
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hexametaphosphate as a potential therapy for the dissolution and prevention of kidney stones†
Thomas E.
Robinson
 *ab,
Erik A. B.
Hughes
a,
Oliver J.
Wiseman
c,
Sarah A.
Stapley
b,
Sophie C.
Cox
a and
Liam M.
Grover
*a
*ab,
Erik A. B.
Hughes
a,
Oliver J.
Wiseman
c,
Sarah A.
Stapley
b,
Sophie C.
Cox
a and
Liam M.
Grover
*a
aSchool of Chemical Engineering, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: TER281@bham.ac.uk; L.M.Grover@bham.ac.uk
bRoyal Centre for Defence Medicine, Birmingham Research Park, Vincent Drive, Edgbaston, B15 2SQ, UK
cDepartment of Urology, Cambridge University Hospital, Cambridge, CB2 0QQ, UK
First published on 21st May 2020
Abstract
The incidence of kidney stones is increasing worldwide, and recurrence is common (50% within 5 years). Citrate, the current gold standard therapy, which is usually given as potassium or sodium salts, is used because it raises urine pH and chelates calcium, the primary component of up to 94% of stones. In this study hexametaphosphate (HMP), a potent calcium chelator, was found to be 12 times more effective at dissolving calcium oxalate, the primary component of kidney stones, than citrate. HMP was also observed to be effective against other common kidney stone components, namely calcium phosphate and struvite (magnesium ammonium phosphate). Interestingly, HMP was capable of raising the zeta potential of calcium oxalate particles from −15.4 to −34.6 mV, which may prevent stone growth by aggregation, the most rapid growth mechanism, and thus avert occlusion. Notably, HMP was shown to be up to 16 times as effective as citrate at dissolving human kidney stones under simulated physiological conditions. It may thus be concluded that HMP is a promising potential therapy for calcium and struvite kidney stones.
Introduction
Kidney stone formation, also termed urolithiasis or urinary/renal calculi, represents one of the most common diseases of the urinary tract, and prevention of their recurrence remains a vital issue.1 Stones can cause intense colicky pain in the loin, obstruction of urine flow, urinary tract infections, and is responsible for 2–3% of end stage renal disease.1,2 The lifetime risk of kidney stones is 10–15% in the Western world, and as high as 25% in the Middle East.3 In the USA, the total prevalence of kidney stones was 8.8% in 2012, compared to 5.2% in 1994, and is higher in men than women (10.6% vs. 7.1%), obese people than those of normal weight (11.2% vs. 6.1%), and in white compared to black or Hispanic people.4 It is also notable that stone formation is a highly recurrent condition, with a rate of recurrence of up to 50% within 5 years.5A key factor in the global growth of kidney stone incidence is changing diet, both directly and indirectly. The increase in stone prevalence correlates strongly with growing total calorie, fat, protein, fruit and vegetable consumption.6 Ingestion of high levels of animal protein, oxalate, sodium and fructose, in addition to low amounts of potassium and citrate, are all indicated in stone formation.7–11 Indirectly, stone formation is strongly associated with obesity and diabetes,4 the prevalence of which are growing worldwide.12,13 Climate change may be another factor increasing the prevalence of kidney stones. Increased temperatures can cause dehydration, leading to decreased urine volumes, raised core temperature and higher salt concentrations in the blood, leading to increased urine supersaturation, while increased vitamin D synthesis may increase calcium excretion.14,15 The population of the USA living in the hot southern ‘stone belt’, where residents are at increased risk of stones due to temperature, is expected to rise from 40% in 2000 to 56% in 2050, and 70% by 2095. This will result in an increase of 1.6 to 2.2 million lifetime cases by 2050 in the USA alone, with an associated cost of around $1 billion annually.16 The cost of kidney stones in the USA was estimated at around $2 billion in 2000, a 50% increase from 1994, and is still increasing.17
Kidney stones can contain several components, and may be composed primarily of one compound or contain a mixture. The vast majority, up to 94%, are calcium based, with 76% of stones being comprised of mostly calcium oxalate (CaOx, Fig. 1A), and 18% primarily calcium phosphate, usually hydroxyapatite (HA, Fig. 1B).18 The remainder are composed of struvite (magnesium ammonium phosphate, 10–15%) (Fig. 1C), uric acid (3–10%) (Fig. 1D) or cystine (<2%) (Fig. 1E), plus a small number of drug induced stones.1 Regardless of composition, kidney stones are formed by supersaturation in the urine leading to nucleation, growth and aggregation, with the latter mechanism leading to the most rapid evolution. Nucleation is usually heterogeneous, and nuclei must anchor to a surface in order to grow before being excreted in the urine stream.19 CaOx stones often form on HA plaques, known as Randall's plaque, which grow in the interstitial tissue, break the urothelium, and promote CaOx aggregation.20 Randall's plaque was found in at least one papilla for 99% of stone formers, and 20% were found to have tubular plugging.21 The precise mechanism of their formation is as yet unclear, but plaque coverage correlates with increasing urinary calcium and decreasing urinary volume.22
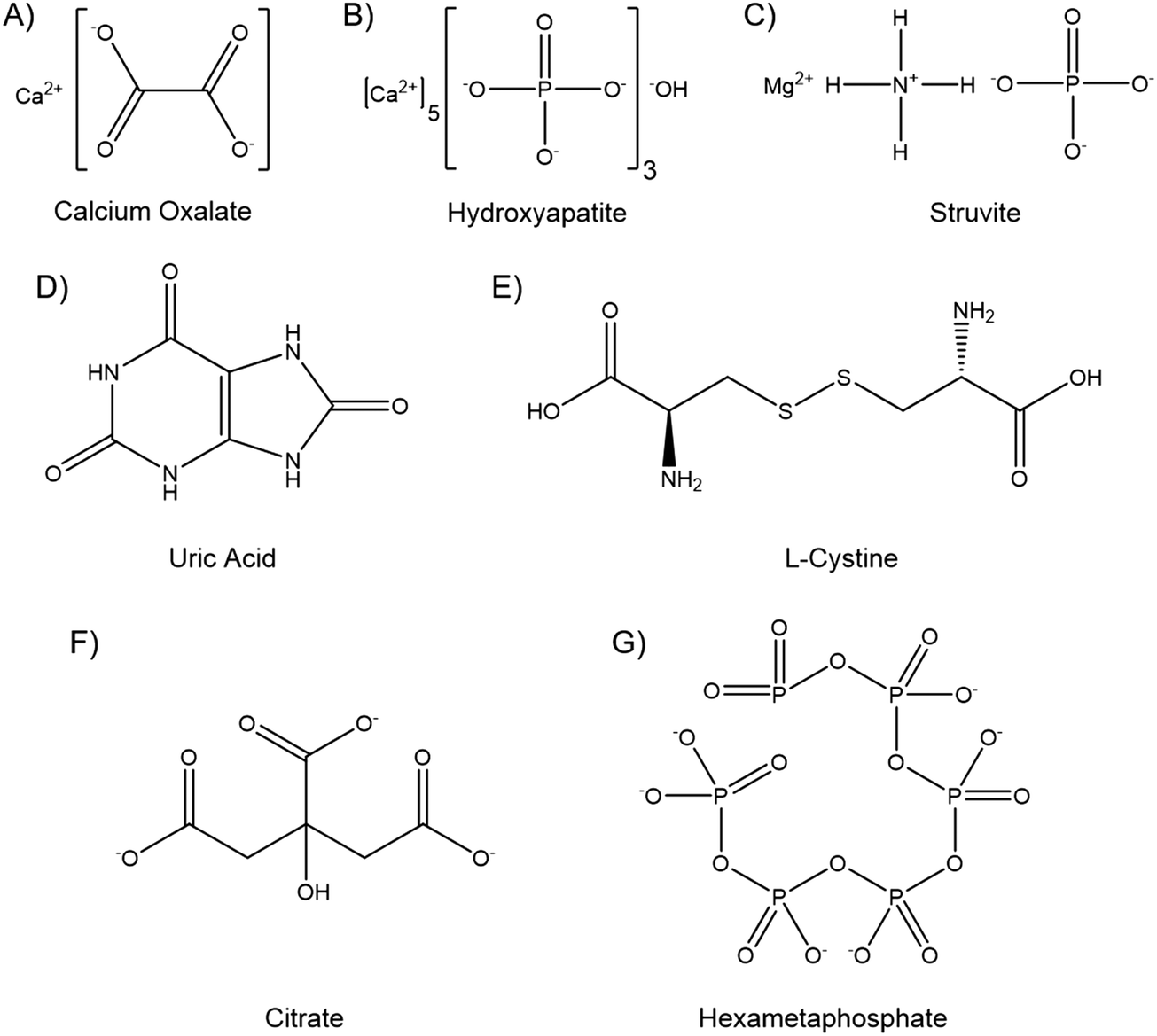 | ||
| Fig. 1 Chemical structures of kidney stone components and dissolution agents. The chemical structure of the kidney stone components (A) calcium oxalate, (B) hydroxyapatite, (C) struvite, (D) uric acid, and (E) L-cystine. The chemical structures of dissolution agents (F) citrate and (G) hexametaphosphate. | ||
