
A versatile, divergent route for the synthesis of ABAC tetraazaporphyrins: molecularly engineered, push–pull phthalocyanine-type dyes†‡
Emre
Güzel
 ab,
Diana-Paola
Medina
a,
María
Medel
a,
Mehmet
Kandaz§
ac,
Tomás
Torres
*ade and
M. Salomé
Rodríguez-Morgade
*ad
ab,
Diana-Paola
Medina
a,
María
Medel
a,
Mehmet
Kandaz§
ac,
Tomás
Torres
*ade and
M. Salomé
Rodríguez-Morgade
*ad
aDepartamento de Química Orgánica Universidad Autónoma de Madrid Cantoblanco, 28049 Madrid, Spain. E-mail: salome.rodriguez@uam.es; tomas.torres@uam.es
bDepartment of Fundamental Sciences, Faculty of Technology Sakarya University of Applied Sciences, Sakarya, Turkey
cDepartment of Chemistry, Sakarya University, TR-54100 Adapazarı, Sakarya, Turkey
dInstitute for Advanced Research in Chemical Sciences (IAdChem) Universidad Autónoma de Madrid Cantoblanco, 28049 Madrid, Spain
eInstituto Madrileño de Estudios Avanzados (IMDEA)-Nanociencia c/Faraday, 9 Cantoblanco, 28049 Madrid, Spain
First published on 17th May 2021
Abstract
A method to prepare key synthetic intermediates of ABAB- and ABAC-phthalocyanines and other tetraazaporphyrin analogues of interest for molecular electronics is described. These intermediates consist of ABAB-tetraazaporphyrins wherein the two B-subunits are endowed with iodine substituents at the meta-positions, whereas the substitution of the A-subunits is not conditioned to sterically demanding ortho-substituents, but rather can include a variety of functional groups at meta- or ortho-positions. The synthetic procedure includes the assembly of an ABAB tetraazaporphyrin by crossover cyclotetramerization of 5(6)-nitro-1,1,3-trichloroisoindolenine with a diiminoisoindoline or diiminopyrroline derivative, followed by a three-step conversion of the two peripheral nitro groups into iodine substituents. The effectiveness of the method is based on two principles. On one hand, the inability of the trichloroisoindolenine derivative to self-condense, together with the mild reaction conditions, favors the preferential formation of an ABAB derivative, without conditioning the substitution at the pyrroline or isoindoline A-precursor. On the other hand, the opposite diiodo-tetraazaporphyrin can be easily functionalized through a variety of metal-catalyzed cross-coupling reactions, thus being a valuable synthetic intermediate for ABAB and ABAC tetraazaporphyrins. A molecularly engineered phthalocyanine-like dye, which features the prototypical structure of a donor–π-bridge–acceptor, was prepared using this method.
Introduction
Non-uniformly substituted tetraazaporphyrins comprise phthalocyanines or porphyrazines bearing different functional groups (represented by A, B, C and D in Fig. 1) at their isoindole or pyrrole moieties.1 Despite the general definition, in practice, this category of porphyrinoids is almost exclusively constituted by tetraazaporphyrins that bear not more than two types of substituents at the pyrrolic subunits (AAAB, AABB and ABAB in Fig. 1). Compounds containing three (A, B and C) or four (A, B, C and D) different subunits are very desirable for manifold applications in fields of molecular electronics, but so far, satisfactory, general synthetic procedures for their preparation have been lacking.2 The term ABAC also implies a specific arrangement of the substituents, referring to a structural isomer bearing the same substitution (A) at two of the opposite pyrrole subunits, while the other two (B and C) contain different functionalizations and all the adjacent units are different to each other (see Fig. 1).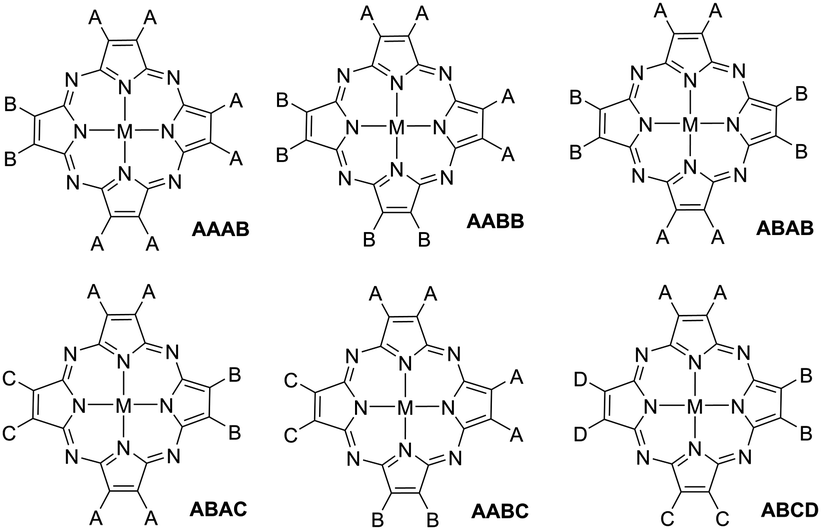 | ||
| Fig. 1 General structures for non-uniformly substituted tetraazaporphyrins. | ||
In the literature, there is a very limited number of methods to prepare non-uniformly substituted tetraazaporphyrins. The main obstacle lies in the scarce number of synthetic intermediates, which often constitute unstable species and offer a very limited variety of substitution patterns. Furthermore, their use does not necessarily provide acceptable yields of unsymmetric macrocycles either.3 This is in stark contrast with porphyrins, for which useful dipyrrolic or tripyrrolic synthetic intermediates have been developed. However, for phthalocyanines or porphyrazines containing two types of substitution A and B, crossover cyclotetramerization of two differently functionalized phthalonitrile, maleonitrile, diiminoisoindoline or diiminopyrroline precursors constitutes the most widely used synthetic procedure. The latter affords a mixture of six compounds wherein four of them constitute non-uniformly substituted tetraazaporphyrins (the so-called AAAB, AABB, ABAB and ABBB tetraazaporphyrins) and the other two are symmetric macrocycles (AAAA and BBBB).3
The yield for each type of macrocycle depends upon statistical considerations,2 the relative reactivity of the two precursors bearing A and B,4,5 the used stoichiometry of reactants and the reaction conditions (i.e. metal template, solvent, temperature, etc.).6 The target unsymmetric compound is obtained in moderate-to-very-low yields (0–40% in the best cases) after a laborious chromatographic separation that is not always possible. Crossover cyclotetramerization is particularly inefficient to prepare the opposite ABAB-isomers as they are statistically unfavoured (theoretically 12.5% against 25% of the adjacent AABB-isomer).3,4,6 These elusive compounds can be prepared with reasonable yields by condensation of a trichloroisoindolenine derivative bearing B, with a diiminoisoindoline or diiminopyrroline derivative bearing A.7,8 Although the method is not fully selective, the trichloroisoindolenine precursor cannot self-condense and, hence, the number of products decreases to three (ABAB, AAAB and AAAA). The limited scope of this method constitutes its major drawback, as trichloroisoindolenine derivatives are very labile,9 and only unsubstituted7 or nitro-substituted compounds8,9b have really shown some utility. The other procedure to prepare the opposite ABAB-tetraazaporphyrins involves the use of phthalonitrile or diiminoisoindolenine precursors with impediments to self-condense, because they are substituted at the ortho-positions with sterically demanding A moieties, such as tert-butyl,6a isopropoxy,5 or benzene rings.6b,10 However, the non-formation of sterically constrained macrocycles (that is, AAAA, AAAB and AABB) depends strongly on the reaction conditions and does not always guarantee acceptable yields of the ABAB compounds.4a,6,10b
It is easy to understand, based in the preceding considerations, that tetraazaporphyrins with triple (A, B and C) or quadruple (A, B, C and D) substitution patterns are almost non-existent, as more than two types of precursors generate very complex mixtures in crossover macrocyclizations (more than 15 compounds for triple and more than 35 compounds for quadruple patterns), which are impossible to separate. To the best of our knowledge, only an ABAC-type phthalocyanine has been prepared by a cyclotetramerization reaction. The compound was obtained in 9% yield by the reaction of two different diiminoisoindolines bearing the B and C substituents, respectively, and a trichloroisoindolenine bearing substituent A.11 The other reported method to prepare ABAC-tetraazaporphyrins is based in the peripheral fabrication of a preformed ABAB macrocycle that contains reactive halogen substituents at the B subunits. This entails garnering important amounts of an ABAB precursor, a compound whose synthesis is problematic in itself. Nonetheless, there are some examples in the literature of preparations using palladium catalysed cross-coupling reactions at the halogen-functionalized opposite-isoindoles to afford the B and C subunits, while the A moieties contain bulky phenyl10a or 3,5-bistrifluoromethylphenyl substituents,4b respectively, at the ortho-positions. The latter proved to be essential for gathering sufficient ABAB precursor to work with. Again, the scope of these methods is very limited, since the design of other ABAC-systems is restricted to compounds containing 3,6-diphenyl- or 3,6-bis(3,5-bistrifluoromethylphenyl)-substituted isoindoles as the A subunits.
