
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-redox assisted nickel catalysis toward stereoselective synthesis of (n+1)-membered cycloalkanes from 1,n-diols with methyl ketones†
Amreen K.
Bains
a,
Abhishek
Kundu
a,
Debabrata
Maiti
 *b and
Debashis
Adhikari
*a
*b and
Debashis
Adhikari
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER)-Mohali, SAS Nagar, Punjab-140306, India. E-mail: amreenbains06@gmail.com
bDepartment of Chemistry, Indian Institute of Technology, Bombay, Powai, Mumbai-400076, India
First published on 5th October 2021
Abstract
A well-defined, bench-stable nickel catalyst is presented here, that can facilitate double alkylation of a methyl ketone to realize a wide variety of cycloalkanes. The performance of the catalyst depends on the ligand redox process comprising an azo-hydrazo couple. The source of the bis electrophile in this double alkylation is a 1,n-diol, so that (n+1)-membered cycloalkanes can be furnished in a stereoselective manner. The reaction follows a cascade of dehydrogenation/hydrogenation reactions and adopts a borrowing hydrogen (BH) method. A thorough mechanistic analysis including the interception of key radical intermediates and DFT calculations supports the ligand radical-mediated dehydrogenation and hydrogenation reactions, which is quite rare in BH chemistry. In particular, this radical-promoted hydrogenation is distinctly different from conventional hydrogenations involving a metal hydride and complementary to the ubiquitous two-electron driven dehydrogenation/hydrogenation reactions.
Introduction
Multisubstituted cycloalkanes including cyclopentane, cyclohexane and cycloheptane rings are found in a wide variety of natural products, pharmaceutical agents and materials.1–5 The earlier approaches towards their synthesis included Diels–Alder cycloaddition followed by reduction,6 hydrogenation of aromatic six-membered rings,7–10 dimerization of two constrained rings,11 the Michael reaction of an enolizable substrate,12 or cyclization of dihalides via Wurtz synthesis.13 The major drawback of these methods is their dependence on the availability of tailor-made precursors, which often requires multistep and challenging synthetic efforts. Furthermore, many of these methods exhibit poor regio- or stereoselectivity and generate copious amounts of hazardous waste. To this end, developing a sustainable, one-step process to furnish these high-value cycloalkane rings from abundantly available building blocks poses a desired, yet challenging task. In this regard, a (n+1) annulation strategy might be very effective where a C-1 building block would appropriately react with a C-n bis-electrophile. The in situ generation of an electrophilic carbonyl species from readily available alcohols has been widely practiced in borrowing hydrogen (BH)-based methods.14–23 Hypothetically, generation of bis-electrophiles from 1,n-diols and their concomitant cyclization to a carbon center should lead to (n+1)-membered cycloalkanes following BH techniques.Catalysis steered by redox active ligands with 3d metals has evolved as an effective strategy as a potential surrogate to precious metal catalysis.24–26 The incorporation of redox responsive ligand motifs can govern multielectron/multiproton processes that are capable of emulating the chemistry which is often done with less abundant, expensive and toxic 4d or 5d transition metals.27–29 Donohoe's group showcased the preparation of cyclohexane by annulation of pentamethyl acetophenone and 1,5-pentanediols employing an iridium catalyst, [Cp*IrCl2]2.30,31 We demonstrate in this report that a nickel catalyst, which is heavily assisted by the 2e−/2H+ process of an azo/hydrazo redox-couple can facilitate such annulation reactions efficiently. The generality of the reaction has been proved by synthesizing a large number of cycloalkanes involving 5-, 6-, and 7-membered rings.
The mechanistic investigation revealed both dehydrogenation and hydrogenation reactions to be mediated by a ligand-centered radical, which is distinctly different from two electron-promoted analogous reactions involving metal hydrides.
