
In situ injection of dual-delivery PEG based MMP-2 sensitive hydrogels for enhanced tumor penetration and chemo-immune combination therapy†
Jianqin
Yan
ab,
Zhuangzhuang
Zhang
b,
Xiaohui
Zhan
b,
Keqi
Chen
c,
Yuji
Pu
 b,
Yan
Liang
*a and
Bin
He
b
b,
Yan
Liang
*a and
Bin
He
b
aDepartment of Pharmaceutics, School of Pharmacy, Qingdao University, Qingdao 266073, China. E-mail: liangyan072@foxmail.com
bNational Engineering Research Centre for Biomaterials, Sichuan University, Chengdu 610064, China
cDepartment of Clinical Laboratory, Qingdao Special Servicemen Recuperation Centre of PLA Navy, Qingdao 266021, China
First published on 23rd April 2021
Abstract
Improving the deep penetration of nanoparticles and realizing the combination of chemotherapy and immunotherapy have become a promising strategy for cancer treatment. Herein, a nuclear-targeted tetrahedral DNA nanostructure (NLS-TDNs, NT) was synthesized to construct matrix metalloproteinase (MMP-2) sensitive hydrogels as delivery vehicles with co-loaded disulfide cross-linked polyethyleneimine (PSP)/nuclear-targeted tetrahedral DNA (NLS-TDNs, NT)/doxorubicin (DOX) nanoparticles (NPs) (PSP/NT/DOX NPs and PNT/DOX NPs) and an immune adjuvant imiquimod (R837) to realize a combination of chemotherapy and immunization for metastatic breast cancer. Due to the membrane-breaking ability of the PNT/DOX NPs, the nanoparticles could effectively achieve deep penetration into tumor tissues, and the in situ generation of tumor-associated antigens by PNT/DOX elicited a strong immune response in the presence of R837, achieving a chemo-immune combination therapy of breast cancer, inducing the maturation of dendritic cells (DCs) and secretion of related cytokines, such as interleukin-6 (IL-6), interleukin-12 (IL-12p70) and tumor necrosis factor (TNF-α) in vitro. The combination significantly promoted the proportions of cytotoxic T cells (CD8+ CTL) and cytotoxic T cells/regulatory T cells (CD8+ CTL/Treg) (5.52% and 11.46%, respectively) and the secretion of cytokines, which cooperatively eradicated primary tumor growth (the tumor growth inhibition (TGI) value was 78.3%) and inhibited the tumor from metastasizing effectively in vivo. Our study provided the basis for activating the antitumor immune system to realize chemo-immunotherapy and tumor metastasis therapy.
Introduction
Cancer immunotherapy by utilizing the patient's own immunological systems to attack cancer cells has emerged as a powerful new generation cancer therapeutic strategy showing tremendous promise in recent years.1–4 Common immunotherapies for cancer, including cytokine therapy,5,6 checkpoint blocking therapy,7,8 and cancer vaccines9–11, have achieved exciting therapeutic results in clinical application. Among them, cancer vaccines could induce antigen-specific immunity against tumors while providing long-term immune memory effects, helping to prevent cancer recurrence.2,12–14 However, the clinical application was limited owing to the complexity of the vaccine production process and unsatisfactory therapeutic effect.15Many research studies have shown that certain doses of chemotherapy drugs such as paclitaxel, doxorubicin, and cisplatin not only trigger cytotoxic effects but also activate the host immune system and tumor immune response.16 Casares et al. in 2005 were the first to demonstrate that the anthracycline antitumor drug doxorubicin (DOX) delivered to the tumor site provides an effective stimulus for immunogenic cell death (ICD).17 The in situ tumor-associated antigens generated by DOX as immunoadjuvants, which enhanced the immune response of antigens, held great potential in combination therapy with DOX. Moreover, imiquimod (R837) was a typical toll-like receptor agonist and could serve as an immunoadjuvant to enhance the immunological stimulation of DOX.18,19
Hydrogels with 3D crosslinked networks could encapsulate bioactive substances and retain them locally within the injected lesion (e.g., organs and tumors) to achieve sustained drug release.20,21 However, for solid tumors, the tumor microenvironment (TME), which includes densely packed heterogeneous tumor cells22 and dense extracellular matrix (ECM),23 constitute extremely difficult barriers for nanoparticle delivery. Hence, the combination of deep drug penetration and long-acting drug release would greatly enhance the advantages of chemotherapy combined with immunotherapy to better cure the cancer.
In this paper, we modified TDNs with nuclear localization signal peptides for improving the drug targeting and loaded the antitumor drug DOX to prepare NLS-TDNs/DOX (NT/DOX). It was known that cationic nanoparticles could bypass endosomes, efficiently penetrate into multiple layers of cells, and access the distal cells in tumors. So, disulfide cross-linked polyethyleneimine (PSP) composite NT/DOX was used to construct and load DOX nanoparticles (PSP/NT/DOX and PNT/DOX) for deep tumor penetration. MMP-2 sensitive PEG hydrogels were synthesized and loaded with PNT/DOX and an immunoadjuvant imiquimod (R837) which is an FDA-approved immunostimulant and a potent toll-like receptor (TLR7) agonist that binds TLR7. The preparation is shown in Scheme 1. In situ administration could improve the drug targeting and achieve long-term controlled drug release. What's more, the integration of chemotherapy and immunotherapy promoted immunostimulation to effectively inhibit the tumor growth.
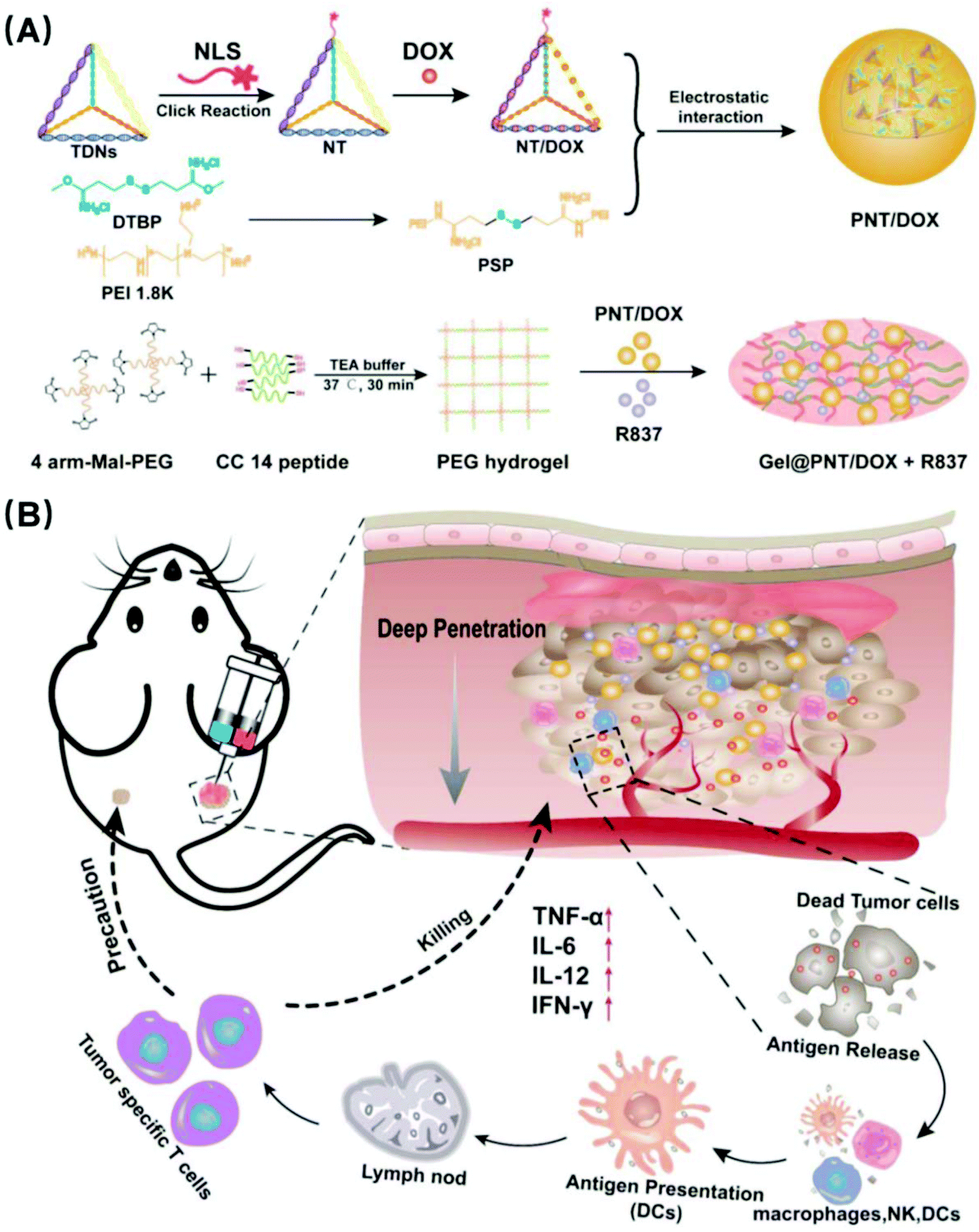 | ||
| Scheme 1 Dual-delivery vehicle based MMP-2 sensitive hydrogel loaded with PNT/DOX nanocomplexes and immunoadjuvant R837 for enhanced tumor penetration and chemo-immune combination therapy. (A) Formation and mechanism of PNT/DOX nanocomplexes and MMP-2 sensitive PEG hydrogel loaded with PNT/DOX and R837 (Gel@ PNT/DOX + R837). (B) Illustrative chemo-immune combination therapy of Gel@ PNT/DOX + R837. | ||
Experimental section
Materials and methods
All DNA oligonucleotides (the sequence is shown in Table S1†) were obtained from by Sangon Biotech (Shanghai) Co. Ltd (Shanghai, China). Nuclear localization signals (NLS, N′-GPKKKRKVEDPY-C′-propargylglycine) and an MMP degradable peptide (Ac-CRD-GPQG↓IWGQ-DRC-NH2, CC-14, 1.7 kDa) were purchased from GL Biochem (Shanghai) Co. Ltd (Shanghai, China). 4 Arm-poly(ethylene glycol)maleimide (4 arm-Mal-PEG, 20 kDa) was purchased from Toyongbio Co. Ltd (Shanghai, China). Recombinant human MMP-2 (62 kDa, 98%) was obtained from PeproTech (USA). Triethanolamine (TEA) buffer (pH 7.4, 4 mM) was purchased from Qincheng BIO Co. Ltd (Shanghai, China). GelRed DNA gel stain solution was purchased from Biotium (USA). DOX hydrochloride (DOX·HCL) was purchased from Zhejiang Hisun Pharmaceutical (Zhejiang, China). Imiquimod (R837) was obtained from Sinopharm Chemical Regent Co. Ltd (Shanghai, China). Redox-sensitive polyethyleneimine (PSP) was prepared according to a previous study.24 L929 fibroblast cells and mice breast cancer (4T1) cells were purchased from the Chinese Academy of Science Cell Bank for Type Culture Collection (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), Roswell Park Memorial Institute 1640 medium (RPMI 1640), fetal bovine serum (FBS), penicillin–streptomycin and click reaction kit were purchased from Life Technologies Co. (Gibco, USA). A calcein-AM/PI double staining kit was purchased from Dojindo Laboratories Co. (Shanghai, China).The sizes and zeta potentials of NT, NT/DOX, PNT and PNT/DOX were determined using a dynamic light scattering (DLS) spectrometer (Zeta sizer Nano ZS, Malvern Instruments, Malvern, UK). Fourier transform infrared (FT-IR) spectra of 4 arm-Mal-PEG and freeze-dried hydrogel were recorded on a FTIR spectrometer (Nicolet 8700; Thermo Fisher Scientific). The morphology of the hydrogel was visualized by scanning electron microscopy (SEM).
