
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHexafluoroisopropanol: the magical solvent for Pd-catalyzed C–H activation
Trisha
Bhattacharya
a,
Animesh
Ghosh
a and
Debabrata
Maiti
 *ab
*ab
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai, Maharashtra 400076, India. E-mail: dmaiti@chem.iitb.ac.in
bTokyo Tech World Research Hub Initiative (WRHI), Laboratory for Chemistry and Life Science, Tokyo Institute of Technology, Tokyo 152-8550, Japan
First published on 10th February 2021
Abstract
Among numerous solvents available for chemical transformations, 1,1,1,3,3,3-hexafluoro-2-propanol (popularly known as HFIP) has attracted enough attention of the scientific community in recent years. Several unique features of HFIP compared to its non-fluoro analogue isopropanol have helped this solvent to make a difference in various subdomains of organic chemistry. One such area is transition metal-catalyzed C–H bond functionalization reactions. While, on one side, HFIP is emerging as a green and sustainable deep eutectic solvent (DES), on the other side, a major proportion of Pd-catalyzed C–H functionalization is heavily relying on this solvent. In particular, for distal aromatic C–H functionalizations, the exceptional impact of HFIP to elevate the yield and selectivity has made this solvent irreplaceable. Recent research studies have also highlighted the H-bond-donating ability of HFIP to enhance the chiral induction in Pd-catalyzed atroposelective C–H activation. This perspective aims to portray different shades of HFIP as a magical solvent in Pd-catalyzed C–H functionalization reactions.
 Trisha Bhattacharya | Trisha Bhattacharya was born in 1993 in the state of West Bengal, India. She obtained her BSc degree in Chemistry from Asutosh College, University of Calcutta in 2015. Then, she completed her MSc in Chemistry from the Indian Institute of Technology, Hyderabad, in 2017 following which she joined Prof. Debabrata Maiti's group at Indian Institute of Technology, Bombay. Presently, she is pursuing her PhD on the topic transition metal catalyzed distal C–H activation. |
 Animesh Ghosh | Animesh Ghosh was born and brought up in Jhargram, West Bengal, India. After completing his graduation from Midnapore College (Autonomous) in 2017, he went to Indian Institute of Technology Guwahati from where he received his Master's degree in Chemistry. Subsequently, he joined Prof. Maiti's group at IIT Bombay as a JRF in July 2019. Currently, he is working on transition metal catalyzed distal C–H bond functionalizations. |
 Debabrata Maiti | Debabrata Maiti received his PhD from Johns Hopkins University (USA) in 2008 under the supervision of Prof. Kenneth D. Karlin. After postdoctoral studies at Massachusetts Institute of Technology (MIT) with Prof. Stephen L. Buchwald (2008–2010), he joined the Department of Chemistry at IIT Bombay in 2011, wherein he is currently an Associate Professor. His research interests focus on the development of new and sustainable catalytic methods. |
1. Introduction
The crucial role of a solvent in governing the pathway of a chemical reaction was realized long ago when the great Greek philosopher Aristotle stated that “…compounds do not react unless fluid or if dissolved”.1 Although solid-state reactions have been well established in recent times, they are bound to certain terms and conditions. A solvent can directly influence the rate of a reaction, the formation of a particular product, the stereochemistry of the intermediate, and hence the product's (recalling the role of solvents in nucleophilic substitution reactions). All of these phenomena are controlled by a few major parameters, which include acidity, ionic strength, boiling point, H-bonding, or other weak interactions, along with supplementary physical properties. While there are plenty of solvents known for their extraordinary assistance in various synthetic transformations, there are a few hidden gems often tagged as “unconventional solvents”.2 1,1,1,3,3,3-Hexafluoro-2-propanol or HFIP is one of them. It could be the corrosive nature (highly burning sensation upon inhalation or skin contact) or inflammability of HFIP that mandates extra care while handling and storage or being expensive (US$130 per kg)3 that holds back researchers across the globe to use the solvent immensely. Despite this quandary, HFIP has earned its eminence in various research domains due to its spectacular physical and chemical properties. Fig. 1 features a comparative statistics of the solvent with isopropanol comprising minute to drastic differences in physical properties, which eventually dictates the mode of a chemical reaction. Unlike other analogues, the presence of two tri-fluoroalkyl groups in HFIP is presumed to remarkably alter the course of a chemical reaction and leverages great opportunities in traditional organic synthesis, electrocatalysis, photocatalysis, biological studies, and even in environmental science. Before we dive into the main part, we take this prospect to describe glimpses of different underprivileged applications of HFIP concisely to highlight its widespread acceptability in various research fields. | ||
| Fig. 1 A comparative overview of different physical characteristics, price, and safety details of HFIP and isopropanol.20,21 | ||
1.1 Traditional organic synthesis
With the incorporation of fluorine, (i) HFIP becomes a befitting excellent H-bond donor and (ii) the polarity of the O–H bond is enhanced, which eventually increases the acidity. In a recent chemical record, Xiao and An have precisely displayed the impact of such substantial properties of HFIP in different genres of synthetic organic chemistry.4 By exploiting the H-bonding capability,5 the solvent can activate key oxidizing agents in extremely useful oxidation reactions (e.g., H2O2 in sulfide to sulfoxide,5aScheme 1A, eqn (1), Baeyer–Villiger oxidation of ketone and epoxidation of olefins5b,c); in other instances such as in a faster Diels–Alder reaction by stimulating the dienophile,6a enabling cyclopropane ring-opening reactions6b and other useful organic transformations.6c In a recent report, Ritter group explains that H-bonding exerted by HFIP often leads to ion–pair separation which indeed promotes [MsO–NH3](OTf), the precursor for arene C–H amination (Scheme 1A, eqn (2)).7 Being non-nucleophilic, the solvent also promotes site-specific ring opening of 1-CF3-substituted epoxy ethers.8 While synthetic chemists often cherish the non-nucleophilic nature of HFIP in different aspects, there are instances where HFIP takes part in nucleophilic addition. One such example is demonstrated by Bonnet-Delpon where a hexafluoropropyloxy acetal can be availed in synthetically useful yield in a HFIP-assisted ion–pair interaction (Scheme 1A, eqn (3)).9 | ||
| Scheme 1 Exposure of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) in different domains of experimental science. | ||
1.2 En route to green chemistry: electro- and photoredox catalysis
The remarkable solvent effect of HFIP is not constrained only to the conventional synthetic platform, rather it is quite impressing in electrocatalysis as well as photocatalysis. Excellent redox stability in combination with a high dielectric constant (ε, 15.7) makes HFIP perfect for photoredox chemistry. The superpolar nature of the solvent [102 factor higher than trifluoroacetic acid (TFA) and by a factor of ∼108 compared to acetonitrile (MeCN)] assists in stabilizing radical cations, which are a frequent intermediate in electro- and photoredox catalysis.10 Probably, due to the same reason, the solvent is found to be way more efficient than any other polar solvents in a recent δ-C–H heteroarylation of protected aliphatic amines via [1,5]HAT (Scheme 1B, eqn (1)).11 Because of the high reactivity of the radical cations, the whole catalysis predominantly depends on the reaction media. It has been observed that non-nucleophilic HFIP can easily solvate the counter anions by H-bonding interactions, thus leaving the radical cations free.12 Recently, Boron Doped Diamond (BDD)-supported electrodes in HFIP medium have emerged as a proficient tool in electrocatalysis. In anodic oxidation of isoeugenol in a 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP)/(BDD) electrode system, HFIP forms a H-bonding network to surround the key radical intermediate. Inside the solvent cage, the intermediary radical species has a specific orientation to attain the highest stereoselectivity (Scheme 1B, eqn (2)).131.3 Biological experiments
Apart from typical synthetic chemical transformations, HFIP is very common in the hands of biologists. The effects of alcoholic solvents on proteins and peptides, to find out the cause of their stability inside the biological system, are a topic of great importance. Seminal studies indicate two major causes of alcohol-mediated protein denaturation (a) disruption of the natural state and (b) introduction of an α-helical motif.14 Polar and protic alcoholic solvents disrupt the distal hydrophobic interactions and elevate the local polar or hydrophilic interactions via weak interactions such as H-bonding. It is worth mentioning that HFIP with six fluorine atoms significantly induces α-helical conformation and imposes local polar interactions to a higher extent, which leads to protein denaturation. Additionally, hexafluoroisopropanol indirectly influences the structural modification of protein and lipid layers of different biological membranes (Scheme 1C).15 Apart from peptide chemistry, HFIP is potential enough to produce lipid bilayer leakage and alter the biochemical properties of the lipid phase.161.4 Deep eutectic solvents
Traditional volatile organic compounds (VOCs) as solvents exert severe adverse impact on the environment such as easy accumulation in the atmosphere (due to low boiling point) causing respiratory trauma, flammability, high-grade toxicity, and most importantly, non-biodegradability.17 Water as a cheap and abundant solvent could be a probable alternative; however, lack of solubility of organic reagents does not meet the benchmark. In this prospect, deep eutectic solvents (DESs) have come up as an impending option to balance the nature and human innovations. A DES generally consists of a eutectic mixture of two or three species capable of self-aggregation where at least one of them is a hydrogen-bond donor (HBD) and the other is a hydrogen-bond acceptor (HBA). Although chlorine-based solvents (e.g., chloroform, tetrachloromethane, etc.) and even imidazole-based ionic liquids (ILs) are well recognized as leading components of DESs, being expensive and having noxious nature often raise severe threat to the environment.18 To bypass such hindrances, 1,1,1,3,3,3-hexafluoro-2-propanol as a HBD is evolving as a green and sustainable DES. HFIP-based aqueous biphasic solvents (ABSs) are now extensively used for the microextraction of components from various beverages, partitioning of dyes, as the green medium for diverse organic synthesis, recycling of food wastes, separation of natural products and so on.19Over years, several reviews20 have come up to render the bigger picture of HFIP in many domains albeit focused reviews on the direct implication of HFIP in transition metal-catalyzed C–H bond functionalization are very scarce.21 It is worth specifying that the reliability of the solvent with palladium catalysts has taken the C–H bond transformations to a greater extent. The scope of this perspective is to highlight an irreplaceable plea for HFIP in Pd-catalyzed C–H activation. As our primary interest, we have emphasized on “how” HFIP is affecting the nature of a reaction. In particular, we describe the direct influence of a weak interaction force like H-bonding to stabilize the metallacycles and/or to interrelate with a suitable substrate to promote stereo- and regioselectivity. While the prime focus of this review is Pd-catalyzed C–H activation reactions, the intimacy of HFIP is not constrained only to Pd, rather its compatibility with other transition metals is also well inculcated in literature.22 For obtaining a better insight, we have sub-classified the entire topic into four major parts commencing with proximal C–H activation.