In this work we have overcome this limitation. Thus, we wish to report a divergent procedure to prepare ABAC-tetraazaporphyrins, wherein the A substituents arise from a diiminoisoindoline or diiminopyrroline derivative, while the B and C substituents originate from the different derivatization of two iodo-substituted, opposite isoindoles, in an ABAB-precursor. As demonstrated by us and others, iodo-substituted phthalocyanines can be used in a variety of palladium catalysed reactions such as carboxylation,12 alkynylation,13–15 amination,13,16 arylation,13,17 vinylation,15,18 or the introduction of phosphinic groups.19 In the present work, the ABAB diiodo-tetraazaporphyrin precursor is prepared by reduction of two peripheral nitro groups, followed by a sequential diazotization reaction and iodide coupling in a preformed opposite isomer. The versatility and effectivity of our approach depends on three important conditions:
(i) The use of trichloroisoindolenine derivatives in the method to prepare the ABAB precursors; this ensures definite reliability to obtain enough amounts of the opposite isomer, precluding the formation of the statistically favoured adjacent isomer, which is also difficult to separate.6b
(ii) The use of 5(6)-nitro-1,1,3-trichloroisoindolenine as a starting material; this compound is more stable and is easier to handle than other trichloroisoindolenine derivatives, such as alkyl- and halogen-substituted compounds, as proved before.9
(iii) There is a wide range of functional groups that can be introduced as the A-type substitution, since the latter arises from a diiminoisoindoline or diiminopyrroline derivative; the synthesis of these precursors by reaction of the corresponding dinitrile with ammonia is well established20 – indeed, just a quick search in Scifinder provides more than 440 of these derivatives, more than 150 of which are commercially available.
Therefore, we report here a convenient synthesis of ABAB-phthalocyanine analogues, endowed with two iodine atoms at the meta-positions of both opposite isoindole units. These compounds are intended to become general key precursors of highly asymmetric ABAC-phthalocyanines and other tetraazaporphyrins of interest in molecular electronics. As a proof of concept, we include the controlled synthesis of a push–pull, ABAC dibenzoporphyrazine, a molecularly engineered phthalocyanine-like dye, which features the prototypical structure of a donor–π-bridge–acceptor, in a linear arrangement. This type of disposition has been shown to be very effective in the design of dyes for dye-sensitized solar cells (DSSCs).4b,21
Results and discussion
Synthesis of the diiodo-substituted ABAB intermediate
3,4-Dipropylpyrroline-2,5-diimine22 was selected as the A moiety as it provides an A substitution pattern that is virtually inaccessible using the previously reported methods for ABAC systems. The opposite isomer of dinitrodibenzoporphyrazine (1, Scheme 1) was prepared following the reported procedure,8a consisting of the reaction of the diiminopyrroline with 5(6)-nitro-1,1,3-trichloroisoindolenine23 at −40 °C in the presence of triethylamine. | ||
| Scheme 1 Synthesis of the ABAB diiododibenzoporphyrazine precursor 3. | ||
This afforded a mixture of free base porphyrazines, with 1 and the A3B benzoporphyrazine as the major compounds. The mixture was subsequently metallated with zinc(II) acetate (Scheme 1). The self-condensation of A to afford the symmetric A4 porphyrazine was almost suppressed24 most probably owing to the low temperature used for the reaction.25
The mixture of 1 and A3B was treated with sodium sulfide, affording the diamine 2 and the corresponding A3B monoamine, which were separated using column chromatography.8a Both 1 and 2 should consist of a cis and trans regioisomeric mixture of porphyrazines, owing to the unsymmetric nature of the 5(6)-nitro-1,1,3-trichloroisoindolenine. Indeed, the 13C NMR spectrum of this precursor (Fig. 2) exhibits eight different double resonances, indicating the presence of both 5-nitro and 6-nitro-isomers. Judging by the similar intensity of the two components for the eight signals, a ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the regioisomers can be inferred. Conversely, the presence of the two regioisomers for 2 could not be confirmed by NMR.
:1 ratio of the regioisomers can be inferred. Conversely, the presence of the two regioisomers for 2 could not be confirmed by NMR.
