
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of crystallinity and dispersity on the self-assembly behavior of block co-oligomers in water†
Marle E. J.
Vleugels
a,
Marnie E.
de Zwart
a,
Jose Rodrigo
Magana
 b,
Brigitte A. G.
Lamers
a,
Ilja K.
Voets
b,
E. W.
Meijer
a,
Katja
Petkau-Milroy
*a and
Anja R. A.
Palmans
*a
b,
Brigitte A. G.
Lamers
a,
Ilja K.
Voets
b,
E. W.
Meijer
a,
Katja
Petkau-Milroy
*a and
Anja R. A.
Palmans
*a
aLaboratory of Macromolecular and Organic Chemistry and Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: k.milroy@gmx.net; a.palmans@tue.nl
bLaboratory of Self-Organizing Soft Matter and Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 23rd October 2020
Abstract
Self-assembly of block copolymers in solution is a topic of great interest in polymer science due to the potential for applications as a drug carrier system. In bulk, fully discrete polymers have been shown to self-assemble in extremely well-defined structures, but the effect of full discreteness on self-assembly in solution is less known. Furthermore, little is known about the effect of molar mass dispersity on crystallization driven self-assembly. Here, we investigate both the effects of dispersity and crystallinity on the self-assembly behavior of low molecular weight poly(lactic acid)-poly(ethylene glycol) block co-oligomers (BCOs) in solution. The results show that the introduction of dispersity and/or crystallinity does not significantly affect spherical and cylindrical morphologies, but vesicular structures are affected. The introduction of dispersity in amorphous vesicle forming BCOs lowers the reproducibility of preparations in solution. For crystalline BCOs, the introduction of dispersity leads to a clear decrease of ordering in bulk and it prevents crystallization of the LLA block in solution. This all arises already at a low dispersity (Đ ≤ 1.06), highlighting the effect of dispersity on assemblies of low MW BCOs. It also underlines the need to take dispersity into account when aiming for homogeneous well-defined structures in solution.
Introduction
Block copolymers (BCPs) are an exciting class of macromolecules that have seized the attention of many polymer chemists. Applications range from the development of drug delivery vehicles in aqueous media to nanolithography in bulk.1–5 Although controlled polymerizations have optimized BCPs, the presence of dispersity is an intrinsic property, both at the level of their molar mass (molar mass dispersity, Đ) and at the level of compositional dispersity. Natural polymers such as DNA and peptides are discrete with a Đ of 1.00 and are sequence-defined. However, achieving such controlled uniformity in synthetic polymers has been a great challenge in polymer science.6,7 Fully discrete low molecular weight oligomers with a Đ of 1.00 have been obtained via iterative synthesis methods,8 using solid-phase9–12 or solution based approaches.13–18 or by combining controlled polymerization techniques with flash chromatography.19–21 In addition, discrete block co-oligomers (BCOs) have been prepared by these methods, affording systems that at low molecular weights form highly-ordered, phase-segregated structures in bulk.22–27 The introduction of dispersity in these low molecular weight BCOs has a dramatic effect on the nanophase separation and long range ordering.28–32In solution the assembly of block copolymers is driven by the difference of lyophobicity between the blocks. Upon solvation in a selective solvent, the lyophobic blocks will rearrange to minimize their contact with the solvent, while the lyophilic blocks will remain solvated. This leads to a segregated structure, in which both a dense lyophobic core, consisting of collapsed chains, and a swollen lyophilic corona are present.33,34 The final morphology in thermodynamic equilibrium is determined by the volume ratio of the lyophobic and the lyophilic blocks according to the theory of Israelachvili.35 In case of disperse lyophobic and lyophilic blocks, the volume ratios throughout the whole population of polymer chains might vary, causing a mixture of morphologies in the final assembly.36 Especially on smaller aggregates of less than 1000 molecules this is a serious drawback, as all aggregates will have different compositions and thus different morphologies. This effect of dispersity and resulting mixture of morphologies has been shown for ABA and AB type block copolymers. Increasing dispersity in either the core or corona forming blocks shifted the morphology from vesicles towards a mixture of vesicles, spheres and worm-like assemblies, or from vesicles to spheres.37–39
In addition to dispersity, crystallinity has shown to be a driving force for morphology change. Crystallization-driven self-assembly of amphiphilic block copolymers has been used to obtain non-spherical morphologies in solution, by using a crystallizable core-forming block. The crystallinity of the core-forming block can be used to switch the morphology from the predicted spherical morphology into cylinders, as the crystalline core of these micelles can act as nuclei after which epitaxial growth can occur.40–44
In previous work of our group, the effect of dispersity on the crystallinity in self-assembled low molecular weight ABA type BCOs was studied in water.45 Similar to the observations in bulk, the introduction of dispersity showed a significant effect on the homogeneity of the obtained particle morphologies in water. To explore the effect of lack of dispersity in these low molecular BCOs, a library of AB type BCOs was synthesized with full control over sequence and molar mass dispersity (Đ = 1.00).46 A discrete L-lactic acid 16-mer was used as a hydrophobic block, and discrete oligo(ethylene glycol) 11-, 17- and 48-mers were used as hydrophilic blocks, to obtain a variety of morphologies upon self-assembly of the BCOs in water. For bilayer morphologies it is known that they can adopt flat, curved, or closed vesicular structures in solution, depending on the ability of the hydrophobic block to bend into the closed vesicular structure. An excellent agreement between theoretically predicted size and morphology was found for all these discrete crystalline BCOs, but it remained unclear to what extent crystallinity and dispersity played a role in the formation of stable and reproducible vesicular structures.
To continue our previous work, we here like to answer the following question: what has a greater influence on the morphology formed by a low MW BCO, crystallinity or dispersity? Therefore, we investigate AB type BCOs consisting out of the same blocks (oligo(lactic acid) and oligo(ethylene glycol)) we previously used,46 but compare discrete and disperse, as well as crystalline and amorphous lactic acid blocks. To this end, we have selected a ratio of the two blocks that ensures the formation of vesicles (LA16EG11). These vesicle forming BCOs were characterized in bulk and in aqueous solution using a combination of scattering, differential scanning calorimetry (DSC) and total internal reflection fluorescence microscopy (TIRF). To shed light on the effect of dispersity and crystallinity on other morphologies, sets of LA16EG17 (cylinders) and LA16EG48 (spherical micelles) were also studied. The packing of the LA16 block in these assemblies in water was analyzed using small angle neutron scattering (SANS) and by assessing the solubilization region of the hydrophobic dye Nile Red (NR).
Results and discussion
Design and synthesis of BCOs
We prepared oligo(lactic acid) of 16 repeat units from L-lactide (LLA16), and selected commercially available oligo(ethylene glycol) of 11 repeat units (EG11). Dispersity is introduced into the LA block (discrete LLA16versus disperse LLA∼16 where the dispersity is indicated by using the tilde symbol) to study the effect of dispersity on crystalline BCOs (LLA16EG11 and LLA∼16EG11). To understand the effect of dispersity without crystallinity present, we synthesized a set of amorphous BCOs starting from the racemic mixture of L-and L-lactide DLLA16 and introduced dispersity in the LA block (DLLA∼16).The synthesis of discrete TBDMS-LLA16-COOH and TBDMS-DLLA16-COOH is based on the previously reported synthetic strategy developed by Hawker's group,13 and later slightly modified by our group.22 Subsequent ligation with commercially available discrete MeO-EG11-OH resulted in discrete BCOs LLA16EG11 and DLLA16EG11 (Fig. 1a). For LLA∼16EG11 ring-opening polymerization (ROP) of L-lactide was performed using MeO-EG11-OH as initiator. For DLLA∼16EG11 first a TBDMS-DLLA∼16-COOH precursor was synthesized via ROP of DL-lactide using benzyl alcohol as initiator and subsequently coupled to MeO-EG11-OH. The TBDMS group was not removed to increase the stability of the BCOs.47,48
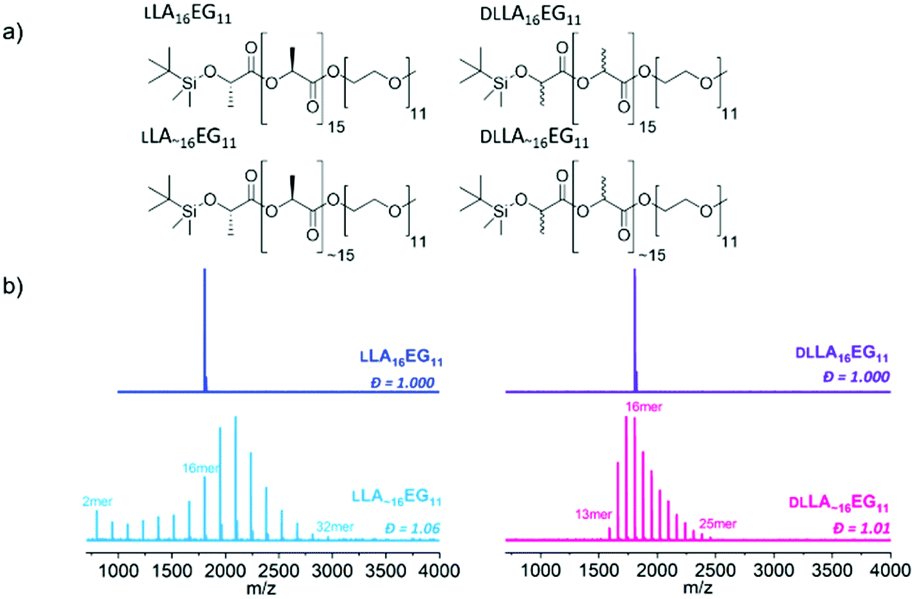 | ||
| Fig. 1 (a) Chemical structures of studied BCOs: crystalline discrete LLA16EG11, crystalline disperse LLA∼16EG11, amorphous discrete DLLA16EG11and amorphous disperse DLLA∼16EG11. (b) MALDI-ToF spectra of all BCOs. | ||
All compounds were purified by automated column chromatography and fully analyzed by 1H NMR, 13C NMR and matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (Fig. S1–S28†). Full synthetic details on the preparation of the BCOs can be found in the ESI.†
While 1H-NMR spectra of all four compounds look identical and give similar degrees of polymerization, MALDI-ToF spectra reveal large differences between the discrete and disperse compounds (Fig. 1b). DLLA∼16EG11 has a narrower distribution than LLA∼16EG11 due to a column purification in the synthetic procedure, but the range of Đ is in both cases low enough to not expect pronounced effects of the difference in dispersity. LLA16EG11 and DLLA16EG11 show only a single peak in MALDI-ToF-MS, underlining the discreteness of these BCOs, while a distribution of peaks is observed for disperse BCOs LLA∼16EG11 and DLLA∼16EG11. We note that the values for the molar mass dispersity, Đ, are narrow, and range between 1.01–1.06 for the disperse compounds. These disperse oligomers are thus a good reference to gain insight into what extent dispersity matters.
In addition to the series LA16EG11 anticipated to form lamellar structures in water, we also synthesized the series LA16EG17 (cylindrical morphologies) and LA16EG48 (micellar morphologies) with discrete/disperse and/or amorphous/crystalline lactic acid blocks. The synthetic details and characterization of these BCOs are given in Schemes S1–S3 and Fig. S1–28.†
Bulk properties of BCOs
Crystalline oligomers LLA16EG11 and LLA∼16EG11 were obtained as waxy solids at room temperature, while amorphous DLLA16EG11 and DLLA∼16EG11 were obtained as viscous oils. Their thermal behavior was investigated using DSC (Table 1, Fig. S29†). For the amorphous BCOs DLLA16EG11 and DLLA∼16EG11 the only visible transition is a glass transition temperature, Tg. For both crystalline BCOs LLA16EG11 and LLA∼16EG11 a clear crystallization transition was observed upon cooling. The enthalpy of crystallization of LLA∼16EG11 is lower than for LLA16EG11, suggesting that the crystalline packing is less defined for the disperse variant. However, disperse LLA∼16EG11 crystallizes at a higher temperature than its discrete counterpart LLA16EG11 (38 °C vs. 22 °C), indicating that the LA chains with DP > 16 in the disperse BCO nucleate crystallization at a higher temperature. Furthermore, by introducing dispersity into the LLA16 block, melting occurs over a broad temperature range. This is in contrast to the sharp melting transition that was observed for the discrete variant (Fig. S29†). These observations are in line with those previously observed in BCOs comprising oligodimethylsiloxanes and LLA.30| Oligomer | Đ | Appearancea | T g [°C] | T c [°C] | ΔH [kJ mol−1] | Phaseb |
d*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c [nm] c [nm] |
|---|---|---|---|---|---|---|---|
| a Physical appearance at room temperature, directly after drying in vacuo. b Bulk morphology determined with SAXS at room temperature. c Domain spacing, calculated as d* = 2π/q*. LAM = lamellar, DIS = disordered. | |||||||
| LLA16EG11 | 1.00 | Wax | — | 22 | 32 | LAM | 8.0 |
| LLA∼16EG11 | 1.06 | Wax | — | 38 | 22 | LAM | 10.1 |
| DLLA16EG11 | 1.00 | Viscous oil | −33 | — | — | DIS | — |
| DLLA∼16EG11 | 1.01 | Viscous oil | −19 | — | — | DIS | — |
The packing and long-range ordering of the oligomers in bulk at room temperature was further investigated with X-ray scattering experiments. The discrete crystalline oligomer LLA16EG11 shows a sharp principal scattering peak and higher order Bragg reflections, indicating a highly structured long-range packing of the chains (Table 1, Fig. 2). The ratios of the Bragg reflections (√4, √9, √16, √25) indicate a lamellar phase (LAM) with an interlayer distance of 8 nm. Furthermore, multiple scattering peaks can be observed in the WAXS region, corresponding to the inter-chain packing of the LLA block, typical for the crystalline packing of lactic acid chains.49 Introducing dispersity in the crystalline oligomer LLA∼16EG11 gives rise to broadening of the primary scattering peak indicating a less ordered structure and smaller crystalline domains. In addition, the maximum of the first-order reflection which arise from the lamellar packing, q*, shifts to smaller q-values, corresponding to longer interlayer distances on the order of 10 nm. This is likely caused by the increasing amount of LLA blocks with DP > 16. At higher q values we can still see the peaks typical for crystalline packing of the lactic acid chain, at the same values as for the discrete block variant. These crystalline features probably arise from the longer lactic acid chains with DP > 16, as the shorter chains are not able to crystallize on short timescales.30 For the amorphous BCOs, DLLA16EG11 and DLLA∼16EG11, the absence of scattering peaks corroborates the thermal data, as no crystallization for the DLLA16 chains is visible.
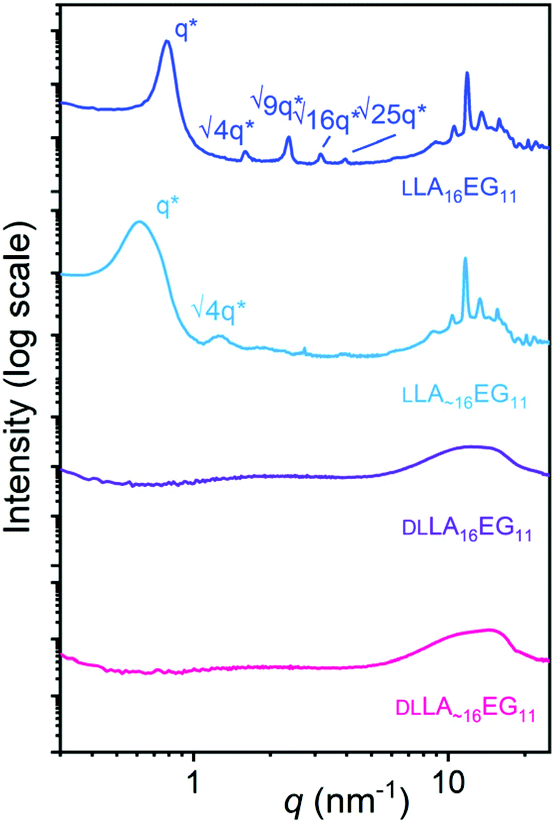 | ||
| Fig. 2 Wide-angle X-ray scattering (WAXS) data for LLA16EG11 (blue), LLA∼16EG11 (light blue), DLLA16EG11 (purple), DLLA∼16EG11 (pink). The data is shifted vertically for clarity. Higher order Bragg reflections are indicated if present. | ||
Self-assembly of BCOs in water
To study the effect of dispersity without any crystallinity present, we first self-assembled the amorphous DLLA16EG11 and DLLA∼16EG11 in water. A slow solvent switch was used, as our previous work showed that dropwise addition of water to a THF stock solution leads to the predicted thermodynamically stable vesicular structures.46 After self-assembly in water, multi-angle light scattering was used to measure the sizes of the morphologies formed and to obtain information on the morphology of the self-assembled structures. The results show that the hydrodynamic radius (RH) is comparable for both DLLA16EG11 and DLLA∼16EG11 (Fig. 3a), and a log–log representation of the corresponding static light scattering data indicate that for both BCOs a vesicular structure is adapted, as I ∝ q−2 (Fig. 3b). These results indicate that introducing dispersity does not lead to differences in the nature of the particles formed by the amorphous BCOs. To visualize the structures in solution formed by DLLA16EG11 and DLLA∼16EG11, the particles were analyzed with TIRF microscopy after addition of Nile Red (Fig. S30†). Nile Red (NR) is a hydrophobic solvatochromic dye, which accumulates into hydrophobic domains where it fluoresces.50 Spherical, non-interacting particles were observed for both discrete and disperse BCOs, corroborating the results obtained from scattering techniques. Overall, the introduction of dispersity into amorphous BCOs does not lead to morphology differences upon particle formation in water. However, the reproducibility of sample preparation is affected by the presence of dispersity (Fig. 3c). After 1 week, the sizes obtained from discrete DLLA16EG11 are comparable, while for several preparations of DLLA∼16EG11 the sizes vary. Even though the difference in dispersity (1.00 vs. 1.01) is small, the consequences for a reproducible sample preparation are significant.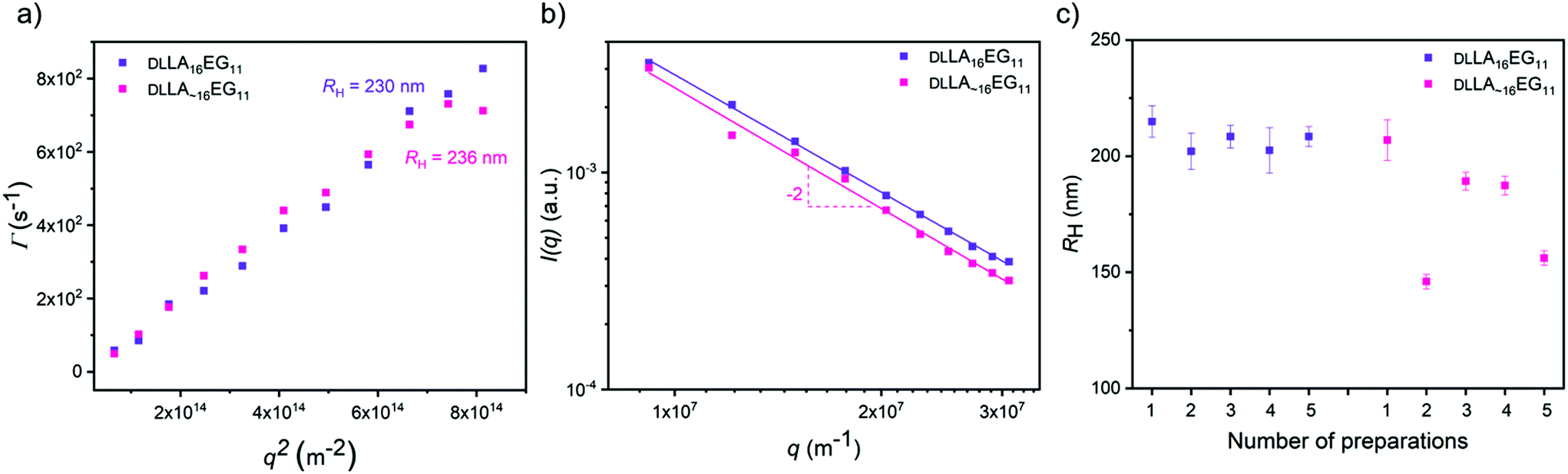 | ||
| Fig. 3 Light scattering results of the amorphous BCOs upon self-assembly in water. (a) Γ vs. q2 plot to determine the RH of the spherical bilayer structures. (b) Scattering intensity I vs. q to probe the shape of the self-assembled structures. (c) Obtained RH of multiple sample preparations. | ||
Our previous work showed that it was possible to obtain spherical vesicular structures from crystalline LLA16EG11 by using a slow solvent switch.46 Important to note here, is that sample preparation and history play an important role in obtaining these vesicular structures. Over time, the BCO crystallizes in bulk, which reduces the solubility of this compound and leads to irreproducible sample preparations in water. Rigorous dissolution in the appropriate solvent and carefully following the dissolution overtime prior to sample preparation in water are required for the sample preparation procedure to be reproducible.
Upon self-assembly of disperse LLA∼16EG11 in water using the slow solvent switch, particles with a spherical shape were obtained (Fig. 4a). To assess the capability of disperse LLA chains to crystallize in solution, LLA∼16EG11 was assembled at higher concentrations and subjected to micro-DSC. In contrast to LLA16EG11, no transitions were visible during the heating run (Fig. 4b). Thus, in solution, the disperse nature of the LLA chains prevents crystallization.
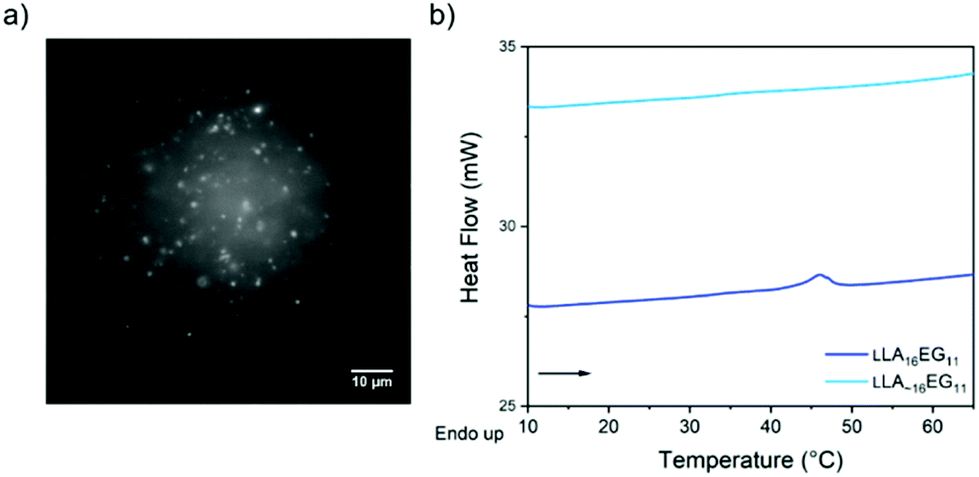 | ||
| Fig. 4 (a) TIRF image of disperse LLA∼16EG11 upon self-assembly in water. (b) Micro-DSC traces of LLA16EG11 and LLA∼16EG11 at 5 mg mL−1 after self-assembly in water. | ||
To further investigate the packing of the LA chains in the assembled structures, SANS experiments were performed for the four BCOs in water (Fig. 5a, Table 2). For LLA16EG11, we previously showed that the small angle X-ray scattering data could be fitted as a flat homogenous lamellar structure with a layer thickness of 5.9 nm, and thus 11.8 nm for the total bilayer. This indicates that the LLA16 chains in the lamellar structures may be packed in a sort of intercalating or collapsed fashion (Fig. 5b), as the lamellar domain spacing of one fully extended LLA16 chain is 6.0 nm.51
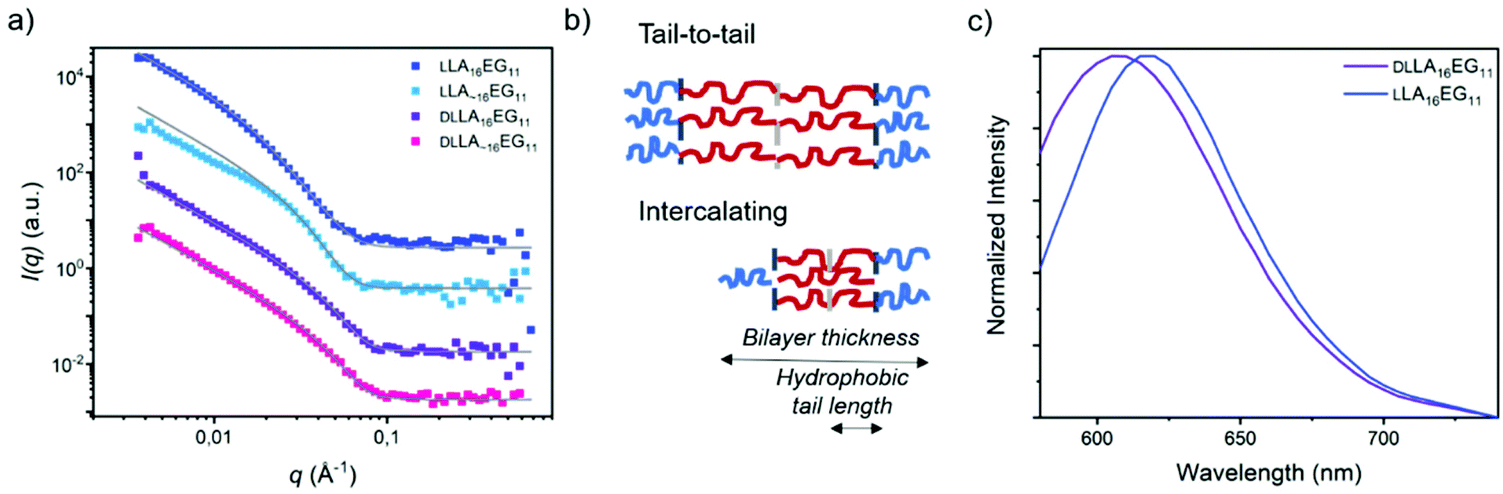 | ||
| Fig. 5 (a) SANS scattering profiles and corresponding fits in D2O, 1 mg mL−1. Data is shifted vertically for clarity. (b) Schematic representation of possible bilayer structures in solution, LA16 in red, EG11 in blue. (c) Normalized emission spectra of the dye NR in self-assembled DLLA16EG11 and DLLA16EG11 in water. | ||
In contrast, the patterns obtained with SANS here were shifted to longer distances. The scattering data could only be described as lamellae if polydispersity in the distribution of the hydrophobic bilayer was considered. Fitting the neutron scattering data with a head/tail lamellar structure (see ESI for details†), gives a bilayer thickness of 14.8 nm for particles formed by LLA16EG11, corresponding with a hydrophobic tail length of the LLA block of 4.3 nm. This larger tail length compared to previously published results likely arises from a different extent of crystallinity present in these structures. Crystalline regions are likely to pack tail-to-tail (Fig. 5b) rather than the intercalating arrangement of more amorphous samples. This would lead to an overall larger hydrophobic tail length (Fig. 5b). As mentioned previously, over time part of the bulk material crystallizes, and it is possible that sample history of the bulk BCOs was not fully removed before making the samples in water. A competing process between self-assembly and crystallization of the aged BCOs upon self-assembly in water can occur, resulting in a mixture of aggregates with a different extent of crystallinity and thus different sizes than observed before. For the scattering profiles in solution, the effect of chain length dispersity was not so pronounced: the scattering pattern and fit results were similar for LLA 16EG11 and LLA∼16EG11, with a smaller bilayer thickness of 12.2 nm for disperse LLA∼16EG11.
The effect of (lack of) crystallinity, however, was clearly visible. For both amorphous DLLA16EG11 and DLLA∼16EG11 the fit results gave a smaller bilayer thickness of 10.6 nm. The hydrophobic tail length of the LA block was 2.3 nm, roughly half the value of the hydrophobic tail length of the crystalline bilayer in LLA 16EG11, indicating a more intercalating type of packing of the DLLA tails. This, together with the crystallization of LLA16EG11 in solution, implies that the ability of the hydrophobic block in LLA16EG11 to crystallize gives rise to a different type of packing in the obtained vesicular structures.
To further assess the nature of the packing of the LA block, we applied the dye NR as a probe. It was recently reported that the emission wavelength, λmax,em, of this dye can be used to assess whether the dye is located in the hydrophobic part of the bilayer or in the corona region after solubilization in self-assembled structures.52 When comparing the λmax,em of NR mixed into DLLA16EG11 or LLA16EG11 (Fig. 5c), a clear difference is observed. The λmax,em is at higher wavelengths for the particles prepared from crystalline LLA16EG11 than for the particles prepared from amorphous DLLA16EG11 (∼620 nm versus ∼605 nm). This indicates that due to the crystallinity of the LLA block, the dye is not able to enter the hydrophobic bilayer and is more located towards the bilayer-corona interface.
Influence of dispersity and crystallinity on other morphologies
The combined effects of dispersity and crystallinity in the BCO series LA16EG11 do not have a significant effect on the type of morphologies formed, as in all cases vesicular structures are formed in water. However, we found pronounced differences in the reliability of sample preparation and on the bilayer thickness of the vesicular structures in water when dispersity and crystallinity were introduced. The question then is, in how far is this effect dependent on morphology? Therefore, we investigated BCOs based on LA16EG17 and LA16EG48, which previously were shown to form cylindrical and spherical micelles, respectively.46 The dissolution process was carefully followed over time to obtain a reproducible sample preparation procedure as these samples are also sensitive to crystallinity and sample history.For the LA16EG17 series, a slow solvent switch by addition of water to the organic stock solution using a syringe pump was used to obtain the particles in solution. SAXS measurements and theoretical calculations have predicted that this BCO composition has the tendency to aggregate into cylindrical micelles.46 The theoretical models, however, do not account for sample crystallinity,33 which may be detrimental to self-assembly.53 SANS profiles for the BCOs of the LA16EG17 series (crystalline, non-crystalline, discrete and disperse) were all characterized by a decay at low q with a slope value of −1, suggesting the presence of rod-like aggregates. Therefore, we speculate that the crystallinity does not significantly influence the morphology of the cylindrical micelles (Fig. 6a). Following this assumption, the data was fit using a core–shell cylinder model. Similar to the LLA16EG11 system, the crystalline variants of LLA16EG17 have a slightly larger cross-sectional radius compared to the non-crystalline ones (Table 3). The packing of the molecules in the cylindrical aggregate may restrict the crystallization of the LLA blocks. It is noteworthy that the maximum concentration that could be achieved with this sample preparation was too low to perform reliable micro-DSC experiments. Therefore, it is unclear whether the LLA block can, in fact, crystallize in the core of the cylindrical micelles.
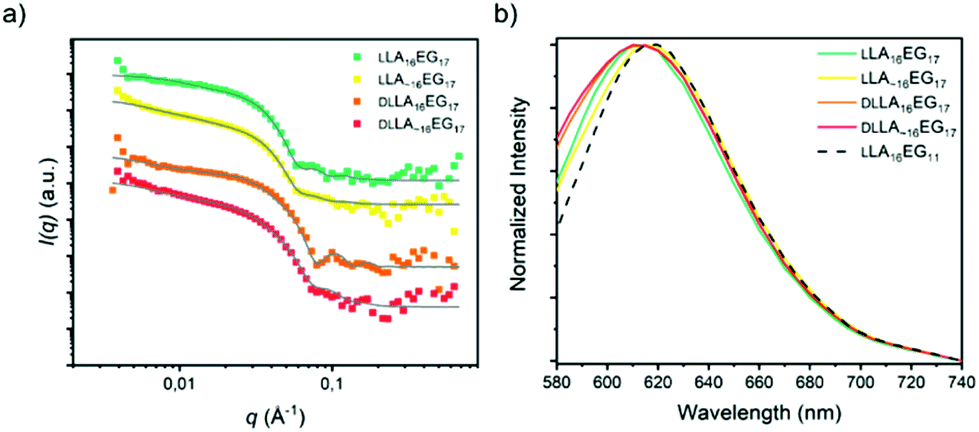 | ||
| Fig. 6 (a) SANS scattering profiles and corresponding fits in D2O, 1 mg mL−1. Data is shifted vertically for clarity. (b) Normalized emission spectra of the dye NR in self-assembled LA16EG17 BCOs in water. | ||
| Oligomer | Đ | Cross-sectional radius [nm] | Core radius [nm] |
|---|---|---|---|
| LLA16EG17 | 1.00 | 6.5 | 2.5 |
| LLA∼16EG17 | 1.04 | 6.5 | 2.5 |
| DLLA16EG17 | 1.00 | 5.0 | 1.0 |
| DLLA∼16EG17 | 1.01 | 5.0 | 1.0 |
Finally, we measured the emission spectra of dye NR in the presence of all four BCOs (Fig. 6b), which are all very similar. Interestingly, the λmax,em is close to that observed for the lamellar morphologies formed by LLA16EG11. This indicates that the dye is not able to enter the core and is more located towards the core–corona interface. The fact that all emission spectra overlay, suggests that the LA block forms a densely packed core, regardless of dispersity and/or crystallinity.
In the case of the series LA16EG48, a fast solvent switch by quick injection of organic stock solution into water was used to obtain self-assembled structures, similar to previously published procedures. The SANS profile overlay (Fig. 7a), which indicates that neither the presence/absence of crystallinity, nor the presence/absence of dispersity significantly affects the size of the structures formed. In addition, the emission spectra of the dye NR are near identical for all BCOs, pointing to a highly similar solubilization region of dye NR (Fig. 7b). The λmax,em of approximately 620 nm indicates a tightly packed core, as this lies close to the value obtained for assemblies of LLA16EG11. Taken together, these results reveal that for BCOs predicted to form spherical micelles, introduction of dispersity or crystallinity does not lead to noteworthy differences in terms of packing of the core LA block.
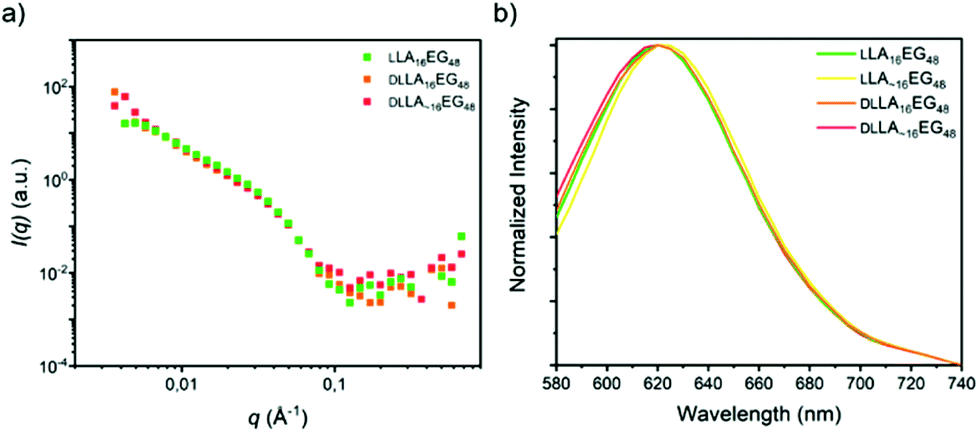 | ||
| Fig. 7 (a) SANS scattering profiles. (b) Normalized emission spectra of the dye NR in self-assembled LA16EG48 BCOs in water. | ||
Conclusion
We successfully synthesized a set of amphiphilic low molecular weight block co-oligomers. By preparing L-lactic acid and DL-lactic acid oligomers in an iterative manner and subsequently ligating to discrete ethylene glycol blocks, fully discrete diblock co-oligomers were obtained. Additionally, by introducing dispersity in crystalline and amorphous lactic acid oligomers, the effect of dispersity with and without crystallinity present was studied. By ligating a hydrophobic LA16 block with EG11, the formation of vesicular structures in solution was ensured. In bulk, discrete crystalline LLA16EG11 formed highly ordered phase separated structures, but upon introduction of dispersity in the LLA block, this ordering was clearly diminished even though the disperse counterpart has a Đ value of only 1.06. In solution, both amorphous BCOs, DLLA16EG11 and DLLA∼16EG11, formed the expected spherical vesicular structure. Particles made from disperse DLLA∼16EG11 showed a lower reproducibility in terms of size, which is quite remarkable as the Đ value is only 1.01, highlighting the effect that dispersity can have on these low molecular weight oligomers in solution. Introducing crystallinity in the BCOs gave rise to a different type of core packing in the obtained lamellar structures. For crystalline BCOs the introduction of dispersity affected the packing of the LLA block, as the LLA chains in LLA∼16EG11 do not show a melting transition upon self-assembly in water and SANS results indicated a smaller bilayer thickness for LLA∼16EG11.Other morphologies (cylindrical and spherical micelles) were also briefly investigated by ligating the LA16 block to different lengths of ethylene glycol. In these morphologies, the combined effects of dispersity and crystallinity were not evident. Only for vesicle forming low MW BCOs, introducing dispersity in crystalline compounds has a noticeable effect on packing of the hydrophobic block, both in bulk and in solution. As the difference in dispersity for this set of BCOs is so small, this highlights the importance of taking dispersity into account in low molecular weight systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035) and the European Research Council (SYNMAT project, ID 788618). I.K.V. acknowledges the Netherlands Organization for Scientific Research (NWO TA Grant No. (31.015.205) for financial support. The SANS experiments were performed at the LARMOR beamline of ISIS, situated at the Rutherford Appleton Laboratory of the Science and Technology Facilities Council, on the Harwell Science and Innovation Campus in Oxfordshire, United Kingdom. We thank Dr Robert Dalgliesh for his assistance acquiring the data and Teun Kleuskens for his work on the synthesis of the disperse building blocks.Notes and references
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- I. W. Hamley, Block Copolymers in Solution: Fundamentals and Applications, Wiley, 2005 Search PubMed.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 628–636 CrossRef CAS.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed, 2013, 52, 13261–13264 Search PubMed.
- S. Martens, J. Van Den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS.
- A. Al Ouahabi, M. Kotera, L. Charles and J. F. Lutz, ACS Macro Lett., 2015, 4, 1077–1080 CrossRef CAS.
- R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237 CrossRef CAS.
- K. Takizawa, H. Nulwala, J. Hu, K. Yoshinaga and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5977–5990 CrossRef CAS.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718–1726 CrossRef CAS.
- S. C. Solleder, K. S. Wetzel and M. A. R. Meier, Polym. Chem., 2015, 6, 3201–3204 RSC.
- S. C. Solleder, D. Zengel, K. S. Wetzel and M. A. R. Meier, Angew. Chem., Int. Ed., 2016, 55, 1204–1207 CrossRef CAS.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS.
- Z. Huang, J. Zhao, Z. Wang, F. Meng, K. Ding, X. Pan, N. Zhou, X. Li, Z. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 13612–13617 CrossRef CAS.
- J. Lawrence, S. H. Lee, A. Abdilla, M. D. Nothling, J. M. Ren, A. S. Knight, C. Fleischmann, Y. Li, A. S. Abrams, B. V. K. J. Schmidt, M. C. Hawker, L. A. Connal, A. J. McGrath, P. G. Clark, W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2016, 138, 6306–6310 CrossRef CAS.
- J. Lawrence, E. Goto, J. M. Ren, B. McDearmon, D. S. Kim, Y. Ochiai, P. G. Clark, D. Laitar, T. Higashihara and C. J. Hawker, J. Am. Chem. Soc., 2017, 139, 13735–13739 CrossRef CAS.
- J. De Neve, J. J. Haven, S. Harrisson and T. Junkers, Angew. Chem., Int. Ed, 2019, 58, 13869–13873 Search PubMed.
- B. Van Genabeek, B. F. M. De Waal, M. M. J. Gosens, L. M. Pitet, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 4210–4218 CrossRef CAS.
- B. Van Genabeek, B. F. M. De Waal, A. R. A. Palmans and E. W. Meijer, Polym. Chem., 2018, 9, 2746–2758 RSC.
- Y. Jiang, M. R. Golder, H. V. T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369–9372 CrossRef CAS.
- J. Sun, A. A. Teran, X. Liao, N. P. Balsara and R. N. Zuckermann, J. Am. Chem. Soc., 2013, 135, 14119–14124 CrossRef CAS.
- J. Sun, A. A. Teran, X. Liao, N. P. Balsara and R. N. Zuckermann, J. Am. Chem. Soc., 2014, 136, 2070–2077 CrossRef CAS.
- Y. Sun, R. Tan, Z. Ma, Z. Gan, G. Li, D. Zhou, Y. Shao, W.-B. Zhang, R. Zhang and X.-H. Dong, ACS Cent. Sci., 2020, 6, 1386–1393 CrossRef CAS.
- B. Oschmann, J. Lawrence, M. W. Schulze, J. M. Ren, A. Anastasaki, Y. Luo, M. D. Nothling, C. W. Pester, K. T. Delaney, L. A. Connal, A. J. McGrath, P. G. Clark, C. M. Bates and C. J. Hawker, ACS Macro Lett., 2017, 6, 668–673 CrossRef CAS.
- B. Van Genabeek, B. F. M. De Waal, B. Ligt, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2017, 6, 674–678 CrossRef CAS.
- B. Van Genabeek, B. A. G. Lamers, B. F. M. De Waal, M. H. C. Van Son, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2017, 139, 14869–14872 CrossRef CAS.
- N. Gangloff, M. Höferth, V. Stepanenko, B. Sochor, B. Schummer, J. Nickel, H. Walles, R. Hanke, F. Würthner, R. N. Zuckermann and R. Luxenhofer, Biopolymers, 2019, 110, e23259 CrossRef.
- R. Tan, D. Zhou, B. Liu, Y. Sun, X. Liu, Z. Ma, D. Kong, J. He, Z. Zhang and X. H. Dong, Chem. Sci., 2019, 10, 10698–10705 RSC.
- G. J. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjes, T. Cosgrove and B. Vincent, Polymers at Interfaces, Springer, Netherlands, 1993 Search PubMed.
- E. B. Zhulina and O. V. Borisov, Macromolecules, 2012, 45, 4429–4440 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, Biochim. Biophys. Acta, 1977, 470, 185–201 CrossRef CAS.
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O. Reilly, Chem. Soc. Rev., 2017, 46, 4119–4134 RSC.
- A. L. Schmitt, M. H. Repollet-Pedrosa and M. K. Mahanthappa, ACS Macro Lett., 2012, 1, 300–304 CrossRef CAS.
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601–5607 CrossRef CAS.
- O. Terreau, C. Bartels and A. Eisenberg, Langmuir, 2004, 20, 637–645 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 644, 644–648 CrossRef.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- N. Petzetakis, D. Walker, P. Dove and R. K. O. Reilly, Soft Matter, 2012, 8, 7408–7414 RSC.
- L. Sun, N. Petzetakis, A. Pitto-Barry, T. L. Schiller, N. Kirby, D. J. Keddie, B. J. Boyd, R. K. O'Reilly and A. P. Dove, Macromolecules, 2013, 46, 9074–9082 CrossRef CAS.
- A. Das, K. Petkau-Milroy, G. Klerks, B. Van Genabeek, R. M. Lafleur, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2018, 7, 546–550 CrossRef CAS.
- K. Petkau-Milroy, A. Ianiro, M. M. L. Ahn, J. R. Magana, M. E. J. Vleugels, B. A. G. Lamers, R. Tuinier, I. K. Voets, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2019, 9, 38–42 CrossRef.
- C. F. Van Nostrum, T. F. J. Veldhuis, G. W. Bos and W. E. Hennink, Polymer, 2004, 45, 6779–6787 CrossRef CAS.
- Y. T. Tam, J. Gao and G. S. Kwon, J. Am. Chem. Soc., 2016, 138, 8674–8677 CrossRef CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS.
- M. C. A. Stuart, J. C. Van De Pas and J. B. F. N. Engberts, J. Phys. Org. Chem., 2005, 18, 929–934 CrossRef CAS.
- B. A. G. Lamers, B. Van Genabeek, J. Hennissen, B. F. M. De Waal, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2019, 52, 1200–1209 CrossRef.
- A. Ianiro, Á. González García, S. Wijker, J. P. Patterson, A. C. C. Esteves and R. Tuinier, Langmuir, 2019, 35, 4776–4786 CrossRef CAS.
- A. Ianiro, J. Patterson, Á. G. García, M. M. J. Van Rijt, M. M. Hendrix, N. A. Sommerdijk, I. K. Voets, A. C. C. Esteves and R. Tuinier, J. Polym. Sci., Part B: Polym. Phys., 2017, 56, 330–339 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) availableExperimental procedures, Schemes S1–3 and Fig. S1–32. See DOI: 10.1039/d0py01161d |
| This journal is © The Royal Society of Chemistry 2020 |