
Importance of multimodal characterization and influence of residual Li2S impurity in amorphous Li3PS4 inorganic electrolytes†
Priyadarshini
Mirmira‡
 a,
Jin
Zheng‡
a,
Peiyuan
Ma
a and
Chibueze V.
Amanchukwu
*ab
a,
Jin
Zheng‡
a,
Peiyuan
Ma
a and
Chibueze V.
Amanchukwu
*ab
aPritzker School of Molecular Engineering, University of Chicago, IL 60637, USA. E-mail: chibueze@uchicago.edu
bChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 6th May 2021
Abstract
Amorphous Li3PS4 (LPS) solid-state electrolytes are promising for energy-dense lithium metal batteries. LPS glass, synthesized from a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ratio of Li2S and P2S5, has high ionic conductivity and can be synthesized by ball milling or solution processing. Ball milling has been attractive because it provides the easiest route to access amorphous LPS with a conductivity of 3.5 × 10−4 S cm−1 (20 °C). However, achieving the complete reaction of precursors via ball milling can be difficult, and most literature reports use X-ray diffraction (XRD) or Raman spectroscopy to confirm sample purity, both of which have limitations. Furthermore, the effect of residual precursors on ionic conductivity and lithium metal cycling is unknown. In this work, we illustrate the importance of multimodal characterization to determine LPS phase and chemical purity. To determine the residual Li2S content in LPS, we show that (1) XRD and 31P solid state nuclear magnetic resonance (ssNMR) are insufficient and (2) Raman loses sensitivity at concentrations below 12 mol% Li2S. Most importantly, we show that 7Li ssNMR is highly sensitive. Using 7Li ssNMR, we investigate the effect of ball milling parameters and develop a robust and highly reproducible procedure for pure LPS synthesis. We find that as the residual Li2S precursor content increases, LPS conductivity decreases and lithium metal batteries exhibit higher overpotentials and poor cycle life. Our work reveals the importance of multimodal characterization techniques for amorphous solid-state electrolyte characterization and will enable better synthetic strategies for highly conductive electrolytes for efficient energy-dense solid-state lithium metal batteries.
:1 mol ratio of Li2S and P2S5, has high ionic conductivity and can be synthesized by ball milling or solution processing. Ball milling has been attractive because it provides the easiest route to access amorphous LPS with a conductivity of 3.5 × 10−4 S cm−1 (20 °C). However, achieving the complete reaction of precursors via ball milling can be difficult, and most literature reports use X-ray diffraction (XRD) or Raman spectroscopy to confirm sample purity, both of which have limitations. Furthermore, the effect of residual precursors on ionic conductivity and lithium metal cycling is unknown. In this work, we illustrate the importance of multimodal characterization to determine LPS phase and chemical purity. To determine the residual Li2S content in LPS, we show that (1) XRD and 31P solid state nuclear magnetic resonance (ssNMR) are insufficient and (2) Raman loses sensitivity at concentrations below 12 mol% Li2S. Most importantly, we show that 7Li ssNMR is highly sensitive. Using 7Li ssNMR, we investigate the effect of ball milling parameters and develop a robust and highly reproducible procedure for pure LPS synthesis. We find that as the residual Li2S precursor content increases, LPS conductivity decreases and lithium metal batteries exhibit higher overpotentials and poor cycle life. Our work reveals the importance of multimodal characterization techniques for amorphous solid-state electrolyte characterization and will enable better synthetic strategies for highly conductive electrolytes for efficient energy-dense solid-state lithium metal batteries.
Introduction
The electrification of transport requires batteries with higher energy densities, lower cost, and improved safety. Lithium-ion batteries are currently state-of-the-art, but their energy densities and cost are currently inadequate for mass market electric vehicle adoption.1 Hence, there is a great need for the development of next generation battery chemistries.2,3 Lithium metal batteries have great promise because lithium metal has an order of magnitude higher gravimetric capacity (3860 mA h g−1) than graphite (372 mA h g−1), which is currently used in lithium ion batteries.1,4 Despite the high energy promise of lithium metal, lithium metal anodes suffer from high reactivity and a propensity to form high surface area deposits during electrodeposition that exacerbate further reaction with the electrolyte.1,5 Liquid electrolytes based on carbonate solvents currently enable Li-ion batteries, but these electrolytes are highly volatile, flammable, and lead to low coulombic efficiencies for lithium metal deposition and stripping.6,7 Several liquid electrolytes such as high and localized concentration electrolytes,8–10 fluorinated ether electrolytes,11–13 and various electrolyte mixtures14,15 have been pursued but challenges remain.Solid state electrolytes have been developed with ionic conductivities that rival those of commercial liquid electrolytes and are nonflammable, nonvolatile, high-energy dense, and safer.16–19 These solid-state electrolytes are primarily inorganic, and they range from families such as oxides (LLZO) and phosphates (LATP) to sulfides (LPS).18 Oxide and sulfide based inorganic solid-state electrolytes are among the most widely studied because they have been shown to enable lithium metal batteries with high energy density.20,21 Among all inorganic solid-state electrolytes, sulfides enable the highest ionic conductivities, have lower Young's Moduli (compared to oxides),22 and are soft enough to enable an intimate contact with a lithium metal anode.23–26 Notably, sulfide electrolytes can be fabricated at room temperature.26–28
The sulfide chemical composition and phase play a significant role in ionic conductivity and electrochemical stability and can be controlled by the synthetic procedure. Some sulfide compounds, such as halogen-doped argyrodite (Li6PS5X, where X = Cl, Br, I), or germanium-doped systems, namely Li10GeP2S12 (LGPS), report very high ionic conductivities, approximately 10−3–10−2 S cm−1 depending on the crystal structure.29–32 In particular, Li3PS4 (LPS) sulfide-type glass is of interest because of its high ionic conductivity and relative ease of synthesis.23,33–35 In particular, LPS electrolytes do not require additional high temperature annealing or sintering steps as required for argyrodite.36 LPS glass has a wide range of reported ionic conductivities depending on the crystal structure. LPS has four reported phases: amorphous and α, β, and γ-crystalline, with the amorphous form reporting the highest conductivity, ranging from 3–4 × 10−4 S cm−1 at 25 °C.37 The β-crystalline form has a conductivity of approximately 1.5 × 10−4 S cm−1, whereas the γ-crystalline form reports a much lower conductivity (10−6 S cm−1).38 Additionally, the α and γ forms of LPS are synthetically more difficult to access than the amorphous or β-crystalline forms, making them less attractive candidates for solid state electrolytes.39,40
LPS can be synthesized using solution processing or ball milling. Solution processing involves mixing Li2S and P2S5 precursors in solvents such as acetonitrile or tetrahydrofuran (THF).41,42 Solution processing is scalable and highly flexible as different solvents can enable morphological, surface area, and phase control. However, solution processing leads to LPS-solvent complexes that require temperatures as high as 100 °C to remove the solvent and form pure LPS.38,41–44 Furthermore, solution processed LPS leads to ionic conductivities that are lower than those obtained using ball milling.44 Finally, LPS stability in solvents is also of concern as recent reports have shown that Li7P3S11 decomposes in polar solvents such as acetonitrile and dimethylformamide (DMF).42 These challenges in solution processing have made ball milling the dominant process for amorphous LPS synthesis.
Ball milling is a popular method for LPS synthesis because it involves the combination of precursors and use of high energy mechanical mixing without any additional heating step. Furthermore, ball milling is the most direct route to access amorphous LPS, which reports the highest conductivity of all LPS phases.38,44 However, it can be difficult to achieve a complete reaction of precursors using ball milling. Often, little information beyond the ball milling speed and time is provided in the literature, making reproducible LPS fabrication more of an art than a science, and stifling further growth in the field. In addition, many literature reports on amorphous LPS synthesis use primarily X-ray diffraction to study phase and product purity even though XRD is a poor tool for amorphous compound characterization.37,43–46 Therefore, the influence of unreacted precursors on amorphous LPS conductivity and electrochemical performance is unknown.
In this work, we illustrate the importance of multimodal characterization to determine LPS product and phase purity, and the influence of unreacted Li2S on ionic conductivity and electrochemical cycling. Using XRD, Raman spectroscopy, and 31P and 7Li solid state NMR (ssNMR), we quantify the amount of Li2S precursor remaining in the sample and assess the limitations of each technique in determining LPS purity. We show that 7Li ssNMR is highly sensitive for LPS purity determination and can detect impurities not observable with Raman, XRD or 31P ssNMR. Equipped with 7Li ssNMR, we evaluate the influence of various ball milling processing parameters and develop a highly reproducible method to consistently obtain pure, amorphous LPS. Using electrochemical impedance spectroscopy, we show that ionic conductivity in the LPS product is a function of Li2S impurity, with conductivity increasing as the Li2S residual content decreases. Finally, we fabricate Li/Li cells using these electrolytes and show that electrolytes with a high residual Li2S content exhibit poor cycling behavior with higher overpotentials and earlier cell death in comparison to pure samples. Our work demonstrates the importance of confirming product purity using a suite of characterization techniques, with 7Li ssNMR a highly sensitive and reliable technique to quantify unreacted Li2S. Although we focus on amorphous Li3PS4, these observations apply to all solid electrolyte processes that involve the formation of amorphous products such as other LPS glasses. These insights will expedite the development and better understanding of sulfide solid electrolytes to enable energy-dense lithium metal batteries that can revolutionize the electrification of transport.
