
Gold and hypervalent iodine(III): liaisons over a decade for electrophilic functional group transfer reactions
Somsuvra
Banerjee
 ab,
Vivek W.
Bhoyare
c and
Nitin T.
Patil
*c
ab,
Vivek W.
Bhoyare
c and
Nitin T.
Patil
*c
aDivision of Organic Chemistry, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
cDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Bhopal-462066, India. E-mail: npatil@iiserb.ac.in
First published on 6th February 2020
Abstract
Over the last two decades, hypervalent iodine(III) reagents have evolved from being ‘bonding curiosities’ to mainstream reagents in organic synthesis, in particular, electrophilic functional group transfer reactions. In this context, gold catalysts have not only emerged as a unique toolbox to facilitate such reactions (especially alkynylations) but also opened new possibilities with their different modes of reactivities for other functional group transfer reactions (acetoxylations and arylations). This feature article critically summarizes hitherto all such Au-catalyzed electrophilic functional group transfer reactions with hypervalent iodine(III) reagents, emphasizing their mechanistic aspects.
1. Introduction
Hypervalent iodine compounds,1 such as λ3-iodanes and λ5-iodanes, have always fascinated chemists because their ‘unique reactivities’ share strong ties with their ‘unique bonding characteristics’. In particular, the molecular orbitals in λ3-iodanes (1) are delocalized over three centres in the linear X–I–Y bond and occupied by two electrons from the (axial) 5p orbital of iodine and one electron each from apical ligand X and Y (Scheme 1a). The presence of such a weak, highly polarized, and three-center-four-electron (3c-4e) ‘hypervalent’2 bond (X–I–Y) in λ3-iodanes (1) successfully validates their special structural features and highly electrophilic reactivity pattern, especially electrophilic functional group transfer reactions.3 In this regard, the pioneering work by Beringer and co-workers has paved the way for the development of diaryliodonium salts (2a) and alkynyliodonium salts (2b), both acyclic λ3-iodanes, as electrophilic aryl- and acetylene synthons, repectively.4 This was an important milestone because iodonium salts (2) were able to deliver alkyne or aryl moieties ‘directly’ in an umpolung fashion and thus tremendously expand the scope of acetylene or aryl transfer reactions beyond the corresponding well-established nucleophilic versions.5 Later on, more methods such as alkenylations, alkylations and halogenations emulated the electrophilic functionalization strategy.6 However, research in this area witnessed a decline after 1995 probably because the high reactivity of acylic λ3-iodanes was accompanied by instability (except for diaryliodonium salts 2a) in the presence of strong bases or transition metals or during heating, which essentially limited their further applications.7a Therefore, striking a proper balance between reactivity and stability was necessary for future advancements in the field of electrophilic functional group transfer reactions.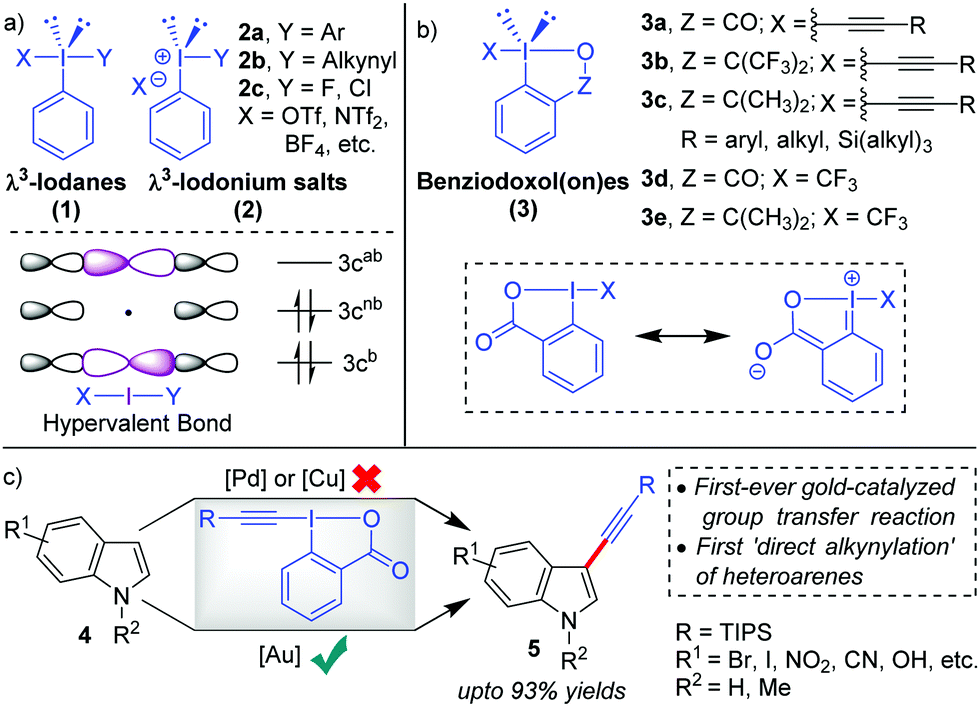 | ||
| Scheme 1 (a) Structure and bondings in λ3-iodanes and λ3-iodonium salts, (b) benziodoxol(on)e (BX) analogues, and (c) C3-alkynylation of indoles: the first Au-catalyzed electrophilic functional group transfer reaction. | ||
With the inception of ethynylbenziodoxol(on)es (EBXs, 3a–c)6a,7 in 1991, by the group of Ochiai and Shiro,8 the stability issue was mitigated9 by bridging the apical and the equatorial positions in a 5-membered heterocycle which introduces rigidity into the backbone (Scheme 1b). In cases where Z = CO, the stability was even more pronounced because of the ‘extended’ conjugative overlap between the lone pairs of electrons on the iodine atom and the π-orbitals of the aromatic nucleus.8 Later in 1996, the Zhdankin group developed a robust scheme for the synthesis of EBXs.10 Along this line, the greater stability of benziodoxol(on)es (BXs) enabled the preparation and isolation of otherwise unstable iodine(III) derivatives with I-CN, I-OOR, I-N3, etc., all of which found practical application as the reagents for oxidative functionalization of organic molecules.1e–i,3 However, the first significant development, regarding the application of benziodoxol(on)e (BX)-based hypervalent iodine(III) reagents in mainstream organic synthesis, was the use of benziodoxol(on)e-derived reagents 3d and 3e for electrophilic trifluoromethylation, by Togni and co-workers in 2006, which became an indispensible method to introduce the –CF3 functionality in the molecule.11 Next, in 2009, the Waser group successfully carried out the challenging alkynylations of indoles (4) with 3a in excellent yields (up to 93%) and thus practically rejuvenated the field of electrophilic alkynylation (Scheme 1c).12,13 Usually, in such oxidative transfer reactions from hypervalent iodine(III) species, palladium and copper are the metals of choice because their well-established two-electron redox transformations enable swift oxidative addition of I(III)–X or I(III)–Y bonds to the low-valent metal centre.5b,11,14 Copper can also initiate SET-induced radical fragmentation to formally transfer X(+) or Y(+).6b,11 In contrast, the key to success for these high-yield alkynylations with EBXs was the application of homogeneous Au-catalysis15 that exploited the nature of Au-catalysts (AuCl in this case) as tunable soft π-acids to activate the alkynyl unit installed in EBXs towards intermolecular attack by nucleophiles. In Waser's own words, “AuCl was uniquely able to activate TIPS-EBX for the C3-alkynylation of indole”.7b
There is indeed a reasonable explanation as to why the application of such cyclic λ3-alkynyliodanes such as EBXs in electrophilic alkynylation took close to two decades to realize although their synthesis was already described in 1991. This is because the superiority of Au(I)-catalysts in carbophilic activation over other late transition metals (Cu, Ag, Pd) was understood only around the beginning of the 21st century.16 By the year 2009, exploration of gold's excellent capability for π-activation had gained pace.16 Conversely, the C–H alkynylation of electron-rich heteroaromatics, which was going to be the first-ever synthetic application of EBXs, returned disappointing results for the Waser group with Pd- and Cu-catalysis (Scheme 1c).12,17 It was at this crucial juncture that the AuCl catalyst was successfully evaluated for the same, which marked the beginning of a new chapter in electrophilic functional group transfer reactions with hypervalent iodine(III) reagents, building upon the liaison between Au-catalysts and EBXs. In the following years, the Waser group was able to extend the scope of electrophilic C–H alkynylation to a range of other electron-rich heterocycles, such as pyrroles,12,13 thiophenes,18 anilines,19 furans20 and benzofurans21 and thus established EBXs as ‘general’ electrophilic acetylene synthons. Since our group had been exclusively committed towards developing novel synthetic methodologies powered by Au-catalysis,22 we were able to contribute significantly to this field of Au-catalyzed alkynylations with EBXs.
As part of our continuous involvement with Au-catalysis, herein, we would like to present an overview of the fascinating, decade-long journey of Au-catalysts and hypervalent iodine(III) reagents. Building on a mechanistic perspective, the review intends to demonstrate how the uniqueness of Au-catalysts has made possible a myriad of electrophilic functional group transfer reactions such as alkynylations, arylations and acetoxylations with the use of hypervalent iodine(III) reagents over the last decade.
2. Gold-catalyzed alkynylations with hypervalent iodine(III) reagents
The current prominence of Au-catalysis in the field of hypervalent iodine(III) chemistry is majorly caused by the excellent liaison between Au and EBXs. To better understand this liaison, it is important to put a mechanistic perspective first, which governs such alkynylation reactions. In addition to their π-activation capability, Au-catalysts can also be ‘forced’ to switch between the +1 and +3 oxidation states under homogeneous catalysis conditions, thereby accessing a Au(I)/Au(III) catalytic cycle which delivers cross-coupled23 products.22a,24 So far as the mechanism of Au-catalyzed alkynylations with EBXs is concerned, different scenarios have been discussed and a mechanistic ambiguity persisted for quite some time.13 To illustrate this, let's consider the case of Au-catalyzed C3-alkynylation of indoles (Scheme 2). The prevailing notion was that π-activation of the alkyne embedded in EBXs 3a triggers the addition of electron-rich (hetero)aromatic nucleus indole (Friedel–Crafts-type reaction) and leads to the formation of vinylgold(I) complexes (7) which, after a Fritsch–Buttenberg–Wiechell-type rearrangement (an α-elimination/1,2-shift sequence), produces the triple bond and the desired alkynylated product 5 (Scheme 2, cycle 1). However, researchers encountered some technical difficulties in garnering conclusive proof in favour of the π-activation mode. For instance, 1,2-silyl shift, which could have been one such conclusive proof, was rejected by 13C-labeling studies, whereas an alternative possibility of 1,2-indole shift could not be probed (Scheme 2b).12 Further, ligated Au(I)(+) complexes were not effective for such C–H alkynylations due to strong poisoning by coordination to the benzoate group of the iodine(III) reagent.25 This left AuCl to be the sole optimum catalyst for alkynylations, which posed additional hurdles13 towards resolving the ambiguity.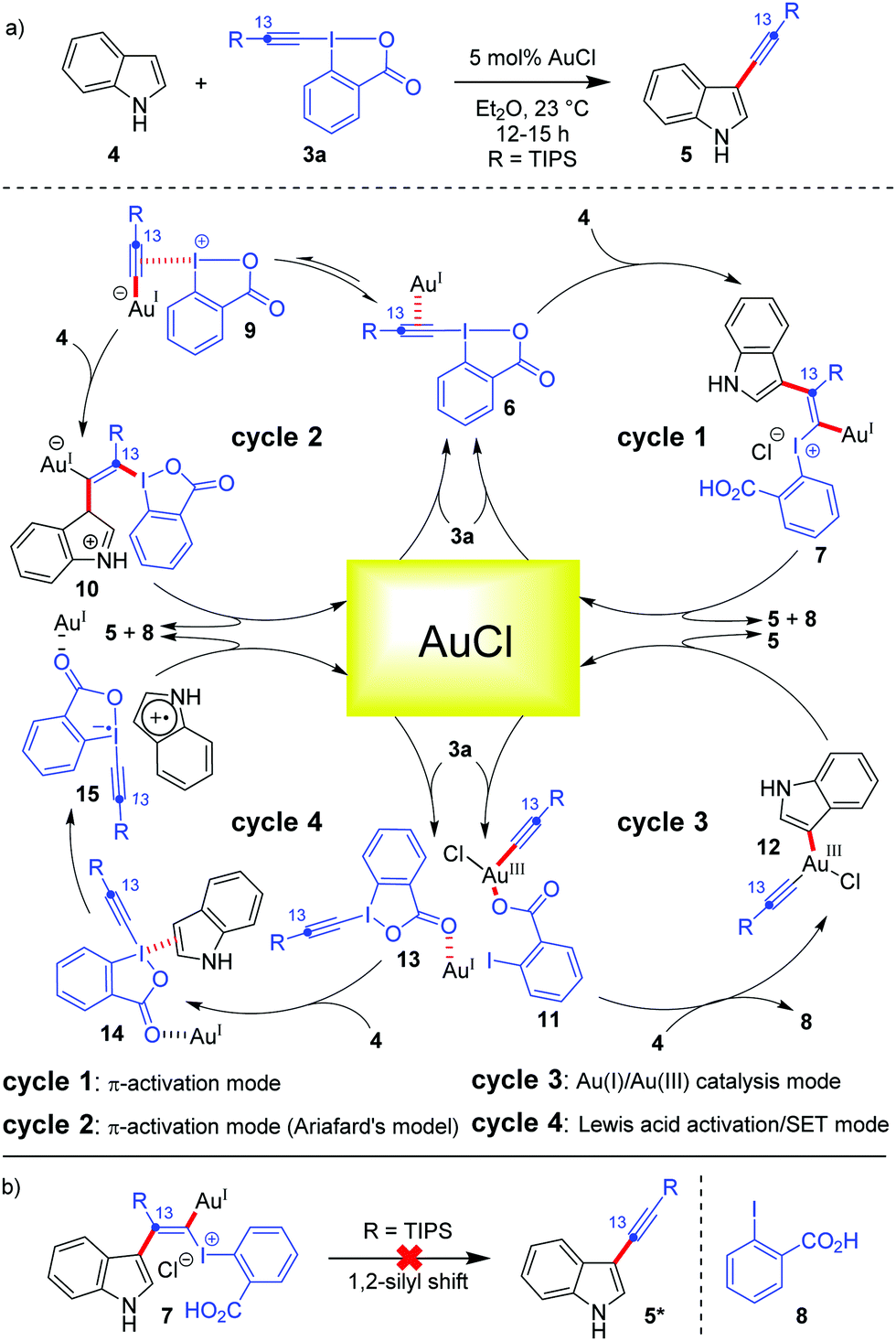 | ||
| Scheme 2 (a) Different mechanistic possibilities depicted for C3-alkynylation of indoles; and (b) 13C labelling studies to probe any 1,2-silyl shift. | ||
A modified version of the π-activation mode was introduced by Ariafard's group who employed DFT studies to suggest that the iodine(III) center in EBXs acts as a Lewis acid for activating the alkyne, even more efficiently than the Au(I)-center25 (Scheme 2, cycle 2). The catalytic reaction starts with the coordination of the alkyne moiety of the iodine(III) reagent to the AuCl catalyst to generate 6, followed by transfer of the alkynyl group from I(III) to Au(I) to access a less stable I(III)-activated alkyne species, 9, which remains in equilibrium with 6. Thereafter, the nucleophilic attack of indole on the I(III)-activated alkyne 9 gives a vinylgold(I) complex, 10, which is the rate-determining step as per the calculations. Complex 10 then undergoes β-elimination to give the desired product 5, regenerating AuCl.