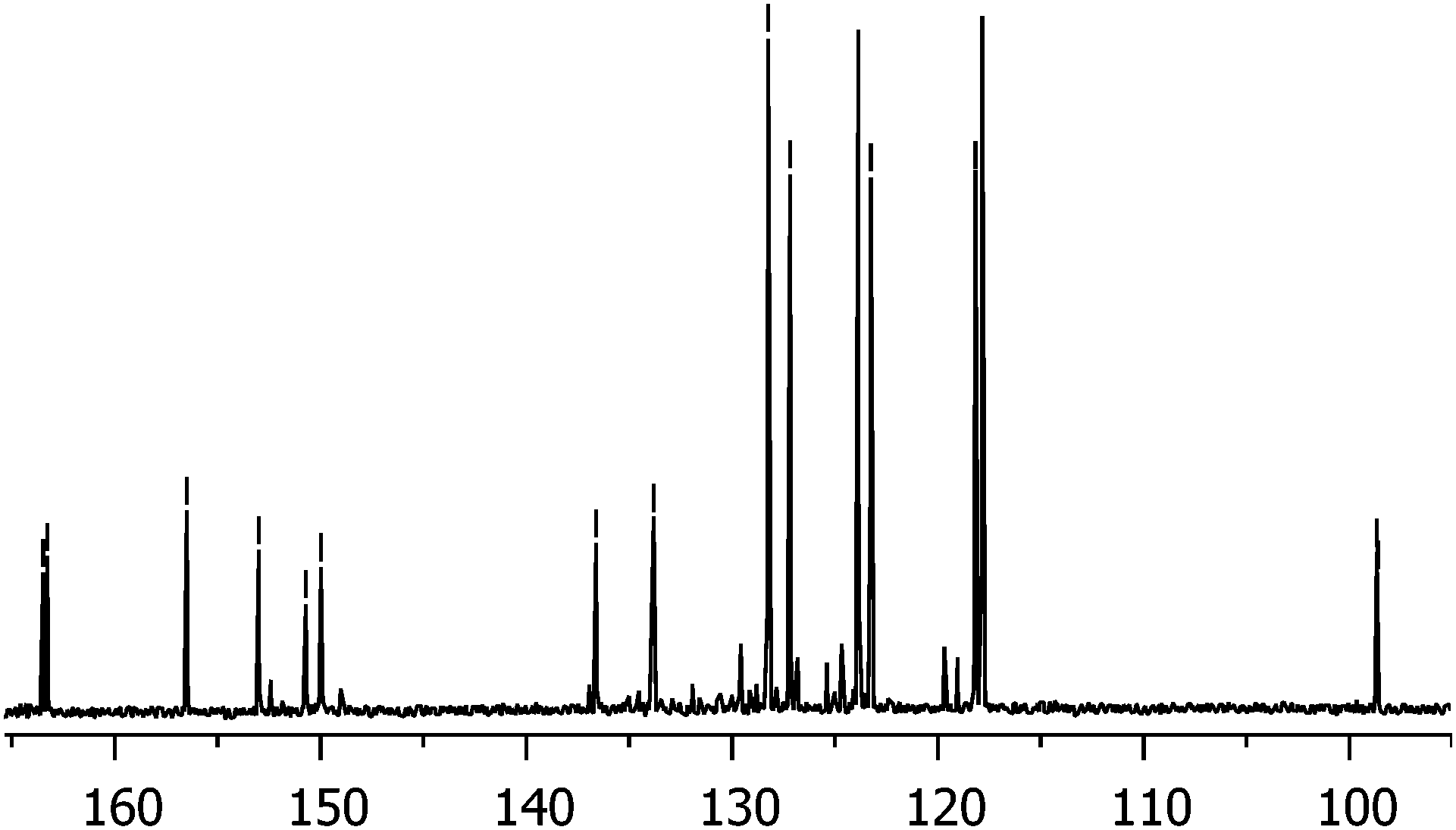 | ||
| Fig. 2 13C NMR spectrum of 5(6)-nitro-1,1,3-trichloroisoindolenine indicating the presence of the regioisomeric mixture. | ||
Porphyrazine 2 was subjected to a diazotization reaction by treatment with NaNO2 in H2SO4, to afford the corresponding bis(diazonium) salt, which was in situ converted into the diiodo-porphyrazine 3 by treatment with KI. The efficiency of this process was very much influenced by the stoichiometry of the reactants and the reaction temperature (Table 1). Thus, the yield of isolated 3 could be optimized up to 36% by using 40 equivalents of KI at a temperature of −15 to −10 °C.
:4:5) ratio
A lower ratio of KI and/or higher temperatures led to different amounts of porphyrazines 4 and 5 (see Fig. 3), arising from the loss of one or both diazonium substituents under the reaction conditions. As indicated in Table 1, this side reaction could be minimized by carrying out the diazotization/substitution reaction under the above-mentioned conditions.
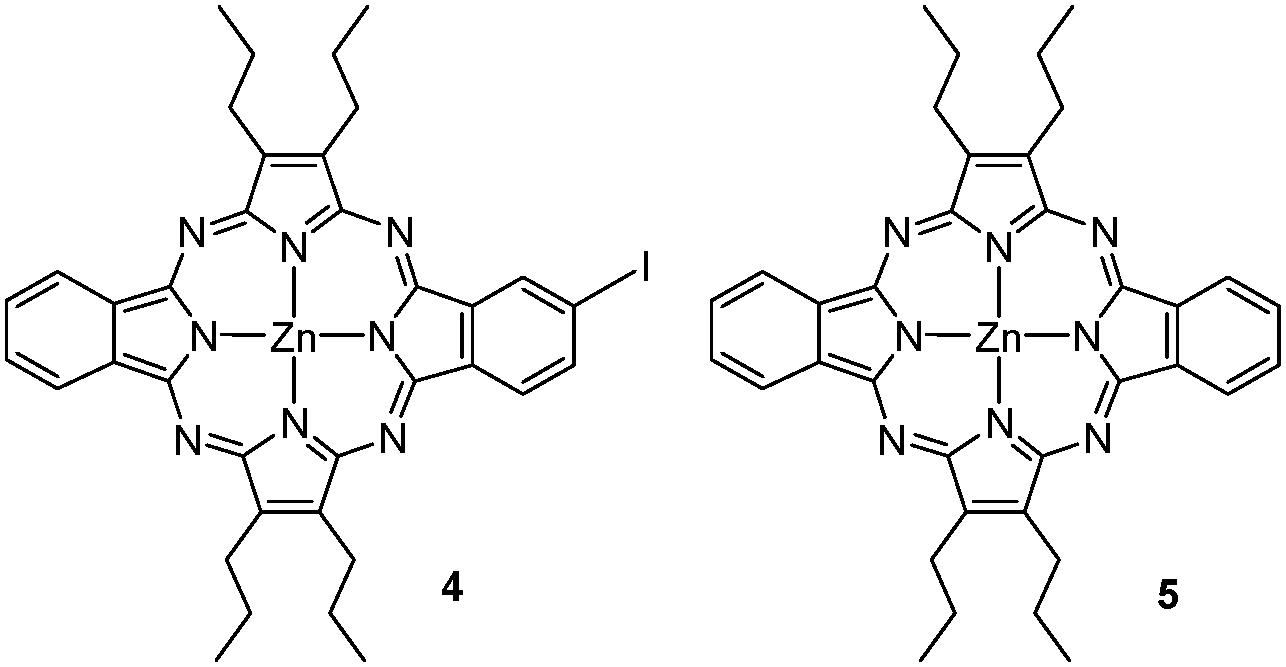 | ||
| Fig. 3 Structures of tetraazaporphyrins 4 and 5. | ||
Compound 3 showed in 1H NMR a doublet of doublets at 8.47 ppm (J = 8.0, 1.6 Hz) that is attributed to HB (see Fig. S1, ESI‡). A second signal centred at 9.55 ppm is constituted by two overlying doublets (J = 1.6 Hz) assignable to HA, while two other doublets (J = 8.0 Hz) centred at 8.97 and 8.98 ppm, respectively, are assigned to HC. The splitting of the signals corresponding to HA and HC arises from the presence of the two cis and trans regioisomers for 3, which seemed to be in a similar ratio. Likewise, all the aliphatic carbons and seven out of the ten non-equivalent aromatic carbons showed split signals in the 13C NMR spectrum of 3 (Fig. S2, ESI‡). Separation of the two regioisomers using chromatographic techniques was not attempted for 3 and nor was it for any of the forthcoming regioisomeric mixtures. Using UV/Vis spectroscopy, the ABAB porphyrazine 3 showed a Q-band at λmax = 626 nm with a full width at half maximum of 32 nm (see Fig. 5 below). For the ABAB and ABAC porphyrazines 5 and 4, the corresponding Q-bands split into the corresponding Qx and Qy components, appearing at 651 and 615 nm for 5 and 643 and 618 nm for 4, respectively (Fig. S13 and S9, ESI‡).