Results and discussion
Multimodal characterization to detect Li2S impurity
Amorphous Li3PS4 can be synthesized by combining Li2S and P2S5 in a stoichiometric molar ratio of 3:1 and ball milling.35,47–49 Ball milling is often favored in research labs because of its ease of use and simplicity where the only two variables to change are the speed and total time. Unfortunately, many literature reports specify only these two variables and omit other considerations such as milling interval or hand milling steps that make the synthetic procedure quite difficult to reproduce. This barrier limits entry to the field and complicates any data science-driven approach to materials synthesis.50,51 Sample 1 was synthesized according to a previously reported procedure52 for LPS (Table 1), and Sample 2 was synthesized with a modification of the same procedure, but two different colors were obtained. Fig. S1† shows that Sample 1 is pale white while Sample 2 has a yellow-green hue that has been reported for LPS.45,52,53 The visual color differences indicate likely unreacted precursors in Sample 1, and X-ray diffraction (XRD) was used to determine the differences.38–41,43 Because Li2S crystallizes in the cubic space group Fm3m, weak reflections within the amorphous LPS spectra corresponding to unreacted Li2S were expected.43 However, as Fig. 1a shows, several broad features are observed in both samples, yielding little insight into the chemical composition of both samples. Nanda et al. and Tatsumisago et al. have previously used XRD to confirm amorphous LPS formation where LPS is reported to exhibit one broad peak around 19° (observed in Sample 2).43,46 The second peak at 30° in Sample 2 corresponds to the Kapton film background (Fig. S2†).43,54 However, the lack of crystalline Li2S peaks in both samples does not indicate a lack of residual Li2S precursors as the ball milling process can amorphosize the crystalline Li2S powder.45,46,54 The Sample 1 pattern exhibited several additional amorphous peaks that are difficult to assign to LPS, Li2S, P2S5 or the Kapton film background. Hence, XRD appears insufficient to differentiate between both samples and to determine the overall product purity. Additional characterization techniques such as Raman and NMR must be used to supplement the XRD data when examining amorphous LPS purity.
:1 molar ratio). 5 min rest per h refers to a 5 minute rest period after an hour of continuous milling. Hand milling refers to mixing in a mortar and pestle before ball-milling. “Break to clean” refers to a stoppage in ball milling to clean the ball mill jar
| Sample number | Mass precursors | Milling media | Speed | Total ball milling time | Milling interval | Hand milling | Break to clean? | Mol% Li2S impurity |
|---|---|---|---|---|---|---|---|---|
| 1 | 5.2 g | 32 g of 5 mm balls | 450 rpm | 65 hours | 5 min rest per h | N | N | 22% |
| 2 | 5.2 g | 64 g of 5 mm balls | 450 rpm | 80 hours | 5 min rest per h | Y, for 10 min | Y, at 65 hours | 0% |
| 3 | 2.5 g | 32 g of 5 mm balls | 450 rpm | 15 hours | No stops | N | N | 30% |
| 4 | 5.2 g | 64 g of 5 mm balls | 450 rpm | 80 hours | 5 min rest per h | Y, for 10 min | Y, at 65 hours | 12% |
| 5 | 2.5 g | 40 g, 10 mm balls | 510 rpm | 15 hours | 3 min rest/5 min | N | N | 89% |
| 6 | 2.5 g | 32 g of 5 mm balls | 500 rpm | 40 hours | 5 min rest per h | Y, for 10 min | N | 20% |
| 7 | 2.5 g | 32 g of 5 mm balls | 450 rpm | 40 hours | 5 min rest per h | Y, for 10 min | N | 19% |
| 8 | 2.5 g | 32 g of 5 mm balls | 350 rpm | 40 hours | 5 min rest per h | Y, for 10 min | N | 30% |
| 9 | 2.5 g | 40 g, 10 mm balls | 510 rpm | 15 hours | 3 min rest/5 min | Y, for 10 min | N | 33% |
| 10 | 5.2 g | 64 g of 5 mm balls | 500 rpm | 20 hours | 3 min rest/5 min | Y, for 10 min | Y, every 4 hours (5×) | 11.1% |
| 11 | 5.2 g | 64 g of 5 mm balls | 500 rpm | 20 hours | 3 min rest/5 min | Y, for 10 min | THF washed Sample 10 | 8% |
| 12 | 2 g | 32 g of 5 mm balls | 450 rpm | 20 hours | 5 min rest per h | Y, for 10 min | N | 15% |
| 13 | 2 g | 32 g of 5 mm balls | 450 rpm | 40 hours | 5 min rest per h | Y, for 10 min after each clean | Y, 20 hours | 0% |
| 14 | 2 g | 32 g of 5 mm balls | 450 rpm | 60 hours | 5 min rest per h | Y, for 10 min after each clean | Y, every 20 hours | 0% |
| 15 | 2 g | 32 g of 5 mm balls | 450 rpm | 80 hours | 5 min rest per h | Y, for 10 min after each clean | Y, every 20 hours | 0% |
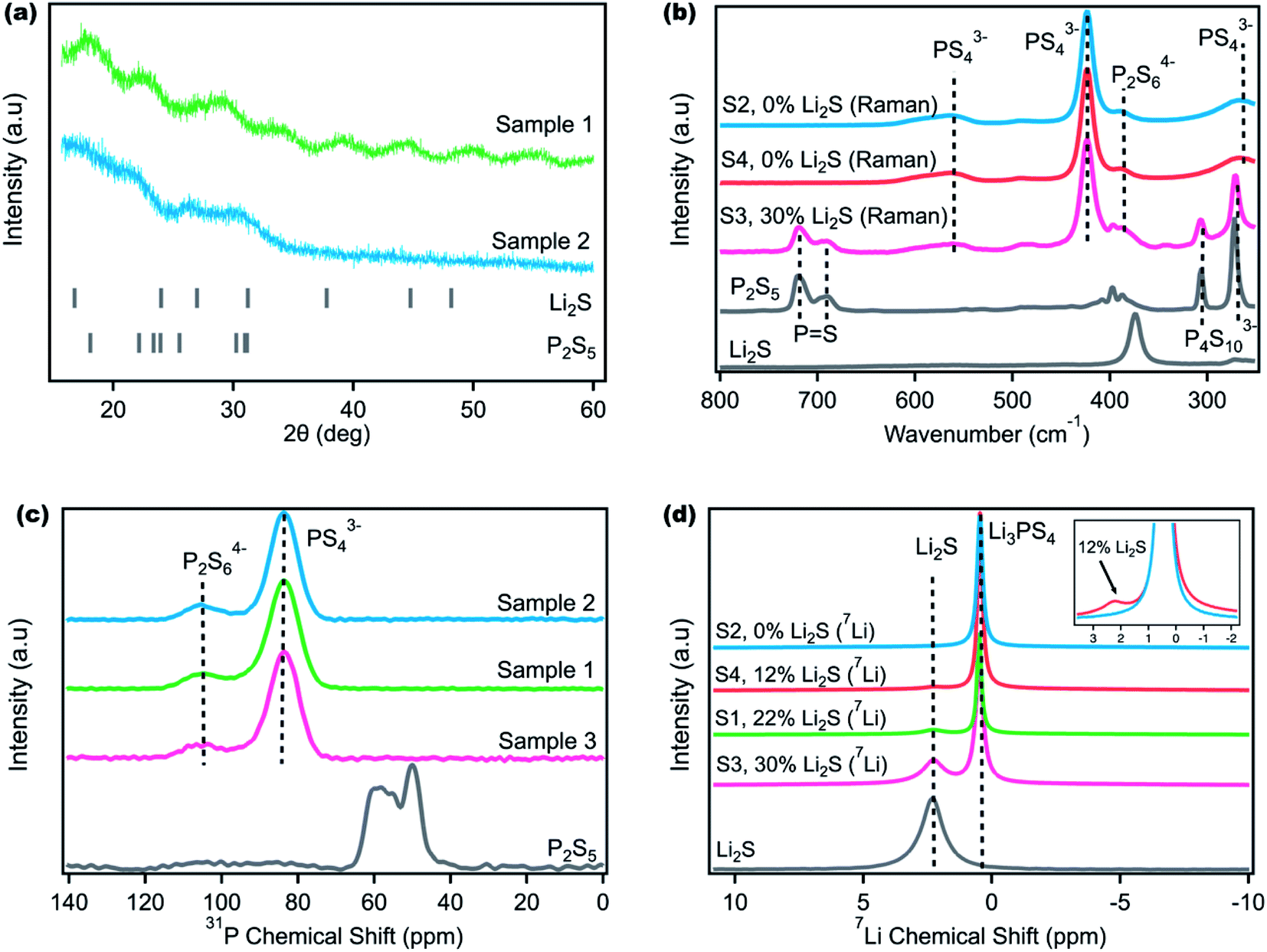 | ||
| Fig. 1 (a) X-ray diffraction (XRD) of Samples 1 and 2 with broad amorphous peaks. (b) Raman spectra of Samples 2–4 and Li2S and P2S5 synthetic precursors. The percent Li2S content listed refers to the Li2S obtained from Raman quantification. Although Samples 2 and 4 show no Li2S content by Raman, NMR data provide further insight in (d). (c) 31P solid state magic angle spinning (ssMAS) NMR spectra of P2S5 and Samples 1–3 that show the absence of any remnant P2S5 despite the Raman data in (b) and NMR data in (d). (d) 7Li ss MAS NMR spectra of Samples 1–5 and the resultant Li2S impurity concentrations determined by peak integration. The inset in (d) demonstrates the visible Li2S peak for the 12 mol% sample (Sample 4) in comparison to the 0 mol% sample (Sample 2), where this peak is clearly not observed. MAS NMR was performed with a spinning speed of 20 kHz and 7Li chemical shift referenced to LiF (−1.0 ppm) and 31P referenced to H3PO4 (0.0 ppm). S1, S2, and S3 refer to Samples 1, 2, and 3. | ||
Raman spectroscopy was used to further study and determine the LPS sample composition and purity. Additional samples were synthesized with Sample 3 as a modification of Sample 1, and Sample 4 an exact replica of Sample 2. Fig. 1b shows that the Raman spectra of Samples 2 and 4 are identical with peaks at 266, 421 and 560 cm−1 attributed to the vibrational modes of the main PS43− anion, and the smaller peak at 387 cm−1 attributed to the minor P2S64− anion.55