Synthesis and characterization of NT and PNT with or without DOX
TDNs and N3-TDNs were synthesized according to a previously reported method.25 NLS-TDNs (NT) were synthesized by using a click reaction kit. In detail, N3-TDNs (2 μM) and NLS modified with propargylglycine (2 μM) were mixed in the click kit buffer at a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and then placed into a shaker overnight (37 °C, 150 rpm). The mixture was transferred to a centrifuge tube (MW > 10 kDa), and the unreacted NLS and the dissociative ions were removed by centrifugation. Finally, the NTs were resuspended in TM buffer (20 mM Tris, 50 mM MgCl2, pH = 8) and distinguished by 12% polyacrylamide gel electrophoresis (PAGE, 1×TBE, 120 V, 1 h). DOX loaded NT (NT/DOX) was prepared as previously reported.25 In brief, DOX·HCL (500 μM) was individually mixed with NT (1 μM) at 37 °C and stirred at 150 rpm for 3 h.
:4 and then placed into a shaker overnight (37 °C, 150 rpm). The mixture was transferred to a centrifuge tube (MW > 10 kDa), and the unreacted NLS and the dissociative ions were removed by centrifugation. Finally, the NTs were resuspended in TM buffer (20 mM Tris, 50 mM MgCl2, pH = 8) and distinguished by 12% polyacrylamide gel electrophoresis (PAGE, 1×TBE, 120 V, 1 h). DOX loaded NT (NT/DOX) was prepared as previously reported.25 In brief, DOX·HCL (500 μM) was individually mixed with NT (1 μM) at 37 °C and stirred at 150 rpm for 3 h.
According to the designed polycation-nitrogen/polyanion-phosphorus ratio (N/P ratio = 30), the PSP solution was added to the NT or NT/DOX solutions and mixed via vortexing, and a correlation analysis was carried out after 30 min of incubation to obtain PSP/NT (PNT) or PSP/NT/DOX (PNT/DOX) nanocomplexes. The formation of PNT was analyzed on 1% (w/v) agarose gel (100 V, 30 min) and the PNT was stained with GelRed for 15 min. DNA bands were imaged by Molecular Imager ChemiDoc XRS+ (Bio-Rad, USA).
Biological evaluation of TDNs and NT
| Cell viability (%) = (ODtreated − ODCCK-8)/(ODcontrol − ODCCK-8) × 100% |
Preparation of MMP-2 sensitive PEG hydrogels
MMP-2 sensitive PEG hydrogels were prepared by the Michael-type addition reaction of a thiol-containing peptide (Ac-GCRD-GPQG↓IWGQ-DGCG-NH2, CC-14 peptide) onto 4 arm-Mal-PEG.26 First, pre-polymer solutions were prepared by dissolving 4 arm-Mal-PEG and thiol-containing peptide (molar ratio of Mal/SH was 1:1.1) into a TEA buffer solution (pH = 7.4, 4 mM) with a specific concentration of 5% or 7.5% (w/v). The pre-polymer solutions were mixed by vortexing and then heated at 37 °C for 30 min until the hydrogel would no longer flow by the force of gravity. DOX- or NP-loaded gels were prepared similarly except for the addition of payload (DOX, NT/DOX, or PNT/DOX) in the 4 arm-Mal-PEG solution.
Rheological study of hydrogels
To investigate the viscoelastic properties of the blank gels, the storage modulus (G′), loss modulus (G′′), and dynamic complex viscosity of the gels (5% or 7.5%) were measured. The angular frequency applied to the gels increased from 0.01 to 10 Hz with a strain of 1.0% at 25 °C. The plots of G′, G′′ and dynamic complex viscosity were obtained directly from the software controlling the rheometer. The rheological property and dynamic complex viscosity of Gel@DOX and Gel@PNT/DOX were also investigated under the above conditions.Measurement of the equilibrium swelling degree
The gel swelling ratio (SR) was measured by weight method. A certain mass (150 mg, m0) of 5% or 7.5% gel was placed in PBS (10 mL) at 37 °C. At the determined time points, the gel mass (mt) was measured after removing free water on the gel surface by using filter paper. Three parallel samples were prepared for each experiment. The SR was calculated using the following formula:| SR (%) = (mt − m0)/m0 × 100%. |
In vitro enzymatic degradation of hydrogels
Recombinant human MMP-2 was first dissolved in PBS buffer to obtain PBS buffer solutions with different MMP-2 concentrations. We then placed 7.5% hydrogels of accurate mass (m0) in different buffer media (PBS, PBS + MMP-2 100 ng mL−1, and PBS + MMP-2 200 ng mL−1) and incubated them at 37 °C. After 5 days, the samples were removed from PBS, washed with distilled water, and then dried under vacuum to obtain the gel mass (mt) after degradation. The gel mass loss ratio was calculated using the following formula: Mass loss ratio (%) = (mt − m0)/m0 × 100%.Live/dead assay
The biocompatibility and toxicity of the gels were studied by a live/dead assay. Gel with flat surface was placed on a 24-well plate and washed with PBS and culture medium for two times. L929 or 4T1 cells (1 mL, 6 × 104 cells per mL) were added and incubated for 72 h, and the medium was changed every 24 h. The medium was removed, and the cells were stained with calcein-AM/PI for 20 min. Then, the gel was carefully transferred over the cover glass and photographed by CLSM.In vitro drug release
FITC-labeled gel was prepared by using FITC-modified PSP, and the in vitro release of PNT NPs from the hydrogel was studied by fluorescence spectroscopy. A vial containing hydrogels and different buffer media (pH = 7.4, 6.8, 6.8 + 200 ng mL−1 MMP-2) was immersed in a shaker shaken at 100 rpm at 37 °C. At predetermined intervals, 1 mL of the released medium was taken out and 1 mL of fresh medium was added. The released PNT was determined using a fluorescence detector (F-7000; Hitachi Co. Ltd, Tokyo, Japan). Three parallel samples were set for each group, and the results were demonstrated as mean ± SD. The DOX release was studied similarly except that the PNT/DOX gel was placed in a dialysis tube (MWCO = 1 kDa) and DTT was added at 24 h to obtain a working concentration of 10 mM.Cellular uptake and drug transportation
4T1 cells were seeded in glass bottom culture dishes (d = 35 mm) at a density of 1 × 104 cells per mL. After the cells were incubated for 24 h, DOX·HCL, NT/DOX and PNT/DOX (DOX concentration = 10 μM) in medium were added, followed by further incubation for 1 and 4 h at 37 °C. Subsequently, the cells were washed with PBS thrice and observed by CLSM.A monolayer of 4T1 cells (1 × 105 cells) was seeded in the Transwell insert (0.4 μm pore, Corning) and incubated for 24 h. Subsequently, the cells were treated with free DOX·HCL, NT/DOX and PNT/DOX for 4 h (DOX concentration: 10 μM). After 4 h, the medium was removed, and the cells were washed with PBS. Fresh medium was added, and the 4T1 cells were co-incubated with 4T1 cells at the lower chamber for 24 h. The lower chamber cells were stained with Hoechst 33342 and observed by CLSM.
Multicellular tumor spheroids
To prepare multicellular tumor spheroids (MTSs) based on 4T1 cells, 1 mL of a sterile 1.5% agarose (w/v) solution was dispensed into 6-well plates and cooled at 4 °C for solidification. A suspension of the 4T1 cells was seeded in the well and incubated at 37 °C for 5 days. When the MTSs reached a diameter of ∼200 μm, the spheroids were transferred to confocal dishes and treated with DOX·HCL, NT/DOX, and PNT/DOX (DOX concentration = 10 μM) for 4 h. The spheroids were then washed with PBS and observed by CLSM in the layer scan mode. Topological 3D view images were obtained by the Image J software.In vitro dendritic cell (DC) activation and cytokine analysis
DCs were isolated from the bone marrow of BALB/c mice, which were purchased from Dashuo (Chengdu) Biotechnology Co. Ltd according to an established method.27 The DCs were treated with medium containing DOX·HCL, R837, DOX + R837, PNT, NT/DOX, PNT/DOX, or PNT/DOX + R837 (DOX and R837 concentrations were 10 μM and 5 μg mL−1, respectively) using a Transwell system. After various treatments, the DCs were stained with anti-mouse CD11c FITC, anti-mouse CD86 PE, and anti-mouse CD80 APC (BioLegend, USA) for flow cytometry analysis. Cytokine analysis was performed by using ELISA kits (Dakewe Biotech Co., Ltd Shenzhen, China) according to the provided protocols.In vivo antitumor activity
Animal experiments were performed in accordance with the Experimental Animal Administrative Committee of Sichuan University and were in line with the American Association for Laboratory Animal Science (AAALAS) guidelines. Male BALB/c mice (initial weight 18–20 g, 6 weeks-of-age) were purchased from Dashuo (Chengdu) Biotechnology Co. Ltd. 4T1 cells suspended in PBS (2 × 106 cells in 100 μL) were subcutaneously injected into the right back of the mice. The drug was administered until the tumor volumes reached ∼150 mm3. The tumor volume (V) was calculated using the length (L) and width (W) as V = L × W2/2. The mice were randomly divided into 6 groups and intratumorally injected with hydrogels loaded with saline (group 1), DOX·HCL (group 2), DOX·HCL + R837 (group 3), NT/DOX (group 4), PNT/DOX (group 5), and PNT/DOX + R837 (group 6). All the groups were injected four times at a 3-day interval at a DOX dose of 5.0 mg kg−1 and/or an R837 dose of 0.35 mg kg−1. After 20 d, the mice were randomly sacrificed. Tumors and major organs were excised for hematoxylin and eosin (H&E) staining and immunohistochemistry analysis.Ex vivo analysis of different groups of T cells and cytokine
After a 3-d treatment, tumors were harvested and homogenized into a single cell suspension. Cells were then stained with anti-mouse CD3, anti-mouse CD4, and anti-mouse CD8a25 (BioLegend, USA) antibodies according to the manufacturer's protocols. To analyze the CD4+ helper T cells, the cells were further stained with anti-Foxp3 (BioLegend, USA) antibodies according to the standard protocol. CD8+ CTLs in the tumor tissues were analyzed by immunofluorescence staining.Serum samples from mice after various treatments were collected for cytokine analysis. Interleukin-6 (IL-6), interleukin-12 (IL-12p70), tumor necrosis factor (TNF-α), and tumor necrosis factor interferon gamma (IFN-γ) in serum were analyzed using ELISA kits according to the vendors’ instructions.
Distant tumor inhibition
When the tumor at the right back reached a volume of 100 mm3, 4T1 cells (2 × 106 cells in 100 μL) were inoculated on the left back to build a distant tumor model. The right back tumors were then treated as described above. The distant tumor volumes were monitored every three days.Statistical analysis
All the data were expressed as mean ± standard deviation (SD). The statistical significance was assessed by Student's t-test. The statistical significance was set at *p < 0.05, and the extreme significance was set at **p < 0.01 and ***p < 0.001.Results and discussion
Synthesis and characterization of MMP-2 sensitive PEG hydrogels
As shown in Fig. S1,† MMP-2 sensitive PEG hydrogels were prepared by crosslinking of 4-arm PEG using dithiol peptide (CC-14) as the crosslinking agent through Michael addition. 4 arm-Mal-PEG (20 kDa) was cross-linked into a hydrogel, which was performed via FTIR and compared with uncrosslinked 4 arm-Mal-PEG. The FTIR spectra of the as-prepared PEG hydrogel are shown in Fig. S1D;† the addition reaction was confirmed by the disappearance of the typical vibration absorption peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1709 cm−1) in 4 arm-Mal-PEG. Two PEG concentrations (5.0% and 7.5%, w/v) were tried to prepare hydrogels, and the gel formation was evaluated by a vial-tilting method. Obviously, 7.5% PEG could efficiently form hydrogel with higher mechanical strength and denser pore structure, which was revealed by SEM (Fig. 1A and S2A†). The porous structure of the hydrogels was expected to facilitate the cell infiltration and the drug release of nanoparticles.
C (1709 cm−1) in 4 arm-Mal-PEG. Two PEG concentrations (5.0% and 7.5%, w/v) were tried to prepare hydrogels, and the gel formation was evaluated by a vial-tilting method. Obviously, 7.5% PEG could efficiently form hydrogel with higher mechanical strength and denser pore structure, which was revealed by SEM (Fig. 1A and S2A†). The porous structure of the hydrogels was expected to facilitate the cell infiltration and the drug release of nanoparticles.
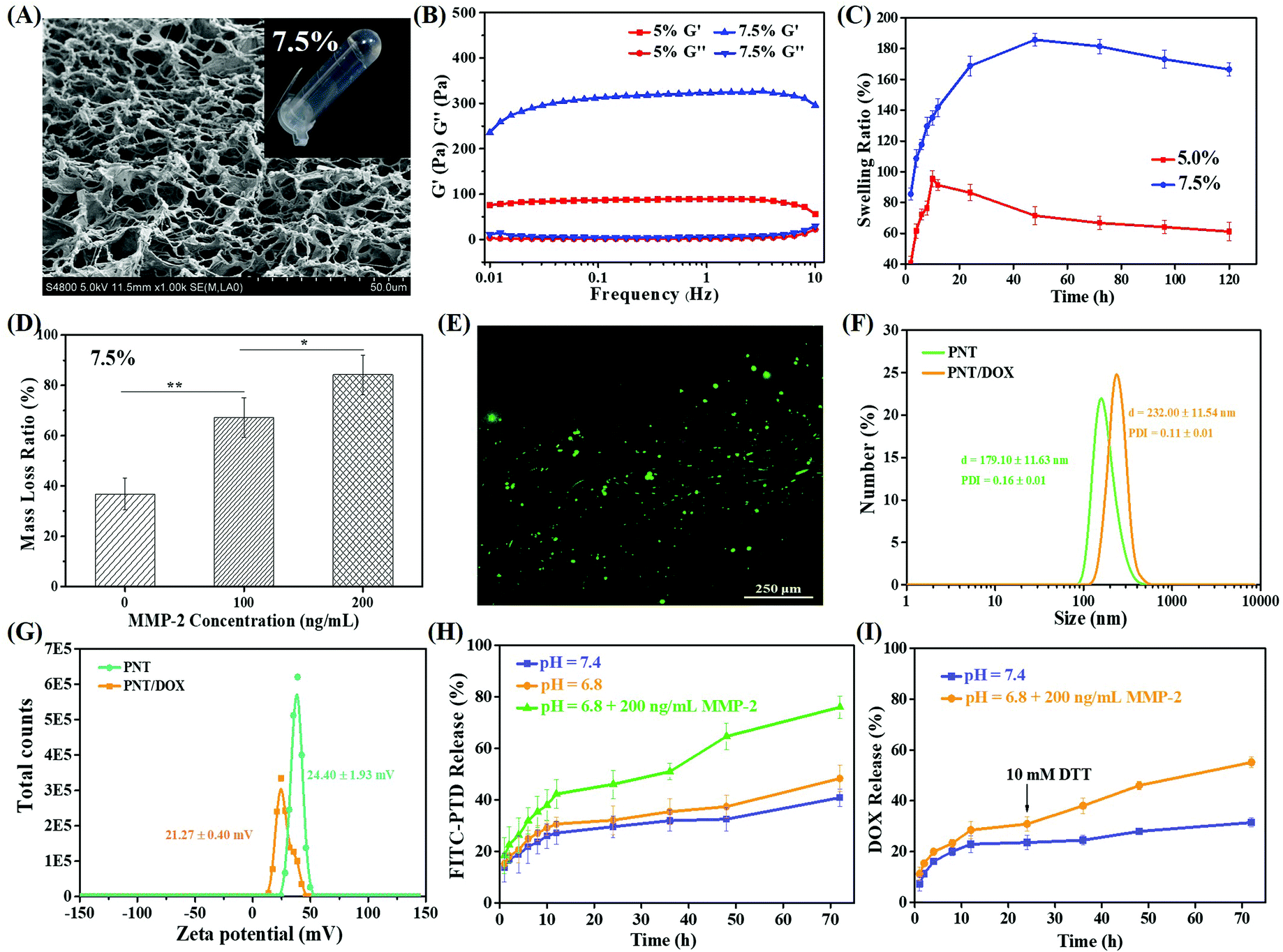 | ||
| Fig. 1 (A) SEM image and photograph (inset) of 7.5% PEG hydrogels. The rheological properties (B) and swelling behavior (C) of 5% and 7.5% PEG hydrogels. (D) Weight loss ratios of 7.5% PEG hydrogels upon treatment with different concentrations of MMP-2. (E) Live/dead assay of 4T1 cells treated with 7.5% PEG hydrogels; live and dead cells were green and red, respectively. The sizes (F) and zeta potentials (G) of PNT and PNT/DOX. The in vitro release profiles of PNT (H) and DOX (I) from 7.5% PEG hydrogels under different conditions at 37 °C. | ||
The viscoelastic properties of both PEG hydrogels (5.0% and 7.5%, w/v) were investigated, and the results are shown in Fig. 1B. Both hydrogels showed a higher storage modulus (G′) than corresponding loss modulus (G′′), indicating the formation of real hydrogels. Besides, 7.5% PEG hydrogels had a G′ value four-fold that of 5.0% PEG hydrogels, indicating more effective intermolecular crosslinking density and higher mechanical properties.28–30 The frequency dependence of the dynamic complex viscosity indicated that both PEG hydrogels had a shear-thinning behavior (Fig. S2B†). Under the same conditions, the complex viscosity of 7.5% PEG hydrogels was higher than that of 5% PEG hydrogels, suggesting that 7.5% hydrogel had higher cross-linking density and more compact gel mesh structure. This observation was consistent with the SEM and rheology results.
