018, 3, 663–668 CrossRef CAS.
- M.-A. Goulet and M. J. Aziz, J. Electrochem. Soc., 2018, 165, A1466–A1477 CrossRef CAS.
- Y. Liu, M.-A. Goulet, L. Tong, Y. Liu, Y. Ji, L. Wu, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, Chemistry, 2019, 5, 1861–1870 CrossRef CAS.
- O. Nolte, P. Rohland, N. Ueberschaar, M. D. Hager and U. S. Schubert, J. Power Sources, 2022, 525, 230996 CrossRef CAS.
- P. Zanello, Inorganic Electrochemistry: Theory, Practice and Application, The Royal Society of Chemistry,Milton Road, Cambridge, UK, 2003 Search PubMed.
- R. S. Nicholson, Anal. Chem., 2002, 37, 1351–1355 CrossRef.
- J. Luo, B. Hu, M. Hu, Y. Zhao and T. L. Liu, ACS Energy Lett., 2019, 4, 2220–2240 CrossRef CAS.
- R. de Levie, J. Electroanal. Chem., 1992, 323, 347–355 CrossRef CAS.
- T. Hagemann, J. Winsberg, M. Grube, I. Nischang, T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, J. Power Sources, 2018, 378, 546–554 CrossRef CAS.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- T. Kusamoto, S. Kume and H. Nishihara, J. Am. Chem. Soc., 2008, 130, 13844–13845 CrossRef CAS PubMed.
- J. D. Milshtein, J. L. Barton, R. M. Darling and F. R. Brushett, J. Power Sources, 2016, 327, 151–159 CrossRef CAS.
- T. Golub and J. Y. Becker, Org. Biomol. Chem., 2012, 10, 3906–3912 RSC.
- A. Fabianska, A. Bialk-Bielinska, P. Stepnowski, S. Stolte and E. M. Siedlecka, J. Hazard. Mater., 2014, 280, 579–587 CrossRef CAS PubMed.
- D. Song, H. Liu, A. Zhang and J. Qu, RSC Adv., 2014, 4, 48426–48432 RSC.
- W. Ben, Y. Shi, W. Li, Y. Zhang and Z. Qiang, Chem. Eng. J., 2017, 327, 743–750 CrossRef CAS.
- A. Acosta-Rangel, M. Sanchez-Polo, M. Rozalen, J. Rivera-Utrilla, A. M. S. Polo, M. S. Berber-Mendoza and M. V. Lopez-Ramon, J. Environ. Manage., 2020, 255, 109927 CrossRef CAS PubMed.
- W. Zhou, W. Liu, M. Qin, Z. Chen, J. Xu, J. Cao and J. Li, RSC Adv., 2020, 10, 21839–21844 RSC.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS PubMed.
- P. P. Pradhan, J. M. Bobbitt and W. F. Bailey, J. Org. Chem., 2009, 74, 9524–9527 CrossRef CAS PubMed.
- K. A. Savin, Writing Reaction Mechanisms in Organic Chemistry, Academic Press – Elsevier Inc, Waltham, USA, 3 edn, 2014 Search PubMed.
- M. S. Puar, J. Chem. Educ., 1970, 47, 473–474 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1ma00663k |
| This journal is © The Royal Society of Chemistry 2022 |