2. Impact of HFIP in Pd-catalyzed proximal C–H functionalizations
It has been a century since C–H bond functionalizations or precisely C–H bond activation has added a new dimension to contemporary organic synthesis. The amplitude of C–H bonds in natural products and pharmaceuticals has compelled the global synthetic chemists to strategize site-selective C–H functionalization as a transformative paradigm to access such valuable motifs. In this section on the perspective, we discuss the influence of HFIP on Pd-catalyzed regiospecific proximal C–H activation reactions (including ortho-C(sp2)–H and α/β-C(sp3)–H bonds). Evidently, transition metal-catalyzed ortho-C(sp2)–H functionalizations are well flourished because of the thermodynamically formed five- or six-membered metallacycles. Since proximal C–H activation is nearly a favorable situation, it occasionally needs any further driving force. Hence, very few cases are known where HFIP is highly essential to attain proximal selectivity or excellent yield.2.1 C(sp2)–H functionalizations
One of the central roles of HFIP in proximal aromatic C–H activation is stimulation of the weakly coordinating directing groups (WCGs) by H-bonding. Intrigued by this observation, Yu and coworkers demonstrated an ether-directed ortho-olefination of aromatic substituents in 2013.23a The strategy was extended thereafter to diolefination and acetoxylation by exploring other weakly coordinating groups such as Weinreb amides, ketones and esters (Scheme 2).23b Enantioselective iodination and acetoxylation of mandelic acid were also reported by the same group using HFIP as the principal solvent.24 Interestingly, HFIP was found to be the appropriate solvent when the reaction followed the Pd(II)/Pd(IV) pathway, whereas tert-amyl-OH appeared to be the best for the Pd(0)/Pd(II) cycle. However, readers should keep in mind that the catalytic cycle may vary from case to case, and HFIP is not always the preeminent choice of solvent in every Pd(II)/Pd(IV) cycle.25 | ||
| Scheme 2 HFIP favoring proximal ortho-C(sp2)–H olefination of weakly coordinating directing groups. | ||
HFIP also serves as the chief solvent in different oxidant-free ortho-functionalizations under mild reaction conditions. Kanai and co-workers reported C–H activation, followed by coupling of oxiranes with 2-phenyl pyridine derivatives to generate ortho-alkylated compounds (Scheme 3).26 This protocol is exceptionally proficient at room temperature and acquires stereoretention with 99% ee in the product. The first report on weak coordination-promoted Pd-catalyzed dehydrogenative cross-coupling reaction of ketones, esters, and carbamates was made by Rao group in HFIP medium (Scheme 3).27 Later on, Su and co-workers reported the arylation of electron-deficient benzoic acid,28a and recently, Li group has also reported a ligand-controlled ortho-arylation of 2-phenylethylamines in HFIP.28b A recent work by Li and Wang shows the remarkable solvent effect by HFIP on a Pd(II)-catalyzed ortho-olefination of arenes directed by a polyfluoroalkylsulfinyl auxiliary.28c In this context, it is worth mentioning that several seminal reports on Pd(II)-catalyzed ortho-C–H functionalizations in recent times have actually evolved employing HFIP as the supreme solvent.29
 | ||
| Scheme 3 Pd-catalyzed C(sp2)–H bond functionalization reactions. | ||
2.2 C(sp3)–H functionalizations
The unique activity of the weakly tethering groups in HFIP can be witnessed in the case of aliphatic β-C(sp3)–H arylation of α-amino acids,30 di-peptides31 (both N-terminus and C-terminus) and Weinreb amides.32 Excluding arylation, other functional transformations like olefination33 and acetoxylation34 of free carboxylic acids are quite sensitive toward HFIP. Other than acid derivatives, HFIP has turned out to be suitable for alcohols also. A recent report on methylene β-arylation of masked aliphatic alcohol by the L, X-type auxiliary has again enlightened HFIP as the optimum solvent (Scheme 4A).35 In 2018, MPAAM (mono-protected aminoethyl amine) ligand-assisted stereoselective β-C(sp3)–H arylation of aliphatic acid was also achieved by the same group with high enantioselectivity (up to 98% ee) in HFIP (Scheme 4B).36 | ||
| Scheme 4 Pd-Catalyzed β-C(sp3)–H functionalization using HFIP as the key solvent. | ||
The extraordinary compatibility of HFIP with aliphatic acids gives leverage to chemists to unfold the other novel possibilities thereafter. For instance, β-lactonization of free acids, followed by ring-opening C–C and C–X (X = O, S, N, halogen, etc.) bond formation (Scheme 4C).37 In both cases, HFIP was found to be the optimum solvent in Pd(II)/Pd(IV) catalysis. While the aforementioned reports justify like dissolves like theory for HFIP, seminal reports suggest the role of hexafluoro propanol in modulating the inert C–H bonds toward oxygenation.38
On the other side, the influence of protic polar nature of HFIP was elegantly demonstrated by Larrosa group through a selective β-arylation method of thiophene and benzo[b]thiophene under mild reaction conditions.39a The key intermediate of this Pd(0)/Pd(II) catalytic cycle was stabilized by H-bonding between two HFIP molecules. The simultaneous kinetic experiment and in silico studies revealed that the reaction follows Heck-type pathway (ΔG‡, 22.4 kcal mol−1) instead of CMD (concerted metalation and deprotonation) (ΔG‡, 24.7 kcal mol−1, higher free energy barrier) as the latter process is thermodynamically formidable (Fig. 2). Even in an Ag-mediated α-arylation of benzo[b]thiophene by the same group, HFIP was used as the prime solvent.39b Along the lines, Xi,40a Li,40b Daugulis41 and Kuninobu42 have independently studied HFIP-promoted arylation of different aromatic and aliphatic systems.