Synthesis of an ABAC donor–π-bridge–acceptor tetraazaporphyrin
Next, we used 3 as the precursor of an ABAC tetraazaporphyrin 12 (Scheme 2), the controlled synthesis of which would be inaccessible under the current state of the art. We selected functionalization at the periphery with these specific electron-donor and -acceptor substituents, as the corresponding porphyrin holding this linear donor–π-bridge–acceptor structure has shown outstanding photovoltaic performance in TiO2-based DSSCs.21 | ||
| Scheme 2 Synthesis of the push–pull, ABAC-tetraazaporphyrin 12. | ||
The preparation of the related push–pull ABAC tetraazaporphyrin 12 is accessible by hydrolysis of the corresponding ester 11 (Scheme 2), which can be assembled using a three-step synthetic sequence from 3. The three-step sequence consists of the introduction of an ethyne linker by Sonogashira coupling on one of the iodo functions of 3, followed by functionalization with the electron-donor moiety 8 by Buchwald–Hartwig reaction on the opposite iodo substituent, and attachment of the electron-acceptor moiety 10 to the ethyne linker (Scheme 2).
The regioisomeric mixture of 3 was first subjected to a copper-free Sonogashira coupling with one equivalent of triisopropylacetylene, using Pd(PPh3)4 as the catalyst, in the presence of Et3N, affording the monoethynyl-substituted macrocycle 6, together with the disubstituted compound 7. Porphyrazine 6 was isolated in 37% yield after column chromatography on silica gel. Fig. 4 displays the aromatic region of the 1H NMR spectrum of 6, which appears as six sets of resonances between 8.1 and 9.2 ppm, corresponding to the six different nuclei, designated as A, B, C, D, E, and F in the figure. The presence of the two regioisomers is clearly evidenced by the splitting of each set of signals. On one side, three broad singlets at 9.13, 9.11 and 9.05 ppm that integrate for two protons correspond to the two A, A′ and D, D′ types of protons (Fig. 4). Two doublets at 8.92 and 8.85 ppm (J = 8Hz) that integrate for one proton are assigned to C and C′, by analogy to the precursor 3. The integration of this signal gives a 45:55 ratio for the cis and trans regioisomers, in line with our previous observations for the tetraazaporphyrin 3 and its trichloroisoindolenine precursor. B and B′ protons are observed at 8.26 ppm as a single doublet (J = 8Hz) integrating for one proton. The two doublets at 8.53 and 8.52 ppm (J = 7.5 Hz) are assigned to the F, F′ protons, while two doublets of doublets at 8.21 and 8.19 ppm (J = 1 Hz and J = 7.5 Hz) correspond to the E, E′ protons. The MS spectrum of 6 showed the cluster corresponding to the expected molecular ion at m/z = 951–957, accompanied by weaker ions at m/z = 1005–1011 and m/z = 790–795, assignable to 7 and deprotected 6, respectively, which could be present as traces, since they were not detected by NMR. HRMS (APCI+) confirmed the structure of 6 with a peak at m/z = 951.2724 corresponding to [M + H]+ (Fig. S18, ESI‡).
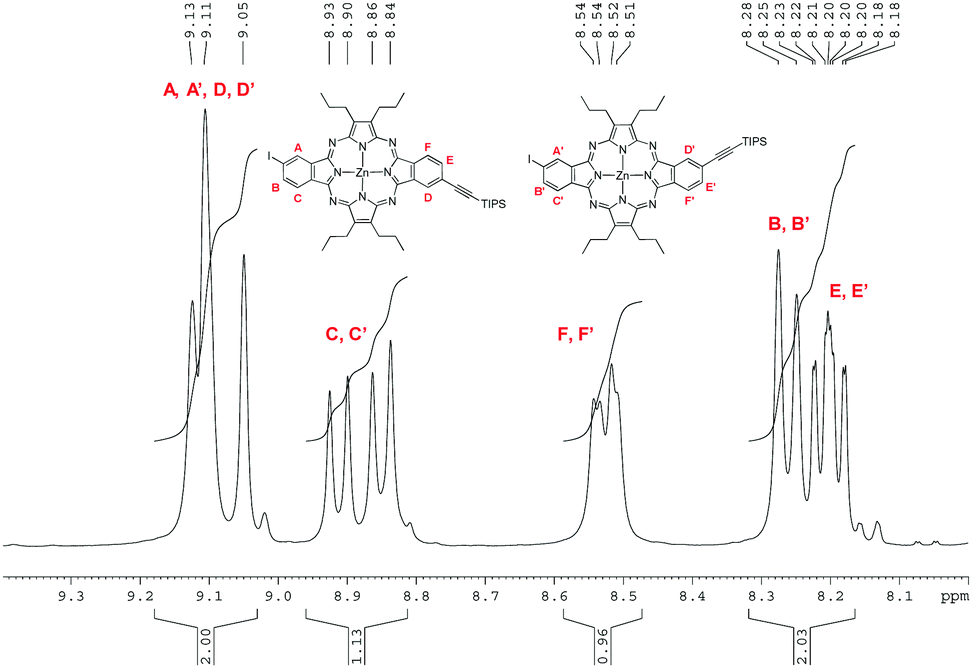 | ||
| Fig. 4 1H NMR spectrum corresponding to the aromatic region of 6. | ||
Compound 7 displays a simpler 1H NMR spectrum, with all the aromatic signals between 7.8 and 7.3 ppm, and a narrower multiplet (3.8–3.6 ppm) corresponding to the methylene protons directly attached to the pyrrole subunits. The molecular ion [M + H]+ appeared at m/z = 1005.5089 in HRMS (APCI+) (Fig. S21, ESI‡).
The donor moiety consisted of the diarylamine 8, which was prepared following reported procedures,26 and attached to the tetraazaporphyrin 6 by Buchwald–Hartwig coupling, giving tetraazaporphyrin 9 in 70% yield. The structure of 9 was assessed by 1H NMR and especially by HRMS (MALDI-TOF), which showed a peak at m/z = 1543.8604 corresponding to [M]+ (Fig. S27, ESI‡).
The benzothiodiazole accepting unit 10,21,27 was incorporated to 9 by deprotection of the ethynyl linker with TBAF, followed by a Sonogashira coupling with the bromo derivative 10. This afforded the push–pull tetraazaporphyrin 11 in 33% yield. Finally, saponification of 11, followed by treatment with hydrochloric acid afforded 12 in 70% yield. Compounds 11 and 12 showed in MS (MALDI-TOF) the clusters corresponding to their molecular ions [M]+ at m/z = 1656–1662 and 1642–1648, respectively. HRMS confirmed the structure of 12, with a value of m/z = 1641.7414, which matches the theoretical value for this compound (1641.7412) (Fig. S34, ESI‡). Based on the ∼1:1 regioisomeric mixture observed for the trichloroisoindolenine precursor and the tetraazaporphyrin intermediates 3 and 6, we assume that both compounds 11 and 12 consist of cis and trans regioisomeric mixtures, although this could not be confirmed spectroscopically.