The swelling ability of the hydrogels played an important role in the drug release rate. Fig. 1C shows the swelling results of both PEG hydrogels within 120 h. Both PEG hydrogels showed fast swelling before 10 h. The highest (or equilibrium) swelling ratios of 5.0% and 7.5% PEG hydrogels were 190% and 100%, which were obtained at 12 h and 48 h, respectively. The swelling behavior of the PEG hydrogels mainly depended on the hydrogen bonding between water molecules and CO of 4 arm-Mal-PEG, so the different PEG concentrations and cross-linking densities of the hydrophilic group in the two PEG hydrogels led to different equilibrium swelling rates. Given that the 7.5% PEG hydrogels showed higher mechanical strength and denser pore structure, they were used for the following study. Meanwhile, the rheological properties and dynamic complex viscosity of 7.5% PEG hydrogels loaded with DOX or PNT/DOX (Gel@DOX or Gel@PNT/DOX) were also tested (Fig. S2F–H†). These results were consistent with those of blank PEG hydrogels.
The biodegradability of 7.5% PEG hydrogels was evaluated in an MMP-2 solution. As shown in Fig. 1D, the PEG hydrogel weight loss followed an MMP-2 concentration-dependent manner; the higher the MMP-2 concentration, the more the weight loss. In addition, about 84% weight loss was observed when the PEG hydrogels were treated with 200 ng mL−1 MMP-2 for 5 days, and a picture of the completely degraded hydrogel is shown in Fig. S2E.† These results clearly manifested the MMP-2-responsiveness of our PEG hydrogels. The cytocompatibility of the PEG hydrogels was evaluated by CCK-8 assay and a live/dead assay. In Fig. S2D,† the cell viabilities of the medium were used as the control, and the cell viabilities were beyond 90% for the gelators’ extracts, even when the concentration of the gelator was as high as 30 mg mL−1. This proved that the gels were non-toxic to the cells. In addition, most L929 and 4T1 cells were alive after 72-h incubation on the PEG hydrogel surface (Fig. 1E and S2C†), indicating the low cytotoxicity and good biocompatibility of the PEG hydrogels.
Synthesis and in vitro biological evaluation of PNT/DOX
TDNs were readily synthesized by the self-assembly of four tailor-made 55-mer ssDNA (Table S1†) via complementary base pairing interactions. The successful synthesis of TDNs was demonstrated by the gel retardation assay (Fig. S3†). A nuclear localization signal peptide (N′-GPKKKRKVEDPY-C′-propargylglycine, NLS) was conjugated to TNDs via alkyne–azide click chemistry. The synthesis of nuclear targeting NLS-TDNs (denoted as NT) was verified by polyacrylamide gel electrophoresis (PAGE). The size and zeta potential of NT and NT/DOX were confirmed by DLS, and the mean diameter and zeta potential of the NT were 13.5 ± 4.4 nm and −4.1 ± 1.8 mV, respectively (Fig. S4†). The cytocompatibility of the TDNs and NT was evaluated by a CCK-8 assay. As shown in Fig. S6A and B,† more than 80% L929 and 4T1 cells were alive after co-incubation with TDNs and NT for 48 h, suggesting that blank DNA nanostructures were safe drug carriers.The anticancer drug doxorubicin (DOX) was then loaded into NT to obtain NT/DOX, which showed a size of 20.5 ± 3.7 nm and surface charge of −12.0 ± 0.9 mV as shown in Fig. S4.† The nuclear targeting ability of the TN was studied by CLSM using non-nuclear targeting TDNs as a control (TDNs were modified with Cy5). Green fluorescence of Cy5-TDNs was mainly distributed in the cytoplasm, and no obvious DOX fluorescence was observed in the nucleus (Fig. S5†) when 4T1 cells were treated with TDNs for 4 h. In contrast, the Cy5 fluorescence of NT was clearly observed in the nuclear compartment. The co-localization of green (Cy5) and blue (Hoechst 33342) fluorescence further indicated the excellent nuclear targeting ability of NT.
The in vitro antitumor efficacy of TDNs/DOX and NT/DOX in 4T1 cells was then investigated by using DOX·HCL as the control. The cells were treated with different DOX-containing formulations for 48 h, and the cell viability results are shown in Fig. S6C.† The half maximal inhibitory concentration (IC50) values of DOX·HCL, TDNs/DOX, and NT/DOX were 1.0, 1.7, and 3.0 μM, respectively. DOX·HCL exhibited the best anticancer efficiency due to its small molecular nature and excellent water solubility, which facilitated quick diffusion into the cells. NT/DOX had a lower IC50 value than TDNs/DOX, and this was probably because NT/DOX could effectively promote the delivery of DOX to the cell nucleus.
Redox-sensitive PEI was then complexed with negatively charged TDNs (or TDNs/DOX) by electrostatic interactions. The as-prepared PSP/NLS-TDNs (PNT) with an optimal N/P ratio of 30 (Fig. S7†) were used for the following study. The mean diameters and zeta potential of PNT were 179.1 ± 11.6 nm and 24.4 ± 1.9 mV, respectively. After DOX was loaded, PNT/DOX showed a size of 232.0 ± 11.5 nm, much larger than that of NT/DOX and PNT. The zeta potential was 21.3 ± 0.4 mV, confirming the generation of PNT/DOX complexes (Fig. 1F and G). The drug loading efficiency of PNT/DOX was 37.6 ± 2.1%.