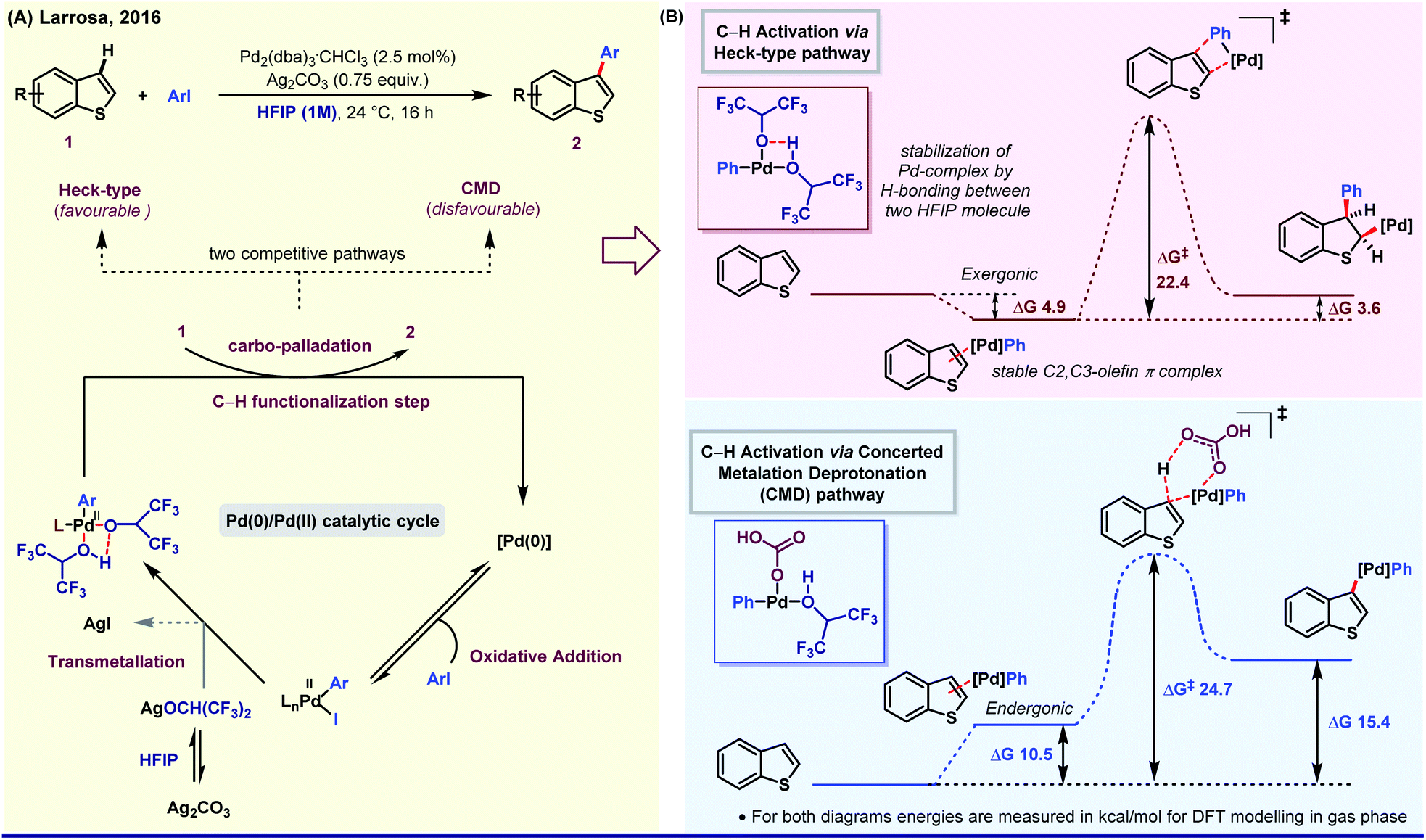 | ||
| Fig. 2 (A) Pd-Catalyzed selective β-arylation of benzo[b]thiophene via a Heck-type pathway. (B) Energy profile diagram of two competitive pathways. | ||
3. HFIP as a key solvent for Pd-catalyzed distal C–H functionalizations
3.1 C(sp2)–H functionalizations
One fundamental challenge to execute site selectivity at the remote positions of arene is overcoming highly strained macrocyclic metallacycle formation. Although different strategies were employed to bypass this issue, most of them ended up with poor regioselectivity and limited substrate variety. HFIP, on the other hand, can stabilize the macrocyclic pre-transition state by its facile H-bond-donating capability. This extra solvent effect remarkably reduces the thermodynamic energy barrier for template-assisted distal C–H functionalizations. In this context, the first breakthrough was attained in 2012 by Yu group when they introduced a ‘U-turned’ weakly coordinating meta-selective nitrile template T1.43 The unique ability of HFIP to enhance the reaction outcome was observed for the meta-olefination of hydrocinnamic acids (Scheme 5). Subsequently, the ‘end-on’ template was successfully employed for the meta-functionalizations of a series of different substrates like phenols, phenylacetic acids, phenyl propionic acids and N-heterocycles, where an excellent enhancement of the product yield, as well as meta-selectivity was observed in HFIP.44 In parallell, we have also introduced a novel and easy to install auxiliary T2 for meta-selective olefination of aryl acetic acids. However, trans-esterification of the substrate diminished the target product formation when trifluoroethanol was used as the solvent. Replacement of TFE by HFIP significantly suppressed the trans-esterification with improved yield and regioselectivity (Scheme 6).45 In other aspects, this simultaneous trans-esterification removed the template in situ without any further step. | ||
| Scheme 5 First instance of Pd(II)-catalyzed meta-C(sp2)–H olefination of hydrocinnamic acids in HFIP medium. | ||
 | ||
| Scheme 6 Remarkable solvent effect by HFIP in Pd(II)-catalyzed meta-C(sp2)–H olefination of aryl acetic acids. | ||
On a different note, the catalytic amount of HFIP in DCE was enough to uplift the yield and selectivity for meta-olefination of arenes by a silyl-tethered nitrile motif by Tan and co-workers in 2013.46 Undoubtedly, apart from just being a solvent, HFIP was found to be extraordinary as an additive too. Stabilization of the electron-deficient metal center by HFIP is a well-known fact; however; experimental evidence of the fact for distal C–H functionalization is rare. Scheme 7 refers to HFIP and metal complex interaction, which is further confirmed by an NMR titration study during meta-acetoxylation of sulfonamides by our group.47 It is worth mentioning that irrespective of modifications in coordination strength (from nitrile to pyrimidine), HFIP has always played a remarkable supportive role to elevate the reaction outcome.48 Interestingly, the same legacy was followed when a carboxylate group was employed for the remote meta-selective olefination of hydrocinnamic amides by Li group in 2019.49 This work describes the predominance of κ2 mode over κ1 mode (which was more facile for proximal ortho-functionalization due to right conformational orientation) of the carboxyl group for any remote C–H functionalization for the very first time.
 | ||
| Scheme 7 HFIP–Pd-ligand interaction in Pd(II)-catalyzed meta-C(sp2)–H acetoxylation of aryl sulfonamides. | ||
With ongoing surges in meta-C–H functionalizations, we thought to reach out to further distal C–H bonds. For the first time, the distal para-C–H bond of toluene was selectively functionalized by a silyl-tethered nitrile template of ‘D’ shape.50 Building on seminal reports, we exploited the H-bond donor nature of HFIP to architect a better para-directing template. It was realized that a rotational restriction might orient the template toward the para-C–H bond completely to improve the para-selectivity (Fig. 3). A di-methoxy substitution was found optimal to induce such H-bonding with HFIP. This revised template (2nd generation template) indeed improved the para-selectivity drastically for other C–H to C–C/C–X functional transformations.51 Viability of the hypothesis was probed by NMR titration study, which reflected a significant substrate–HFIP interaction.51a However, it is not always the polarity or H-bond-donating ability, rather the exceedingly acidic character of HFIP also manipulates the mechanistic pathway of a reaction. For instance, when HFIP was substituted with its higher pKa variants like d2-HFIP or isopropanol in the Pd-catalyzed p-ketonization reaction, no corresponding ketonized product was formed.51b This indeed proved the role of HFIP in protonating the vinyl ethers for generating a reactive intermediate for ketonization. These seminal insights made us even more enthusiastic to explore other influences of HFIP in distal C–H activation. In our recent study, the crucial role of HFIP in Pd-catalyzed meta-allylation was monitored by NMR titration analysis.52 In contrast to other meta-selective templates, in T3, the pyrimidine moiety acts as a π-acceptor in addition to being a good σ-donor (Fig. 4A). Additionally, the weak H-bonding interaction between HFIP and pyrimidine ‘N’s as well as with ‘O’ in ether linker eventually makes the template statistically more approachable toward the meta-C–H bond. This study further ensures the stabilization of the key ion pairs by the polar protic nature of HFIP (Fig. 4B). A systematic NMR study clearly indicates a downfield shift of substrate protons, which further confirms the presence of an electronegative moiety, and thus, justifies the HFIP–substrate interaction and the role of the solvent (Fig. 4C). Very recently, our group has strategized to reach out the distal para-C–H bond of arene by Pd–norbornene (Pd/NBE) dual cooperative catalysis precluding the exact need of a para-selective template.53 With this, we report the first synthetic route to synthesize para-selective biaryls by C–H activation. Importantly, this strategy also involves HFIP as the sole solvent. The extraordinary effect of the solvent on the yield and regioselectivity is observed here as well. The compatibility of HFIP with Pd/NBE catalysis can also be seen in a recent work by Yu for meta-arylation of fluoroarenes.54