These spectra are consistent with reported Raman peaks for LPS.55–57 Furthermore, no additional precursor peaks from P2S5 or Li2S were observed. In contrast, Sample 3 (a modification of Sample 1) shows the presence of peaks from the P2S5 precursor. Peaks at 272 and 305 cm−1 are attributed to the P4S103− anion and peaks at 690 and 714 cm−1 attributed to P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S stretch, modes that are present in P2S5, but not in LPS.57–59 The presence of unreacted P2S5 supports the presence of unreacted Li2S as both precursors react in a stoichiometric manner. The Li2S peak overlaps with the P2S64− peak present in LPS, and it is difficult to isolate. To quantify the percent P2S5 impurity, the PS peak at 714 cm−1 and the PS43− peak at 421 cm−1 were integrated and the ratio of the two integrals was computed. The mol% Li2S content can then be obtained knowing the original stoichiometric ratio added. Although it is difficult to directly compute the Li2S content using Raman, the distinct P2S5 peak can be observed in impure LPS samples, showing that Raman is a better tool to quantify LPS product purity compared to XRD. However, as will be discussed in the following paragraph, the sensitivity of Raman is limited as it is unable to distinguish between Samples 2 and 4, despite NMR showing that Li2S is still present in Sample 4 (discussed later). The limit of Raman sensitivity has been discussed in other studies as it depends on the functional group being probed.40,55
S stretch, modes that are present in P2S5, but not in LPS.57–59 The presence of unreacted P2S5 supports the presence of unreacted Li2S as both precursors react in a stoichiometric manner. The Li2S peak overlaps with the P2S64− peak present in LPS, and it is difficult to isolate. To quantify the percent P2S5 impurity, the PS peak at 714 cm−1 and the PS43− peak at 421 cm−1 were integrated and the ratio of the two integrals was computed. The mol% Li2S content can then be obtained knowing the original stoichiometric ratio added. Although it is difficult to directly compute the Li2S content using Raman, the distinct P2S5 peak can be observed in impure LPS samples, showing that Raman is a better tool to quantify LPS product purity compared to XRD. However, as will be discussed in the following paragraph, the sensitivity of Raman is limited as it is unable to distinguish between Samples 2 and 4, despite NMR showing that Li2S is still present in Sample 4 (discussed later). The limit of Raman sensitivity has been discussed in other studies as it depends on the functional group being probed.40,55
Solid state MAS NMR provides a powerful technique to independently probe the residual P2S5 and Li2S precursors and determine their content in the final LPS product. Firstly, 31P NMR was performed as has been done in the literature.40,56,60,61Fig. 1c shows that all the samples are identical and they do not show any unreacted P2S5 peaks. The peaks at 83 ppm and 105 ppm correspond to the major PS43− and minor P2S64− anions reported for LPS, respectively.40,60 Despite the Raman data in Fig. 1b showing the presence of unreacted P2S5 in Sample 3, no P2S5 was observed in the 31P NMR spectra. Again, the color of Samples 1 and 2 varies significantly (Fig. S1†), but 31P NMR was unable to differentiate between them. Therefore, while 31P NMR can indicate LPS formation (and phase purity), its sensitivity for unreacted P2S5 appears worse than Raman and it is unable to determine the overall product purity.
7Li MAS NMR was performed to provide further information on phase and product purity (Fig. 1d). Li2S has a chemical shift of 2.3 ppm while LPS has a reported shift of 0.5 ppm that is also observed in Fig. 1d.60 Interestingly, one sample that was deemed to be ‘pure’ with Raman (Sample 4) and the sample that was difficult to decipher with XRD (Sample 1) show high amounts of unreacted Li2S. The mol percent of residual Li2S was calculated by integrating the Li2S and LPS peaks and taking the ratio of the respective integrals. Sample 3 shows the same Li2S content in both Raman and 7Li NMR, illustrating that when an impurity such as Li2S or P2S5 is visible in Raman, it can be quantified with reasonable accuracy. However, the lack of Li2S or P2S5 in Raman does not indicate purity as observed when comparing Sample 4 in Fig. 1b and d. Additional data in Fig. S3† show that 7Li NMR can detect Li2S impurities as low as 4 mol% that cannot again be seen through Raman. 7Li NMR appears sensitive enough to provide quantitative data regarding product purity for amorphous LPS in a manner that supersedes data obtained using XRD, Raman, and 31P NMR.
Influence of ball milling parameters on Li2S impurity in Li3PS4 synthesis
Multimodal characterization and especially 7Li NMR can be pivotal in determining product purity; however sample reproducibility can still be a concern. This challenge is illustrated by Samples 2 and 4 that were synthesized using the same procedure but yielded different Li2S impurity contents. Now equipped with 7Li MAS NMR, different ball milling strategies were then pursued to determine the best procedure to reproducibly obtain pure LPS. The first parameter studied was the effect of the milling speed (Fig. 2a). The total sample mass (2.5 g), mixing media (32 g of 5 mm balls), and ball mill time (40 hours) were kept constant, and the precursors were hand-milled in a mortar and pestle for approximately ten minutes prior to transferring to a 45 mL Zirconia jar.52Fig. 2a shows that as the ball milling speed is increased from 350 rpm to 450 rpm, there is a decrease in the residual Li2S content from 30 to 19 mol percent; however increasing it further to 500 rpm does not lead to any improvement.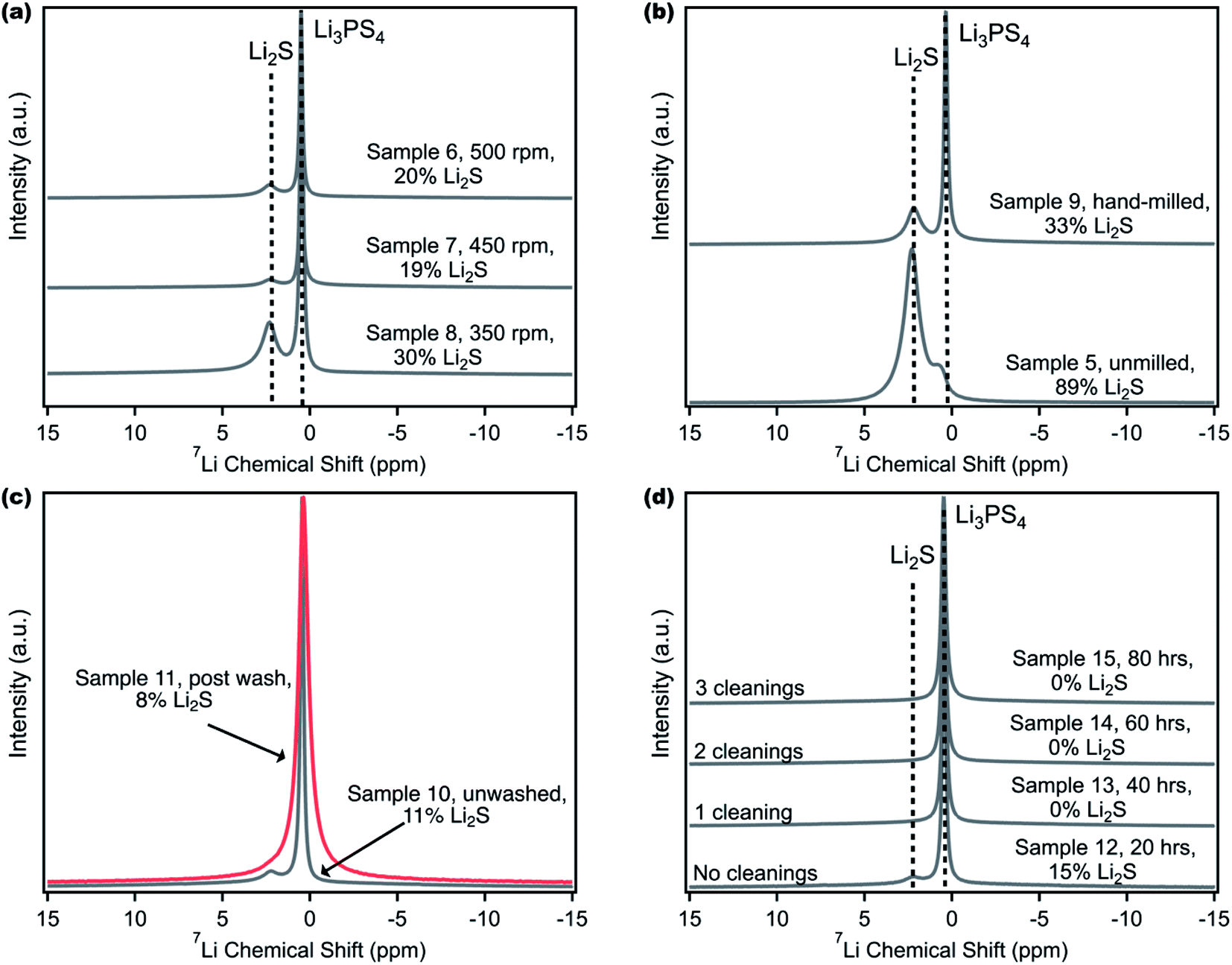 | ||
| Fig. 2 Influence of ball milling processing parameters. (a) 7Li MAS NMR spectra of Samples 6–8, ball milled at different speeds showing that the ball mill speed can decrease the residual Li2S content until a certain percentage. (b) 7Li MAS NMR spectra of Samples 5 and 9, without and with ten minutes of hand-milling prior to ball milling. (c) Normalized 7Li MAS NMR spectra of Samples 10 and 11, pre- and post-washing with tetrahydrofuran (THF). The amorphous Li3PS4 peak broadens after THF washing. (d) 7Li MAS NMR spectra of Samples 12–15, ball milled with a stop to clean the ball mill jar in 20 hour intervals. Reported residual Li2S content in all figures was obtained using 7Li NMR. | ||
Ball milling between Li2S and P2S5 leads to an increase in the temperature of the ball mill jar which facilitates the reaction between these two precursors;37,62–64 therefore, a higher ball mill speed than 350 rpm would be required for this synthesis. However, at speeds such as 500 rpm, the caking of the precursors in the ball mill jar becomes prominent, hence limiting the mixing that is required between Li2S and P2S5. Numerous research papers have used 450 rpm as the ball milling speed25,52,53 and we settled on 450 rpm, noting that it does improve LPS synthesis, but changing the ball milling speed alone does not appear sufficient to obtain pure, amorphous LPS.
