
Microfluidic-enabled magnetic labelling of nanovesicles for bioanalytical applications†
Cornelia A.
Hermann
 ,
Michael
Mayer
,
Christian
Griesche
,
Franziska
Beck
and
Antje J.
Baeumner
*
,
Michael
Mayer
,
Christian
Griesche
,
Franziska
Beck
and
Antje J.
Baeumner
*
Institute for Analytical Chemistry, Chemo- and Biosensors, University of Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: antje.baeumner@ur.de
First published on 26th November 2020
Abstract
Bearing multiple functionalities dramatically increases nanomaterial capabilities to enhance analytical assays by improving sensitivity, selectivity, sample preparation, or signal read-out strategies. Magnetic properties are especially desirable for nanoparticles and nanovesicles as they assist in negating diffusion limitations and improving separation capabilities. Here, we propose a microfluidic method that reliably labels functional nanovesicles while avoiding the risk of crosslinking that would lead to large conglomerates as typically observed in bulk reactions. Thus, the carboxy groups of bi-functional biotinylated fluorescent liposomes were activated in bulk. They were then covalently bound to amino group presenting magnetic beads immobilized through a magnetic field within microfluidic channels. Microfluidic design and coupling strategy optimization led to a 62% coupling efficiency when using 1 μm magnetic beads. The yield dropped to 13% with 30 nm magnetic nanoparticles (MNPs) likely due to crowding of the MNPs on the magnet. Finally, both populations of these tri-functional liposomes were applied to a biological binding assay demonstrating their superior performance under the influence of a magnetic field. The microfluidic functionalization strategy lends itself well for massively parallelized production of larger volumes and can be applied to micro- and nanosized vesicles and particles.
Introduction
Liposomes are commonly applied labels in analytical assays due to their inherent signal enhancement capability by encapsulating marker molecules in their inner cavity and presenting binding molecules on their outer surface.1 Signal amplification factors of more than three order of magnitude can be achieved in comparison to single molecule, enzymatic or nanoparticle labelling.2 This is of special interest in trace analysis such as the detection of minute amounts of biomarkers, environmental pollutants or foodborne diseases, but also in scenarios where only small volumes can be analysed such as microchip strategies.3 Traditionally, liposomes are generated in bulk, producing large quantities of the same liposome population. Miniaturized, microfluidic-based approaches have also been demonstrated enabling more refined particular size and size distributions. By varying parameters like flow dynamics, channel design, lipid composition or concentrations, the resulting liposome characteristics can also be tailored toward encapsulation efficiency and outer surface compositions.4 Other strategies demonstrate the on-chip combination of characterization and synthesis. For example, dynamic vesicle formation can be monitored directly by integrating asymmetric flow field-flow fractionation combined with quasi-elastic light scattering and multiangle laser-light scattering; or by propane jet-freezing and in situ cryo electron microscopy.5 Also, the influence of controlled mixing can be investigated by adjusting the ratio of volumetric flow rates of different solvent solutions.6Like other nanoparticle and nanovesicle signalling entities, liposomes suffer from diffusion limitations when compared to molecular entities which is caused primarily by diffusion coefficients that are easily two orders of magnitude worse (see ESI chapter 1†). Here, magnetic labelling is deemed an ideal solution as it allows direct manipulation of the liposomes within a magnetic field without affecting their binding and signalling abilities, respectively. This was demonstrated successfully by incorporating magnetic nanoparticles (MNPs) into the lipid bilayer7 and applying it to DNA binding assays. Synthesis of these liposomes is more complicated and limited as the MNPs can easily lead to membrane disruption and distortion. Larger MNPs, and hence stronger magnetic properties can be achieved by encapsulating MNPs into the inner cavity.8 The major drawback here is low colloidal stability of MNPs in aqueous solution which in turn leads to low MNP concentrations and hence low encapsulation efficiency. A third, highly attractive approach is the labelling of liposomes on their outer surface done post synthesis.9 This has the advantage that particles of different size and different coverage can be bound. Furthermore, since the same liposome batch can be used during optimization experiments, high comparability and reproducibility can be achieved. However, obviously, magnetic labelling in bulk is impossible as it leads to cross linking and agglomeration. Thus, the inherent laminar flow properties of microfluidics to precisely control the convergence and mixing of components can be used to overcome this challenge. Fig. 1 shows this strategy. Here, magnetic particles are immobilized at the bottom of the channel and only one side of them is facing the solution on top. Thus, liposomes can only bind from this side and no other magnetic beads can crosslink. By deactivation of the active carboxy groups prior to magnetic release, further crosslinking after release can be prevented. Similar strategies have already been employed for the formation of multifunctional particles, e.g. by combination of quantum dots and magnetic particles to single Janus cores inside acrylate droplets,10 or by fusing liposomes of different composition by the use of microelectrodes,11 or by coupling magnetic particles and liposomes to nanochains via solid supports.12 We developed a new strategy for a directed coupling of bi-functional biotinylated, fluorescent liposomes to magnetic particles to result in tri-functional nanovesicles. Exploiting their multiple functions, these are then further studied as labels in bioanalytical assays.