Results and discussion
We have recently designed a nickel catalyst 1 (Scheme 1), comprising an azophenolate ligand, which can successfully dehydrogenate alcohols, facilitate alkylation of amines and form multiple heterocycles.32–35 Discovering the capability of the azo moiety to reversibly store the extracted hydrogen atom from an alcohol, we wondered whether this molecule can catalyze a cascade (de)hydrogenation cycle to synthesize (n+1)-membered cycloalkanes from 1,n-diols. Notably, such a cascade of (de)hydrogenation reactions en route to cycloalkane formation is challenging and only two base metal catalysts comprising manganese and iron are known.36–38 Nickel catalysts have so far been successful in monoalkylation of ketones or secondary alcohols but cascade (de)hydrogenations to form annulated products by using nickel are unknown.39,40 Gratifyingly, 1,5-pentane diol can dialkylate pentamethyl acetophenone to afford substituted cyclohexanes in very high yield (87%) when the reaction was conducted at 135 °C. A series of optimization studies revealed toluene as the most suitable solvent for this protocol and 2 equiv. of KOtBu was found to be crucial for the reaction. Other bases such as KOH, NaOH, and K2CO3 were ineffective in steering this alkylation procedure. A catalyst loading of 7 mol%, reaction time of 24 h and temperature of 135 °C turned out to be optimum for this reaction (see Tables S1–S4 in the ESI† for the optimization). Further control experiments indicated that both the nickel catalyst and the base were essential for this reaction.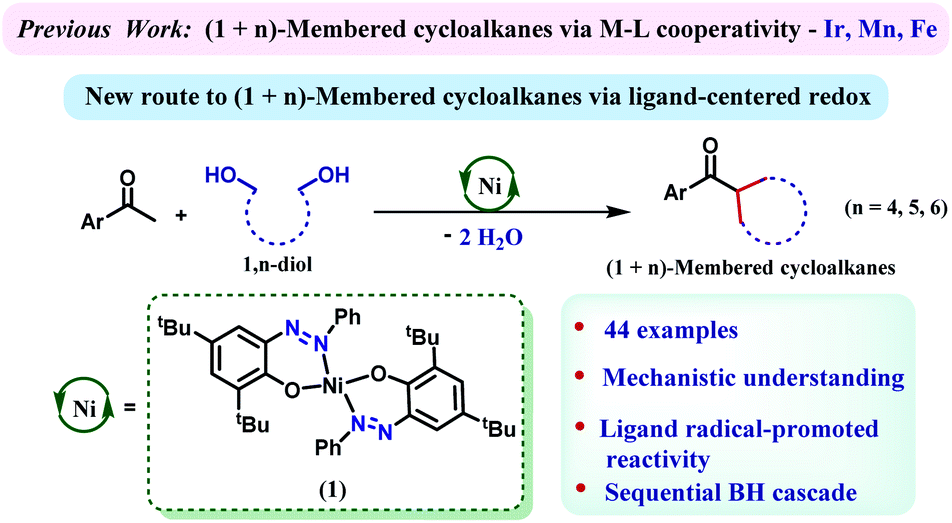 | ||
| Scheme 1 Ni-catalyzed cycloalkane formation via ligand-centric redox. | ||
Importantly, at the end of the reaction, we did not observe any precipitation, and a test of homogeneity in the presence of excess mercury proved the catalysis to be homogeneous in nature (Section 5.6, ESI†).
Notably, the steric protection at the carbonyl carbon of acetophenone with the use of a 2,6-disubstituted aryl group is required for the reaction. Previous work of Donohoe elegantly established that the steric encumbrance at the α,α′-positions of the aryl ring in an aryl ketone is crucial for a successful reaction.41,42
The lack of this protection triggers many unwanted condensation reactions, thwarting the successful cycloalkane formation. Notably, such protection can be easily removed by a retro-Friedel–Crafts reaction, and further derivatization of the ketone functionality can be realized.30,43 The bench-stability of our nickel catalyst renders the process operationally very simple and loading of all the reaction components under air is possible. A quick purging with nitrogen to prohibit aerobic oxidation of the hydrogen stored in the form of a hydrazo motif ensures the redelivery of the hydrogen to follow BH reactions.
Having the optimized reaction conditions in hand, we focused on understanding the scope of the reaction. Initially we chose 1,5-pentanediols as the alcohol substrate, and reacted with a variety of sterically protected ketones to afford the desired products in good to excellent yields.
With unsubstituted 1,5-diols, pentamethyl-, 1,3,5-triethyl- and mesityl acetophenones afforded the corresponding cyclohexane products (4aa–4ac) in 77–87% yields. Then the diol partner was switched to 2,2-dimethyl 1,5-pentanediol. Pentamethyl acetophenone reacted smoothly to offer the product 4ad in 85% yield. Very similar yield (86%) of the product 4ag was also obtained with mesityl acetophenone as the ketone substrate.
High yields of cyclohexane products (4ae and 4af) were also isolated with 1,2,4,5-tetramethyl- and 1,3,5-triethyl acetophenone substrates. Similarly, geminal methyl groups at the 3,3-position of the 1,5-diol did not hamper the reaction and afforded the products (4ah–4aj) in very good yield (71–74%) with diverse sterically hindered ketones (Table 1). Furthermore, a 1,5-diol was selected where 3,3-pentamethylene substituents were present. Such a diol resulted in a variety of spirocyclic molecules (4ak–4an) with a variety of acetophenones in 71–83% yields. Interestingly, the 1,5-diol derived from (+)-camphoric acid underwent the dialkylation reaction smoothly to give the bicyclic product 4ao in 61% yield with a high diastereomeric ratio (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). Importantly, when 3-methyl-1,5-pentanediol was reacted with pentamethyl acetophenone, the cyclohexane product 4ap was isolated in 75% yield as an exclusive diastereomer. Analogously, 3-(p-tolyl)-1,5-pentanediol was reacted with pentamethylaryl and mesityl ketones to afford the respective cyclohexane products 4aq–4ar in 69–74% yields, as a single diastereomer. A similar level of diastereocontrol was observed in the products 4as–4at when 3-(p-methoxyphenyl)-1,5-pentanediol was used. Finally, 3-(m-bromophenyl)-1,5-pentanediol was cyclized to afford the respective cyclohexane products 4au–4aw maintaining high yields and exclusive diastereoselectivity.