The intracellular internalization of DOX loaded nanocomplexes was studied by using CLSM in the 4T1 cells (Fig. 2A and Fig. S8†). With the incubation time increased from 1 to 4 h, a large amount of DOX was successfully internalized into the cytoplasm. DOX·HCL and PNT/DOX exhibited higher fluorescence intensity compared to NT groups in the nucleus for 1 and 4 h. DOX·HCL exhibited higher fluorescence intensity due to its water solubility, allowing for quick diffusion into the cells. Notably, the PNT/DOX group resulted in a sharp increase in fluorescent signals in the 4T1 cells, indicating that the highly cationic nanocomplexes could improve the permeability of cell membranes and facilitate the internalization.31
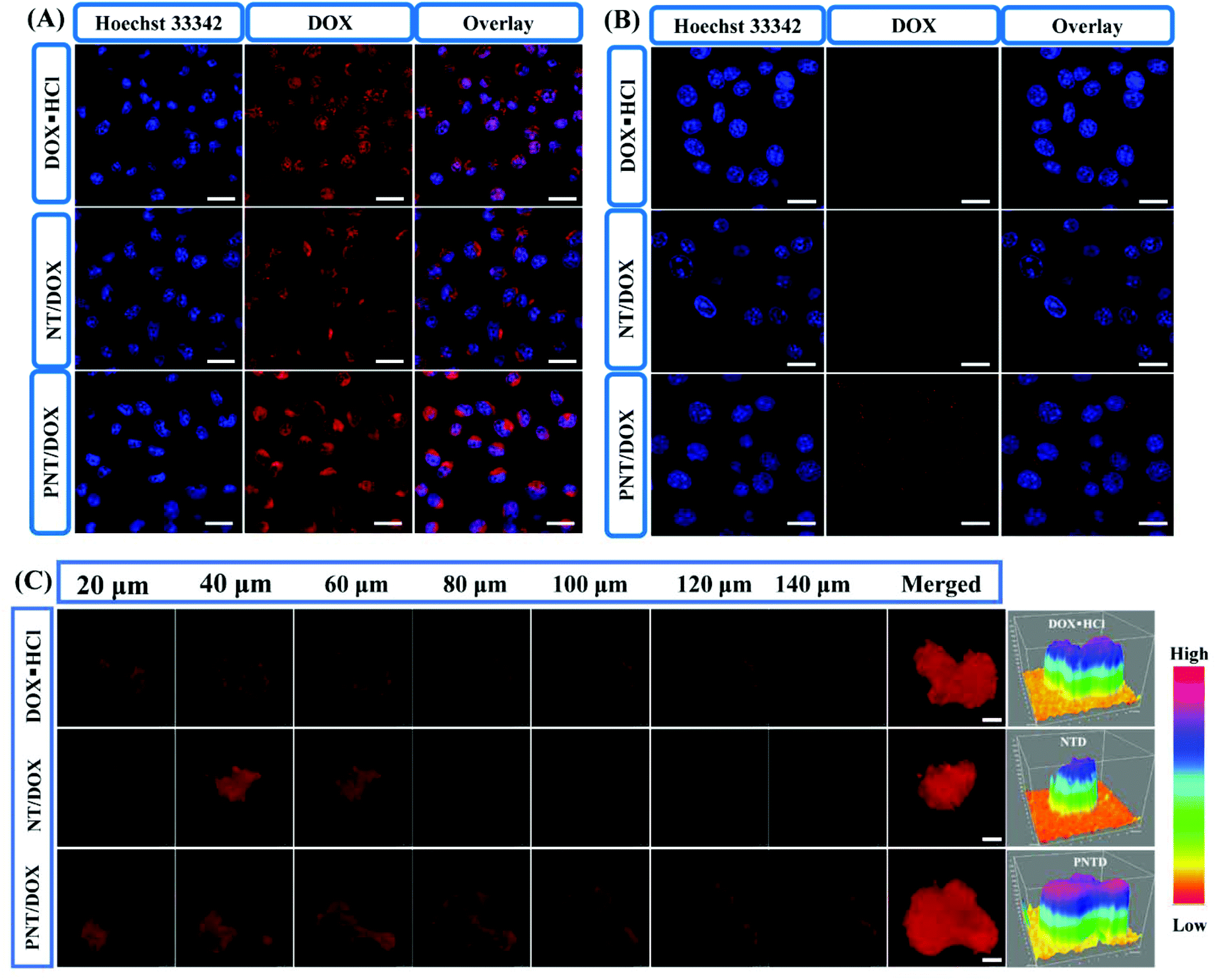 | ||
| Fig. 2 (A) The CLSM images of 4T1 cells after treatment with free DOX, NT/DOX, and PNT/DOX for 4 h. (B) CLSM images of 4T1 cells in the low chamber of Transwell. (C) The CLSM images of 4T1 MTS after 4 h incubation with free DOX, NT/DOX, and PNT/DOX (DOX concentration = 10 μM, the scale bar in CLSM images represented 25 μm). | ||
We used a Transwell model to evaluate whether PNT/DOX could “infect” different batch cells or not (Fig. 2B). The 4T1 cells in the insert were treated with free DOX·HCL, NT/DOX and PNT/DOX for 4 h. The pretreated 4T1 cells in the insert were co-incubated with 4T1 cells at the bottom chamber for 24 h. As expected, the PNT/DOX group showed the strongest DOX signal, indicating that PNT/DOX had penetrated into the 4T1 cells in the lower chamber. The result suggested that PNT/DOX had the potential to penetrate into deep tumor cells through “infection” cells, attributed to the size change of PNT/DOX under the biomimetic tumor intracellular conditions (GSH-contained solution) for efficient deep tumor penetration.31
To further investigate the tumor-penetrating ability of PNT/DOX, multi-cellular tumor spheroids (MTSs) were constructed as 3D tumor models to make in vitro models. After treatment with DOX·HCL and NT/DOX, weak fluorescence signals were detected in the 4T1 MTSs (Fig. 2C). In contrast, the PNT/DOX group showed a higher fluorescence intensity in each optical section (>140 μm). The reason for this result may be that the presence of PSP leads to cell perforation, which in turn promoted cell internalization and enhanced cell penetration.32–34 Topological 3D view images more visually reflected the total fluorescence intensity in the 4T1 MTSs. The highest fluorescence intensity was observed in the PNT/DOX group, which was consistent with the CLSM results. The deep penetration property of PNT/DOX in the tumor site was of a great significance for drug enrichment and tumor treatment, which resolved the difficulties of poor drug distribution and accumulation in drug-resistance tumors.
In vitro drug release profiles
The in vitro release of PNT from the PEG gel containing FITC-labeled PNT was studied to simulate the PNT/DOX release behavior. As shown in Fig. 1H, the PNT release was pH independent; about 50% PNT was released at 72 h under the conditions of pH 7.4 and 6.8. The PNT release was significantly accelerated in the presence of 200 ng mL−1 MMP-2, and the cumulative release amount was 80% at 72 h. The MMP-2 enzyme-enhanced drug release suggested the potential of tumor-specific activation of drug release.In addition, the DOX release behavior from Gel@ PNT/DOX was studied, and the results are shown in Fig. 1I. The DOX release was slightly faster at pH 6.8 than that at pH 7.4 within 24 h. Upon addition of 10 mM DTT at 24 h, the release rate of DOX significantly increased. This was because DTT could cleave the S–S bond in PSP and disassociate nanoparticles, thereby accelerating the release of DOX. Together, given the MMP-2- and GSH-accelerated nanoparticles and DOX release, PNT/DOX-Gel held the potential for tumor-specific inhibition.
In vitro dendritic cell activation and cytokine secretion
We envisioned that Gel@PNT/DOX + R837 could induce ICD and release tumor-associated antigens after the intratumoral administration, which could further be captured by APCs for immunotherapy in the presence of immunoadjuvants. Dendritic cells (DCs), an important type of antigen-presenting cells, were responsible for activating and regulating the innate and adaptive immunities.35 Immature DCs could be activated by antigens and/or immunoadjuvants, releasing important immune-related cytokines,36 including interleukin-6 (IL-6), interleukin-12 (IL-12) and tumor necrosis factor α (TNF-α). To mimic the in vitro activation of PNT/DOX + R837, a Transwell system was designed and is shown in Fig. 3A. We evaluated the maturation of the DCs upon treatment with cancer vaccines that were generated by 4T1 cells after treatment with PNT/DOX + R837. The mature DCs were quantitatively determined by the measurement of co-stimulatory molecules CD11c, CD80 and CD86 by using flow cytometry. The CD11c positive cells were firstly gated, and as shown in Fig. 3B and C, DOX·HCL, R837, NT/DOX, and PNT/DOX could lead to an obvious improvement in DC maturation relative to the control group. The PNT/DOX + R837 group induced the highest DC maturation of 72.4%. Besides, PEI also exerted an adjuvant effect on BMDCs, which was consistent with a previous study reported by David J. Mooney et al.37 The significant immune response of DCs to PNT/DOX + R837 was also confirmed by the level of cytokines (Fig. 3D–F). DOX·HCL induced a moderate release of cytokines. R837, as an immunoadjuvant, significantly promoted the release of IL-6, IL-12, and TNF-α. As expected, PNT/DOX + R837 induced a significant release of all these three cytokines, much higher than those elicited by R837 or DOX alone. The results were mainly derived from the PNT/DOX-mediated enhancement of ICD, as well as the adjuvant effect of PSP and R837.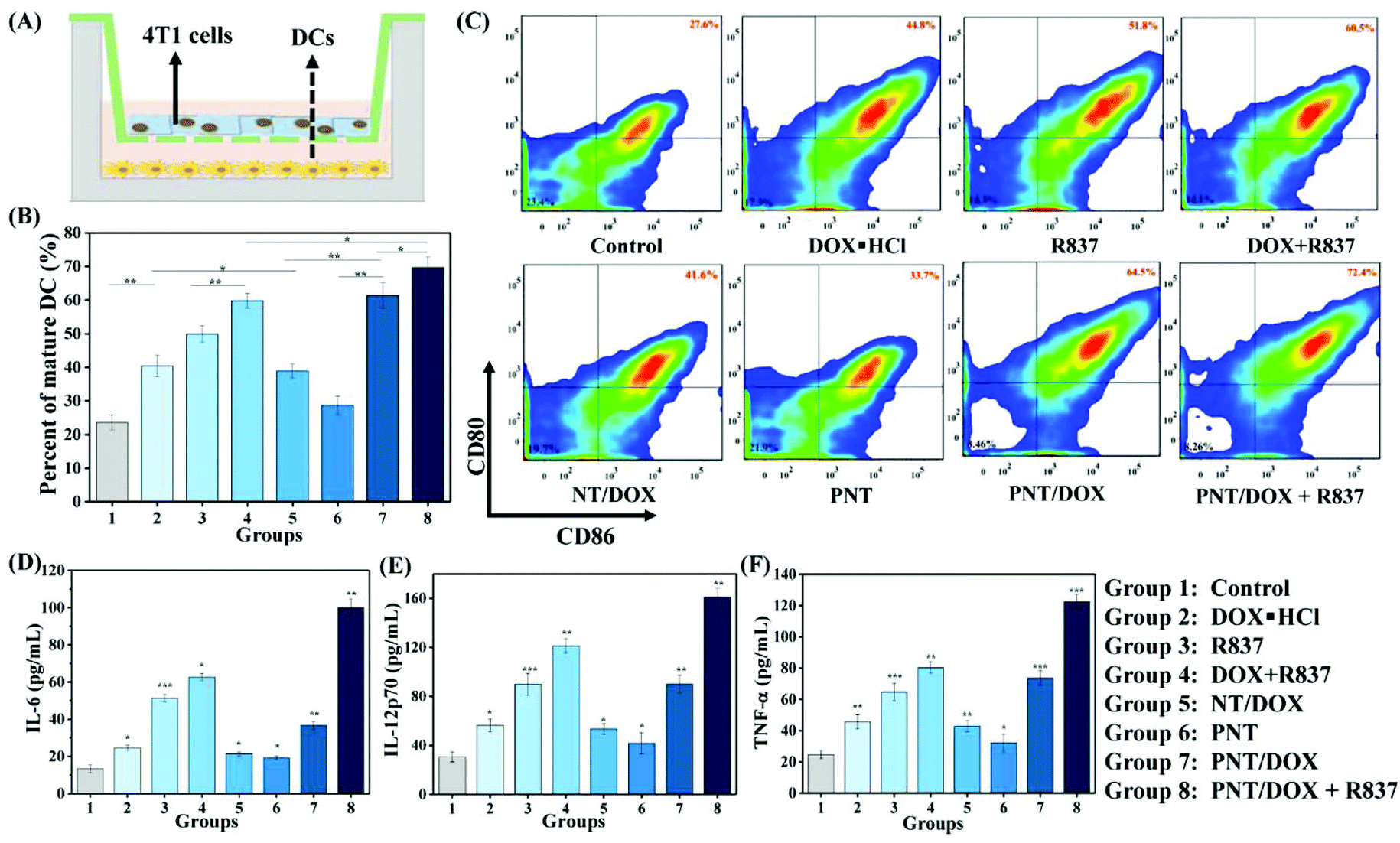 | ||
| Fig. 3 Maturation of bone-marrow-derived DCs. (A) A scheme illustrating the transwell co-culture system. (B) The statistical data of mature DCs (CD11c+ CD80+ CD86+) after different treatments. (C) In vitro DC maturation after various treatments for assessment by flow cytometry after staining with CD11c, CD80 and CD86. ELISA detection of (D) IL-6, (E) IL-12p70 and (F) TNF-α (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 vs. control). | ||
In vivo antitumor efficacy of Gel@PNT/DOX + R837
Encouraged by the excellent in vitro anticancer efficacy and immune response efficiency, we then performed an in vivo antitumor study in a 4T1 mouse tumor model. Balb/c male mice were randomly divided into six groups and intratumorally injected with Gel@saline, Gel@DOX·HCL, Gel@DOX + R837, Gel@NT/DOX, Gel@PNT/DOX and Gel@PNT/DOX + R837. The weight and tumor volume changes were monitored, and the results are shown in Fig. 4A and B. The maximum weight loss of the mice in the Gel@DOX·HCL and Gel@DOX + R837 groups was about 8%, indicating the relatively low systemic toxicity of local DOX delivery.38 The Gel@PNT/DOX + R837 group exhibited a negligible weight loss, and the weight increase trend was similar to that of the Gel@saline group (Fig. 4A). The low toxicity of Gel@PNT/DOX + R837 was probably due to the tumor environment-activated DOX release. As shown in Fig. 4B, Gel@saline could not inhibit the tumor growth, and the tumor volume rapidly increased (12V0 at the 18th day; V0 is the tumor volume at day 0). All the treatment groups could somehow efficiently suppress the tumor burdens. Gel@PNT/DOX + R837 had the most significant tumor inhibition effect, better than those of Gel@DOX + R837 and Gel@PNT/DOX. The result indicated the vital role of redox-sensitive NPs and immunoadjuvant R837. Additionally, all the tumors were excised from the mice at the end of therapy and weighed to obtain the tumor growth inhibition (TGI). The Gel@PNT/DOX + R837 group had the lightest tumor weight of 0.247 ± 0.08 g and the highest TGI value of 78.3% (Fig. 4C). The survival rate of the mice in the Gel@PNT/DOX + R837 group was as high as 60% at day 50, much higher than those for the other treatment groups (Fig. 4D). Ultimately, Gel@PNT/DOX + R837 exhibited the best tumor inhibition effect and low systemic toxicity.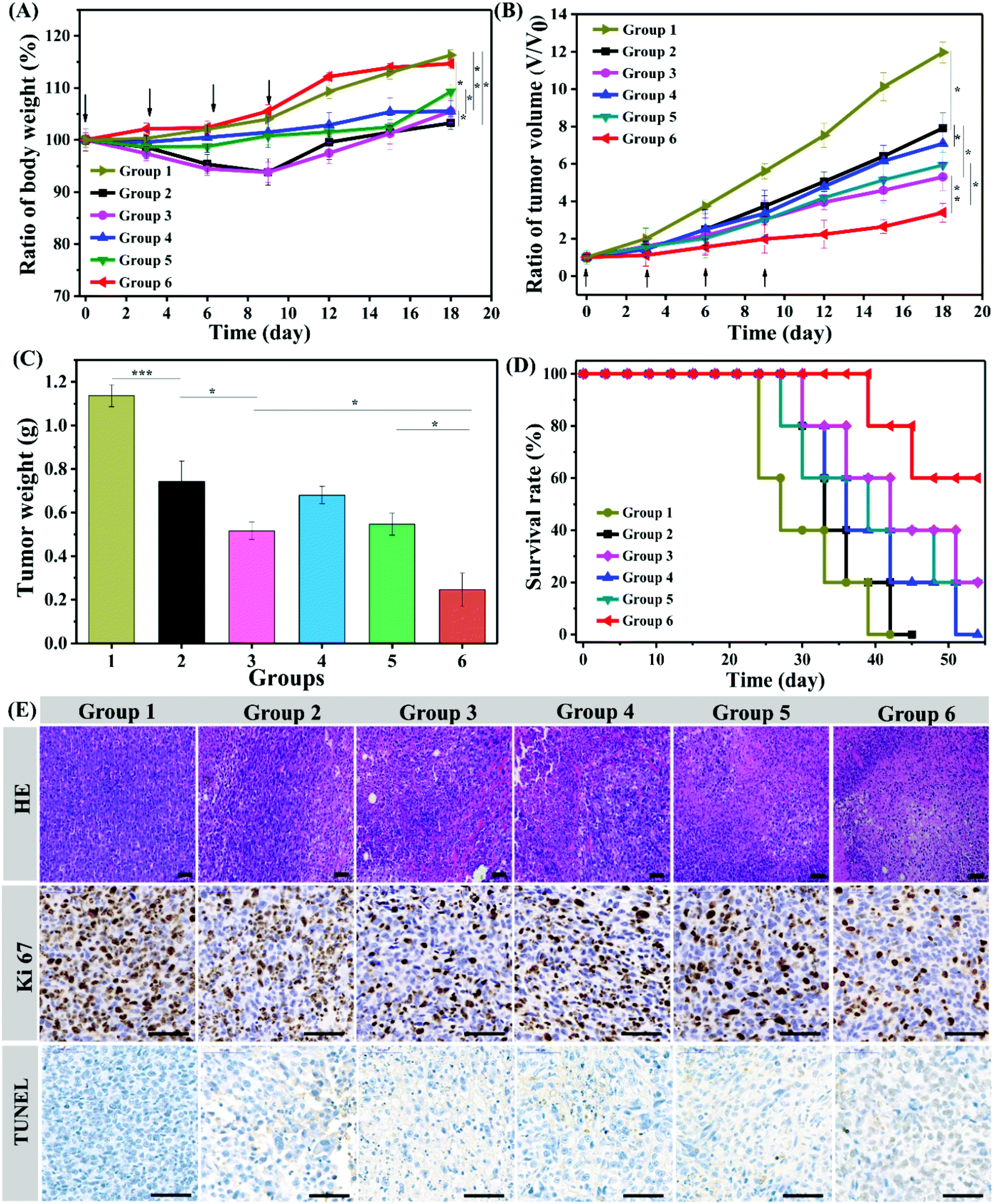 | ||
| Fig. 4 In vivo antitumor evaluation of PEG hydrogel formulations in 4T1 tumor-bearing BALB/c mice (DOX dose: 5 mg kg−1; R837 dose: 0.35 mg kg−1). Body weight (A) and tumor volume (B) changes over time. (C) Tumor weights on the 18th day. (D) Survival rates of mice in different treatment groups. (E) Histological analysis of tumor tissues that were immunohistochemically stained with H&E, Ki67, and TUNEL (n = 5. *p < 0.05, **p < 0.01, and ***p < 0.001). Group 1: Gel@saline, group 2: Gel@DOX·HCL, group 3: Gel@DOX + R837, group 4: Gel@NT/DOX, group 5: Gel@PNT/DOX, group 6: Gel@PNT/DOX + R837. | ||
The antitumor efficacy of the PEG hydrogels was further studied by hematoxylin and eosin (H&E) staining of tumor slices (Fig. 4E). Gel@PNT/DOX + R837 caused the most severe damage and necrosis in the tumor tissues. Meanwhile, an immunohistochemical study manifested that few Ki-67-positive proliferating cells and many TUNEL-positive apoptotic cells were clearly observed in the tumor tissues of the Gel@PNT/DOX + R837 group, in contrast to the proliferating cells and fewer apoptotic cells in the other control groups. These results were in good agreement with the tumor-volume-based tumor inhibition results. In addition, histological analysis of different organs presented that the Gel@DOX and Gel@DOX + R837 groups resulted in mild local bleeding with a degree of cardiotoxicity. Simultaneously, the Gel@saline, Gel@DOX and Gel@DOX + R837 groups exhibited different degrees of renal interstitial hemorrhage and more granulocyte infiltration, while the other groups did not exhibit obvious toxic side effects on tissues and organs (Fig. S10†).
To explore whether Gel@PNT/DOX + R837 could induce in vivo immune activation and immunotherapy of cancer, immune-fluorescence staining of tumor-infiltrating T cells was carried out. As shown in Fig. 5A, Gel@PNT/DOX + R837 significantly enhanced the infiltration of CD8+ T cells in comparison with the other treatments. CD3+ cells were firstly gated, and the percentage of CD8+ T cells in tumor was further quantified by flow cytometry (Fig. 5B and C). Obviously, the Gel@PNT/DOX + R837 group showed the highest percentage of CD8+ T cells (5.52%), higher than those of the Gel@DOX + R837 (3.69%) and Gel@PNT/DOX (3.51%) groups. Meanwhile, for the evaluation of ICD, ATP production in 4T1 cells and the expression of CRT and HMGB1 were demonstrated. As shown in Fig. S11,† the results showed that the Gel@PNT/DOX + R837 group caused a higher level of ATP production, and calreticulin (CRT) exposure and high mobility group box 1 (HMGB1) on the 4T1 tumor were also the highest. Although the elicitation of ICD was favorable for antitumor immune responses, there existed immune-suppressive mechanisms within the tumor to compensate for antitumor immunities.39,40 For example, helper T cells (CD3+ CD4+ CD8−) played important roles in regulating the adaptive immunities. It was known that CD4+ helper T cells could be classified into effector T cells (CD3+ CD4+ Foxp3−, Teff) and were in favor of immune responses, together with regulatory T cells (CD3+ CD4+ Foxp3+, Treg), which played positive and negative roles in the antitumor immune response, respectively.41,42 In Fig. 5D and E, an obvious reduction in the Treg level was observed in the Gel@PNT/DOX + R837 group, which had the highest CD8+/Treg ratio of 11.5%. Moreover, the cytokine level in serum was analyzed to further verify the immune response. Four proinflammatory cytokines including IL-6, IL-12p70, IFN-γ, and TNF-α were detected, and all the treatment groups were able to increase the secretion of these cytokines. As expected, Gel@PNT/DOX + R837 caused the most secretion of cytokines (Fig. 5F–I), implying the strongest immune response. In summary, Gel@PNT/DOX + R837 showed excellent tumor inhibition and effectively activated the anti-tumor immune response in vivo, achieving a synergistic chemo-immunotherapy.
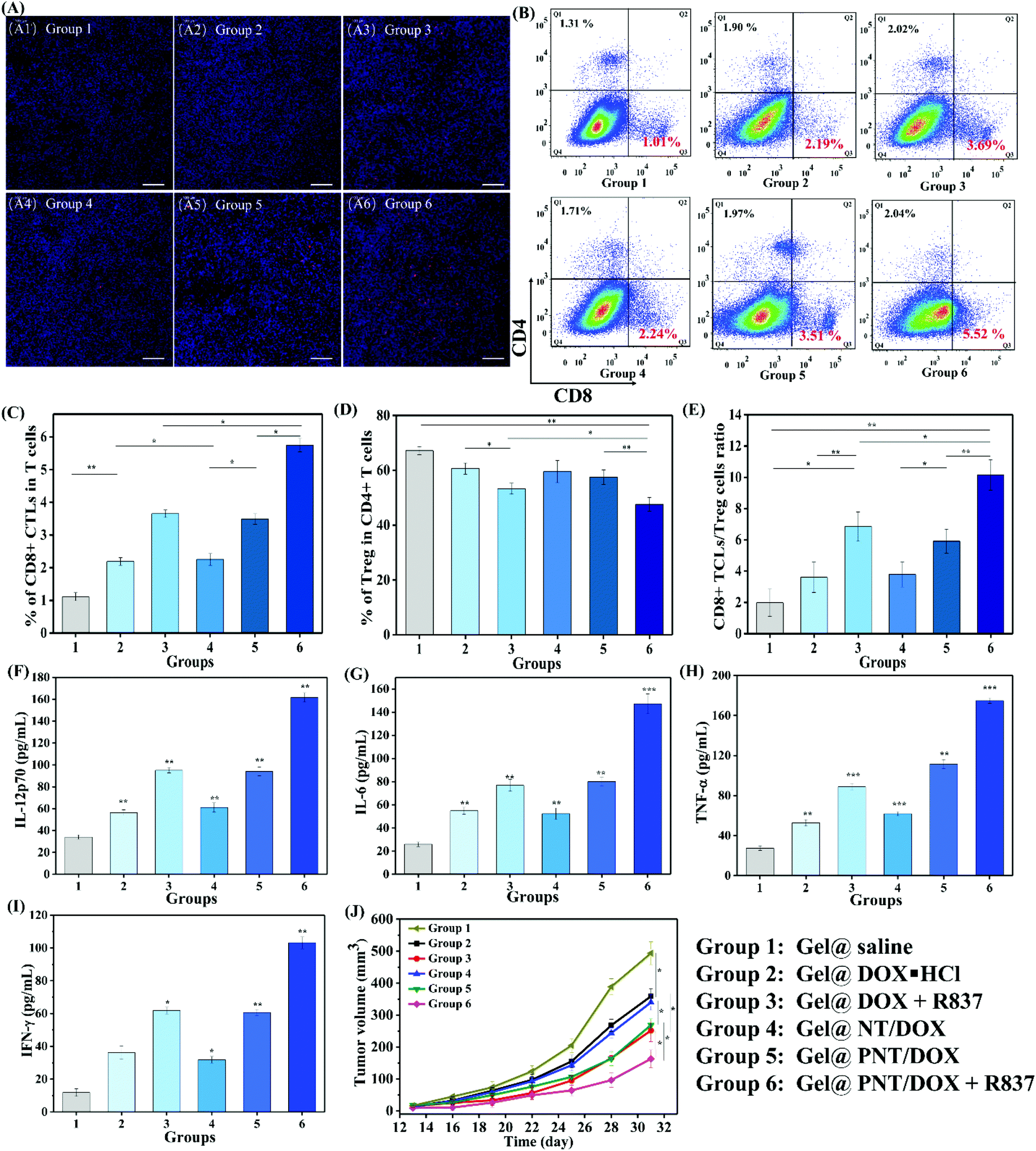 | ||
| Fig. 5 (A) The CLSM images of CD8+ CTL staining of tumors; the scale bars represented 100 μm. (B) The different types of T cells in tumors by flow cytometry. (C) Proportions of CD8+ killer T cells. (D) Proportions of FoxP3+ Tregs in CD4+ T cells, and (E) CD8+ CTL/Treg ratios in tumors for different groups (n = 5). IL-12p70 (F), IL-6 (G), TNF-α (H), and IFN-γ (I) levels in mouse serum (n = 3). (J) The tumor volume change curves of the secondary tumors (n = 5, *p < 0.05; **p < 0.01; ***p < 0.001). | ||
To further confirm the immune response and immunotherapy effect, a distant tumor model was established (Fig. S9†) by subcutaneous injection of 4T1 cells until the primary tumor volume reached a volume of 100 mm3 (day 7). The distant tumor volume changes are shown in Fig. 5J. Gel@DOX + R837 and Gel@PNT/DOX induced better distant tumor inhibition than Gel@DOX·HCL and Gel@NT/DOX, indicating better immune response. This result was in agreement with the serum cytokine results. Importantly, the Gel@PNT/DOX + R837 group showed the best inhibition of distant tumor growth where the distant tumor volume was 164 mm3 at day 31, much smaller than that of the Gel@saline group (493 mm3).
Conclusions
In summary, we developed dual-delivery PEG based MMP-2 sensitive hydrogels for enhanced tumor penetration and chemo-immune combination therapy by co-delivering DOX-loaded nanoparticles (PNT/DOX) and an immunoadjuvant (R837). PNT/DOX could be released from the PEG hydrogels by MMP-2 activation and liberate DOX under the condition of high intracellular GSH concentration, realizing an enhanced chemotherapy effect and tumor penetration. The combination of PNT/DOX and R837 significantly induced the maturation of DCs and secretion of the related cytokines both in vitro and in vivo. Gel@PNT/DOX + R837 induced the most severe cancer cell apoptosis and necrosis and the most intense immune response, thereby presenting the most significant antitumor effect in both primary and secondary tumors. Meanwhile, the PEG hydrogel delivery system exhibited excellent biocompatibility and low toxicity to normal organs. The hydrogel-based in situ delivery system presents a robust platform for efficient cancer synergistic therapy and low systemic toxicity.Author contributions
Yan Liang conceived and designed the research. Jianqin Yan developed methods, analyzed data, and organized figures. Jianqin Yan and Yan Liang wrote the manuscript. Yuji Pu and Bin He reviewed and revised the manuscript. Yan Liang and Jianqin Yan performed most of the experiments. Zhuangzhuang Zhang, Xiaohui Zhan and Keqi Chen participated in the animal experiments. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (51803098, 51873121 and 51773130) and the Department of Science and Technology of Sichuan Province (2019YJ0057). The authors also thank the Centre of Testing and Analysis, Sichuan University, for FTIR measurements, and Life Science Core Facilities, College of Life Sciences, Sichuan University, for FCM measurements.Notes and references
- S. A. Rosenberg, N. P. Restifo, J. C. Yang, R. A. Morgan and M. E. Dudley, Nat. Rev. Cancer, 2008, 8, 299–308 CrossRef CAS PubMed.
- S. A. Rosenberg, J. C. Yang and N. P. Restifo, Nat. Med., 2004, 10, 909–915 CrossRef CAS PubMed.
- L. Tang, Q. Yin, Y. X. Xu, Q. Zhou, K. M. Cai, J. Yen, L. W. Dobruckic and J. J. Cheng, Chem. Sci., 2015, 6, 2182–2186 RSC.
- Z. H. Xu, Y. H. Wang, L. Zhang and L. Huang, ACS Nano, 2014, 8, 3636–3645 CrossRef CAS PubMed.
- Y. Matsuo, H. Takeyama and S. Guha, Curr. Pharm. Des., 2012, 18, 2416–2419 CrossRef CAS PubMed.
- G. R. Zhang, W. Li, L. Holle, N. Y. Chen and W. Y. Chen, Clin. Cancer Res., 2002, 8, 1196–1205 CAS.
- M. M. Gubin, X. L. Zhang, H. Schuster, E. Caron, J. P. Ward, T. Noguchi, Y. Ivanova, J. Hundal, C. D. Arthur, W. J. Krebber, G. E. Mulder, M. Toebes, M. D. Vesely, S. S. K. Lam, A. J. Korman, J. P. Allison, G. J. Freeman, A. H. Sharpe, E. L. Pearce, T. N. Schumacher, R. Aebersold, H. G. Rammensee, C. J. M. Melief, E. R. Mardis, W. E. Gillanders, M. N. Artyomov and R. D. Schreiber, Nature, 2014, 515, 577–581 CrossRef CAS PubMed.
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed.
- B. M. Carreno, V. Magrini, M. Becker-Hapak, S. Kaabinejadian, J. Hundal, A. A. Petti, A. Ly, W. R. Lie, W. H. Hildebrand, E. R. Mardis and G. P. Linette, Science, 2015, 348, 803–808 CrossRef CAS PubMed.
- K. Palucka and J. Banchereau, Nat. Rev. Cancer, 2012, 12, 265–277 CrossRef CAS PubMed.
- T. Tanimoto, A. Hori and M. Kami, N. Engl. J. Med., 2010, 363, 1966–1966 CrossRef CAS PubMed.
- O. J. Finn, Nat. Rev. Immunol., 2003, 3, 630–641 CrossRef CAS PubMed.
- P. P. Lee, C. Yee, P. A. Savage, L. Fong, D. Brockstedt, J. S. Weber, D. Johnson, S. Swetter, J. Thompson, P. D. Greenberg, M. Roederer and M. M. Davis, Nat. Med., 1999, 5, 677–685 CrossRef CAS PubMed.
- P. L. Lollini, F. Cavallo, P. Nanni and G. Forni, Nat. Rev. Cancer, 2006, 6, 204–216 CrossRef CAS PubMed.
- L. Cicchelero, H. de Rooster and N. N. Sanders, Expert Rev. Vaccines, 2014, 13, 721–735 CrossRef CAS PubMed.
- B. Cerbelli, A. Pernazza, A. Botticelli, L. Fortunato, M. Monti, P. Sciattella, D. Campagna, F. Mazzuca, M. Mauri, G. Naso, P. Marchetti, G. d’Amati and L. Costarelli, BioMed Res. Int., 2017, 2017, 1–7 CrossRef PubMed.
- N. Casares, M. O. Pequignot, A. Tesniere, F. Ghiringhelli, S. Roux, N. Chaput, E. Schmitt, A. Hamai, S. Hervas-Stubbs, M. Obeid, F. Coutant, D. Metivier, E. Pichard, P. Aucouturier, G. Pierron, C. Garrido, L. Zitvogel and G. Kroemer, J. Exp. Med., 2005, 202, 1691–1701 CrossRef CAS PubMed.
- I. Le Mercier, D. Poujol, A. Sanlaville, V. Sisirak, M. Gobert, I. Durand, B. Dubois, I. Treilleux, J. Marvel, J. Vlach, J. Y. Blay, N. Bendriss-Vermare, C. Caux, I. Puisieux and N. Goutagny, Cancer Res., 2013, 73, 4629–4640 CrossRef CAS PubMed.
- J. Wei, Y. Long, R. Guo, X. Liu, X. Tang, J. Rao, S. Yin, Z. Zhang, M. Li and Q. He, Acta Pharm. Sin. B, 2019, 9, 819–831 CrossRef PubMed.
- Z. Q. Meng, F. Wei, R. H. Wang, M. G. Xia, Z. G. Chen, H. P. Wang and M. F. Zhu, Adv. Mater., 2016, 28, 245–253 CrossRef CAS PubMed.
- J. Y. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- G. Alexandrakis, E. B. Brown, R. T. Tong, T. D. McKee, R. B. Campbell, Y. Boucher and R. K. Jain, Nat. Med., 2004, 10, 203–207 CrossRef CAS PubMed.
- M. F. Flessner, J. Choi, K. Credit, R. Deverkadra and K. Henderson, Clin. Cancer Res., 2005, 11, 3117–3125 CrossRef CAS PubMed.
- M. A. Gosselin, W. J. Guo and R. J. Lee, Bioconjugate Chem., 2001, 12, 989–994 CrossRef CAS PubMed.
- J. Yan, J. Chen, N. Zhang, Y. Yang, W. Zhu, L. Li and B. He, J. Mater. Chem. B, 2020, 8, 492–503 RSC.
- E. A. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. A. Sulchek, T. H. Barker and A. J. Garcia, Adv. Mater., 2012, 24, 64–70 CrossRef CAS PubMed.
- J. Helft, J. Bottcher, P. Chakravarty, S. Zelenay, J. Huotari, B. U. Schraml, D. Goubau and C. R. E. Sousa, Immunity, 2015, 42, 1197–1211 CrossRef CAS PubMed.
- M. A. Daniele, A. A. Adams, J. Naciri, S. H. North and F. S. Ligler, Biomaterials, 2014, 35, 1845–1856 CrossRef CAS PubMed.
- Q. S. Yang, L. H. Ma and J. J. Shang, Int. J. Solids Struct., 2013, 50, 2437–2448 CrossRef CAS.
- C. Y. Cha, S. R. Shin, X. G. Gao, N. Annabi, M. R. Dokmeci, X. W. Tang and A. Khademhosseini, Small, 2014, 10, 514–523 CrossRef CAS PubMed.
- J. Yan, N. Zhang, Z. Zhang, W. Zhu, B. Li, L. Li, Y. Pu and B. He, J. Controlled Release, 2020, 329, 36–49 CrossRef PubMed.
- S. P. Hong, A. U. Bielinska, A. Mecke, B. Keszler, J. L. Beals, X. Y. Shi, L. Balogh, B. G. Orr, J. R. Baker and M. M. B. Holl, Bioconjugate Chem., 2004, 15, 774–782 CrossRef CAS PubMed.
- S. P. Hong, P. R. Leroueil, E. K. Janus, J. L. Peters, M. M. Kober, M. T. Islam, B. G. Orr, J. R. Baker and M. M. B. Holl, Bioconjugate Chem., 2006, 17, 728–734 CrossRef CAS PubMed.
- H. Moroson, Cancer Res., 1971, 31, 373–380 CAS.
- C. A. Janeway and K. Bottomly, Cell, 1994, 76, 275–285 CrossRef CAS PubMed.
- I. Mellman and R. M. Steinman, Cell, 2001, 106, 255–258 CrossRef CAS PubMed.
- A. W. Li, M. C. Sobral, S. Badrinath, Y. Choi, A. Graveline, A. G. Stafford, J. C. Weaver, M. O. Dellacherie, T. Y. Shih, O. A. Ali, J. Kim, K. W. Wucherpfennig and D. J. Mooney, Nat. Mater., 2018, 17, 528–534 CrossRef CAS PubMed.
- J. Li, X. Zhang, M. Zhao, L. Wu, K. Luo, Y. Pu and B. He, Biomacromolecules, 2018, 19, 3140–3148 CrossRef CAS PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- G. Kroemer, L. Galluzzi, O. Kepp and L. Zitvogel, Annu. Rev. Immunol., 2013, 31, 51–72 CrossRef CAS PubMed.
- J. Duraiswamy, K. M. Kaluza, G. J. Freeman and G. Coukos, Cancer Res., 2013, 73, 3591–3603 CrossRef CAS PubMed.
- A. Facciabene, G. T. Motz and G. Coukos, Cancer Res., 2012, 72, 2162–2171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr01155c |
| This journal is © The Royal Society of Chemistry 2021 |