 | ||
| Fig. 1 Scheme of the directed coupling of liposomes (purple spheres, 135 nm in diameter) with magnetic nanoparticles (brown spheres, 30 nm in diameter) inside a microfluidic channel. The particles are captured on a magnet while liposomes bind to the particles on their way through the channel (flow from back to front). | ||
Experimental
Chemicals and instruments
All chemicals are of analytical grade unless otherwise specified and either obtained from Avanti Polar Lipids, Inc. (http://www.avantilipids.com), Roth (http://www.carlroth.com), Sigma Aldrich (http://www.sigmaaldrich.com), Fisher Scientific (http://www.fishersci.com), Merck (http://www.merckmillipore.com) or VWR (de.vwr.com).HEPES buffer (10 mM HEPES, 200 mM NaCl, 0.01% (w/v) NaN3, pH 7.5), phosphate buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and washing buffer (0.05% (v/v) Tween 20, 0.01% (w/w) bovine serum albumin in PBS) are prepared with double distilled water.
An extruder equipped with syringes, filter supports and membranes for extrusion of liposomes was obtained from Avanti Polar Lipids, Inc. (http://www.avantilipids.com).
Fluorescence measurements are performed with a BioTek SYNERGY neo2 (http://www.biotek.com). For more information on chemicals and instruments view ESI.†
Liposome synthesis
For liposome synthesis, reverse phase evaporation according to an established procedure from Edwards et al.13 is employed.Briefly, 20 μmol DPPC (33%), 26 μmol cholesterol (43%), 10 μmol DPPG (16%), 1.5 μmol Biotin-DPPE (2%) and 3.5 μmol N-glutaryl-DPPE (6%) are dissolved in 3 mL chloroform and 0.5 mL methanol, 2 mL sulforhodamine B (SRB) solution (10 mM SRB, 2 mM HEPES, pH 7.5) are added and the mixture is sonicated. Organic solvents are vaporized at a rotary evaporator at 60 °C under reduced pressure. 2 mL SRB solution are added and any remaining organic solvent is evaporated at the rotary evaporator. The liposomes are extruded each 21 times at 60 °C through polycarbonate membranes (1.0 and 0.4 μm). Medium and high concentrated liposome containing fractions are collected ater size exclusion column chromatography (1.5 × 20 cm, Sephadex G50) and transferred to dialysis (MWCO 12–14 kDa, spectrumlab.com) against HEPES buffer.