:5). Importantly, when 3-methyl-1,5-pentanediol was reacted with pentamethyl acetophenone, the cyclohexane product 4ap was isolated in 75% yield as an exclusive diastereomer. Analogously, 3-(p-tolyl)-1,5-pentanediol was reacted with pentamethylaryl and mesityl ketones to afford the respective cyclohexane products 4aq–4ar in 69–74% yields, as a single diastereomer. A similar level of diastereocontrol was observed in the products 4as–4at when 3-(p-methoxyphenyl)-1,5-pentanediol was used. Finally, 3-(m-bromophenyl)-1,5-pentanediol was cyclized to afford the respective cyclohexane products 4au–4aw maintaining high yields and exclusive diastereoselectivity.
| a Reaction conditions: 1 (7 mol%, with respect to 2a), 2a (0.5 mmol), substituted-1,5-pentanediol (1.5 mmol), KOtBu (1 mmol), toluene (5 mL), 135 °C, 24 h (isolated yield). |
|---|
|
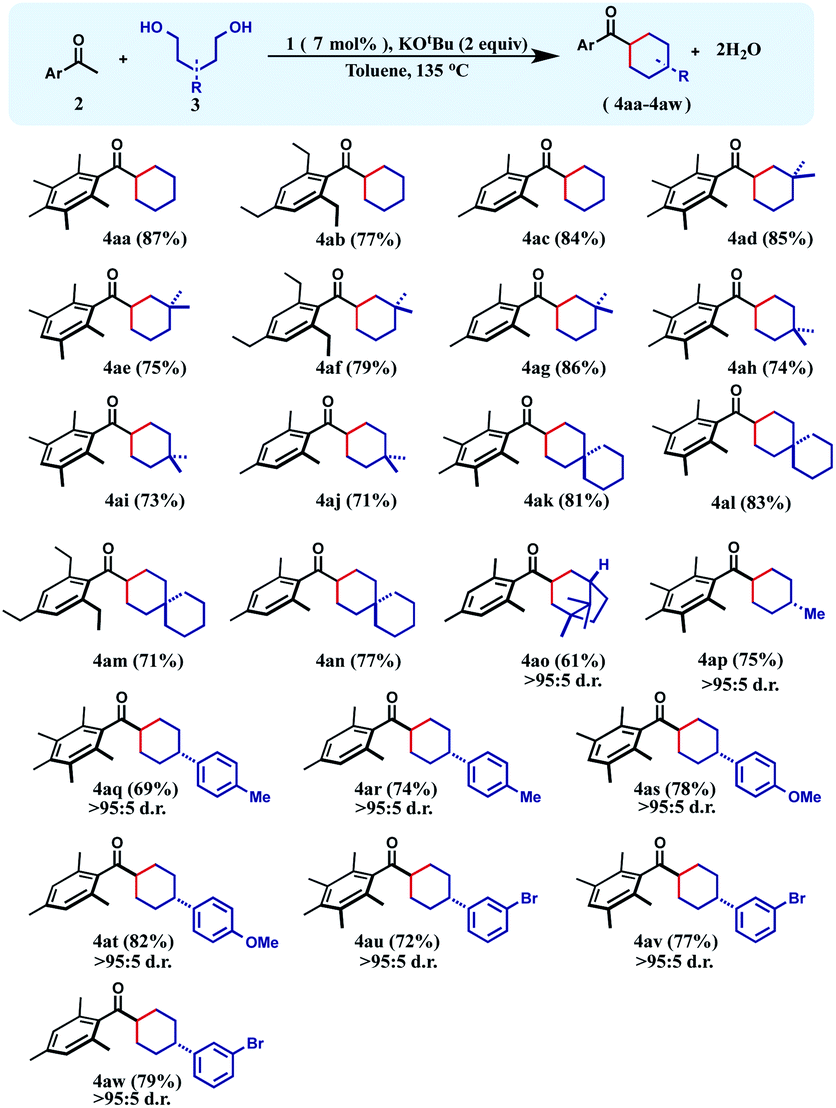
The successful preparation of a series of cyclohexane products inspired us to test the reaction over 1,4-diols so that cyclopentanes can also be synthesized in a similar manner. In this direction, when unsubstituted 1,4-butanediol was chosen as the substrate, cyclopentane products (4ba–4bc) were isolated in moderate yields (54–60%) during the reaction with a variety of differently substituted acetophenones. We next turned our focus to the construction of more complex cyclopentanes from substituted diols. Accordingly, when 2-methyl-1,4-pentanediol was reacted with a group of ketones, substituted cyclopentanes (4bd–4bh) were obtained in moderate to high yields with very high cis-diastereoselectivity (see the ESI†). Similarly, 2,5-hexanediol reacted smoothly with a variety of acetophenones affording the substituted cyclopentyl products (4bi–4bl) in good yields along with high diastereoselectivity (Table 2).
| a Reaction conditions: 1 (7 mol%, with respect to 2a), 2a (0.5 mmol), substituted-1,4-butanediol (1.5 mmol), KOtBu (1 mmol), toluene (5 mL), 135 °C, 24 h (isolated yield). |
|---|
|
After successfully synthesizing substituted cyclopentanes, we planned to extend the scope of the protocol towards preparing cycloheptanes.