Among these mechanistic conundrums, two other modes of activation regarding Au-catalyzed cyclizations with EBXs were also speculated.13 In the Au(I)/Au(III) catalysis mode, the Au(I) species is oxidized to form a Au(III)-complex, 11, followed by indole metalation and reductive elimination to furnish the desired alkynylated product (Scheme 2, cycle 3). In the Lewis acid activation/SET mode, AuCl acts as a Lewis acid and increases the electrophilicity of the iodine atom (Scheme 2, cycle 4). The activated hypervalent iodine 13 forms a charge transfer complex, 14, with the electron-rich indole. Thereafter, the SET process takes over and subsequently produces the desired alkynylated product 5. Though the Lewis acid activation/SET mode (Scheme 2, cycle 4) was dismissed at the earliest by the Waser group by a series of control experiments,13 the distinction between the π-activation mode (cycle 1 or 2) and the Au(I)/Au(III) catalysis mode (cycle 3) for Au-catalyzed alkynylations became arduous. In this context, based on the computational study by the Ariafard group26 on the Au-catalyzed domino cyclization/alkynylation approach20 to 3-alkynyl furans from keto-allenes, two very important rationales can be given regarding the general mechanism of alkynylation:
(A) If the yields of the reaction concerned are below par with ‘naked’ Au(I)- or Au(III)-catalysts (typical π-activation), it is likely that the reaction ‘demands’ a Au(I)/Au(III) catalysis mode. To serve this purpose, a Au(III)-stabilizing ligand in the reaction (e.g. N- and O-coordination of picolinic acid in this case) needs to be introduced, which has been observed to improve the yields substantially.
(B) In such cases, another mode of reactivity is also feasible which involves the interplay of both the reactivities (i.e., both π-activation and Au(I)/Au(III) catalysis: the ‘interplay’ mode).
Both rationales A and B were later substantiated with experimental evidence by the groups of Hashmi27 and Patil28 in their direct alkynylation of cyclopropenes and 1,2-oxyalkynylation of N-allenamides, respectively. Therefore, the Au-catalyzed alkynylations with EBXs discussed herein are categorized into three broad classes based on the activation mode of Au-catalysts.
1. Alkynylations via the π-activation mode in which Au(I)-catalysts activate the alkyne system of EBXs or that of the partner nucleophile.
2. Alkynylations via the Au(I)/Au(III) catalysis mode where EBX oxidizes Au(I) to Au(III) in situ and the final alkynylated product results from a reductive elimination from the alkynylgold(III) intermediate.
3. Alkynylations via the ‘interplay’ mode wherein Au-catalysts activate the π-system embedded in the partner nucleophile and are also oxidized to Au(III) by EBXs (not necessarily in that order). Further, the final alkynylated product results from a reductive elimination from the alkynylgold(III) intermediate.
The reports are segregated according to the mechanism proposed originally by the authors unless new mechanistic insights came to the fore. Furthermore, if we have assigned the report to a particular category according to the original mechanism but sincerely believed that the actual mechanism might differ based on our rationale A, we have included our perspective there. On grounds of the same rationale A, in case of unresolved mechanistic ambiguities mentioned in the article concerned between a π-activation mode and a Au(I)/Au(III) catalysis mode, it is categorized as a π-activation mode.
2.1 Alkynylations via the π-activation mode
Theoretical studies by the Ariafard group25 currently suggest that the C3-alkynylation of indoles (4) leading to 5 follows cycle 2 (Scheme 2), which, a priori, also corroborates with our rationale A that no Au(III)-stabilizing factor (ligand coordination in TS) is required in the reaction to increase the yield to the desired level. Hence, C3-alkynylation of indoles is categorized as ‘alkynylations via the π-activation mode’. In fact, all the Au-catalyzed alkynylations with EBXs following the π-activation mode should, in principle, follow cycle 2, Scheme 2. However, while describing this section, we have considered the mechanisms originally proposed by the authors.Further, there were subsequent improvements and modifications being reported for the construction of C3-alkynylated indoles (5). First, the original Au-catalyzed, direct C–H alkynylation of indoles was made more efficient by the Bolm group by conducting the reaction under mechanochemical conditions (Scheme 3a).29 In comparison to Waser's method, Bolm's ball milling method at 30 Hz sports a solvent-free approach, a lower AuCl-loading (2 mol% vs. 5 mol%) and a shorter reaction (milling) time (1.5 h vs. 12 h), although the yields of alkynylated products were moderate (up to 82%). Later on, the Waser group combined the Au(III)-catalyzed cyclization of 2-alkynylanilines (16) with the Au(I)-catalyzed C3-selective direct alkynylation of indoles (4) using EBXs (3a) in a one-pot sequential fashion to give straightforward access to 2-substituted-3-alkynylindoles (5) (up to 96% yields, Scheme 3b).30 This remains to be the first example of a one-pot process combining a Au(III)-catalyst and a Au(I)-catalyst. Of note, AuCl was unable to catalyze step 1 of the sequential process due to the ‘stochastical’ degradation of AuCl under these conditions and NaAuCl4 was observed to fail the alkynylation step.
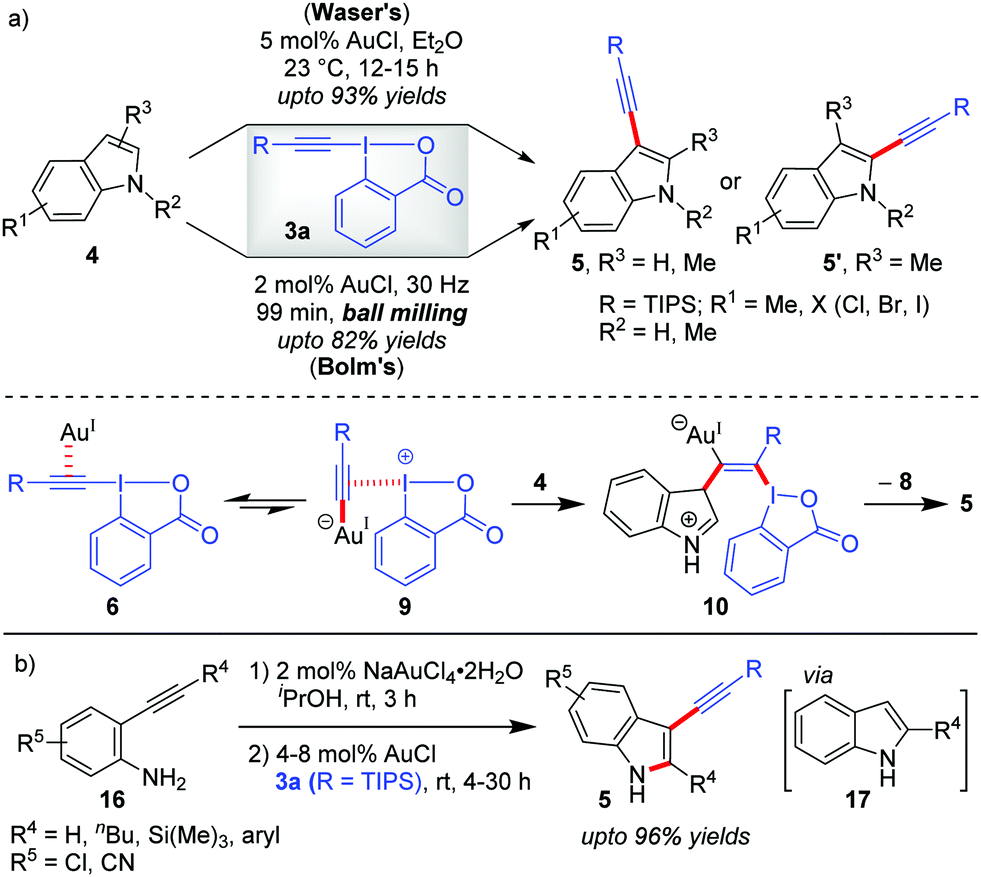 | ||
| Scheme 3 (a) Comparison of Waser's and Bolm's methods for C3-alkynylation of indoles; and (b) one-pot sequential route to 2-substituted-3-alkynylindoles from unprotected ortho-alkynylanilines. | ||
After the maiden success of the Au-catalyzed C–H alkynylation of electron-rich heteroarene indoles (Scheme 2),12,13 the Waser group further extended the scope of direct alkynylation reaction to other heteroarenes like pyrroles,12,13 thiophenes18 and benzofurans21 (19a–19d, Scheme 4). While the same reaction conditions leave unsatisfactory outcomes in the case of less electron-rich heteroarenes such as benzofurans and thiophenes, the activation of EBXs (3a) with either TFA or Zn(OTf)2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with respect to EBXs) proved to be a highly successful strategy. The role of such Lewis acid/Brønsted acid is supposedly to activate the EBXs by complexing with the carboxylate group of the hypervalent iodine(III) reagent, enhancing their electrophilic reactivity for efficient acetylene transfer. Along this line, further modulation of the reactivity of EBXs via trans-influence31 of ligands in the hypervalent X–I–Y bond and substituent effects on the aromatic ring were also studied by the Waser group.13 In the same report, the low-yield issue in the case of alkynylation of pyrroles (18a) was also addressed by using stoichiometric amounts of pyridine as an additive. The pyridine was able to suppress the competitive degradation of pyrroles by removing adventitious HCl generated from AuCl during the reaction. However, use of excess EBX led to dialkynylation of pyrroles.
:1 ratio with respect to EBXs) proved to be a highly successful strategy. The role of such Lewis acid/Brønsted acid is supposedly to activate the EBXs by complexing with the carboxylate group of the hypervalent iodine(III) reagent, enhancing their electrophilic reactivity for efficient acetylene transfer. Along this line, further modulation of the reactivity of EBXs via trans-influence31 of ligands in the hypervalent X–I–Y bond and substituent effects on the aromatic ring were also studied by the Waser group.13 In the same report, the low-yield issue in the case of alkynylation of pyrroles (18a) was also addressed by using stoichiometric amounts of pyridine as an additive. The pyridine was able to suppress the competitive degradation of pyrroles by removing adventitious HCl generated from AuCl during the reaction. However, use of excess EBX led to dialkynylation of pyrroles.
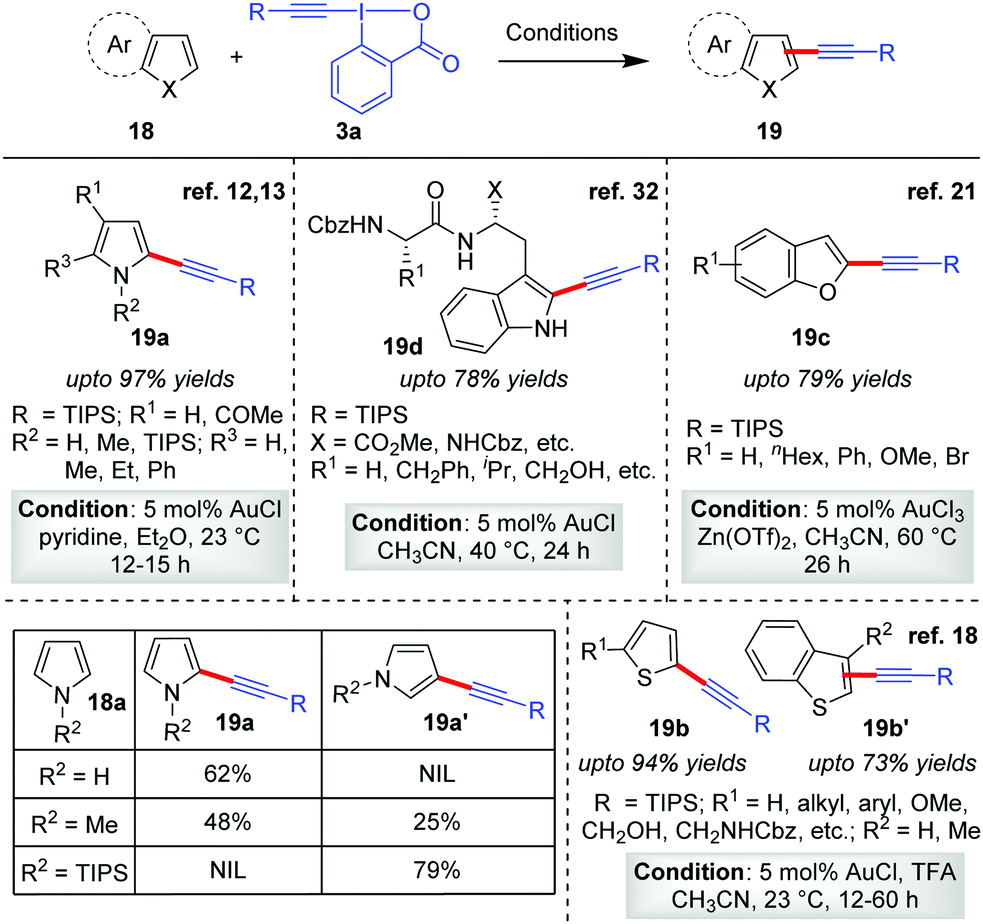 | ||
| Scheme 4 Alkynylation of electron-rich heterocycles: pyrroles, thiophenes, benzofurans and tryptophan. | ||
For all the heteroarenes, the regioselectivity of the alkynylation reaction happens to be in exclusive agreement with electrophilic aromatic substitution (SEAr); that is, it is reliant on the relative electron richness of the respective sites. The regioselectivity can also be predicted by considering the thermodynamic stability of the intermediates obtained by the Friedel–Crafts addition of heteroarenes onto intermediate 9 (Scheme 3a). In case the site concerned is occupied, the alkynylation would proceed to the next available site. For instance, the formation of C2-alkynylated products (5′) were observed exclusively for C3-substituted indoles (Scheme 3a), whereas for pyrroles the regioselectivity was sensitive to the steric bulk on the nitrogen atom12 (Scheme 4, table). Therefore, by applying easily removable protecting groups such as TIPS, the regioselectivity could be easily controlled. The robustness of the C–H alkynylation was further demonstrated in the direct alkynylation of tryptophan in peptides (19d, up to 78% yields, Scheme 4),32 which is indeed highly useful for bio-conjugating tryptophan under mild conditions without the installation of non-natural amino acids. Of note, the mechanism for obtaining alkynylated products 19a–19d follows cycle 1, Scheme 2, as originally proposed by the authors.