Fig. 5 displays the absorption spectra in CHCl3 of tetraazaporphyrin 12, and its synthetic intermediates 3, 6, 9 and 11. Despite their non-symmetric ABAB or ABAC structures, all the compounds exhibit non-split, though broad, Q-bands.28 As a general feature, compounds 9, 11, and 12, containing the amine function (red and light- and dark-green lines, respectively, in Fig. 5), exhibit broader (full width at half maximum of 41–44 nm) and red-shifted Q-bands (λmax = 650–657 nm vs. λmax = 626–632 nm for macrocycles 3 and 6). This reflects the extension of the conjugation through the amine group.
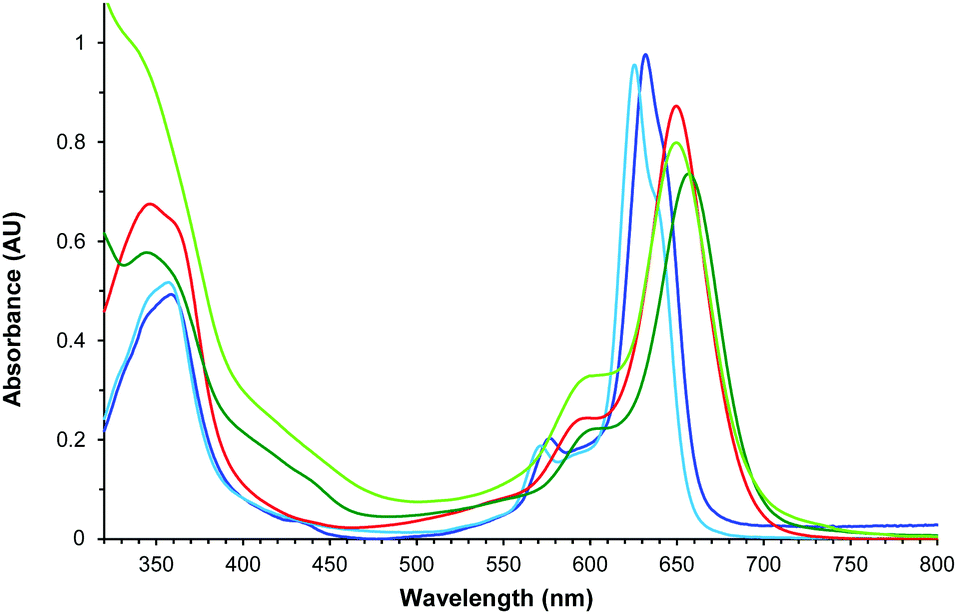 | ||
| Fig. 5 Normalized UV/Vis spectra of 3 (navy blue), 6 (light blue), 9 (red), 11 (dark green) and 12 (light green) in CHCl3. | ||
DFT calculations for the trans isomer of 12 were performed at the B3LYP/6-31G(d,p) level, using the GAUSSIAN 09 package.29 The frontier orbitals estimated for 12 are represented in Fig. 6.
 | ||
| Fig. 6 HOMO and LUMO orbitals of the trans isomer of 12 calculated using DFT (B3LYP/6-31G (d,p)). | ||
As for the parallel porphyrin,21 compound 12 shows a high degree of orbital partitioning. Thus, the HOMO orbital is predominantly localized on the macrocycle and the donor moiety. By contrast, the LUMO orbital is fully localized on the accepting unit, showing a high density in the BTD ring. Hence, the HOMO–LUMO transition should have a considerable CT character for this regioisomer.
Experimental
Instrumentation and materials
Commercially available solvents and reagents were used without further purification unless otherwise mentioned. For anaerobic and/or anhydrous reactions, solvents were dried by distillation with Na/benzophenone (THF), or by treatment with previously activated molecular sieves (3 or 4 Å), or with an Innovative Technology Inc. MD-4-PS system. The air-sensitive reactions were carried out using standard Schlenk techniques. Column chromatography was carried out on silica gel Merck-60 (230–400 mesh, 60 Å), and TLC on aluminum sheets precoated with silica gel 60 F254 (E. Merck). Size exclusion chromatography was performed using Bio-BeadsTM S-X1 apparatus. UV-vis spectra were recorded using a JASCO V-660 spectrometer. MS experiments were performed by the Servicio Interdepartamental de Investigación (SIdI) at the Autónoma University of Madrid. EI spectra were obtained using a Waters GCT Agilent Technologies 6890N spectrometer, FAB spectra were obtained with a Waters VG AutoSpec spectrometer and MALDI-TOF-MS spectra were obtained using a BRUKER ULTRAFLEX III instrument equipped with a nitrogen laser operating at 337 nm. NMR spectra were recorded using BRUKER AC-300 (300 MHz) or Bruker XRD-500 (500 MHz) instruments. The temperature was actively controlled at 298 K. Chemical shifts are measured in ppm relative to the corresponding deuterated solvent. Carbon chemical shifts are measured downfield from TMS using the resonance of the deuterated solvent as the internal standard. Infrared spectra (IR) were recorded using a Bruker Vector 22 instrument. Porphyrazines 1 and 2,8a diarylamine 826 and thiadiazole 1021,27 were prepared following the reported procedures.Zn(II) [2,3-12,13-bis(benzo-4′(5′)-iodo)-7,8,17,18-tetrapropylporphyrazinato] (3)
A suspension of porphyrazine 28a (100 mg, 0.15 mmol) in 2.5 M H2SO4 (2 mL) and dry THF (1 mL) was cooled at −15 °C and a solution of NaNO2 (180 mg, 2.59 mmol) in water was added dropwise under stirring. After total addition, the mixture was further stirred for 30 min at −10 °C and then poured over a solution of KI (2 g, 12.05 mmol) in cold water (1 mL). The resulting mixture was stirred for 45 min at the same temperature, and after that the blue solid was filtered, washed with water, and dissolved in THF (20 mL). The solution was subsequently washed with a saturated solution of Na2S2O3 (10 mL), water (15 mL) and dried over Na2SO4. After filtration of the drying agent, the solvent was rotary evaporated and the blue solid purified by column chromatography on silica gel using a 50:1 mixture of toluene/pyridine. The first fraction was collected affording a blue solid that was washed with methanol to give 46 mg (36%) of 3. 1H NMR (300 MHz, THF-d8): δ (ppm) = 9.55 (2d, J = 1.6 Hz, 2H), 8.98, 8.97 (2d, J = 8.0 Hz, 2H), 8.47 (dd, J = 8.0, 1.6 Hz, 2H), 3.9–3.8 (m, 8H), 2.4–2.3 (m, 7H), 1.3–1.2 (m, 12H). 13C NMR (75.5 MHz, THF-d8): δ (ppm) = 158.46, 158.38, 158.30, 158.21, 153.49, 153.40, 152.63, 152.51, 144.69, 144.64, 139.20, 138.69, 132.56, 124.95, 96.09, 29.09, 28.97, 26.57, 26.34, 15.31, 15.24. FTIR (KBr): ν (cm−1) = 3063 (Ar-H), 2956, 2865 (R-H), 1597, 1454, 1365, 1291, 1261, 1134, 1103, 1002, 897, 822, 760, 750. UV-vis (THF): λmax (nm) (logε (dm3 mol−1 cm−1)) = 639 (sh), 626 (3.90), 571 (3.19), 357 (3.63). MS (MALDI-TOF, DCTB): m/z = 896–901 [M]+. HRMS: m/z = 896.0248; calculated for [C36H34I2N8Zn]: 896.0282.
Zn(II) [2,3(benzo-4′-iodo)-12,13-benzo-7,8,17,18-tetrapropylporphyrazinato] (4)
When the above-described diazotization and substitution reactions were performed using 4 equivalents of KI at a temperature range of −10 to +5 °C, three fractions were collected by column chromatography. The second fraction afforded 4 in 18% yield. 1H NMR (300 MHz, THF-d8): δ (ppm) = 9.63 (d, J = 3Hz, 1H), 9.34–9.31 (m, 2H), 9.06 (d, 6Hz, 1H), 8.45 (dd, J = 6Hz, 2Hz, 1H), 8.16–8.13 (m, 2H), 4.0–3.9 (m, 8H), 2.5–2.4 (m, 8H), 1.4–1.3 (m, 12H). FTIR (KBr): ν (cm−1) = 3050 (Ar-H), 2954, 2927, 2867 (R-H), 1598, 1477, 1454, 1366, 1329, 1291, 1132, 1102, 1005, 952, 893, 819, 761, 746, 704. UV-vis (THF): λmax (nm) (logε (dm3 mol−1 cm−1)) = 643 (3.61), 618 (3.76), 592 (sh), 566 (3.05), 352 (3.54). MS (MALDI-TOF, DCTB): m/z = 770–775 [M]+.
Zn(II) [2,3-12,13-dibenzo-7,8,17,18-tetrapropylporphyrazinato] (5)
The third eluted fraction afforded 5 in ∼18% yield. This compound was contaminated with 4. The spectroscopic data are taken from the mixture of the two compounds. 1H NMR (300 MHz, THF-d8): δ (ppm) = 9.31–9.28 (m, 4H), 8.13–8.11 (m, 4H), 3.94 (t, J = 9Hz, 8H), 2.4–2.3 (m, 8H), 1.3–1.1 (m, 12H). FTIR (KBr): ν (cm−1) = 3081, 3054 (Ar-H), 2957, 2926, 2869 (R-H), 1716, 1645, 1597, 1527, 1477, 1455, 1337, 1289, 1102, 1004, 758, 744, 702. UV-vis (THF): λmax (nm) (logε (dm3 mol−1 cm−1)) = 651 (3.52), 632 (sh), 615 (3.48), 595 (sh), 578 (sh), 559 (sh), 349 (3.53). MS (MALDI-TOF, DCTB): m/z = 644–649 [M]+.
Zn(II) [2,3-(benzo-4′-ethynyltriisopropylsilane)-12,13-(benzo-4′′(5′′)-iodo)-7,8,17,18-tetrapropylporphyrazinato] (6)
A mixture of 3 (100 mg, 0.1 mmol), triisopropylacetylene (25 μL, 0.1 mmol), Pd(PPh3)4 (6.4 mg, 0.05 eq.), dry THF (10 mL) and dry Et3N (2 mL) wa gently refluxed under Ar for 4 h. Afterwards, the solvent was rotary evaporated and the residue was chromatographed on Biobeads using toluene as the eluent. The first fraction contained compound 6 (34 mg, 37%), which was obtained as a blue-greenish solid. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 9.13–9.05 (m, 2H), 8.92–8.84 (m, 1H), 8.54–8.51 (m, 1H), 8.26–8.24 (m, 1H), 8.21–8.19 (m, 1H), 3.23 (m, 8H), 2.02 (m 8H), 1.26–1.16 (m, 30H), 0.93–0.81 (m, 3H). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 143.48, 143.42, 143.41, 143.20, 138.30, 137.74, 137.48, 137.17, 133.00, 131.49, 131.43, 131.36, 129.87, 129.74, 127.67, 127.51, 126.24, 124.37, 124.34, 123.67, 123.63, 122.36, 108.58, 95.49, 92.82, 32.09, 29.86, 29.52, 27.98, 27.91, 27.79, 25.41, 22.85, 19.15, 15.06, 15.02, 14.27, 11.85. FTIR (KBr): ν (cm−1) = 3049, 3025 (Ar-H), 2956, 2928, 2866 (R-H), 1720, 1670, 1580, 1457, 1375, 1290, 1264, 1217, 1087, 1042, 803, 738, 697. UV-vis (CHCl3): λmax (nm) (logε (dm3 mol−1 cm−1)) = 644 (sh), 632 (3.87), 577 (3.18), 440 (sh), 358 (3.57). HRMS (APCI+): m/z = 951.2724; calculated for [C47H56IN8SiZn]: 951.2728 [M + H]+.
Zn(II) [2,3-12,13-bis(benzo-4′(5′)-ethynyltriisopropylsilane)-7,8,17,18-tetrapropylporfirazinato] (7)
The disubstituted compound 7 (5 mg, 5%) was isolated from the second fraction of the column chromatography. 1H-NMR (300 MHz, CHCl3): δ (ppm) = 7.8–7.3 (m, 6H), 3.8–3.6 (m, 8H), 2.2–2.1 (m, 36H), 1.9–1.8 (m 8H), 1.3–1.2 (m, 12H), 0.9–0.8 (m, 6H). UV-vis (CHCl3): λmax (nm) (logε (dm3 mol−1 cm−1)) = 6.34 (3.98), 5.79 (3.32), 355 (3.68), 3.42 (sh). HRMS (APCI+): m/z = 1005.5089; calculated for [C58H77N8Si2Zn]: 1005.5096 [M + H]+.
Zn(II) [2,3-(benzo-4′(5′)-ethynyltriisopropylsilane)-12,13-(benzo-4′′-bis(2′,4′′′-dihexoxybiphenyl-4-yl)amine)-7,8,17,18-tetrapropylporphyrazinato] (9)
To a deaerated Schlenk tube charged with 6 (20 mg, 0.02 mmol), diarylamine 8 (19.5 mg, 1.5 eq.), Pd2(dba)3 (0.17 mg, 0.01 eq.), tri-t-butylphosphine (0.11 mg, 0.03 eq.) and t-BuONa (4.3 mg, 2.5 eq.), deaerated, anhydrous toluene (2 mL) was added. The suspension was refluxed for 30 minutes and, after filtration over Celite, the solution was rotary evaporated. Gel permeation chromatography of the residue using toluene as the eluent afforded 9 as a greenish-blue solid (19 mg, 70%). 1H NMR (300 MHz, CDCl3): δ (ppm) = 9.4–9.0 (3 m, 4H), 8.1–7.1 (5 m, 12H), 6.6 (m, 4H), 4.3–3.9 (m, 16H), 2.5–2.1 (m, 8H), 1.8–1.7 (m, 8H), 1.5–1.1 (m, 54H), 0.9–0.8 ppm (m, 15H). DEPT-13C NMR (75 MHz, CDCl3): δ (ppm) = 130.9, 130.5, 130.4, 130.3, 127.7, 126.0, 125.2, 124.0, 122.1, 117.2, 105.4, 100.6, 68.0, 68.0, 34.0, 29.9, 26.8, 25.0, 24.7, 22.9, 18.9, 14.9, 14.8, 14.8, 14.0, 14.0, 11.6. FTIR (KBr): ν (cm−1) = 2962, 2919, 2850 (R-H), 1722, 1668, 1608, 1463, 1374, 1273, 1118, 1103, 727. UV-vis (CHCl3): λmax (nm) (logε (dm3 mol−1 cm−1)) = 650 (3.79), 598 (3.23), 362 (sh), 346 (3.67). MS (MALDI-TOF, DCTB), m/z: 1544–1550 [M]+. HRMS: m/z = 1543.8604; calculated for [C95H121N9O4SiZn]: 1543.8597.
Tetraazaporphyrin 11
To a solution of 9 (18 mg, 0.012 mmol) in dry THF (2 mL), a TBAF solution (0.03 mL, 1 M in THF, 2.5 eq.) was added and the resulting solution was stirred at room temperature for 30 min. After addition of water (5 mL), the solution was extracted using DCM (2 × 5 mL). The organic phase was dried over Na2SO4, the solution was rotary evaporated, and the residue was dried. The resulting solid was transferred to a Schlenk tube, together with the thiadiazole 10 (6.1 mg, 1.5 eq.), Pd(PPh3)4 (0.67 mg, 0.01 eq.) and the mixture was deaerated. A deaerated benzene/triethylamine 1:1 solution (2 mL) was added, and the mixture was heated at 60 °C until all the starting material had reacted (4 h, monitored by TLC). After evaporation of the solvent at reduced pressure, water was added to the residue and the solution was extracted using CH2Cl2. The organic extracts were washed with water and brine and dried over MgSO4. After evaporation of the solvent, gel permeation chromatography of the residue, using toluene as the eluent, afforded 11 (6 mg, 33%) as a greenish-blue solid. UV-vis (CHCl3): λmax (nm) (logε (dm3 mol−1 cm−1)) = 657 (3.79), 600 (sh), 441 (sh), 414 (sh), 344 (3.68). MS (MALDI-TOF, DCTB), m/z: 1656–1662 [M]+.
Tetraazaporphyrin 12
To a solution of 11 (5 mg, 0.003 mmol) in a 3:2 mixture of THF/MeOH (2 mL), a solution of NaOH in water (1 mL, 20%) was added and the mixture was heated at 40 °C for 2 h. After cooling to rt, diethyl ether (5 mL) was added, and the solution was washed using HCl and water. The organic solution was dried (MgSO4) and the solvent was rotary evaporated. The residue was chromatographed on silica gel using DCM, followed by a 1:99 mixture of MeOH/DCM, affording 12 (4 mg, 75%) as a green solid. FTIR (KBr): ν (cm−1) = 2958, 2928, 2866 (R-H), 1726, 1716, 1683, 1668, 1558, 1456, 1374, 1230, 1064, 1038. UV-vis (CHCl3): λmax (nm) (logε (dm3 mol−1 cm−1)) = 650 (3.86), 600 (3.48), 4.52 (sh), 418 (sh), 346 (3.94). MS (MALDI-TOF, DCTB): m/z = 1642–1648 [M]+. HRMS: m/z = 1641.7414; calculated for [C99H107N11O6SiZn]: 1641.7412.
Conclusions
A method to prepare ABAB-phthalocyanines and tetraazaporphyrin analogues, wherein A is a pyrrole or isoindole ring that can be substituted with a variety of functional groups, and the B-subunit consists of an isoindole ring functionalized with one iodine atom at the meta-position, has been developed. The procedure uses the crossover condensation of 5(6)-nitro-1,1,3-trichloroisoindolenine with a diiminopyrroline or diiminoisoindoline derivative as the key step, to assemble the ABAB macrocycle in a controlled fashion. The two nitro-functional groups are subsequently converted into iodo substituents in three synthetic steps. The manifold reactivity of iodo-substituents in different palladium catalyzed chemical transformations converts these diiodo-tetraazaporphyrins into valuable synthetic intermediates of ABAB- and ABAC-tetraazaporphyrins. As the B-subunit arises from a trichloroisoindolenine precursor that is unable to self-condense, the functionalization of the A-subunits is not conditioned to bulky moieties attached to the ortho-positions of isoindole rings. ABAB-phthalocyanines find applications in nonlinear-optics,7d,30 the photodynamic inactivation of bacteria,31 PDT,32 organic field-effect transistors,33 or as precursors of linear34 and ladder polymers.9b,35As a proof of concept, the controlled synthesis of a push–pull, ABAC dibenzoporphyrazine, containing a linear arrangement of the donor and acceptor fragments, has been prepared. This molecularly engineered phthalocyanine-type dye, which features the prototypical structure of a donor–π-bridge–acceptor, improves light-harvesting properties, and opens a route to push–pull phthalocyanine-like compounds of interest, for example, in DSSCs,10a,21 electrocatalysis,36 nonlinear optics,37 or amphiphilic chromophores for biological applications.38 We are currently applying the developed method to the preparation of regioisomerically pure compounds in related ABAC systems by examining the separation of regioisomers, in order to study their properties as photosensitizers in DSSCs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Spanish MICINN (CTQ2017-85393-P, PID2020-116490GB-I00) is acknowledged. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant No. SEV2016-0686). DPM thanks “Convocatoria 617 – Doctorados en el exterior”, COLCIENCIAS, Colombian government, for a doctoral fellowship.Notes and references
- J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281–321 Search PubMed.
- V. N. Nemykin, S. V. Dudkin, F. Dumoulin, C. Hirel, A. G. Gürek and V. Ahsen, ARKIVOC, 2014,(i), 142–204 Search PubMed.
- For major reviews on the different procedures for the synthesis of tetraazaporphyrins and analogues see (a) M. S. Rodríguez-Morgade and T. Torres, 17.9.24 Phthalocyanines and Related Compounds, Sci. Synth., Knowl. Updates, 2017, 2, 1–210 Search PubMed; (b) N. B. McKeown, Product Class 9: Phthalocyanines and Related Compounds, Sci. Synth., 2004, 17, 1237–1368 Search PubMed.
- (a) T. G. Linßen and M. Hanack, Chem. Ber., 1994, 127, 2051–2057 Search PubMed; (b) E. Fazio, J. Jaramillo-García, M. Medel, M. Urbani, M. Grätzel, M. K. Nazeerudin, G. de la Torre and T. Torres, ChemistryOpen, 2017, 6, 121–127 Search PubMed.
- T. P. Forsyth, D. B. G. Williams, A. G. Montalban, C. L. Stern, A. G. M. Barrett and B. M. Hoffman, J. Org. Chem., 1998, 63, 331 Search PubMed.
- (a) S. Rodríguez-Morgade and M. Hanack, Chem. – Eur. J., 1997, 3, 1042–1050 Search PubMed; (b) T. Fukuda, S. Homma and N. Kobayashi, Chem. – Eur. J., 2005, 11, 5205–5216 Search PubMed.
- (a) W. J. Youngblood, J. Org. Chem., 2006, 71, 3345–3356 Search PubMed; (b) J.-D. Wang, M.-J. Lin, S.-F. Wu and Y. Lin, J. Organomet. Chem., 2006, 691, 5074–5076 Search PubMed; (c) S. Dong, C. Bao, H. Tian, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2013, 25, 1165–1169 Search PubMed; (d) M. M. Ayhan, A. Singh, C. Hirel, A. G. Gürek, V. Ahsen, E. Jeanneau, I. Ledoux-Rak, J. Zyss, C. Andraud and Y. Bretonnière, J. Am. Chem. Soc., 2012, 134, 3655–3658 Search PubMed; (e) M. M. Ayhan, A. Singh, E. Jeanneau, V. Ahsen, J. Zyss, I. Ledoux-Rak, A. Gül Gürek, C. Hirel, Y. Bretonniere and C. Andraud, Inorg. Chem., 2014, 53, 4359–4370 Search PubMed; (f) M.-J. Lin and J.-D. Wang, J. Mol. Struct., 2007, 837, 284–289 Search PubMed.
- (a) M. Kandaz, S. L. Michel and B. M. Hoffman, J. Porphyrins phthalocyanines, 2003, 7, 700–712 Search PubMed; (b) S. B. Ekren, F. Dumoulin, E. Musluoglu, V. Ahsen and O. Guengoer, J. Porphyrins phthalocyanines, 2019, 23, 1448–1454 Search PubMed; (c) Y. Gök, H. Kantekin and İ. Değirmencioğlu, Supramol. Chem., 2003, 15, 335–343 Search PubMed; (d) A. Altindala, Z. Z. Öztürk, S. Dabak and Ö. Bekaroğlu, Sens. Actuators, B, 2001, 77, 389–394 Search PubMed.
- (a) M. M. Ayhan, Y. Zorlu, Ö. Gökdemir, A. Gül Gürek, F. Dumoulin, V. Ahsen and C. Hirel, CrystEngComm, 2014, 16, 6556–6563 Search PubMed; (b) M. Hanack and P. Stihler, Eur. J. Org. Chem., 2000, 303–311 Search PubMed.
- (a) S. Yamamoto, S. Mori, P. Wagner, A. J. Mozer and M. Kimura, Isr. J. Chem., 2016, 56, 175–180 Search PubMed; (b) N. Kobayashi, H. Miwa and V. N. Nemykin, J. Am. Chem. Soc., 2002, 124, 8007–8020 Search PubMed.
- F. Dumoulin, Y. Zorlu, M. M. Ayhan, C. Hirel, Ü. Isci and A. Ahsen, J. Porphyrins phthalocyanines, 2009, 13, 161–165 Search PubMed.
- I. Aguirre de Carcer García, A. M. Sevim, A. de la Escosura and T. Torres, Org. Biomol. Chem., 2013, 11, 2237–2240 Search PubMed.
- H. Ali, S. Ait-Mohand, S. Gosselin, J. E. van Lier and B. Guérin, J. Org. Chem., 2011, 76, 1887–1890 Search PubMed.
- (a) W. Seitz, A. J. Jiménez, E. Carbonell, B. Grimm, M. S. Rodríguez-Morgade, D. M. Guldi and T. Torres, Chem. Commun., 2010, 46, 127–129 Search PubMed; (b) A. J. Jiménez, M. Sekita, E. Caballero, M. L. Marcos, M. S. Rodríguez-Morgade, D. M. Guldi and T. Torres, Chem. – Eur. J., 2013, 19, 14506–14514 Search PubMed.
- H. Ali, O. St-Jean, J.-P. Tremblay-Morin and J. E. van Lier, Tetrahedron Lett., 2006, 47, 8275–8278 Search PubMed.
- M. Kloz, S. Pillai, G. Kodis, D. Gust, T. A. Moore, A. L. Moore, R. van Grondelle and J. T. M. Kennis, J. Am. Chem. Soc., 2011, 133, 7007–7015 Search PubMed.
- M. Ince, F. Cardinali, J.-H. Yum, M. V. Martínez-Díaz, M. K. Nazeeruddin, M. Grätzel and T. Torres, Chem. – Eur. J., 2012, 18, 6343–6348 Search PubMed.
- A. de la Escosura, M. V. Martínez-Díaz, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2006, 128, 4112–4118 Search PubMed.
- I. López-Duarte, M. Wang, R. Humphry-Baker, M. Ince, M. V. Martínez-Díaz, M. K. Nazeeruddin, T. Torres and M. Grätzel, Angew. Chem., Int. Ed., 2012, 51, 1895–1898 Search PubMed.
- W. M. Sharman and J. E. van Lier, Synthesis of Phthalocyanine Precursors in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, 2003, Vol. 15, pp. 45–47 Search PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 Search PubMed.
- H. Nie, A. G. M. Barrett and B. M. Hoffman, J. Org. Chem., 1999, 64, 6791–6796 Search PubMed.
- J. G. Young and W. Onyebuagu, J. Org. Chem., 1990, 55, 2155–2159 Search PubMed.
- The A4 macrocycle was only detected by MS of the reaction crude.
- P. Stihler, B. Hauschel and M. Hanack, Chem. Ber., 1997, 130, 801–806 Search PubMed.
- (a) C. Yi, F. Giordano, N. L. Cevey-Ha, H. N. Tsao, S. M. Zakeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 Search PubMed; (b) C. Yi, F. Giordano, N. L. Cevey-Ha, H. N. Tsao, S. M. Zakeeruddin and M. Grätzel, ChemSusChem, 2014, 7, 1107–1113 Search PubMed.
- M. D. Hylarides, D. Scott Wilbur, S. W. Hadley and A. R. Fritzberg, J. Organomet. Chem., 1989, 367, 259–265 Search PubMed.
- Y. Rio, M. S. Rodríguez-Morgade and T. Torres, Org. Biomol. Chem., 2008, 6, 1877–1894 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- W. Cao, K. Wang, I. Ledoux-Rak and J. Jiang, Inorg. Chem. Front., 2016, 3, 1146–1151 Search PubMed.
- M. A. Revuelta-Maza, P. Gonzalez-Jimenez, C. Hally, M. Agut, S. Nonell, G. de la Torre and T. Torres, Eur. J. Med. Chem., 2020, 187, 111957 Search PubMed.
- M. A. Revuelta-Maza, M. Mascaraque, P. González-Jiménez, A. González-Camuñas, S. Nonell, Á. Juarranz, G. de la Torre and T. Torres, Molecules, 2020, 25, 213 Search PubMed.
- S. Dong, C. Bao, H. Tian, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2013, 25, 1165–1169 Search PubMed.
- A. Kumar Mutyala, S.-M. Hong, J. W. Namgoong, J. P. Kim and J. S. Park, Dyes Pigm., 2017, 142, 237–242 Search PubMed.
- (a) Y. Yamada, T. Kubota, M. Nishio and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 6505–6509 Search PubMed; (b) M. Suzuki, K. Yokota, H. Tsuchida, K. Yamoto, M. Nagata, S. Matsuoka, K. Takagi, K. Johmoto and H. Uekusa, ACS Macro Lett., 2015, 4, 247–250 Search PubMed.
- R. Nkhahle and T. Nyokong, Electroanalysis, 2021, 33, 11–22 Search PubMed.
- A. Khadria, Y. de Coene, P. Gawel, C. Roche, K. Clays and H. L. Andersonm, Org. Biomol. Chem., 2017, 15, 947–956 Search PubMed.
- K. Zheng, X. Liu, H. Liu, D. Dong, L. Li, L. Jiang, M. Huang and C. Ding, ACS Appl. Mater. Interfaces, 2021, 13, 10674–10688 Search PubMed.
Footnotes |
| † Dedicated to Professor Jaume Veciana and Professor Concepció Rovira on the occasion of their retirement. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc00990g |
| § M. Kandaz passed away in October 2017. |
| This journal is © The Royal Society of Chemistry 2021 |