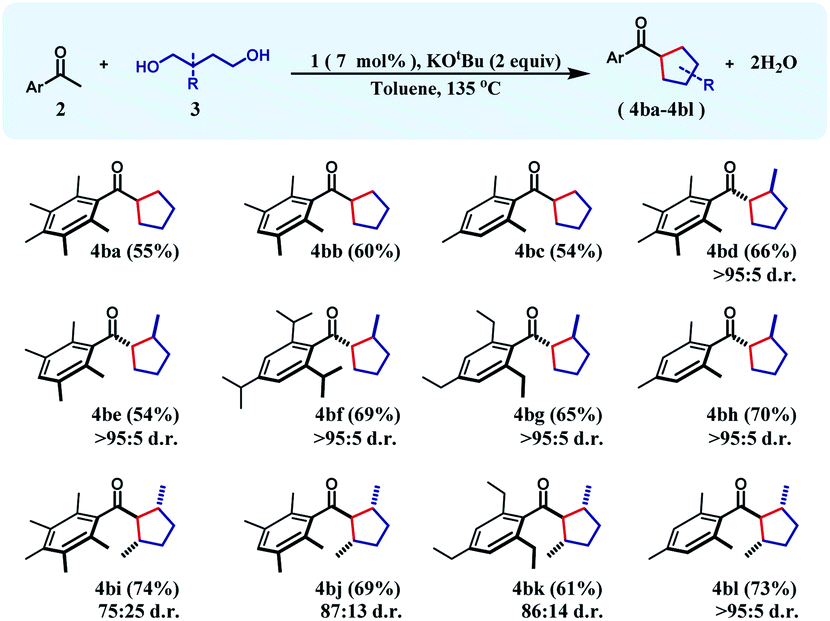
To achieve this 1,6-hexanediol was chosen as the starting alkylating substrate and annulation reactions were conducted under identical reaction conditions. With an array of acetophenones, 1,6-hexanediol reacts smoothly to give substituted cycloheptanes (4ca–4cd) in 59–75% yields (Table 3). Similar reactions of 3-methyl-1,6-hexane diol with three different acetophenones afforded the respective cycloheptane products (4ce–4cg) in good yields (60–69%) albeit with poor diastereoselectivity.
| a Reaction conditions: 1 (7 mol%, with respect to 2a), 2a (0.5 mmol), substituted-1,6-hexanediol (1.5 mmol), KOtBu (1 mmol), toluene (5 mL), 135 °C, 24 h (isolated yield). |
|---|
|
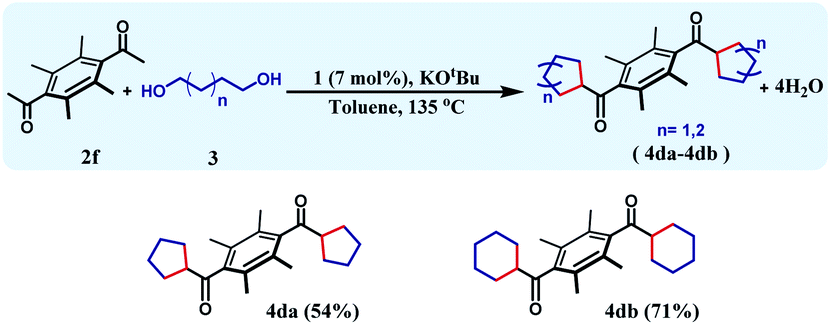
The reaction can be further extended to a set of substrates, where bis-cycloalkanes can be synthesized. Along this direction, bis(ethane-1-one), 2f was chosen as the diketone substrate, and cycloalkanes were formed with 1,4-butanediol and 1,5-pentanediol (Table 4). Under the identical reaction conditions, the first reaction afforded 4da in 54% yield, whilst the bis cyclohexane containing product 4db was formed in 71% yield. The formation of three different cycloalkane (5-, 6-, and 7-membered rings) in good yields and high diastereoselectivity ascertains the general efficiency and versatility of this synthetic methodology.
| a Reaction conditions: 1 (7 mol%, with respect to 2f), 2f (0.25 mmol), 1,4-butanediol/1,5-pentanediol (1.5 mmol), KOtBu (1 mmol), toluene (5 mL), 135 °C, 24 h (isolated yield). |
|---|
|
It is intuitive to understand that there are two consecutive dehydrogenation/hydrogenation cycles starting from a diol substrate that can lead to the desired product. To illustrate further, in the first cycle, a diol is oxidized and one end of the resulting carbonyl engages in aldol condensation to produce an enone, 5. A selective hydrogenation reduces the olefin of the enone 5 (Scheme 2). During the dehydrogenation of an alcohol, the azo-arm of the ligand drives the process by a radical mechanism and stores the extracted hydrogen in the form of hydrazo.32
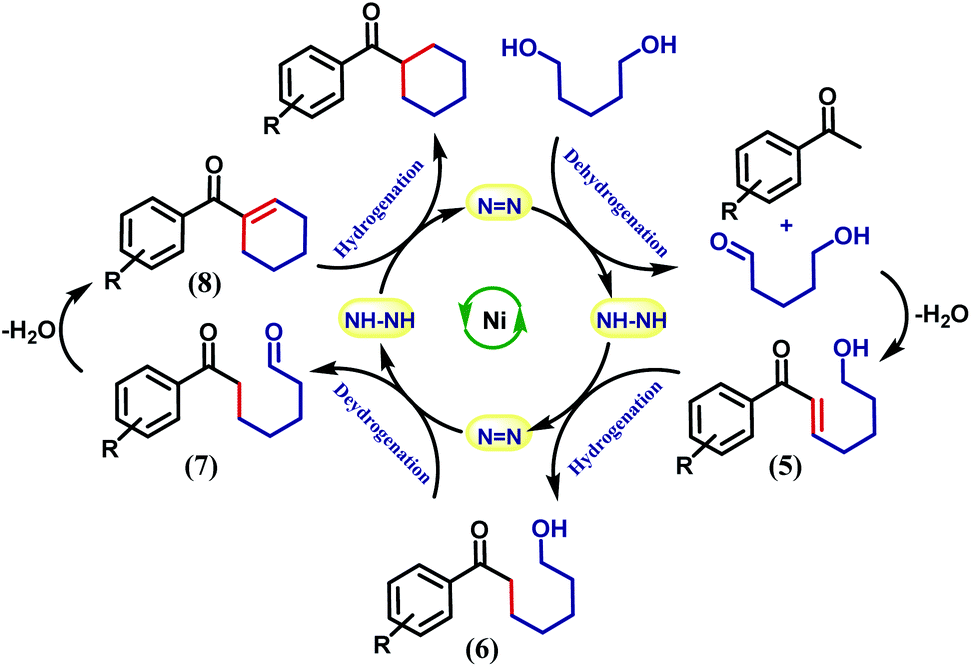 | ||
| Scheme 2 Cascade dehydrogenation and hydrogenations required for cycloalkane formation. | ||
The selective hydrogenation of the olefinic bond in the in situ-generated enone will lead to the ketone 6, where one of the ketone arms is an aliphatic alcohol. As a model enone, 5a was synthesized separately and hydrogenated under identical catalytic conditions using 1,5-pentanediol resulting in the formation of the hydrogenated products in 74% yield (Scheme 3a). The details of the ligand-promoted redox pathway to drive the hydrogenation will be elucidated below. The formation of 6 completes the first cycle following the BH method. During the intramolecular cyclization of an alcohol in 5, we did not observe the formation of any by-product. Perhaps, this fact indicates much faster hydrogenation of the enone compared to the cyclization process.
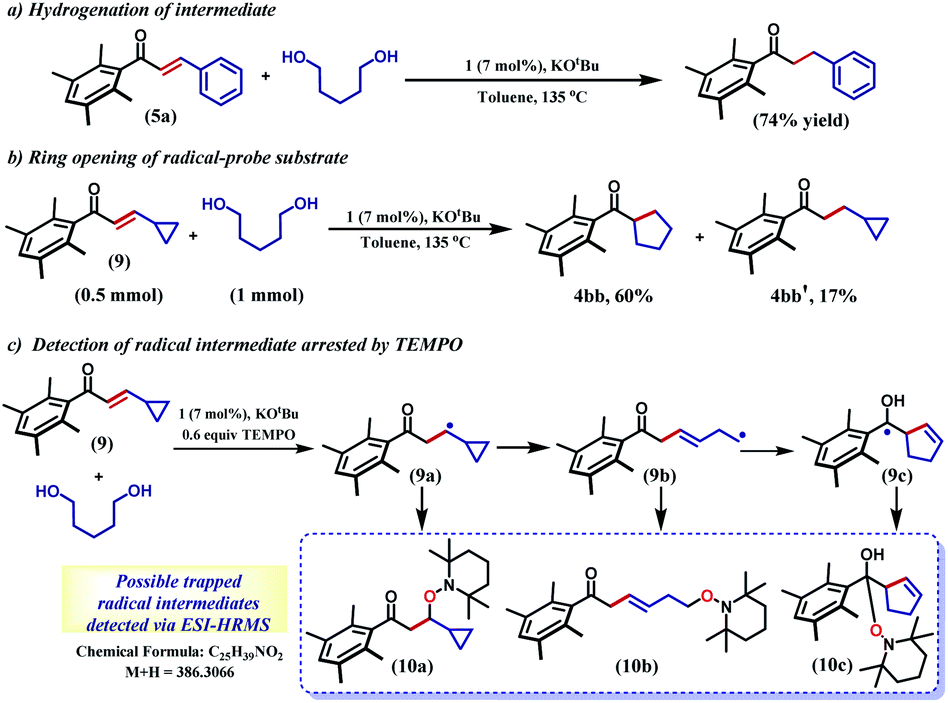 | ||
| Scheme 3 Control reactions and detection of radical intermediates. | ||
In the second cycle, the same sequence of steps is repeated. The ketonic alcohol 6 becomes further oxidized to 7, after which it undergoes an intramolecular aldol condensation followed by dehydration to result in the formation of a ketone with cycloalkene, the cyclic enone 8. This putative cycloalkene 8 has been isolated from the reaction mixture, owing to its presence to incomplete hydrogenation. We assume that the double C-alkylation thus occurs in a step-wise fashion via a one-pot reaction to overcome the problem of enolate-based consecutive alkylation processes. Final hydrogenation selectively reduces the olefinic bond of the cyclic enone, 8 to yield the desired cycloalkane product. Indeed, hydrogenation of an isolated cyclic enone using our catalyst afforded 78% yield of the desired cycloalkane product (see Section 5.1, ESI†). The catalyst is thus efficient in executing cascade hydrogenation and dehydrogenation cycles to accomplish this challenging transformation.
The dehydrogenation mechanism of an alcohol by using our nickel catalyst has been described elaborately in our previous work (see Fig. S4, ESI† for the dehydrogenation catalytic cycle). To reiterate this, the azo backbone is monoreduced by KOtBu, which leads to a crucial hydrogen atom transfer (HAT) step from a bound alcohol to the nickel center. Such a radical mediated process generates the ketyl radical, which upon quick rearrangement liberates the oxidized carbonyl product and leaves the electron in the second azo arm of the nickel catalyst. To delve into the details of the hydrogenation mechanism, we have performed a systematic study with a series of control reactions, and the conclusions are further corroborated by theoretical calculations through DFT methods. The dehydrogenation of a substrate alcohol will lead to the formation of II (Fig. 1), where one of the azo arms of the catalyst has been converted to hydrazo, whilst the second azo arm remains mono reduced. In the present context, we attempt to investigate the details of olefin hydrogenation of the enone 5 and show that hydrogenated product 6 forms by a radical mechanism.
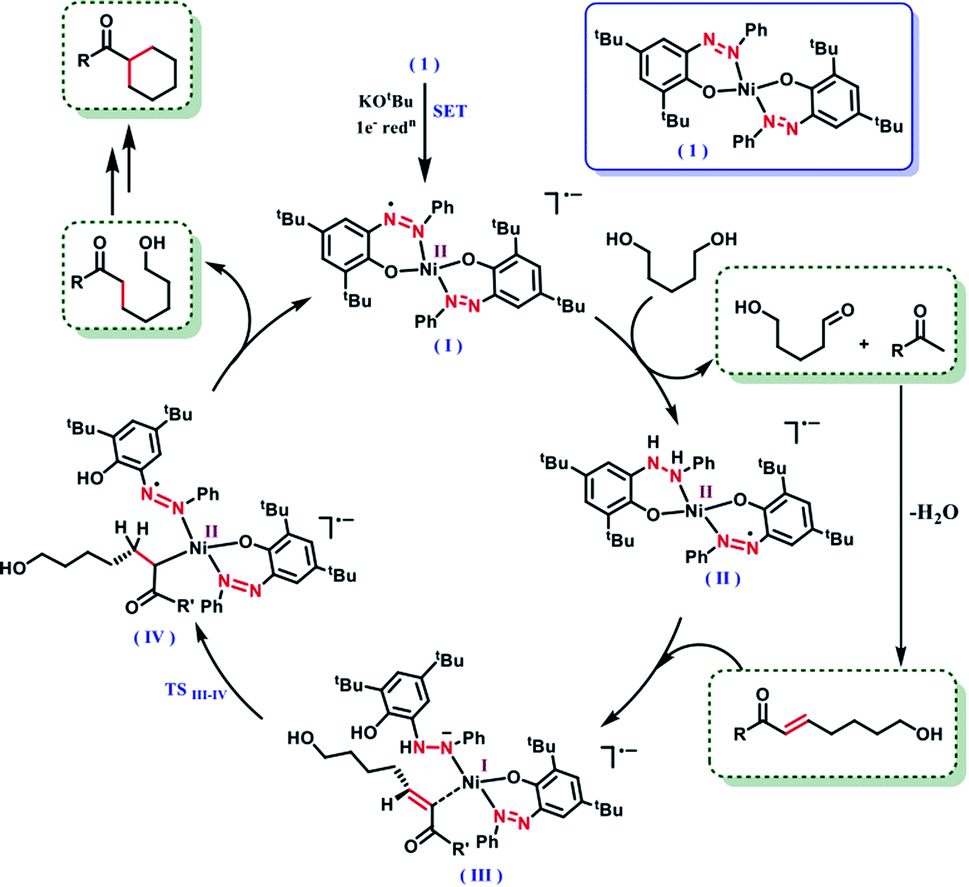 | ||
| Fig. 1 Plausible mechanism for the radical-promoted selective hydrogenation of a double bond in an enone. | ||
Our proposal of radical-mediated enone hydrogenation is indirectly supported by the failure of the reaction in the presence of a radical inhibitor like TEMPO (Section 5.2, ESI†). We gather collective evidence from control reactions and try to delineate the hydrogenation mechanism, which is driven by radical intermediates. To start the hydrogenation of the olefinic bond in enone 5, it binds to nickel, at the expense of one of the phenolate arms being detached upon protonation. In the olefin-bound form, the mono-reduced azo passes the electron to reduce the olefin in intermediate III (Fig. 1). Such a reduction is facilitated by the nickel being a mediator, which can also change its redox state when required. The oxidation state of nickel in the catalyst is +2, which supposedly becomes reduced to +1 in the intermediate III.
In the intermediate III, a carbon-based radical character develops that facilitates the further hydrogen atom transfer (HAT) event. We undertook a series of studies to accumulate unambiguous evidence for such a carbon-based radical generation during the sequence of reactions. To amass compelling evidence, we selected a radical-probe substrate. In the enone 9, a cyclopropyl group is connected, and during the generation of a carbon-centred radical this ring is expected to open leaving the signature of radical formation.
Indeed, the hydrogenation reaction of the radical-probe substrate revealed a rearranged product after ring-opening to form 4bb predominantly (60%). The formation of 4bb further proves that the ring-opening of the cyclopropyl ring in 9 is completely mediated by a radical, as opposed to a metal-hydride promoted process.44 Conversely, only 17% of the isolated product showed cyclopropyl ring retention (4bb′, Scheme 3b). Moreover, the carbon-based radical from a substrate enone was intercepted by TEMPO and detected by ESI-mass spectrometry at 386.3066 amu (Section 5.4, ESI†). Notably, this mass can be representative of any of the trapped forms of the radical 10a–10c, while the pathways for their formation have been described in the ESI (Scheme S1†).
Taken together, these experiments demonstrate a radical mechanism for hydrogenation. A HAT from N–H happens in intermediate III, to form an azo-centred radical in the intermediate IV. The intermediate IV also gains stability from its nickel coordination. Finally, the alkyl end coordinated to nickel requires protonation to complete the hydrogenation step, where the source of protons can be the phenol arm. Upon release of the hydrogenated product phenolate regains coordination to the nickel. It can be assumed that the same hydrogenation would be followed for the cyclic enone to release the final cycloalkyl ketone product.
The discrete involvement of radicals in the hydrogenation step intrigued us to study the mechanism of the hydrogenation by theoretical methods that can shed light on the nature of intermediate species in the catalytic cycle. To focus on the hydrogenation part of the reaction, we conducted calculations on a model enone using a meta hybrid functional M06-2X and 6-311G*/(lanl2dz for nickel) basis set.45,46 The hydrogenation starts with olefin binding of the enone to nickel in catalytic intermediate III. In this computed intermediate III, the N–H in the embedded hydrazo is properly aligned to the carbon, where its migration is necessary to pave the hydrogenation. Computationally, we investigated the alternative possibility of nickel binding to the beta-carbon of the enone, but such attempts resulted in non-converged intermediates. This is suggestive of very high energy intermediates in such binding modes compared to our proposed intermediate III. In such a situation, HAT is fully feasible where, the H migrates from the nitrogen to the putative carbon. Encouragingly, such a transition state (TS) for HAT can be located in the potential energy surface which poses a barrier of 26.12 kcal mol−1, when computed at 298 K. In the TS, the pseudo-tetrahedral geometry of the nickel is maintained and the respective N…H and C…H distances are found to be 1.37 and 1.29 Å, respectively (Fig. 2). The portion of the alkyl chain in this enone substrate can be placed outside, so that steric clash with the substituents in the ligand backbone can be avoided. Upon this HAT, a nitrogen-centered radical is generated, which is further stabilized via the nickel coordination. Indeed, the developing covalency between the ligand radical and the metal center confers stability to an otherwise rather unstable intermediate. As expected, in this computed intermediate, the spin density is observed in the azo fragment and nickel.
 | ||
| Fig. 2 Potential energy diagram of olefin hydrogenation of α,β-unsaturated carbonyl via HAT (* marked transition states are for illustration and were not determined). All hydrogens except chemically important ones have been removed for clarity. | ||
The resulting intermediate III from HAT possesses an energy of 7.09 kcal mol−1 with respect to the reference state. At this stage, adjacent carbon in the olefin is strongly bound to nickel which requires further protonation to facilitate the liberation of the hydrogenated product. Complete release of the hydrogenated product makes this process close to thermoneutral.
Notably, this mode of hydrogenation is distinctly different from conventional hydrogenations involving a metal hydride.47–50 Indeed, in metal–ligand cooperativity51 driven hydrogenation reactions, a metal-hydride has been often invoked and usually an insertion of the olefin to such a metal hydride follows. Our findings showcase that such a pathway can be completely avoided by one-electron HAT processes,52 which are predominantly steered by redox-active ligands appropriately placed in the metal coordination sphere.
Conclusion
In conclusion, we present the first example of a nickel catalyst that can furnish a host of cycloalkanes in a stereoselective manner from a ketone and 1,n-diol substrates under low catalyst loading. The reaction follows a BH-method, where the extracted hydrogen from the diol is stored in the azo ligand backbone, fully avoiding a metal hydride formation. A sequence of dehydrogenation and hydrogenation reactions is conducted that is fully dictated by the radical pathway, thus distinguishing this completely from two-electron chemistry. Multiple radical isolation and radical-probe substrates support the mechanistic proposal and promise this pathway to be complementary to the ubiquitous two-electron driven (de)hydrogenation reactions.Data availability
Experimental and computational data associated with this work have been included in the ESI.†Author contributions
AKB, AK, DA conceptualized the project. AKB performed all experimental work with some help from AK. AK carried out the computational part under the guidance of DA. All authors analyzed the data. DA and DM wrote this paper with inputs from other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank SERB (DST), India (Grant No. ECR/2017/001764 to DA) and (CRG/2018/003951 to DM) for financial support and IISER Mohali for seed grant. AKB thanks IISER Mohali for a research fellowship. AK thanks CSIR for a senior research fellowship.References
- R. A. Holton, C. Somoza, H. B. Kim, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith and S. Kim, J. Am. Chem. Soc., 1994, 116, 1597–1598 CrossRef CAS.
- S. Goudedranche, W. Raimondi, X. Bugaut, T. Constantieux, D. Bonne and J. Rodriguez, Synthesis, 2013, 45, 1909–1930 CrossRef CAS.
- J. A. Dabrowski, D. C. Moebius, A. J. Wommack, A. F. Kornahrens and J. S. Kingsbury, Org. Lett., 2010, 12, 3598–3601 CrossRef CAS PubMed.
- G. Xia, X. Han and X. Lu, Org. Lett., 2014, 16, 2058–2061 CrossRef CAS PubMed.
- A. Banerjee, S. Sarkar and B. K. Patel, Org. Biomol. Chem., 2017, 15, 505–530 RSC.
- S. Danishefsky and T. Kitahara, J. Am. Chem. Soc., 1974, 96, 7807–7808 CrossRef CAS.
- B. K. Peters, J. Liu, C. Margarita, W. Rabten, S. Kerdphon, A. Orebom, T. Morsch and P. G. Andersson, J. Am. Chem. Soc., 2016, 138, 11930–11935 CrossRef CAS PubMed.
- D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed.
- A. Kumar, V. Goyal, N. Sarki, B. Singh, A. Ray, T. Bhaskar, A. Bordoloi, A. Narani and K. Natte, ACS Sustainable Chem. Eng., 2020, 8, 15740–15754 CrossRef CAS.
- W. Ma, J. Fang, J. Ren and Z. Wang, Org. Lett., 2015, 17, 4180–4183 CrossRef CAS PubMed.
- M. E. Jung, Comprehensive Oranic Synthesis, Pergamon, Oxford, 1991 Search PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, 2006 Search PubMed.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC.
- A. Quintard and J. Rodriguez, ChemSusChem, 2016, 9, 28–30 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed.
- P. Chakraborty, N. Garg, E. Manoury, R. Poli and B. Sundararaju, ACS Catal., 2020, 10, 8023–8031 CrossRef CAS.
- R. Qu, Y. Cheng, S. Yang, C. Zhao, H. Liu and X. Huang, ChemistrySelect, 2021, 6, 4089–4097 CrossRef CAS.
- N. S. Lawal, H. Ibrahim and M. D. Bala, Monatsh. Chem., 2021, 152, 275–285 CrossRef CAS.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- B. Sarkar, D. Schweinfurth, N. Deibel and F. Weisser, Coord. Chem. Rev., 2015, 293–294, 250–262 CrossRef CAS.
- W. Kaim, Eur. J. Inorg. Chem., 2020, 2020, 875–878 CrossRef CAS.
- D. L. J. Broere, B. Q. Mercado, E. Bill, K. M. Lancaster, S. Sproules and P. L. Holland, Inorg. Chem., 2018, 57, 9580–9591 CrossRef CAS PubMed.
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed.
- W. M. Akhtar, R. J. Armstrong, J. R. Frost, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2018, 140, 11916–11920 CrossRef CAS PubMed.
- R. J. Armstrong, W. M. Akhtar, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, Tetrahedron, 2019, 75, 130680 CrossRef.
- A. K. Bains, A. Kundu, S. Yadav and D. Adhikari, ACS Catal., 2019, 9, 9051–9059 CrossRef CAS.
- A. K. Bains and D. Adhikari, Catal. Sci. Technol., 2020, 10, 6309–6318 RSC.
- A. K. Bains, D. Dey, S. Yadav, A. Kundu and D. Adhikari, Catal. Sci. Technol., 2020, 10, 6495–6500 RSC.
- A. K. Bains, A. Biswas and D. Adhikari, Chem. Commun., 2020, 56, 15442–15445 RSC.
- A. Jana, K. Das, A. Kundu, P. R. Thorve, D. Adhikari and B. Maji, ACS Catal., 2020, 10, 2615–2626 CrossRef CAS.
- L. Bettoni, S. Gaillard and J.-L. Renaud, Chem. Commun., 2020, 56, 12909–12912 RSC.
- A. Kaithal, L.-L. Gracia, C. Camp, E. A. Quadrelli and W. Leitner, J. Am. Chem. Soc., 2019, 141, 17487–17492 CrossRef CAS PubMed.
- J. Das, K. Singh, M. Vellakkaran and D. Banerjee, Org. Lett., 2018, 20, 5587–5591 CrossRef CAS PubMed.
- R. Babu, M. Subaramanian, S. P. Midya and E. Balaraman, Org. Lett., 2021, 23, 3320–3325 CrossRef CAS PubMed.
- W. M. Akhtar, C. B. Cheong, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2017, 139, 2577–2580 CrossRef CAS PubMed.
- R. J. Armstrong and T. J. Donohoe, Tetrahedron Lett., 2021, 74, 153151 CrossRef CAS.
- C. B. Cheong, J. R. Frost and T. J. Donohoe, Synlett, 2020, 31, 1828–1832 CrossRef CAS.
- T. G. Attig, Inorg. Chem., 1978, 17, 3097–3102 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theo. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- S. Chiodo, N. Russo and E. Sicilia, J. Chem. Phys., 2006, 125, 104107 CrossRef CAS PubMed.
- R. Xu, S. Chakraborty, S. M. Bellows, H. Yuan, T. R. Cundari and W. D. Jones, ACS Catal., 2016, 6, 2127–2135 CrossRef CAS.
- P. Chakraborty, M. K. Gangwar, B. Emayavaramban, E. Manoury, R. Poli and B. Sundararaju, ChemSusChem, 2019, 12, 3463–3467 CrossRef CAS PubMed.
- M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 14967–14971 CrossRef PubMed.
- K. Tokmic, C. R. Markus, L. Zhu and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 11907–11913 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401–12422 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectroscopic data, coordinates of computed structures and NMR spectra of products. See DOI: 10.1039/d1sc04261k |
| This journal is © The Royal Society of Chemistry 2021 |