The second parameter investigated was the effect of hand-milling the Li2S and P2S5 precursors in a mortar and pestle prior to ball milling (Fig. 2b). This detail is not often reported in the literature for the synthesis of LPS, and it can be difficult to standardize the procedure for hand-milling a sample. Samples 5 and 9 were synthesized according to a previously reported procedure from Balsara et al.65 and Tatsumisago et al.66 where 2.5 g of precursors were milled at 510 rpm, with eight, 10 mm ZrO2 balls (equivalent to approximately 40 g of media), for a total ball mill time of 15 hours. It was found that Sample 9, which included ten minutes of hand milling, demonstrated a nearly two-thirds reduction in residual Li2S. The effect of hand milling prior to ball milling can also be seen with Samples 17 and 18, described in Table S1† (Fig. S4†) which were ball milled at 450 rpm for 20 hours. One explanation for this dramatic reduction in impurity concentration is that hand-milling allows for increased homogeneity of the sample prior to undergoing the actual reaction of the precursors to form LPS in the ball mill jar. We must note that it is also possible that hand milling may yield some LPS product even before ball milling. Again, the hand milling parameter alone is insufficient in obtaining a pure product.
One alternative to removing Li2S impurity from LPS could involve a post-processing step. Fig. 2c displays the effect of washing an impure LPS sample with THF to remove any residual Li2S. Solvent selection is particularly important as LPS has been shown to be unstable in common polar solvents such as acetonitrile.44,55,67 Furthermore, the solvent choice can affect the LPS phase, as β-crystalline LPS is typically obtained during solution synthesis of a 3:1 molar ratio of Li2S to P2S5.41,43,54,55,63 The suspension and reaction of Li2S and P2S5 in THF have been reported to produce amorphous LPS,38 and while Li2S has limited solubility in ether solvents, LPS is not soluble in these solvents.39 Therefore, THF was chosen as the solvent to wash Sample 10, which contained 11 mol% Li2S impurity (with 7Li NMR). The sample was suspended in THF, allowed to settle, and the yellow-green supernatant was removed three times, leaving behind a pale white powder. The sample was then heated at 80 °C to remove any THF that has complexed with LPS.41,43,68 The resultant spectrum in Fig. 2c shows one broad peak at approximately 0.5 ppm. The full width at half-maximum (FWHM) of the peak in the washed sample (Sample 11) was computed to be 0.68 ppm (106 Hz), compared to the unwashed sample (Sample 10), where the FWHM was 0.22 ppm (34 Hz). After deconvolution, there was a slight reduction in the Li2S content from 11 to 8 mol%, but it was not eliminated. The three-fold increase in the FWHM was attributed to the formation of the LPS·THF complex. Additionally, the broadening of peaks in 7Li NMR may indicate an increase in the lithium chemical environments or a decrease in lithium mobility within LPS due to LPS-solvent complexation.61 To confirm the reproducibility of this potential complex, an additional sample containing 22 mol% Li2S (Sample 25, detailed in the ESI†) was washed with THF using the same procedure and 1H and 7Li spectra were taken (Fig. S5†). The 1H spectrum of Sample 25 showed several strong, broad peaks that were consistent with literature reports for LPS·THF complexation.69 To complement the 1H spectrum, the 7Li spectrum of Sample 25 also shows broadening of the peak at 0.5 ppm in comparison to the unwashed sample. Introducing a solvent wash in addition to ball milling appears counterproductive as the high temperature solvent removal step as well as possible changes to the LPS phase eliminates the benefits of ball milling and leads to a different LPS compound.
Through the numerous attempted ball milling strategies, it was observed that the caking of the precursors on the sides of the milling jar was significant and could hamper reaction completion. Samples that were hand milled and ball milled at 450 rpm still contained unreacted Li2S, demonstrating that these two parameters alone were not enough to achieve pure LPS. Recently, Kundu et al. reported a cleaning procedure in between ball milling runs for LPS synthesis.70 Although they only used XRD to determine crystalline product purity, we investigated the impact of cleaning. For this experiment, the sample was milled for a total of 80 hours, but after every 20 hours, the sample was scraped from the sides of the jar and the jar and balls were cleaned (Samples 12–15). The resultant 7Li NMR spectra (Fig. 2d) show that after 20 hours of milling, Sample 12 had a 15 mol% Li2S impurity content; however, after a total of 40 hours of milling with one break to clean the jar, the same sample (now labeled Sample 13) contained 0 mol% Li2S. As a result, samples 14 and 15 also contained 0 mol% Li2S. The removal of powder, remixing, and systematic cleaning of the ball mill jar appear vital to reproducibly obtaining pure LPS. For pure LPS, ball milling for 60 hours total, as compared to 40 hours, was the most robust and reproducible procedure (Fig. S6†). To confirm the reproducibility of the new procedure, five separate additional samples were synthesized for 60 hours total with cleaning after every 20 hours, and Fig. S7† shows no observed residual Li2S. If it is of interest to synthesize LPS with some Li2S impurity, as it could be for a solid-state lithium–sulfur battery cathode where Li2S is the active material and LPS is the ionic conductor, reproducing a specific Li2S content will be difficult because of variability in hand milling from one day to another or from one researcher to another. Although we have reported the best procedure for reproducibly synthesizing LPS, the long times (60 hours) and small amount (2 g) show significant limitations in ball milling as a technique for scaling the production of pure, amorphous LPS. Therefore, it is important to pursue scalable solution-based processes that maintain high LPS conductivity when compared to ball milled samples. Recent work by Uchimoto et al. has shown a promising path forward using acetate solvents with low polarity to synthesize solution-processed LPS with high ion conductivity.44 Regardless of the synthetic steps followed, 7Li NMR as a primary mode of characterization is paramount in determining the success of LPS synthesis.