Elevated ionic concentrations and lower urinary volumes contribute to the supersaturation responsible for urolithiases. However, the CaOx concentration found in normal urine is four-fold greater than its solubility would suggest is possible, while nucleation occurs at between seven and eleven times solubility.19 This supersaturation without precipitation is termed metastability, and is permissible due to molecules in the urine that inhibit the nucleation, growth and aggregation of crystals.23 Citrate (Fig. 1F), osteopontin, Tamm–Horsfall glycoprotein, calgranulin, prothrombin F1 fragment, albumin, glycosaminoglycans, and fragments of DNA and RNA have all been identified as crystallisation inhibitors in the urine.24 These molecules are, or include, polyanionic chains that bind to calcium to prevent growth and aggregation, and raise the metastable limit of supersaturation, which is lower in stone formers than healthy individuals.24,25
Small precipitations that do not anchor or grow may be excreted asymptomatically. Small stones <5 mm have a high chance of passage, stones 5–7 mm have around a 50% chance, while stones >7 mm are likely to require intervention.24 Current interventions for formed stones include disruption and break down with shock waves, lasers applied via an endoscope, or a small surgical procedure, which all have varying degrees of success.26–28 However, given the recurrent nature of kidney stones, secondary prophylaxis is often employed. Patients are often advised to increase fluid intake, maintain calcium but limit sodium and oxalate consumption, and to eat less animal protein, in an effort to reduce supersaturation of the urine and increase the limit of metastability.29 In addition to dietary augmentation, common therapeutics include thiazide diuretics, to lower urine calcium and pH, and citrate therapy. Citrate forms soluble complexes with calcium, and is reported to inhibit crystal nucleation, growth and aggregation, in addition to stabilising polynuclear complexes and amorphous CaOx phases.30,31 However, in the clinic high rates of stone recurrence are seen even with citrate treatment, particularly in patients with low citrate levels.32 In randomised controlled trials of citrate therapies, one in three patients dropped out, primarily due to side effects that include eructation, bloating, gaseousness, upper gastrointestinal tract disturbance, rashes and diarrhoea.32,33
Hexametaphosphate (HMP, Fig. 1G) is an inorganic oligophosphate, commonly used as a deflocculant and sequestering agent in the food and minerals industries.34 HMP chelates and forms soluble complexes with divalent metal cations, such as calcium and magnesium.35 HMP has been shown to prevent crystallisation in saturated calcium phosphate solutions even in small quantities, and to dissolve HA.36,37 HMP has also been investigated biologically, and has been shown to inhibit chick femur mineralisation and to dissolve bone mineral ex vivo.38,39 While calcium phosphates have been most widely investigated, HMP has also been shown to dissolve CaOx and prevent struvite formation by sequestering the Ca2+ and Mg2+ cations, respectively.40,41 HMP thus appears to be a potent inhibitor of crystallisation and able to dissolve the vast majority of kidney stone components. Further, the mechanism of action, divalent cation chelation, appears to be similar to the currently used citrate therapy, and hydroxycitrate, which is currently being researched as an alternative.42 The hypothesis of this study is thus that HMP may be a more potent alternative to citrate therapy, imparting greater therapeutic action to curb kidney stone recurrence, and potentially with fewer deleterious side effects.
Experimental
Calcium salt dissolution
Calcium salt dissolution experiments were performed in deionised water from a Milli-Q system (Millipore) in order to probe the chemistry precisely and elucidate the chemical ratios. A light extinction approach was used so that several precipitate concentrations could be probed simultaneously. Absorbance of 1 mL of suspension in a 24 well CellStar suspension culture plate (Greiner Bio-One) was measured at 600 nm after 1 hour, using a Spark plate reader (Tecan).Concentrations were initially probed so as to give a wide range in the plate reader, i.e. an undissolved absorbance of around 1. The following concentrations were used which gave this range. For oxalate, 5 mL of 5 mM calcium chloride dihydrate (Sigma) was added to 5 mL of 5 mM sodium oxalate, 99% (Alfa Aesar). X-ray diffraction (XRD) confirmed the formation of calcium oxalate monohydrate (Fig. S1A–C, ESI†). For phosphate, 5 mL of 50 mM CaCl2 was added to 3 mL of 50 mM sodium phosphate, 96% (Aldrich), in order to give the correct stoichiometry for HA formation. XRD confirmed the formation of HA (Fig. S1D and E, ESI†). For carbonate, 5 mL of 50 mM CaCl2 was added to 5 mL of 50 mM sodium carbonate decahydrate, 99+% (Alfa Aesar). Calibration curves (Fig. S2, ESI†) were produced by serial dilution of these solutions in deionised water, so that absorbance values could be mapped to a concentration for each species. Concentration and absorbance showed a linear correlation for all salts in the concentration range studied.
For dissolution experiments, 5 mL of calcium chloride was added to 5 mL of sodium salt, with 1 mL of HMP (sodium hexametaphosphate, general purpose grade, Fisher) solution of varying concentration added to give the desired Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HMP molar ratios. To simulate prevention, the HMP solution was added to the sodium salt solution prior to the addition of the calcium chloride. To simulate dissolution, the HMP solution was added after the precipitation.
:HMP molar ratios. To simulate prevention, the HMP solution was added to the sodium salt solution prior to the addition of the calcium chloride. To simulate dissolution, the HMP solution was added after the precipitation.