The concentration of liposome solutions is determined with a Spectroflame-EOP inductively coupled plasma optical emission spectrometer (ICP-OES) from Spectro (http://www.spectro.com) or an ELAN 9000 (ICP-MS) from PerkinElmer (http://www.perkinelmer.com), whereas hydrodynamic diameters and ζ-potentials are measured by dynamic light scattering (DLS) at 20 °C with a Malvern Zetasizer Nano-ZS (http://www.malvern.com) in disposable poly(methyl methacrylate) (PMMA) cuvettes (semi-micro) and disposable PMMA capillary cells, respectively.14
Microfluidic setup and coupling procedure
The microfluidic chip is produced with a laser scriber VLS2.30 from Universal Laser Systems (http://www.ulsinc.com). The channel is cut into double-sided adhesive tape (type 415 from 3 M (http://www.3M.com), 100 μm in thickness), then a PMMA slide (1.6 mm in thickness) is glued on top and inlet holes are cut with the laser scriber. Afterwards a second PMMA slide is clued to the bottom to seal the channel. The PMMA slides are treated directly before mounting with UV/ozone treatment for 5 min with a Model 42 UV–ozone–cleaner from Jelight Company Inc. (http://www.jelight.com) to increase the hydrophilicity of the channel. The overall dimensions of the microfluidic chip are 3.3 × 60 × 30 mm, with a channel height of 100 μm and width from 1.5 to 6.87 mm (Fig. 2B). Then the chip is inserted into a chip holder with underlying magnet holder plate, as represented in Fig. S1,† and tubing is attached to connect the inlet and outlet holes with syringes mounted in Legato 180 syringe pumps from kd Scientific (http://www.kdscientific.com).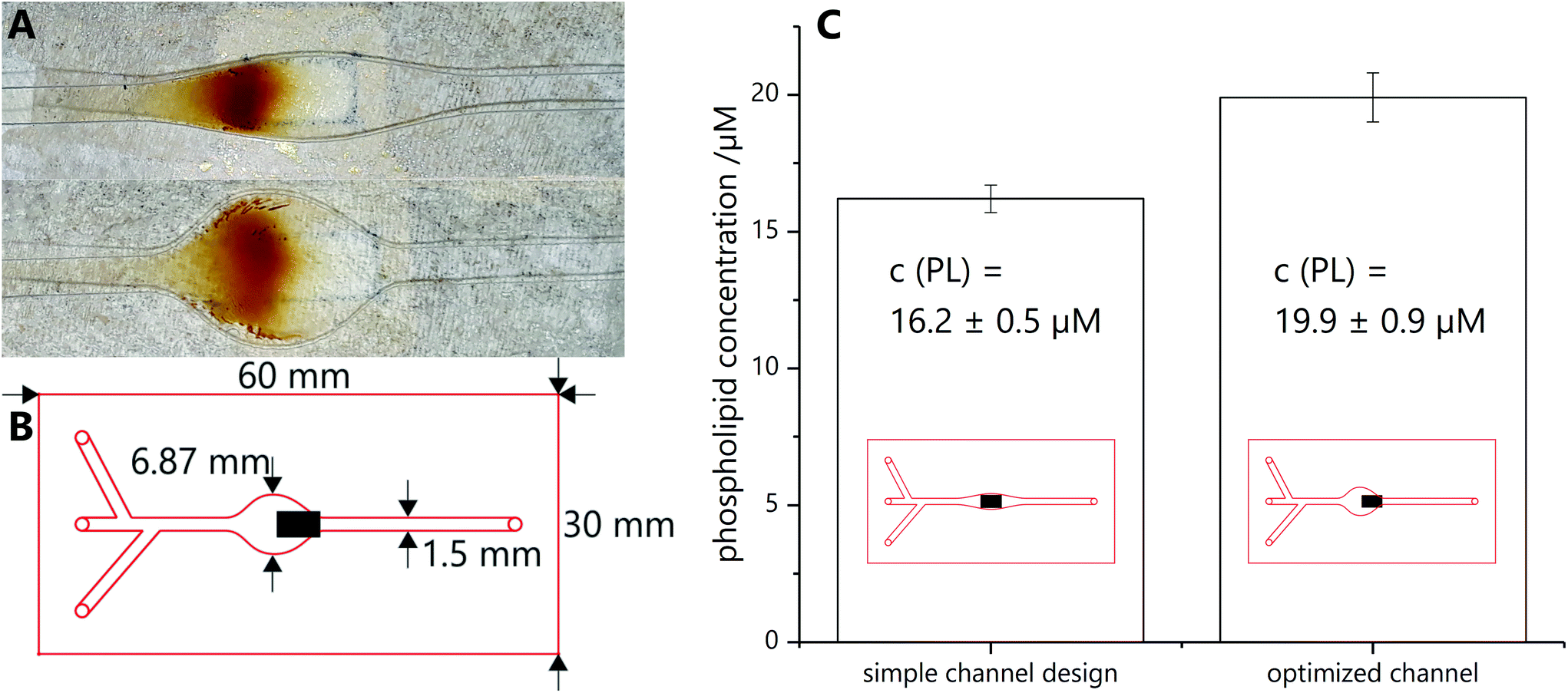 | ||
| Fig. 2 (A) Same particle amount caught on simple (top) and optimized (bottom) channel. The underlying magnet is visible as faint grey square (flow direction from left to right). (B) Shape and dimensions of the optimized microfluidic chip with three inlets and one outlet. The black box represents the underlying magnet with a size of 5 × 3 mm. Its magnetic field lines lead to the capture of the magnetic beads as shown in (A). (C) Comparison of phospholipid concentration measured by ICP-OES for simple and optimized channel layout (4 samples, 3 times measured). | ||
Before coupling can take place, carboxy modified liposomes have to be activated by EDC-sNHS coupling with a modified procedure by Bogdanov et al.15 Activated liposomes solution are prepared freshly each day and always used for three or four consecutive couplings. A typical activation solution contains 60 μM carboxy groups on the surface of liposomes and each 15 mM EDC and sNHS. Activation is performed for 20 min at 23 °C in a ThermoMixer C (online-shop.eppendorf.de).