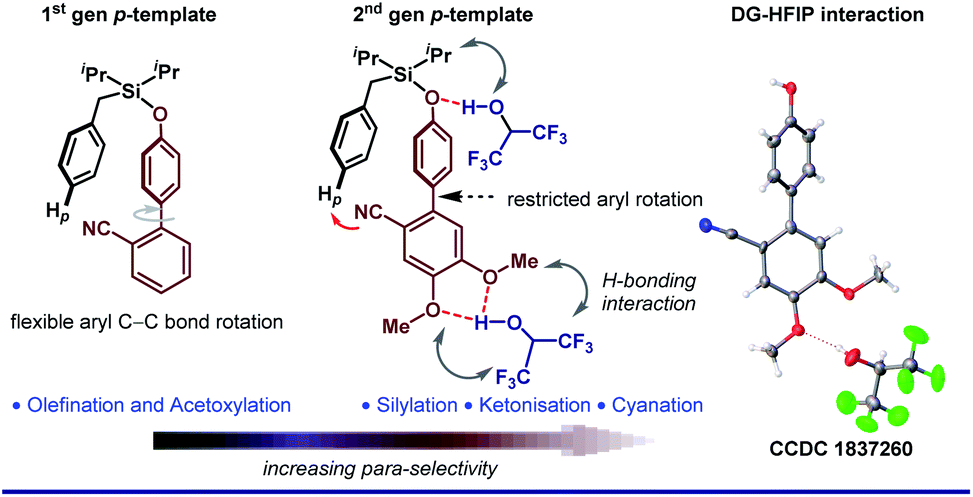 | ||
| Fig. 3 HFIP–Substrate interaction in elevating para-selectivity. | ||
 | ||
| Fig. 4 (A) Inside out of meta-selective pyrimidine DG; (B) stepwise meta-allylation of arenes; and (C) NMR titration analysis depicting the role of HFIP in tuning substrate-DG orientation. | ||
3.2 C(sp3)–H functionalizations
Although the role of HFIP in Pd(II)-catalyzed distal arene C–H functionalizations is acknowledged enough, it is a different story for aliphatic C–H activations where HFIP directly influences the reaction intermediates. One of the major problems associated with distal C–H functionalization is the involvement of biased metallation, which often limits its way to achieve homologous functionalizations.Distal γ-C–H functionalization of aliphatic alcohols demands the formation of a six-membered metallacycle over the thermodynamically accessible five-membered ring. In 2019, Yu and co-workers proposed a strategy for γ-arylation of aliphatic alcohols using 3-nitro-5-chloro-2-pyridone (L) as an external ligand via Pd(II)/Pd(IV) catalysis (Scheme 8).55 In this reaction, the alcohol was masked with an auxiliary T4 in such a way that the internal ring strain in the transition state allows [5,6]-fused bicyclic palladacycles over the [5,5]-bicylic one. The use of HFIP as the key solvent maintained the optimal solubility of the reagents and eventually stabilized the metallacyclic intermediate. In a subsequent work, the same group came up with γ-carbonylation of aliphatic alcohols in the form of a hemilabile benzyl ether.56 While one end of the ether linker conceals the hydroxyl group of alcohol, the other end is linked with a bidentate ligand T5 which orients the palladium toward the target γ-C–H bond.
 | ||
| Scheme 8 Distal γ-arylation of masked aliphatic alcohols. | ||
Upon migratory insertion of CO in int A, HFIP replaces the ligand from the metal coordination site. Eventually, the carbonylated product is obtained in the form of hexafluoroisopropanoate ester (Scheme 9). Nevertheless, chelation-assisted distal C(sp3)–H functionalizations require pre-installation and post-synthetic removal of the directing template, which make the entire protocol sluggish and step-consuming. Alternatively, directing the ability of native functional groups has remained restricted to the proximal site.
 | ||
| Scheme 9 HFIP stabilizing intermediate palladacycle for γ-carbonylation of aliphatic alcohols. | ||
The reaction is well compatible with a wide range of aryl coupling partners and exceptionally sensitive toward the nature of the solvent. Apart from the high altitude of solubility in HFIP, the protic nature of the solvent plays a significant role in maintaining the optimal pH of the reaction medium.57
Recently, van Gemmeren58 and Yu59 group independently extended the scope of the protocol. Unlike arylation, γ-olefination demands an external ligand, which is often a mono-N-protected amino acid (Scheme 10). Indeed, in this case also, HFIP attributes for the highest experimental efficiency. While two different groups highlighted different aliphatic acids separately, our group simultaneously discovered an unusual seven-membered ε-lactone formation by utilizing maleimide as an unconventional source of olefin (Scheme 11).60
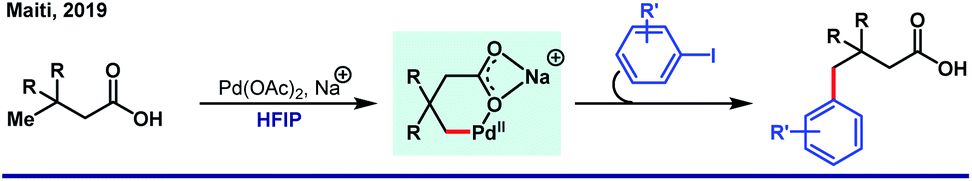 | ||
| Scheme 10 Pd(II)-catalyzed γ-arylation of free carboxylic acids in HFIP. | ||
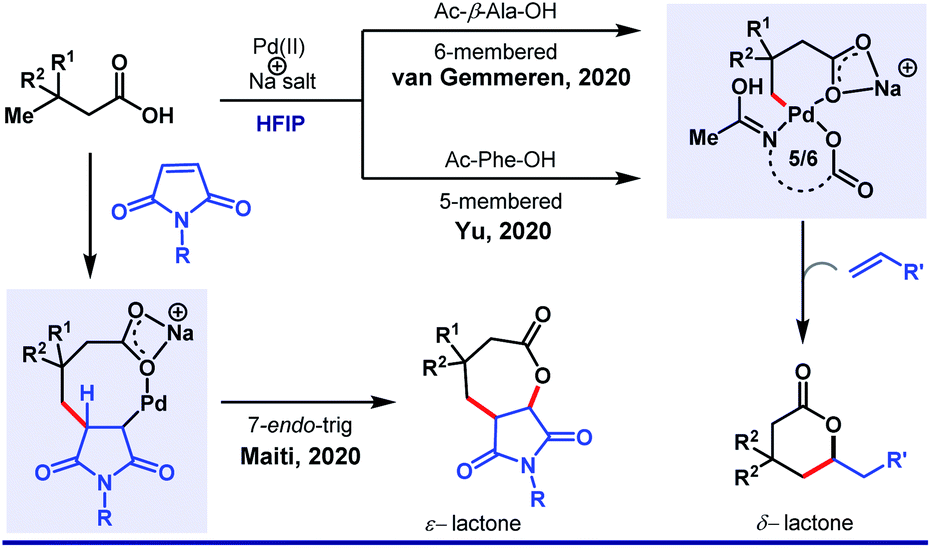 | ||
| Scheme 11 HFIP as the sole solvent in Pd(II)-catalyzed γ-olefination of free carboxylic acids to generate six-membered lactones. | ||
4. HFIP in Pd-catalyzed transient directing group-assisted C–H functionalizations
HFIP as a solvent and/or a co-solvent has played an immense role in different aspects of directing group (DG)-assisted C–H functionalization, as we have already mentioned earlier. However, transient directing group (TDG)-assisted C–H functionalization has emerged as a promising approach to circumvent the existing complications associated with the DG approach. TDGs have added a new dimension for synthetic organic chemists because of its several advantages. First, only a catalytic amount of use of TDGs; second, being economical and less tedious i.e., installation of TDGs, functionalization of C(sp2/sp3)–H bonds, and removal of TDGs, all were done in a single-pot reaction.61,62 Like other sub-domains of C–H activation reactions, HFIP is equally effective as a solvent and sometimes as a co-solvent in a cocktail of solvents (a mixture of two or three solvents with different proportions) to execute TDG-assisted C–H activation. However, the actual role of HFIP in different Pd-catalyzed transient auxiliary mediated C–H activation has not been well discussed significantly.In this context, the first example of using HFIP was put forth by Yu and co-workers in 2016 where they described enantioselective benzylic C(sp3)–H arylation of aldehydes enabled by a chiral TDG 1L-tert-leucine in HFIP (Scheme 12A).62 A better productivity (up to 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 er) was obtained when a catalytic amount of AcOH was added in HFIP (HFIP:AcOH = 9:1, v/v). Recently, the same group has demonstrated an enantioselective β-C(sp3)–H arylation of cyclobutyl ketones utilizing a similar concept.63 Enhanced acidity was the foremost requirement to pursue the functionalization using different TDGs, thereby delivering the best possible result. Recently, Gou et al. depicted site-selective C(sp3)–H arylation of phenylacetaldehydes centered on a similar idea.64 Later, in 2017, the same group reported ortho-C(sp2)–H arylation of substituted benzaldehydes, where HFIP was used as the key solvent (Scheme 12B).65 α-Substituted amino acids were found to be extremely proficient as a transient moderator (TDG 2) for such transformation. With the same perception, Sorensen and co-workers explored the synthesis of substituted fluorenones from benzaldehydes and aryl iodide through the Pd(II)-catalyzed C(sp2)–H functionalization reaction (Scheme 12C).66 However, here authors found 2-amino benzoic acid (TDG 3) as the most promising transient auxiliary. Interestingly, HFIP played a prominent role in the isolation of [5,6]-fused palladacycle (int B) albeit being used as an additive (Scheme 12C).