Effect of residual Li2S on ionic conductivity and electrochemical cycling performance
The influence of impure solid state LPS electrolytes on ionic conductivity and electrochemical performance was investigated. Fig. 3 shows ionic conductivity as a function of temperature for samples with different Li2S contents. Indium foil blocking electrodes were used, and electrochemical impedance spectroscopy (EIS) was performed. As discussed previously, the Li2S content indicated in Fig. 3 was obtained using 7Li NMR. Fig. 3a shows that ionic conductivity increases as the residual Li2S content decreases, illustrating the importance of synthetic procedures. Li2S is a well-known ionic and electronic insulator.68,71 Furthermore, the lack of appropriate characterization tools to confirm product purity may help explain the disparities that have been noted for LPS conductivities.35,52,65 At 0 mol% Li2S, the measured conductivity of 3.5 × 10−4 S cm−1 at 20 °C (Fig. 3b) is consistent with reported conductivities.52,53Fig. 3b shows that the room temperature conductivity as a function of Li2S content follows a sigmoidal pattern, and interestingly the conductivity plateaus when the Li2S concentration reaches 12 mol% and below. We speculate that at a lower Li2S content, lithium ion transport is not inhibited as LPS remains the continuous phase. Additionally, the conductivity curves in Fig. 3a were fit according to the Arrhenius equation: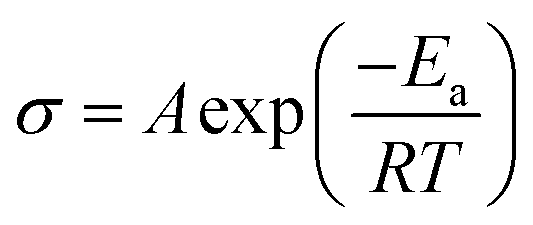 | (1) |
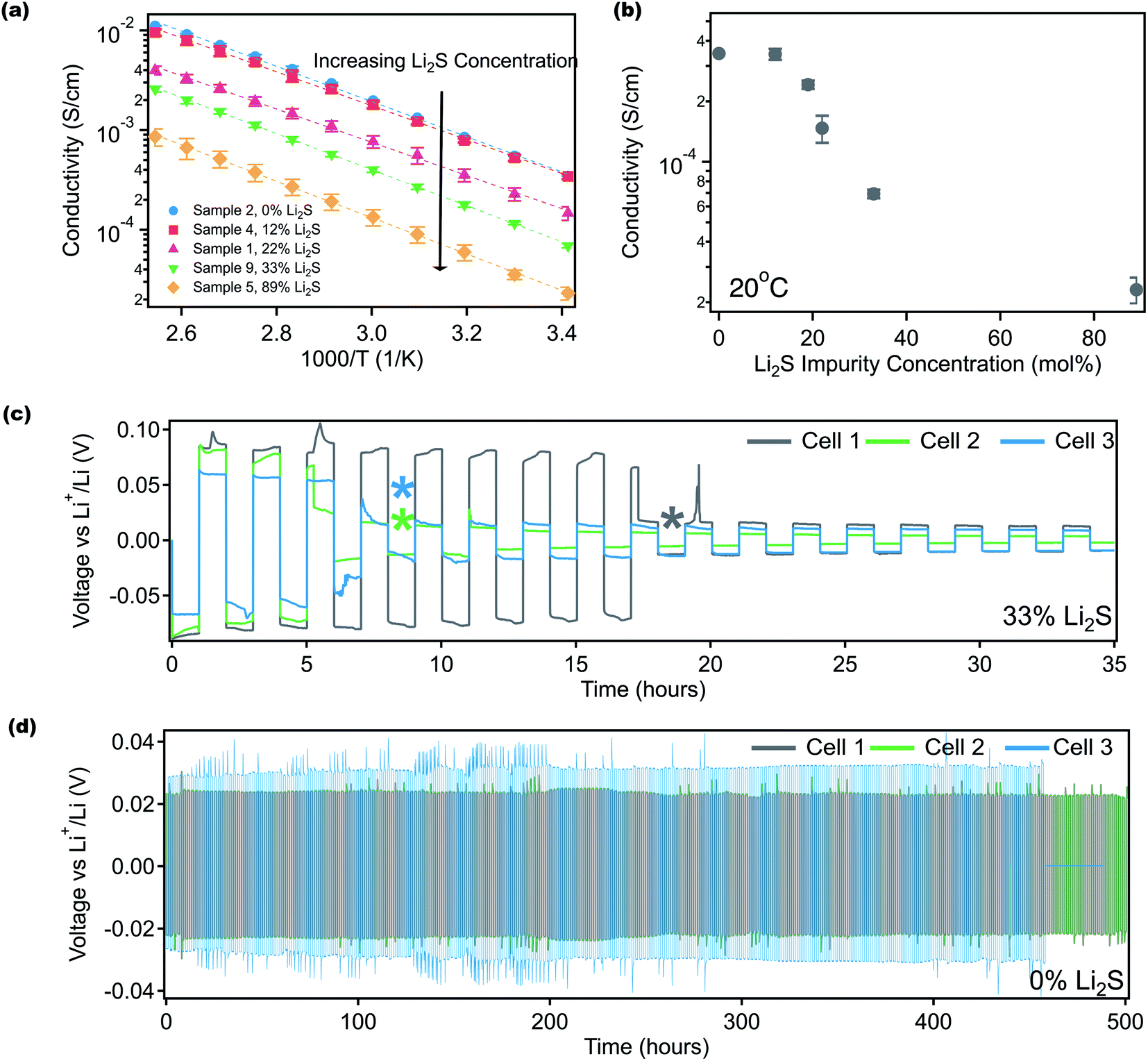 | ||
| Fig. 3 Effect of residual Li2S impurity on LPS electrochemistry. (a) Ionic conductivity as a function of temperature for samples with different concentrations of Li2S impurity. (b) Ionic conductivity as a function of Li2S impurity at 20 °C. Li/Li symmetric cell cycling for (c) Sample 9, containing 33 mol% Li2S and (d) Sample 15, containing 0 mol% Li2S. All cells were cycled at a current rate of 0.05 mA cm−2 to 0.05 mA h cm−2 after a 10 hour rest. The asterisks indicate the point at which each cell shorted. The average overpotential for 33 mol% impure cells was 76 mV and 0 mol% cells was 28 mV, and the average time-to-death for these cells was approximately 10.3 hours and 480 hours, respectively. | ||
Symmetric lithium/lithium half cells containing LPS electrolytes with either 33 mol% (Fig. 3c) or 0 mol% Li2S (Fig. 3d) were cycled at a current density of 0.05 mA cm−2 to 0.05 mA h cm−2 to investigate the impact of residual Li2S on long-term cycling performance. Electrolyte thicknesses were approximately 0.9–1.2 mm. Three replicate cells were run for each electrolyte, and Fig. 3c shows that impure LPS cells had a significantly shorter lifetime than those containing pure LPS. The average lifetime of these cells was approximately 10.3 hours (5.1 cycles) for cells containing 33 mol% Li2S while the lifetime was over 480 hours (240 cycles) for pure LPS. In addition, the overpotential for lithium deposition and stripping was 76 mV for impure LPS (33 mol% Li2S) and 28 mV for pure LPS. The overpotential for pure LPS cells is similar to the reported overpotential for lithium deposition and stripping for LPS cycled at this rate.23,41,43,72–75 Prior to Li/Li cycling, EIS was performed, and the bulk and the interfacial impedance of the cells were recorded (Fig. S9†). The average interfacial impedance (Table S2†) of the impure LPS cells was found to be about 2047 ohms, which is about 400 ohms larger than the interfacial impedance of the pure LPS cells (about 1640 ohms). The interfacial impedance for pure LPS is about 600 ohms higher than the impedance reported in the literature for this electrolyte.23,72–77 One possible explanation for the larger interfacial impedance in this study is the differences attributed to cylindrical cell measurements versus coin cell measurements. Several studies use cylindrical cells to apply constant pressure to the electrode and electrolyte during cycling and improve the interfacial impedance, a setup that is infeasible for coin cells.72–75,78
Additionally, upon contact with lithium metal, it has been reported that LPS decomposes to form Li2S in the solid electrolyte interface,62,68,79,80 which is a self-passivating interface as it is both ionically and electronically insulating. However, it appears that when significant Li2S is present in the bulk electrolyte, the electrochemical performance struggles significantly. We speculate that this trend could be due to the poor pellet quality that results from a large Li2S concentration in LPS electrolytes. The impure pellets fractured easily in comparison to the pure LPS samples and were therefore more likely to short earlier than the pure samples. These experiments demonstrate that unreacted Li2S plays a role in both measured ionic conductivity and electrochemical cycling, illustrating the importance of multimodal characterization to measure the Li2S content, and developing synthetic strategies to ensure high LPS product purity.
Conclusions
In summary, Li3PS4 sulfide-type electrolytes were synthesized according to previously reported ball milling procedures, and the resultant concentration of unreacted Li2S was characterized using XRD, Raman, and 31P and 7Li solid state NMR. We demonstrate that a suite of characterization techniques is necessary to confirm both the phase and chemical purity of the resultant amorphous LPS electrolyte. We show that 7Li solid state NMR is a powerful and reliable tool for characterizing unreacted Li2S at low concentrations, when other techniques, such as Raman, XRD, and 31P, are unable to distinguish the chemical environments. Using 7Li NMR, we investigate different ball milling synthetic parameters such as the ball milling speed and time to determine the most reproducible process for LPS synthesis. Intermittent cleaning of the ball mill jar between runs was found to be the best procedure for obtaining pure LPS. Additionally, the impact of the Li2S impurity content on ionic conductivity was explored and LPS ionic conductivity increased with decreasing residual Li2S content. However, there was a sigmoidal behavior as ionic conductivity plateaued at impurity concentrations below 12 mol%. Finally, we fabricate Li/Li half cells and show that impure samples lead to higher lithium overpotentials and faster cell death compared to pure LPS samples. In this work, we report the importance of multimodal characterization to determine both product and phase purity in LPS electrolytes, and the impact of unreacted precursors on LPS solid state cycling performance. Although this study focuses on amorphous Li3PS4, the conclusions and techniques will apply to all amorphous solid-state electrolytes especially LPS glass synthesized using Li2S and P2S5 precursors.Experimental section
Materials
Li2S was purchased from Sigma Aldrich (99.98%) and from MSE Supplies (99%). P2S5 (99%) was purchased from Sigma Aldrich. Both Li2S and P2S5 were stored in an Argon-filled glovebox (H2O and O2 <1 ppm). Ball milling balls (5 mm and 10 mm) were made of Yttria-stabilized ZrO2 (YSZ) and were purchased from MSE Supplies. Lithium metal (0.75 mm, 99.9% metals basis) and indium ingots (99.999%) were purchased from Alfa Aesar. For THF washes, anhydrous THF (99.9%, inhibitor-free) was purchased from Sigma Aldrich, and dried using 4 Å molecular sieves overnight in an Argon glovebox before use.Ball milling
A Retsch PM-100 ball mill was used for all experiments. A 45 mL ZrO2 jar (Retsch) with a screw-top clamp for sealing was used for ball milling. All sample preparation was performed in an Argon-filled glovebox (Vigor Tech, O2 and H2O <1 ppm) solely dedicated to sulfide work. The jar was placed into the ball mill and milled at a specified amount of time, clockwise. After the milling interval, the jar was allowed to rest for a set time (see Table 1 for details), and then milled again, counterclockwise. The direction of the jar rotation changed with each milling interval.Synthesis of LPS samples
Sample 1: in an Argon glovebox, Li2S and P2S5 precursors were weighed in a 3:1 mole ratio, for a total mass of 5.2 g of sample. The precursors were then transferred to a 45 mL ZrO2 jar, and 32 g of 5 mm ZrO2 milling balls were added. The sample and milling balls were mechanically stirred for about five minutes to coat the sample onto the balls. The jar was then sealed and transferred out of the glovebox and into the ball mill. The sample was then milled at a speed of 450 rpm for 65 hours, with a milling interval of a 5 minute rest every hour.
Sample 2: Li2S and P2S5 precursors were weighed in a 3:1 mole ratio, for a total mass of 5.2 g of sample. The precursors were hand-milled in a mortar and pestle for approximately 10 minutes before being transferred to a 45 mL ZrO2 jar, and 64 g of 5 mm ZrO2 milling balls were added. The sample and milling balls were mechanically stirred for about five minutes to coat the sample onto the balls and then milled at a speed of 450 rpm for 65 hours, with an interval of a 5 minute rest every hour. After 65 hours, the sample was removed from the ball miller and transferred back into the glovebox, and powder was scraped off the sides of the jar. The sample was then removed, and the jar and mixing balls were then cleaned with ethanol. After cleaning, the sample was then placed back into the jar and milled again for an additional 15 hours under the same conditions.
Sample 3: a total of 2.5 g of precursors were weighed and placed inside the milling jar along with 32 g of 5 mm ZrO2 balls. The sample and milling balls were mechanically stirred for about five minutes to coat the sample onto the balls and then milled at a speed of 450 rpm for 15 hours, continuously.
Sample 4: the procedure for Sample 2 was repeated for this sample with no changes.
Sample 5: a total of 2.5 g of precursors were weighed and placed inside the milling jar along with 8 and 10 mm ZrO2 balls. The sample and milling balls were mechanically stirred for about five minutes to coat the sample onto the balls and then milled at a speed of 510 rpm for 15 hours. The milling interval was set for a 3 minute rest for every 5 minutes of milling.
Sample 6: a total of 2.5 g of precursors were weighed and hand-milled in a mortar and pestle for approximately ten minutes. The sample was then placed inside the milling jar along with 32 g of 5 mm ZrO2 balls. The sample and milling balls were mechanically stirred for about five minutes to coat the sample onto the balls and then milled at a speed of 500 rpm for a total of 40 hours. The milling interval was set for a 5 minute rest every hour.
Sample 7: the procedure for Sample 6 was repeated except that the milling speed was set to 450 rpm.
Sample 8: the procedure for Sample 6 was repeated except that the milling speed was set to 350 rpm.
Sample 9: the procedure for Sample 5 was repeated except that the precursors were hand-milled in a mortar and pestle for ten minutes prior to transferring into the jar.
Sample 10: a total of 5.2 g of precursors were weighed and hand-milled in a mortar and pestle for approximately ten minutes. The sample was then placed inside the milling jar with 64 g of 5 mm ZrO2 balls and the sample and balls were mechanically stirred for about five minutes. The sample was then placed inside the ball miller at 500 rpm, with a milling interval of a 3 minute rest every 5 minutes. After 192 minutes (a total milling time of 2 hours), the jar was removed from the ball miller, the sample was scraped off the walls, and the jar was cleaned. The sample was then hand-milled again for another ten minutes before transferring back to the jar and milled again for another 192 minutes. This procedure was repeated 10 times for a total milling time of 20 hours.
Sample 11: Sample 10 was suspended in approximately 5 mL of anhydrous THF inside the glovebox. The sample was mechanically shaken using a vortex for about five minutes, and then left stationary so that LPS could settle at the bottom. After about an hour, the supernatant was decanted, and this washing procedure was repeated three times. After the third wash, the sample was allowed to heat on a hot plate at 80 °C overnight, and then transferred to a vacuum oven, where it was allowed to dry under vacuum at the same temperature overnight.
Sample 12: a total of 2 g of precursors were weighed and then hand-milled in a mortar and pestle for approximately ten minutes. The sample was then placed inside the milling jar with 32 g of 5 mm ZrO2 balls, and the sample and balls were mechanically stirred for about five minutes. The sample was then ball milled at a speed of 450 rpm for 20 hours. The milling interval was set to a 5 minute rest per hour.
Sample 13: Sample 12 was removed from the miller and transferred back into the glovebox after 20 hours of milling. The sample was then scraped from the sides of the jar, the jar and balls were then cleaned, and the sample was hand-milled in a mortar and pestle for about ten minutes. The sample was placed back in the jar and milled again at 450 rpm for an additional 20 hours, making the total milling time 40 hours.
Sample 14: the procedure for Sample 13 of cleaning the jar and hand-milling the resultant sample was repeated. The sample was milled again for an additional 20 hours, making the total milling time 60 hours.
Sample 15: the procedure for Sample 13 of cleaning the jar and hand-milling the resultant sample was repeated. The sample was milled again for an additional 20 hours, making the total milling time 80 hours.
Samples 16–24: these samples (described in Table S1†) were synthesized according to the same procedure as Samples 12–15. About 2 g of precursors were milled with 32 g of 5 mm ZrO2 balls for either 20, 40, or 60 hours. Samples 20–25 were exact replicates of Sample 14 to test the reproducibility of achieving pure LPS after 60 hours of milling.
XRD measurements
XRD patterns were collected on a Bruker D8 Advance X-ray diffractometer with λ = 1.54060 Å. Prior to measurements, powder samples were placed on poly(methyl methacrylate) (PMMA) sample holders and sealed under Kapton films in an Argon-filled glovebox (O2, and H2O <1.0 ppm). The samples were spun at two rotations per minute.Raman spectroscopy
Raman spectra were taken by using a HORIBA LabRAM HR Evolution Confocal Raman Microscope using a 532 nm ULF laser as the light source. The sample was prepared by sealing LPS powder in a glass chamber inside an argon filled glovebox (O2 and H2O <1 ppm). The glass chamber was assembled using glass slides and silicone isolators purchased from Grace Bio-Labs.Magic angle spinning NMR spectroscopy
All 7Li and 31P magic angle spinning NMR experiments were carried out on a Bruker Avance III wide-bore 400 MHz solid-state NMR spectrometer under a field of 9.5 Tesla. Samples were packed into a 1.9 mm zirconia rotor (Brüker) in an argon-filled glovebox and spun at 20 kHz. 7Li and 31P NMR spectra were collected corresponding to a 7Li Larmor frequency of 155.5 MHz and a 31P Larmor frequency of 162.0 MHz. For 7Li, the 90° pulse length was 0.9 μs and the recycle delay was 20 s. Fig. S10† shows the 7Li NMR comparison of 20 s and 100 s recycle delays, of which the spectra overlap well. The recycle delay of 20 s is long enough to quantify Li2S impurity. For 31P, the 90° pulse length was 8.6 μs and the recycle delay was 50 s. The 7Li chemical shift was referenced to solid LiF at −1.0 ppm. The 31P chemical shift was referenced to 85 wt% H3PO4 at 0.0 ppm.Coin cell fabrication
The samples were pressed into pellets and placed into coin cells for electrochemical measurements. Coin cell parts were purchased from Xiamen TOB New Energy Technology. All sample preparation was performed in an Argon-filled glovebox (Vigor Tech, O2 and H2O <1 ppm). Approximately 100 mg of sample powder was loaded into a pellet chamber with a 10 mm diameter (MTI Corporation, DIE10B), and a piston was placed inside the chamber. The chamber was then placed into a 15T hydraulic press (MTI Corporation, YLJ-15L) and pressed at a pressure of 100 bar for about 2 minutes to form a pellet. Prior to coin cell assembly, the thickness of the pellet was measured. Pellet thicknesses ranged from approximately 0.9–1.2 mm. Pellets had a tendency to break after removal from the chamber, which caused thickness variations. Typically, pellets would break in such a way that small chunks of material would fall off the bottom of the pellet, but not disturb the overall shape. If the pellets fractured such that they were not perfectly circular (i.e. shattered), they were not used for electrochemical experiments.Indium or lithium foil was placed inside a metal bag with a polymer coating and rolled into long strips, using the chamber of the pellet press as a rolling pin, to a thickness of approximately 0.25 mm. Then, 8 mm-diameter electrodes were cut out from the foil. Coin cells (CR 2032) were assembled in the following order: positive case, spring, stainless-steel spacer, electrode, sample pellet, electrode, and negative case. The coin cells were then crimped at a pressure of 750 kg.
Electrochemical impedance spectroscopy
Impedance measurements were taken using a BioLogic VSP-300 potentiostat with a frequency range of 7 MHz to 1 Hz. Coin cells were assembled using the following configuration: SS‖In (8 mm)‖Pellet (10 mm)‖In (8 mm). No second stainless steel spacer was used in between the second electrode and negative case. This was done to allow for additional room so the coin cell could be crimped at a high pressure (and ensure an air-tight seal) without breaking the pellet. For temperature-dependent measurements, “cooling” scans were conducted. The samples were heated to 120 °C and held at that temperature for 45 minutes. Three impedance measurements were then taken after the thermal equilibration step, and then samples were cooled, in 10-degree intervals, back to 20 °C, with a 45 minute equilibration step at each temperature. After the measurements were completed, the coin cells were then taken back into the glovebox and de-crimped, and the thickness of the pellet and indium foil was measured. The pellet thickness was recorded after subtracting the thickness of the indium foil and compared with the thickness measured prior to assembling the coin cell. If the two thicknesses did not match, the thickness after the cooling scan was used for conductivity calculations. The resistance (R) was computed by fitting the resultant Nyquist plots to an equivalent circuit model (Fig. S11†) and extracting the bulk resistance (R1 + R2). Conductivity was then calculated according to the equation σ = L/(RA), where L is the thickness of the sample, R is the extracted resistance, and A is the electrode area.Symmetric Li/Li cycling
Symmetric Li/Li cycling measurements were taken using a Neware BTS4000 battery tester. Coin cells were fabricated according to the following configuration: SS‖Li (8 mm)‖Pellet (10 mm)‖Li (8 mm). After 10 hours of resting, the coin cells were cycled from 0.05 mA cm−2 to 0.05 mA h cm−2. The cycling was performed at 20 °C, and the cutoff voltages were set to be 1 V and −2 V vs. Li/Li+.Prior to cycling, interfacial impedance measurements were taken using the BioLogic VSP-300 potentiostat using a frequency range of 7 MHz to 1 Hz at 20 °C. The Nyquist plots were fit according to the equivalent circuit model (Fig. S9†), and R3 was taken as the interfacial resistance (Table S2†).