Magnetic beads (1 μm in diameter, MB) are diluted with HEPES buffer to 0.11 mg mL−1, equalling 44 nmol mL−1 amino groups, while magnetic nanoparticles (30 nm in diameter, MNP) are diluted to 0.17 mg mL−1 (no number given for concentration of amine groups by producer). Ethanolamine is diluted to 25% (4.2 mM) with HEPES buffer.
Four syringes are loaded with liposome solution, ethanolamine, HEPES buffer and MB/MNP solution, respectively. Liposomes are connected to the top inlet in Fig. 2B, MBs/MNPs to the bottom inlet and ethanolamine and HEPES buffer are added through the middle inlet, syringes connected by a T-piece. Channels are filled by hand to ensure that no bubble remain. Flow speeds and volumes are given in Table 1. Liposomes are added in two steps: first faster (10 μL min−1) to ensure total coverage of the magnetic particle spot with liposomes, than with very slow flow speed (1 μL min−1) to ensure enough time for sufficient coupling. Based on previous studies, low flow rates enable best chemical interactions with liposomes.16 Therefore, the lowest flow rate possible of 24 μm per second was applied to promote the chemical reaction between liposomes and magnetic beads. After this procedure, the magnetic plate is removed and the coupling product is flushed out by hand and collected (in the following called ‘microfluidic product).
| Solution | Flow speed/μL min−1 | Linear velocity/mm s−1 | Infused volume μL−1 | Infused amount | ||
|---|---|---|---|---|---|---|
| 1.50 mm | 3.95 mm | 6.87 mm | ||||
| Three different linear velocities are given as the channel has different widths over its length (see Fig. 2 and Fig. S1†). | ||||||
| MB/MNPs | 10 | 1.1 | 0.42 | 0.24 | 150 | 6.6 nmol amine groups/∼25 μg particles |
| Liposomes | 10 | 1.1 | 0.42 | 0.24 | 10 | 0.6 nmol carboxy groups |
| 1 | 0.11 | 0.042 | 0.024 | 70 | 4.2 nmol carboxy groups | |
| Ethanolamine | 10 | 1.1 | 0.42 | 0.24 | 150 | 630 nmol amine groups |
| HEPES buffer | 15 | 1.7 | 0.63 | 0.36 | 100 | |
Concentration determination
Concentration determination is performed with three different methods: fluorescence measurements, ICP-MS and ICP-OES measurements and the Bartlett Assay.13 For details view ESI.†Determination of coupling efficiency
Consecutive washing steps in black half area 96-well microtiter plates (http://www.gbo.com) are performed to determine fluorescence intensity of unlysed and lysed microfluidic product before and after magnetic separation. SRB, a fluorescent dye, is encapsulated into the liposomes’ inner cavity in high concentration, inducing self-quenching. By lysis of the liposomes with a surfactant (n-octyl-β-D-glucoside, OG) the dye is released and a fluorescent signal can be detected.Prior to measurement, all wells are blocked with washing buffer (2 × 50 μL per well) and washed with HEPES buffer (2 × 50 μL per well). Then the microfluidic product (each 30 μL per well) is added. Analysis is performed with consecutive washing steps with fluorescence measurement after each step. First, the unaltered sample as collected from the microfluidic chip diluted in HEPES buffer (pure in Fig. 3) as well as an equally diluted sample in OG solution for lysis (pure lysed) are measured. After measurement, the microtiter plate is positioned on a magnetic plate (96S Super Ring Magnet Plate with NdFeB magnets, http://www.alpaqua.com) and the supernatant of the pure sample solution in HEPES buffer is taken off. The remaining beads are resuspended in HEPES buffer and measured again (mag. sep.). This step is repeated, but with addition of OG solution instead of HEPES buffer (mag. sep. lysed).
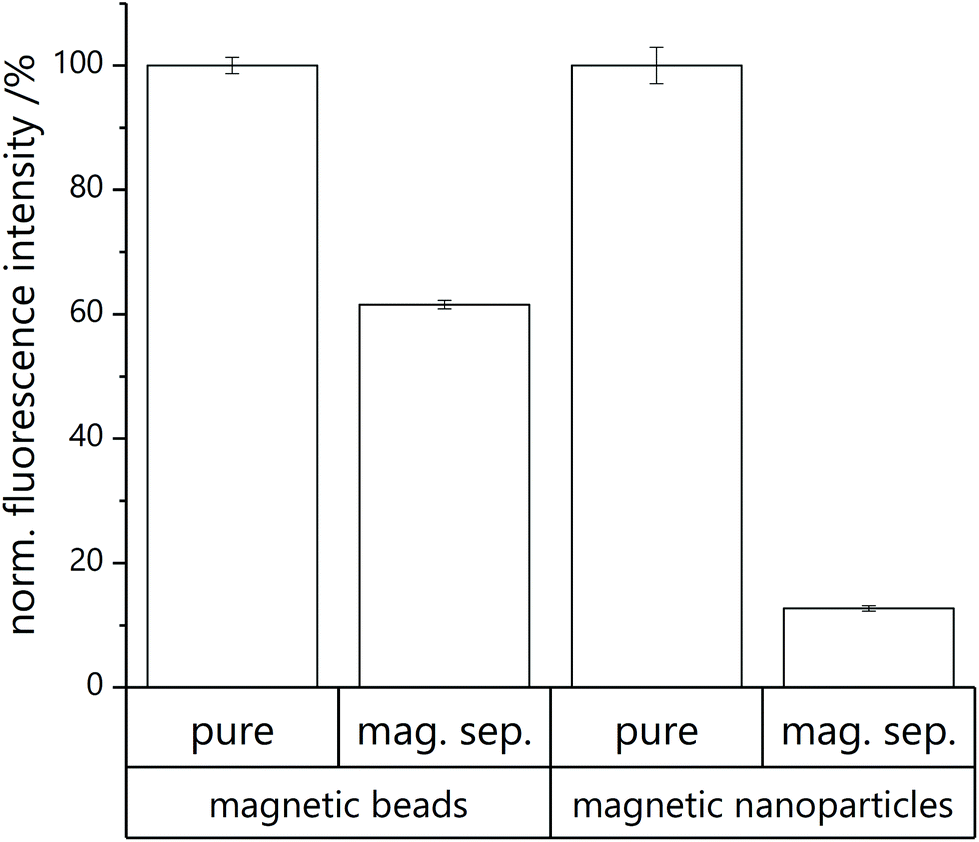 | ||
| Fig. 3 Coupling efficiency determined by fluorescence measurement of unlysed (fluorescence background) vs. lysed product (fluorescence of released SRB) with and without magnetic separation for coupling of liposomes to MBs and MNPs. Normalized fluorescence values after background subtraction are provided (3 samples, 3 times measured). | ||
Biotin–streptavidin-binding-assay
Streptavidin coated microtiter plates (MTPs) are used for the following assay. All steps are performed in parallel on two identical MTPs, with the difference that during liposome incubation, one of the plates is positioned on a permanent magnet while the other one is positioned outside the magnetic field.Before incubating the streptavidin-coated wells with biotin-containing liposome sample, each well is washed with washing buffer (2 × 200 μL per well) and HEPES buffer (2 × 200 μL per well). Then the microfluidic product (varying concentrations, 100 μL per well) is added and incubated for 60 min at room temperature. In between, both MTPs are positioned three times on a ThermoMixer C and shaken for 10 s. Unbound liposomes are removed and the wells are washed with HEPES buffer (2 × 200 μL per well).
Background fluorescence is measured in 100 μL HEPES buffer once. Afterwards, the supernatant is removed and OG (30 mM, 100 μL per well) is added to lyse the liposome bilayer. After 5 min incubation, fluorescence is measured again.
Results and discussion
Liposome and magnetic particle characterization
Liposomes were synthesized using standard protocols13 and then characterized regarding their hydrodynamic diameter (135 nm, PdI of 0.116) and zeta potential ((−25.2 ± 1.2) mV) by DLS (Fig. S2†), which are typical values for liposomes and yields dispersions with high colloidal stability. Two differently dimensioned particles were investigated: MBs with a diameter of 1 μm and MNPs with 30 nm in diameter, both modified with amino groups at the surface. Together with the carboxy-modified liposomes a simple covalent coupling strategy can be realized in which liposomes are activated with a linker (EDC and sNHS), bonded to the MNPs/MBs, and remaining active binding sites are deactivated by ethanolamine before release off the magnet and hence out of the channel. The pure magnetic particles, mixed with inactivate liposomes and the coupling product were studied using DLS supporting expected values prior and post microfluidic coupling (compare ESI chapter 5†). Also, prior to coupling in the microfluidic channels, the coupling chemistry was confirmed in bulk experiments. The formation of cross linked conglomerates as a predominant peak around 5000 nm in the DLS spectrum could be easily observed in comparison to the separate peaks of free liposomes and MNPs (after sonication) (Fig. S3†).Channel optimization
Various microfluidic channel designs were investigated to promote best spreading of magnetic particles on an external magnet, prevent clogging of the channel and consequently create a large reactive magnetic particle surface. In the end, a straight channel with a widened area above the external magnet was chosen. Here, depending on the widened channel cross section, magnetic bead collection can be tailored and reproducibly carried out (Fig. 2A), whereas care has to be taken to pair particle concentration and channel width in order to avoid clogging. Also a mismatch of channel width and external magnet can cause hotspot capturing which should be avoided. It should be pointed out that the magnetic field lines will heavily influence coverage distribution, i.e. complete coverage of the widened area is not possible with one external magnet (compare ESI chapter 6†). However, interestingly, even simple designs lead to efficient liposome coupling when compared to those carefully optimized with respect to channel/magnet dimensions and hence optimized surface coverage (1.2 fold improvement) (Fig. 2C). Minimal optimization of the microfluidic design was done, which focused mainly on enabling magnetic bead flow, capture and release. In the future, further optimization strategies will be used in order to enhance mass transport within the channels and hence increase the percentage of liposomes getting into contact with the active magnetic bead layer. Here, microfluidic mixers as designed in earlier studies will be used.17Conjugation product characterization
The amount of non-conjugated liposomes in the microfluidic elute was determined by measuring the fluorescence of lysed and non-lysed microfluidic product before and after magnetic separation. In the case of MBs, (61.6 ± 0.7)% of the liposomes were effectively conjugated with magnetic particles (Fig. 3A). This substantial coupling efficiency is hence by far superior to other strategies studied, considering that bilayer integration or encapsulation of MNPs yield in maximum a 15% magnetic labelling.18In the case of microfluidic MNP coupling, a lower coupling efficiency of only (12.7 ± 0.4)% was observed (Fig. 3B). While this could be due to MNPs forming multiple layers hence effectively lowering the surface available for coupling, it may also represent the fact that more than one MNP per liposome is needed to achieve sufficient magnetic labelling. It should be noted that some loss of liposomes is observed in all coupling processes due to lysis of the liposomes, as indicated by the free SRB dye available in the solution leading to a smaller increase in fluorescence signal in the untreated microfluidic product when compared to the purified product.
Previous research using liposomes has shown, that an inherent size distribution vs. monodisperse systems does not negatively affect the analytical performance with respect to reproducibility and accuracy. Since hundreds to thousands of liposomes are involved in the signal generation of such bioanalytical assays, we assume that their size distribution does not have an effect on the assay itself.
Biotin–streptavidin-binding assays
The analytical performance of the newly generated tri-functional liposomes was evaluated by conducting biotin–streptavidin binding assays. Specifically, liposomes were synthesized bearing both a carboxy group and a biotin moiety on their outer surface prior to microfluidic magnetic labelling. This moiety can obviously be exchanged for other bioreceptors for varying recognition purposes in the future.Lipid concentration of the product was determined with fluorescence measurements, by ICP-MS/-OES and with the Bartlett assay. The total lipid concentration of the microfluidic product is determined to be between 34 and 48 μM (Table S3, Fig. S9†). For the following assays, a concentration of the undiluted microfluidic product of 40 μM was assumed.
All magnetic tri-functional liposomes demonstrated superior bioassay performance in comparison to non-magnetic conditions (Fig. 4A), i.e. for optimal comparison, the same liposomes were used with and without magnet in the bioassay. Finally, when concentrating liposomes coupled to MBs and MNPs via magnetic separation prior to conducting the bioassay and hence separating out non-magnetic liposomes, they performed even better (Fig. 4B and C), although their concentration was significantly lowered (from 40 μM to a maximum of 25 and 5 μM total lipid, respectively).
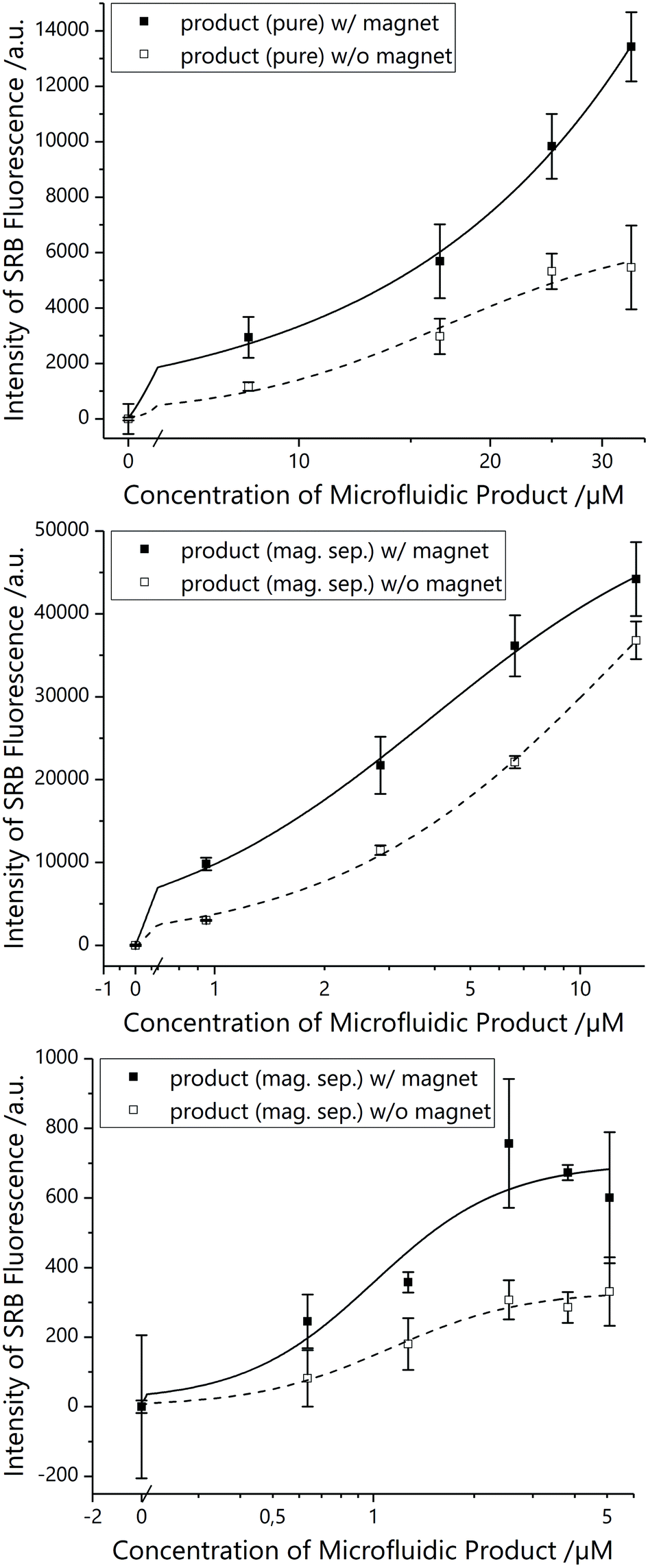 | ||
| Fig. 4 Biotin–streptavidin-binding assay with microfluidic coupling product between magnetic particles and liposomes bearing biotin on the surface, performed in parallel in the absence and presence of an external magnetic field. top: Pure product with 1 μm MBs without any purification. middle: Product with 1 μm MBs after magnetic separation (mag.sep.), concentration given in percent of pure MB product (undiluted product after magnetic separation equalling 61.6% of pure MB product). bottom: Product with 30 nm MNPs after magnetic separation (mag.sep.), concentration given in percent of pure MNP product (undiluted product after magnetic separation equalling 12.7% of pure MNP product) (3 samples). | ||
Conclusions
Simple microfluidic strategies can be used for magnetic labelling of nanovesicles and nanoparticles. Through controlled convergence in a microchannel the risk of conglomeration as observed in bulk coupling strategies can be effectively avoided. Here, bi-functional biotinylated, fluorescent liposomes were labelled with either magnetic microbeads or magnetic nanoparticles. Their superior performance as signalling label in biological binding assays was demonstrated, because their manipulated movement in a magnetic field could overcome diffusion limitations. This microfluidic magnetic modification strategy is not limited to liposomes, but can be used also for other colloidally stable nanoparticles and vesicles such as quantum and carbon dots, upconversion nanoparticles, silica particles, and Raman labels like gold or silver nanomaterials. In addition, through mass parallelization and by expansion of the location and size of the magnetic field, large scale production is possible. These multifunctional conjugates can then be applied for sensing, imaging, directed drug transport etc. In analytical chemistry as well as life science and biomedicine they can serve as smart components in lab-on-a-chip systems,19 as barcode elements for multianalyte detection10 or as combined separation tools and labels.20Author contributions
Cornelia A. Hermann and Antje J. Baeumner designed the research plan; Cornelia A. Hermann carried out experiments; Christian Griesche and Michael Mayer assisted in strategic discussions; Franziska Beck assisted in magnetic setup optimization; Cornelia A. Hermann and Antje J. Baeumner wrote this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Vanessa Tomanek for drawing the 3D graphic for the introduction.References
- (a) K. A. Edwards and A. J. Baeumner, Talanta, 2006, 68, 1421–1431 CrossRef CAS; (b) Q. Liu and B. J. Boyd, Analyst, 2013, 138, 391–409 RSC.
- (a) T. Suita and T. Kamidate, Anal. Sci., 1999, 15, 349–352 CrossRef CAS; (b) A. K. Singh, P. K. Kilpatrick and R. G. Carbonell, Biotechnol. Prog., 1996, 12, 272–280 CrossRef CAS; (c) M. Lee, R. A. Durst and R. B. Wong, Anal. Chim. Acta, 1997, 354, 23–28 CrossRef CAS.
- (a) J. T. Connelly, S. Kondapalli, M. Skoupi, J. S. L. Parker, B. J. Kirby and A. J. Baeumner, Anal. Bioanal. Chem., 2012, 402, 315–323 CrossRef CAS; (b) M. B. Esch, L. E. Locascio, M. J. Tarlov and R. A. Durst, Anal. Chem., 2001, 73, 2952–2958 CrossRef CAS; (c) Y. Ishimori and K. Rokugawa, Clin. Chem., 1993, 39, 1439–1443 CrossRef CAS; (d) E. Canova-Davis, C. T. Redemann, Y. P. Vollmer and V. T. Kung, Clin. Chem., 1986, 32, 1687–1691 CrossRef CAS; (e) A. S. Janoff, S. Carpenter-Green, A. L. Weiner, J. Seibold, G. Weissmann and M. J. Ostro, Clin. Chem., 1983, 29, 1587–1592 CrossRef CAS.
- (a) T. A. Balbino, A. R. Azzoni and L. G. de La Torre, Colloids Surf., B, 2013, 111, 203–210 CrossRef CAS; (b) D. Carugo, E. Bottaro, J. Owen, E. Stride and C. Nastruzzi, Sci. Rep., 2016, 6, 25876 CrossRef CAS.
- A. Jahn, F. Lucas, R. A. Wepf and P. S. Dittrich, Langmuir, 2013, 29, 1717–1723 CrossRef CAS.
- A. Jahn, W. N. Vreeland, D. L. DeVoe, L. E. Locascio and M. Gaitan, Langmuir, 2007, 23, 6289–6293 CrossRef CAS.
- E. Amstad, J. Kohlbrecher, E. Muller, T. Schweizer, M. Textor and E. Reimhult, Nano Lett., 2011, 11, 1664–1670 CrossRef CAS.
- K. A. Edwards and A. J. Baeumner, Anal. Chem., 2014, 86, 6610–6616 CrossRef CAS.
- C. Men, C. H. Li, X. M. Wei, J. J. Liu, Y. X. Liu, C. Z. Huang and S. J. Zhen, Analyst, 2018, 143, 5764–5770 RSC.
- Y. Zhao, H. C. Shum, H. Chen, L. L. A. Adams, Z. Gu and D. A. Weitz, J. Am. Chem. Soc., 2011, 133, 8790–8793 CrossRef CAS.
- A. Strömberg, A. Karlsson, F. Ryttsén, M. Davidson, D. T. Chiu and O. Orwar, Anal. Chem., 2001, 73, 126–130 CrossRef.
- P. M. Peiris, L. Bauer, R. Toy, E. Tran, J. Pansky, E. Doolittle, E. Schmidt, E. Hayden, A. Mayer, R. A. Keri, M. A. Griswold and E. Karathanasis, ACS Nano, 2012, 6, 4157–4168 CrossRef CAS.
- K. A. Edwards, K. L. Curtis, J. L. Sailor and A. J. Baeumner, Anal. Bioanal. Chem., 2008, 391, 1689–1702 CrossRef CAS.
- B. Maherani, E. Arab-tehrany, A. Kheirolomoom, V. Reshetov, M. J. Stebe and M. Linder, Analyst, 2012, 137, 773–786 RSC.
- A. A. Bogdanov, A. L. Klibanov and V. P. Torchilin, FEBS Lett., 1988, 231, 381–384 CrossRef CAS.
- V. N. Goral, N. V. Zaytseva and A. J. Baeumner, Lab Chip, 2006, 6, 414–421 RSC.
- K. P. Nichols, J. R. Ferullo and A. J. Baeumner, Lab Chip, 2006, 6, 242–246 RSC.
- C. A. Hermann, C. Hofmann, A. Duerkop and A. J. Baeumner, Anal. Bioanal. Chem., 2020, 412, 6295–6305 CrossRef CAS.
- X.-T. Sun, M. Liu and Z.-R. Xu, Talanta, 2014, 121, 163–177 CrossRef CAS.
- N. Duan, M. Shen, S. Wu, C. Zhao, X. Ma and Z. Wang, Microchim. Acta, 2017, 184, 2653–2660 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an02027c |
| This journal is © The Royal Society of Chemistry 2021 |