Artificial urine
To more closely simulate physiological conditions, all further experiments were carried out in artificial urine (AU). This was prepared by the method of Parra et al.,43 dissolving the components in 1 L of deionised water (Table 1). The final pH of AU at 20 °C was 5.8.| Chemical name | Formula | Amount (g L−1) | Supplier |
|---|---|---|---|
| Calcium chloride dihydrate >99% | CaCl2·H2O | 1.103 | Sigma |
| Sodium chloride >99% | NaCl | 2.925 | Sigma |
| Sodium sulphate anhydrous >99% | Na2SO4 | 2.25 | Fisher |
| Potassium phosphate monobasic ACS >99% | KH2PO4 | 1.40 | Sigma-Aldrich |
| Potassium chloride, ACS >99% | KCl | 1.60 | Alfa Aesar |
| Ammonium chloride >98% | NH4Cl | 1.00 | Alfa Aesar |
| Urea >98% | CO(NH2)2 | 25.00 | Alfa Aesar |
| Creatinine 98% | C4H7N3O | 1.10 | Alfa Aesar |
Struvite, uric acid and cystine dissolution
The effect of HMP on the other most common components of kidney stones, struvite, uric acid and cystine, was tested by mass because all three components sediment quickly, making absorbance measurements unsuitable. 5 g of ammonium magnesium phosphate hexahydrate 98% (struvite), uric acid 99%, or L-cystine 99%, all purchased from Alfa Aesar, were added to centrifuge tubes with 40 mL of AU with or without 0.2 M HMP. A pH probe (Metler Toledo) was used to confirm that HMP did not alter the pH of the AU. The tubes were agitated with a shaker plate to keep the stone component powders suspended for 3 days. Following this, the tubes were centrifuged at 4700 rpm for 10 minutes and then the fluid was removed. The powder was resuspended in deionised water, centrifuged and fluid removed three times in order to wash the precipitates. The tubes were then dried at 60 °C and 500 mbar for 24 hours and weighed to ascertain the mass change.Comparison to citrate
As AU contains 7.5 mM calcium ions, CaOx suspensions were prepared by adding 2.5 mM equivalent of sodium oxalate to the solution. Varying amounts of HMP or trisodium citrate dihydrate ACS grade >99% (Alfa Aesar) were then added to study their effect on the dissolution of the CaOx. Concentration was measured by absorbance as described above, and converted to concentration using a calibration curve (Fig. S3, ESI†).Zeta and size analysis
Zeta potential was measured via electrophoretic light scattering, using a Zetasizer Nano ZS (Malvern), which calculated the zeta potential after measuring the electrophoretic mobility via laser Doppler velocimetry, at a wavelength of 633 nm. For each measurement, 10–100 runs were taken at 25 °C. Size was measured on the same instrument, using 173° scattered light, and reported size is the Z-average. However, only particles dissolved in HMP or citrate for 4 hours were of an appropriate size for this methodology. The particle size of the remaining suspensions was measured using a Mastersizer 3000 (Malvern), and the reported size is the D50.Kidney stone analysis
Approval for this study was granted by the STEM Ethical Review Committee at the University of Birmingham (ERN_19-1249). Kidney stones, which would otherwise have been discarded, were received from Cambridge University Hospital, UK, following percutaneous nephrolithotomy, with patients’ consent.XRD was performed on a D8 Advance Diffractometer (Bruker), with a copper X-ray beam (0.15418 nm). Spectra were collected between a 2θ of 10 and 50°, with a step size of 0.02° and a step time of 0.3 s. Intensity was normalised for each scan and spectra were compared to standards from the International Centre for Diffraction Data (ICDD); hydroxyapatite (PDF 01-074-9761), brushite (PDF 00-009-0077) and calcium oxalate monohydrate (PDF 00-020-0231). XRD was used to establish the precise chemical species present in the stones.
Micro X-ray fluorescence (XRF) data was acquired with an M4 Tornado (Bruker) with a rhodium X-ray tube at 50 kV and 300 μA, and under reduced pressure (20 mbar). Maps show the relative intensity of each element at each pixel, normalised to the maximally intense pixel for that element. As elements with a low atomic weight, such as carbon and oxygen, cannot be detected by XRF, calcium in the absence of phosphate can be taken to be oxalate. XRF was used as a screening tool, in addition to XRD, to give further confidence that all phases in the stone were identified.
Micro computed tomography (microCT) scans were carried out before and after dissolution on a Skyscan 1172 (Bruker), with the following settings: 0.5 mm aluminium filter, beam voltage 80 kV and current 100 μA, exposure time 400 ms, pixel size 9.73 μm, rotation step 0.4° and a frame averaging of 6. Reconstruction, numerical analysis and 3D rendering were carried out in NRecon, CTAn and CTVox (Bruker), respectively. The same parameters were used for all scans. Total stone volume was established by performing 3D analysis with a low binary threshold to separate stone from air. Volumetric component ratios were found by finding the volume of each component separately, utilising different binary thresholding parameters, and calculating a ratio.
Kidney stone dissolution
All stones were initially weighed, and a selection of stones within a tight mass range (18.8–22.8 mg) were chosen for the study. These stones then underwent stratified randomisation into groups of 3, so that each group had one relatively large, medium and small stone. This method was chosen because a preliminary study (data not shown) revealed that initial stone mass had a significant effect on the rate of dissolution. Stones were then placed in 5 mL of AU with a given concentration of HMP or citrate, and incubated at 37 °C. This volume was used as a conservative estimate for the amount of fluid a stone may be exposed to in vivo; the total volume of the kidney is around 200 mL in men and 150 mL in women.44 A static, rather than flow, model was used in this proof of concept study for simplicity and to reduce confounding factors. Every 24 hours, the stones were carefully removed from solution and dried with absorbent paper. Their mass was recorded and they were placed back into solution.Results
Hexametaphosphate can dissolve CaOx, HA and struvite
The dissolution capability of HMP was tested against the two calcium salts found in kidney stones and Randall's plaque, oxalate and phosphate in the form of HA. HMP was added before or after the precipitation of the calcium salts, to simulate prevention and dissolution, respectively. Increasing HMP was able to both dissolve and prevent formation of both calcium salts (Fig. 2A and B). Notably, it was observed that HMP was more effective in prevention than in dissolution, up to 70% after one hour for CaOx, likely due to a lack of kinetic boundaries. In both cases, and also for calcium carbonate (Fig. S4, ESI†), complete prevention was found to occur at a HMP:Ca ratio above 1:3. This suggests that the mechanism of calcium chelation is ubiquitous, without dependence on specific salt, and that each HMP molecule can bind up to 3 calcium cations. This is consistent with the idea that each calcium ion is bound between two negatively charged oxygen atoms in the phosphoric acid groups (Fig. 2C).
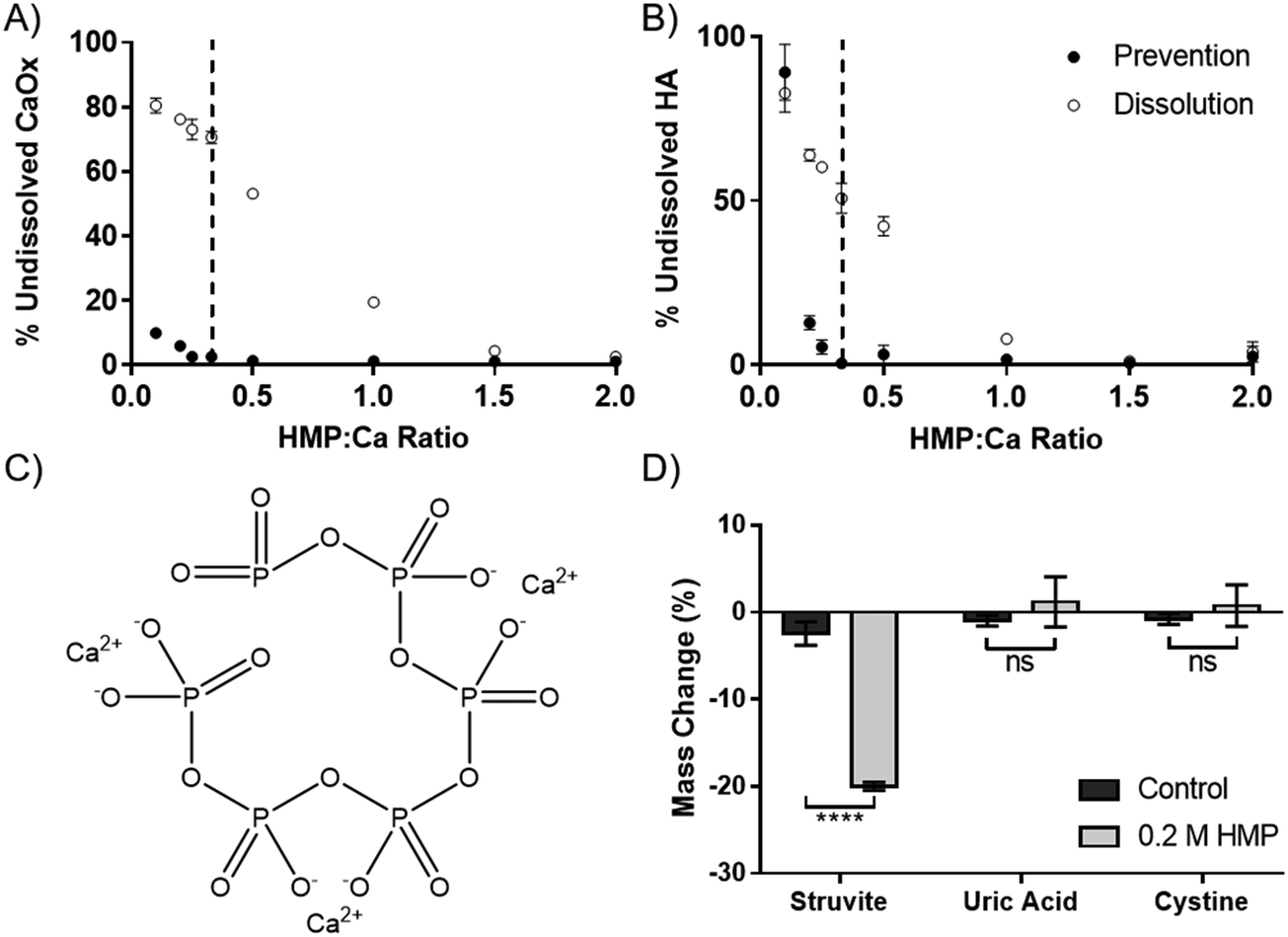 | ||
| Fig. 2 Hexametaphosphate dissolves calcium and magnesium salts. The prevention of precipitation (solid circles) and dissolution (open circles) of (A) calcium oxalate and (B) hydroxyapatite by HMP. Dashed line indicates a HMP:Ca ratio of 1:3. (C) Proposed mechanism by which HMP chelates calcium ions. (D) The change in mass after 3 days of struvite, uric acid and cystine in artificial urine with and without 0.2 M HMP. (A), (B) and (D) Show mean ± SD (n = 3). (D) Shows the result of multiple unpaired t-tests. | ||
In addition to calcium salts, kidney stones may also contain struvite, uric acid and cystine. Struvite, a magnesium salt (Fig. 1C), was significantly dissolved by HMP, with a 20% mass reduction over 72 hours (p < 0.0001). However, neither uric acid nor cystine were affected (Fig. 2D). This shows that HMP is also able to chelate magnesium, another divalent cation, but had no effect on the tested covalently bonded molecules.
Hexametaphosphate is a more potent dissolution agent than citrate, and prevents reprecipitation
HMP was tested against citrate, the current gold standard, to compare its efficacy at dissolving CaOx. Experiments were carried out in artificial urine (AU), to more closely replicate the physiological environment. Interestingly, HMP did not affect the pH of the AU, which was 5.8, but citrate raised the pH to 6.8. HMP has twice as many chelating groups as citrate (Fig. 1), thus direct comparisons were made between citrate and half the concentration of HMP. Neither compound was effective in this system below 1 mM, however at higher concentrations HMP was significantly more effective at dissolving CaOx than citrate after one hour, which was entirely dissolved in 50 mM HMP (Fig. 3A).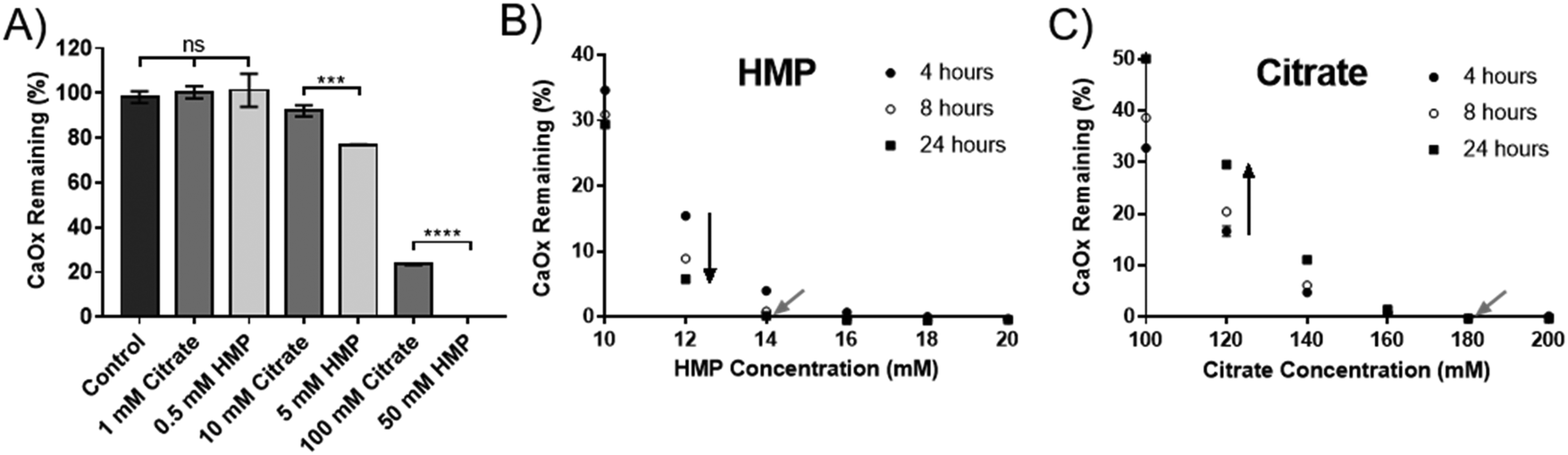 | ||
| Fig. 3 HMP is a more potent dissolution agent than citrate. (A) The percentage of CaOx in AU remaining after one hour in different concentrations of HMP or citrate. (B) The percentage of CaOx in AU remaining at different times and concentrations of HMP. (C) The percentage of CaOx in AU remaining at different times and concentrations of citrate. Mean ± SD (n = 3). (A) shows the results of an ordinary 1 way ANOVA with a post hoc Tukey's multiple comparisons test. | ||
To quantify this, the minimum concentration of citrate or HMP required to completely dissolve 2.5 mM CaOx in AU (the critical dissolution concentration) was identified. This was approximately 14 mM for HMP (Fig. 3B – grey arrow), and 180 mM for citrate (Fig. 3C – grey arrow) after 24 hours, suggesting that HMP is more than 12 times as effective a dissolution agent of CaOx as citrate, on a molar basis. As expected, HMP dissolved calcium oxalate in a time dependant manner, dissolving more of the salt over time (Fig. 3B – black arrow). Interestingly, this was not the case for citrate. Unless completely dissolved in 4 hours, there appeared to be a rebound effect whereby the concentration of undissolved CaOx increased at 8 and 24 hours (Fig. 3C – black arrow). This reprecipitate was confirmed to be CaOx by XRD (Fig. S1C, ESI†).
To elucidate the apparent reprecipitation with citrate, but not HMP, the zeta potential and size of the CaOx particles were measured. A concentration just below the critical dissolution concentration at four hours was used; 140 mM for citrate and 14 mM for HMP. Without addition of either dissolving agent the zeta potential was −15.4 mV, showing that the CaOx particles carry a negative charge in AU (Fig. 4A). Citrate did not significantly alter the zeta potential from the control after immediate addition or 4 hours (p > 0.05). HMP, on the other hand, made the zeta potential significantly more negative (p < 0.0001), with an average of −34.6 mV observed. This enhanced surface charge confers greater electrostatic repulsion between particles and decreases their propensity to aggregate.
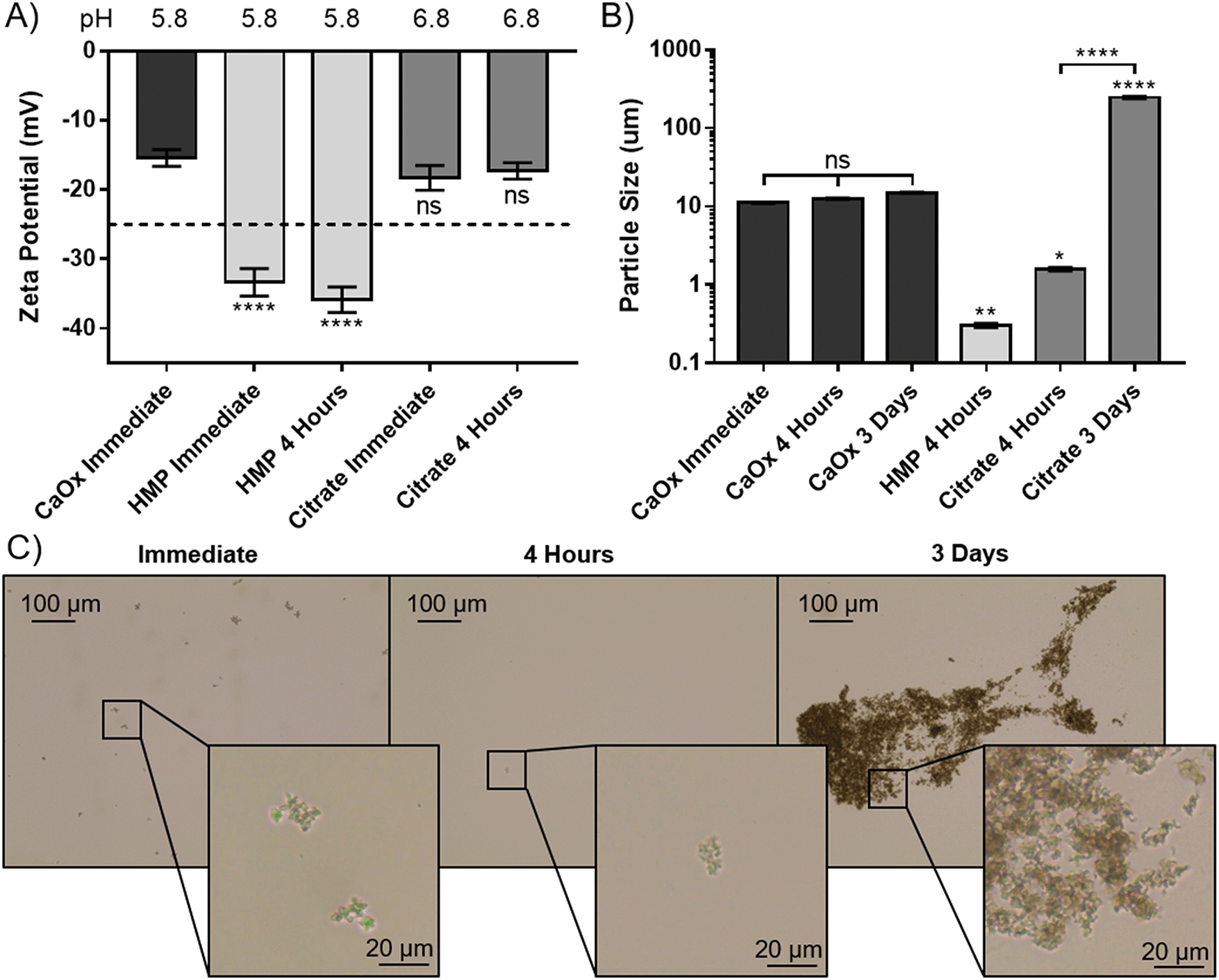 | ||
| Fig. 4 HMP prevents CaOx from aggregating. (A) Zeta potential measurements of a CaOx suspension treated with 14 mM HMP or 140 mM citrate. (B) The average particle size of precipitated CaOx treated with 14 mM HMP or 140 mM citrate. (C) Light microscopy images of CaOx suspensions treated with 140 mM citrate at different time points. (A) and (B) Show mean ± SD (n = 5). (A) and (B) Show the results of ordinary 1-way ANOVAs with post hoc Tukey's multiple comparisons tests. Stars above columns indicate comparison to untreated CaOx suspension at the same time point. | ||
The size of the CaOx particles formed in this system, without addition of either chelating agent was approximately 11 μm initially, and did not increase significantly within the 72 hours testing period (Fig. 4B). HMP was observed to significantly reduce the average CaOx particle size to 0.3 ± 0.018 μm within 4 hours, and they were entirely dissolved within 3 days. In this period, citrate was also shown to dissolve the particles, with a significant decrease to an average particle size of 1.6 μm. However, the particle size then significantly increased over time, to an average size of 237 μm at 3 days. Light microscopy images were taken over this period, in order to ascertain whether this increase was due to crystal growth or aggregation (Fig. 4C). These images suggest the CaOx particles initially form aggregates, with individual crystals ranging between 1 and 5 μm in diameter. After only 4 hours, the particles treated with HMP cannot be seen by light microscopy. Citrate appears to initially reduce the size of the crystals to 1 μm or less, or dissolve them completely, in four hours. Over time, however, the crystals regrow and aggregate; at 3 days, individual crystals appear to be between 1 and 3 μm, but are massed together in large aggregates several hundreds of microns in size, accounting for the significant increase in measured particle size.
Hexametaphosphate dissolves human kidney stones
Human kidney stones were obtained from patients following percutaneous nephrolithotomy. XRD of the stones showed that they were principally CaOx monohydrate (Fig. 5A). Additional compositional analysis using XRF was able to identify a calcium phosphate phase present in the stones (Fig. 5B). The small peak at 2θ = 11.7° in the XRD pattern suggests this phase may be brushite, as does the shoulder at 30.5°, though the peak height at 39.8° may indicate HA. In either case, the phosphate appears to be a minority component.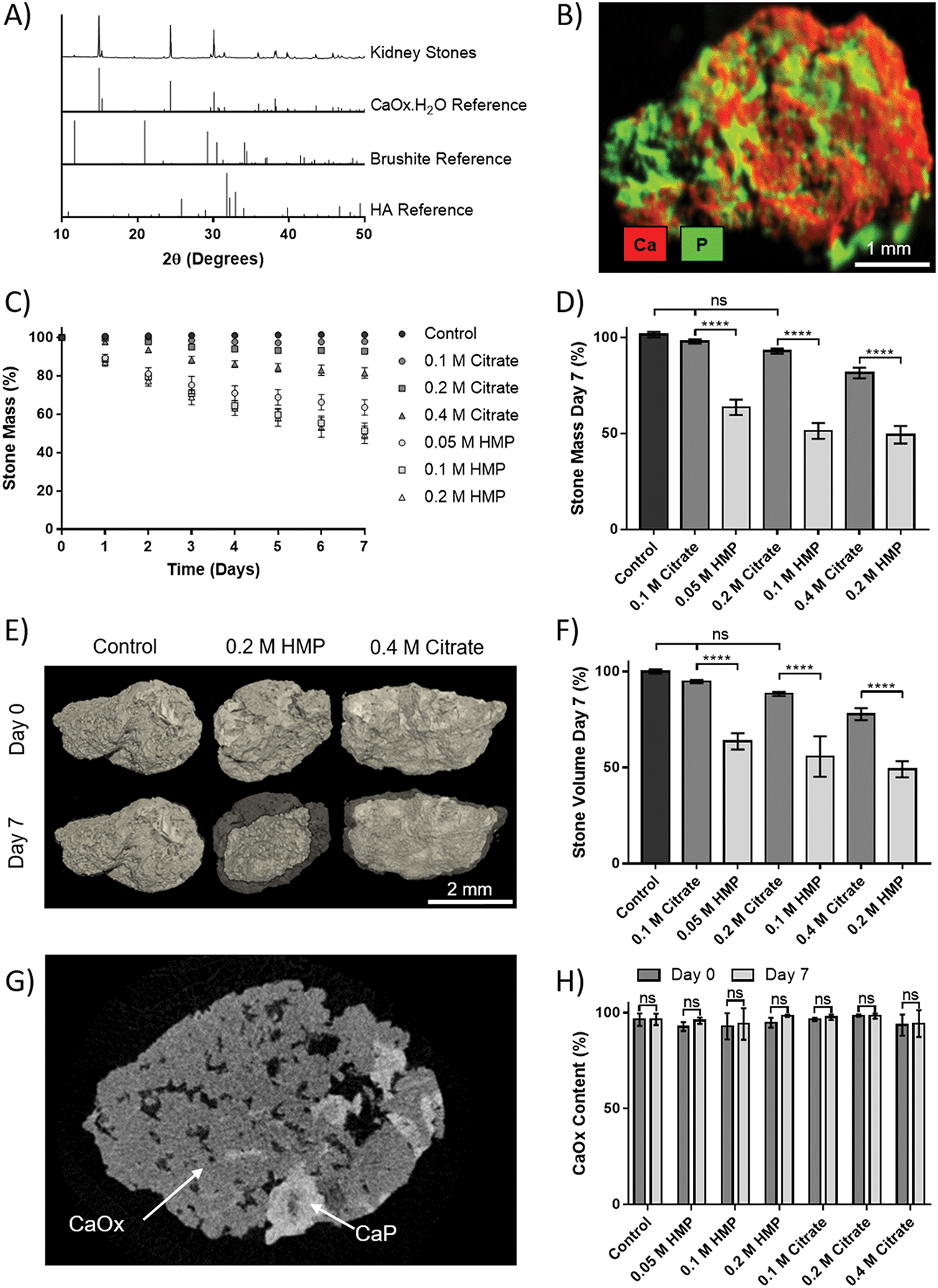 | ||
| Fig. 5 HMP is more potent than citrate at dissolving calcium stones derived from human patients. (A) X-ray diffraction patterns of powdered kidney stones and reference patterns. (B) X-ray fluorescence image of a stone prior to dissolution. (C) Mass of stones in solutions of citrate or HMP at various concentrations over time. (D) Change in mass of stones after 7 days. (E) 3D microCT rendering of stones before and after dissolution. (F) Change in volume of stones after 7 days. (G) MicroCT slice showing the distinct oxalate and phosphate phases (untreated stone). (H) CaOx content of stones at day 0 and day 7. (C), (D), (F) and (H) Show mean ± SD (n = 3). (D) and (F) Show the results of an ordinary 1-way ANOVA with a post hoc Tukey's multiple comparisons test. (H) Shows multiple paired t-tests. | ||
To assess dissolution capability, stones that initially weighed 18.8–22.8 mg were placed in AU containing different concentrations of HMP or citrate, and their mass was tracked over 7 days (Fig. 5C). The relative mass of stones in HMP was significantly lower than in a comparative citrate concentration at every time point. After seven days, the relative mass of stones in HMP was reduced by up to 55%, and their masses were significantly lower (32–42%, p < 0.0001) than those in a corresponding citrate concentration (Fig. 5D). In contrast, the mass of those in 0.1 or 0.2 M citrate did not significantly decrease in mass over 7 days, compared to controls. 3D volume rendering via microCT showed that mass loss correlated to a visible reduction in stone volume (Fig. 5E). This was confirmed via quantitative analysis of the microCT data, which revealed that stone volume followed the same trend as mass (Fig. 5F). Notably, two phases could be clearly discerned via grey scale values in microCT data (Fig. 5G and Fig. S5, ESI†). The ratio of these phases was calculated and revealed that the stones were on average 95% calcium oxalate by volume, which is in strong agreement with the XRD analysis. Interestingly, this ratio did not change significantly following dissolution, suggesting that HMP dissolves both phases with equal efficacy (Fig. 5H).
Discussion
Kidney stones affect a substantial fraction of the world's population, and incidence is increasing across all demographics. While techniques for stone removal are improving worldwide, prophylaxis remains fundamental for patient care, especially given the recurrent nature of kidney stones.45 Citrate is commonly prescribed for kidney stone prophylaxis, however recurrence remains frequent, and side effects may lead patients to discontinue treatment.32 There is thus a clear need for new, more effective therapies. A recent study discussing the multiple growth, aggregation and dissolution steps by which stones form describe the need for therapies that inhibit crystal growth and aggregation while also promoting dissolution.46 The findings in this study suggest that HMP is a promising therapeutic candidate that meets all of these requirements.Commonly used for its ability to chelate divalent metal cations, HMP was expected to be able to dissolve calcium and magnesium salts; indeed it is demonstrated in Fig. 2 and suggested in previous studies that HMP can dissolve calcium oxalate, phosphate and struvite, the most common stone components.37,40,41 However, HMP had no effect on uric acid or cystine (Fig. 2), which are both stones commonly treated by alkalizing the urine.47 This highlights the need for stratification of treatment, based on stone type and aetiology. HMP may thus be a suitable therapeutic for calcium and magnesium stones, which make up the vast majority of cases. Use of HMP to prevent precipitation of CaOx was shown to be more effective than dissolution (Fig. 2A and B). This is likely due to higher availability of cations and a lack of kinetic limitations to chelation when cations are in solution, compared to when they are locked into a solid mineral. It may thus be concluded that HMP might be more effective as a prevention than a treatment for formed stones, although notably it is capable of both.
When directly compared to citrate, HMP was shown to be more than 12 times as effective at dissolving CaOx on a molar basis, and thus more than 6 times as effective for the same number of potential binding sites (Fig. 3). This may be because the Ca-HMP complex is far more stable than Ca-citrate.48,49 Even among phosphate species, HMP forms the most stable complex with calcium, with a stability constant twice that of tripolyphosphate or phytate, which form the next most stable calcium complexes.48 In addition to being a more potent chelator, it was found that HMP continues to dissolve CaOx over time, while it appears to reprecipitate when treated with citrate (Fig. 3B and C). HMP was shown to significantly increase the negativity of CaOx particle zeta potential in AU to −34.6 mV. This is well beyond the ±25 mV often taken as the minimum for a high surface charge that can confer long-term electrostatic stability.50 HMP has been studied previously for its ability to disperse HA suspensions by binding to the particle surface and providing both electrostatic and steric hindrance to aggregation.51 Prevention of particle aggregation is critical to avoid kidney stones; crystal growth is not sufficient for nuclei to become large enough to anchor or occlude a lumen in the 5–7 minutes it takes for them to pass through the nephrons, however they can reach such a size within a minute via aggregation.19 Increasing repulsion between particles to prevent aggregation may be a key advantage of HMP compared with existing approaches for kidney stone prevention. Conversely, citrate did not significantly alter the zeta potential, but facilitated aggregation of particles, which was highlighted through an increase in particle size over time (Fig. 4B and C).
Finally, the efficacy of HMP was tested against human kidney stones. XRD and XRF analyses showed that the stones were a mixture of calcium oxalate and phosphate, and thus susceptible to dissolution by HMP and citrate. These ex vivo experiments confirmed that HMP was significantly more effective than citrate at dissolving kidney stones, and this was observed for all tested concentrations (0.05–0.4 M) and time points (up to 7 days). It is also notable that citrate was ineffective below 0.4 M while the mass of stones in 0.05 M HMP decreased by 36.4% in 7 days. When comparing the mass loss in this treatment group to stones placed in 0.4 M citrate, it may be concluded that HMP is up to 16 times more effective on a molar basis. This finding is in line with the results obtained for CaOx precipitate (Fig. 3). Future dissolution experiments on majority phosphate or struvite stones should be undertaken to establish whether a similar relationship exists between in vitro and ex vivo dissolution. Assessment of the human stones with microCT after 7 days revealed a correlation between mass and volume loss, which is of clinical significance since size dictates whether the patient can pass the stone or whether it will cause a blockage leading to more severe symptoms.24
This study has examined HMP as a potential new therapy for kidney stones, and has shown that it is highly efficacious at dissolving calcium and magnesium salts in vitro and human stones ex vivo. Future work in this area must therefore be aimed at translating this material chemistry into a biological environment, and foremost on how HMP might be delivered to the kidney. HMP is a common food additive, generally recognised as safe by the US Food and Drug Administration, and has very low oral toxicity.52,53 Oral delivery would be optimal for patient compliance, however there is some debate on how much condensed phosphate is absorbed. Some studies suggest that polyphosphates are broken down in the bowel into orthophosphates, and only small amounts of the condensed phosphate are absorbed.54 However, a recent study found that orally administered pyrophosphate, a shorter condensed phosphate, was readily absorbed and available to inhibit calcification.55 This needs to be confirmed for HMP. Chemical modification of HMP, for example the addition of P–C–P bonds, may act to render the molecule more resistant to hydrolytic and enzymatic break down, as is the case for bisphosphonates.56 Similar effects may be achieved by loading the HMP into a delivery vehicle, in order to protect it until it reaches the kidney. However, both of these approaches may make the therapy considerably more expensive. Alternatively, administration via injection or implantation may serve to bypass the digestive system entirely, however this will likely reduce patient compliance, as would direct delivery to the kidney via a nephrostomy tube.
In addition to delivery, more in-depth studies on the effects of HMP on tissues and organs may be required. The effect on urological tissues must be established, especially if HMP is to be administered directly, and in high concentrations. Any deleterious effects on other mineralised tissues such as bones and teeth should also be identified. However, delocalisation in sufficient quantity to cause pathology is unlikely if delivered directly to the urinary system, especially since the dose required to prevent and dissolve kidney stones it likely to be much less than that to dissolve bone. Aiding this is the fact that HMP is far more effective at lower pH,39 and will therefore its efficacy will be specifically increased in the urinary tract, typically pH 6, compared to the rest of body at pH 7.4. Further, HMP is degraded by alkaline phosphatase,39 an enzyme found ubiquitously in the body, which will work to break the HMP down before it can act away from the kidney. However, these questions need to be answered in more complex in vitro models, which introduce biological components and flow,57–59 then animal models,60,61 and eventually human trials, as the next step in making HMP a therapy for kidney stones.
Conclusions
This study demonstrates that HMP is able to dissolve they key stone components calcium oxalate, calcium phosphate, and struvite, and is more potent than citrate at doing so. It also highlights that HMP is capable of increasing the zeta potential of CaOx in solution, which may be exploited to prevent particle aggregation and thus avert the most rapid mechanism of stone growth. Promisingly, ex vivo testing confirmed the potent ability of HMP to dissolve human kidney stones further advocating that this compound may be an answer to this growing clinical need. The ability of HMP to maintain a low urine pH is also notable since it makes it especially suited for calcium phosphate and struvite stones, where acidifying the urine is recommended.47 HMP may therefore also be suitable for decalcifying Randall's plaque, which is predominantly calcium phosphate, though further work is needed to confirm this. The findings of this study therefore demonstrate that further research is warranted and should focus on addressing the delivery of HMP to the kidney and examining efficacy in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Kasra Saeb-Parsy for his assistance in stone procurement. This work was funded by the EPSRC CDT for Formulation Engineering in the School of Chemical Engineering at the University of Birmingham, UK, Grant reference EP/L015153/1, and the Royal Centre for Defence Medicine.References
- T. Alelign and B. Petros, Adv. Urol., 2018, 3068365 Search PubMed
.
- M. Courbebaisse, C. Prot-Bertoye, J. P. Bertocchio, S. Baron, G. Maruani, S. Briand, M. Daudon and P. Houillier, Rev. Med. Interne, 2017, 38, 44–52 CrossRef CAS PubMed
- O. W. Moe, Lancet, 2006, 367, 333–344 CrossRef CAS
- C. D. Scales, A. C. Smith, J. M. Hanley and C. S. Saigal, Eur. Urol., 2012, 62, 160–165 CrossRef PubMed
- S. R. Khan, M. S. Pearle, W. G. Robertson, G. Gambaro, B. K. Canales, S. Doizi, O. Traxer and H.-G. Tiselius, Nat. Rev. Dis. Primers, 2016, 2, 16008 CrossRef PubMed
- S. K. De, X. Liu and M. Monga, Urology, 2014, 84, 1030–1033 CrossRef PubMed
- I. P. Heilberg and D. S. Goldfarb, Adv. Chronic Kidney Dis., 2013, 20, 165–174 CrossRef PubMed
- P. M. Ferraro, E. N. Taylor, G. Gambaro and G. C. Curhan, Clin. J. Am. Soc. Nephrol., 2013, 8, 1389–1395 CrossRef PubMed
- E. N. Taylor and G. C. Curhan, Kidney Int., 2008, 73, 207–212 CrossRef CAS PubMed
- P. M. Ferraro, E. I. Mandel, G. C. Curhan, G. Gambaro and E. N. Taylor, Clin. J. Am. Soc. Nephrol., 2016, 11, 1834–1844 CrossRef CAS PubMed
- T. Mitchell, P. Kumar, T. Reddy, K. D. Wood, J. Knight, D. G. Assimos and R. P. Holmes, Am. J. Physiol., 2019, 316, F409–F413 Search PubMed
- M. Agha and R. Agha, Int. J. Surg. Oncol., 2017, 2, e17 CrossRef PubMed
- N. H. Cho, J. E. Shaw, S. Karuranga, Y. Huang, J. D. da Rocha Fernandes, A. W. Ohlrogge and B. Malanda, Diabetes Res. Clin. Pract., 2018, 138, 271–281 CrossRef CAS PubMed
- R. J. Johnson, L. G. Sánchez-Lozada, L. S. Newman, M. A. Lanaspa, H. F. Diaz, J. Lemery, B. Rodriguez-Iturbe, D. R. Tolan, J. Butler-Dawson, Y. Sato, G. Garcia, A. A. Hernando and C. A. Roncal-Jimenez, Ann. Nutr. Metab., 2019, 74, 38–44 CrossRef CAS PubMed
- K. A. Barraclough, G. A. Blashki, S. G. Holt and J. W. M. Agar, Kidney Int., 2017, 92, 526–530 CrossRef PubMed
- T. H. Brikowski, Y. Lotan and M. S. Pearle, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9841–9846 CrossRef CAS PubMed
- M. S. Pearle, E. A. Calhoun and G. C. Curhan, J. Urol., 2005, 173, 848–857 CrossRef PubMed
- P. Singh, F. T. Enders, L. E. Vaughan, E. J. Bergstralh, J. J. Knoedler, A. E. Krambeck, J. C. Lieske and A. D. Rule, Mayo Clin. Proc., 2015, 90, 1356–1365 CrossRef PubMed
- F. L. Coe, J. H. Parks and J. R. Asplin, N. Engl. J. Med., 1992, 327, 1141–1152 CrossRef CAS PubMed
- E. Letavernier, D. Bazin and M. Daudon, C. R. Chim., 2016, 19, 1456–1460 CrossRef CAS
- M. P. Linnes, A. E. Krambeck, L. Cornell, J. C. Williams, M. Korinek, E. J. Bergstralh, X. Li, A. D. Rule, C. M. McCollough, T. J. Vrtiska and J. C. Lieske, Kidney Int., 2013, 84, 818–825 CrossRef CAS PubMed
- R. L. Kuo, J. E. Lingeman, A. P. Evan, R. F. Paterson, J. H. Parks, S. B. Bledsoe, L. C. Munch and F. L. Coe, Kidney Int., 2003, 64, 2150–2154 CrossRef PubMed
- C. Y. C. Pak and K. Holt, Metabolism, 1976, 25, 665–673 CrossRef CAS PubMed
- F. L. Coe, A. Evan and E. Worcester, J. Clin. Invest., 2005, 115, 2598–2608 CrossRef CAS PubMed
- J. R. Asplin, J. H. Parks, Y. Nakagawa and F. L. Coe, Kidney Int., 2002, 61, 1821–1829 CrossRef PubMed
- A. C. Lopes Neto, F. Korkes, J. L. Silva, R. D. M. Amarante, M. H. E. Mattos, M. Tobias-Machado and A. C. L. Pompeo, J. Urol., 2012, 187, 164–168 CrossRef PubMed
- A. R. El-Nahas, H. M. Ibrahim, R. F. Youssef and K. Z. Sheir, BJU Int., 2012, 110, 898–902 CrossRef PubMed
- A. Srisubat, S. Potisat, B. Lojanapiwat, V. Setthawong and M. Laopaiboon, Cochrane Database Syst. Rev., 2014, 2014(11), 1–40 Search PubMed
- M. S. Pearle, D. S. Goldfarb, D. G. Assimos, G. Curhan, C. J. Denu-Ciocca, B. R. Matlaga, M. Monga, K. L. Penniston, G. M. Preminger, T. M. T. Turk and J. R. White, J. Urol., 2014, 192, 316–324 CrossRef PubMed
- J. D. Rimer, K. Sakhaee and N. M. Maalouf, Curr. Opin. Nephrol. Hypertens., 2019, 28, 130–139 CrossRef CAS PubMed
- E. Ruiz-Agudo, A. Burgos-Cara, C. Ruiz-Agudo, A. Ibañez-Velasco, H. Cölfen and C. Rodriguez-Navarro, Nat. Commun., 2017, 8, 768 CrossRef PubMed
- R. Phillips, V. S. Hanchanale, A. Myatt, B. Somani, G. Nabi and C. S. Biyani, Cochrane Database Syst. Rev., 2015, 2015(10), 1–41 Search PubMed
- D. Mattle and B. Hess, Urol. Res., 2005, 33, 73–79 CrossRef CAS PubMed
- S. Hesaraki, A. Zamanian and F. Moztarzadeh, J. Biomed. Mater. Res., Part A, 2009, 88, 314–321 CrossRef CAS PubMed
- F. Andreola, E. Castellini, T. Manfredini and M. Romagnoli, J. Eur. Ceram. Soc., 2004, 24, 2113–2124 CrossRef CAS
- H. Fleisch and W. F. Neuman, Am. J. Physiol., 1961, 200, 1296–1300 CrossRef CAS PubMed
- C. McGaughey, Caries Res., 1983, 17, 229–241 CrossRef CAS PubMed
- H. Fleisch, F. Straumann, R. Schenk, S. Bisaz and M. Allgöwer, Am. J. Physiol., 1966, 211, 821–825 CrossRef CAS PubMed
- N. Eisenstein, R. Williams, S. Cox, S. Stapley and L. Grover, J. Mater. Chem. B, 2016, 4, 3815–3822 RSC
- R. T. Thomson, Analyst, 1936, 61, 320–323 RSC
-
R. E. Martin, E. P. Carter, J. F. J. George and L. M. Davis, Marine and Freshwater Products Handbook, Technomic Pub. Co., 2000 Search PubMed
- J. Chung, I. Granja, M. G. Taylor, G. Mpourmpakis, J. R. Asplin and J. D. Rimer, Nature, 2016, 536, 446–450 CrossRef CAS PubMed
- K.
N. Parra, S. Gul, J. M. Aquino, D. W. Miwa and A. J. Motheo, J. Solid State Electrochem., 2016, 20, 1001–1009 CrossRef CAS
- B. Cheong, R. Muthupillai, M. F. Rubin and S. D. Flamm, Clin. J. Am. Soc. Nephrol., 2007, 2, 38–45 CrossRef PubMed
- J. D. Denstedt, Asian J. Urol., 2018, 5, 203–204 CrossRef PubMed
- M. Sivaguru, J. J. Saw, J. C. Williams, J. C. Lieske, A. E. Krambeck, M. F. Romero, N. Chia, A. L. Schwaderer, R. E. Alcalde, W. J. Bruce, D. E. Wildman, G. A. Fried, C. J. Werth, R. J. Reeder, P. M. Yau, R. A. Sanford and B. W. Fouke, Sci. Rep., 2018, 8, 13731 CrossRef PubMed
- L. Frassetto and I. Kohlstadt, Am. Fam. Physician, 2011, 84, 1234–1242 Search PubMed
- R. E. Gosselin and E. R. Coghlan, Arch. Biochem. Biophys., 1953, 45, 301–311 CrossRef CAS PubMed
- R. P. Singh, Y. D. Yeboah, E. R. Pambid and P. Debayle, J. Chem. Eng. Data, 1991, 36, 52–54 CrossRef CAS
- A. Doostmohammadi, A. Monshi, R. Salehi, M. H. Fathi, Z. Golniya and A. U. Daniels, Ceram. Int., 2011, 37, 2311–2316 CrossRef CAS
- R. R. Rao and T. S. Kannan, J. Am. Ceram. Soc., 2001, 84, 1710–1716 CrossRef CAS
- M. L. Weiner, W. F. Salminen, P. R. Larson, R. A. Barter, J. L. Kranetz and G. S. Simon, Food Chem. Toxicol., 2001, 39, 759–786 CrossRef CAS PubMed
- Food and Drug Administration, Code of Federal Regulations Title 21, Chapter 1, Subchapter B, Part 182, Subpart G, Section 182.6760 Sodium Hexametaphosphate, https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=182.6760, accessed 5 December 2017.
- R. S. Lanigan, Int. J. Toxicol., 2001, 20, 75–89 CrossRef PubMed
- D. Dedinszki, F. Szeri, E. Kozák, V. Pomozi, N. Tőkési, T. R. Mezei, K. Merczel, E. Letavernier, E. Tang, O. Le Saux, T. Arányi, K. Wetering and A. Váradi, EMBO Mol. Med., 2017, 9, 1463–1470 CrossRef CAS PubMed
- H. Fleisch, Drugs, 1991, 42, 919–944 CrossRef CAS PubMed
- M. M. Tunney, M. C. Bonner, P. F. Keane and S. P. Gorman, Biomaterials, 1996, 17, 1025–1029 CrossRef CAS PubMed
- B. Subramanian, D. Rudym, C. Cannizzaro, R. Perrone, J. Zhou and D. L. Kaplan, Tissue Eng., Part A, 2010, 16, 2821–2831 CrossRef CAS PubMed
- T. M. DesRochers, E. Palma and D. L. Kaplan, Adv. Drug Delivery Rev., 2014, 69–70, 67–80 CrossRef CAS PubMed
- S. R. Khan, World J. Urol., 1997, 15, 236–243 CrossRef CAS PubMed
-
S. R. Khan, Animal Models for the Study of Human Disease, Elsevier Inc., 2013, pp. 483–498 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00343c |
| This journal is © The Royal Society of Chemistry 2020 |