:2 er) was obtained when a catalytic amount of AcOH was added in HFIP (HFIP:AcOH = 9:1, v/v). Recently, the same group has demonstrated an enantioselective β-C(sp3)–H arylation of cyclobutyl ketones utilizing a similar concept.63 Enhanced acidity was the foremost requirement to pursue the functionalization using different TDGs, thereby delivering the best possible result. Recently, Gou et al. depicted site-selective C(sp3)–H arylation of phenylacetaldehydes centered on a similar idea.64 Later, in 2017, the same group reported ortho-C(sp2)–H arylation of substituted benzaldehydes, where HFIP was used as the key solvent (Scheme 12B).65 α-Substituted amino acids were found to be extremely proficient as a transient moderator (TDG 2) for such transformation. With the same perception, Sorensen and co-workers explored the synthesis of substituted fluorenones from benzaldehydes and aryl iodide through the Pd(II)-catalyzed C(sp2)–H functionalization reaction (Scheme 12C).66 However, here authors found 2-amino benzoic acid (TDG 3) as the most promising transient auxiliary. Interestingly, HFIP played a prominent role in the isolation of [5,6]-fused palladacycle (int B) albeit being used as an additive (Scheme 12C).
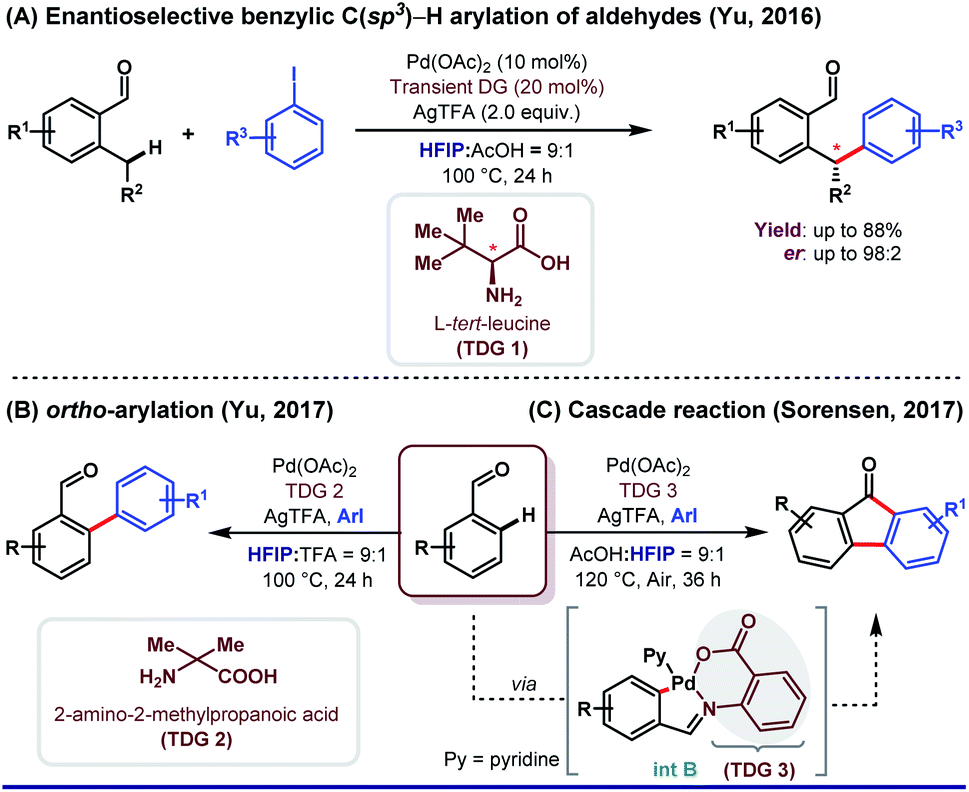 | ||
| Scheme 12 Different roles of HFIP in combination with Pd catalysis using transient auxiliaries (A) and (B) as the major solvent and (C) as additives in ortho-arylation of benzaldehydes. | ||
Besides aromatic aldehydes, HFIP is also well attuned with aliphatic aldehydes, ketones, and primary amines. The polar protic nature of HFIP immensely manipulates the solubility of the substrates apart from maintaining the optimum pH of reaction media. In 2016, Ge and co-workers reported the β-C(sp3)–H arylation of aliphatic aldehyde enabled by 3-aminopropionic acid (TDG 4).67 The cocktail of HFIP and AcOH in a volumetric ratio of 5:1 showed a dramatic improvement in yield (from 29% to 75%) compared to that of only HFIP. Another interesting result of site-selective C(sp3)–H γ-arylation of primary aliphatic amines was published by the same research group employing glyoxylic acid as a TDG (Scheme 14A).68 They discovered AcOH as the optimal solvent for arylation with better yield and excellent site selectivity, yet HFIP played an unprecedented role in isolating the palladacycle as previously observed.
Although the authors have not provided detailed computational and mechanistic studies regarding the exact role of HFIP in the formation of intermediate (int C), it is very likely that HFIP induces an H-bonding interaction with the ‘O’ center of glyoxylic acid TDG, which further accelerates the reaction in the forward direction. Later, Bull and co-workers reported the β-C(sp3)–H arylation of tertiary aldehydes, facilitated by a transient imine directing group (Scheme 13B).69 Only HFIP was ineffective for this reaction, where a 1:1 mixture of HFIP and AcOH provided the best result. This solvent combination has increased not only the overall yield but also the amount of mono-arylated products compared to di- and tri-arylated products.
 | ||
| Scheme 13 Pd-catalyzed TDG-assisted C(sp3)–H arylation. (A) HFIP as an optimum solvent in the synthesis of intermediate; (B) solvent effect on the overall yield and selectivity. | ||
Until 2018, aliphatic TDG-assisted C(sp3)–H functionalizations were mostly restricted to the β-position of free amines, ketones, and aldehydes. HFIP was suitable for the γ-C(sp3)–H (hetero)arylation of ketones using 2,2-dimethyl aminooxyacetic acid auxiliary enabled by 2-pyridone ligand (Scheme 14A).70 The role of HFIP is attributed to its H-bonding interaction with the substrate for the formation of a six-membered palladacycle intermediate and in the successful removal of DG. Mechanistic studies revealed how ligand exchanges its position with HFIP, followed by an oxidative addition to further accelerate the Pd(II)/Pd(IV) catalytic cycle and stabilize the Pd(IV) intermediate (Scheme 14B). Second, they described a method of ligand directed selective γ- and δ-C(sp3)–H arylation of free amines by a bidentate imine and a carboxylate L, X-type directing group.71 This result was the outcome of the cooperative effect between the transient directing group and external pyridone ligands in HFIP solvent medium. In addition, other research groups like Jin,72 Ge and Li,73 and Chen in collaboration with He,74 Wei,75 and Zhang76 also contributed with their valuable results to enrich this area. However, the exact role of HFIP in the aforementioned reactions is still unknown, presumably due to the lack of computational and mechanistic studies.
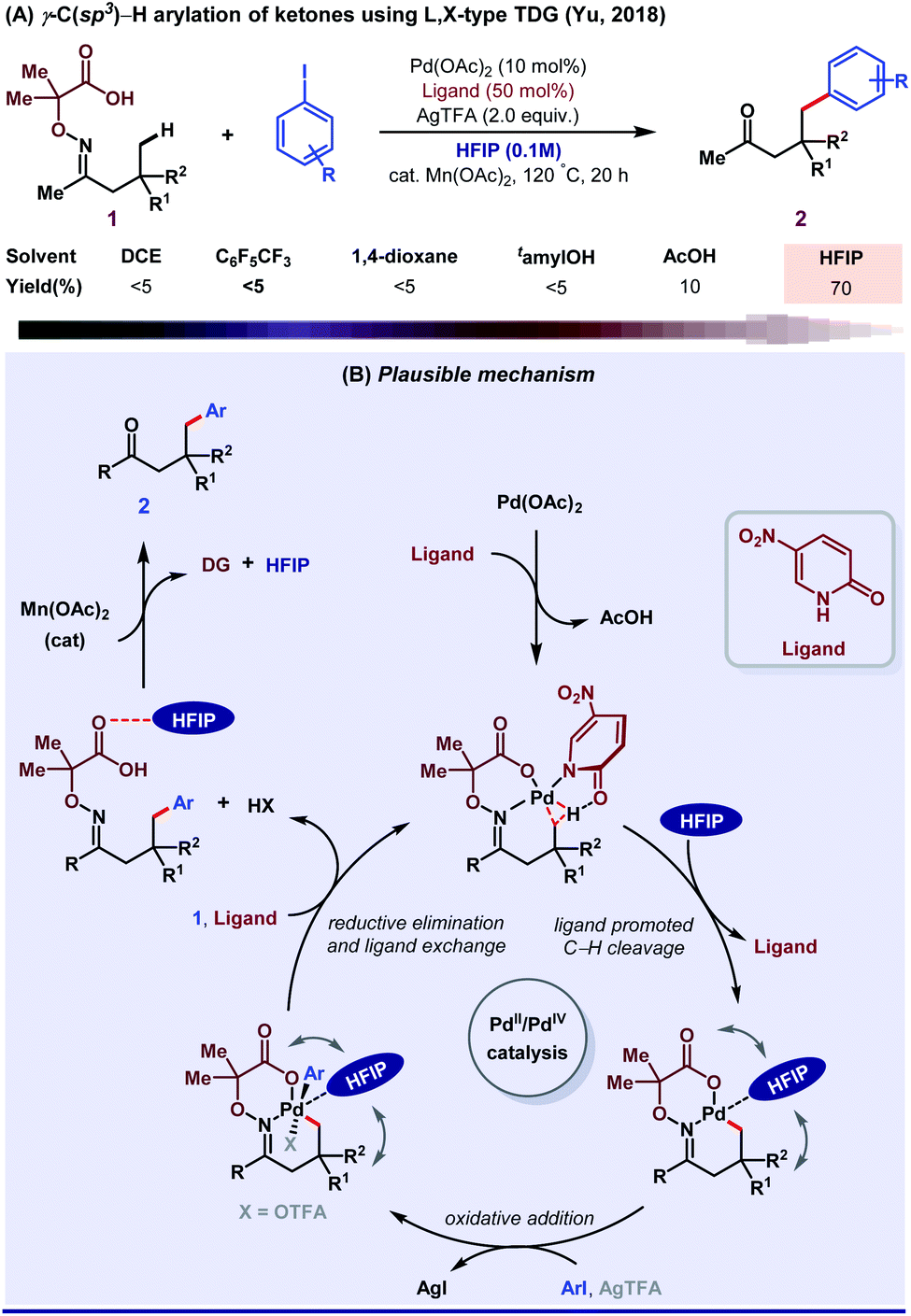 | ||
| Scheme 14 (A) γ-C(sp3)–H arylation of ketones; (B) mechanistic blueprint displaying the detailed role of HFIP. | ||
5. HFIP: boosting stereoselectivity in Pd-catalyzed asymmetric C–H activation
Irrespective of different modes of action (as a solvent or an additive or a chiral auxiliary), fluorinated alcohols (TFE and HFIP) have gained enough acceptance for their remarkable impact on elevating stereoselectivity. Before its direct influence in asymmetric C–H activation, the unprecedented impact of HFIP as a solvent in a Pd-catalyzed asymmetric hydrogenation reaction was realized by Uneyama's group long back.77Despite its uninterrupted application in other areas of homogeneous catalysis,78 HFIP has just started its journey as a game changer in stereoselective C–H functionalization.21a It is hypothesized that the weak coordination of fluorinated alcohols stabilizes the active catalyst. Moreover, being a strong H-bond donor, HFIP can easily manipulate its adjacent C–H bond or other H-bond acceptors to stabilize or destabilize a particular intermediate or substrate. Realizing the crucial role of HFIP in C–H activation, Colobert and Wencel-Delord re-discovered a Pd-catalyzed atroposelective olefination of a biaryl sulfoxide motif in HFIP solvent. The reaction previously performed in DCE led to the desired olefinated product with a compromised yield and enantioselectivity at the cost of superstochiometric use of AgOAc (6 equiv.) and elevated reaction temperature (Scheme 15A).79 A dramatic improvement in yield and enantioselectivity was observed when HFIP was employed, whereas other polar and/or acidic solvents failed or took a longer time to deliver the desired product at room temperature (Scheme 15B). Based on several seminal reports, the authors suggested that the sulfoxide moiety present in the substrate is highly prone to H-bonding with HFIP which lengthens or alternatively weakens the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond which alters the electronic environment of the sulfoxide group and consequently affects the rate of C–H activation step. Moreover, such H-bonding also adds an additional steric factor in the palladacycle, preferably allowing a less hindered atropoisomeric intermediate to react further (Scheme 15C). Another significant benefit of using HFIP as a solvent is reduction in excess use of AgOAc (6 to 2 equiv.) for the reaction, which could be possible by a HFIP facilitated re-oxidation of Pd(0) along with the AgI oxidant.
O bond which alters the electronic environment of the sulfoxide group and consequently affects the rate of C–H activation step. Moreover, such H-bonding also adds an additional steric factor in the palladacycle, preferably allowing a less hindered atropoisomeric intermediate to react further (Scheme 15C). Another significant benefit of using HFIP as a solvent is reduction in excess use of AgOAc (6 to 2 equiv.) for the reaction, which could be possible by a HFIP facilitated re-oxidation of Pd(0) along with the AgI oxidant.
 | ||
| Scheme 15 Pd(II)-catalyzed atroposelective olefination of biaryls. (A) Overall optimization of the reaction; (B) effects of different solvents at room temperature on yield; and (C) steric induction by HFIP favoring one diastereoisomer. | ||
Subsequently, Shi and co-workers elegantly described the possibility of utilizing HFIP as a solvent in another Pd-catalyzed atroposelective olefination of bi-aryl aldehydes.80 In this work, a stark improvement in yield and enantioselectivity was observed when a 4:1 (v/v) combination of HFIP and acetic acid was employed (Scheme 16).
 | ||
| Scheme 16 Effect of HFIP in enhancing the reaction yield. | ||
Although this observation indicates the necessity of a low pH reaction media to improve the reaction outcome, the authors did not specify the actual role of HFIP in this reaction. Based on a similar concept, the authors came up with several new atroposelective functionalization reactions with HFIP being the ‘best suitable solvent’.81 In addition to the aforementioned works, several asymmetric C–H activation reactions regardless of substrate diversity have been performed using HFIP as the solvent.82 Thorough optimization led the authors to choose HFIP over other solvents for better sustainability. Nevertheless, the actual role of HFIP has remained uninvestigated in a majority of cases.
6. Conclusions and outlook
Over the centuries, solvents have manipulated the direction of different chemical conversions by their steric and electronic features. For any catalytic reaction, it is not the catalyst that always dictates the competence of the protocol; rather it also depends on the reaction media. While we discuss the solvent effect in different aspects of organic synthesis, we cannot overlook their influence on biological systems and environment. In that perspective, 1,1,1,3,3,3-hexafluoro-2-propanol or HFIP is emerging as a solvent of choice. Especially, due to its multidirectional application in transition metal-catalyzed C–H functionalizations, the urge for HFIP is almost inimitable now. Although HFIP is a renowned solvent, this perspective portrays other deprived roles of HFIP (e.g. as additive, catalyst, activator, etc.) synchronously. As of our primary focus, we have precisely discussed three foremost impacts of HFIP in Pd-catalyzed C–H activation reactions. First, as we move from proximal (ortho-) to distal (meta- and para-) positions of an arene, HFIP helps in harnessing the transition metal catalyst with the target C–H bond through H-bonding interactions. Moreover, being a strong H-bond donor, HFIP is able to introduce steric crowding in the transitional steps and uplift stereoselectivity in many asymmetric C–H activations. Second, the polar protic environs of HFIP stabilizes macrocyclic cyclophanes like metallacycles (especially when metals are in a high oxidation state), ion pairs and radical cations involved in proximal and distal C–H activation. Third, as an additive, HFIP lessens the pH of the media, which eventually helps in solubilizing acidic and/or polar substrates (e.g., acids, free amines and alcohols). In addition, this review embraces every other subtle aspects of HFIP in Pd-catalyzed C–H activation reactions in detail along with their mechanistical intricacies.Despite its excellence, a pervasive limitation of using HFIP as a sustainable reagent could be its low cost efficiency and corrosive nature. While an extra layer of cautiousness can diminish the second concern, the other issue is also addressable by recycling the solvent through distillation (low bp of HFIP, 58.6 °C). Nevertheless, very few works have highlighted a detailed study to investigate the actual role of HFIP (whether solvent, co-solvent and/or additives) in Pd-catalyzed C–H activation reactions. Hence, more experimental and computational analyses will further amplify the other unaddressed aspects of HFIP in palladium catalysis in near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by SERB India (CRG/2018/003951). Financial support from UGC (T. B.) and CSIR (A. G.), India, is gratefully acknowledged.Notes and references
- Selected reviews on solvent effects in chemical transformations. (a) C. Reichardt, Org. Process Res. Dev., 2007, 11, 105–113 CrossRef CAS; (b) P. J. Dysona and P. G. Jessopb, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- C. Yu, J. S.-Orduna, F. W. Patureau and M. H. Pérez-Temprano, Chem. Soc. Rev., 2020, 49, 1643–1652 RSC.