Conflicts of interest
There are no conflicts to declare.Author contributions
P. M. performed all the inorganic synthesis and electrochemical characterization. J. Z. obtained all solid-state NMR spectra and performed subsequent deconvolution and quantification. P. Y. M carried out Raman spectroscopy. The project was conceived and the manuscript was written by P. M., J. Z., and C. V. A.Acknowledgements
The authors thank Zachary Hood at the Argonne National Lab for obtaining the XRD patterns. This work was partially supported by the University of Chicago Materials Research Science and Engineering Center (MRSEC), which is funded by the National Science Foundation under award number DMR-2011854. Raman measurements were performed at the UChicago MRSEC and solid state NMR experiments were performed at the University of Illinois at Chicago NMR facility (NIH award 1S10RR025105-01). The authors thank Daniel McElheny for help in setting up some of the solid-state NMR parameters.References
- D. Lin, Y. Liu and Y. Cui, Reviving the lithium metal anode for high-energy batteries, Nat. Nanotechnol., 2017, 12, 194–206 Search PubMed.
- C. V. Amanchukwu, et al., One-Electron Mechanism in a Gel−Polymer Electrolyte Li−O 2 Battery, Chem. Mater., 2016, 28, 7167–7177 Search PubMed.
- C. V. Amanchukwu, The Electrolyte Frontier: A Manifesto, Joule, 2020, 4, 281–285 Search PubMed.
- M. Keller, A. Varzi and S. Passerini, Hybrid electrolytes for lithium metal batteries, J. Power Sources, 2018, 392, 206–225 Search PubMed.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Transition of lithium growth mechanisms in liquid electrolytes, Energy Environ. Sci., 2016, 9, 3221–3229 Search PubMed.
- M. Keller, et al., Electrochemical performance of a solvent-free hybrid ceramic-polymer electrolyte based on Li7La3Zr2O12 in P(EO)15LiTFSI, J. Power Sources, 2017, 353, 287–297 Search PubMed.
- K. Xu, Electrolytes and Interphases in Li-Ion Batteries and Beyond, Chem. Rev., 2014, 114, 11503–11618 Search PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Advances and issues in developing salt-concentrated battery electrolytes, Nat. Energy, 2019, 4, 269–280 Search PubMed.
- S. Chen, et al., High-Efficiency Lithium Metal Batteries with Fire-Retardant Electrolytes, Joule, 2018, 2, 1548–1558 Search PubMed.
- X. Ren, et al., Localized High-Concentration Sulfone Electrolytes for High-Efficiency Lithium-Metal Batteries, Chem, 2018, 4, 1877–1892 Search PubMed.
- C. V. Amanchukwu, et al., A New Class of Ionically Conducting Fluorinated Ether Electrolytes with High Electrochemical Stability, J. Am. Chem. Soc., 2020, 142, 7393–7403 Search PubMed.
- Z. Yu, et al., Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries, Nat. Energy, 2020, 5, 526–533 Search PubMed.
- X. Cao, et al., Monolithic solid-electrolyte interphases formed in fluorinated orthoformate-based electrolytes minimize Li depletion and pulverization, Nat. Energy, 2019, 4, 796–805 Search PubMed.
- C. V. Amanchukwu, X. Kong, J. Qin, Y. Cui and Z. Bao, Nonpolar Alkanes Modify Lithium-Ion Solvation for Improved Lithium Deposition and Stripping, Adv. Energy Mater., 2019, 9, 1902116 Search PubMed.
- J. Zheng, et al., Electrolyte additive enabled fast charging and stable cycling lithium metal batteries, Nat. Energy, 2017, 2, 17012 Search PubMed.
- N. U. Day, C. C. Wamser and M. G. Walter, Porphyrin polymers and organic frameworks types of porphyrin polymers and preparative methods, Polym. Int., 2015, 64, 833–857 Search PubMed.
- Y.-C. Jung, M.-S. Park, C.-H. Doh and D.-W. Kim, Organic-inorganic hybrid solid electrolytes for solid-state lithium cells operating at room temperature, Electrochim. Acta, 2016, 218, 271–277 Search PubMed.
- D. H. S. Tan, A. Banerjee, Z. Chen and Y. S. Meng, From nanoscale interface characterization to sustainable energy storage using all-solid-state batteries, Nat. Nanotechnol., 2020, 15, 170–180 Search PubMed.
- J. Janek and W. G. Zeier, A solid future for battery development, Nat. Energy, 2016, 1, 16141 Search PubMed.
- Z. Gao, et al., Promises, Challenges, and Recent Progress of Inorganic Solid-State Electrolytes for All-Solid-State Lithium Batteries, Adv. Mater., 2018, 30, 1–27 Search PubMed.
- Y.-G. Lee, High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes, Nat. Energy, 2020, 5, 299–308 Search PubMed.
- F. P. McGrogan, et al., Compliant Yet Brittle Mechanical Behavior of Li2S–P2S5 Lithium-Ion-Conducting Solid Electrolyte, Adv. Energy Mater., 2017, 7, 1602011 Search PubMed.
- T. Yamada, et al., All Solid-State Lithium–Sulfur Battery Using a Glass-Type P 2 S 5 –Li 2 S Electrolyte: Benefits on Anode Kinetics, J. Electrochem. Soc., 2015, 162, A646–A651 Search PubMed.
- M. Tatsumisago, F. Mizuno and A. Hayashi, All-solid-state lithium secondary batteries using sulfide-based glass–ceramic electrolytes, J. Power Sources, 2006, 159, 193–199 Search PubMed.
- M. V. Reddy, C. M. Julien, A. Mauger and K. Zaghib, Sulfide and oxide inorganic solid electrolytes for all-solid-state li batteries: A review, Nanomaterials, 2020, 10, 1–80 Search PubMed.
- A. Gupta, et al., Evaluating the Effects of Temperature and Pressure on Li/PEO-LiTFSI Interfacial Stability and Kinetics, J. Electrochem. Soc., 2018, 165, 2801–2806 Search PubMed.
- J. Wolfenstine, et al., A preliminary investigation of fracture toughness of Li7La 3Zr2O12 and its comparison to other solid Li-ionconductors, Mater. Lett., 2013, 96, 117–120 Search PubMed.
- A. Gupta and J. Sakamoto, Controlling ionic transport through the PEO-LITFSi/LLZto interface, Electrochem. Soc. Interface, 2019, 28, 63–69 Search PubMed.
- Z. Ding, J. Li, J. Li and C. An, Review—Interfaces: Key Issue to Be Solved for All Solid-State Lithium Battery Technologies, J. Electrochem. Soc., 2020, 167, 070541 Search PubMed.
- Y. Zhao, et al., A new solid polymer electrolyte incorporating Li10GeP2S12 into a polyethylene oxide matrix for all-solid-state lithium batteries, J. Power Sources, 2016, 301, 47–53 Search PubMed.
- S. Wang, et al., High-conductivity free-standing Li6PS5Cl/poly(vinylidene difluoride) composite solid electrolyte membranes for lithium-ion batteries, J. Mater., 2020, 6, 70–76 Search PubMed.
- F. J. Simon, M. Hanauer, A. Henss, F. H. Richter and J. Janek, Properties of the Interphase Formed between Argyrodite-Type Li6PS5Cl and Polymer-Based PEO10:LiTFSI, ACS Appl. Mater. Interfaces, 2019, 11, 42186–42196 Search PubMed.
- T. Yersak, J. R. Salvador, R. D. Schmidt and M. Cai, Hot Pressed, Fiber-Reinforced (Li2S)70(P2S5)30 Solid-State Electrolyte Separators for Li Metal Batteries, ACS Appl. Energy Mater., 2019, 2, 3523–3531 Search PubMed.
- K. Hamabe, F. Utsuno and T. Ohkubo, Lithium conduction and the role of alkaline earth cations in Li2S–P2S5–MS (M = Ca, Sr, Ba) glasses, J. Non-Cryst. Solids, 2020, 538, 120025 Search PubMed.
- F. Mizuno, A. Hayashi, K. Tadanaga and M. Tatsumisago, New, Highly Ion-Conductive Crystals Precipitated from Li2S–P2S5 Glasses, Adv. Mater., 2005, 17, 918–921 Search PubMed.
- J. Christopher Bachman, et al., Inorganic Solid-State Electrolytes for Lithium Batteries: Mechanisms and Properties Governing Ion Conduction, Chem. Rev., 2016, 116, 140–162 Search PubMed.
- A. Hayashi, S. Hama, T. Minami and M. Tatsumisago, Formation of superionic crystals from mechanically milled Li2S-P2S5 glasses, Electrochem. Commun., 2003, 5, 111–114 Search PubMed.
- A. Matsuda, H. Muto and N. H. H. Phuc, Preparation of Li 3 PS 4 Solid Electrolyte by Liquid-Phase Shaking Using Organic Solvents with Carbonyl Group as Complex Forming Medium, J. Jpn. Soc. Powder Powder Metall., 2016, 63(11), 976–980 Search PubMed.
- L. Pan, et al., Revisiting the ionic diffusion mechanism in Li3PS4 via the joint usage of geometrical analysis and bond valence method, J. Mater., 2019, 5, 688–695 Search PubMed.
