
Semifluorinated, kinked polyarylenes via direct arylation polycondensation†
Fabian
Kempe
 a,
Felix
Riehle
b,
Hartmut
Komber
c,
Rukiya
Matsidik
a,
Michael
Walter
d and
Michael
Sommer
*a
a,
Felix
Riehle
b,
Hartmut
Komber
c,
Rukiya
Matsidik
a,
Michael
Walter
d and
Michael
Sommer
*a
aChemnitz University of Technology, Straße der Nationen 62, 09111 Chemnitz, Germany. E-mail: michael.sommer@chemie.tu-chemnitz.de
bInstitute for Macromolecular Chemistry, University of Freiburg, Stefan-Meier-Str. 31, 79104 Freiburg, Germany
cLeibniz Institute of Polymer Research, Hohe Str. 6, 01069 Dresden, Germany
dFreiburg Center for Interactive Materials and Bioinspired Technologies (FIT), University of Freiburg, Georges-Koehler-Allee 105, 79110 Freiburg, Germany
First published on 7th October 2020
Abstract
Semifluorinated, amorphous polyarylenes PmmpF4 with kinked backbone structure were prepared from a meta-substituted, biphenol-based monomer with varying alkoxy substituents R and 1,2,4,5-tetrafluorobenzene (pF4) via direct arylation polymerization (DAP). The chemistry employed is simple, scalable and does not rely on tedious purification techniques. Polycondensation occurs cleanly without major side reactions. Despite the clean polycondensation reaction, very high molar mass materials are difficult to obtain, which is ascribed to an unusual solubility behavior compared to non-fluorinated analogs, and similar, yet more linear tetrafluorobenzene copolymers based on fluorene or carbazole. In order to investigate this phenomenon further, the side chain-dependent properties PmmpF4 are investigated using linear, branched and cyclic side-chains. While the glass transition temperature of PmmpF4 is a strong function of R and can be varied between 35 °C and 197 °C for constant backbone structure and molecular weight, solubility cannot be improved by using longer linear or branched side chains. Density functional theory calculations suggest significant polarization-type non-covalent interactions between tetrafluorobenzene and the biphenol-based monomer as origin for the observed limited solubility, which guide the design of both kinked and straight conjugated polymers with high molar mass and solubility.
Introduction
Polyarylenes offer unique thermal and chemical stability compared to most aliphatic polymers.1,2 Other aromatic polymers like aromatic polyesters, polyaramides or aromatic polycarbonates contain heteroatom-bonding in their backbone and therefore lack the inherent stability of aryl–aryl bonds. Aromatic carbon bonds are stable against common acids, bases and redox agents that other polymers may suffer from due to their backbones containing functional groups such as esters, carbonates, amides and ethers.3,4 The presence of aromatic rings in the repeat unit also causes high glass transition temperatures in polyarylenes, as generally observed when cyclic units are incorporated into polymer backbones.5 High glass transition temperatures are caused by the lower degree of freedom of planar (aromatic) rings in which individual atoms can only move simultaneously by rotation as a single unit.6 This low degree of freedom is often accompanied by liquid crystallinity or even fully crystalline properties.2,5 However, high Tgs and high crystallinity may cause brittleness of materials. This may be mitigated by addition of aliphatic side chains to an aromatic polymer backbone. Thereby solubility improves due to addition of degrees of freedom in the flexible side chain while maintaining a durable polymer backbone. Nonetheless, para-polyarylenes are brittle materials due to their low entanglement density caused by their rigidity and rod-like shape.7,8 However, pioneering work by Schlüter et al. on polyarylenes with meta-comonomers proved that sufficient entanglement could be generated at very high molecular weights.7,9,10 These poly(meta-/para-polyarylenes) (PmpP) are tough, amorphous materials on par with aromatic polycarbonates. Yet improved mechanical properties can be achieved by incorporating a “double meta-” motif developed by our group.11 Here the resulting poly(meta-/meta-/para-polyarylenes) (PmmpP) are tough materials without prior purification starting from affordable and scalable building blocks.The typical cross-coupling variant for the transition metal-catalyzed polycondensation of phenylene-based monomers is the Suzuki–Miyaura reaction.12 Monomers are typically aryl halides coupled with a main group metal coupling partner. Polyarylenes prepared by direct arylation polymerization (DAP) are especially efficient with respect to atom economy compared to other cross coupling reactions.13–16 Aryl halides are coupled directly with C–H activated arylenes without the need for additional functional groups. 1,2,4,5-Tetrafluorobenzene (pF4) has been successfully employed in this regard in order to synthesize copolymers of fluorene or carbazole.17,18 We were interested in kinked, semifluorinated polyarylenes as the envisaged chemistry involving DAP appears highly attractive in terms of improved atom economy, simple monomers and selective regiochemistry. Usage of F4 as the nucleophile in DAP is also expected to lead to less degradation of functional groups during polycondensation such as protio- or oxidative deborylation.19–23 Regarding properties, backbone fluorination is generally expected to significantly change material properties in terms of chain rigidity, solubility and order, and thus novel property profiles may become accesible.24–26 We therefore investigated the copolymerization of commercially available F4 with a series of 2,2′-biphenol bromide-based monomers having a double meta motif and different side chains developed by our group. The monomer synthesis employed here includes three simple steps only, does not rely on chromatography or distillation and is easily scalable. The molecular and thermal properties of the resulting polymers PmmpF4 are investigated. Density functional theory calculations are finally employed to understand and explain the unexpected, rather limited solubility of PmmpF4, and these result may guide the design of other polyarylenes or conjugated polymers in general as well.
Results and discussion
Synthesis
We have screened the copolymerization of F4 with differently substituted 2,2′-biphenol monomers with respect to the solvent, concentration and temperature. The catalytic system itself was Pd2dba3/P(o-anisyl)3/pivalic acid, a system that has emerged as a universal combination for the synthesis of many conjugated polymers via DAP.13 The side chains R used were isopropyl (3i), sec-butyl (4s), cyclopentyl (5c), n-hexyl (6n), n-octyl (8n), n-decyl (10n) and 2-ethylhexyl (EH) (Scheme 1). All entries showed gelation or precipitation at moderate molecular weight. This is in stark contrast to PmmpP, which is the non-fluorinated analog that shows excellent solubility for Mw up to ∼100 kg mol−1. We prefer to report Mw values here as Mn values are strongly influenced by oligomer content (see size exclusion chromatography (SEC) curves shown as Fig. S1 and S2†).11 Therefore, molecular weight is indicative of solubility of the copolymer at the temperature of the reaction. Interestingly, longer side chains did not generally improve solubility. The introduction of branched side chains was beneficial to improve solubility in some cases (4s vs. 5c and 3i). Interestingly, usage of EH instead of 6n or also 8n did not improve solubility significantly neither. These observations are in stark contrast to conjugated polymers where side chain branching is key to increase solubility significantly and for many examples to enable processing at all.27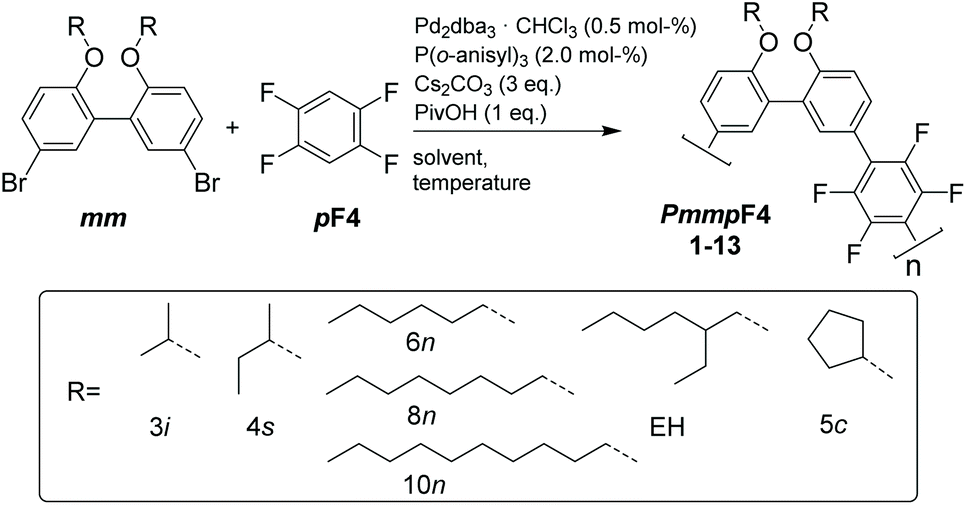 | ||
| Scheme 1 Synthesis of kinked, semifluorinated PmmpF4 via DAP. | ||
Good solvents for PmmpF4 are chloroform, dichloromethane and 1,2-dichloroethane, all of which are unsuitable for cross coupling reactions. Chlorobenzene (CB) is a good solvent for PmmpF4 but leads to phenyl-endcapping (see Fig. S14†). The best results were achieved for toluene (Tol) as solvent, 100 °C reaction temperature and a monomer concentration of 0.8 M (Table 1). Despite screening for solvents, side chains and temperature, molecular weights were moderate with values up to Mw,SEC ∼30 kg mol−1, which sometimes could be slightly increased by fractionation (entries 2a and 8). In general, meta-substituted polyarylenes show better solubility compared to para-substituted ones.28 To check whether a substantial increase in solubility of PmmpF4 could be achieved by backbone modification, F4 was replaced by 1,2,3,5-tetrafluorobenzene (mF4) resulting in “all-meta” semifluorinated polyarylenes PmmmF4 (Table 1, entry 14 and Scheme 2). However, molar masses of PmmmF4 were lower compared to the best conditions of PmmpF4, hence further optimization was not attempted.
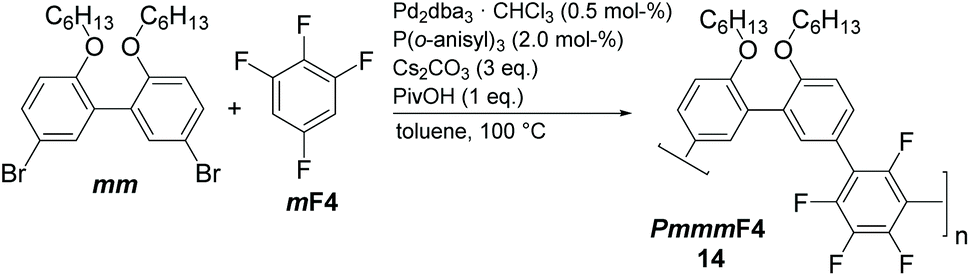 | ||
| Scheme 2 Synthesis of an all-meta semi-fluorinated polyarylene PmmmF4. | ||
| Entry # | M1 | M2 | Solvent | Temp./°C | Conc./M | M n/kg mol−1 | M w/kg mol−1 |
|---|---|---|---|---|---|---|---|
| a After Soxhlet extraction with ethyl acetate. b A small excess of 0.1 eq. of F4 was used. | |||||||
| 1 | 6n | pF4 | Tol | 70 | 1.0 | 3.9 | 8.7 |
| 2 | 6n | pF4 | Tol | 100 | 0.8 | 10.8 | 21.5 |
| 2aa | 6n | pF4 | Tol | 100 | 0.8 | 15.8 | 31.2 |
| 3 | 6n | pF4 | Tol | 120 | 0.8 | 3.9 | 5.5 |
| 4 | 6n | pF4 | THF | 100 | 0.8 | 7.9 | 16.8 |
| 5 | 6n | pF4 | Dioxane | 100 | 0.5 | 6.4 | 15.0 |
| 6 | EH | pF4 | Tol | 100 | 1.2 | 10.2 | 14.8 |
| 7 | 4s | pF4 | Tol | 100 | 0.8 | 10.1 | 17.1 |
| 8b | 4s | pF4 | Tol | 100 | 0.8 | 14.4 | 28.5 |
| 9 | 3i | pF4 | Tol | 100 | 0.8 | 1.9 | 2.5 |
| 10 | 5c | pF4 | Tol | 100 | 0.8 | 6.6 | 11.8 |
| 11 | 8n | pF4 | Tol | 100 | 1 | 6.1 | 12.6 |
| 12 | 8n | pF4 | CB | 90 | 1 | 6.2 | 13.0 |
| 13 | 10n | pF4 | Tol | 100 | 0.8 | 11.7 | 20.6 |
| 14 | 6n | mF4 | Tol | 100 | 0.8 | 8.9 | 18.8 |
PmmpF4 was characterized by 1H and 19F NMR spectroscopy (Fig. 1a and Fig. S10–S16†). Additionally, the 13C NMR spectrum was recorded for the hexyl derivative (entry 2, Fig. 1b). The rather clean 1H NMR spectra without intense end group signals suggest good molecular weights. To shine light on termination reactions, an end group investigation exemplified by entry 13, was carried out. A comparison of the aromatic protons’ region, the monomer mm (10n) containing the Br end group and 2,2′-(hexyloxy)biphenyl as model compound for H end groups reveals dehalogenation29 as a major cause for functional group degradation (Fig. S17†). An –OCH3 signal at 3.55 ppm, visible in most polymers, indicates anisyl end groups that arise from the covalent attachment of the ligand P(o-anisyl)3.17 The identification of F4-based end groups from the 19F NMR spectra is straightforward. Proven end groups are –C6F4-H and –C6F4-Ph, which result in characteristic low intensity signals in the 19F NMR spectra (Fig. S10b–S16b†). The –C6F4-Ph end group is only observed when CB was used as solvent for polymerization (Fig. S14b†). The herein found end groups are thus typical for cross coupling reactions and DAP, and thus PmmpF4 is not an exception in this regard. Quantification of end group intensities with good accuracy is very challenging, and hence not attempted here. In general, despite a closer look at typical end groups as well as the use of model compounds, the estimation of absolute molecular weights Mn,NMR is not possible for PmmpF4. What can be concluded from the synthesis and NMR spectroscopic investigations is that the monomer couple mm/F4 is a simple comonomer system that undergoes clean chemistry with typical side/termination reactions. Entries that reached Mw,SEC ∼20–30 kg mol−1 precipitated or gelated independent of the side chain, a property that apparently is inherent to PmmpF4.
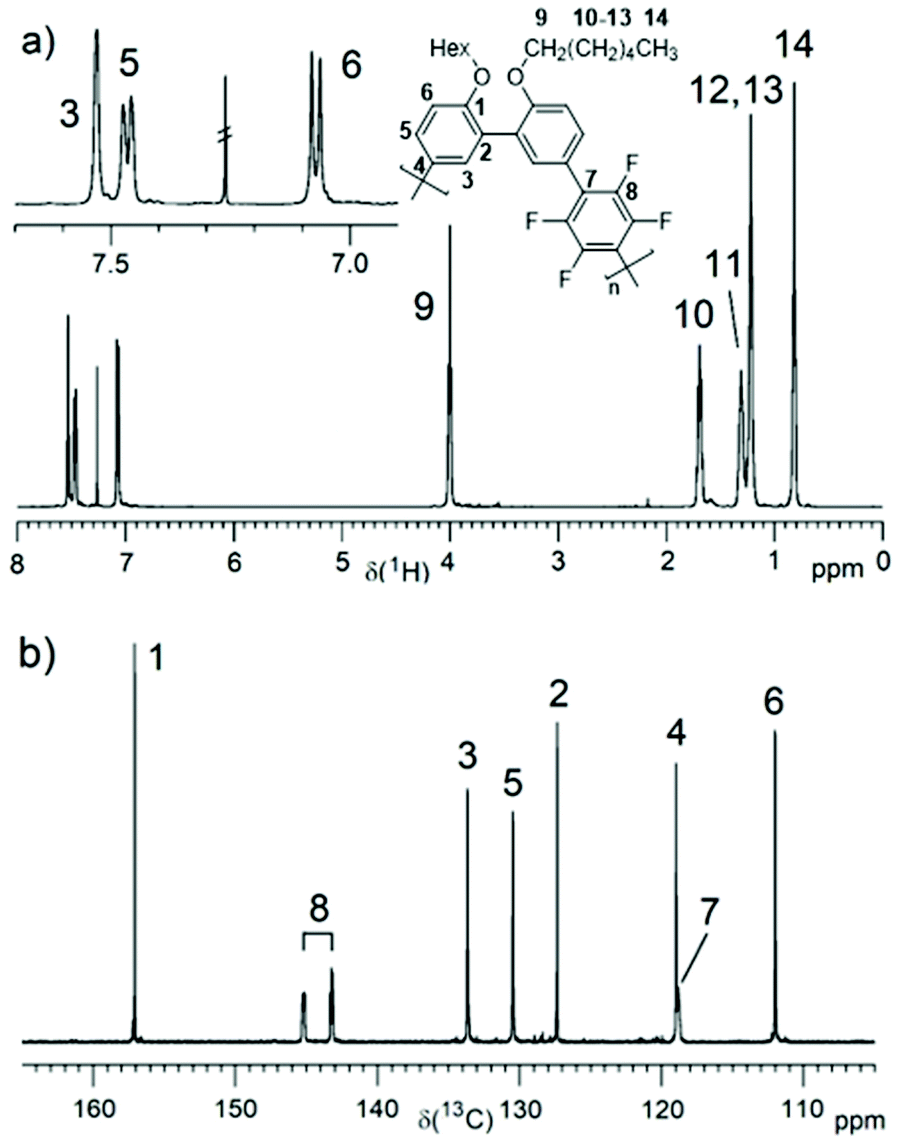 | ||
| Fig. 1 1H (a) and 13C (b) NMR spectra of PmmpF4 (entry 2) in CDCl3 with assignments. | ||
Thermal properties
The thermal properties were investigated next (Fig. 2). Fig. 2a shows the dependence of Tg of PmmpF4 with n-hexyl side chains on molecular weight. A trend showing saturation at 90 °C for Mn/Mw ∼10/20 kg mol−1 is seen, which is expected for conjugated polymers (Table 2).30 The dependence of the Tg of PmmpF4 on the side chain for similar molecular weight is shown in Fig. 2b and Table 2. Usually, longer side-chains decrease the Tg of conjugated polymers.30–33 This is also seen here, with the Tg of PmmpF4 with 10n being lower (35 °C) than of PmmpF4 with 6n (83 °C), as expected.33 PmmpF4 with 4s and EH side chains exhibits higher Tgs (173 and 68 °C, respectively) compared to 6n and 8n (83 and 50 °C, respectively, Fig. 2a, Table 2). The increase from Tg for linear to branched side chains at a constant number of carbons in the side chain is again similar to e.g. polythiophenes, where for poly(3-octylthiophene) and poly(3-(2-ethylhexyl)thiophene) glass transition temperatures of −13 °C and 24 °C, respectively, have been reported.31,34 The Tg of PmmpF4 further increases more strongly when moving to cyclic side chains. Upon attaching 5c side chains a Tg of 197 °C is measured. Clearly, cyclic side chains increase Tg much stronger than linear ones. While cyclic side chains are rather uncommon for conjugated polymers, the comparison of the Tgs of poly(1-hexene) (−63 °C)35 and polyvinylcyclohexane (80 °C)36 also indicates a strong increase in Tg for linear versus cyclic side chains at constant side chain fraction. We also attempted to install cyclohexyl side chains, which failed at the monomer synthesis stage where mono-substituted products were obtained (not shown). Thus, the Tg of PmmpF4 can be varied by as much as ∼160 K for the herein investigated range of side chains.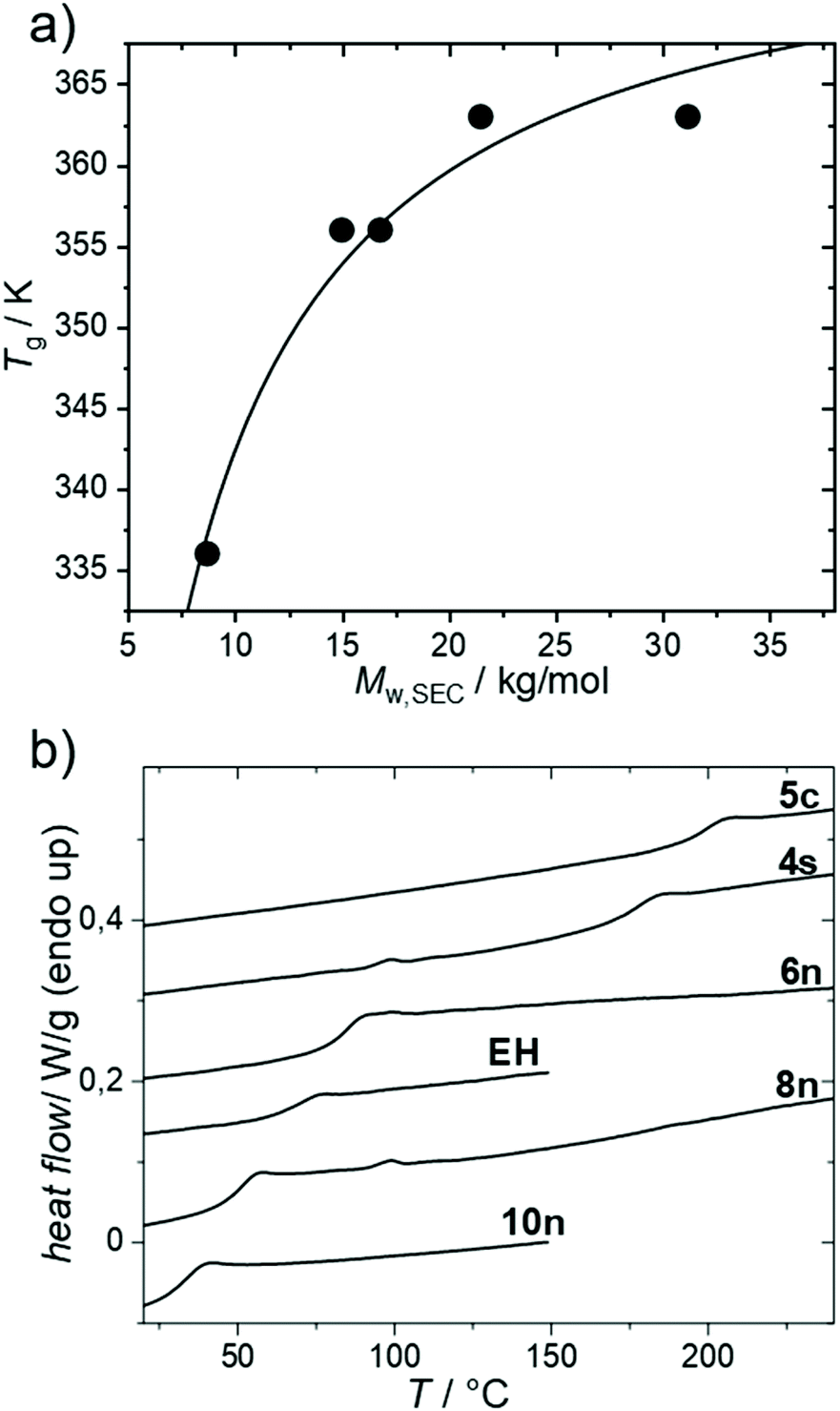 | ||
| Fig. 2 Glass transition temperature of PmmpF4 from differential scanning calorimetry with 6n as a function of molecular weight (a) and for varying side chains including entry 5 (b). Conditions: 2nd heating, 10 K min−1, N2. | ||
| Entry # | R= | T g/°C | M w/kg mol−1 | Đ |
|---|---|---|---|---|
| 2a | n-Hexyl | 90 | 31.2 | 1.5 |
| 2 | n-Hexyl | 90 | 21.5 | 2.0 |
| 4 | n-Hexyl | 83 | 16.8 | 2.1 |
| 5 | n-Hexyl | 83 | 15.0 | 2.3 |
| 1 | n-Hexyl | 63 | 8.7 | 2.2 |
| 7 | s-Butyl | 173 | 17.1 | 1.7 |
| 10 | Cyclopentyl | 197 | 11.8 | 1.8 |
| 12 | n-Octyl | 50 | 13.0 | 2.1 |
| 6 | (2-Ethyl) hexyl | 68 | 14.8 | 1.5 |
| 13 | n-Decyl | 35 | 20.6 | 1.8 |
With PmmpP being highly soluble,11 it is tempting to explain the limited solubility of PmmpF4 by the replacement of Ph by F4. However, the mere presence of an F4 moiety in the backbone alone cannot explain the limited solubility. Other alternating F4 copolymers with fluorene (PF8F4) or carbazole show excellent solubility with molecular weights of PF8F4 of up to Mn,SEC = 347 kg mol−1.17 With PF8F4 exhibiting stiff and coplanar dioctylfluorene units in combination with pF4, it appears counterintuitive that the herein investigated twisted and kinked PmmpF4 with a similar amount of side chains exhibits a lower solubility than PF8F4 for the same range of solvents. We therefore assumed attractive interactions between segments of PmmpF4 to be present. Possible interactions anticipated include oxygen/lone pair-F4 interactions, which may be comparable to anion/lone pair-π-interactions, and π–π interactions (Fig. 3a and b).37,38
 | ||
| Fig. 3 (a and b) Possible chain chain interactions of PmmpF4 and (c) model compounds used for DFT calculations. | ||
However, F4-oxygen interactions as depicted in Fig. 3a are difficult to prove. The fact that the solubility of PmmpF4 in THF was even lower than in Tol did also not point to such a motif. Interactions including π–π-stacked configurations as depicted in Fig. 3b were therefore investigated theoretically.
DFT calculations
In order to further investigate a possible origin for the reduced solubility of PmmpF4, we have performed an extensive theoretical study of the weak interactions between its building blocks. The absence of effects due to the side chains suggests that strong interactions between different parts of the PmmpF4 backbone might be responsible for the decreased solubility relative to PmmpP observed. We were particularly interested in the effect of fluorination of the phenyl-based comonomer and the role of substitution of the biphenyl comonomer on the aggregation behavior. We therefore studied the interaction of building blocks of PmmpF4 as well as differently substituted derivatives. Fig. 3c shows chemical structures of all compounds involved, namely C6H6, C6H2F4, C6F6, C15H14, C16H18 and C14H14O2.The density functional theory (DFT) calculations were performed in the projector augmented wave method39 as implemented in the GPAW package.40,41 The smooth Kohn–Sham wave functions were represented on real space grids with a grid spacing of 0.2 Å and the electron density on grids of 0.1 Å spacing. The exchange–correlation energy was approximated as devised by Perdew, Burke and Ernzerhof (PBE)42 and the corrections proposed by Tkatchenko and Scheffler (TS09)43 were included to describe dispersive interactions. The results were cross checked using the vdW-DF2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 functional. The structures were set up using the atomic simulation environment41 and the simulation box was ensured to contain at least 4 Å of space around each atom. Molecules were placed in random relative orientations and structures subsequently relaxed to the next local minimum. This resulted in 190–650 relaxed structures for each pairing from which we have obtained the configurations of lowest energy. The configurations were accepted if at least two lowest energy structures were found within the range of 4 kJ mol−1 representing chemical accuracy (see Fig. S19†).45 Interactions involving C6H2F4 required considerably more pairs than those containing C6H6 or C6F6 to achieve this goal. This effect can be attributed to the lower symmetry of C6H2F4, which results in a larger structural entropy in the pairs.
44 functional. The structures were set up using the atomic simulation environment41 and the simulation box was ensured to contain at least 4 Å of space around each atom. Molecules were placed in random relative orientations and structures subsequently relaxed to the next local minimum. This resulted in 190–650 relaxed structures for each pairing from which we have obtained the configurations of lowest energy. The configurations were accepted if at least two lowest energy structures were found within the range of 4 kJ mol−1 representing chemical accuracy (see Fig. S19†).45 Interactions involving C6H2F4 required considerably more pairs than those containing C6H6 or C6F6 to achieve this goal. This effect can be attributed to the lower symmetry of C6H2F4, which results in a larger structural entropy in the pairs.
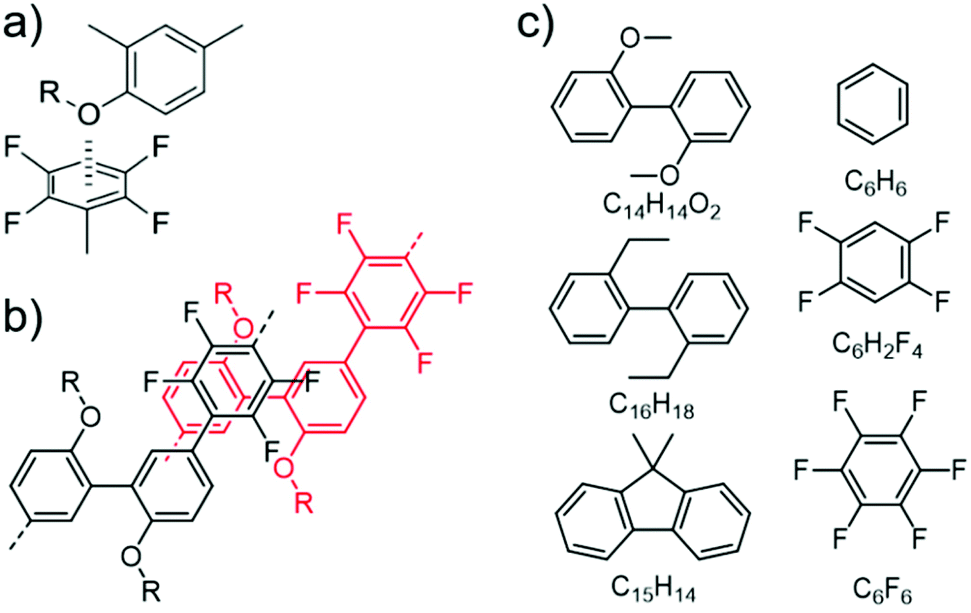
Fig. 4a displays the binding energies (BEs, positive values indicate attraction) of the energetically best pairs found. There is a clear trend of enhanced attraction between fluorinated species as compared to benzene. The lowest BE is found for the benzene pair with 15 kJ mol−1 in the parallel-displaced configuration (see Fig. S20†). This value is slightly higher than the CCSD(T) value of 11–12 kJ mol−1 (ref. 46 and 47) and the difference can be assigned to the tendency of the TS09 approximation to overestimate the interactions,48 while keeping the relative ordering correct.49 We find the C6H6–C6F6 pair to bind with 42 kJ mol−1 in good agreement to MP2 value of 39 kJ mol−1.50 The overall highest BE between benzene and its fluorinated versions is found for the C6F6 pair with 52 kJ mol−1. All combinations involving C6H6, C6H2F4 and C6F6 prefer parallel-displaced configurations.
 | ||
| Fig. 4 (a) Scheme of binding energies between the model fragments of PmmpF4, for chemical structures see Fig. 3c. (b) Selected lowest energy structures. (c) Binding energies in vdW-DF2 and PBE in the TS09 relaxed structure. | ||
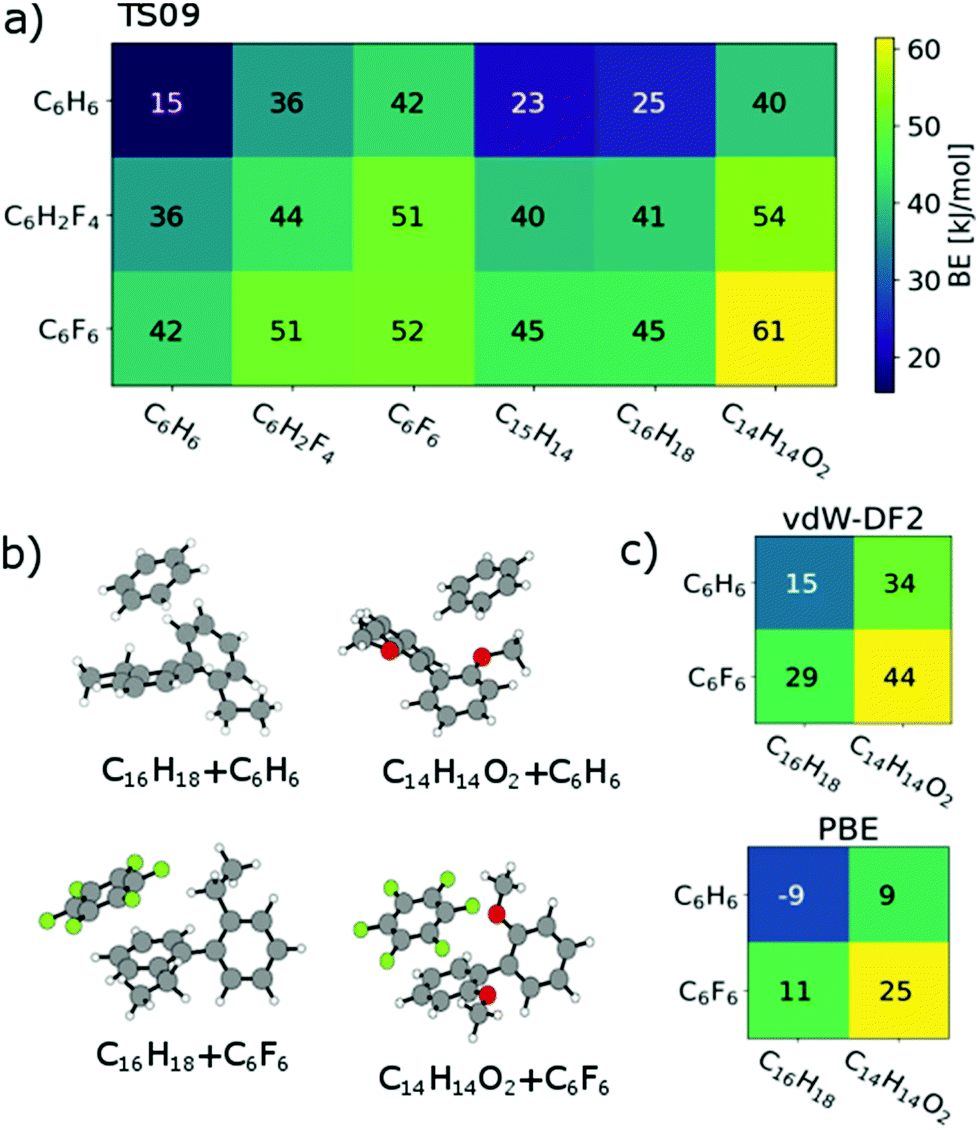
Regarding relaxed structures between C6H6, C6H2F4 and C6F6 with C15H14, C16H18 and C14H14O2, there is a large variety of relative configurations with the tendency to parallel stacked configurations of the fluorinated benzenes with the biphenyl model compounds (see Fig. 3b, 4b and S16†). Also here, the BEs follow clear trends of stronger interactions with increasing number of fluorine substituents for each biphenyl derivative. Pairs involving C14H14O2 in combination with C6H6 and C6H2F4 are found to form CH-O hydrogen bonds (see Fig. S20†) with bond lengths between 2.7 Å and 2.9 Å for the two best structures of each pair, which obviously are not relevant for the polymer under scrutiny. However, from a general perspective, this structure factor is remarkable despite of the weakness of this bond that should be less than 10 kJ mol−1.51,52 Most importantly, however, is the finding that all three benzene derivatives exhibit the strongest interactions with C14H14O2. A higher BE is found for C14H14O2 and C6H6, C6H2F4 as well as C6F6 in comparison to the coplanar fluorene derivative C15H14. C16H18 shows nearly identical BEs indicating the presence of oxygen to be the decisive factor for enhanced interaction. A cross check with the conceptually different description of dispersive interactions by the vdW-DF2 functional leads to slightly lower binding energies, but retains the same relative ordering as seen in Fig. 4c. A similar behaviour is already obtained at the PBE level (evaluated for TS09 relaxed structures), which indicates that the effect is rather due to polarization than dispersion.53 Bader analysis54,55 did not reveal any significant charge transfer in these structures.
The larger BE values of the couple C14H14O2/C6F6 compared to C15H14/C6F6 reflect both the rather good solubility of PF8F417 as well as the limited solubility of PmmpF4. Furthermore, these polar backbone interactions help to understand the rather small influence of side chain length on solubility, which is uncommon for aromatic polymers. From a structural design point of view, the combination of the F4 motif with alkoxylated biphenyls apparently makes a large difference, as the increase of chain–chain interactions leads to reduced solubility. A further interesting observation is that the clear trends in BEs as shown in Fig. 4 are only found when a trans-conformation of the biphenyl model structure of C14H14O2 (Fig. 3c) is used as starting point. Starting from the corresponding cis-conformation also yields somewhat larger BE values for the fluorinated phenyls, but the trends are less clear and the relative differences in BE values lower (Fig. 5).
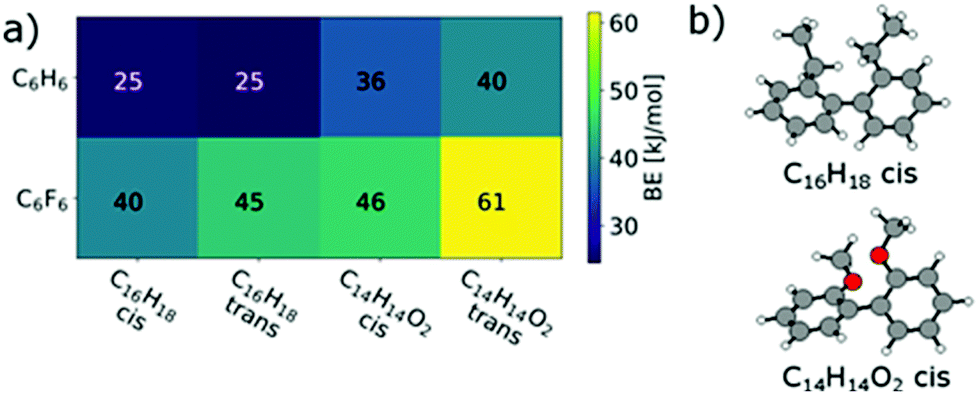 | ||
| Fig. 5 (a) cis/trans effect in C16H18 and in C14H14O2 on the binding energies to C6H6 and C6F6. (b) cis configurations of C16H18 and C14H14O2. | ||
The cis/trans effect is nearly vanishing for C16H18. This may indicate that such trans-conformations are present in the copolymer and present an important contribution to the observed solubility behaviour. Restricting the presence of trans-conformations by conformational locking may therefore address the limited solubility of PmmpF4.
Conclusions
We have successfully prepared semifluorinated, kinked polyarylenes PmmpF4 by a straightforward and simple direct arylation approach with high atom economy. The glass transition temperature of these semifluorinated aromatic copolymers can be varied between 35 and 197 °C depending on type and size of the side chain. Molar mass is limited by solubility and not by termination reactions, which is surprising considering the kinked backbone structure and the exceptionally good solubility of the non-fluorinated analog PmmpP. Density functional theory calculations on model systems reveal strong attractive interactions between the fluorinated and dialkoxylated comonomers, which continuously increase with the degree of fluorination. This effect is caused mainly by an increased polarization interaction rather than changes in dispersion or charge transfer. It is thus the combination of the alkoxy side chains with the F4 unit that causes the experimentally observed limited solubility, which should be avoided if copolymers with high molar mass and solubility are the target.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. and F. K. acknowledge funding from the DFG (SO 1213/8-1). M. W. acknowledges computational resources provided by the state of Baden-Württemberg through bwHPC via the JUSTUS cluster.Notes and references
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- M. Friedman and G. Walsh, Polym. Eng. Sci., 2002, 42, 1756–1788 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y.-K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS.
- J. A. Reglero Ruiz, M. Trigo-López, F. C. García and J. M. García, Polymers, 2017, 9, 414 CrossRef.
- I. A. Ronova and S. S. A. Pavlova, High Perform. Polym., 1998, 10, 309–329 CrossRef CAS.
- R. Kandre, K. Feldman, H. E. H. Meijer, P. Smith and A. D. Schlüter, Angew. Chem., Int. Ed., 2007, 46, 4956–4959 CrossRef CAS.
- S. Jakob, A. Moreno, X. Zhang, L. Bertschi, P. Smith, A. D. Schlüter and J. Sakamoto, Macromolecules, 2010, 43, 7916–7918 CrossRef CAS.
- R. Kandre and A. D. Schlüter, Macromol. Rapid Commun., 2008, 29, 1661–1665 CrossRef CAS.
- B. Hohl, L. Bertschi, X. Zhang, A. D. Schlüter and J. Sakamoto, Macromolecules, 2012, 45, 5418–5426 CrossRef CAS.
- F. Kempe, O. Brügner, H. Buchheit, S. N. Momm, F. Riehle, S. Hameury, M. Walter and M. Sommer, Angew. Chem., Int. Ed., 2018, 57, 997–1000 CrossRef CAS.
- J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schlüter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS.
- M. Wakioka and F. Ozawa, Asian J. Org. Chem., 2018, 7, 1206–1216 CrossRef CAS.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS.
- J. Kuwabara and T. Kanbara, Bull. Chem. Soc. Jpn., 2018, 92, 152–161 CrossRef.
- R. M. Pankow and B. C. Thompson, Polym. Chem., 2020, 11, 630–640 RSC.
- M. Wakioka, Y. Kitano and F. Ozawa, Macromolecules, 2013, 46, 370–374 CrossRef CAS.
- W. Lu, J. Kuwabara and T. Kanbara, Macromolecules, 2011, 44, 1252–1255 CrossRef CAS.
- A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864–883 CrossRef.
- M. Sommer, H. Komber, S. Huettner, R. Mulherin, P. Kohn, N. C. Greenham and W. T. S. Huck, Macromolecules, 2012, 45, 4142–4151 CrossRef CAS.
- S. Kappaun, M. Zelzer, K. Bartl, R. Saf, F. Stelzer and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2130–2138 CrossRef CAS.
- T. Kirschbaum, R. Azumi, E. Mena-Osteritz and P. Bäuerle, New J. Chem., 1999, 23, 241–250 RSC.
- M. Jayakannan, J. L. J. van Dongen and R. A. J. Janssen, Macromolecules, 2001, 34, 5386–5393 CrossRef CAS.
- K. D. Deshmukh, R. Matsidik, S. K. K. Prasad, N. Chandrasekaran, A. Welford, L. A. Connal, A. C. Y. Liu, E. Gann, L. Thomsen, D. Kabra, J. M. Hodgkiss, M. Sommer and C. R. McNeill, ACS Appl. Mater. Interfaces, 2018, 10, 955–969 CrossRef CAS.
- X. Liao, F. Wu, Y. An, Q. Xie, L. Chen and Y. Chen, Macromol. Rapid Commun., 2017, 38, 1600556 CrossRef.
- A. Nitti, F. Debattista, L. Abbondanza, G. Bianchi, R. Po and D. Pasini, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1601–1610 CrossRef CAS.
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- R. Pinal, Org. Biomol. Chem., 2004, 2, 2692–2699 RSC.
- F. Lombeck, R. Matsidik, H. Komber and M. Sommer, Macromol. Rapid Commun., 2015, 36, 231–237 CrossRef CAS.
- C. Müller, Chem. Mater., 2015, 27, 2740–2754 CrossRef.
- S. Pankaj, E. Hempel and M. Beiner, Macromolecules, 2009, 42, 716–724 CrossRef CAS.
- Z. Qian, Z. Cao, L. Galuska, S. Zhang, J. Xu and X. Gu, Macromol. Chem. Phys., 2019, 220, 1900062 CrossRef.
- R. Xie, A. R. Weisen, Y. Lee, M. A. Aplan, A. M. Fenton, A. E. Masucci, F. Kempe, M. Sommer, C. W. Pester, R. H. Colby and E. D. Gomez, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- L. Yu, E. Davidson, A. Sharma, M. R. Andersson, R. Segalman and C. Müller, Chem. Mater., 2017, 29, 5654–5662 CrossRef CAS.
- M. H. Jandaghian, A. Soleimannezhad, S. Ahmadjo, S. M. M. Mortazavi and M. Ahmadi, Ind. Eng. Chem. Res., 2018, 57, 4807–4814 CrossRef CAS.
- J. S. Grebowicz, Polym. Eng. Sci., 1992, 32, 1228–1235 CrossRef CAS.
- N. J. Singh, H. M. Lee, I.-C. Hwang and K. S. Kim, Supramol. Chem., 2007, 19, 321–332 CrossRef CAS.
- S. Kozuch, Phys. Chem. Chem. Phys., 2016, 18, 30366–30369 RSC.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035109 CrossRef.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dulak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- K. Lee, É. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- J. A. Pople, Rev. Mod. Phys., 1999, 71, 1267–1274 CrossRef CAS.
- Y. C. Park and J. S. Lee, J. Phys. Chem. A, 2006, 110, 5091–5095 CrossRef CAS.
- E. Miliordos, E. Aprà and S. S. Xantheas, J. Phys. Chem. A, 2014, 118, 7568–7578 CrossRef CAS.
- A. Tkatchenko, R. A. DiStasio, R. Car and M. Scheffler, Phys. Rev. Lett., 2012, 108, 236402 CrossRef.
- M. Hassan, M. Walter and M. Moseler, Phys. Chem. Chem. Phys., 2013, 16, 33–37 RSC.
- S. Tsuzuki, T. Uchimaru and M. Mikami, J. Phys. Chem. A, 2006, 110, 2027–2033 CrossRef CAS.
- S. Scheiner, T. Kar and J. Pattanayak, J. Am. Chem. Soc., 2002, 124, 13257–13264 CrossRef CAS.
- D. Ž. Veljković, G. V. Janjić and S. D. Zarić, CrystEngComm, 2011, 13, 5005–5010 RSC.
- J. Badorrek, M. Walter and M.-P. Laborie, J. Renewable Mater., 2018, 6, 325–335 CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford England: New York, 1st Paperback edn, 1994 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00973c |
| This journal is © The Royal Society of Chemistry 2020 |