- This market value of HFIP is taken from Synquest Laboratories. The market price for HFIP according to European currency is £99 per kg (from Fluorochem). While handling HFIP safety precautions as per standard MSDS has to be thoroughly maintained.
- X.-D. An and J. Xiao, Chem. Rec., 2019, 19, 1–21 CrossRef.
- (a) K. S. Ravikumar, Y. M. Zhang, J.-P. Begue and D. Bonnet-Delpon, Eur. J. Org. Chem., 1998, 2937–2940 CrossRef CAS; (b) K. Neimann and R. Neumann, Org. Lett., 2000, 2, 2861–2863 CrossRef CAS; (c) O. Hollóczki, A. Berkessel, J. Mars, M. Mezger, A. Wiebe, S. R. Waldvogel and B. Kirchner, ACS Catal., 2017, 7, 1846–1852 CrossRef; (d) A. Berkessel and J. A. Adrio, J. Am. Chem. Soc., 2006, 128, 13412–13420 CrossRef CAS; (e) A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS.
- (a) C. Cativiela, J. I. García, J. A. Mayoral and L. Salvatella, Can. J. Chem., 1994, 72, 308–311 CrossRef CAS; (b) E. Richmond, J. Yi, V. D. Vuković, F. Sajadi, C. N. Rowley and J. Moran, Chem. Sci., 2018, 9, 6411–6416 RSC; (c) A. Lücht, P. G. Jones and D. B. Werz, Eur. J. Org. Chem., 2019, 5254–5260 CrossRef.
- E. M. D'Amato, J. Börgel and T. Ritter, Chem. Sci., 2019, 10, 2424–2428 RSC.
- I. Rodrigues, D. Bonnet-Delpon and J. P. Bégué, J. Org. Chem., 2001, 66, 2098–2103 CrossRef CAS.
- A. Di Salvo, M. David, B. Crousse and D. Bonnet-Delpon, Adv. Synth. Catal., 2006, 348, 118–124 CrossRef CAS.
- S. Ayata, A. Stefanova, S. Ernst and H. Baltruschat, J. Electroanal. Chem., 2013, 701, 1–6 CrossRef CAS.
- Z. Deng, G.-X. Li, G. He and G. Chen, J. Org. Chem., 2019, 84, 15777–15787 CrossRef CAS.
- N. Shida, Y. Imada, S. Nagahara, Y. Okada and K. Chiba, Chem. Commun., 2019, 24–30 CrossRef.
- T. Yamamoto, B. Riehl, K. Naba, K. Nakahara, A. Wiebe, T. Saitoh, S. R. Waldvogel and Y. Einaga, Chem. Commun., 2018, 54, 2771–2773 RSC.
- N. Hirato, Y. Goto and K. Mizuno, Protein Sci., 1997, 6, 416–421 CrossRef.
- P. Mandal and A. R. Molla, Protein Pept. Lett., 2020, 27, 538–550 CrossRef CAS.
- M. Zhang, T. Peyear, I. Patmanidis, D. V. Greathouse, S. J. Marrink, O. S. Andersen and H. I. Ingólfsso, Biophys. J., 2018, 115, 679–689 CrossRef CAS.
- D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. J. Ramon, Eur. J. Org. Chem., 2016, 612–632 CrossRef CAS.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS.
- (a) P. Liu, J.-W. Hao, L.-P. Mo and Z.-H. Zhang, RSC Adv., 2015, 5, 48675–48704 RSC; (b) K. Xua, P. Xub and Y. Wang, Talanta, 2020, 213, 120839–120849 CrossRef.
- For selected reviews on HFIP in organic synthesis (a) J.-P. Bégué, D. Bonnet-Delpon and B. Crousse, Synlett, 2004, 18–29 Search PubMed; (b) I. A. Shuklov, N. V. Dubrovina and A. Börner, Synthesis, 2007, 2925–2943 CrossRef CAS; (c) T. Dohi, N. Yamaoka and Y. Kita, Tetrahedron, 2010, 66, 5775–5785 CrossRef CAS; (d) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 1–12 CrossRef; (e) X.-D. An and J. Xiao, Chem. Rec., 2020, 20, 142–161 CrossRef CAS; (f) V. Pozhydaiev, M. Power, V. Gandon, J. Moran and D. Lebœuf, Chem. Commun., 2020, 56, 11548–11564 RSC.
- For selected reviews on HFIP in TM catalyzed C–H functionalizations- (a) J. Wencel-Delord and F. Colobert, Org. Chem. Front., 2016, 3, 394–400 RSC; (b) S. K. Sinha, T. Bhattacharya and D. Maiti, React. Chem. Eng., 2019, 4, 244–253 RSC.
- HFIP in different transition metal catalyzed C–H activation reactions: (a) E. Erbing, A. S. Marco, A. V. Romero, J. Malmberg, M. J. Johansson, E. Gómez-Bengoa and B. Martín-Matute, ACS Catal., 2018, 8, 920–925 CrossRef CAS; (b) F. Li, Y. Zhou, H. Yang, D. Liu, B. Sun and F.-L. Zhang, Org. Lett., 2018, 20, 146–149 CrossRef CAS; (c) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4684–4687 CrossRef CAS; (d) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688–4690 CrossRef CAS; (e) K. Shin, S.-W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584–8592 CrossRef CAS; (f) E. Gaster, Y. Vainer, A. Regev, S. Narute, K. Sudheendran, A. Werbeloff, H. Shalit and D. Pappo, Angew. Chem., Int. Ed., 2015, 54, 4198–4202 CrossRef CAS; (g) A. Libman, H. Shalit, Y. Vainer, S. Narute, S. Kozuch and D. Pappo, J. Am. Chem. Soc., 2015, 137, 11453–11460 CrossRef CAS; (h) S. Narute, R. Parnes, F. D. Toste and D. Pappo, J. Am. Chem. Soc., 2016, 138, 16553–16560 CrossRef CAS; (i) H. Wang, M. Moselage, M. J. González and L. Ackermann, ACS Catal., 2016, 6, 2705–2709 CrossRef CAS; (j) F. Romanov-Michailidis, K. F. Sedillo, J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2015, 137, 8892–8895 CrossRef CAS; (k) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735–2738 CrossRef CAS; (l) S. Yu, N. Lv, C. Hong, Z. Liu and Y. Zhang, Org. Lett., 2020, 22, 8354–8358 CrossRef CAS; (m) Y. Jiang, P. Li, J. Zhao, B. Liu and X. Li, Org. Lett., 2020, 22, 7475–7479 CrossRef CAS; (n) E. Weis, M. J. Johansson and B. M.-Matute, Chem.–Eur. J., 2020, 26, 10185–10190 CrossRef CAS; (o) N. Wang, Q. Yang, Z. Deng, X. Mao and Y. Peng, ACS Omega, 2020, 5, 14635–14644 CrossRef CAS; (p) J. Chen, T. Huang, X. Gong, Z.-J. Yu, Y. Shi, Y.-H. Yan, Y. Zheng, X. Liu, G.-B. Li and Y. Wu, Adv. Synth. Catal., 2020, 362, 2984–2989 CrossRef CAS.
- (a) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245–1247 CrossRef CAS; (b) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef CAS.
- N. Dastbaravardeh, T. Toba, M. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 9877–9884 CrossRef CAS.
- (a) L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712–733 RSC; (b) P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824–889 CrossRef CAS.
- Z. Wang, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 6140–6143 CrossRef CAS.
- C. Zhang and Y. Rao, Org. Lett., 2015, 17, 4456–4459 CrossRef CAS.
- (a) C. Zhu, Y. Zhang, J. Kan, H. Zhao and W. Su, Org. Lett., 2015, 17, 3418–3421 CrossRef CAS; (b) Y. Ding, S. Fan, X. Chen, Y. Gao, S. Li and G. Li, Org. Lett., 2019, 21, 4224–4228 CrossRef CAS; (c) Y.-H. Chen, N.-Q. Shao, C. Li and D.-H. Wang, Org. Lett., 2020, 22, 5880–5884 CrossRef CAS.
- (a) R. S. Thombal, T. Feoktistova, G. A. Gonzalez-Montiel, P. H.-Y. Cheong and Y. R. Lee, Chem. Sci., 2020, 11, 7260–7265 RSC; (b) X. Huo, J. Han, X. Yan, H. Zhang, J. Xiong, J. Liu, X. Wang, H. Li and L. Huo, Asian J. Org. Chem., 2020, 9, 2087–2091 CrossRef CAS.
- (a) G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Miller, M. A. Poss, P. M. Scola, K.-S. Yeung and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 3338–3351 CrossRef CAS; (b) G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 1506–1509 CrossRef CAS.
- W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS.
- H. Park, N. Chekshin, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 9292–9297 CrossRef CAS.
- (a) Z. Zhuang, C.-B. Yu, G. Chen, Q.-F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 10363–10367 CrossRef CAS; (b) H. Park, Y. Li and J.-Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 11424–11428 CrossRef CAS.
- Z. Zhuang, A. N. Herron, Z. Fan and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 6769–6776 CrossRef CAS.
- G. Xia, Z. Zhuang, L.-Y. Liu, S. L. Stuart, L. Schreiber, B. Melillo and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 7783–7787 CrossRef CAS.
- P.-X. Shen, L. Hu, Q. Shao, K. Hong and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545–6549 CrossRef CAS.
- Z. Zhuang and J.-Q. Yu, Nature, 2020, 577, 656–659 CrossRef CAS.
- V. Dantignana, M. Milan, O. Cussó, A. Company, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 1350–1358 CrossRef CAS.
- (a) C. Colletto, S. Islam, F. Julia-Hernandez and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 1677–1683 CrossRef CAS; (b) C. Colletto, A. Panigrahi, J. Fernández-Casado and I. Larrosa, J. Am. Chem. Soc., 2018, 140, 9638–9643 CrossRef CAS.
- (a) J. Peng, C. Chen and C. Xi, Chem. Sci., 2016, 7, 1383–1387 RSC; (b) R. Wang, Y. Y. Ding and G. Li, Org. Biomol. Chem., 2017, 15, 4966–4970 RSC.
- (a) S. H. Kwak, N. Gulia and O. Daugulis, J. Org. Chem., 2018, 83, 5844–5850 CrossRef CAS; (b) K. K. A. Le, H. Nguyen and O. Daugulis, J. Am. Chem. Soc., 2019, 141, 14728–14735 CrossRef CAS.
- H.-L. Li and Y. Kuninobu, Adv. Synth. Catal., 2020, 362, 2637–2641 CrossRef CAS.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS.
- A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS.
- M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS.
- S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS.
- A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147–3153 RSC.
- Selective papers on meta-functionalizations involving HFIP as key solvents with different directing groups: (a) L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056–18059 CrossRef CAS; (b) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS; (c) S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595–5600 RSC; (d) A. Modak, A. Mondal, R. Watile, S. Mukherjee and D. Maiti, Chem. Commun., 2016, 52, 13916–13919 RSC; (e) M. Bera, S. K. Sahoo and D. Maiti, ACS Catal., 2016, 6, 3575–3579 CrossRef CAS; (f) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443–10451 CrossRef; (g) S. Bag, R. Jayarajan, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 3182–3186 CrossRef CAS; (h) M. Brochetta, T. Borsari, S. Bag, S. Jana, S. Maiti, A. Porta, D. B. Werz, G. Zanoni and D. Maiti, Chem.–Eur. J., 2019, 25, 10323–10327 CrossRef CAS.
- S. Li, H. Wang, Y. Weng and G. Li, Angew. Chem., Int. Ed., 2019, 58, 18502–18507 CrossRef CAS.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS.
- Selective papers on para-C–H functionalizations using HFIP: (a) A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 14903–14907 CrossRef CAS; (b) A. Maji, A. Dahiya, G. Lu, T. Bhattacharya, P. Liu, G. Zanoni and D. Maiti, Nat. Commun., 2018, 9, 3582–3592 CrossRef; (c) S. Pimparkar, T. Bhattacharya, A. Maji, A. Saha, R. Jayarajan, U. Dutta, G. Lu, D. W. Lupton and D. Maiti, Chem.–Eur. J., 2020, 26, 11558–11564 CrossRef CAS.
- S. Bag, K. Surya, A. Mondal, R. Jayarajan, U. Dutta, S. Porey, R. B. Sunoj and D. Maiti, J. Am. Chem. Soc., 2020, 142, 12453–12466 CrossRef CAS.
- U. Dutta, S. Porey, S. Pimparkar, A. Mandal, J. Grover, A. Koodan and D. Maiti, Angew. Chem., Int. Ed., 2020, 59, 20831–20836 CrossRef CAS.
- L.-Y. Liu, J. X. Qiao, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 13831–13939 CrossRef CAS.
- G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J.-Q. Yu, Nat. Chem., 2019, 11, 571–577 CrossRef CAS.
- K. Tanaka, W. R. Ewing and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 15494–15497 CrossRef CAS.
- P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773–13777 CrossRef CAS.
- (a) K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12848–12852 CrossRef CAS; (b) F. Ghiringhelli, A. Uttry, K. K. Ghosh and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 23127–23131 CrossRef CAS.
- H. S. Park, Z. Fan, R. Y. Zhu and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853–12859 CrossRef CAS.
- J. Das, P. Dolui, W. Ali, J. P. Biswas, H. B. Chandrashekar, G. Prakash and D. Maiti, Chem. Sci., 2020, 11, 9697–9702 RSC.
- Advantages of use of Transient Directing Groups (TDGs) over covalently tethered Directing Groups (DGs) see: (a) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS; (b) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456–19464 RSC.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS.
- X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS.
- B.-B. Gou, H.-F. Liu, J. Chen and L. Zhou, Org. Lett., 2019, 21, 7084–7088 CrossRef CAS.
- (a) R.-Y. Zhu, L.-Y. Liu, H. S. Park, K. Hong, Y. Wu, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 16080–16083 CrossRef CAS; (b) L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 9594–9600 CrossRef CAS.
- X.-Y. Chen, S. Ozturk and E. J. Sorensen, Org. Lett., 2017, 19, 1140–1143 CrossRef CAS.
- K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS.
- Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26–32 CrossRef CAS.
- S. S. John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840–4847 RSC.
- R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564–3568 CrossRef CAS.
- Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884–17894 CrossRef CAS.
- J. Xu, Y. Liu, Y. Wang, Y. Li, X. Xu and Z. Jin, Org. Lett., 2017, 19, 1562–1565 CrossRef CAS.
- L. Pan, K. Yanga, G. Li and H. Ge, Chem. Commun., 2018, 54, 2759–2762 RSC.
- D. Mu, G. He and G. Chen, Chem.–Asian J., 2018, 13, 2423–2426 CrossRef CAS.
- C. Dong, L. Wu, J. Yao and K. Wei, Org. Lett., 2019, 21, 2085–2089 CrossRef CAS.
- M. Ding, W. Hua, M. Liu and F. Zhang, Org. Lett., 2020, 22, 7419–7423 CrossRef CAS.
- H. Abe, H. Amii and K. Uneyama, Org. Lett., 2001, 3, 313–315 CrossRef CAS.
- T. Sugiishi, M. Matsugi, H. Hamamoto and H. Amii, RSC Adv., 2015, 5, 17269–17282 RSC.
- Q. Dherbassy, G. Schwertz, M. Chessé, C. K. Hazra, J. Wencel-Delord and F. Colobert, Chem.–Eur. J., 2015, 22, 1735–1743 CrossRef.
- Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS.
- (a) G. Liao, B. Li, H.-M. Chen, Q. J. Yao, Y. N. Xia, J. Luo and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 17151–17155 CrossRef CAS; (b) J. Fan, Q.-J. Yao, Y.-H. Liu, G. Liao, S. Zhang and B.-F. Shi, Org. Lett., 2019, 21, 3352–3356 CrossRef CAS.
- Selected literatures on asymmetric C–H activation reactions employing HFIP as sole solvent: (a) H. Wang, Y. Park, Z. Bai, S. Chang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 7194–7201 CrossRef CAS; (b) J. Zhang, Q. Xu, J. Wu, J. Fan and M. Xie, Org. Lett., 2019, 21, 6361–6365 CrossRef CAS; (c) J. Xu, Y. Liu, J. Zhang, X. Xu and Z. Jin, Chem. Commun., 2018, 54, 689–692 RSC.
| This journal is © The Royal Society of Chemistry 2021 |