- C. Dietrich, et al., Lithium ion conductivity in Li2S-P2S5 glasses-building units and local structure evolution during the crystallization of superionic conductors Li3PS4, Li7P3S11 and Li4P2S7, J. Mater. Chem. A, 2017, 5, 18111–18119 Search PubMed.
- H. Wang, Z. D. Hood, Y. Xia and C. Liang, Fabrication of ultrathin solid electrolyte membranes of β-Li3PS4 nanoflakes by evaporation-induced self-assembly for all-solid-state batteries, J. Mater. Chem. A, 2016, 4, 8091–8096 Search PubMed.
- D. H. S. Tan, A. Banerjee, Z. Deng, E. A Wu, H. Nguyen, J. M. Doux, X. Wang, J. Cheng, S. P. Ong, Y. S. Meng and Z. Chen, Enabling Thin and Flexible Solid-State Composite Electrolytes by the Scalable Solution Process, ACS Appl. Energy Mater, 2019, 2(9), 6542–6550 Search PubMed.
- E. C. Self, et al., Solvent-mediated synthesis of amorphous Li3PS4/polyethylene oxide composite solid electrolytes with high Li+ conductivity, Chem. Mater., 2020, 32, 8789–8797 Search PubMed.
- K. Yamamoto, et al., High Ionic Conductivity of Liquid-Phase-Synthesized Li 3 PS 4 Solid Electrolyte, Comparable to That Obtained via Ball Milling, ACS Appl. Energy Mater., 2021, 4(3), 2275–2281 Search PubMed.
- A. Hayashi, S. Hama, H. Morimoto, M. Tatsumisago and T. Minami, Preparation of Li2S–P2S5 Amorphous Solid Electrolytes by Mechanical Milling, J. Am. Ceram. Soc., 2001, 84, 477–479 Search PubMed.
- H. Muramatsu, A. Hayashi, T. Ohtomo, S. Hama and M. Tatsumisago, Structural change of Li2S-P2S5 sulfide solid electrolytes in the atmosphere, Solid State Ionics, 2011, 182, 116–119 Search PubMed.
- K. Minami, A. Hayashi and M. Tatsumisago, Characterization of Solid Electrolytes Prepared from Li2S–P2S5 Glass and Ionic Liquids, J. Electrochem. Soc., 2010, 157, A1296–A1301 Search PubMed.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Sulfide solid electrolyte with favorable mechanical property for all-solid-state lithium battery, Sci. Rep., 2013, 3, 2261 Search PubMed.
- M. Tatsumisago, M. Nagao and A. Hayashi, Recent development of sulfide solid electrolytes and interfacial modification for all-solid-state rechargeable lithium batteries, J. Asian Ceram. Soc., 2013, 1, 17–25 Search PubMed.
- E. Kim, et al., Materials Synthesis Insights from Scientific Literature via Text Extraction and Machine Learning, Chem. Mater., 2017, 29, 9436–9444 Search PubMed.
- E. A. Olivetti, et al., Data-driven materials research enabled by natural language processing and information extraction, Appl. Phys. Rev., 2020, 7, 041317 Search PubMed.
- M. B. Dixit, et al., In Situ Investigation of Chemomechanical Effects in Thiophosphate Solid Electrolytes, Matter, 2020, 3, 1–22 Search PubMed.
- M. B. Dixit, et al., Nanoscale Mapping of Extrinsic Interfaces in Hybrid Solid Electrolytes, Joule, 2020, 4, 207–221 Search PubMed.
- Z. D. Hood, et al., The ‘filler effect’: A study of solid oxide fillers with β-Li3PS4 for lithium conducting electrolytes, Solid State Ionics, 2015, 283, 75–80 Search PubMed.
- M. Ghidiu, J. Ruhl, S. P. Culver and W. G. Zeier, Solution-based synthesis of lithium thiophosphate superionic conductors for solid-state batteries: A chemistry perspective, J. Mater. Chem. A, 2019, 7, 17735–17753 Search PubMed.
- H. Stöffler, et al., Amorphous versus Crystalline Li3PS4: Local Structural Changes during Synthesis and Li Ion Mobility, J. Phys. Chem. C, 2019, 123, 10280–10290 Search PubMed.
- B. Cherry, J. W. Zwanziger and B. G. Aitken, The structure of GeS2-P2S5 glasses, J. Phys. Chem. B, 2002, 106, 11093–11101 Search PubMed.
- S. S. Berbano, I. Seo, C. M. Bischoff, K. E. Schuller and S. W. Martin, Formation and structure of Na2S + P2S5 amorphous materials prepared by melt-quenching and mechanical milling, J. Non-Cryst. Solids, 2012, 358, 93–98 Search PubMed.
- J. O. Jensen and D. Zeroka, Theoretical studies of the infrared and Raman spectra of P 4 S 10, J. Mol. Struct., 1999, 487, 267–274 Search PubMed.
- H. Stöffler, et al., Li+-Ion Dynamics in β-Li3PS4 Observed by NMR: Local Hopping and Long-Range Transport, J. Phys. Chem. C, 2018, 122, 15954–15965 Search PubMed.
- K. Hayamizu and Y. Aihara, Lithium ion diffusion in solid electrolyte (Li2S) 7(P2S5)3 measured by pulsed-gradient spin-echo 7Li NMR spectroscopy, Solid State Ionics, 2013, 238, 7–14 Search PubMed.
- J. Lau, et al., Sulfide Solid Electrolytes for Lithium Battery Applications, Adv. Energy Mater., 2018, 8, 1800933 Search PubMed.
- Q. Zhang, et al., Sulfide-Based Solid-State Electrolytes: Synthesis, Stability, and Potential for All-Solid-State Batteries, Adv. Mater., 2019, 31, 1901131 Search PubMed.
- A. Miura, et al., Liquid-phase syntheses of sulfide electrolytes for all-solid-state lithium battery, Nat. Rev. Chem., 2019, 3, 189–198 Search PubMed.
- I. Villaluenga, et al., Compliant glass–polymer hybrid single ion-conducting electrolytes for lithium batteries, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 52–57 Search PubMed.
- A. Hayashi, T. Harayama, F. Mizuno and M. Tatsumisago, Mechanochemical synthesis of hybrid electrolytes from the Li 2 S-P 2 S 5 glasses and polyethers, J. Power Sources, 2006, 163, 289–293 Search PubMed.
- D. D. S. Tan, et al., Enabling Thin and Flexible Solid-State Composite Electrolytes by the Scalable Solution Process, ACS Appl. Energy Mater., 2019, 2, 6542–6550 Search PubMed.
- H. Jiang, et al., Li2S–Li3PS4 (LPS) Composite Synthesized by Liquid-Phase Shaking for All-Solid-State Lithium–Sulfur Batteries with High Performance, Energy Technol., 2020, 8, 2000023 Search PubMed.
- D. J. Morales and S. Greenbaum, NMR investigations of crystalline and glassy solid electrolytes for lithium batteries: A brief review, Int. J. Mol. Sci., 2020, 21, 3402 Search PubMed.
- C. Hänsel, P. V. Kumar and D. Kundu, Stack Pressure Effect in Li3PS4 and Na3PS4 Based Alkali Metal Solid-State Cells: The Dramatic Implication of Interlayer Growth, Chem. Mater., 2020, 32, 10501–10510 Search PubMed.
- F. Han, et al., High electronic conductivity as the origin of lithium dendrite formation within solid electrolytes, Nat. Energy, 2019, 4, 187–196 Search PubMed.
- J. Liang, et al., In Situ Li3PS4 Solid-State Electrolyte Protection Layers for Superior Long-Life and High-Rate Lithium-Metal Anodes, Adv. Mater., 2018, 30, 1804684 Search PubMed.
- A. Kato, et al., High-Temperature Performance of All-Solid-State Lithium-Metal Batteries Having Li/Li 3 PS 4 Interfaces Modified with Au Thin Films, J. Electrochem. Soc., 2018, 165, A1950–A1954 Search PubMed.
- X. Xu, et al., Li7P3S11/poly(ethylene oxide) hybrid solid electrolytes with excellent interfacial compatibility for all-solid-state batteries, J. Power Sources, 2018, 400, 212–217 Search PubMed.
- L. Sang, et al., Understanding the Effect of Interlayers at the Thiophosphate Solid Electrolyte/Lithium Interface for All-Solid-State Li Batteries, Chem. Mater., 2018, 30, 8747–8756 Search PubMed.
- Y. Seino, et al., Analysis of the structure and degree of crystallisation of 70Li2S–30P2S5 glass ceramic, J. Mater. Chem. A, 2015, 3, 2756–2761 Search PubMed.
- R. Garcia-Mendez, J. G. Smith, J. C. Neuefeind, D. J. Siegel and J. Sakamoto, Correlating Macro and Atomic Structure with Elastic Properties and Ionic Transport of Glassy Li2S-P2S5 (LPS) Solid Electrolyte for Solid-State Li Metal Batteries, Adv. Energy Mater., 2020, 10, 2000335 Search PubMed.
- A. Gupta, et al., Evaluating the Effects of Temperature and Pressure on Li/PEO-LiTFSI Interfacial Stability and Kinetics, J. Electrochem. Soc., 2018, 165, A2801–A2806 Search PubMed.
- F. Han, et al., High-performance all-solid-state lithium-sulfur battery enabled by a mixed-conductive Li2S nanocomposite, Nano Lett., 2016, 16, 4521–4527 Search PubMed.
- T. Hakari, M. Nagao, A. Hayashi and M. Tatsumisago, All-solid-state lithium batteries with Li3PS4 glass as active material, J. Power Sources, 2015, 293, 721–725 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02754a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |