
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plasmonic catalysis with designer nanoparticles
Anderson G. M.
da Silva
 a,
Thenner S.
Rodrigues
b,
Jiale
Wang
c and
Pedro H. C.
Camargo
*d
a,
Thenner S.
Rodrigues
b,
Jiale
Wang
c and
Pedro H. C.
Camargo
*d
aDepartamento de Engenharia Química e de Materiais-DEQM, Pontifícia Universidade Católica do Rio de Janeiro (PUC-Rio), Rua Marquês de São Vicente, 225 – Gávea 22453-900, Rio de Janeiro, RJ, Brazil
bNanotechnology Engineering Program, Alberto Luiz Coimbra Institute for Graduate Studies and Research in Engineering, COPPE, Federal University of Rio de Janeiro, Av. Horácio Macedo, 2030, 21.941-972, Rio de Janeiro, RJ, Brazil
cCollege of Science, Donghua University, Shanghai 201620, P. R. China
dUniversity of Helsinki, Department of Chemistry, A.I. Virtasen aukio 1, Helsinki, Finland. E-mail: pedro.camargo@helsinki.fi
First published on 12th January 2022
Abstract
Catalysis is central to a more sustainable future and a circular economy. If the energy required to drive catalytic processes could be harvested directly from sunlight, the possibility of replacing contemporary processes based on terrestrial fuels by the conversion of light into chemical energy could become a step closer to reality. Plasmonic catalysis is currently at the forefront of photocatalysis, enabling one to overcome the limitations of “classical” wide bandgap semiconductors for solar-driven chemistry. Plasmonic catalysis enables the acceleration and control of a variety of molecular transformations due to the localized surface plasmon resonance (LSPR) excitation. Studies in this area have often focused on the fundamental understanding of plasmonic catalysis and the demonstration of plasmonic catalytic activities towards different reactions. In this feature article, we discuss recent contributions from our group in this field by employing plasmonic nanoparticles (NPs) with controllable features as model systems to gain insights into structure–performance relationships in plasmonic catalysis. We start by discussing the effect of size, shape, and composition in plasmonic NPs over their activities towards LSPR-mediated molecular transformations. Then, we focus on the effect of metal support interactions over activities, reaction selectivity, and reaction pathways. Next, we shift to the control over the structure in hollow NPs and nanorattles. Inspired by the findings from these model systems, we demonstrate a design-driven strategy for the development of plasmonic catalysts based on plasmonic-catalytic multicomponent NPs for two types of molecular transformations: the selective hydrogenation of phenylacetylene and the oxygen evolution reaction. Finally, future directions, challenges, and perspectives in the field of plasmonic catalysis with designer NPs are discussed. We believe that the examples and concepts presented herein may inspire work and progress in plasmonic catalysis encompassing the design of plasmonic multicomponent materials, new strategies to control reaction selectivity, and the unraveling of stability and reaction mechanisms.
 Anderson G. M. da Silva | Anderson da Silva received his PhD from University of São Paulo (with Professor Pedro Camargo) in 2017, MSc degree from University of Ouro Preto (under supervisor of Prof. Patricia Alejandra Robles) in 2013, and BSc degree in Industrial Chemistry from University of Ouro Preto in 2010. From 2017, he was a postdoc at the University of São Paulo. In 2019, he became an Associate Professor at the Department of Chemical and Materials Engineering at Pontifical Catholic University, Rio de Janeiro, Brazil. His interests include the synthesis of controlled nanomaterials for applications in nanocatalysis, plasmonic nanocatalysis, and electrochemistry. |
 Thenner S. Rodrigues | Thenner Rodrigues received his BSc and MSc in Industrial Chemistry in 2010 and 2013, respectively, from Federal University of Ouro Preto. He obtained his PhD in Chemistry from the University of São Paulo in 2017, working with Prof. Pedro Camargo. He was a post-doctorate fellow at the Institute of Energy and Nuclear Research until October 2018. He was then hired as an Assistant Professor at Federal University of Rio de Janeiro and promoted to Adjunct Professor in 2021. His research focuses on the synthesis of metal and oxide nanostructures for applications in sustainable energy, catalysis, and adsorptive processes. |
 Jiale Wang | Jiale Wang got his BSc degree in Physics in 2006, from Shanghai Jiaotong University, Shanghai, China. In 2009, he got his MSc in Condensed Matter Physics from Fudan University, Shanghai, China. He received his PhD degree from Université Pierre et Marie Curie – Paris VI, France, in 2013. He then spent 2 years at the Universidade de São Paulo as a postdoc. Since 2016, he has been a professor at Donghua University, Shanghai, China. His research focuses on the fabrication of nanomaterials and their application in physics, chemistry, and biology. |
 Pedro H. C. Camargo | Pedro Camargo received a BSc and MSc in Chemistry in 2003 and 2005, respectively, from Federal University of Paraná. In 2009, he obtained his PhD in Biomedical Engineering from Washington University in St. Louis. In 2011, he was hired as an Assistant Professor at the University of São Paulo. He was promoted to Associate and Full Professor in 2015 and 2018, respectively. In 2019, he relocated to a Professor position at the University of Helsinki, Department of Chemistry. His research interests include designer nanomaterials for nanocatalysis and plasmonic catalysis. He serves as an Editor of the Journal of Materials Science. |
1. Introduction
Catalysis represents a crucial discipline when it comes to providing the scientific and technological foundation for making cleaner, more efficient, and economically viable chemical production processes.1–4 Since catalysis is involved in the synthesis of most chemicals, its developments will result in a huge effect on addressing several worldwide challenges of technological, economic, and environmental impacts.5,6 Catalysis can and will play a central role in enabling a more sustainable future and a circular economy.5,6 This can be achieved by using more environmentally friendly conditions to reduce energy consumption, i.e. lower temperatures and pressures, transformations with higher selectivity, less generation of waste, better utilization of renewable materials, among others.7–9 Catalysis can also contribute to mitigating climate change by turning CO2 from a waste into a commodity, in which CO2 can be converted to liquid fuels and other value-added molecules.10 Another area of impact by catalysis is the generation of H2 by reforming or water-splitting processes.11If the energy to drive catalytic processes could be harvested directly from sunlight, the possibility of replacing contemporary industrial processes based on terrestrial fuels by the conversion of radiant energy into chemical energy could become a step closer to reality.12 Common photocatalysts based on wide bandgap metal oxides can only be excited at the ultraviolet (UV) range (3–5% of the total solar energy) and may require electrical or thermal energy input.13 Visible light-harvesting semiconductors, such as sulfides and selenides, are unstable under photoreaction conditions.13 Finally, processes display low efficiencies and a lack of product selectivity.14 Due to these limitations, the search for photocatalysts with better performances and higher selectivity in the visible (VIS) and near-infrared (near-IR) ranges remains an important challenge. The diffuse nature of the solar flux and the negative dependence of photocatalytic rates on operating temperature are recognized as important obstacles that lead to low rates of photocatalytic reactions per material volume (mass), even for relatively efficient semiconductors.15
Plasmonic catalysis exhibits different behavior as compared with photocatalysis with semiconductors. The rates of photocatalytic reactions on excited plasmonic NPs show a super-linear power-law dependence on light intensity (rate is proportional to intensityn, with n > 1). Consequently, catalytic reactions on plasmonic NPs may display a positive relationship between quantum efficiency and photon flux (semiconductors exhibit lower quantum efficiency at higher light intensity).16 Also, the rate and quantum efficiency of plasmonic catalytic reactions may increase with operating temperature.16,17 This positive dependence provides the possibility that plasmonic NPs can effectively couple thermal and photonic stimuli to drive molecular transformations.16,18 Consequently, plasmonic catalysis allow integration of strong visible-wavelength plasmonic excitation with surface activation of molecules.19
In plasmonic nanoparticles (NPs), incident photons resonantly interact with the collective motion of electrons.12,20 This phenomenon is referred to as localized surface plasmon resonance (LSPR) and takes place at a characteristic frequency that is dependent on size, shape, structure, composition of the NPs, and the dielectric constant of the surroundings.21,22 For plasmonic metals such as silver (Ag), gold (Au), copper (Cu), and aluminum (Al), the LSPR excitation can take place at the visible and near-infrared ranges, opening up possibilities for the utilization of solar radiation to excite their LSPR.18,23 Concerning catalysis, it has been established that the LSPR excitation in plasmonic NPs can accelerate the rates of several chemical transformations, the so-called plasmonic catalysis or plasmonic photocatalysis.17,24–29 Therefore, plasmonic NPs have been attracting strong interest due to their ability to accelerate and control molecular transformations due to the LSPR excitation under milder conditions relative to traditional catalysis based on external heating.30–34 This field enables, at least in principle, to combine optical and catalytic properties at the nanoscale as well as to use VIS or near-IR light as a sustainable and abundant energy input to drive and control molecular transformations.18,26,35 Importantly, plasmonic catalysis allows one to overcome some of the intrinsic limitations of conventional wide bandgap semiconductor photocatalysts.16 In fact, plasmonic NPs are currently at the forefront of enhancing photocatalytic efficiencies.18,29,31,32,36,37 While plasmonic NPs have been extensively employed as sensitizers in conjunction with semiconductor photocatalysts, it is important to clarify that this is not the focus of this feature article.38–41
Two main mechanisms have been proposed for the observed activities in plasmonic catalysis: activation of molecules at the metal–molecule interface by LSPR-generated charge carriers via direct and indirect pathways and photothermal localized heating.17,18,42 Upon light LSPR excitation (Fig. 1A), the LSPR oscillation decays via non-radiative and radiative pathways. Non-radiative damping (Landau damping) leads to the generation of a non-thermal distribution of hot carriers (Fig. 1B).43 Further redistribution of their energy leads to a Fermi–Dirac-like distribution of charge carriers (Fig. 1C), which also further decays leading to the dissipation of heat to the surroundings via thermal conduction (Fig. 1D).43 Regarding the enhancement mechanism via LSPR-excited hot carriers, two potential microscopic pathways, indirect and direct, can take place (Fig. 1E and F). In the indirect pathway, following the generation of LSPR-excited distribution of hot carriers, charge carriers of appropriate energy from this distribution can scatter through the adsorbate states (Fig. 1E). This results in the nonthermal vibrational excitation of a bond in the adsorbate, which is then much more likely to undergo further reaction than one would expect from the equilibrium temperature.17,18,42,44 Conversely, the direct transfer pathway involves an LSPR-induced electron excitation from occupied to unoccupied orbitals of the metal–molecule or metal–adsorbate complex (Fig. 1F).43 Therefore, LSPR excited charge carriers can participate in the enhancement of rating or mediating molecular transformations via vibrational or electronic activation of adsorbed molecules at the surface.18,26,45,46
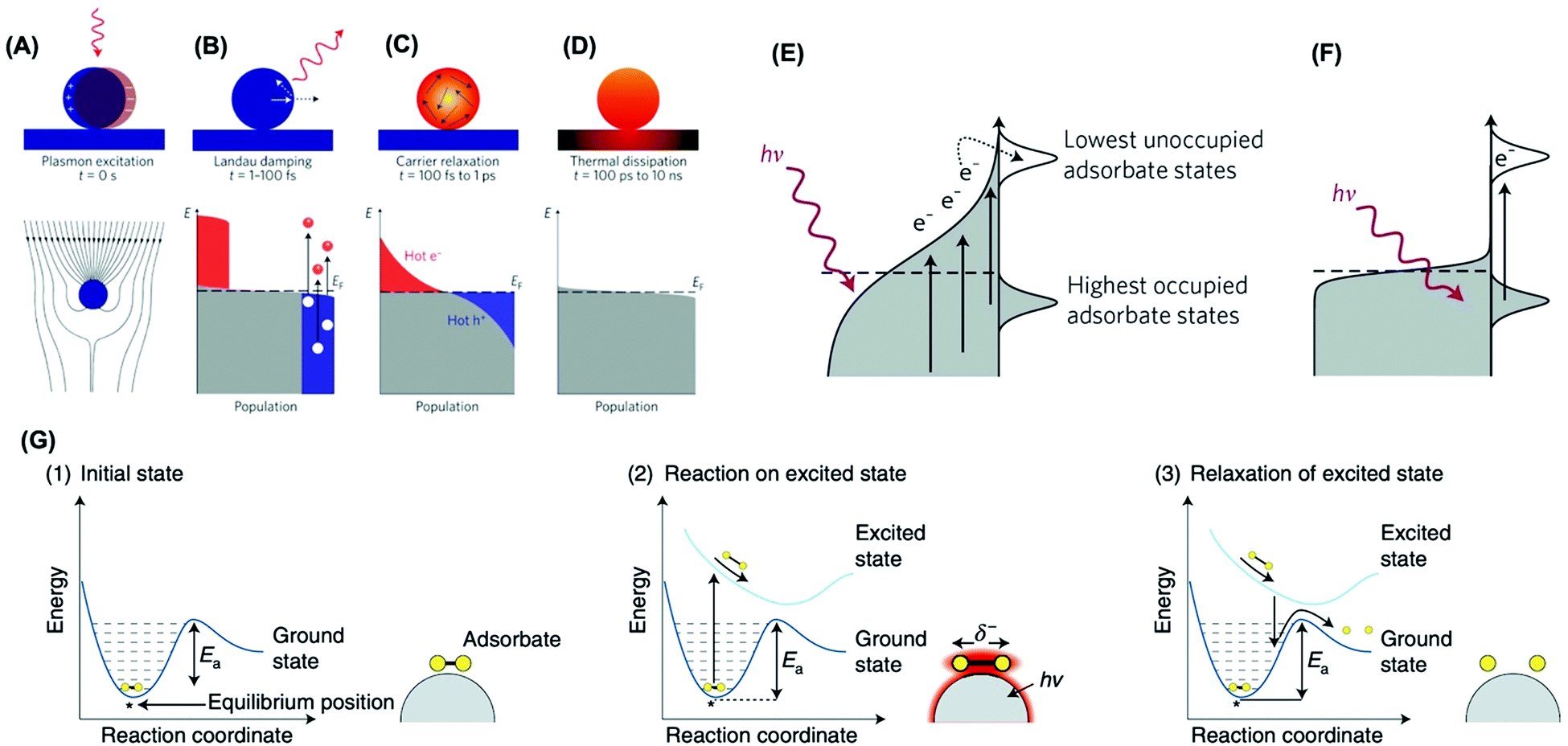 | ||
| Fig. 1 The LSPR excitation (A) and (B–D) population of the electronic states (grey) following LSPR excitation: hot electrons are represented by the red areas above the Fermi energy EF and hot hole distributions are represented by the blue area below EF; (B) In the first 1–100 fs following Landau damping, the thermal distribution of electron–hole pairs decays either through re-emission of photons or through carrier multiplication due to electron–electron interactions; (C) the hot carriers will redistribute their energy by electron–electron scattering processes on a timescale ranging from 100 fs to 1 ps; (D) heat is transferred to the surroundings on a timescale ranging from 100 ps to 10 ns.43 (E and F) Mechanisms of plasmon-mediated energy transfer to reactants: indirect (E) and direct (F) transfer mechanisms.17 (G) Mechanism of plasmon-mediated bond activation. Photoexcitation of the initial state (1) deposits energy into the adsorbate and elevates it to an excited potential energy surface. The adsorbate then moves along the excited potential energy surface, gaining kinetic energy and reacting in the excited state (2). If the adsorbate does not react in the excited state, it decays to the ground-state potential energy surface in a vibrationally excited state lowering the barrier for dissociation (3).18 (A–D) Reproduced by permission from Springer Nature, Nat. Nanotechnol., ref. 43. Copyright 2015. (E and F) Reproduced by permission from Springer Nature, Nat. Mater., ref. 17. Copyright 2015. (G) Reproduced by permission from Springer Nature, Nat. Catal., ref. 18. Copyright 2018. | ||
Fig. 1G depicts the mechanism for the plasmonic activation of the reactants. The molecule in its initial state (panel 1) is excited and then evolves along a charged (or excited) potential energy surface (panel 2).18 This molecular evolution can lead to a chemical transformation on the charged (or excited) potential energy surface, or once the charged (or excited) state decays to the ground-state potential energy surface but with additional vibrational energy (panel 3). This leads to a transient electronic exchange between the metal and reactant (an electronic excitation in the adsorbate–metal complex), yielding transient adsorbate ions (or excited states localized on the reactant). These adsorbate ions survive on metal surfaces tens of femtoseconds before the relaxation, which is sufficient to induce chemical transformation or add vibrational energy to the reactant to undergo a subsequent reaction.18 In addition to hot electrons, it has been shown hot holes can also participate in vibrational or electronic activation processes at the metal–molecule interface.43 Interestingly, the LSPR-generated hot charge carriers can contribute not only to enhanced reaction rates but also control over reaction selectivity. For example, the formation of reaction intermediates can be tuned, bond activation can be selective, and novel reaction pathways may arise relative to transformations employing external heating as the energy input.18,46,47 It is noteworthy that localized heating following LSPR relaxation can also contribute to increased rates under light excitation.17,26,42,43,45 This has been a topic of recent debate.48–51 While it is challenging to decouple this effect from the role played by hot charge carriers, recent studies have suggested that hot charge carriers have played a major contribution in plasmonic catalysis.18,48,52–54
A variety of plasmonic NPs and hybrid plasmonic-catalytic components have been employed as plasmonic catalysts towards a variety of transformations including oxidations, reductions, and coupling.29 Generally, studies have focused on the fundamental understanding of plasmonic catalysis and the demonstration of plasmonic catalytic activities, which also include plasmonically enhanced electrocatalysis.18,28,29,55 The main goal of this feature article is to discuss the recent contributions from our group in this field by employing plasmonic nanoparticles with well-defined and controllable physicochemical features as model systems for plasmonic catalysis. Controlled plasmonic NPs allow us to unravel how activity varies as a function of size, shape, composition, and structure (solid vs. hollow interiors) and get insights into structure–performance relationships in plasmonic catalysis. We believe that the establishment of structure performance relationships, by unraveling key compositions and morphologies as well as how and why plasmonic catalysis work under light excitation is important not only to the optimization of performances but also to enable the rational design of plasmonic catalysts with desired activities and selectivity for a transformation of interest (Fig. 2A). This would enable a transition to a design-driven approach in plasmonic catalysis, which can pave the way for widespread applications of solar-driven chemistry. We start by discussing the effect of size, shape, and composition in plasmonic NPs over their activities towards a model LSPR-mediated molecular transformations (Fig. 2B). Then, we focus on the effect of metal support interactions in supported plasmonic NPs over activities, reaction selectivity, and reaction pathways in plasmonic catalysis both in terms of charge transfer and the generation of active sites at the metal–support interface (Fig. 2B). Next, we shift to the control over the structure in hollow NPs and plasmonic nanorattles, which contain a plasmonic core inside a plasmonic shell separated by a void (Fig. 2B). This system is particularly interesting as it enables the generation of electromagnetic hot spots in a controllable fashion, therefore not relying on the uncontrolled aggregation of NPs, and the role of these electromagnetic hot spots in plasmonic catalysis. Based on these findings from these fundamental studies, we demonstrate a design-driven strategy for the development of plasmonic catalysis based on plasmonic-catalytic multicomponent NPs for two types of molecular transformations: the selective hydrogenation of phenylacetylene and the oxygen evolution reaction. Finally, we will discuss future directions, challenges, and perspectives in plasmonic catalysis with designer NPs.
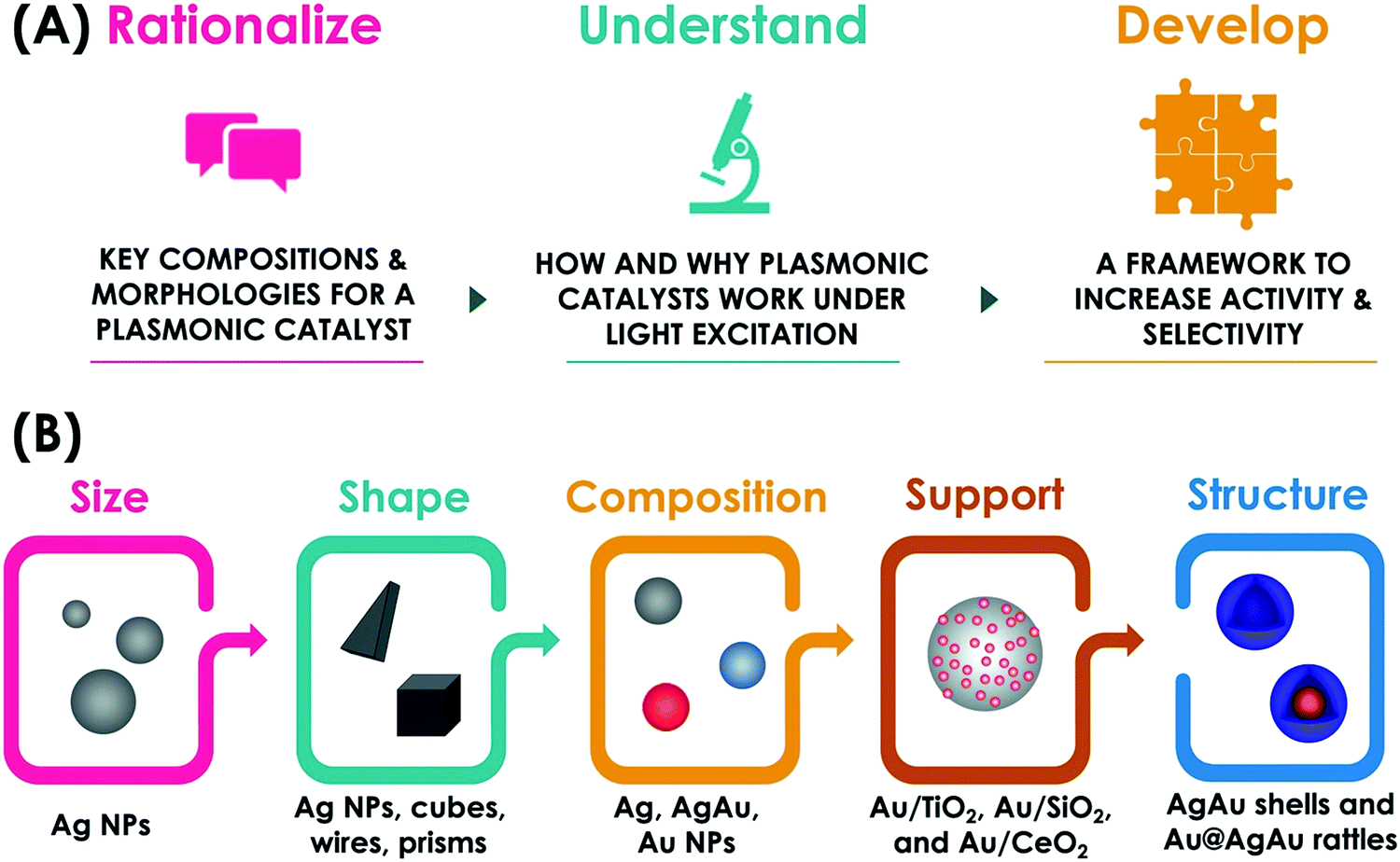 | ||
| Fig. 2 Conceptual figure of this feature article: (A) The use of designer nanoparticles as model systems in plasmonic catalysis enables the correlation on how key morphologies and compositions influence how and why a plasmonic catalyst work. This knowledge can provide the basis to transition towards a design-driven approach in plasmonic catalysis to generate systems with desired activities and selectivity. (B) This feature article covers efforts towards this goal by investigating the effect of size, shape, composition, metal–support interactions, and structure in LSPR-driven molecular transformations. | ||
It is important to mention that, despite the progress, plasmonic catalysis still presents some limitations and challenges. For example, research has often been focused on Ag and Au NPs. Ag and Au NPs, while displaying outstanding optical properties, are not very catalytic towards a variety of catalytic reactions. Moreover, the important NPs physical and chemical parameters required for the optimization of optical properties (sizes that maximize absorption relative to scattering) are not necessarily the same required for the maximization of catalytic properties. Thus, NPs designs that enable the maximization of both optical and catalytic properties are needed. Moreover, several metals that are catalytically important in catalysis, such as Pd and Pt, do not support plasmonic excitation in the visible or near-infrared ranges. This limits the scope of reactions that can be addressed by plasmonic catalysis. In this context, bimetallic plasmonic-catalytic NPs in the form of core–shell, core-island, and alloys have been proposed and represent the state-of-the-art in the field. Nevertheless, these systems are still limited in terms of catalytic metal use (which is pivotal for platinum group metals): NPs morphologies that allow for the maximum catalytic metal use efficiency are needed. In terms of optical properties, the formation of alloys or the deposition of catalytic metal layers at the surface of a plasmonic material can lead to a decrease in the absorption efficiencies and E-fields because of LSPR excitation (plasmon is damped). Thus, it is essential to develop plasmonic-catalytic materials in which the optical properties can be maximized and are not damped.
2. Size, composition, and shape
Size can play an important role in activities in plasmonic catalysis. Firstly, as established in nanocatalysis, a decrease in NPs size leads to a higher surface-area-to-volume ratio and therefore higher activities.56,57 Secondly, it is known that the absorption, relative to scattering, is strongly dependent on size.58,59 As absorption is related to the generation of LSPR excited charge carriers, i.e., hot electrons and holes, it is expected that the relative ratio of absorption and scattering is an important parameter that would influence performances in plasmonic catalysis.60 To precisely study the effect of size in plasmonic catalysis, NPs with controlled sizes in a reasonable range that is interesting for plasmonic catalysis are required. Nevertheless, a systematic control over plasmonic NPs size is not trivial. To address this challenge, we developed a strategy for the synthesis of Ag NPs in the 50–12 nm range.60 The approach was based on the use of Ag NPs 50 nm in size as starting materials to a controlled etching process in the presence of HNO3 (Fig. 3A). By controlling the HNO3 concentration and keeping the etching time constant at 40 min, the extent of chemical etching, that leads to a decrease in size, can be tuned. This enabled us to control the Ag NPs size from 50 to 12 nm in diameter as a function of the amount of HNO3 employed as etchant (Fig. 3B). Then, we employed the LSPR-mediated oxidation of p-aminothiophenol (PATP) to p,p′-dimercaptoazobenzene (DMAB) at the surface of the NPs as a model transformation.61 This transformation can be investigated in a Raman spectrophotometer, in which the incoming laser can work both as the LSPR excitation source and monitor the formation of the product by the SERS effect, as PATP and DMAB molecules have Raman signatures in different spectral positions.61,62 In this reaction, the LSPR excitation leads to the formation of hot electrons and holes, which can activate adsorbed O2 molecules, generating activated oxygen that subsequently drives the PATP oxidation at the surface of the NPs.28,63–65 In this transformation, the plasmonic NPs are deposited over a Si substrate and functionalized with PATP, and then the SERS spectra are acquired. Therefore, the DMAB to PATP conversion is monitored by measuring the relative areas of the SERS bands assigned to these molecules (in different wavenumber positions). Thus, the number/concentration of NPs in the measurements reported on the studies over the effect of size, composition, and shape are not expected to affect these relative ratios (a higher number of particles should increase the intensity of both PATP and DMAB peaks when conversion is not 100%). It is also noteworthy that this does not represent an example of catalysis as both reactant and product molecules are functionalized at the surface of the NPs via M-S bonds. Instead, this model reaction can be regarded as an example of LSPR-driven transformation. So, the use of this model reaction is limited in terms of catalytic oxidation as the removal of molecules from the surface for quantification/practical catalysis is challenging. Hence, one important limitation is not able to provide classical catalytic parameters such as TON or TOF. Therefore, it is important to mention that a direct comparison of the performance based on the intensity ratios is challenging due to the limitations of this model transformation as this is depended on several experimental parameters, such as the laser intensity, during the Raman measurements. Nevertheless, the use of the intensity ratio of DMAB/(DMAB + PATP) has allowed us and other groups in this field to probe the LSPR-driven transformation of PATP to DMAB in several plasmonic NPs.65–69 To address this issue, we also performed an analogous transformation in solution, i.e., the oxidation of aniline, for some NPs systems (nanorattles and Au/CeO2 NPs as discussed in Sections 3 and 4). Due to the similarity of these transformations, it can be inferred that the PATP oxidation would be in the solution too if monitored by isolated yield or other non-SERS techniques.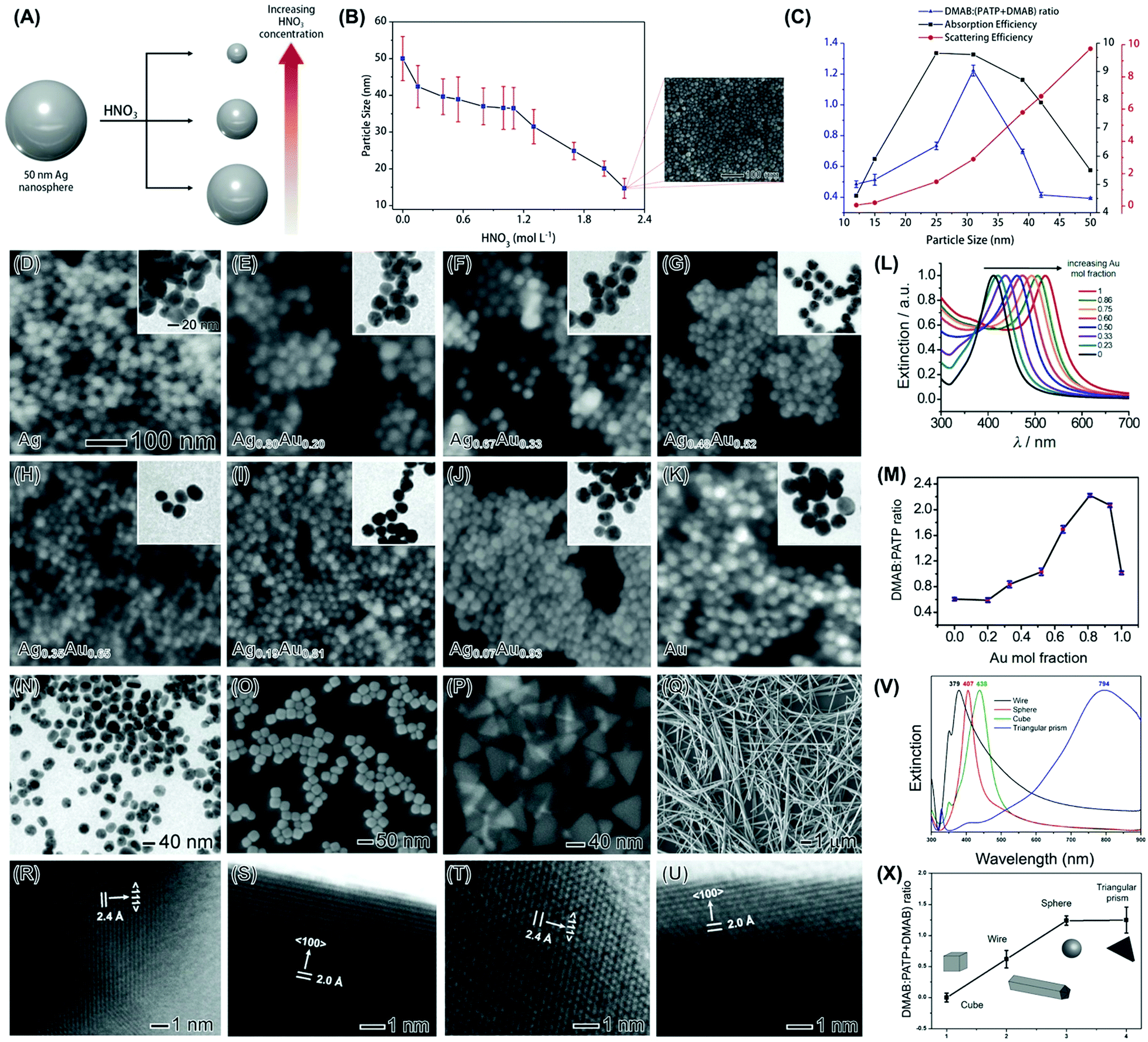 | ||
Fig. 3 (A) Top-down strategy for the synthesis of Ag NPs displaying controllable sizes.60 Ag NPs 50 nm in size were used as starting material in an etching process with HNO3 to generate Ag NPs of smaller sizes. As the HNO3 concentration increases, the Ag NPs decrease from 50 to 12 nm.60 (B) Ag NPs sizes was plotted as a function of the HNO3 concentration employed during the chemical etching procedure. In all cases, the etching time corresponded to 40 min. The inset shows the SEM image of 12 nm Ag NPs.60 (C) Volcano plot of absorption efficiencies (black trace), scattering efficiencies (red trace), and the DMAB:(PATP + DMAB) intensity ratios (blue trace) as a function of Ag NPs size.60 (D-K) SEM and TEM (insets) images for (D) Ag, (E) Ag0.80Au0.20, (F) Ag0.67Au0.33, (G) Ag0.48Au0.52, (H) Ag0.35Au0.65, (I) Ag0.19Au0.81, (J) Ag0.07Au0.93, and (K) Au NPs. The scale bars in (D) apply to all images/insets.70 (L) UV-VIS extinction spectra as a function of the Au content (Au mol fraction) in the NPs. (M) Obtained DMAB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(DMAB + PATP) 1433 cm−1:1081 cm−1 intensity ratios as a function of the Au content. The spectra were recorded under 633 nm excitation.70 (N–U) TEM (N), SEM (O–Q), and HRTEM (R–U) images for Ag nanospheres (N and R), nanocubes (O and S), triangular nanoprisms (P and T), and nanowires (Q and U).72 The nanospheres, nanocubes, nanoprisms, and nanowires were 36 ± 4 nm in diameter, 35 ± 5 nm in edge length, 64 ± 9 nm in edge length, and 153 ± 15 nm in width, respectively. (V) UV-VIS extinction spectra from aqueous suspensions containing Ag nanowires, nanospheres, nanocubes, and triangular nanoprisms (black, red, green, and blue traces, respectively).72 (X) DMAB:(DMAB + PATP) 1433 cm−1:1081 cm−1 intensity ratios as a function of the Ag shape. The spectra were recorded under 633 nm excitation.72 (A–C) Reproduced from ref. 60 with permission from Wiley-VCH, copyright 2018. (D–M) Reprinted (adapted) with permission from reference 70. Copyright 2014 American Chemical Society. (N–X) Reprinted (adapted) with permission from reference 72. Copyright 2015 American Chemical Society. :(DMAB + PATP) 1433 cm−1:1081 cm−1 intensity ratios as a function of the Au content. The spectra were recorded under 633 nm excitation.70 (N–U) TEM (N), SEM (O–Q), and HRTEM (R–U) images for Ag nanospheres (N and R), nanocubes (O and S), triangular nanoprisms (P and T), and nanowires (Q and U).72 The nanospheres, nanocubes, nanoprisms, and nanowires were 36 ± 4 nm in diameter, 35 ± 5 nm in edge length, 64 ± 9 nm in edge length, and 153 ± 15 nm in width, respectively. (V) UV-VIS extinction spectra from aqueous suspensions containing Ag nanowires, nanospheres, nanocubes, and triangular nanoprisms (black, red, green, and blue traces, respectively).72 (X) DMAB:(DMAB + PATP) 1433 cm−1:1081 cm−1 intensity ratios as a function of the Ag shape. The spectra were recorded under 633 nm excitation.72 (A–C) Reproduced from ref. 60 with permission from Wiley-VCH, copyright 2018. (D–M) Reprinted (adapted) with permission from reference 70. Copyright 2014 American Chemical Society. (N–X) Reprinted (adapted) with permission from reference 72. Copyright 2015 American Chemical Society. | ||
According to our data, a volcano type variation between PATP conversion and size was detected (Fig. 3C, blue trace).60 Here, the highest activities were obtained when the diameter of the NPs was around 30 nm. This result shows that the correlation between LSPR mediated activity and size is not obvious, and there is an intermediate size in which the conversion was optimized.60 This highlights the importance of fine-tuning size to maximize conversions in LSPR mediated transformations. When the PATP conversion was compared with the calculated absorption (Fig. 3C, black trace) and scattering (Fig. 3C, red trace) efficiencies as a function of size, we can observe that the variations in conversion followed the trend on the increase of absorption relative to scattering. In other words, better PATP to DMAB conversions are achieved for sizes that maximize the contribution of absorption, relative to scattering, in the NPs. Here, as absorption is related to the generation of LSPR excited charge carriers, the increased absorption relative to scattering will lead to higher conversions driven by the LSPR excitation.60 It is important to note that a correlation of absorption and scattering efficiencies at the excitation wavelength, rather than at the LSPR maximum, could represent a more precise description of how NPs activity is impacted by extinction, and this was investigated in the liquid phase transformations as described in the following sections. However, for the studies on NPs size, that were performed on a solid substrate, our data indicate that the correlations of absorption and scattering efficiencies at the LSPR maximum may represent a good approximation and match relatively well the experimental data. Moreover, we have compared the calculated and experimental spectra as a function of size for the NPs in suspension. Our results indicated that both calculated and experimental spectra were comparable. While the experimental LSPR for 50, 25, and 12 nm were located at 425, 405, and 400 nm, respectively, the calculated LSPR for 50, 25, and 12 nm were located at 411, 393, and 390 nm, respectively.60
We then turned our attention to the effect of composition towards LSPR-driven chemistry. We focused on bimetallic AgAu NPs as both Ag and Au display plasmonic properties in the visible range as we aimed to correlate bimetallic compositions for two metals that are plasmonic with the detected LSPR-mediated conversions.70 We also employed the LSPR-driven PATP oxidation to DMAB as a model transformation and bimetallic AgAu prepared via co-reduction as bimetallic plasmonic NPs,61,62 as shown in Fig. 3D–K. The co-reduction approach for the synthesis of AgAu NPs employed AgNO3 and HAuCl4 as the Ag and Au precursors, respectively, and sodium citrate as both the reducing agent and stabilizer, and 100 °C as the reaction temperature. The composition was controlled by manipulating the Ag+/AuCl4− molar ratios.70 In these NPs, the Au molar fraction gradually increased from 0 (Ag NPs) to 1 (Au NPs) as indicated by the labels in Fig. 3D–K (Ag, Ag0.80Au0.20, Ag0.67Au0.33, Ag0.48Au0.52, Ag0.35Au0.65, Ag0.19Au0.81, Ag0.07Au0.93, and Au NPs).70 The nanoparticles displayed spherical shape and well-defined compositions as determined by FAAS (flame atomic absorption spectroscopy). Their optical properties were strongly dependent on composition, in which a redshift in the extinction band was observed with the increased ratio of Au (Fig. 3L). These variations agree with the formation of AgAu bimetallic NPs and varied from 410 to 520 nm as the Au molar fraction increased from 0 to 1. Interestingly, the PATP conversion was strongly dependent on the compositional variations of the NPs, in which a volcano-type variation of conversion as a function of composition was detected (Fig. 3M).70 While some bimetallic compositions displayed higher catalytic activities relative to their monometallic counterparts, a peak in conversion was observed when the composition of the NPs corresponded to Ag0.19Au0.81. The observed variations in activity could be explained by two factors that influence LSPR excitation: as the Au content was increased from 0 up to 0.81 in the NPs, the increased matching between the excitation wavelength (633 nm) and the LSPR position led to stronger plasmon excitation and thus improved catalytic activities.70 As the Au content was further increased and the LSPR position further red-shifted, interband transitions above 500 nm contributed to a decrease in catalytic activity.71 This data suggested that the control over composition also plays a pivotal role in the optimization of activities in plasmonic catalysis, which does not simply follow the trend that would be expected based on the better matching between LSPR excitation wavelength and extinction band.
Next, we investigated the effect of shape by employing Ag nanospheres, nanocubes, triangular prisms, and nanowires as model systems (Fig. 3N–U).72 The nanospheres, nanocubes, and nanowires were prepared by the polyol synthesis while the triangular nanoprisms were synthetized by a visible-light-photoinduced conversion route.72 They are interesting to study shape-dependent behavior as they enable the exposure of different fractions of surface facets: Ag nanospheres (obtained from polyol synthesis) present a mix of various facets,73 nanocubes are enclosed by {100} facets,74 triangular prisms contain a twin plane parallel to the {111} top and bottom surfaces,75 and nanowires contain a mixture of {100} facets on the sides and {111} at the ends/tips.76 Therefore, the fraction of {111} relative to {100} surface facets increase in the order: nanocubes < nanowires < nanospheres < nanoprisms.72 The UV-VIS extinction spectra for the Ag nanospheres, nanocubes, triangular prisms, and nanowires are shown in Fig. 3V. It can be observed that the LSPR band was centered at 379, 407, 438, and 794 nm (black, red, green, and blue traces, respectively) for the nanowires, nanospheres, nanocubes, and nanoprisms, respectively, demonstrating that the optical properties were strongly dependent on shape as expected.72
The LSPR mediated oxidation of PATP to DMAB employing 633 nm as the excitation wavelength for the different Ag nanocrystals is shown in Fig. 3X. A strong shape dependency can be observed, in which the LSPR-mediated conversion increased in the order nanocubes < nanowires < nanospheres < nanoprisms. This indicates that this increase in conversion also followed the order in which the fraction of {111} relative to {100} surface facets increased in the nanocrystals. It is important to note that these variations cannot be solely explained based on the matching between the LSPR position and the wavelength of the incoming light excitation (633 nm).72 While it may explain the higher activity of the triangular prisms, it does not correlate with the LSPR band position for the nanowires, nanospheres, and nanocubes. Here, it is plausible that the shape-dependent behavior could be related to the stronger O2 adsorption energy on Ag{100} relative to Ag{111} facets.72,77 As activation of adsorbed O2 followed by their desorption represents a crucial step for the DMAB formation, the weaker adsorption of O2 on Ag{111} surface facets can favor the O2 desorption step after activation, and thus contributes to better catalytic performances.72
3. Metal–support interactions
The examples discussed so far focused on the utilization of the unsupported plasmonic NPs as model systems to get insights into how size, composition, and shape influence performances in a transformation at the surface of the NPs that is driven by the LSPR excitation. However, for the practical use of plasmonic catalysis in liquid and gas-phase transformations, it is imperative to use supported NPs, i.e., materials in which the NPs (active phase in plasmonic catalysis) are dispersed at the surface of support material.78,79 In analogy with nanocatalysis, supported systems enable a good dispersion of the active phase, can contribute to an increase in the stability, avoid agglomeration during reaction conditions, and facilitate recovery after the reaction.80 In addition, when metal–support interactions can take place, the formation of active sites at the metal–molecule interface and bifunctional mechanisms may also arise, in which both the NPs and the support contribute with catalytic sites for a target reaction.81,82 While plasmonic NPs have been employed as sensitizers to improve photocatalytic performances of semiconductors, in the context of plasmonic catalysis, charge transfer between the plasmonic NPs and semiconductor support (injection at the conduction band, for example) can also influence plasmonic catalytic activities of the plasmonic component. Although these concepts have been established in nanocatalysis and photocatalysis with semiconductors, their exploration had remained limited in terms of plasmonic catalysis. Recently, we have focused on the synthesis of several model-supported plasmonic NPs to understand the effect of metal–support interactions over reaction selectivity and reaction pathways dependence (based on charge transfer between plasmonic NPs and support), and activity (based on the activation of species at the metal–support interface).The first system that was investigated was based on Au NPs supported on TiO2 (Au/TiO2 NPs).83 This material is attractive to study metal–support interactions based on charge transfer as it has been established that the LSPR excitation of Au NPs generates hot electrons that can be injected into the TiO2 conduction band.83 Our group has developed a strategy for the controlled deposition of Au NPs at the surface of TiO2 colloidal spheres.84 The synthesis was based on seed growth of Au at the surface of TiO2 without the need for any pre-treatment or functionalization by employing HAuCl4 as the Au precursor, PVP as a stabilizer, TiO2 microspheres as physical templates, ascorbic acid as the reducing agent, and water as the solvent.84 The Au surface coverage and size could be controlled by performing successive Au reduction and deposition steps (Fig. 4A). This approach enabled the control over the Au NPs size while enabling a uniform distribution of NPs at the surface of TiO2 (Fig. 4B).84
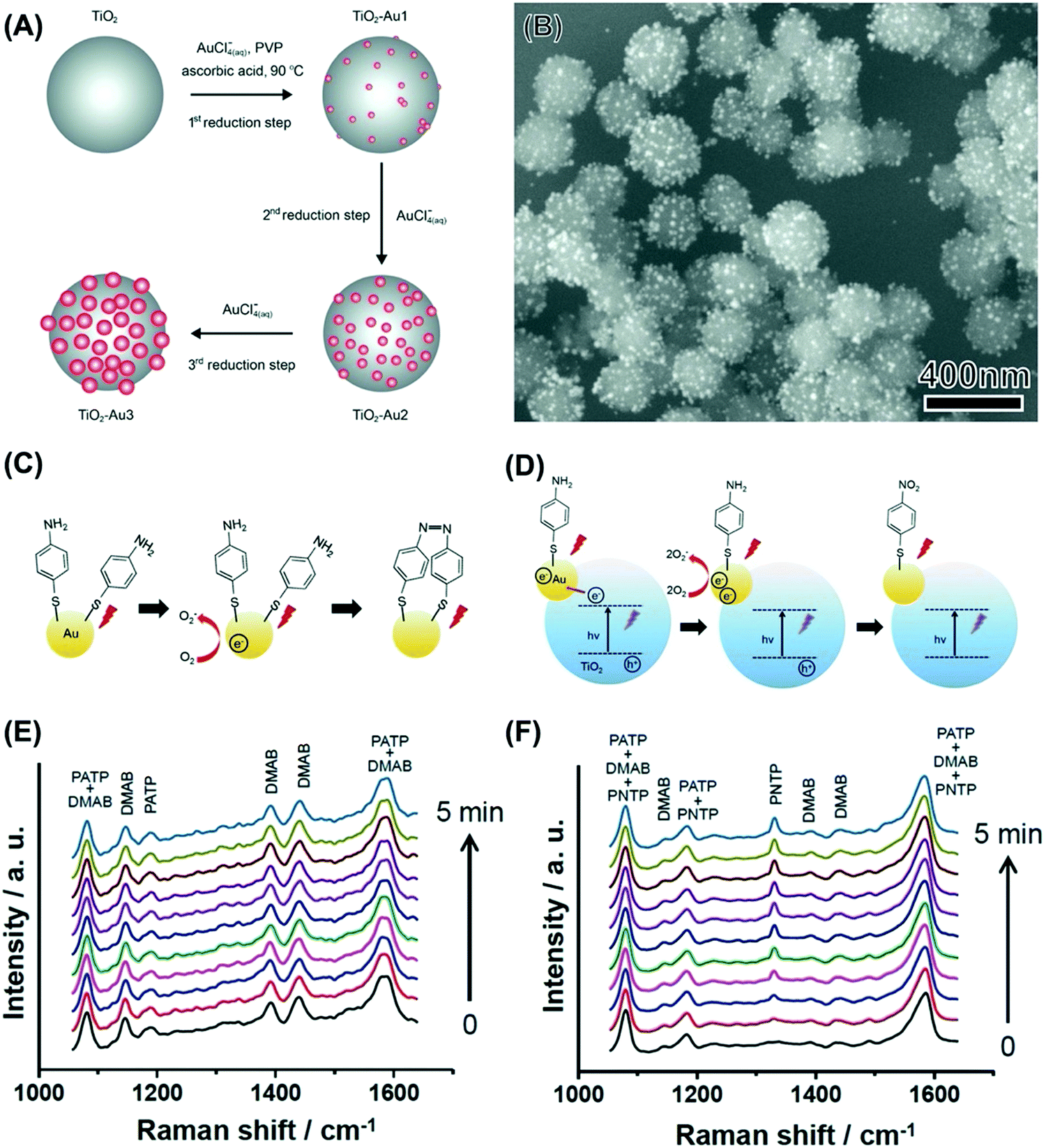 | ||
| Fig. 4 (A) Strategy for the synthesis of TiO2 colloidal spheres decorated with Au NPs having controlled sizes and uniform NPs distribution.84 This strategy was based on successive reduction steps employing AuCl4−(aq) as the Au precursor, PVP as the stabilizer, ascorbic acid as the reducing agent, and water as the solvent.84 (B) SEM images of Au/TiO2 NPs employed for the LSPR-mediated oxidation of PATP.83 (C and D) Proposed mechanism for the oxidation of PATP to DMAB over Au NPs (C) and the oxidation of PATP to PNTP over Au/TiO2 NPs (D).83 In Au NPs (C), LSPR-excited hot electrons contribute to the PATP oxidation to DMAB. When the Au/TiO2 NPs were excited both by the LSPR and UV (D), LSPR-excited hot electrons and electrons transferred from TiO2 to Au contributed to the formation of PNTP rather than DMAB.83 (E and F) Time-dependent SERS spectra for Au (E) and Au/TiO2 (F) NPs that had been functionalized with PATP. The initial spectra (0 s) were recorded without UV illumination. After this initial spectrum was registered, the UV light was turned on and the subsequent SERS spectra were recorded in 30 s intervals (bottom to top). All spectra were acquired at 632.8 nm excitation.83 (A) Reprinted (adapted) with permission from ref. 84. Copyright 2013 American Chemical Society. (B–F) Reproduced from ref. 33 with permission from Wiley-VCH, copyright 2015. | ||
We started by employing the LSPR mediated oxidation of PATP as a model transformation.61 As discussed previously, the LSPR excitation of unsupported Au NPs leads to the formation of DMAB as schematically shown in Fig. 4C and the SERS spectra in Fig. 4E).70,72 No conversion was observed when the Au/TiO2 NPs were excited in the visible range as LSPR excited hot electrons in Au are injected into the TiO2 conduction band, hampering the O2 activation steps and thus the PATP to DMAB oxidation. Interestingly, it was found that, when Au/TiO2 NPs were excited both in the visible and the ultraviolet region, the LSPR mediated PATP oxidation leads to the formation of p-nitrothiopenol (PNTP), as schematically shown in Fig. 4D and in the SERS spectra (Fig. 4F).83 Here, the excitation of Au/TiO2 NPs in the visible and ultraviolet region leads to the excitation of electrons in TiO2 across the bandgap. These electrons can be transferred to Au, increasing the number of hot electrons that can activate O2 molecules, which in turn contribute to the PATP oxidation to PNTP. The transfer of electrons from the conduction band from TiO2 to Au has been confirmed by photoluminescence, in which its intensity was significantly weaker for the samples containing Au NPs relative to pure TiO2. This was assigned to electron injection into the Au NPs reducing recombination at the band edge of TiO2. This was further shown by ultrafast charge dynamics studies and supported by DFT simulations as later described in Fig. 5G.85 These observations agree with reported photocatalytic systems based on TiO2 and Au.86 Therefore, these results show that the control over the charge transfer process in plasmonic–support (Au–TiO2) materials can be employed to control the reaction selectivity in LSPR-driven molecular transformations.83 Our results indicate that under visible light excitation (no UV) and various experimental conditions, hot electrons from Au in Au/TiO2 are transferred to the TiO2 conduction band rather than activating oxygen or favoring PNTP reduction. Therefore, transfer to the TiO2 conduction band is favored and the activation of oxygen or the reduction of PNTP by hot electrons under visible light excitation takes place only in the absence of TiO2 (pure Au NPs). In Au/TiO2, oxygen activation or the reduction of PNTP was observed only under UV + visible light irradiation.