It was quite obvious from alkynylation of heteroarenes that the electronic effects play a major role in determining site selectivity. In particular, the directing effect of the heteroatom functional group (e.g. ‘NR’) is the most effective amongst all, functioning as an embedded enamine-type system. Along this line, the Patil33 group and the Li group,34 in 2016, independently reported a Au-catalyzed site-selective C4-alkynylation of isoquinolones/2-pyridones (20), respectively, giving 21 (up to 92% yields, Scheme 5). As usual, the intrinsic electronic bias of the isoquinolones/2-pyridone rings allowed the alkynylation at the most electron-rich C4 site. A theoretical investigation on the operating mechanism in this reaction by the Yu and Liu group35 reinforced Ariafard's proposal25 that the iodine(III) centre, instead of the Au(I)-centre, acts as a strong Lewis acid to activate the alkyne moiety efficiently (cf.11). In addition, they reasoned that the C4-alkynylation product is preferred over the C8-alkynylation product for isoquinolone systems mainly because of the higher steric effects in 23′ (TS) compared with that in 23 (TS). Of note, the reactions in Scheme 5 were originally a part of catalyst-controlled regiodivergent alkynylation programs featuring Au(I)- and Rh(III)-catalysts. It is the coordination with the amide carbonyl group that directs Rh(III)-catalysts to effect C–H activation at a different site.
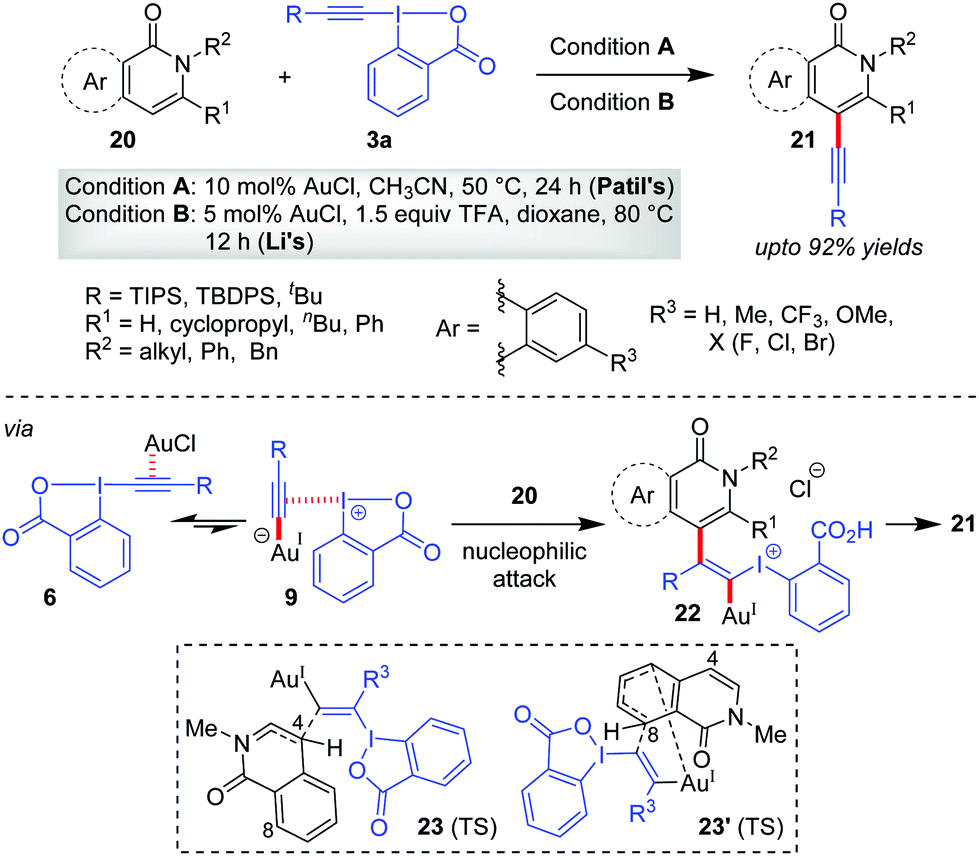 | ||
| Scheme 5 Alkynylation of isoquinolones and 2-pyridones at the C4 site. | ||
Alkynylation promoted by pre-formed acyclic enamine systems was first evaluated by the Patil group where tandem alkynylation of ortho-hydroxyarylenaminones (24) followed by intramolecular cyclization was performed to generate a diverse array of 3-alkynylchromones (25, up to 96% yields, Scheme 6).36 Control experiments suggested that enaminone 24 would undergo alkynylation first to produce intermediate 27 (via26) which after intramolecular cyclization would afford intermediate 28. Finally, the spontaneous loss of N,N-dimethylamine would occur to generate 3-alkynylchromones 25.
 | ||
| Scheme 6 Alkynylation/cyclization cascade of ortho-hydroxyarylenaminones to 3-alkynylchromones. | ||
Exercising cooperation between amine catalysis and Au-catalyzed alkynylation via EBXs, the Huang group disclosed the first direct α-vinylidenation of aliphatic aldehydes 29 (Scheme 7a).37 The authors reasoned that the addition of enamine 33 to the Au-activated π-system in EBXs (3a) would be followed by an α-elimination/1,2-shift to lead to 34 which, after tautomerization, gives the key ynenamine intermediate 35. Intermediate 35 would be further hydrolyzed by γ-protonation to yield α-allenyl aldehydes (30). The competing α-protonation, which leads to α-alkynyl aldehydes (31), was assumed to be less favoured by the authors. Unfortunately, the selectivity of the reaction towards α/γ-protonation was not satisfactory enough and the two constitutional isomers 30 and 31 were observed to be inseparable (combined yields up to 88%). The methodology can be further extended to affect a relay38 α-vinylidenation/γ-alkynylation of aldehydes 29 with excess equivalents of EBXs through a subsequent second electrophilic alkynylation of the ynenamine intermediate 35 preferably at the γ-carbon atom (Scheme 7b). The obtained γ-alkynyl allenyl aldehydes (37, up to 79% yields) were rather unstable and were smoothly converted into 2-alkynyl-3-silylfurans (38, up to 93% yields) using AuCl3, thus rendering a highly efficient two-step synthesis of trisubstituted furans 38 from aldehydes 29. Further, an attempt towards developing the enantioselective version of the one-pot α-vinylidenation/γ-alkynylation cascade with a chiral primary amine, 36, led to 17% ee of the resulting allene 37.
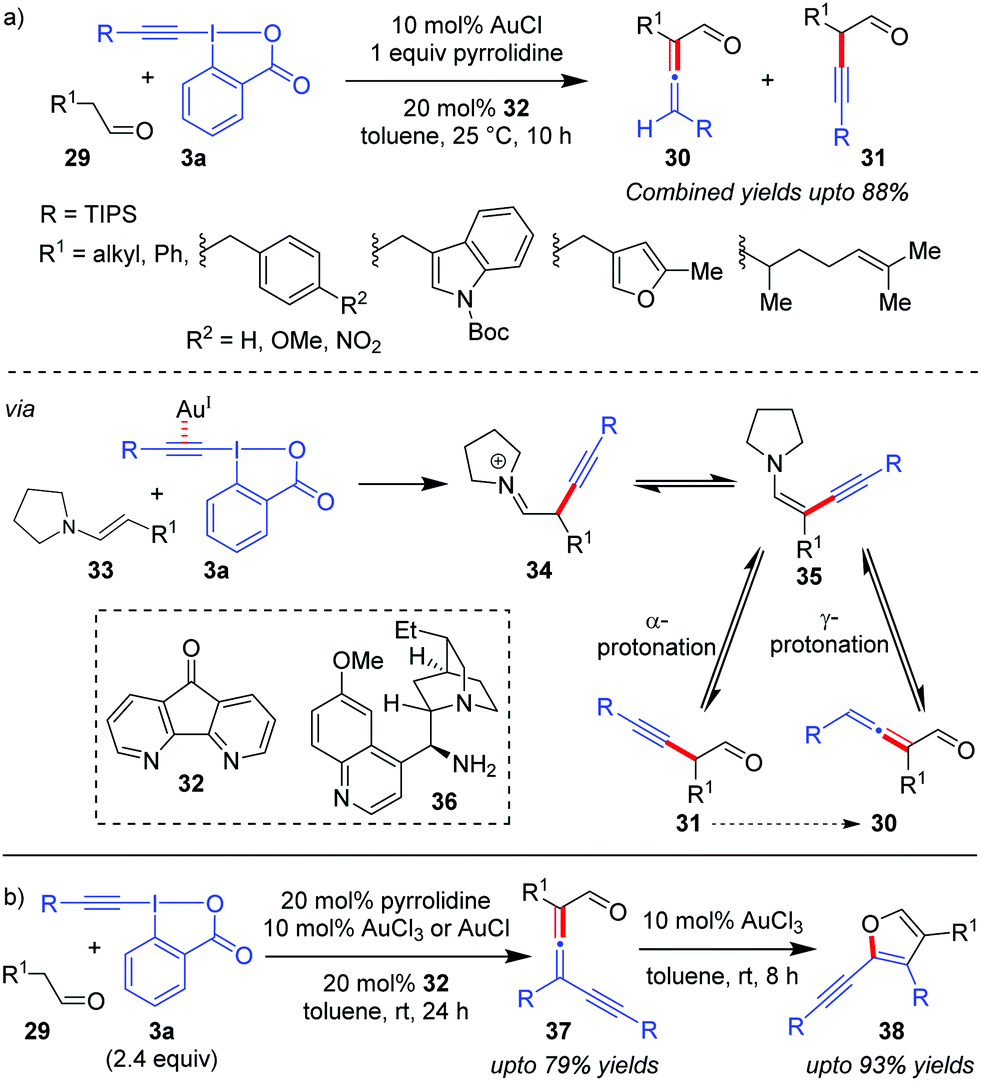 | ||
| Scheme 7 (a) α-Vinylidenation of aliphatic aldehydes using a Au/pyrrolidine catalyst system; and (b) one-pot relay of α-vinylidenation/γ-alkynylation of aliphatic aldehydes. | ||
An interesting extension to the Huang group's α-vinylidenation can be seen in their later work where ynenamine 35, generated in situ from the α-allenyl aldehyde 30 and the secondary amine pyrrolidine, can undergo a subsequent in situ oxidative cleavage of the C–C bond with aerobic molecular oxygen to furnish ynones 39 (up to 75% yields, Scheme 8).39 This became the first direct synthesis of various ynones 39 from readily available aldehydes 29. The isolation of the byproduct pyrrolidine-1-carbaldehyde 42 in good yield supports the assumption that the aerobic oxidation of 35 occurs through the 1,2-dioxetane intermediate 41. Further, since this oxidative cleavage step demands the presence of a Au-catalyst and adopts a free radical pathway as evidenced by control experiments, it is surmised that Au(III) activates molecular oxygen and facilitates single electron transfer from the electron-rich ynenamine 35 to O2 (cf.40).
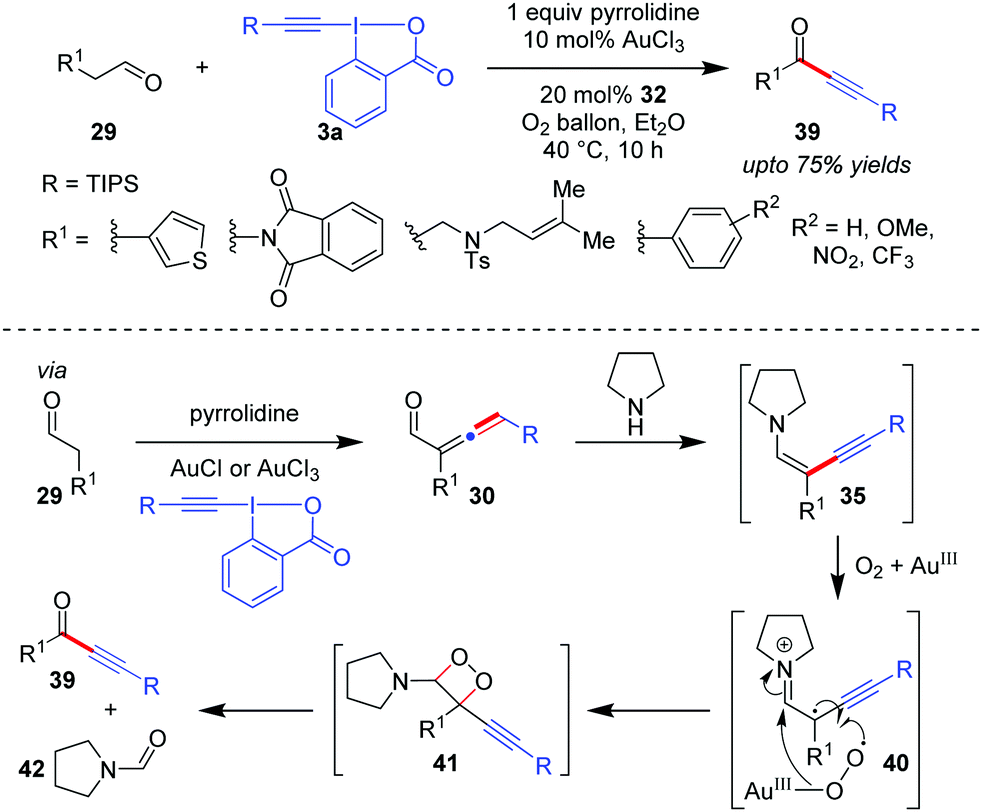 | ||
| Scheme 8 Direct access to ynones from aldehydes via oxidative C–C bond cleavage of intermediate ynenamine under aerobic conditions. | ||
Similar to the formation of ynenamine 35 from aliphatic aldehydes 29 by the action of a Au/secondary amine catalyst system, Huang's group in 2018 isolated ynedienamines 44 from ynones 43 in a near-quantitative yield (Scheme 9).40 Products 44 were subsequently involved in an asymmetrical fluorination by Selectfluor/chiral phosphoric acid (48), yielding enantioenriched ynones 47 in high yields (up to 93%), boasting an α-quaternary carbon which contains a fluorine atom. To this end, the current methodology became the first streamlined synthetic protocol for asymmetric α-difunctionalization of ynones. Herein, the secondary amine was a recyclable chiral amine auxiliary, 45, which, in combination with 48, eventually increased the enantioselectivity of the fluorination step.
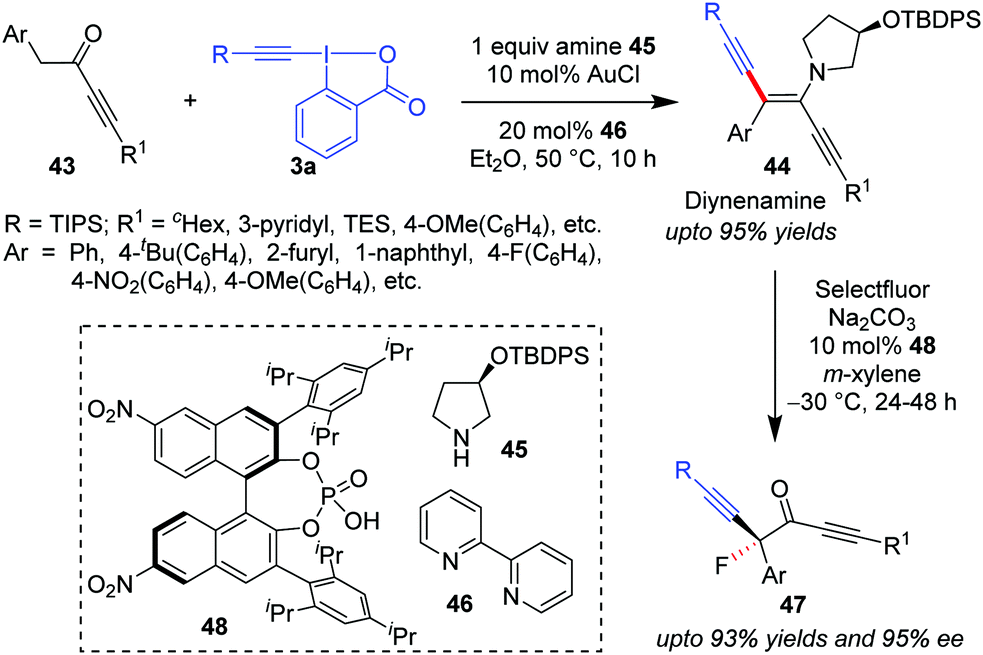 | ||
| Scheme 9 Synthetic protocol for asymmetric α-difunctionalization of ynones. | ||
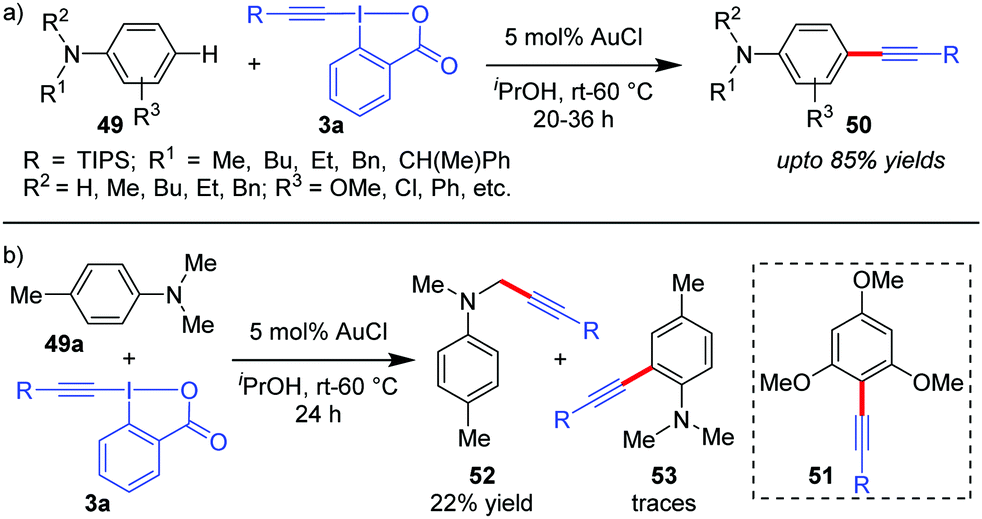
The reaction conditions for C–H alkynylation of heteroarenes were also well tolerated for electron rich arenes like anilines (49) and 1,3,5-trimethoxybenzene to obtain 50 (up to 85% yields) and 51 (50%), respectively (Scheme 10).19 The reaction was observed to be para-selective, which is complementary to the existing directing group-assisted transition metal-catalyzed ortho-alkynylation41 of anilines 49 and the mechanism is similar to cycle 1 of Scheme 2, as originally proposed by the authors. With para-substituted 4-methyl-N,N-dimethyl aniline (49a), C(sp3)-H alkynylated products (52) were obtained as the major product (22%) due to the relatively high oxidation capability of 3a with traces of ortho-alkynylated products (53).
 | ||
| Scheme 10 para-Selective direct alkynylation of anilines. | ||
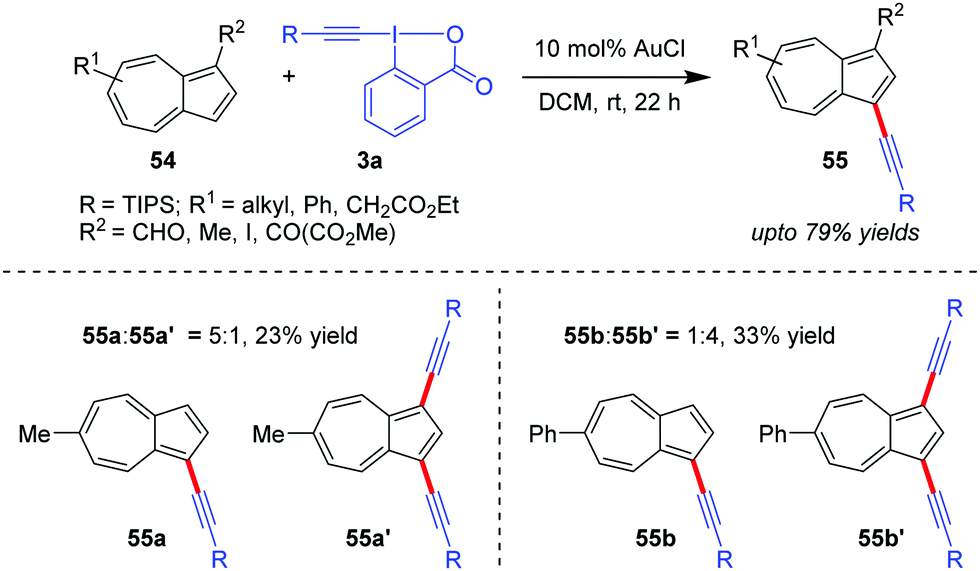
Azulenes (54), whose special [7+5] membered aromatic ring system contributes to the dipolar nature of the molecule with a high dipole moment (1.08 D) and hence increased reactivity, were efficiently alkynylated at the C3 site to obtain 55 (up to 79% yields) by the Novák group using Au-catalyzed alkynylation with EBXs (Scheme 11).42 However, in cases where R2 = H, a mixture of the corresponding mono- and dialkynylated products is formed (cf.55a:55a′ and 55b:55b′). The authors originally proposed the mechanism to be similar to cycle 1 of Scheme 2.
 | ||
| Scheme 11 Direct alkynylation of azulenes. | ||
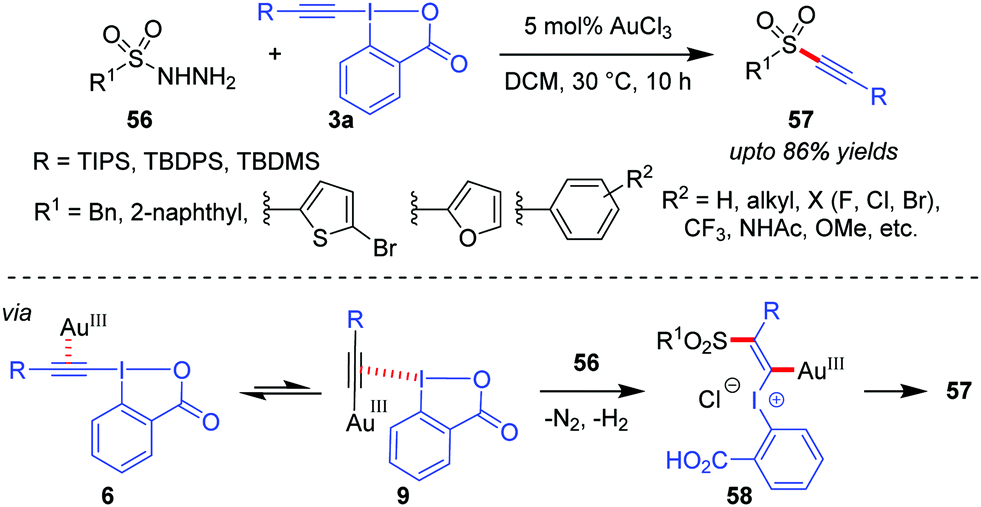
A Au(III)-catalyzed dehydrazinative C(sp)–S coupling reaction between 3a and arylsulfonyl hydrazides (56) was developed by the Patil group for the synthesis of a variety of alkynyl aryl sulfones (57, up to 86% yields, Scheme 12).43 The current reaction describes a unique way for the use of sulfonyl hydrazides as sulfur nucleophiles in Au-catalyzed reactions. Interestingly, the performance of Au(III)-catalysts in the current alkynylation reaction is better than that of Au(I), which is generally uncharacteristic of such alkynylations.
 | ||
| Scheme 12 Dehydrazinative C(sp)–S cross-coupling reactions of arylsulfonyl hydrazides. | ||
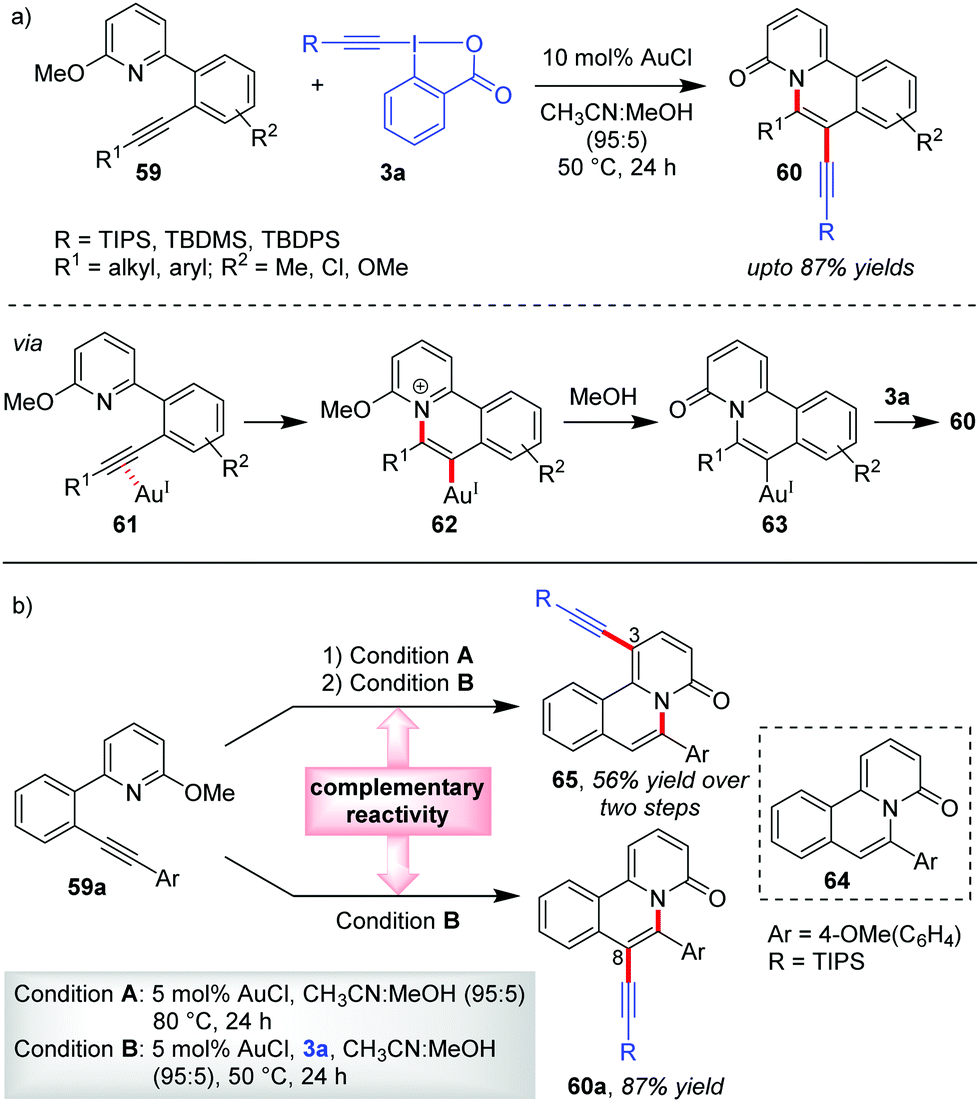
Unlike the previous cases which involve the activation of only 3a, Patil's report on the Au-catalyzed aminoalkynylation of alkynes leading to the formation of quinalizinones 60 (up to 87% yields) from pyridinoalkynes 59 involves the activation of both 3a and its coupling partner 59 (Scheme 13a).44 The vinylgold(I) complex 63 results from an intramolecular addition of a tethered nucleophile to the Au-activated π-system incorporated into 59. Thereafter, a direct nucleophilic attack of such organogold(I) intermediates 63 onto EBXs can also lead to the desired alkynylated products.
 | ||
| Scheme 13 (a) Aminoalkynylation of alkynes to access alkynylated quinalizinones; and (b) C3-alkynylation vs. C8-alkynylation: complementary reactivity of pyridinoalkynes. | ||
During control experiments, when 6-aryl-2-pyridone 64, obtained from 59a with conditions A, was subjected to the standard reaction conditions, the substrate underwent a ‘direct’ C3-alkynylation (65, Scheme 13b), analogous to the alkynylation previously developed by Patil33 and Li.34 This implies that a complementary reactivity can be developed out of these pyridinoalkynes (e.g., 59a), wherein the current domino cyclization/alkynylation approach (conditions B) would lead to C8-alkynylated product 60a (87% yield) and performing the same in a sequential manner (conditions A and then conditions B) would lead to a C3-alkynylated product, 65 (56% yield over two steps). Of note, this report constitutes the standalone example of Au-catalyzed aminoalkynylation of alkynes.
2.2 Alkynylations via the Au(I)/Au(III) catalysis mode
Gold-catalyzed C–C cross-coupling reactions using hypervalent iodine(III) reagents as sacrificial oxidants have been existing for quite some time.45 But developing Au-catalyzed, sacrificial oxidant-free C–C cross-coupling reactions24i requires a single entity OxC (67) that could serve a dual role as an oxidant and a coupling partner (Scheme 14a). EBXs (3a–c), by design, can function as OxC (67) by giving researchers the opportunity to oxidize Au(I) (cf.66) to Au(III) (cf.68) in situ as well as the concomitant transfer of an alkynyl group. This premise of EBXs (3a) serving the dual role of an oxidant and an alkyne surrogate greatly assisted the development of Au-catalyzed, alkynylative C–C cross-coupling reactions and is discussed in this section.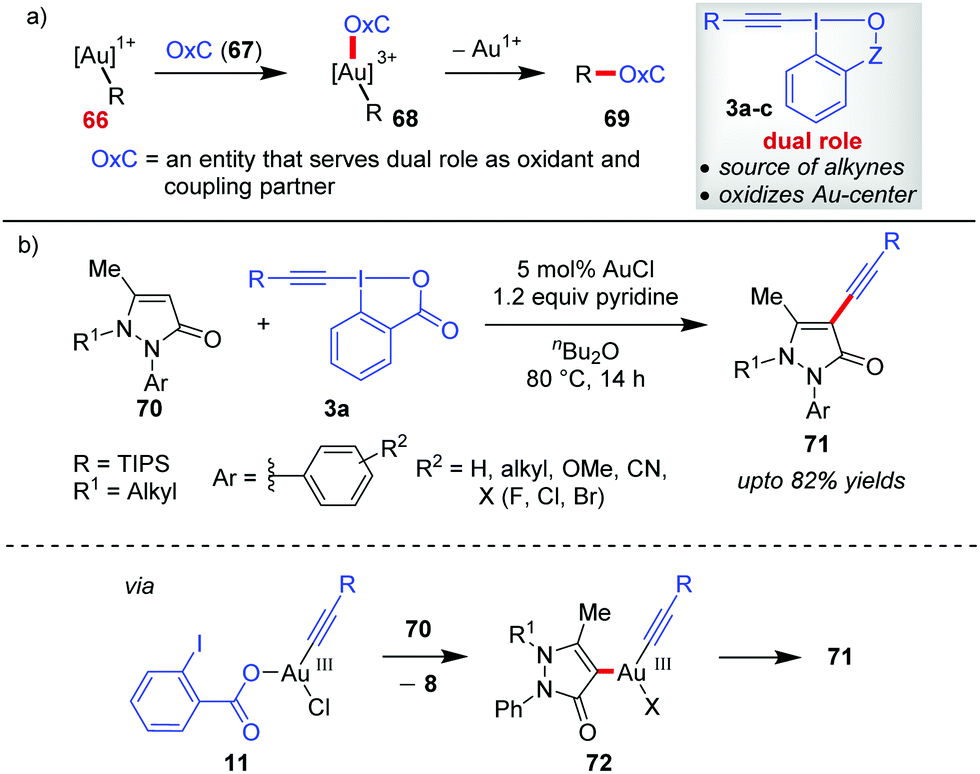 | ||
| Scheme 14 (a) New coupling partner‚ ‘Oxc’ for executing Au-catalyzed, redox-neutral C–C cross-coupling; and (b) alkynylation of N-alkyl pyrazolone at the C4-site. | ||
In continuation of the regiodivergent alkynylations as mentioned in the discussion pertaining to Scheme 5, the Xia group reported a Au(I)-catalyzed alkynylation of 2-aryl-3-pyrazolones (70) at the most electron-rich C4 site to give 71 (up to 82% yields, Scheme 14).46 On the basis of the H/D exchange at the C4 position of the pyrazolone (70), the authors proposed an alkynylative cross-coupling pathway involving a Au(I)/Au(III) catalytic cycle where the oxidative addition of Au(I) with 3a leads to the formation of an alkynylgold(III) intermediate (11), which after an heteroarene metalation (cf.72) and reductive elimination affords the final product 71 and regenerates the Au(I)-catalyst. However, the evidence in favour of the Au(I)/Au(III) catalytic cycle is apparently not concrete. As we shall see later in this Section (2.2) and Section 2.3, such a Au(I)/Au(III) catalytic cycle warrants the use of a weaker bidentate π-acceptor ligand like phen (76) as a Au(III)-stabilizing agent, which plays an important role in promoting the oxidation of the Au(I)-complex by EBXs and facilitates a rapid reductive elimination from the Au(III)-complex. Nevertheless, the role of pyridine acting as a ligand in facilitating oxidative addition to such a Au(I) complex cannot be neglected.27b
In 2017, Au-catalyzed C(sp)–C(sp) cross-coupling of terminal alkynes 73 with alkynyl hypervalent iodine(III) reagents 3 to furnish unsymmetrical 1,3-butadiynes 75 was independently developed by Liu and Patil with up to 98% and 86% yields, respectively (Scheme 15).47 The initial sacrificial oxidant-empowered, Au-catalyzed version of C(sp)–C(sp) cross-coupling of two terminal alkynes developed by the Shi group48 overcame the selectivity issues posed by conventional Pd-/Cu-/Ni-catalyzed cross-couplings (Glaser–Hay49 coupling and Cadiot–Chodkiewicz50 coupling) via the ability of Au(I) to promote the preferential formation of arylgold acetylide (77) over alkylgold acetylide (78) for electronically-biased terminal alkynes (up to 93% yields of 75, Scheme 15a). The current report is a reasonable modification to Shi's work where the use of EBXs, which can act as oxidants as well as alkyne transfer agents, not only eliminated the need for an external oxidant (e.g. PhI(OAc)2) but also helped the preferential formation of one metal acetylide exclusively over the other without the need for electronic bias among the alkynes concerned. If we draw a comparison between the two sets of reaction conditions set by the respective groups, the principal shortcoming in Liu's reaction conditions, would be the use of a Ag(I)(+) additive and sub-stoichiometric amounts of ligand 76 (0.5 equiv.) to achieve the desired yield and heteroselectivity of the reaction, which is not the case with Patil's reaction conditions (no Ag(I)(+), 0.15 equiv. of 76). Conversely, the substrate scope in Liu's work, as compared to that in Patil's, accommodates alkyl-EBXs (3b, R = alkyl) and aryl-EBXs (3b, R = aryl) as coupling partners and thus truly delivers on the promise of affording unsymmetrical 1,3-butadiynes (75) having alkynes with even similar structures or electronic properties in high yields.
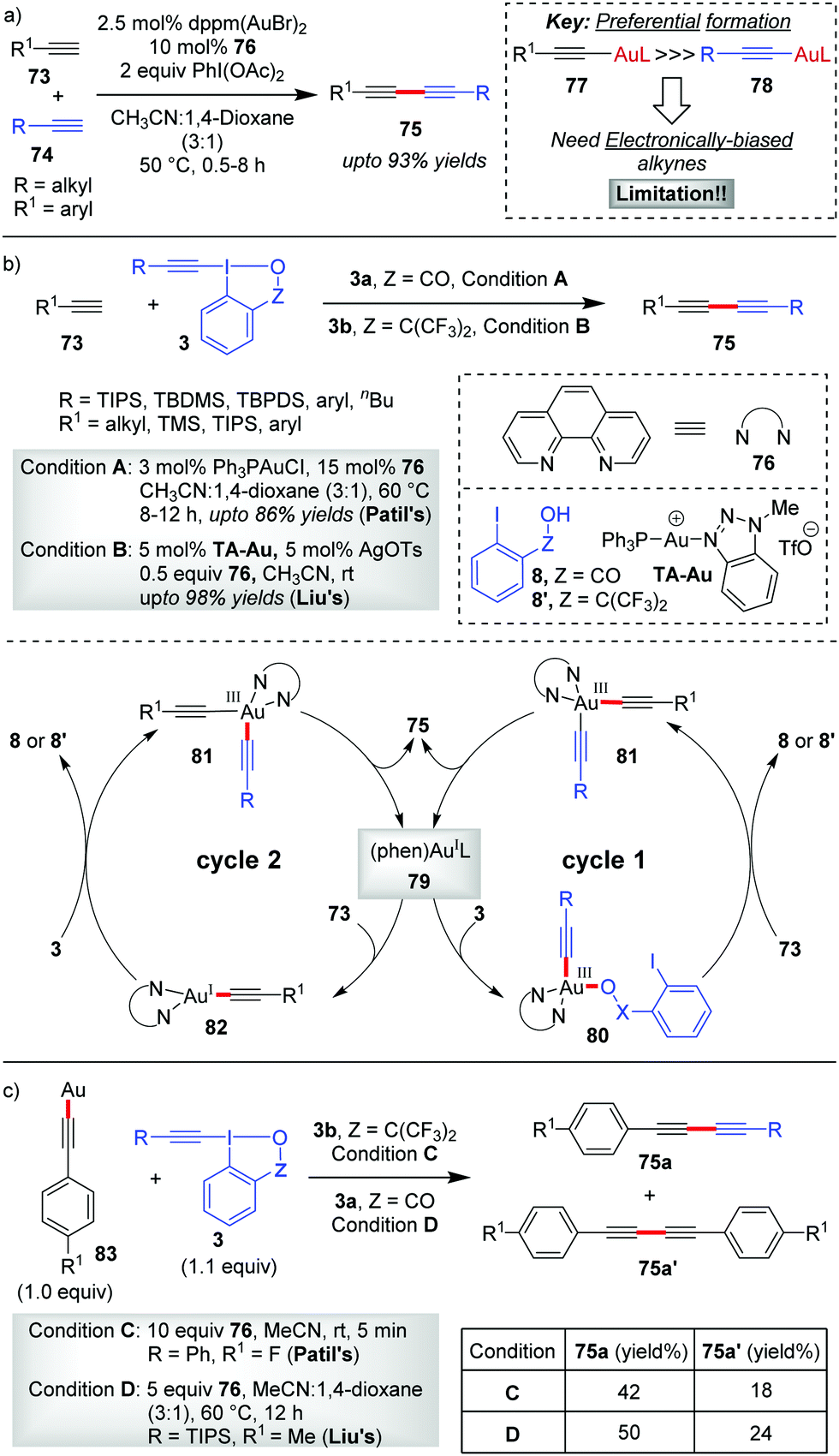 | ||
| Scheme 15 Au-Catalyzed C(sp)–C(sp) cross-coupling reactions: (a) Shi's method; (b) Liu and Patil's method; and (c) control experiments with ligand-free polymeric Au(I)-acetylide by Liu and Patil. | ||
The reaction may follow two different pathways (cycle 1 or 2) to form common intermediate 81 based on the sequence of oxidation and transmetalation. A direct oxidation of 79 to 80, followed by coordination with the terminal alkyne, leads to the dialkynylgold(III) species 81 (cycle 1). Alternatively, the oxidation could occur preferentially on phen-ligated Au(I)-acetylide 82 generated in situ, thus forming the common intermediate 81 (cycle 2). Once formed, the Au(III)-species 81 undergoes reductive elimination to give the desired cross-coupled product 75 with the regeneration of the active catalyst 79. In a control experiment, the reaction involving a stoichiometric amount of the ligand-free polymeric Au-acetylide (83), which was supposed to generate 82in situ under the standard reaction conditions, demonstrated commendable heteroselectivity (Scheme 15c). Apparently, it is suggested that the intermediacy of a phen-ligated acetylide (82) is highly likely and even the mechanistic pathway depicted in cycle 2 might be a favoured pathway over that of cycle 1.
However, new mechanistic insights from Hashmi's group27b provided strong experimental evidence in favour of oxidative addition of 3 to the tri-coordinate Au(I)-species 79 to generate 80, which essentially supports cycle 1.
The Hashmi group in 2019 reported the direct alkynylation of cyclopropenes 84via Au/Ag cooperative-catalysis,38 affording alkynyl-substituted cyclopropenes (85, up to 92% yields, Scheme 16).27a The involvement of (phen)Au(I)(+) (86) as the catalytically active species for all alkynylative cross-couplings with EBXs was widely speculated before. Using 94 as a pre-catalyst for 86, when the corresponding reaction rate was compared to that of other precatalysts (PPh3AuNTf2 and AuCl), it revealed a higher initial reaction rate in case of 94 which further confirmed the role of 86 as the active catalyst for the reaction. It is formed as a result of ligand exchange (between PPh3AuNTf2 and 76) at the beginning of the alkynylation reaction, which typically takes 20 minutes as per the observation of induction time for the alkynylation reaction. This ligand exchange process is notably similar to Liu and Patil's case as discussed in Scheme 15. Further, the essential role of Ag(I)(+) is manifested in the C–H metalation of the cyclopropenes 84 rather than being a halogen scavenger as judged by H/D exchange experiments with 84. The available mechanistic data indicate an oxidative catalytic cycle involving an alkynylgold(III) complex, 88, formed by the oxidative addition of the hypervalent iodine(III) reagent 3b to (phen)Au(I)(+) (86). In fact, the putative alkynylgold(III) intermediate 88 was stoichiometrically generated in situ (cf.88a), characterized and engaged in a successful alkynylation of cyclopropene 84a, which further validated its intermediacy (Scheme 16b). After the formation of 88 in the mechanistic cycle, the alkoxy anion was then transferred from Au(III) to Ag(I) (cf.92), followed by C–H metalation to afford the cyclopropenylsilver 93. Next, a transmetalation occurred between Ag(I) species 93 and Au(III)-species 89 to obtain another Au(III)-species, 90, followed by reductive elimination to form the alkynylated cyclopropene 85 with the regeneration of (phen)AuL 86. Recent mechanistic studies by the same group depicted a clearer picture of the formation of 88 which now consists of two steps of oxidative additions and one step of reductive elimination (Scheme 16c).27b The true identity of 86 is also revealed as (phen)Ph3PAuNTf2 (96), wherein the two Au–N bonds are unsymmetric. The first oxidative addition to the unsymmetric tri-coordinate Au(I)-species 96 proceeds with alkynyl hypervalent iodine 3b through a tetra-coordinate Au(I)-transition state 97 (TS), affording 98. After that, carbon–phosphorus reductive elimination of Au(III)-species (98) gives the trigonal Au(I)-complex (100) along with 99, which has been observed experimentally. Finally, the Au(III)-compound 88a forms after a second oxidative addition of 3b to 100.
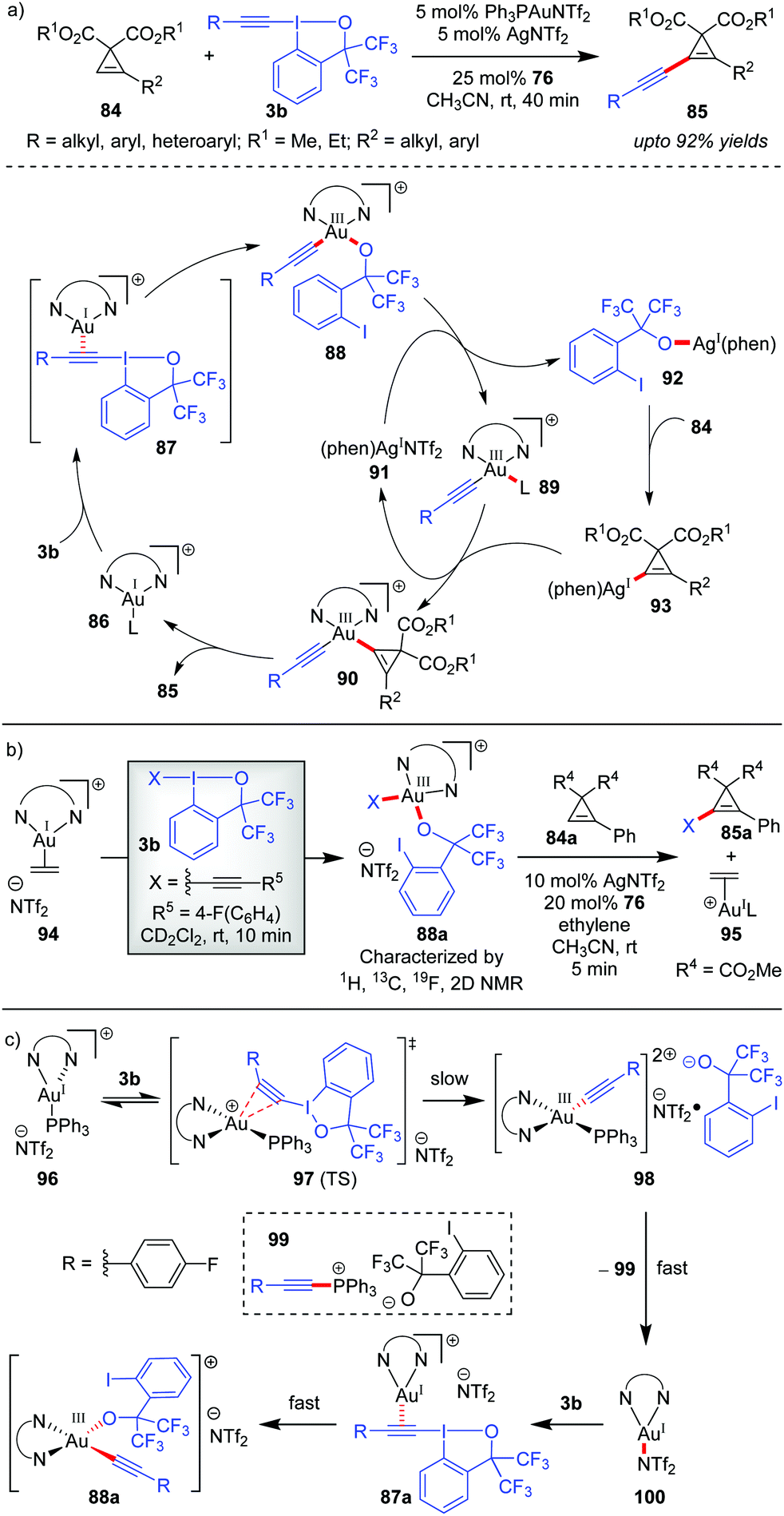 | ||
| Scheme 16 (a) Direct alkynylation of cyclopropenes via Au/Ag cooperative-catalysis; (b) control experiment to prove (phen)Au(I)(+) as the active catalyst and the intermediacy of Au(III); and (c) new mechanistic insights into the formation of 88. | ||
2.3 Alkynylations via ‘interplay’ mode
According to our rationale B (cf. Section 2), there exists a third mode of reactivity of Au-catalysts in alkynylation reactions with EBXs which involves the interplay of both the π-activation mode and Au(I)/Au(III) catalysis mode. The first instance of this ‘interplay’ mode was observed in the Waser group's report in 2013 wherein they established a domino cyclization/alkynylation approach to C3-alkynylated furans 104 (up to 97% yields) from allenic ketones 102 and EBXs 3b in the presence of a Au(III)-catalyst, 105 (Scheme 17).20 This is indeed complementary to the C2-alkynylated furans 103 (up to 90% yields), obtained via direct C–H alkynylation of furans (101) with 3a and AuCl. Interestingly, they noticed that AuCl did not promote the former reaction at all, which is atypical of such alkynylation reactions. As the reaction mechanism was not understood yet, it was very difficult to rationalize the differences between the two catalytic processes (C3- vs. C2-alkynylation). In the new mechanistic insights provided by the Ariafard group,26 it was so disclosed that the used Au(III)-catalyst 105 is a precursor of an active Au(I)-catalyst, 107, which activates allene 102 towards 5-exo-dig cyclization. Thereafter, deprotonation gives the organogold(I) intermediate 109 which after oxidative addition of 3b offers 111via transition state 110. Here, the DFT calculations further predict that the picolinic acid ligand (106) is responsible for stabilizing the Au(III)-centre and thus essentially lowering the energy of the transition state 110. The resulting Au(III)-complex 111 then undergoes reductive elimination to give C3-alkynylated furans 104 and regenerates the active Au(I)-catalyst 107. In contrast, the C2-alkynylation leading to 103 proceeds via the π-activation mode only (similar to cycle 1, Scheme 2), as originally proposed by the authors. The complementary reactivity depicted here can be correlated with that of Scheme 13b, highlighting the unique alliance of Au-catalysis and EBXs in electrophilic alkynylation reactions.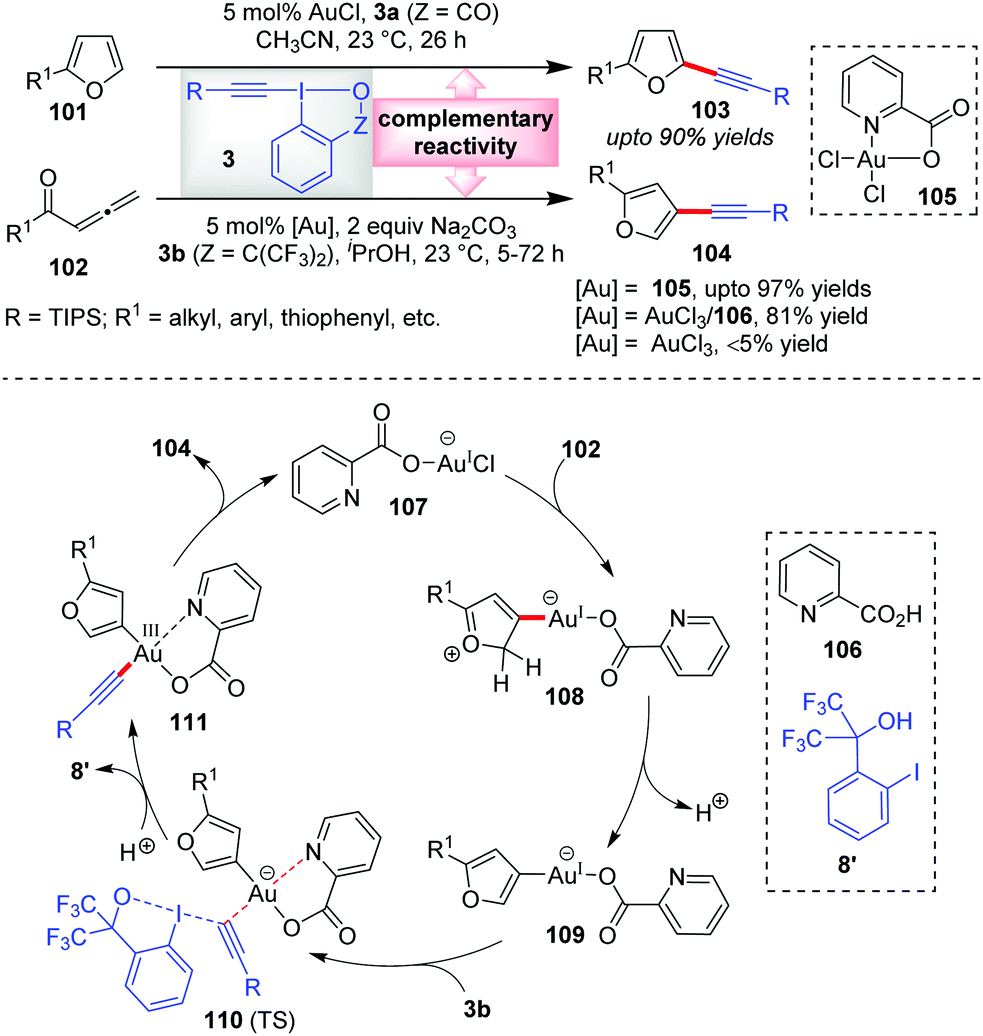 | ||
| Scheme 17 π-Activation mode and interplay mode of Au-catalysis giving rise to regioselective approaches towards C2- and C3-alkynyl furans, respectively. | ||
Of note, control experiments regarding alternate catalyst systems reveal that the AuCl3/106 catalyst system results in 81% yield, whereas bare AuCl3 gives less than 5% yield. This is similar to the observation reported by Huang's group in their α-vinylidation (cf.Scheme 7) that the performance of AuCl3 is significantly dependent on the presence of bidentate ligand 32 (31% vs. 91% yield). Similar is the case with AuCl (36% vs. 93% yield). The latter outcome may be rationalized in the way that 32 serves as a Au(III)-stabilizing agent during the oxidative addition of 3a to Au(I), lowering the energy of the key transition state for oxidative addition.27,28 In the case of the former outcome with AuCl3, it is highly possible that the Au(III)-complex, which forms in situ in coordination with ligand 32, is in fact a precursor of an active Au(I)-catalyst, in line with Ariafard's proposal,26 which requires the support of ligand 32 for oxidative addition. This leaves a reasonable speculation that the α-vinylidation of aldehydes might proceed via a Au(I)/Au(III) catalysis mode as opposed to the π-activation mode proposed originally by Huang's group.
In another domino cyclization/alkynylation approach, where such interplay of activation modes is seen would be the highly stereoselective synthesis of (E)-alkynyloxazolines (113, up to 67% yields) by N-propargylcarboxamides 112 and benziodoxole reagents 3b (Scheme 18).51 Mechanistically, the carbonyl oxygen atom attacks the alkyne, which is π-coordinated to Au(I), in a 5-exo-dig fashion to form vinylgold(I) intermediate 115. Next, the oxidative addition of intermediate 115 to 3b affords intermediate 116, which then undergoes ligand dissociation and reductive elimination to give the final product (E)-113 with the regeneration of the active Au(I)-catalyst. The authors suggested that the oxidative addition step is facilitated because intermediate 115 is a negatively charged ate-complex of Au(I). Further, a comparison of the computed energies of the (Z)- and (E)-isomers of product 113 suggests kinetic control as the cause of the observed selectivity. Additionally, the acetic acid additive accelerates the catalytic cycle by protonating the alkoxide in complex 116, which assists the formation of 8′. Notably, there are three such Au-catalyzed cyclization/alkynylation cascades with EBXs reported so far (cf.Schemes 13, 17 and 18) and only Waser's work in Scheme 17 has received proper mechanistic investigation.
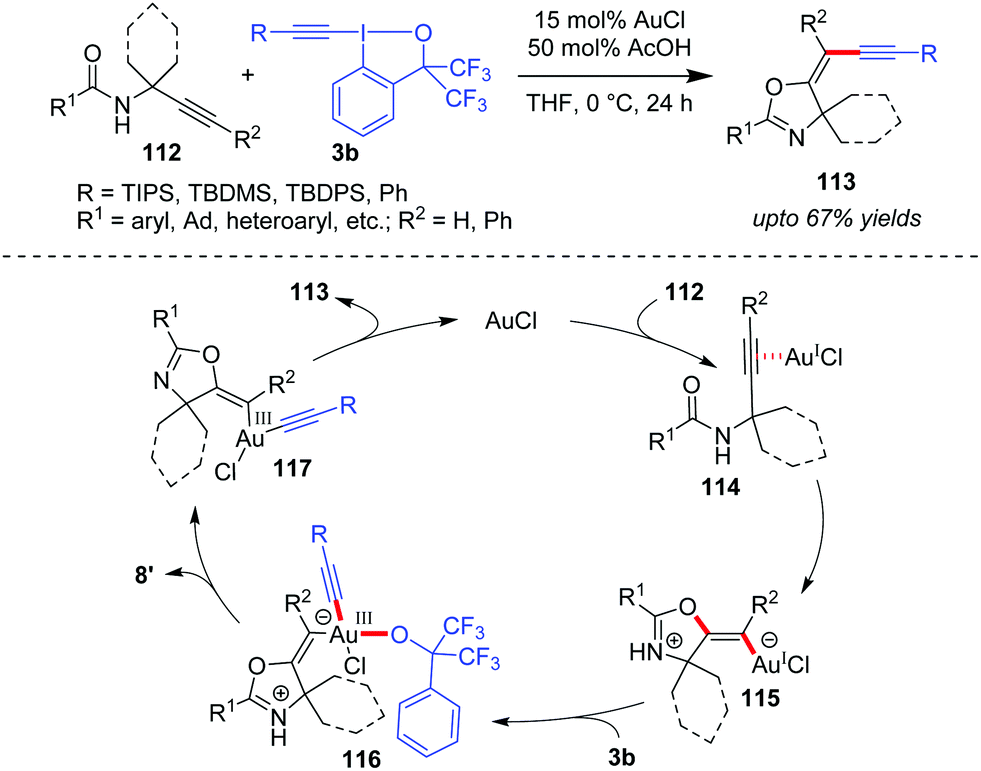 | ||
| Scheme 18 Stereoselective domino cyclization/alkynylation of N-propargylcarboxamides. | ||
Despite their amazing forte of alkyne transfer, EBXs suffer a drawback in terms of the reactivity portfolio since 2-iodobenzoic acid (8) is generated as a stoichiometric byproduct. By introducing Cu-catalyzed oxyalkynylations of diazo compounds with 3a,52 the Waser group resolved this issue by putting 8, the nucleophilic carboxylate part of EBXs, to appropriate use and became one of the pioneers in developing atom-economical53 use of 3a. Along similar lines, the Patil group recently disclosed the first Au-catalyzed 1,2-oxyalkynylation of N-allenamides (118) with appropriate atom-economical use of 3a, offering direct access to 1,3-enynes 119 (up to 83% yields, Scheme 19).28 Mechanistically, AuCl undergoes oxidative addition to EBXs (3a) to give the key tetra-coordinated Au(III)-species 120, which is stabilized with phen (76) and ion-paired with aryl carboxylate ligand 8. As an explanation of how the reaction takes place without a Cl(−) scavenger which is supposed to vacate a coordinating site of Au(III),22a,24l,m the authors surmised that the labile nature of bidentate ligand phen (76) allows the cationic Au(III) center of 120 to accommodate N-allenamide (118) in its coordination sphere, leading to 121. Thereafter, intermediate 121 upon reductive elimination affords species 122 and regenerates AuCl. Species 122 further undergoes conjugate attack by the laid-over carboxylate ligand 8 to afford the oxyalkynylated product 119. Both intermediates 120 and 122 are evidenced by tandem mass spectrometry analysis, which unequivocally supports a Au(I)/Au(III) catalysis mode.
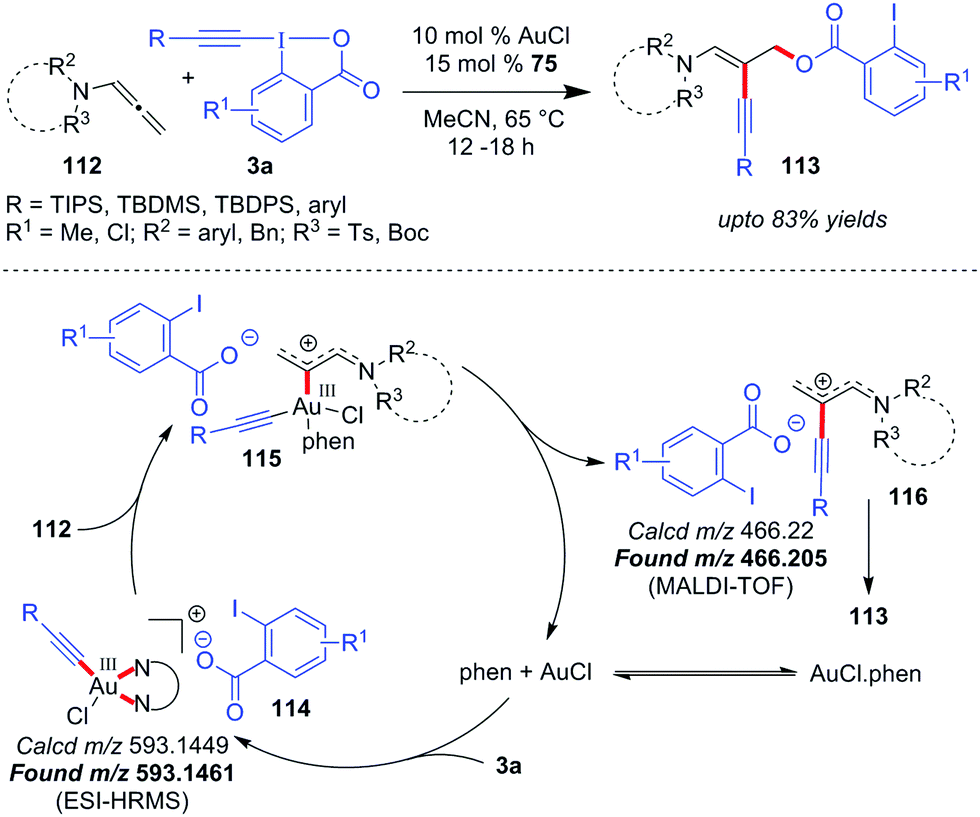 | ||
| Scheme 19 1,2-Oxyalkynylation of N-allenamides. | ||
Since the formation of allyl cationic Au(III)-species 121 requires the carbophilic activation of N-allenamide 118 by species 120, this report constitutes another example of ‘interplay’ of activation modes for Au-catalyzed alkynylations. The detection of species 122 attests to the fact that reductive elimination precedes the conjugate nucleophilic attack of carboxylate anion 8 to ultimately provide the desired product. It is also interesting to notice that performing the title reaction without ligand phen (76) leaves no desirable outcome which, to some extent, resonates with the observations made by Huang37 and Hashmi.27a
3. Gold-catalyzed acetoxylations and arylations with hypervalent iodine(III) reagents
Direct acetoxylation of arenes with transition metal catalysis, without employing the ‘directing group’ strategy, can become a challenging task because of regioselectivity issues.41b Based on the well-established fact that Au(III) can also electrophilically metalate an aromatic C–H bond to generate arylgold(III) species,54 the Wang group was interested in investigating whether Au-catalyzed direct C–H functionalization could become an alternative approach towards maintaining the regioselectivity, high efficiency and generality of the reaction. Towards this end, the Wang group developed a AuCl3-catalyzed direct acetoxylation of electron-rich aromatic compounds (123) with iodobenzene diacetate (1a) as the acetoxylation reagent to obtain 124 in high yields (up to 82%, Scheme 20).55 In this case, the overacetoxylation problem (leading to di- and triacetoxylation products) has been overcome by employing an excess amount (2.5 equiv.) of aromatic substrates. In the presence of a strong electron-withdrawing group –CN or –CO2Me as a substituent, a slightly higher catalyst loading (5%) was required. The reaction also demonstrated commendable regioselectivity, which is evident from the fact that 123a (R1 = OMe) underwent para-acetoxylation only and in the case of other substituted anisole substrates, the methoxy substituent dictates the regioselectivity of the acetoxylation.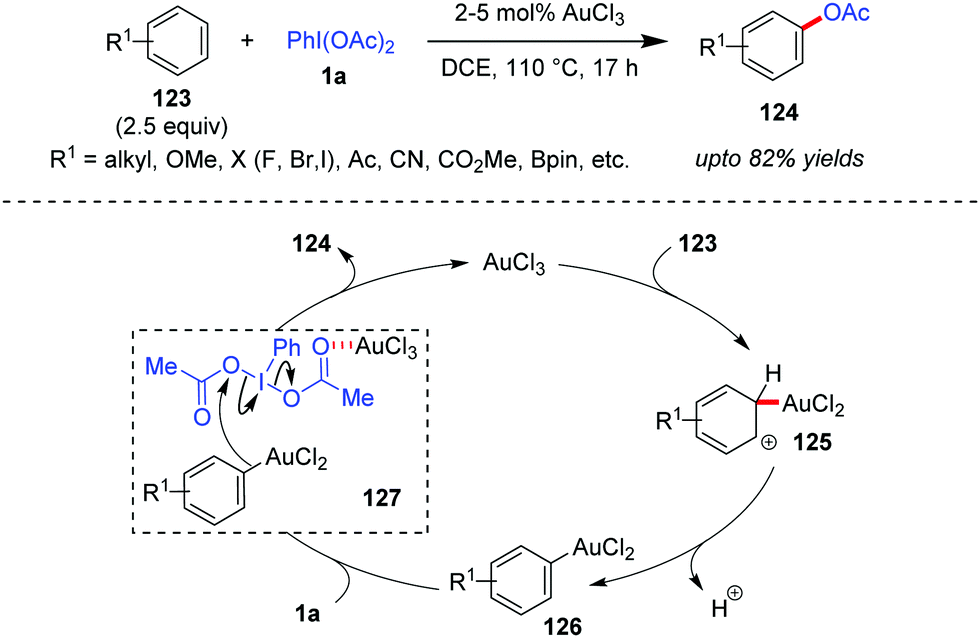 | ||
| Scheme 20 Direct acetoxylation of arenes. | ||
Mechanistically, it has been proposed that the arylgold(III) species 126 forms after a C–H auration/deprotonation sequence (cf.125) from 123. Thereafter, species 126 attacks the Au-activated iodobenzene diacetate (cf.127) to afford the acetoxylation product (124). Here the high catalytic efficiency of AuCl3 over other Lewis acid or protonic acid catalysts can be explained in terms of its dual activation mode of direct auration of 123 as well as the activation of 1a through complexation. Kinetic isotope effect studies show the formation of the Wheland intermediate 125 to be the rate-limiting step. An alternative mechanistic possibility in which an intermediate iodonium salt of the electron-rich aromatic substrate (123) could form and undergo a subsequent Au-mediated process leading to the final product (124) could not be ruled out by the Wang group.
The suitability of diaryliodonium salts (2a) as aryl sources is well-established in Pd- or Cu-catalyzed cross-coupling reactions.5b,56 In this context, the Glorius group described the first use of 2a as a coupling partner with a Au/photocatalyst system in the intermolecular multicomponent oxyarylation of simple, non-activated alkenes (128) with alcohol (129) under blue LEDs, leading to the desired arylated ether product 130 at room temperature (up to 91% yields, Scheme 21).57 The stability and ready accessibility of diaryliodonium salts (2a) over aryldiazonium salts makes them viable, alternative aryl-coupling partners in cross-coupling reactions using photoredox strategies.58 Mechanistically, coordination of the alkene (128) to the cationic Au(I)-catalyst, followed by intermolecular nucleophilic attack of the alcohol (129), affords an alkylgold(I) complex of type 131. The aryl radical generated through SET reduction of a diaryliodonium salt (2a) by Ir(III)* adds to complex 131 to furnish a Au(II)-species of type 132, which is further oxidized by Ir(IV) to generate a Au(III)-complex of type 133, with the completion of the photocatalytic cycle. Finally, the Au(III)-complex 133 undergoes reductive elimination to yield the product (130) with regeneration of the Au(I)-catalyst. The use of Ir(III) as a photocatalyst over Ru(II) is preferred because excited state Ir(III)* has a higher reduction potential than excited state Ru(II).
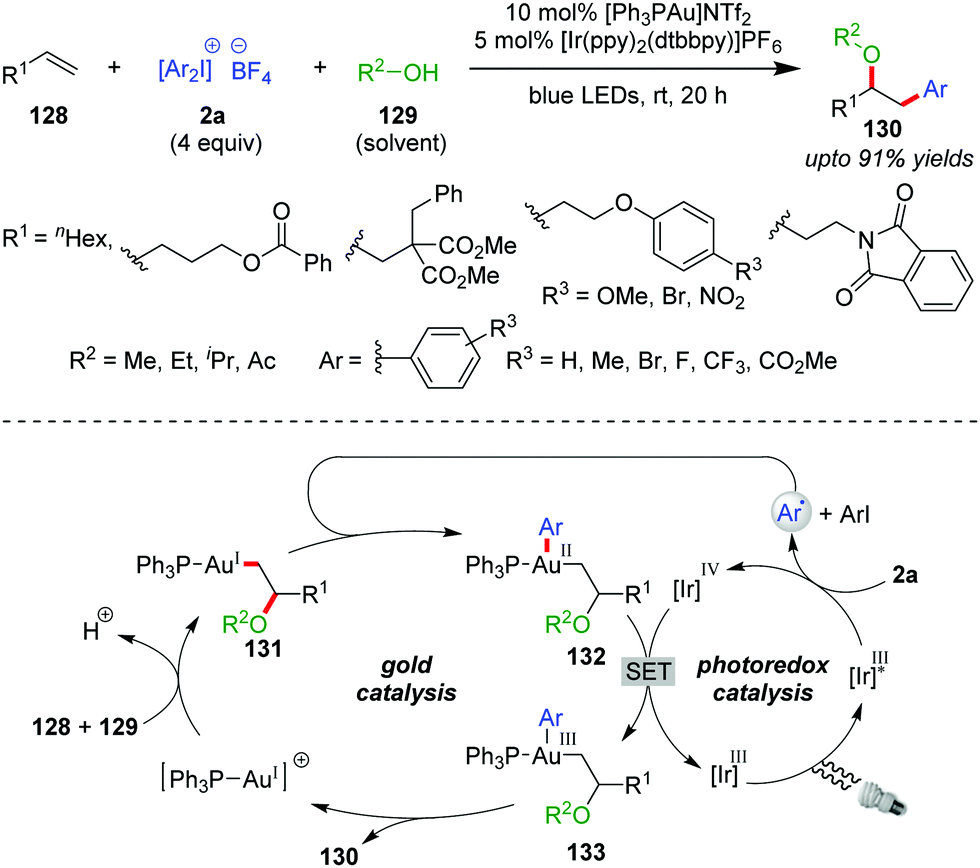 | ||
| Scheme 21 Multicomponent oxyarylation of non-activated alkenes. | ||
A single-step chemoselective arylsulfonylation of boronic acids (134) by potassium metabisulfite (K2S2O5, 135) and diaryliodonium salts (2a) to access substituted diarylsulfones (136) was accomplished by the Tu group under (NHC)Au(I)-catalysis (137) (up to 98% yields, Scheme 22).59 Even in the case of unsymmetrical diaryliodonium salts, the fact that a more electron-rich and bulky sulphone (containing Ar3) is formed over the less electron-rich and less bulky one (containing Ar2) showcases the excellent chemoselectivity of the reaction. Mechanistically, this is explained on the basis of the stability of the intermediate (141) formed in situ from bulky and electron-rich aryl groups (Ar3) over that of less bulky and electron-poor aryl groups (Ar2). Meanwhile, sulfur dioxide, generated in situ by heating K2S2O5, is inserted into the Au–CAr bond of intermediate 138, which has resulted from an initial transmetalation of 137 with aryl boronic acid (134). Consequently, sterically congested sulfonyl Au(I)-complex 139 produces the key sulfinate intermediate 140 and regenerates the NHC–Au(I)-species 137 under basic conditions. The crucial sulphinate intermediate 140 can react with the more stable, bulky and electron-rich 141 formed from diaryliodonium salts (2a) and thus afford the diarylsulfones (136). The current selectivity is in direct contrast to Pd-catalyzed C–CAr bond formation reactions, in which the less bulky aryl groups are usually transferred.60
 | ||
| Scheme 22 Arylsulfonylation of boronic acids. | ||
4. Conclusions and future outlook
From just being a passing infatuation for chemists for their unique structural characteristics to becoming well-established reagents in mainstream synthetic chemistry ensuring delivery of functional groups with reversed polarity, hypervalent iodine(III) reagents have come a long way. The exceptional reactivity of hypervalent iodine(III) reagents, in combination with the extra stability and modularity of the cyclic system, is primarily behind their popularity amongst the scientific community. If we closely scrutinize the development of such functional group transfer reactions with hypervalent iodine(III) reagents, especially alkynylations with EBXs, we are bound to find an undeniable correlation with the development of Au-catalysis. With their two different modes of activation (i.e. π-activation mode or Au(I)/Au(III) catalysis mode), which can also interplay under certain conditions (‘interplay mode’), Au-catalysts have formed a strong liaison with EBXs to achieve a wide array of alkynylations. Mechanistically, it is now understood that in the presence of a stabilizing agent in the reaction, which is often bidentate N-coordinating ligands like phen (76) or picolinic acid (106), the alkynylation reaction is more likely to proceed via Au(I)/Au(III) catalysis. Despite this, in some reports where a mechanistic ambiguity still exists as described by the authors, the intervention of computational chemists to gain proper insights into the mechanism is necessary. Personally, we believe that the mechanism for gold-catalyzed alkynylations with EBXs would vary depending on the reaction.It is, however, true that the number of gold-catalysts that are well-established with hypervalent iodine(III) reagents concerning functional group transfer reactions is limited. In such cases, tuning the reactivity of hypervalent iodine(III) reagents via modulation of the ‘ligand-sphere’ around the iodine atom can be one of the solutions (e.g.3avs.3b) which was elegantly utilized by Waser (Scheme 17). In fact, many variants of such cyclic λ3-alkynyliodanes have been reported so far with ‘tuned’ reactivity7b,52c and are waiting to be screened with gold-catalysts. Furthermore, enantioselective electrophilic alkynylations with EBXs under Au-catalysis would be a much-anticipated accomplishment in the near future. In particular, conducting the alkynylation of alkenes/allenes and the C(sp3)–H alkynylations of unactivated systems in regio-, diastereo- and enantioselective manners would be particularly desirable for future applications of EBXs.61 One way to achieve this is to incorporate a suitable combination of a Au(I)/chiral hypervalent iodine(III)62 system in the transformation. Alternatively, since hypervalent iodine(III) reagents can oxidize Au(I) to Au(III) in situ and chiral Au(III)-complexes63 can effect better chiral induction than Au(I)-complexes, the chiral Au(I)/hypervalent iodine(III) system can also be explored along this line. On the other hand, while the report on the progress of Au-catalyzed electrophilic alkynylation over the past decade is impressive and noteworthy, the available literature on other such Au-catalyzed functional group transfer processes such as arylation56 and trifluoromethylation6b is still trailing behind. In particular, in arylations under gold/photoredox catalysis, the potential of diaryliodonium salts (2a) as alternative aryl-sources hasn’t been exploited much. Nevertheless, as a consequence of the strong alliance between the π-activation capability of Au-catalysts and the dual-functioning capability of EBXs as alkyne coupling partners as well as oxidants that can effortlessly overcome the high oxidation potential of the Au(I)/Au(III) redox couple,64 the Au-catalyzed cross-coupling reactions are currently undergoing a paradigm shift in terms of generating new reactivities and selectivities under redox-neutral conditions. Therefore, the authors strongly believe that a paradigm shift awaits us where Au-catalysts, with their different modes of activation as discussed in the current article, are going to make remarkable contribution to promoting new reactivities (e.g. Au-carbenes with cyclic λ3-alkynyliodanes), which would further underscore the liaison of Au-catalysts with hypervalent iodine(III) reagents in electrophilic functional group transfer reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support by the Science and Engineering Research Board (SERB), New Delhi (File No. EMR/2016/007177 and DIA/2018/000016), is gratefully acknowledged for related research in our laboratory. SB and VWB thank UGC and CSIR, respectively, for providing Senior Research Fellowships (SRF).Notes and references
- (a) Iodine Chemistry and Applications, ed. T. Kaiho, John Wiley & Sons, Inc., New York, 2015 Search PubMed; (b) V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Application of Polyvalent Iodine Compounds, John Wiley & Sons Ltd., New York, 2014 Search PubMed; (c) M. Ochiai, in Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis, ed. T. Wirth, Springer, New York, 2003 Search PubMed; (d) Chemistry of Hypervalent Compounds, ed. K. Y. Akiba, Wiley-VCH, New York, 1999. Selected reviews on the application of hypervalent iodine(III) reagents Search PubMed; (e) X. Wang and A. Studer, Acc. Chem. Res., 2017, 50, 1712 CrossRef CAS PubMed; (f) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed; (g) L. Wang and J. Liu, Eur. J. Org. Chem., 2016, 1813 CrossRef CAS; (h) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed; (i) T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656 CrossRef CAS PubMed; (j) V. V. Zhdankin, Curr. Org. Synth., 2005, 2, 121 CrossRef CAS; (k) V. V. Zhdankin, Rev. Heteroat. Chem., 1997, 17, 133 CAS.
- Although the concept of hypervalency of polycoordinated main-group elements like iodine has been the subject of controversy, it has been widely accepted by synthetic chemists to describe the structural features and special reactivity of such elements. See: R. J. Gillespie and B. Silvi, Chem. Rev., 2002, 233–234, 53 CAS.
- J. P. Brand, D. F. González, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102 RSC.
- (a) F. M. Beringer and S. A. Galton, J. Org. Chem., 1965, 30, 1930 CrossRef CAS; (b) F. M. Beringer, P. S. Forgione and M. D. Yudis, Tetrahedron, 1960, 8, 49 CrossRef CAS.
- (a) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed.
- (a) D. P. Hari, S. Nicolai and J. Waser, Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd, Chichester, UK, 2018, DOI:10.1002/9780470682531.pat0951; (b) D. Katayev, B. Jelier and A. Togni, Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd, Chichester, UK, 2018, DOI:10.1002/9780470682531.pat0949.
- For general reviews on EBXs, see: (a) D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212 CrossRef CAS PubMed; (b) J. Waser, Synlett, 2016, 2761 CrossRef CAS; (c) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS PubMed; (d) J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2015, 54, 8876 CrossRef CAS PubMed.
- M. Ochiai, Y. Masaki and M. Shiro, J. Org. Chem, 1991, 56, 5511 CrossRef CAS.
- R. L. Amey and J. C. Martin, J. Org. Chem., 1979, 44, 1779 CrossRef CAS.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed.
- J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed.
- J. P. Brand, C. Chevalley, R. Scopelliti and J. Waser, Chem. – Eur. J., 2012, 18, 5655 CrossRef CAS PubMed.
- G. L. Tolnai, Z. Gonda and Z. Novák, Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd., Chichester, UK, 2018, DOI:10.1002/9780470682531.pat0961.
- (a) Gold Catalysis: An Homogeneous Approach (Catalytic Science Series), ed. F. D. Toste and M. Veronique, Imperial College Press, Singapore, 2014, vol. 13 Search PubMed; (b) Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and F. D. Toste, Wiley-VCH, 2012 Search PubMed.
- For conceptual reviews on the superiority of gold-catalysis, see: (a) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed; (b) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed . For general reviews on gold-catalysis, see: ; (c) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (d) N. T. Patil, Curr. Sci., 2013, 104, 1671 CAS; (e) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (f) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC; (g) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC; (h) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536 RSC; (i) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (j) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed; (k) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (l) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS PubMed; (m) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed; (n) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed.
- The idea was greatly inspired by the pioneering work of Sanford and Gaunt on the arylation of indoles with aryliodonium salts. (a) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS PubMed; (b) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972 CrossRef CAS PubMed . During this investigation, palladium catalysts gave only traces of C3 alkynylated indoles, albeit with very high C2 selectivity, see: ; (c) G. L. Tolnai, S. Ganss, J. P. Brand and J. Waser, Org. Lett., 2013, 15, 112 CrossRef CAS PubMed.
- J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 7304 CrossRef CAS PubMed.
- J. P. Brand and J. Waser, Org. Lett., 2012, 14, 744 CrossRef CAS PubMed.
- Y. Li, J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 6743 CrossRef CAS PubMed.
- Y. Li and J. Waser, Beilstein J. Org. Chem., 2013, 9, 1763 CrossRef PubMed.
- Recent reports on gold catalysis from our lab: (a) M. O. Akram, A. Das, I. Chakrabarty and N. T. Patil, Org. Lett., 2019, 21, 8101 CrossRef CAS PubMed; (b) C. C. Chintawar, M. V. Mane, A. G. Tathe, S. Biswas and N. T. Patil, Org. Lett., 2019, 21, 7109 CrossRef CAS PubMed; (c) R. D. Mule, A. C. Shaikh, A. B. Gade and N. T. Patil, Chem. Commun., 2018, 54, 11909 RSC; (d) I. Chakrabarty, M. O. Akram, S. Biswas and N. T. Patil, Chem. Commun., 2018, 54, 7223 RSC; (e) A. C. Shaikh, D. S. Ranade, P. R. Rajamohanan, P. P. Kulkarni and N. T. Patil, Angew. Chem., Int. Ed., 2017, 56, 757 CrossRef CAS PubMed; (f) M. O. Akram, P. S. Mali and N. T. Patil, Org. Lett., 2017, 19, 3075 CrossRef CAS PubMed; (g) P. N. Bagle, M. V. Mane, K. Vanka, D. R. Shinde, S. R. Shaikh, R. G. Gonnade and N. T. Patil, Chem. Commun., 2016, 52, 14462 RSC; (h) A. B. Gade and N. T. Patil, Org. Lett., 2016, 18, 1844 CrossRef CAS PubMed.
- For an overview on gold catalysis in cross-coupling reactions, see: (a) A. Nijamudheen and A. Datta, Chem. – Eur. J., 2020, 26, 1442 CrossRef CAS PubMed; (b) M. Livendahl and A. M. Echavarren, Chim. Oggi, 2012, 30, 19 CAS.
- For reviews on sacrificial oxidant-aided oxidative gold catalysis, see: (a) K. Chen and S. Zhu, Synlett, 2017, 640 CAS; (b) S. Kramer, Chem. – Eur. J., 2016, 22, 1558 CrossRef PubMed; (c) Z. Zheng, Z. Wang, Y. Wang and L. Zhang, Chem. Soc. Rev., 2016, 45, 4448 RSC; (d) J. Miró and C. del Pozo, Chem. Rev., 2016, 116, 11924 CrossRef PubMed; (e) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS PubMed; (f) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236 CrossRef CAS PubMed; (g) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248 CrossRef CAS PubMed; (h) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493 CrossRef CAS . For reviews on oxidant-free oxidative gold catalysis, see: ; (i) M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069 RSC . For reviews on oxidative gold-catalysis by employing a visible-light photoredox strategy, see: ; (j) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193 RSC; (k) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed . For reports on oxidative gold catalysis effected by rational ligand designs, see: ; (l) J. Rodriguez, N. Adet, N. Saffon-Merceron and D. Bourissou, Chem. Commun., 2020, 56, 94 RSC; (m) A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed.
- A. Ariafard, ACS Catal., 2014, 4, 2896 CrossRef CAS.
- H. Ghari, Y. Li, R. Roohzadeh, P. Caramenti, J. Waser and A. Ariafard, Dalton Trans., 2017, 46, 12257 RSC.
- (a) Y. Yang, P. Antoni, M. Zimmer, K. Sekine, F. Mulks, L. Hu, L. Zhang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 5129 CrossRef CAS PubMed; (b) Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc, 2019, 141, 17414 CrossRef CAS PubMed.
- S. Banerjee, B. Senthilkumar and N. T. Patil, Org. Lett., 2019, 21, 180 CrossRef CAS PubMed.
- G. N. Hermann, M. T. Unruh, S.-H. Jung, M. Krings and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 10723 CrossRef CAS PubMed.
- J. P. Brand, C. Chevalley and J. Waser, Beilstein J. Org. Chem., 2011, 7, 565 CrossRef CAS PubMed.
- (a) P. K. Sajith and C. H. Suresh, Inorg. Chem., 2013, 52, 6046 CrossRef CAS PubMed; (b) P. K. Sajith and C. H. Suresh, Inorg. Chem., 2012, 51, 967 CrossRef CAS PubMed; (c) M. Ochiai, T. Sueda, K. Miyamoto, P. Kiprof and V. V. Zhdankin, Angew. Chem., Int. Ed., 2006, 45, 8203 CrossRef CAS PubMed.
- G. L. Tolnai, J. P. Brand and J. Waser, Beilstein J. Org. Chem., 2016, 12, 745 CrossRef CAS PubMed.
- A. C. Shaikh, D. R. Shinde and N. T. Patil, Org. Lett., 2016, 18, 1056 CrossRef CAS PubMed.
- Y. Li, F. Xie and X. Li, J. Org. Chem., 2016, 81, 715 CrossRef CAS PubMed.
- F. Zhao, B. Xu, D. Ren, L. Han, Z. Yu and T. Liu, Organometallics, 2018, 37, 1026 CrossRef CAS.
- M. O. Akram, S. Bera and N. T. Patil, Chem. Commun., 2016, 52, 12306 RSC.
- Z. Wang, X. Li and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 14219 CrossRef CAS PubMed.
- N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211 RSC.
- Z. Wang, L. Li and Y. Huang, J. Am. Chem. Soc., 2014, 136, 12233 CrossRef CAS PubMed.
- S. Peng, Z. Wang, L. Zhang, X. Zhang and Y. Huang, Nat. Commun., 2018, 9, 375 CrossRef PubMed.
- (a) M. Tobisu, Y. Ano and N. Chatani, Org. Lett., 2009, 11, 3250 CrossRef CAS . For a general review on palladium-catalyzed ligand-directed C–H functionalization, see: ; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- A. Székely, Á. Péter, K. Aradi, G. L. Tolnai and Z. Novák, Org. Lett., 2017, 19, 954 CrossRef PubMed.
- P. S. Shinde and N. T. Patil, Eur. J. Org. Chem., 2017, 3512 CrossRef CAS.
- P. S. Shinde, A. C. Shaikh and N. T. Patil, Chem. Commun., 2016, 52, 8152 RSC . A relatable Pt(II)-catalyzed version can be found is Waser's report, see: Y. Li and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 5438 CrossRef CAS PubMed.
- Selected reports: (a) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636 CrossRef CAS PubMed; (b) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed; (c) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS PubMed.
- X. Wang, X. Li, Y. Zhang and L. Xia, Org. Biomol. Chem., 2018, 16, 2860 RSC.
- (a) X. Li, X. Xin, N. Sun and Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 6994 CrossRef CAS PubMed; (b) S. Banerjee and N. T. Patil, Chem. Commun., 2017, 53, 7937 RSC.
- H. Peng, Y. Xi, N. Ronaghi, B. Dong, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2014, 136, 13174 CrossRef CAS PubMed.
- (a) A. S. Hay, J. Org. Chem., 1962, 27, 3320 CrossRef CAS; (b) A. S. Hay, J. Org. Chem., 1960, 25, 1275 CrossRef CAS; (c) C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422 CrossRef.
- W. Chodkiewicz and P. Cadiot, Compt. Rend., 1955, 241, 1055 CAS.
- X. Zhao, B. Tian, Y. Yang, X. Si, F. F. Mulks, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2019, 361, 3155 CrossRef CAS.
- (a) D. P. Hari and J. Waser, J. Am. Chem. Soc, 2017, 139, 8420 CrossRef CAS PubMed; (b) D. P. Hari and J. Waser, J. Am. Chem. Soc., 2016, 138, 2190 CrossRef CAS PubMed . For a recent report on oxyalkynylation of C–S bonds, see: ; (c) J. Borrel, G. Pisella and J. Waser, Org. Lett., 2020, 22, 422 CrossRef CAS PubMed . For a related reaction with ethynylbenziodazolones (EBZs), see: ; (d) D. P. Hari, L. Schouwey, V. Barber, R. Scopelliti, F. Fadaei-Tirani and J. Waser, Chem. – Eur. J., 2019, 25, 9522 CrossRef CAS PubMed.
- Atom-economical use of 3a was originally discovered by Yoshikai's group: (a) J. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2015, 54, 11107 CrossRef CAS PubMed; (b) B. Lu, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 11598 CrossRef CAS PubMed.
- (a) Y. Fuchita, Y. Utsunomiya and M. Yasutake, J. Chem. Soc., Dalton Trans., 2001, 2330 RSC; (b) P. W. de Graaf, J. Boersma and G. J. M. van der Kerk, J. Organomet. Chem., 1976, 105, 399 CrossRef CAS; (c) K. S. Liddle and C. Perkin, J. Chem. Soc., Chem. Commun., 1972, 26 RSC; (d) M. S. Kharasch and H. S. Isbell, J. Am. Chem. Soc., 1931, 53, 3053 CrossRef CAS.
- D. Qiu, Z. Zheng, F. Mo, Q. Xiao, Y. Tian, Y. Zhang and J. Wang, Org. Lett., 2011, 13, 4988 CrossRef CAS PubMed.
- (a) B. Olofsson, in Topics in Current Chemistry, ed. T. Wirth, Springer International Publishing, 2016, 373, 135 Search PubMed; (b) P. Villo and B. Olofsson, in Patai's Chemistry of Functional Groups: The Chemistry of Hypervalent Halogen Compounds, ed. B. Olofsson, I. Marek, Z. Rappoport, 2018, DOI:10.1002/9780470682531.pat0950.
- M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794 CrossRef CAS.
- K. Jia and Y. Chen, Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd., Chichester, UK, 2018, DOI:10.1002/9780470682531.pat0958.
- H. Zhu, Y. Shen, D. Wen, Z.-G. Le and T. Tu, Org. Lett., 2019, 21, 974 CrossRef CAS PubMed.
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed.
- J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 6046 CrossRef CAS PubMed.
- Recent reviews on chiral hypervalent iodine(III) reagents: (a) A. Parra, Chem. Rev., 2019, 119, 12033 CrossRef CAS PubMed; (b) H. Liang and M. A. Ciufolini, Angew. Chem., Int. Ed., 2011, 50, 11849 CrossRef CAS PubMed; (c) A. Parra and S. Reboredo, Chem. – Eur. J, 2013, 19, 17244 CrossRef CAS PubMed.
- (a) J.-J. Jiang, J.-F. Cui, B. Yang, Y. Ning, N. C.-H. Lai and M.-K. Wong, Org. Lett., 2019, 21, 6289 CrossRef CAS PubMed; (b) P. T. Bohan and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 11016 CrossRef CAS PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |