
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic spatial and structural organization in artificial cells regulates signal processing by protein scaffolding†
Bastiaan C.
Buddingh'
 ,
Antoni
Llopis-Lorente
,
Loai K. E. A.
Abdelmohsen
* and
Jan C. M.
van Hest
*
,
Antoni
Llopis-Lorente
,
Loai K. E. A.
Abdelmohsen
* and
Jan C. M.
van Hest
*
Department of Chemical Engineering and Chemistry, Institute for Complex Molecular Systems, Department of Biomedical Engineering, Eindhoven University of Technology, PO Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: J.C.M.v.Hest@tue.nl; L.K.E.A.Abdelmohsen@tue.nl
First published on 5th November 2020
Abstract
Structural and spatial organization are fundamental properties of biological systems that allow cells to regulate a wide range of biochemical processes. This organization is often transient and governed by external cues that initiate dynamic self-assembly processes. The construction of synthetic cell-like materials with similar properties requires the hierarchical and reversible organization of selected functional components on molecular scaffolds to dynamically regulate signaling pathways. The realization of such transient molecular programs in synthetic cells, however, remains underexplored due to the associated complexity of such hierarchical platforms. In this contribution, we effectuate dynamic spatial organization of effector protein subunits in a synthetic biomimetic compartment, a giant unilamellar vesicle (GUV), by associating in a reversible manner two fragments of a split luciferase to the membrane. This induces their structural dimerization, which consequently leads to the activation of enzymatic signaling. Importantly, such organization and activation are dynamic processes, and can be autonomously regulated – thus opening up avenues toward continuous spatiotemporal control over supramolecular organization and signaling in an artificial cell.
Introduction
Versatility and adaptability of biological systems arise from well-defined chemical pathways that control their structural and spatial organizational states. Such states are often dynamic and play a significant role in controlling key biological processes. For example, so-called ‘supramolecular organizing centers’ (e.g. inflammasomes) are localized higher-order signaling complexes that utilize dynamic protein scaffolding processes to organize downstream operations and integrate upstream signaling events.1 Enzyme activation by protein dimerization is a particularly powerful concept in such supramolecular regulation – for instance, caspase-1 forms an active heterodimer upon assembly on cytosolic receptors, which initiates an inflammatory cellular response.1 Inspired by this biochemical logic, scientists from various disciplines have undertaken to mimic such systems and behaviors in synthetic analogs.2–7 In this regard, self-assembled structures based upon peptides8,9 or DNA10–14 have been exemplary in how to program synthetic systems the way biology does; however, these systems do not display autonomous functionality. Contrary to most synthetic materials, living systems exhibit self-regulating and adaptable behavior that is maintained by dynamic (and transient) processes, where spatiotemporal regulation and organization of the flow of chemical information is critical.15 Successful programming of dynamic behavior in synthetic materials thus requires implementation of regulatory mechanisms that exert control over the resulting functional ensemble.In a first approximation, it is possible to identify two general, yet crucial sub-categories to the area of mimicking biological dynamic behavior: (i) the development of synthetic compartmentalized systems that mimic structural aspects of biological entities (i.e. cells),16–20 and (ii) reconstitution of biochemical processes such as protein expression and enzymatic networks to mimic functional aspects of cellular behavior.21–24 Although either category has been indispensable in building up our knowledge and capacity in this discipline, the merger of these two important properties is required to create cell-like entities that display dynamic behavior. Toward this, our group has previously developed an approach to spatially organize proteins inside a synthetic cell mimic.25 Although this protein localization process was dynamic, both the structure and function of the protein remained static. Capitalizing on this, we herein demonstrate a self-regulated and dynamic system in which its spatial organization induces a biochemical response inside an artificial cell.
Despite great efforts to incorporate regulatory mechanisms into artificial structures, integration of functional responses with self-regulated spatio-structural organization within a compartmentalized system remains underexplored. The reason for this shortfall is the physicochemical complexity associated with integrating regulatory and responsive elements in one compartmentalized system. In this paper we combine these elements in compartmentalized systems that are capable of engaging in dynamic assembly processes and activating signaling responses as a consequence of an internal enzymatic program. We induce spatio-structural organization in a synthetic cell-like compartment, by organizing effector proteins at the lipid membrane in response to external environmental cues (Fig. 1). Upon assembly, the complex is activated and promotes a signaling response. This concept is effectuated by recruiting two fragments of a split protein to the membrane (spatial organization), which induces their dimerization (structural organization) and activates enzymatic signaling (functional response). Importantly, this organization and protein activation is dynamic, providing continuous spatiotemporal control over signaling in an artificial cell. The incorporation of a regulatory internal enzymatic program showed that these artificial cells could potentially operate as autonomous life-like entities that respond to external molecular information through programmable enzymatic machinery.
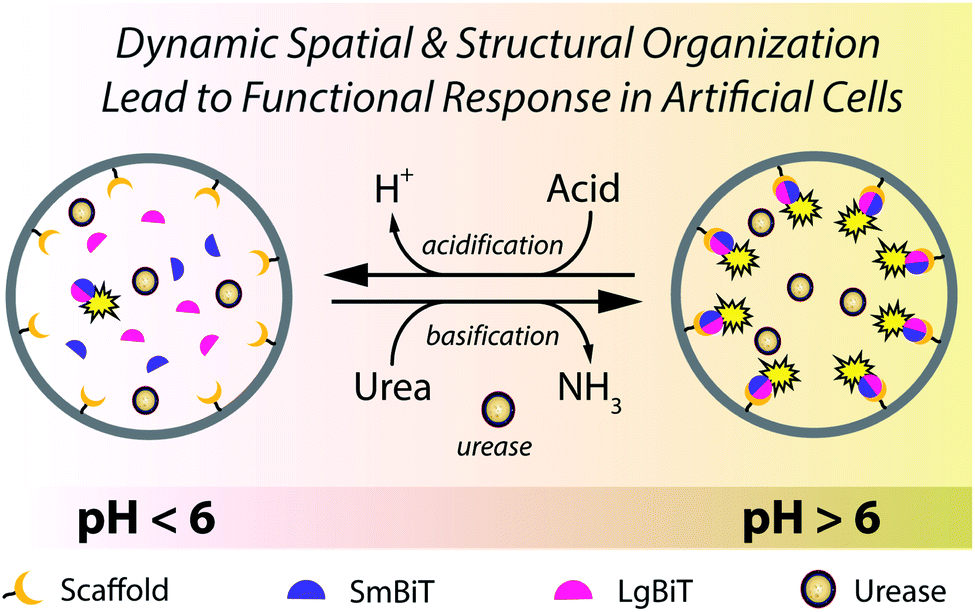 | ||
| Fig. 1 A giant vesicle responds to environmental cues through a pre-programmed enzymatic machinery that modulates the pH to induce spatio-structural organization and activation of effector proteins at the lipid membrane. Addition of urea increases the pH by virtue of encapsulated urease, which converts urea into ammonia. This pH increase triggers spatial organization of two protein fragments (SmBiT and LgBiT) at the membrane, followed by their structural organization and subsequent enzymatic activation. Upon addition of acid, the active membrane-bound complex dissociates and enzymatic signaling is extinguished until complex formation is reinitiated by addition of urea. | ||
Results and discussion
Spatial organization of a split protein at the vesicle membrane
Dynamic spatial organization to induce protein structural organization was effectuated by directing the association of a split protein to the inner leaflet of the membrane of giant unilamellar vesicles (GUVs). GUVs were engineered to display a nickel-nitrilotriacetic acid (NTA) headgroup, whereas the split protein fragments were synthesized with N-terminal polyhistidine fusion peptides (His-tags). The strong affinity of the His-tagged proteins for the NTA–lipid headgroups drove the association of the split protein fragments to the GUV membrane. Furthermore, the non-covalent nature of the His/Ni–NTA interactions allowed dissociation of the complex by treatment with EDTA or imidazole, or by decreasing the pH of the solution, which protonated the histidine residues.26,27 Such controlled spatial organization at the GUVs' membrane was translated into protein structural organization and consequent enzymatic signaling by employing the split luciferase protein ‘NanoBiT’.28,29 This split luciferase is composed of two subunits (LgBiT and SmBiT); when both protein fragments were His-tagged they could be collectively recruited to the membrane of the GUVs, where proximity-driven subunit complementation restored their activity. In the presence of its substrate this yielded a bright bioluminescent signal. As such, the enzymatic activity was regulated by the spatial and structural organization of the split luciferase at the GUV membrane. The pH-dependency of the His/Ni–NTA interactions further provided a mechanism to regulate this organization through prevalent biochemical reactions.To load GUVs with the LgBiT and SmBiT fragments we employed the inverted emulsion technique, as it ensures high encapsulation efficiencies and stable vesicles under physiological buffer conditions.30 First, a water-in-oil emulsion was prepared containing both protein fragments in the water phase and phospholipids in the oil phase. Next, the droplets were passed through a water/oil interface covered by phospholipids. This generated GUVs loaded with both luciferase fragments in the desired ratio and concentration. Furthermore, a phospholipid with a Ni–NTA moiety conjugated to its head group (DOGS–NTA–Ni) was incorporated into the GUV membrane at concentrations of up to 2 mol%. LgBiT and SmBiT binding to the membrane through the programmed His/Ni–NTA interactions and subsequent formation of the luminescent complex was monitored by bioluminescence microscopy and spectroscopy.
When DOGS–NTA–Ni was incorporated into the GUVs, the formation of a ring of bioluminescence originating from the GUV membrane indicated formation of the active luciferase complex at the membrane (Fig. 2a). Some Ni–NTA-independent formation of the active luciferase was also detected and attributed to low levels of spontaneous dimerization of LgBiT and SmBiT. In control experiments, we confirmed that no bioluminescence was produced in the absence of luciferase substrate (ESI Fig. 1†). In addition, we determined that 5 μM of encapsulated LgBiT and SmBiT was appropriate for bioluminescence microscopy imaging (ESI Fig. 2†), and that the amount of DOGS–NTA–Ni in the GUV membrane controls LgBiT and SmBiT binding (ESI Fig. 3†). The role of the His/Ni–NTA interaction was further verified using EDTA to chelate nickel, which effectively inhibited the recruitment of the luciferase fragments to the GUV membrane from the external solution (ESI Fig. 4†). To exclude any contribution of the externally facing ligands to the reconstitution of the split luciferase, 2 mM EDTA was added to the external solution, which blocked recruitment of both fragments from the external solution, yet not from the lumen of the vesicles (ESI Fig. 5†). The Ni–NTA moieties thus constituted selective ligands that facilitated the localization of a His-tagged protein to the vesicle membrane through recruitment of its hexahistidine domain.
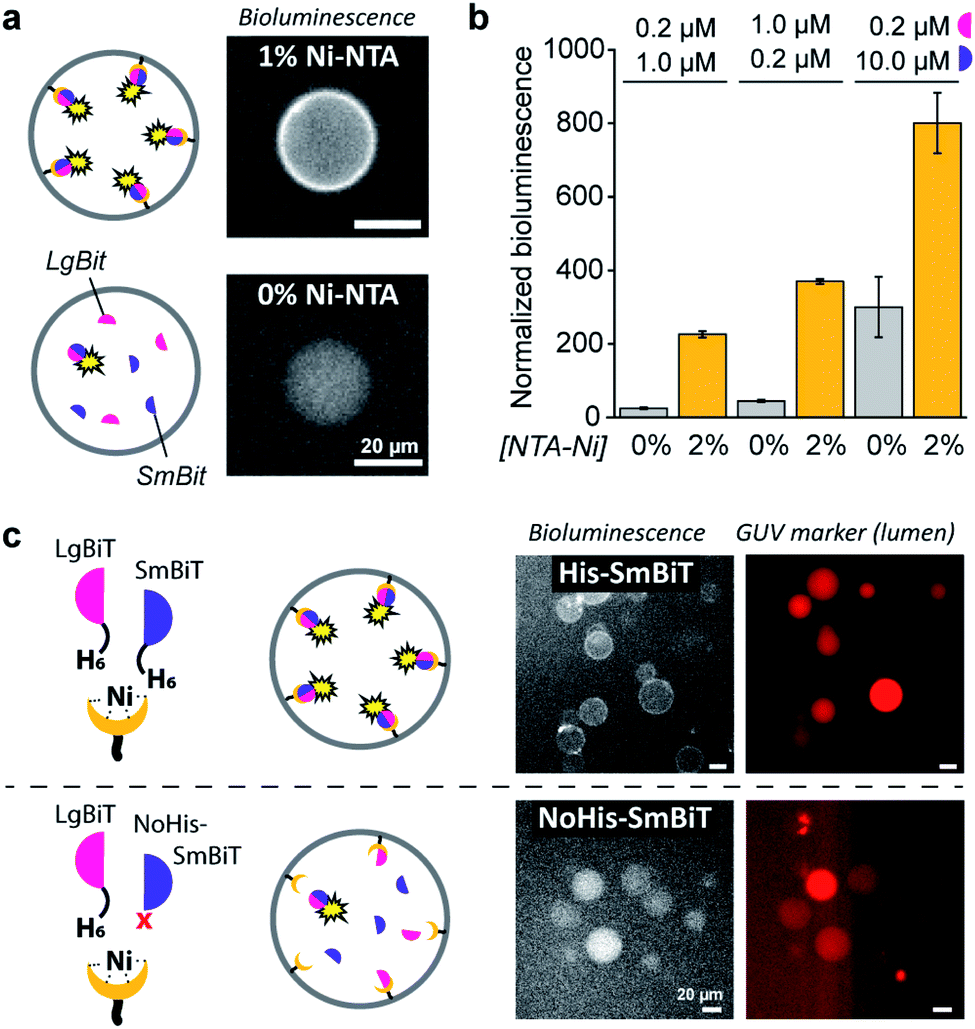 | ||
| Fig. 2 Ni–NTA-dependent spatial organization and activation of NanoBiT in GUVs. (a) Bioluminescence micrographs of GUVs loaded with SmBit and LgBiT (5 μM each) that were functionalized with 1% DOGS–NTA–Ni or left unfunctionalized. (b) Bioluminescence of GUVs with or without 2% DOGS–NTA–Ni and varying concentrations of encapsulated LgBiT and SmBit. The bioluminescence was normalized to the number of GUVs. (c) Bioluminescence micrographs of GUVs loaded with LgBiT (5 μM) and either His-SmBit or NoHis-SmBiT (5 μM). The GUVs contained 2% DOGS–NTA–Ni, and sulforhodamine (red) for tracking. Global bioluminescence quantification of corresponding samples is shown in ESI Fig. 8.† 2 mM EDTA was added to the external solution and all experiments were performed at pH 7.4. | ||
Spatio-structural organization controls enzymatic activation
As demonstrated, these His/Ni–NTA interactions are a powerful instrument to induce spatial organization of hierarchical complexes of nanoscopic components inside artificial cells. Yet, recruitment of proteins to the GUV membrane can also promote their structural organization (mediated by dimerization) and – in case of enzymes – subsequent activation, which is a common regulatory process in natural cells. Here, membrane localization increases the local concentration of LgBiT and SmBiT (spatial organization), which is expected to enhance their dimerization (structural organization).To investigate the enhancement of signaling responses by both spatial and structural organization of the luciferase fragments, the bioluminescence output of populations of vesicles was monitored in bulk measurements, allowing a more quantitative assessment of the overall luciferase reconstitution than that offered by microscopy. To correct for the concentration of vesicles in each sample, the bioluminescence was normalized to the fluorescence of an inert fluorophore loaded into the GUVs. Indeed, the total bioluminescence of vesicles that contained DOGS–NTA–Ni was markedly higher than that of vesicles without Ni–NTA groups (Fig. 2b). The absolute bioluminescent signal depended on the concentration of both fragments inside the vesicles; the stoichiometry of the interaction was unimportant, but – as expected – higher concentrations of LgBiT and SmBiT produced higher enzymatic activity. At low concentrations of both fragments (0.2 and 1.0 μM), the bioluminescent signal in artificial cells without DOGS–NTA–Ni-mediated spatio-structural organization was negligible, whereas organization of the complex at the membrane induced an increase of the signal of up to one order of magnitude. This indicated that membrane recruitment did not only spatially redistribute the enzymatic activity, but also resulted in enhanced activation of signaling. Therefore, spatial organization led to structural organization and enzymatic activation; processes highly resembling protein signaling in biology.
To study if spatial organization of both fragments was required for NanoBiT reconstitution, a SmBiT fragment without His-tag (NoHis-SmBiT) was synthesized, and its ability to form the active complex was evaluated. In a first set of experiments, bioluminescence microscopy was used to assess the relative membrane-to-lumen signal ratio and therefore the spatial localization of the fragments (NanoBiT reconstitution) at the membrane. Micrographs showed that the active luciferase is preferentially formed at the membrane only if both LgBiT and SmBiT are His-tagged (Fig. 2c). The GUVs loaded with His-tagged LgBiT and NoHis-SmBiT showed some spontaneous reconstitution of the active luciferase in the lumen, yet spatial organization of the luciferase at the membrane was not observed; thus indicating that both SmBiT and LgBiT should be His-tagged to induce effective complex assembly at the membrane. This was supported by the global luminescence signal (quantified by bulk bioluminescence spectroscopy and normalized to the signal of encapsulated marker to account for the total number of GUVs in the sample), which was higher when both fragments were His-tagged (due to effective dimerization at the membrane) than for NoHis-SmBiT (ESI Fig. 8†). When the GUVs did not contain Ni–NTA on their membrane, His-SmBiT and NoHis-SmBiT samples showed no notable differences in global bioluminescence (originating from spontaneous complementation in the GUV lumen) (ESI Fig. 8†). This also suggested that the intrinsic LgBiT–SmBiT interactions were not significantly altered by the His-tag, as confirmed in control experiments (ESI Fig. 18†).
Altogether, these results demonstrated that spatial organization of both fragments at the GUV membrane and subsequent dimerization of the two fragments promotes subsequent reconstitution of enzymatic activity through formation of a higher-order signaling complex.
Reversible organization of protein complexes regulates enzymatic signaling
Having established Ni–NTA-mediated spatial organization of proteins in compartmentalized synthetic cells as a strategy to organize protein dimerization and induce signaling, we sought to control such processes reversibly. Such an approach aims to mimic the dynamic self-assembly in biology, where reversible organization of responsive elements produces the complex behavior apparent in living systems. To this end, we utilized the non-covalent nature of the His-tag/Ni–NTA interaction to enable reversible formation and dissociation of the luciferase fragments, which, in turn, provides control over the activation of enzymatic signaling. The strength of the His-tag/Ni–NTA interaction can be modulated by tuning the pH of the environment; thus, this offers a general framework to interface the internal luciferase signaling with programmable self-regulating or adaptive processes that modulate the internal pH. The pKa of the imidazole side chains in the His-tag is 6.0; consequently, acidification of a neutral solution decidedly weakens the His/Ni–NTA interactions. Therefore, by alternatingly increasing and decreasing the pH the reversibility of the system can be demonstrated.To test the responsiveness of the system to pH, GUVs were treated with small doses of acid or base, followed by 20 min of incubation and subsequent recording of the resulting enzymatic activity. Complexation between His-tagged proteins and Ni–NTA moieties has been reported to occur in a timescale of seconds.32 In addition, the relatively high diffusion coefficient of small proteins (>200 μm2 s−1) should allow their rapid assembly/disassembly from the membrane.33 To ensure rapid equilibration of the internal and external solutions, the vesicles were incubated with alpha-hemolysin, a self-inserting membrane pore.31
At pH 7.4 LgBiT and SmBiT formed the active luciferase at the membrane, as expected (Fig. 3). When the pH was decreased to 5.5 by addition of dilute HCl, the membrane-associated bioluminescence disappeared and a somewhat weaker signal originating from the lumen was observed. Not all GUVs emitted a detectable luminescence; this was attributed to the lower activity of the luciferase at pH 5.5 (a previous report found a reduction of ca. 50% at pH 5.5 as compared to pH 7.5 and we found a similar decrease in bulk LgBiT–SmBit solutions as shown in ESI Fig. 18†),28 and possibly partial loss of SmBiT from some vesicles with high αHL content. Gratifyingly, when the pH was reversed to 7.5 the bioluminescent ring reappeared, which indicated that both protein fragments had reassociated with the membrane and reconstituted the active luciferase anew. In control experiments, the pH-responsive binding of His-tagged proteins to the Ni–NTA-containing liposomal membrane was confirmed by confocal microscopy using fluorescent tdTomato (ESI Fig. 19†) and by quantifying the global luciferase activity at different pH values (ESI Fig. 20†). Accordingly, pH-controlled reversible switching of the enzyme activity through reversible organization of NanoBiT at the membrane was demonstrated.
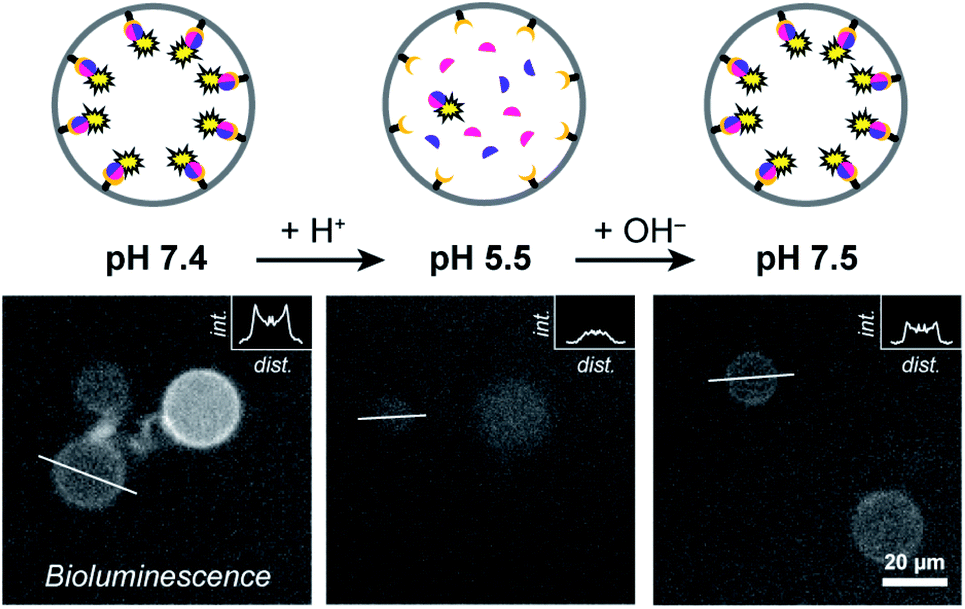 | ||
| Fig. 3 pH-dependent spatial organization of NanoBiT at the membrane. The bioluminescence of different GUVs was recorded following acidification and subsequent basification of the external solution. GUVs contained DOGS–NTA–Ni (1%), LgBiT and SmBiT (5 μM each). α-Hemolysin (20 μg ml−1) facilitated equilibration of the pH over the membrane. The insets display the luminescence intensities along a cross section of the GUVs (indicated by white bars). Note the absence of membrane-associated peaks at acidic pH. | ||
Enzymatic signaling in artificial cells in response to environmental cues
In a final set of experiments, we aimed at integrated pre-programmed autonomous control over such spatio-structural organization processes by incorporating regulatory enzymatic machinery. This, when viewed in the context of dynamic self-assembly, is a step toward emulating organizational processes in a more biomimetic fashion. To this end, urease – an enzyme that converts urea into ammonia, thereby increasing the pH – was integrated into the system. Urea functioned as environmental cue to steer the controlled assembly of the system. A ratiometric pH probe was also incorporated in the GUV lumen, and time-series data showed the possibility to upregulate the pH according to urease and urea concentration (ESI Fig. 21†).Spatial organization of SmBiT and LgBiT at the liposome membrane was initiated at pH 7.5 (Fig. 4a). Decreasing the pH to 5.5 by addition of HCl led to spontaneous disassembly of the scaffolded luciferase and consequent disappearance of bioluminescence. Upon addition of urea the pH increased and bioluminescence was restored, confirming the successful association of SmBiT and LgBit at the membrane (Fig. 4a–c). A fraction of vesicles, however, did not recover their enzymatic signaling; this is attributed to content release of some vesicles during the pH switch. In control experiments where urea was not added to the liposomes after they had been treated with HCl, the pH remained low and bioluminescence was not observed. These results confirm that the spatio-structural organization process was controlled by urease, and not an inherent property of the system.
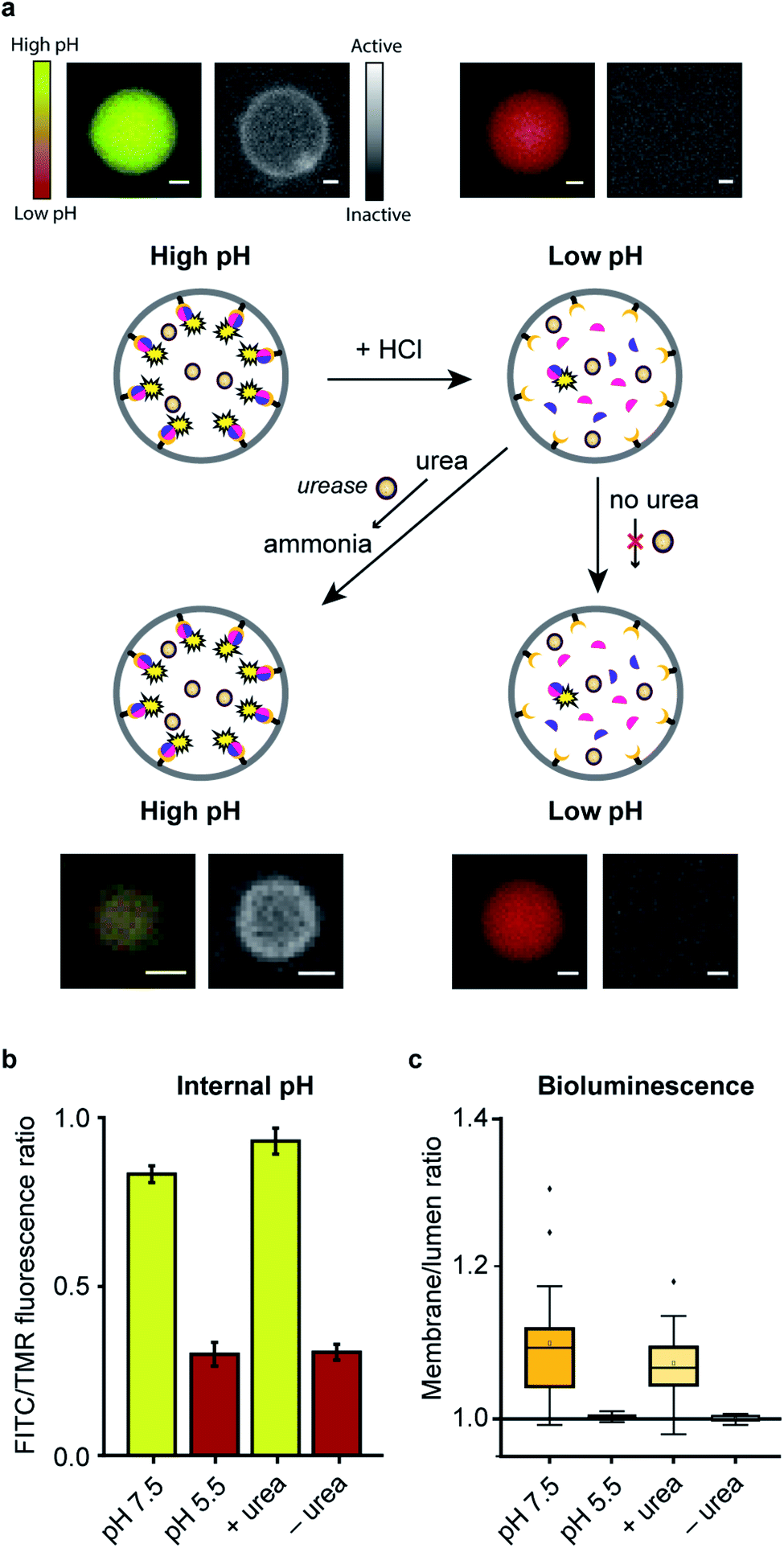 | ||
| Fig. 4 Integrated pre-programmed control over spatio-structural organization processes in GUVs. (a) Spatial-structural organization and enzymatic signaling of LgBiT and SmBiT (5 μM each) is promoted at pH 7.5. Addition of HCl dissociated the complex. Next, urea served as an environmental cue to restore enzymatic signaling at the membrane. Fluorescence micrographs of representative GUVs display internal pH (left panels) and bioluminescence micrographs indicate enzymatic signaling (right panels). Scale bars represent 5 μm. (b) The internal pH of >40 GUVs was tracked using a fluorescent ratiometric pH probe (dextran-FITC-TMR). (c) The bioluminescence localization was quantified by comparing the integrated signal at the membrane and inside the lumen (nGUVs = 18). | ||
From a broader perspective, although this particular system (reconstitution of luciferase in response to urea) has no apparent utility in biological terms, artificial cells that produce an output signal in response to molecular cues could potentially be used for diverse applications such as sensing and information processing.
Conclusions
In summary, we have engineered an artificial cell equipped with dynamic spatial and structural organization processes. Spatial organization was achieved by the localization of two fragments of a split luciferase through specific interactions between the hexahistidine domains on the protein fragments and a Ni–NTA moiety in the lipid membrane. This resulted in the fragments' structural dimerization, and subsequent functional signaling response. Due to the non-covalent, dynamic nature of these interactions, the supramolecular complex could be reversibly assembled and disassembled. Importantly, the signaling process was controlled by an enzymatic program in response to environmental cues. Our work thus constitutes a significant advancement in the creation of synthetic cell models that approach natural systems in terms of functional complexity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Anniek den Hamer and Lenne Lemmens are thanked for providing the LgBiT fragment. Ardjan van der Linden and Pascal Pieters are acknowledged for microscopy support. The Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035) and the ERC Advanced grant Artisym 694120 are acknowledged for funding.References
- J. C. Kagan, V. G. Magupalli and H. Wu, Nat. Rev. Immunol., 2014, 14, 821–826 CrossRef CAS.
- S. Loescher and A. Walther, Angew. Chem., Int. Ed., 2020, 59, 5515–5520 CrossRef CAS.
- C. Love, J. Steinkühler, D. T. Gonzales, N. Yandrapalli, T. Robinson, R. Dimova and T. Y. D. Tang, Angew. Chem., Int. Ed., 2020, 59, 5950–5957 CrossRef CAS.
- H. Zhao, V. Ibrahimova, E. Garanger and S. Lecommandoux, Angew. Chem., Int. Ed., 2020, 59, 11028–11036 CrossRef CAS.
- V. Mukwaya, P. Zhang, H. Guo, A. Y. Dang-i, Q. Hu, M. Li, S. Mann and H. Dou, ACS Nano, 2020, 14, 7899–7910 CrossRef CAS.
- E. Magdalena Estirado, A. F. Mason, M. Á. Alemán García, J. C. M. van Hest and L. Brunsveld, J. Am. Chem. Soc., 2020, 142, 9106–9111 CrossRef CAS.
- M. G. F. Last, S. Deshpande and C. Dekker, ACS Nano, 2020, 14, 4487–4498 CrossRef CAS.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2018, 3, 17068 CrossRef CAS.
- E. Winfree, F. Liu, L. A. Wenzler and N. C. Seeman, Nature, 1998, 394, 539–544 CrossRef CAS.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- B. J. H. M. Rosier, A. J. Markvoort, B. Gumí Audenis, J. A. L. Roodhuizen, A. den Hamer, L. Brunsveld and T. F. A. de Greef, Nat. Catal., 2020, 3, 295–306 CrossRef CAS.
- T. Pawson, Science, 2003, 300, 445–452 CrossRef CAS.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS.
- A. F. Mason, N. A. Yewdall, P. L. W. Welzen, J. Shao, M. van Stevendaal, J. C. M. van Hest, D. S. Williams and L. K. E. A. Abdelmohsen, ACS Cent. Sci., 2019, 5, 1360–1365 CrossRef CAS.
- S. Deshpande, F. Brandenburg, A. Lau, M. G. F. Last, W. K. Spoelstra, L. Reese, S. Wunnava, M. Dogterom and C. Dekker, Nat. Commun., 2019, 10, 1800 CrossRef.
- T. Trantidou, M. Friddin, Y. Elani, N. J. Brooks, R. V. Law, J. M. Seddon and O. Ces, ACS Nano, 2017, 11, 6549–6565 CrossRef CAS.
- R. J. Brea, A. K. Rudd and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8589–8594 CrossRef CAS.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669–17674 CrossRef CAS.
- A. M. Tayar, E. Karzbrun, V. Noireaux and R. H. Bar-Ziv, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11609–11614 CrossRef CAS.
- S. N. Semenov, A. S. Y. Wong, R. M. van der Made, S. G. J. Postma, J. Groen, H. W. H. van Roekel, T. F. A. de Greef and W. T. S. Huck, Nat. Chem., 2015, 7, 160–165 CrossRef CAS.
- J. W. Sadownik, E. Mattia, P. Nowak and S. Otto, Nat. Chem., 2016, 8, 1–6 CrossRef.
- R. J. R. W. Peters, M. Nijemeisland and J. C. M. van Hest, Angew. Chem., Int. Ed., 2015, 54, 9614–9617 CrossRef CAS.
- J. A. Bornhorst and J. J. Falke, Methods Enzymol., 2000, 326, 245–254 CAS.
- S. Knecht, D. Ricklin, A. N. Eberle and B. Ernst, J. Mol. Recognit., 2009, 22, 270–279 CrossRef CAS.
- M. P. Hall, J. Unch, B. F. Binkowski, M. P. Valley, B. L. Butler, M. G. Wood, P. Otto, K. Zimmerman, G. Vidugiris, T. Machleidt, M. B. Robers, H. A. Benink, C. T. Eggers, M. R. Slater, P. L. Meisenheimer, D. H. Klaubert, F. Fan, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2012, 7, 1848–1857 CrossRef CAS.
- A. S. Dixon, M. K. Schwinn, M. P. Hall, K. Zimmerman, P. Otto, T. H. Lubben, B. L. Butler, B. F. Binkowski, T. Machleidt, T. A. Kirkland, M. G. Wood, C. T. Eggers, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2016, 11, 400–408 CrossRef CAS.
- S. Pautot, B. J. Frisken and D. A. Weitz, Langmuir, 2003, 19, 2870–2879 CrossRef CAS.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859–1865 CrossRef CAS.
- G. Raghunath and R. B. Dyer, Langmuir, 2019, 35, 12550–12561 CrossRef CAS.
- J. F. Torres, A. Komiya, J. Okajima and S. Maruyama, Defect Diffus. Forum, 2012, 326, 452–458 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details, as well as additional experimental data. See DOI: 10.1039/d0sc03933k |
| This journal is © The Royal Society of Chemistry 2020 |