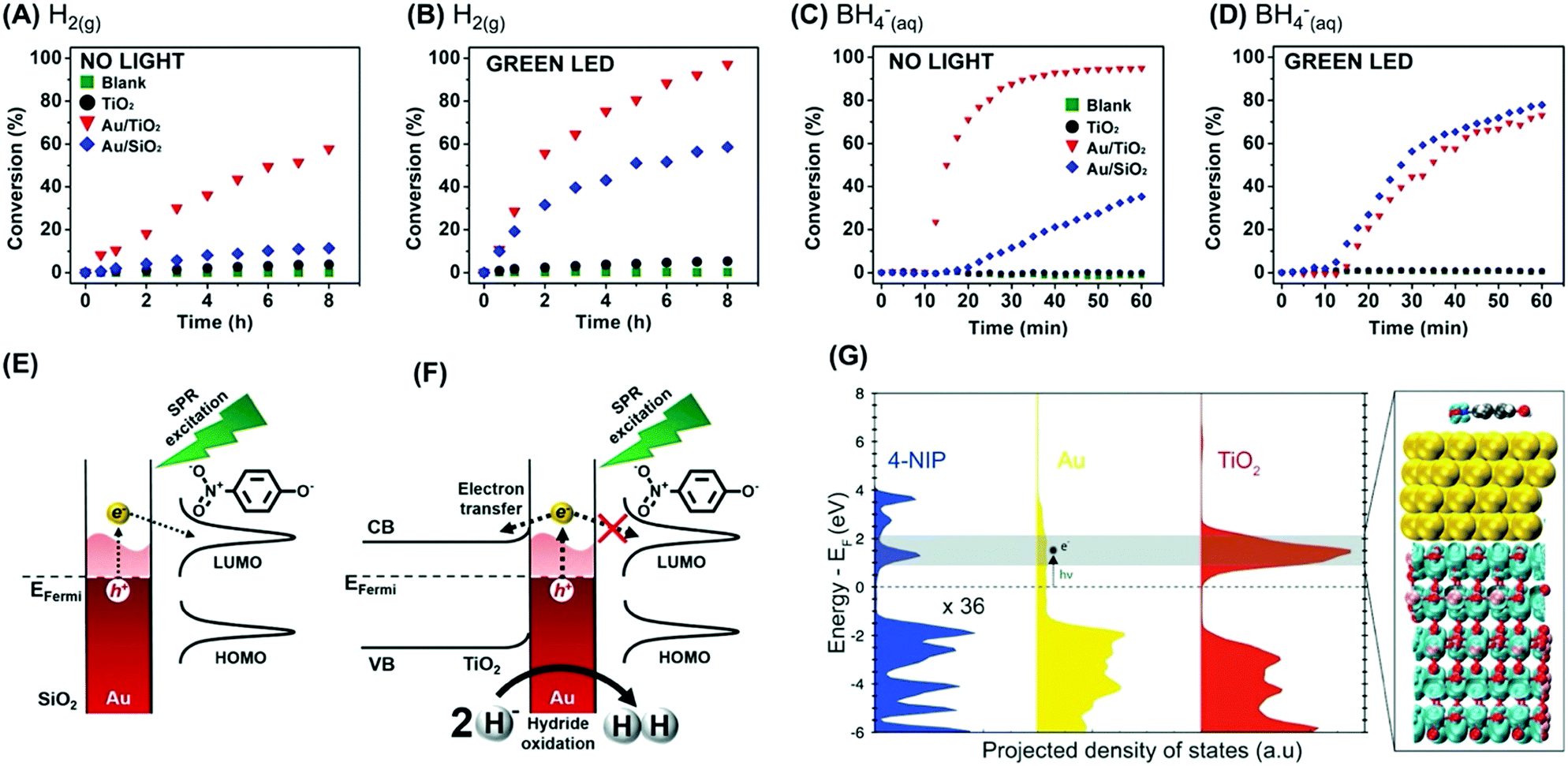 | ||
| Fig. 5 (A–D) conversion % as a function of time for the 4-nitrophenol hydrogenation by H2(g) (A and B) or BH4−(aq) (C and D) catalyzed by TiO2 (black trace), Au/TiO2 (red trace), and Au/SiO2 (blue trace).87 A blank reaction (in the absence of any catalyst) is also shown (green trace). The reaction was performed in the absence (A and C) and presence (B and D) of visible light excitation by a green LED. (E and F) Scheme for the detected activities towards the 4-nitrophenol hydrogenation by BH4−(aq) catalyzed by (E) Au/SiO2 and (F) Au/TiO2 under LSPR excitation.87 (G) DFT-calculated electronic structure of 4-nitrophenol adsorption on an Au/TiO2 surface. The projected density of states of each component (4-nitrophenol, Au, TiO2) is shown for comparison. The charge density plot shows the degenerate states of the conduction band of TiO2 and the LUMO of 4-nitrophenol.87 Reproduced from ref. 87 with permission from Wiley-VCH, copyright 2018. | ||
Next, these Au/TiO2 NPs were employed as model systems to investigate how metal support interactions (due to charge transfer) influence the performances in plasmonic catalysis as a function of the reaction pathways.87 To this end, we focused on two types of liquid phase transformations: the hydrogenation of nitrophenol employing H2(g) or BH4−(aq). This is interesting because the use of these different reducing agents occurs through distinct reaction pathways.87 When BH4−(aq) is employed as a reducing agent, the formation of M–H bonds at the NPs surface takes place.88 The transfer of this H species to substrate molecules at the nanoparticle surface represents the rate-limiting step according to the Langmuir–Hinshelwood mechanism.88 In the case of H2(g), the rate-limiting step represents the cleavage of molecular hydrogen leading to the formation of M–H bonds as they interact with the metal surface.89
Fig. 5A and B show the conversion % as a function of time for the nitrophenol hydrogenation employing H2(g) as a reactant under dark and light illumination conditions, respectively. The data for the blank reaction (in the absence of catalyst), TiO2, and Au/SiO2 NPs that were employed as a control (in which charge transfer from Au does not take place) is also shown. The conversion percentages for Au/TiO2 NPs (and Au/SiO2 NPs) increased under light illumination, which agrees with the LSPR enhancement of the reaction rate. In this reaction, the LSPR excitation leads to the generation of hot electrons that can activate the H2 antibonding orbitals, facilitating the H2 cleavage at the surface and thus increasing the reaction rate.87 When BH4−(aq) was employed as a reactant, a different behavior was detected (Fig. 5C and D for reactions under dark and light illumination conditions, respectively). While the activity of Au/SiO2 NPs increased under light illumination, a decrease in activity was detected for Au/TiO2 NPs. In Au/SiO2 NPs, hot electrons can be injected into the LUMO orbital of adsorbed 4-nitrophenolate species, leading to the acceleration of the reaction under light excitation (Fig. 5E).87 Conversely, for Au/TiO2 NPs it is proposed that LSPR excited hot electrons are preferentially injected into the conduction band of TiO2 instead (Fig. 5F). In this case, hot holes can contribute to oxidize and removing M–H species from the surface via oxidation and generation of H2(g), leading to a decrease in activity under light illumination. Control experiments in which the amount of H2(g) generated from the reaction was measured both in the absence and in the presence of visible light excitation showed that the generation of H2 from the reaction increased under green light excitation. These observations are supported by DFT calculations, that show a higher DOS for the conduction band of TiO2 closer to the Fermi energy level of Au relative to the LUMO of 4-nitrophenol (Fig. 5G).87 The built-in electronic field at the Au/TiO2 interface drives electrons into TiO2, whereas the weak interaction between the molecule and Au hinders the electron injection into the molecular orbital. This result indicates that the reaction pathway also is an important parameter to consider when designing a plasmonic catalyst, and charge transfer processes that result from metal support interactions affect reactions differently as a function of the reaction pathway. It is noteworthy that an induction period has been often observed for the 4-nitrophenol reduction over metal NPs.88 The origin of this induction period has been debated and assigned to different processes such as the diffusion time required for 4-nitrophenol l to be adsorbed onto the catalyst's surface before the reaction could start, surface restructuring necessary to render the metal nanoparticles as active catalysts, and the rapid conversion of 4-aminophenolate back into 4-nitrophenolate by a side reaction that terminates when the dissolved oxygen is consumed.90
In addition to the plasmonic NPs as the active phase, metal–support interactions can contribute to the improvements of performances due to the formation of active species at the metal–support interface. For example, CeO2 is a particularly interesting support material towards oxidation reactions in nanocatalysis to its oxygen mobility and storage properties, which can be further enhanced at the metal–support interface due to metal–support interactions that lead to the formation of Ce3+ as well as adsorbed oxygen species (O2− or O−).91,92 These species, in principle, can be activated by the LSPR excitation and lead to an increase in activities in plasmonic catalysis, such as towards oxidations.93 Intriguingly, this has remained unexploited in plasmonic catalysis. This requires an understanding of the role played by the LSPR-mediated activation of surface oxide species in CeO2 supports at the metal–support interface relative to the contribution/activation of O2 from the air is required. To bridge this gap, we developed a simple approach to isolate and quantify the contribution from surface oxide species at the metal–support interface in Au/CeO2 materials (relative to O2 from the air) to the plasmonic catalytic activity towards oxidation reactions.93 In these systems, the metal–support interactions were probed by temperature-programmed reduction (TPR), Raman, and XPS. In Au/CeO2, the presence of Au lowered the temperature for the reduction of CeyOx oxides at the surface (this peak shifted from 620 to 410 °C). In the Raman, the strong band centered at 467 cm−1 can be attributed to the symmetric stretching vibration mode of oxygen atoms around the Ce4+ ions. In addition, the Raman mode near 625 cm−1, corresponding to the presence of oxygen vacancies, can be observed in the Au/CeO2 material. Finally, XPS spectra for the O 1s showed that the OS/OL (surface/lattice oxygen species) ratios increases for Au/CeO2 relative to CeO2.
The plasmonic catalytic activity was investigated employing transformations that rely on the activation of O2via LSPR excitation. We employed both the oxidation of (PATP) functionalized at the surface and the liquid phase oxidation of aniline as proof-of-concept transformations.93 To assess the contribution from surface oxide species, the plasmonic catalytic performances of Au/CeO2 were compared to Au/SiO2. While in Au/CeO2 the LSPR excitation can activate the oxygen species from both metal–support interactions and the O2 atmosphere (Fig. 6A), the LSPR excitation can activate only atmospheric O2 in Au/SiO2 due to the absence of the active oxygen species originating from metal–support interactions (Fig. 6B).93 Moreover, we probed the performances of Au/CeO2 in the presence and absence of an O2 atmosphere.
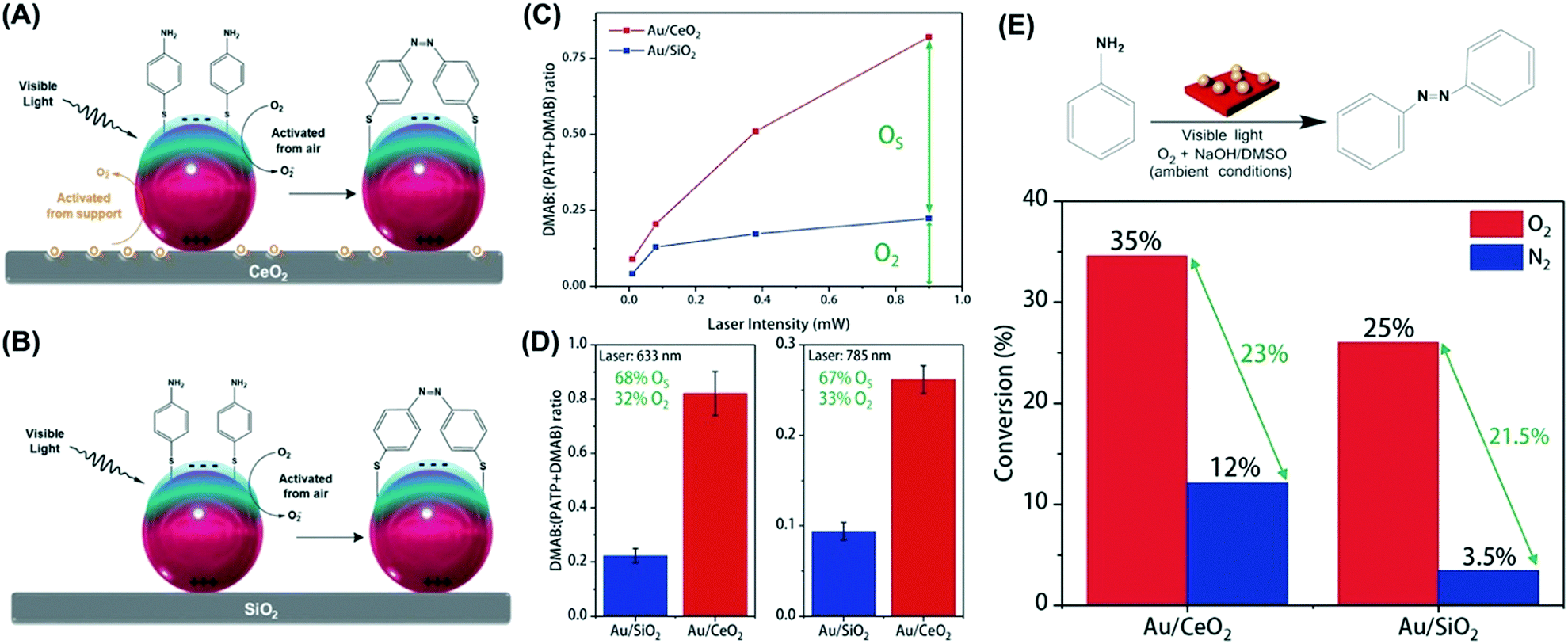 | ||
| Fig. 6 Scheme for the LSPR-mediated oxidation of PATP to DMAB promoted by activated oxygen under visible light employing (A) Au/CeO2 and (B) Au/SiO2 NPs.93 In (A) Au/CeO2, the activation of atmospheric oxygen (O2, black trace) and surface oxygen species (OS, orange trace) take place and contribute to the PATP to DMAB conversion. In (B) Au/SiO2, only the activation of atmospheric oxygen takes place. (C and D) Laser-power-dependent (C) and laser-wavelength-dependent (D) oxidation of PATP, expressed as the DMAB:(PATP + DMAB) ratio, for Au/CeO2 and Au/SiO2 (red and blue lines/bars, respectively). (E) Oxidation of aniline catalyzed by Au/CeO2 and Au/SiO2 materials under O2 and N2 atmospheres (red and blue bars, respectively). Conversion % are expressed for 12 h of reaction time. The reaction was carried out under ambient conditions.93 Reproduced from ref. 93 with permission from the Royal Society of Chemistry, copyright 2018. | ||
By first probing the LSPR-mediated oxidation of PATP at the surface, the data showed that the conversion as a function of the laser power (633 nm) was much higher for Au/CeO2 relative to Au/SiO2, in which the difference could be assigned to the activation of surface species formed at the metal–substrate interface (Fig. 6C).93 These differences were observed both at 633 and 785 nm as the excitation wavelengths, indicating that around 68% of the conversion arises from the activation of species at the metal–substrate interface (Fig. 6D).93 Then, the plasmonic catalytic activities towards the liquid phase oxidation of aniline were investigated (Fig. 6E). In this case, a 300 W tungsten lamp was the excitation source under ambient conditions (room temperature, 1 atm of O2 or N2). Fig. 6E shows the conversion (%) for the oxidation of aniline after 12 hours by employing Au/CeO2 and Au/SiO2 as catalysts under O2 (red bars) and N2 (blue bars). In both cases, the selective oxidation of aniline to azobenzene was detected. Similar to what was observed for the PATP oxidation, Au/CeO2 led to higher conversions under light excitation in both cases.93 The differences in conversion (∼10%) between the Au/CeO2 and Au/SiO2 NPs under O2 and N2 atmosphere can be assigned to the activation of oxygen species from the metal–support interface, representing a contribution of 28.6% to the detected conversion percentage. This result demonstrates that in oxidations promoted by activated O2, metal–support interactions can not only be put to work towards the optimization of LSPR-mediated transformations but also can play a significant role in the detected performances.93 This is especially attractive in the context of the utilization of plasmonic catalysis towards green catalytic oxidations under ambient conditions.
4. Structure (solid vs. hollow interiors)
The synthesis of NPs displaying hollow interiors and thin, porous walls is attractive for various catalysis applications.94–96 Hollow NPs enable higher surface-to-volume ratios relative to their solid counterparts.94,95 Hollow plasmonic NPs also display tunable optical properties, which combined with the higher surface-to-volume ratios than conventional solid NPs can contribute to achieving the optimization of activities in plasmonic catalysis.94,95 Taking hollow NPs based on Ag and Au as an example of hollow plasmonic NPs, it has been shown that AgAu nanoshells and nanorings can be prepared by a galvanic replacement reaction between Ag NPs as sacrificial templates and AuCl4−(aq).97,98The galvanic replacement reaction between Ag and AuCl4−(aq) proceeds according to eqn (1). The galvanic reaction is a redox process, in which the electrical reduction potential difference between a sacrificial template (Ag in this case) and metal ions in solution (AuCl4−(aq)) provide the driving force for the reaction, i.e., the oxidation and dissolution of the template together with the reduction of metal ions from solution and their deposition over the template's surface. As 3Ag0 atoms are oxidized for each AuCl4−(aq) that is reduced to Au0, this leads to the hollowing of the nanostructure. The overall composition and structure can thus be tailored by controlling the extent of the reaction (the molar ratio between a sacrificial template and metal ions in solution).
| 3Ag(s) + AuCl4−(aq) → 3Ag+(aq) + Au(s) + 4Cl−(aq) | (1) |
:AuCl4− ratios during the synthesis enables the control over the composition, structure (extent of hollowing), and optical properties.97,98 For instance, it has been observed that the LSPR band was gradually red-shifted and broadened in the range from around 400 nm to 800 nm as a function of the formation of bimetallic compositions and hollow interiors (extent of the galvanic replacement reaction).97,98
While several studies in plasmonic catalysis involve the use of lasers as excitation sources, the use of inexpensive LEDs and commercial white lamps is highly desirable in practical applications.97 If the use of commercial white lamps is considered, developing plasmonic NPs having LSPR extinction that closely matches the emission spectra of commercial lamps represents an intuitive strategy to maximize performance.97 Nevertheless, studies employing white light as the excitation sources still focus on Ag and Au NPs whose LSPR extinction does not necessarily match the lamp emission. In this context, we prepared AgAu nanorings by a controlled galvanic replacement reaction between Ag NPs and AuCl4−(aq) (Fig. 7A).97 The produced AgAu nanorings had ultrathin walls and uniform shapes and sizes (Fig. 7B). Interestingly, this approach allowed us to obtain an LSPR extinction spectrum that closely matches the emission spectra of a commercial and inexpensive halogen–tungsten lamp as shown in Fig. 7C.97 By employing the liquid phase oxidation of methylene blue as a model transformation, it was demonstrated that this approach was indeed effective in the maximization of activities (Fig. 7D). The plasmonic catalytic activity for the AgAu nanorings was 4.3 and 4.7-fold higher relative to the Au and Ag nanospheres (and solid AgAu NPs) under similar metal loading conditions.97
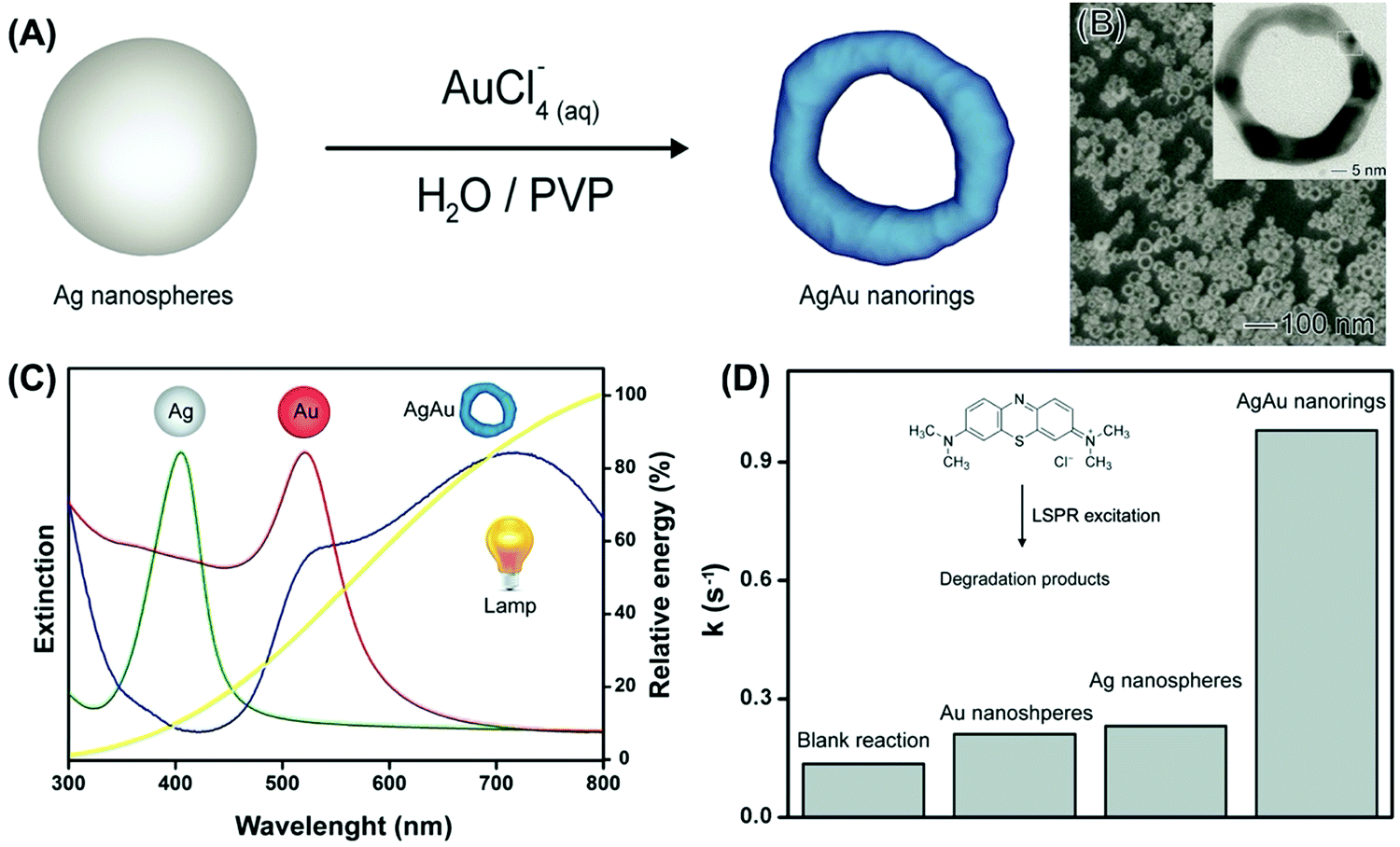 | ||
| Fig. 7 (A) Synthesis of AgAu nanorings by galvanic replacement reaction between Ag NPs and AuCl4−(aq).97 (B) SEM and TEM (inset) image for the AgAu nanorings. (C) UV-VIS extinction spectra were recorded from aqueous suspensions containing Ag NPs (green trace), Au NPs (red trace), and AgAu nanorings (blue trace).97 The yellow trace shows the emission spectrum for the commercial tungsten–halogen lamp employed as the excitation source in the LSPR-mediated catalytic experiments.97 (D) Pseudo-first order rate constants (k) calculated from C/C0 and ln(C/C0) profiles as a function of time for Ag NPs, Au NPs, and AgAu nanorings (a blank reaction is also shown).97 Reproduced from ref. 97 with permission from the Royal Society of Chemistry, copyright 2016. | ||
As discussed so far in this feature article and as reported in the literature, most studies on LSPR-mediated transformations and plasmonic catalysis have focused on the utilization of individual Ag and Au NPs as catalysts, including shape-controlled systems such as nanocubes, nanowires, nanoshells, nanowires, and triangular nanoprisms.60,72 These NPs can be regarded as first-generation plasmonic catalysts.64 Still, we were interested in pushing plasmonic catalysis further based on morphology, composition, and structure control to generate plasmonic catalysts with improved catalytic activities relative to these individual nanoparticles.64 In this context, we proposed the utilization of metallic nanorattles, comprised of a nanosphere inside of a nanoshell, as a next-generation of plasmonic catalysts by taking advantage of the plasmon hybridization concept.64 Our simulations indeed demonstrated that the E-Field enhancements for nanorattles are significantly higher as compared to nanoshells and NPs counterparts, showing that this material has the potential for improved plasmonic catalytic activities (Fig. 8A–D). The higher E-field enhancements can contribute to higher absorption efficiencies, therefore increasing the generation of LSPR-excited charge carriers.64,99 To experimentally investigate how the increased E-field enhancements observed for the nanorattles can be employed to achieve higher plasmonic catalytic performances relative to a nanoshell and a nanosphere, we synthesized nanorattles comprised of an Au sphere inside an AgAu nanoshell. They were prepared by a two-step approach (Fig. 8E).64 First, Au NPs were employed as physical templates for the deposition of Ag at their surface. This led to the formation of an Au@Ag core–shell system. Then, these Au@Ag core–shell NPs were employed as chemical templates in a galvanic replacement reaction with AuCl4−(aq). This leads to the formation of an AgAu shell at the surface of the nanoparticle, which becomes separated from the Au NPs by a void generating Au@AgAu nanorattles (Fig. 8F).64
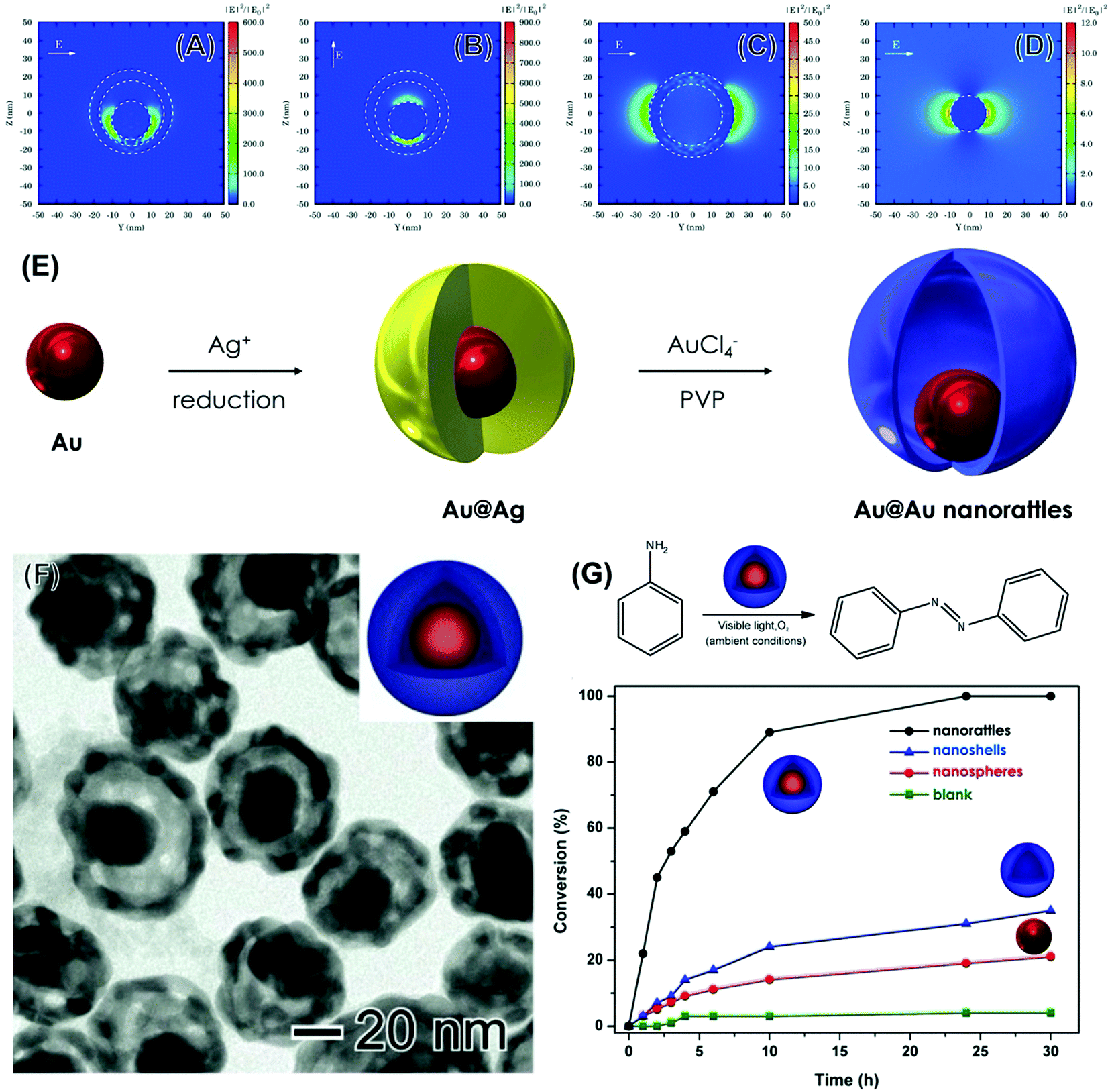 | ||
| Fig. 8 (A–D) Electric field enhancement contours calculated by the DDA method for AgAu nanorattles 40 nm in outer diameter, 5 nm in shell thickness, and having an Au nanosphere 20 nm inside its core (A and B), AgAu nanoshells 40 nm in diameter and 5 nm in shell thickness (C), and Au nanospheres 20 nm in diameter (D).64 In all cases, the wavelength of the incoming electromagnetic wave corresponded to 633 nm. The incident light was along the x-axis and the E-field was along the y- and z-axis [010] and [011] direction in (A) and (B), respectively, and along the y-axis in (C) and (D). (E) Approach for the synthesis of Au@AgAu nanorattles. Firstly, Au nanoparticles were employed as physical templates for Ag growth in presence of citrate, leading to the formation of Au@Ag core–shell NPs.64 Then, the Au@AgAu nanorattles were obtained by a galvanic replacement reaction between Au@Au core–shell and AuCl4−(aq). (F) TEM image for obtained Au@AgAu nanorattles. (G) LSPR-mediated oxidation of aniline catalyzed by the nanorattles and conversion (%) of aniline as a function of time catalyzed by Au@AgAu nanorattles, AgAu nanoshells, and Au NPs (black, blue, and red traces, respectively). A blank reaction is also shown (green trace).64 Reproduced from ref. 64 with permission from Wiley-VCH, copyright 2016. | ||
The Au@AgAu nanorattles were employed as plasmonic catalysts for the liquid phase oxidation of aniline under ambient conditions and using visible light as the only energy input (Fig. 8E).64Fig. 8E shows the conversion (%) for the oxidation of aniline as a function of time by employing the Au@AgAu nanorattles (black trace), AgAu nanoshells (blue trace), and Au NPs. The utilization of the nanorattles as plasmonic catalysts led to much better conversion percentages relative to their counterparts. For example, >90% of conversion after 10 h was obtained when the nanorattles were employed as catalysts. Here, all the catalytic experiments were carried out under the same loading of nanoparticles.64 The conversion (%) for the nanorattles was ∼3-fold higher relative to the sum of nanoshells and nanoparticles. This result indicates that the plasmon hybridization in Au@AgAu nanorattles is put to work to improve the performances in plasmonic catalysis and that nanorattles may represent a next generation of plasmonic catalysts towards a variety of transformations.64 As the nanorattles allow the formation of electromagnetic hot spots without relying on the uncontrolled aggregation of nanostructures they become attractive in plasmonic catalysts towards both liquid and gas phase transformations under mild conditions.
5. Design-controlled synthesis of plasmonic catalysts
Plasmonic catalysis offers unique opportunities to bring solar-driven chemistry to the synthesis of fuels and value-added chemicals under mild conditions closer to reality.29 However, the widespread use of plasmonic catalysis remains challenging due to several key factors. For example, most of the research has often been focused on Ag and Au NPs. These systems, while displaying outstanding optical properties, are not very catalytic towards a variety of transformations.100 Moreover, the important NPs physical and chemical parameters required for the optimization of optical properties (sizes that maximize absorption relative to scattering) are not necessarily the same required for the maximization of catalytic properties.100 Therefore, NPs designs that enable the maximization of both optical and catalytic properties are needed. In addition, several important catalytic metals, such as Pd and Pt, do not support plasmonic excitation in the visible or near-infrared ranges.101 This limits the scope of chemical reactions that can be addressed by plasmonic catalysis. One way to overcome this limitation relies on the development of multicomponent plasmonic-catalytic NPs.102,103 Plasmonic-catalytic NPs are currently at the forefront of plasmonic catalysis.29 In these systems, the plasmonic component harvests energy from light in the form of LSPR-generated hot carriers, that can flow towards the non-plasmonic but catalytic sites, enabling the acceleration and control of chemical reactions beyond those for which plasmonic NPs are catalytically active.18 In this context, bimetallic plasmonic-catalytic NPs in the form of core–shell, core-island, and alloys have been proposed and represent the state-of-the-art in the field.104 However, these systems are still limited in terms of catalytic metal use (which is pivotal for platinum group metals). Thus, nanoparticle morphologies that allow for the maximum catalytic metal use efficiency are needed. In terms of optical properties, the formation of alloys or the deposition of catalytic metal layers at the surface of a plasmonic material can lead to a decrease in the absorption efficiencies and E-fields because of LSPR excitation (plasmon is damped).64 This leads to detrimental effects on optical properties and plasmonic catalytic performances.64 Here, it is essential to develop plasmonic-catalytic materials in which the optical properties can be maximized and are not damped. Based on the knowledge acquired using controlled NPs systems in the previous sections, we turned our attention to the design-controlled synthesis of plasmonic-catalytic NPs that can respond to these challenges.In the first example, we focused on multimetallic plasmonic-catalytic nanorattles containing Pt as the catalytic component.100 They were comprised of an Au plasmonic core and an AgPt plasmonic-catalytic shell, in which the core and shell are separated by a void (Au@AgPt nanorattles).100 We focused on the nanorattle morphology inspired by our findings that plasmonic nanorattles enabled the controlled generation of electromagnetic hot spots,64 which in turn can lead to much higher plasmonic catalytic activities than their individual NPs counterparts. To rigorously evaluate and benchmark the plasmonic catalytic Au@AgPt nanorattles, we employed the equivalent plasmonic-catalytic core–shell Au@Ag@Pt as a reference NPs morphology. Both these systems present a plasmonic core and Pt as the catalytic component at the shells. These NPs could be prepared by the seeded growth of Ag over Au NPs seeds (leading to Au@Ag core–shell NPs) followed by a galvanic replacement approach between Ag in the shells and PtCl62−(aq) as illustrated in Fig. 9A.100 We found that by tuning the PtCl62−(aq) concentration employed in the galvanic replacement reaction, the NPs morphology could be precisely maneuvered to generate either Au@Ag@Pt core–shell NPs or Au@AgPt nanorattles (Fig. 9A). More specifically, the use of 0.1 M PtCl62−(aq) led to the deposition of a thin Pt shell (<3 nm) at the surface of Au@Ag NPs, leading to the Au@Ag@Pt core–shell morphology as confirmed by the STEM-HAADF (Fig. 9B), STEM-EDX (Fig. 9C) and the STEM-EDX line scan (Fig. 9D) along with points I and II in Fig. 9C.100 In this case, the extend of the galvanic replacement reaction was low, and no significant hollowing of the Ag layer took place during the deposition of Pt at the surface, thus leading to a core–shell morphology. On the other hand, the use of 0.2 M PtCl62−(aq) in the synthesis led to the formation of Au@AgPt nanorattles as shown by the STEM-HAADF (Fig. 9E), STEM-EDX (Fig. 9F), and the STEM-EDX line scan (Fig. 9G) along with points III and IV in Fig. 9F.100 Here, with an increase in the PtCl62−(aq) contend added to the reaction, galvanic replacement took place to a larger extent leading to significant hollowing of the Ag layer and therefore the nanorattle morphology. Importantly, both NPs displayed ultrathin Pt-based shells with similar sizes, making them ideal for comparing their Pt catalytic properties.
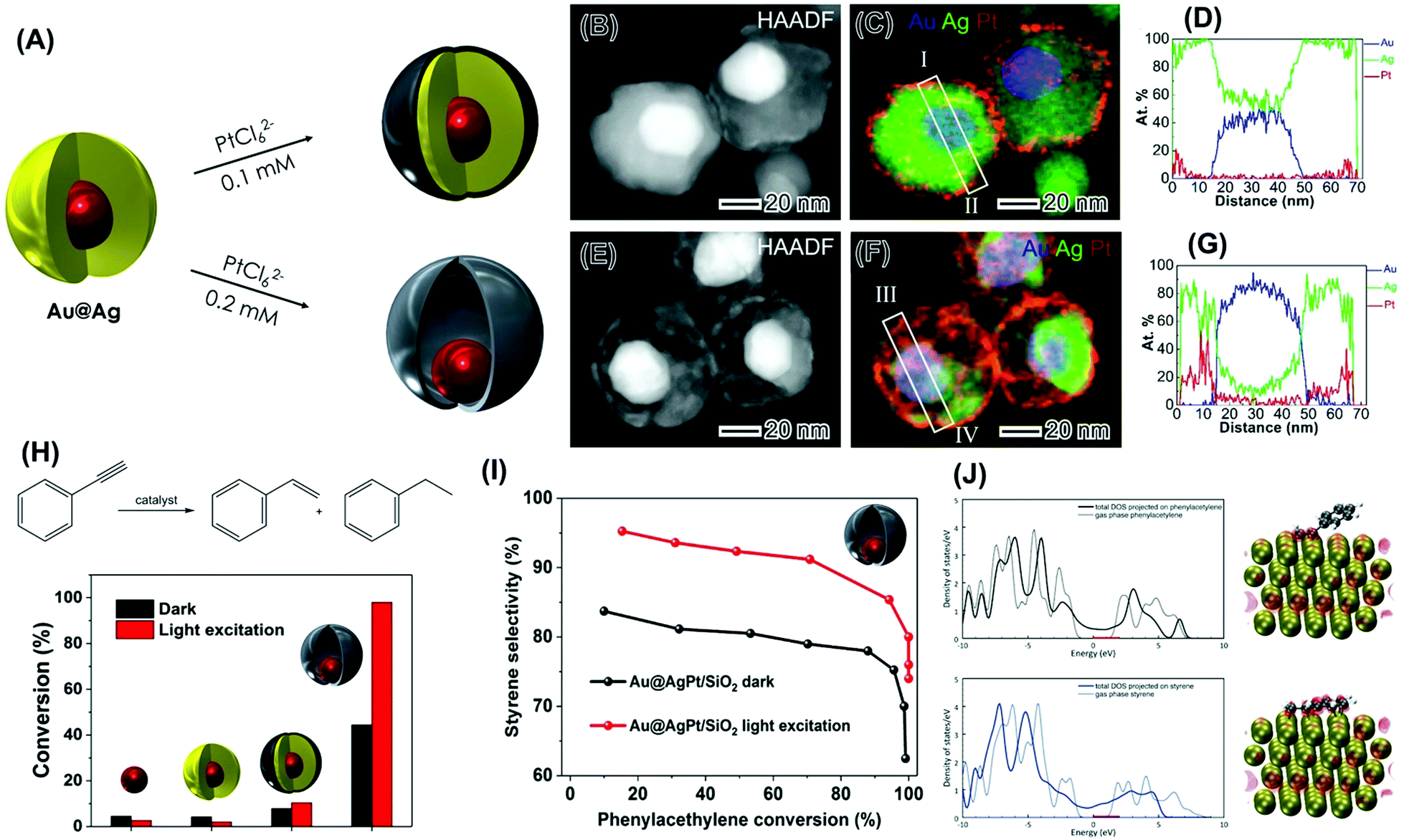 | ||
| Fig. 9 (A) Synthesis of plasmonic-catalytic Au@Ag@Pt core–shell (top) and Au@AgPt nanorattles (bottom) via the galvanic replacement reaction between PtCl62−(aq) and Ag in Au@Ag core–shell NPs.100 (B–G) STEM analysis for Au@Ag@Pt core–shell (B–D) and Au@AgPt nanorattles (E–G).100 (B and E) HAADF image in which the brightest contrast from Au core results from its higher Z (atomic number) as ZAu > ZPt > ZAg. (C and F) Corresponding composite elemental map EDS elemental maps of Au (blue), Ag (green), and Pt (red). (D and G) Atomic-percent distribution from quantified EDS line scan along the line I–II (D) marked in (C) and along line III–IV (G) marked in (F). (H) Scheme for the phenylacetylene hydrogenation reaction leading to the formation of styrene and ethylbenzene and conversion (%) in the dark and under visible light excitation (black and red bars, respectively) employing supported Au, Au@Ag, Au@Ag@Pt core–shell NPs, and Au@AgPt nanorattles as catalysts. (I) Styrene selectivity as a function of conversion percentages for Au@AgPt nanorattles under dark (black trace) and visible light excitation conditions (red trace).100 (J) DFT-calculated projected density of states of phenylacetylene (top panel) and styrene (bottom panel) in the gas phase (dotted trace) and adsorbed on Pt (solid trace). The optimized structure and integrated state from the Fermi Level (0 eV) to 2 eV are also shown.100 Reprinted (adapted) with permission from ref. 100. Copyright 2018 American Chemical Society. | ||
To study how the optical properties of the plasmonic component influence the activities of the catalytic metal in these NPs via plasmonic catalysis, we focused on a model reaction that is catalyzed only by Pt under our employed conditions: the hydrogenation of phenylacetylene as described in Fig. 9H.100 In these studies, all the syntheses of the NPs were scaled up and the materials were supported into SiO2 to generate supported plasmonic catalysts. Au NPs and Au@Ag NPs were also investigated as control systems.
Fig. 9H shows the conversion (%) for the phenylacetylene hydrogenation both under dark (black bars) and light excitation conditions from a white lamp (red trace). While no significant conversions were detected for Au and Au@Ag NPs, the control over the morphology (nanorattles vs. core–shell NPs) enabled the optimization of catalytic activities both in the dark and under light excitation conditions. In the dark, the higher activity of the nanorattles may be assigned to their hollow interiors, which enabled higher surface-to-volume ratios relative to the solid core–shell systems.100 Under LSPR excitation, while only a slight increase in the phenylacetylene conversion was observed for the core−shell Au@Ag@Pt NPs, a remarkable increase was observed for the Au@AgPt nanorattles (from 44 to 98%). This highlights the importance of the morphology control in which the plasmonic-catalytic nanorattles can enable much higher enhancement in catalytic activity under LSPR excitation relative to core–shell systems. It is important to note that the Pt loading in the employed plasmonic catalysis comprising Au@AgPt nanorattles supported on SiO2 corresponded to 0.1 wt%. This indicates that plasmonic catalysis (light irradiation) and morphology/composition optimization represent an attractive approach to make these transformations proceed under lower loadings of Pt, which is often reported under higher loadings (1–10 wt%) for this transformation.
In addition to the optimization of conversion (%), it was found that the LSPR excitation also enabled the control over the reaction selectivity towards the formation of styrene in the plasmonic-catalytic nanorattles (Fig. 9I).100 In this case, for all conversion % values, there was an increase in the styrene selectivity under light excitation relative to dark conditions. Thus, nanorattles were not only more active toward the phenylacetylene conversion under dark conditions but also displayed a higher selectivity for the semi hydrogenation reaction relative to the core–shell architecture. These results indicate that plasmonic catalysis can lead not only to improvements in reaction rates but also in reaction selectivity. DFT calculations Fig. 9J showed that phenylacetylene prefers to adsorb in a tilted configuration to maximize the d–π interaction between the triple bond and the metal d electrons (top panel).100 Moreover, the charge analysis suggested that DOS within the range from the Fermi level (0 eV) up to 2 eV are localized at the metal and the C–C triple bond. For styrene (Fig. 9J, bottom panel), adsorption on Pt takes place flat configuration to maintain the conjugation and to maximize the d–π interaction, in which the DOS energy states between 0 and 2 eV delocalized in the molecule (both the double bond and the ring due to the resonance) and the metal. Therefore, the mechanism for the increased selectivity was proposed based on the selective activation of the triple bond under LSPR excitation due to the localized interaction between the triple bond and the surface.100
In the last example, we illustrate how the knowledge and design principles established so far can then be used to, based on a reaction of interest, design nanomaterials with desired plasmonic and catalytic properties. To this end, we wanted to harvest plasmonic effects to the improvement of the oxygen evolution reaction (OER). It has been established that IrO2 is among the best electrocatalysts for the OER,105 but this material does not support LSPR excitation in the visible or near-infrared ranges.101 Therefore, plasmonic-catalytic combinations are needed. Regarding the morphology, we were interested in the synthesis of NPs that enable the formation of electromagnetic hot spots in a controlled manner so that the optical properties of the plasmonic metal could be employed to enhance the electrocatalytic performances of the catalytic component (IrO2).106,107 To this end, we developed nanoflowers based on Au as the plasmonic component and IrO2.108 More specifically, we targeted Au–IrO2 plasmonic-catalytic nanoparticles displaying a tortuous dendritic morphology, in which several branches around 5 nm in diameter made up of Au are closely spaced to each other as shown in Fig. 10A.108 This enables the plasmon hybridization between the branches, which can lead to high plasmonic catalytic activities because of high electric fields (electromagnetic hot spots, Fig. 10B) and the high contribution of absorption relative to scattering to the optical efficiency in this system (Fig. 10C).108 In principle, by covering these branches with an ultrathin (1 nm) and incomplete IrO2 shell (Fig. 10D), the catalytic component can efficiently be exposed to the energy that is harvested from the light because of LSPR excitation, paving the way to improved activities. Here, the ultrathin and incomplete IrO2 shell at the surface of each branch maximizes the light-harvesting by Au and the subsequent flow of charge carriers from Au to the Ir-based shell, while also maximizing the IrO2 surface area and utilization of Ir.108
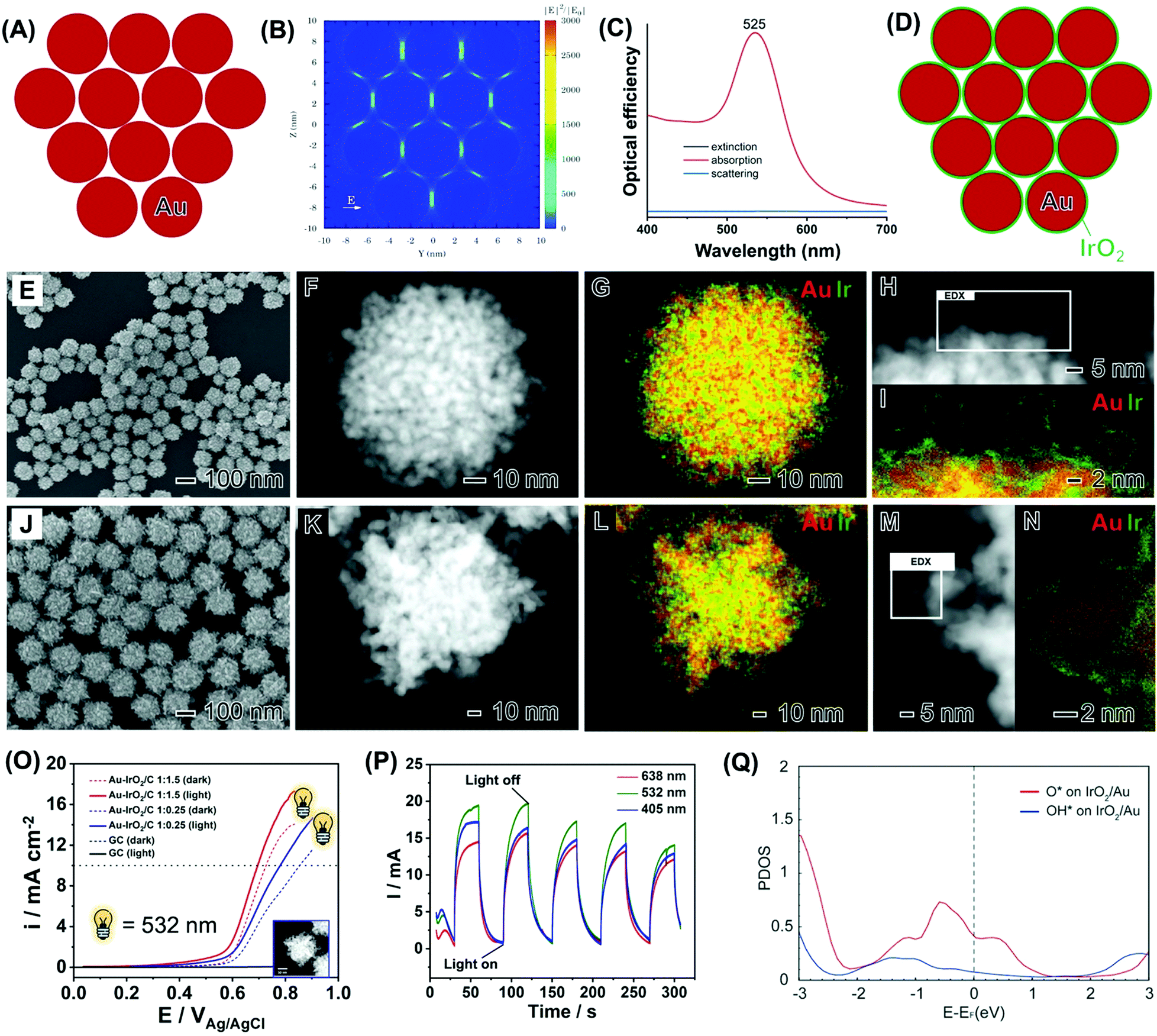 | ||
| Fig. 10 (A) Ensemble of 12 Au nanospheres 5 nm in diameter separated by a 1 nm gap in a close-packed arrangement employed to simulate the arrangement of Au branches in nanoflowers and the corresponding calculated electric field enhancement contours |E|2/|E0|2.108 (B) and extinction (black trace), absorption (red trace) and scattering (blue trace) optical efficiencies (C).108 (D) Scheme illustrating the deposition of a thin IrO2 shell over the surface of each Au branch.108 (E–I) SEM (E), HAADF-STEM (F and H), and STEM-EDX (G and I) images of Au–IrO2 nanoflowers obtained by co-reduction of AuCl4− and IrCl3·xH2O precursors in a 1:1.5 molar ratio. The Au and Ir at% in the samples corresponded to 85 and 15, respectively.108 (J–N) SEM (J), HAADF-STEM (K and M), and STEM-EDX (L and N) images for Au–IrO2Au–IrO2 nanoflowers obtained by co-reduction of AuCl4− and IrCl3·xH2O precursors in a 1:0.25 molar ratio. The Au and Ir at% in the samples corresponded to 96 and 4, respectively. The elemental distributions for Au and Ir are shown in red and green, respectively, in the STEM-EDX maps. Where both green and red signals are overlapping the color, the map appears yellow.108 (O) OER linear scanning voltammetries recorded at a scan rate = 10 mV s−1 in a 0.1 M KOH electrolyte with (solid line) and without (dashed line) LSPR excitation at 532 nm for the Au–IrO2 nanoflowers obtained by co-reduction employing AuCl4− and IrCl3·xH20 as precursors in a 1:1.5 (red trace) 1:0.25 (blue trace) molar ratios.108 (P) Light wavelength I–t curves recorded at 0.65 V (vs. Ag/AgCl) with light irradiation on/off for Au–IrO2 nanoflowers obtained by co-reduction employing AuCl4− and IrCl3·xH20 as precursors in a 1:1.5 molar ratios under 405 (blue trace), 532 (green trace), and 638 nm (red trace) excitation.108 (Q) Projected density of states of O and OH adsorbed on Au–IrO2.108 Reproduced from ref. 108 with permission from the Royal Society of Chemistry, copyright 2020. | ||
We then performed the synthesis of Au–IrO2 nanoflowers depicting these compositional and morphological features by a co-reduction approach as shown in Fig. 10E–I. The molar ratio between AuCl4−(aq) and IrCl3·xH2O precursors employed during the synthesis corresponded to AuCl4−:IrCl3·xH2O 1:1.5. The SEM image (Fig. 10E) shows that the nanoflowers displayed an overall spherical morphology, monodisperse sizes (93.2 ± 9 in diameter), and a highly tortuous branched surface structure.108 The branched morphology composed of individual closely spaced Au branches around 5 nm in size is more clearly visualized in the HAADF-STEM images shown in Fig. 1F and H. The STEM-EDX elemental maps (Fig. 10G and I) show that the nanoflowers are composed of a core–shell morphology with an Au core covered by an ultrathin (1 nm or thinner) Ir rich surface layer.108 The high-resolution elemental map (Fig. 10I) demonstrates that the Ir-based ultrathin layer does not completely cover the surfaces of the Au branches. A similar nanoflower, core–shell morphology, although with different overall size and more elongated branches, was also observed when the molar ratio between the precursors in the synthesis changed to AuCl4−:IrCl3·xH2O 1:0.25 (Fig. 10J–N).
Linear sweep voltammetry (LSV) tests for the nanoflowers in the presence of light excitation (solid lines) and the absence of light excitation are shown in Fig. 10O. It can be observed that the nanoflowers with a higher IrO2 content (red traces) displayed higher activities. The activities could be significantly increased under light excitation at 532 nm. In this case, a significant decrease in onset potential and increase in the detected current densities were detected under visible light illumination, assigned to the plasmonic enhancement of the OER. Fig. 10P shows the I–t curve at 0.65 V (vs. Ag/AgCl) for the Au–IrO2 nanoflowers shown in Fig. 10E–I under chopped light illumination at 405 (blue trace), 523 (green trace), and 638 nm (red trace).108 The samples displayed fast and reproducible current responses to on–off illumination cycles. The current densities were wavelength-dependent, being greatest for 532 nm excitation, which is the wavelength that better matches the LSPR spectrum of the nanoflowers. In this system, an overpotential of just 286 mV was detected for the plasmonically assisted OER. It is noteworthy that reported best values of overpotential for IrO2 and RuO2 electrocatalysts are typically in the 300–400 mV range. Therefore, an overpotential of just 286 mV for the plasmonically assisted OER activity represents beyond those reported for IrO2 electrocatalysts and also matches the most active reported electrocatalysts. This indicates that control over the morphology and composition of the nanoflowers together with the LSPR effect via visible light excitation was an effective strategy to reduce the overpotential in this reaction, making the process more efficient and opening possibilities for milder conditions and reducing IrO2 loadings.
Finally, DFT calculations have shown that the projected density of states (DOS) for O* and OH* adsorbed on IrO2 supported on Au NPs indicate that the states between −2.33 eV and the Fermi level are more pronounced than the states above the Fermi level.108 This high population of states below the Fermi level may indicate a more pronounced mechanism based on the effect of the hot holes generated under LSPR excitation towards the enhanced OER activities. It is plausible that, in this case, hot holes can be generated at the Au NPs and flow to the IrO2, where they enhance the OER process.108
6. Conclusions and outlook
There has been a huge interest in the harvesting of plasmonic effects to enhance photocatalytic performances. This field, plasmonic catalysis, has emerged and consolidated in the last decades as an important branch of photocatalysis, offering many advantages relative to “classical” photocatalysis with semiconductors for enabling solar-driven chemistry. The use of plasmonic catalysis towards a variety of molecular transformations (reductions, oxidations, and couplings) has been demonstrated in the past years. This also includes reactions that are relevant in the context of green energy generation via nanocatalysis or electrocatalysis such as water splitting, CO2 reduction,36 and methane dry reforming.37 Importantly, the last decade also witnessed significant progress and systematic studies on the understanding of the mechanisms by which plasmonic catalysis takes place, although the unraveling of the precise and relative contributions of hot charge carriers and localized heating remain challenging. In addition to mechanistic understanding, the establishment of structure–performance relationships in plasmonic catalysis is essential to move the field forward. This will contribute not only to the optimization of activities but also enable a transition to a design-driven approach in response to a target activity and selectivity for a transformation of interest.This feature article discussed the recent efforts from our group towards the understanding of structure–performance relationships in LSPR driven or accelerated molecular transformations. We employed a variety of well-defined plasmonic and plasmonic-catalytic nanomaterials as model systems to investigate how LSPR-driven transformations or plasmonic catalysis is influenced by several physical and chemical parameters that define these nanomaterials. We started by discussing the effect of size, shape, and composition in exclusively plasmonic NPs over a proof-of-concept LSPR mediated transformation. In analogy to what has been established in nanocatalysis, LSPR-mediated activities were strongly dependent on these parameters. It was unraveled that the maximization of absorption relative to scattering was important when investigating the size-dependent activities, which resulted in intermediate sizes having the maximum activity. While the shape effect could be related to the strength of interaction with substrate/reactants (as in nanocatalysis), the overlap between LSPR extinction and excitation wavelength, electronic effects, as well as the occurrence of interband vs. intraband transitions in bimetallic AgAu NPs dictated the performances. We also investigated the effect of metal–support interactions in supported plasmonic NPs. This is important as practical applications in catalysis require supported NPs and the fact that metal–support interactions have been employed to maximize performances or to lead to bifunctional mechanisms in nanocatalysis. By employing Au NPs supported on CeO2, it was demonstrated that metal–support interactions can lead to significant improvements in performances. This was assigned to the activation, by the plasmonic excitation, of species at the metal–support interface (surface oxygen species) that can contribute to enhancing catalytic activity. Interestingly, it was also found that metal–support interactions because of charge transfer can also influence performances, and this effect was dependent on the reaction pathway. These findings open additional pathways to optimize the performance that goes beyond the plasmonic NP by itself.
The last examples on model plasmonic NPs were focused on the effect of structure, encompassing hollow plasmonic NPs and plasmonic nanorattles. Plasmonic nanorattles represent a specially interesting class of nanomaterials and this morphology enables the plasmonic hybridization between the plasmonic core and plasmonic shell components. This hybridization leads to much higher electric field enhancements relative to their individual NPs counterparts as a result of the formation of electromagnetic hot spots, while not relying on the random aggregation of individual NPs. We have found that this can be translated to much higher plasmonic catalytic activities than individual NPs, therefore plasmonic nanorattles hold enormous potential as the next generation of plasmonic catalysts. Overall, the data obtained from these model plasmonic catalysts suggest that the desirable features for plasmonic catalysts are: (i) NPs that maximize absorption relative to scattering; (ii) compositions and shapes (electronic and geometric effects) that provide the optimum interaction between the surface and adsorbates as well as the attributes of the optical properties. In this case, NPs having sharp corners (cubes and prisms) may enable higher electric field enhancements relative to round counterparts, for example. On the other hand, the formation of bimetallic NPs between a plasmonic and catalytic metal may lead to the damping of absorption of the plasmonic metal, leading to a decrease in plasmonic catalytic activity; and (iii) NPs that enable maximization of electric field enhancements by the formation of electromagnetic hot spots.
Based on these principles for high plasmonic catalytic activity, we discussed the designed controlled synthesis of NPs having both plasmonic and catalytic components: multimetallic nanorattles containing a plasmonic core and a plasmonic-catalytic shell (Pt as the catalytic component) and nanoflowers based on several Au branches that are closely spaced among each other (leading to electromagnetic hot spots) that are covered by an incomplete, sub 1 nm layer of an IrO2 shell. The plasmonic-catalytic multimetallic nanorattles enabled not only high catalytic activities towards the hydrogenation of phenylacetylene, but also control over reaction selectivity towards the formation of styrene under light excitation. In the case of the Au–IrO2 nanoflowers, this approach enabled optimization over the IrO2 performances towards the oxygen evolution reaction, demonstrating that the design-controlled synthesis of plasmonic catalytic NPs from the principles established by the model systems represented an efficient strategy to optimize activities and control reaction selectivity.
While this feature article was focused on a few model transformations, NPs with well-defined physical–chemical features, and the conclusions/trends in reactivity may depend on a variety of different factors, the observed trends discussed herein in plasmonic catalysis agree with other reports and different classes of transformations. The literature has established, as described herein, that better performances are achieved for NPs sizes and morphologies that enable higher absorption relative to scattering,109,110 shapes that provide more reactive surface facets (optimization of absorption and desorption energies) for a transformation of interest,34 compositions that enable both maximum absorptions by the plasmonic component and best electronic effect enabled by the surface composition,110 and NPs morphologies that enable the generation of electromagnetic hot spots.99,111 Moreover, studies performed under SERS experimental conditions using model transformations have provided insights into plasmonic catalysis, and we have discussed one example in which is a good match for studies performed in the liquid phase.64,112
Although a lot of progress has been achieved in plasmonic catalysis, several challenges remain, offering great opportunities for future work in the field.33 Some examples include studies and the unraveling of the durability/stability of plasmonic catalysts as well as the understanding of shape and composition changes under reaction conditions. Another important aspect in this context in terms of activity and stability is the in situ and operando characterization of plasmonic catalysis and metal–adsorbates interactions. This is crucial in terms of understanding mechanisms and stability, which can have important implications to improve our understanding of plasmonic catalysis. Finally, we believe that there is a lot of untapped potential for plasmonic catalysis for the control over reaction selectivity.113 The use of light to control reaction pathways and control the outcome of molecular transformations is intriguing and can represent a change in paradigm in synthetic chemistry. Nevertheless, the observation and control of reaction selectivity in plasmonic catalysis remain limited. Finally, future work could also include supporting various plasmonic-catalytic NPs onto a variety of oxides to improve the harvesting of light from the solar spectrum as well as to facilitate recovery and re-use.
Whereas various plasmonic and plasmonic-catalytic NPs have been demonstrated in the literature to be effective in accelerating various molecular transformations under visible light excitation, the results described in this feature article for shape, composition, and size-controlled NPs evidence the importance of the control over these parameters to optimize and understand plasmonic catalytic activities and selectivity control. We believe that the examples and concepts presented in this feature article may inspire future work and progress in the field of plasmonic catalysis encompassing the design-controlled synthesis of plasmonic and plasmonic-catalytic multicomponent materials, new strategies to the control over reaction selectivity with visible or near-IR light, and the unraveling of stability and reaction mechanisms in plasmonic catalysis. This would enable a broader impact of solar-driven chemistry in the molecular sciences, which is the stepping stone towards a more sustainable and circular future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Jane and Aatos Erkko Foundation, start-up funds from the University of Helsinki, Academy of Finland, seed funding from the Helsinki Institute of Sustainability Science (HELSUS). J. W. Was supported by the Shanghai Natural Science Foundation (20ZR1401700), and the National Natural Science Foundation of China (21703031).References
- P. Losch, W. Huang, E. D. Goodman, C. J. Wrasman, A. Holm, A. R. Riscoe, J. A. Schwalbe and M. Cargnello, Nano Today, 2019, 24, 15–47 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. Corma, Angew. Chem., Int. Ed., 2016, 55, 6112–6113 CrossRef CAS PubMed.
- Y. Shi, Z. Lyu, M. Zhao, R. Chen, Q. N. Nguyen and Y. Xia, Chem. Rev., 2021, 121, 649–735 CrossRef CAS PubMed.
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- A. S. Sharma, H. Kaur and D. Shah, RSC Adv., 2016, 6, 28688–28727 RSC.
- J. C. Védrine, ChemSusChem, 2019, 12, 577–588 CrossRef PubMed.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- S. Peiris, J. McMurtrie, H.-Y. Zhu, Sarina, X. Wang, H. Su, X. Fu, M.-J. Cho, S. D. Henam, R. S. Varma and P. S. Branco, Catal. Sci. Technol., 2016, 6, 320–338 RSC.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CrossRef CAS PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- N. N. Vu, S. Kaliaguine and T. O. Do, ChemSusChem, 2020, 13, 3967–3991 CrossRef CAS PubMed.
- J. G. Smith, J. A. Faucheaux and P. K. Jain, Nano Today, 2015, 10, 67–80 CrossRef CAS.
- B. J. Wiley, S. H. Im, Z.-Y. Y. Li, J. McLellan, A. Siekkinen and Y. N. Xia, J. Phys. Chem. B, 2006, 110, 15666–15675 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- M. Kim, M. Lin, J. Son, H. Xu and J. M. Nam, Adv. Opt. Mater., 2017, 1700004 CrossRef.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- M. Sun and H. Xu, Small, 2012, 8, 2777–2786 CrossRef CAS PubMed.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- Y. Kim, D. Dumett Torres and P. K. Jain, Nano Lett., 2016, 16, 3399–3407 CrossRef CAS PubMed.
- C. Zhan, X.-J. Chen, J. Yi, J.-F. Li, D.-Y. Wu and Z.-Q. Tian, Nat. Rev. Chem., 2018, 2, 216–230 CrossRef.
- A. Gellé, T. Jin, L. De La Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2020, 986–1041 CrossRef PubMed.
- H. Robatjazi, J. L. Bao, M. Zhang, L. Zhou, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Catal., 2020, 3, 564–573 CrossRef CAS.
- L. Zhou, M. Lou, J. L. Bao, C. Zhang, J. G. Liu, J. M. P. Martirez, S. Tian, L. Yuan, D. F. Swearer, H. Robatjazi, E. A. Carter, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022109118 CrossRef CAS PubMed.
- D. Devasia, A. J. Wilson, J. Heo, V. Mohan and P. K. Jain, Nat. Commun., 2021, 12, 2612 CrossRef CAS PubMed.
- E. Cortés, L. V. Besteiro, A. Alabastri, A. Baldi, G. Tagliabue, A. Demetriadou and P. Narang, ACS Nano, 2020, 14, 16202–16219 CrossRef PubMed.
- M. J. Landry, A. Gellé, B. Y. Meng, C. J. Barrett and A. Moores, ACS Catal., 2017, 7, 6128–6133 CrossRef CAS.
- N. Jiang, X. Zhuo and J. Wang, Chem. Rev., 2018, 118, 3054–3099 CrossRef CAS PubMed.
- S. Yu and P. K. Jain, Nat. Commun. DOI:10.1038/s41467-019-10084-5.
- L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- E. Kowalska, O. O. P. Mahaney, R. Abe and B. Ohtani, Phys. Chem. Chem. Phys., 2010, 12, 2344 RSC.
- E. Kowalska, M. Janczarek, L. Rosa, S. Juodkazis and B. Ohtani, Catal. Today, 2014, 230, 131–137 CrossRef CAS.
- E. Kowalska, R. Abe and B. Ohtani, Chem. Commun., 2009, 241–243 RSC.
- A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2012, 134, 14526–14533 CrossRef CAS PubMed.
- G. Baffou and R. Quidant, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- P. K. Jain, J. Phys. Chem. C, 2019, 123, 24347–24351 CrossRef CAS.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS PubMed.
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 14542 CrossRef CAS PubMed.
- L. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Zhao, L. Henderson, L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- L. Zhou, D. F. Swearer, H. Robatjazi, A. Alabastri, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2019, 364, DOI:10.1126/science.aaw9545.
- Y. Sivan, J. Baraban, I. W. Un and Y. Dubi, Science, 2019, 364, DOI:10.1126/science.aaw9367.
- Y. Sivan, I. W. Un and Y. Dubi, 2019, arXiv:1902.03169.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 10545 CrossRef CAS PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2012, 13, 240–247 CrossRef PubMed.
- P. K. Jain, J. Phys. Chem. C, 2019, 123, 24347–24351 CrossRef CAS.
- J. M. Nam, L. Liz-Marzán and N. Halas, Acc. Chem. Res., 2019, 52, 2995–2996 CrossRef CAS PubMed.
- A. Janz, A. Köckritz, L. Yao and A. Martin, Langmuir, 2010, 26, 6783–6789 CrossRef CAS PubMed.
- N. G. Bastús, J. Piella and V. Puntes, Langmuir, 2016, 32, 290–300 CrossRef PubMed.
- K. Li, N. J. Hogan, M. J. Kale, N. J. Halas, P. Nordlander and P. Christopher, Nano Lett., 2017, 17, 3710–3717 CrossRef CAS PubMed.
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed.
- R. S. Geonmonond, A. G. M. Da Silva, T. S. Rodrigues, I. C. De Freitas, R. A. Ando, T. V. Alves and P. H. C. Camargo, ChemCatChem, 2018, 10, 3447–3452 CrossRef CAS.
- Y.-F. Huang, D.-Y. Wu, H.-P. Zhu, L.-B. Zhao, G.-K. Liu, B. Ren and Z.-Q. Tian, Phys. Chem. Chem. Phys., 2012, 14, 8485–8497 RSC.
- D.-Y. Wu, L.-B. Zhao, X.-M. Liu, R. Huang, Y.-F. Huang, B. Ren and Z.-Q. Tian, Chem. Commun., 2011, 47, 2520–2522 RSC.
- A. Zaleska and D. Glass, Chem. Commun., 2019, 55, 7127–7133 RSC.
- A. G. M. da Silva, T. S. Rodrigues, V. G. Correia, T. V. Alves, R. S. Alves, R. A. Ando, F. R. Ornellas, J. Wang, L. H. Andrade and P. H. C. Camargo, Angew. Chem., Int. Ed., 2016, 55, 7111–7115 CrossRef CAS PubMed.
- Y.-F. F. Huang, M. Zhang, L.-B. Bin Zhao, J.-M. M. Feng, D.-Y. Y. Wu, B. Ren and Z.-Q. Q. Tian, Angew. Chem., Int. Ed., 2014, 53, 2353–2357 CrossRef CAS PubMed.
- P. Xu, L. Kang, N. H. Mack, K. S. Schanze, X. Han and H.-L. Wang, Sci. Rep., 2013, 3, 2997 CrossRef PubMed.
- L.-B. Bin Zhao, M. Zhang, Y.-F. F. Huang, C. T. Williams, D.-Y. Y. Wu, B. Ren and Z.-Q. Q. Tian, J. Phys. Chem. Lett., 2014, 5, 1259–1266 CrossRef PubMed.
- Y. Fang, Y. Li, H. Xu and M. Sun, Langmuir, 2010, 26, 7737–7746 CrossRef CAS PubMed.
- S. Li, P. Miao, Y. Zhang, J. Wu, B. Zhang, Y. Du, X. Han, J. Sun and P. Xu, Adv. Mater., 2021, 33, 2000086 CrossRef CAS PubMed.
- J. L. Wang, R. A. Ando and P. H. C. C. Camargo, ACS Catal., 2014, 4, 3815–3819 CrossRef CAS.
- M. K. Kumar, S. Krishnamoorthy, L. K. Tan, S. Y. Chiam, S. Tripathy and H. Gao, ACS Catal., 2011, 1, 300–308 CrossRef CAS.
- A. G. M. Da Silva, T. S. Rodrigues, J. Wang, L. K. Yamada, T. V. Alves, F. R. Ornellas, R. A. Ando and P. H. C. Camargo, Langmuir, 2015, 31, 10272–10278 CrossRef CAS PubMed.
- P.-Y. Silvert, R. Herrera-Urbina, N. Duvauchelle, V. Vijayakrishnan and K. T. Elhsissen, J. Mater. Chem., 1996, 6, 573–577 RSC.
- Y. Wang, Y. Zheng, C. Z. Huang and Y. Xia, J. Am. Chem. Soc., 2013, 135, 1941–1951 CrossRef CAS PubMed.
- J. E. Millstone, S. J. Hurst, G. S. Metraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS PubMed.
- C. C. S. de Oliveira, R. A. Ando and P. H. C. Camargo, Phys. Chem. Chem. Phys., 2013, 15, 1887–1893 RSC.
- Q. Wang, X. Cui, W. Guan, L. Zhang, X. Fan, Z. Shi and W. Zheng, J. Power Sources, 2014, 269, 152–157 CrossRef CAS.
- A. G. M. da Silva, T. S. Rodrigues, L. S. K. Taguchi, H. V. Fajardo, R. Balzer, L. F. D. Probst and P. H. C. Camargo, J. Mater. Sci., 2015, 51, 603–614 CrossRef.
- T. S. Rodrigues, A. G. M. da Silva, M. C. Gonçalves, H. V. Fajardo, R. Balzer, L. F. D. Probst and P. H. C. Camargo, ChemNanoMat, 2015, 1, 46–51 CrossRef CAS.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 5302–5304 RSC.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS PubMed.
- J. Wang, R. A. Ando and P. H. C. Camargo, Angew. Chem., Int. Ed., 2015, 54, 6909–6912 CrossRef CAS PubMed.
- T. C. Damato, C. C. S. de Oliveira, R. A. Ando and P. H. C. Camargo, Langmuir, 2013, 29, 1642–1649 CrossRef CAS PubMed.
- A. Al-Otaify, M. A. Leontiadou, F. V. E. Dos Reis, T. C. Damato, P. H. C. Camargo and D. J. Binks, Phys. Chem. Chem. Phys., 2014, 16, 14189–14194 RSC.
- A. Kumar, P. Choudhary, A. Kumar, P. H. C. Camargo and V. Krishnan, Small, 2021, 2101638 Search PubMed.
- E. C. M. Barbosa, J. L. Fiorio, T. Mou, B. Wang, L. M. Rossi and P. H. C. Camargo, Chem. – Eur. J., 2018, 24, 12330–12339 CrossRef CAS PubMed.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC.
- T. Fujitani, I. Nakamura, T. Akita, M. Okumura and M. Haruta, Angew. Chem., Int. Ed., 2009, 48, 9515–9518 CrossRef CAS PubMed.
- S. Pal, A. Dutta, M. Paul and A. Chattopadhyay, J. Phys. Chem. C DOI:10.1021/acs.jpcc.9b08523.
- K. Mudiyanselage, S. D. Senanayake, L. Feria, S. Kundu, A. E. Baber, J. Graciani, A. B. Vidal, S. Agnoli, J. Evans, R. Chang, S. Axnanda, Z. Liu, J. F. Sanz, P. Liu, J. A. Rodriguez and D. J. Stacchiola, Angew. Chem., Int. Ed., 2013, 52, 5101–5105 CrossRef CAS PubMed.
- A. G. M. da Silva, H. V. Fajardo, R. Balzer, L. F. D. Probst, N. T. Prado, P. H. C. Camargo and P. A. Robles-Dutenhefner, Chem. Eng. J., 2016, 286, 369–376 CrossRef CAS.
- R. S. Geonmonond, J. Quiroz, G. F. S. R. Rocha, F. E. Oropeza, C. J. Rangel, T. S. Rodrigues, J. P. Hofmann, E. J. M. Hensen, R. A. Ando and P. H. C. Camargo, Nanoscale, 2018, 10, 8560–8568 RSC.
- A. G. M. da Silva, T. S. Rodrigues, S. J. Haigh and P. H. C. Camargo, Chem. Commun., 2017, 53, 7135–7148 RSC.
- T. S. Rodrigues, A. G. M. Da Silva and P. H. C. Camargo, J. Mater. Chem. A, 2019, 7, 5857–5874 RSC.
- T. S. Rodrigues, A. H. M. da Silva, A. G. M. da Silva, D. G. Ceara, J. F. Gomes, J. M. Assaf and P. H. C. Camargo, Catal. Sci. Technol., 2016, 6, 2162–2170 RSC.
- T. S. Rodrigues, A. G. M. da Silva, A. B. L. de Moura, I. G. Freitas and P. H. C. Camargo, RSC Adv., 2016, 6, 62286–62290 RSC.
- T. S. Rodrigues, A. G. M. Silva, A. Macedo, B. W. Farini, R. da, S. Alves and P. H. C. Camargo, J. Mater. Sci., 2015, 50, 5620–5629 CrossRef CAS.
- S. Lee, H. Hwang, W. Lee, D. Schebarchov, Y. Wy, J. Grand, B. Auguié, D. H. Wi, E. Cortés and S. W. Han, ACS Energy Lett., 2020, 5, 3881–3890 CrossRef CAS.
- J. Quiroz, E. C. M. M. Barbosa, T. P. Araujo, J. L. Fiorio, Y.-C. C. Wang, Y.-C. C. Zou, T. Mou, T. V. Alves, D. C. De Oliveira, B. Wang, S. J. Haigh, L. M. Rossi and P. H. C. C. Camargo, Nano Lett., 2018, 18, 7289–7297 CrossRef CAS PubMed.
- J. H. Weaver, Phys. Rev. B: Solid State, 1975, 11, 1416–1425 CrossRef CAS.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2015, 137, 948–957 CrossRef CAS PubMed.
- Q. Xiao, S. Sarina, A. Bo, J. Jia, H. Liu, D. P. Arnold, Y. Huang, H. Wu and H. Zhu, ACS Catal., 2014, 4, 1725–1734 CrossRef CAS.
- T. P. Araujo, J. Quiroz, E. C. M. Barbosa and P. H. C. Camargo, Curr. Opin. Colloid Interface Sci., 2019, 39, 110–122 CrossRef CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- H. Huang, J.-H. Wang, W. Jin, P. Li, M. Chen, H.-H. Xie, X.-F. Yu, H. Wang, Z. Dai, X. Xiao and P. K. Chu, Small, 2014, 10, 4012–4019 CrossRef CAS PubMed.
- N. Zou, G. Chen, X. Mao, H. Shen, E. Choudhary, X. Zhou and P. Chen, ACS Nano, 2018, 12, 5570–5579 CrossRef CAS PubMed.
- I. C. De Freitas, L. S. Parreira, E. C. M. Barbosa, B. A. Novaes, T. Mou, T. V. Alves, J. Quiroz, Y. C. Wang, T. J. Slater, A. Thomas, B. Wang, S. J. Haigh and P. H. C. Camargo, Nanoscale, 2020, 12, 12281–12291 RSC.
- H. Tada, Dalton Trans., 2019, 48, 6308–6313 RSC.
- U. Aslam, S. Chavez and S. Linic, Nat. Nanotechnol., 2017, 12, 1000–1005 CrossRef CAS PubMed.
- D. Liu and C. Xue, Adv. Mater., 2021, 33, 2005738 CrossRef CAS PubMed.
- L. Papa, I. C. De Freitas, R. S. Geonmonond, C. B. De Aquino, J. C. Pieretti, S. H. Domingues, R. A. Ando and P. H. C. Camargo, J. Mater. Chem. A, 2017, 5, 11720–11729 RSC.
- E. Cortés, Adv. Opt. Mater., 2017, 5, 1700191 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |