
Gold nanostars as a colloidal substrate for in-solution SERS measurements using a handheld Raman spectrometer†
Ahmed Y. F.
Mahmoud‡
,
Casey J.
Rusin‡
 and
Mark T.
McDermott
*
and
Mark T.
McDermott
*
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: mark.mcdermott@ualberta.ca
First published on 29th January 2020
Abstract
The evolution of Raman spectroscopy into a useful analytical technique has been due, in part, to the development of inexpensive, compact instrumentation and advancements in methodologies that enhance Raman intensities. Surface enhanced Raman scattering (SERS) is a primary methodology for quantitative and low detection limit measurements. While a broad array of applications using solid SERS substrates have been demonstrated, in-solution SERS measurements are not as widely pursued. This work seeks to optimize the synthesis of gold nanostars (AuNS) as a colloidal SERS substrate for in-solution measurements using handheld instrumentation. The types and concentrations of two buffers typically used for AuNS synthesis are examined to optimize the SERS intensity of a chemisorbed Raman probe. The observed SERS intensity primarily depends on conditions that allow higher surface coverage of the probe. Conditions that result in AuNS aggregates are found to be most optimal for SERS, similar to other nanoparticle shapes. A method to quantitate methimazole, an anti-hormone pharmaceutical, in urine is developed and reported. The primary impact of this work is the demonstration of the combination of water dispersible substrates and handheld instrumentation for rapid and sensitive analytical measurements.
Introduction
Raman spectroscopy has evolved from an instrument intensive, high-cost research lab-based methodology to an inexpensive, routine analysis technique.1–3 One main driving force behind this evolution was the development of high-performance yet lower-cost, compact instrument components such as lasers, wavelength selectors and detectors.1 This has led to commercial bench top Raman systems with a small footprint as well as handheld Raman devices.1,3 Another driving force of analytical measurement applications has been advances in platforms that enhance Raman signals, specifically surface enhanced Raman scattering (SERS).4 A significant area of advancement has been in the synthesis of noble metal nanoparticles (NP) of controlled size and shape.5 Computational models and experiments have both opened pathways for the application of plasmonic metal NPs in quantitative SERS measurements.6,7 Further introduction of SERS into a wider range of analytical measurements will rely on the pairing of appropriate SERS substrates with compact instrumentation.Anisotropic gold nanostructures have been synthesized in many different shapes such as rods,8,9 cubes,10 bipyramids,11 cages,12 prisms,13 nanostars,14–16etc. The localized surface plasmon resonance (LSPR) of these structures is tunable over a wide wavelength range when compared to spherical nanoparticles.17–20 Previous reports have shown that plasmonic excitation of anisotropic nanoparticles result in intense electric fields characteristically localized at their sharp edges.17–20 This phenomenon is called the “lightning rod” effect21 and can significantly improve SERS enhancement provided that the shape and the size of these particles are uniform.17–20
Gold nanostars (AuNS) are examples of anisotropic nanoparticles which have attracted interest as a SERS substrate because of their multiple branches. Byrne and co-workers have shown that the SERS enhancement of R6G adsorbed to AuNS was more pronounced than that of nanotriangles and nanospheres.22 Rodríguez-Lorenzo et al. reported a zeptomole detection limit of 1,5-naphtalenedithiol by sandwiching the molecule between a gold substrate and the tips of AuNS.23 Indrasekara et al. attached AuNS to a thin gold film using a short amine terminated alkanethiol.24 The substrate had an enhancement factor up to 5 orders of magnitude higher than that of gold nanospheres and achieved femtomolar level of detection of 4-mercaptobenzoic acid.24 These nanostructures have also been integrated into sandwich lateral flow SERS assays for the detection of human immunoglobulin G, Zika and dengue biomarkers.25,26 All these SERS applications of AuNS have been in the solid state, where the spectra were collected in air at a substrate surface. Haes and co-workers have used AuNS functionalized with carboxylic acid terminated alkanethiols for uranyl detection.27,28 The initial study in this series reported the use of AuNS in colloidal form with the measurements being made in solution.
AuNS are synthesized via two main approaches, seed-mediated and seedless growth protocols. The seed-mediated protocol is a two-step approach that requires the synthesis of isotropic gold nanoseeds, where anisotropic structures are grown on the seeds.22–24,29–31 For example, the nanoseeds are grown into AuNS by the reduction of HAuCl4 using ascorbic acid in the presence of cetyltrimethylammonium bromide and AgNO3.22,30 In addition, nanoseeds can be grown into AuNS when HAuCl4 is reduced using N,N-dimethylformamide in the presence of poly(vinylpyrrolidone).23,29 Gold nanoseeds can also be grown into AuNS by the reduction of HAuCl4 using ascorbic acid in the presence of HCl and AgNO3.24,31
The seedless protocol is a single step one-pot synthesis approach. In this approach, AuNS are synthesized by the reduction of HAuCl4 using Good's buffers. 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)14,16,32,33 and 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (EPPS)32–34 are the most commonly used Good's buffer to synthesize AuNS. 3-(N-Morpholino)propanesulfonic acid (MOPS)32 has also been used to synthesize AuNS. The tertiary amines from the piperazine group form cationic free radicals and acts as the reducing agent for Au ions.14,32,35,36 The terminal alkanesulfonate group acts as a shaping-directing agent and promote bilayer formation on the AuNS.32,33,37 The terminal hydroxyl groups promote the bilayer formation and shape stability via hydrogen bonding.32,33 The hydrophilic nature of the hydroxyl groups also provide water dispersibility and colloidal stability of the AuNS.33 The simplicity of the one-pot seedless synthesis approach of AuNS using Good's buffers has attracted attention for various applications.25–28,34,38 These nanostars have been applied in many SERS applications using different synthesis conditions.25–28 The plasmonic behaviour of AuNS has been systematically optimized by varying experimental conditions such as the concentration ratio of Good's buffer to HAuCl4, choice of Good's buffer and pH of the reaction.32 However, the effect of these various conditions on the SERS performance has not been investigated.32
Herein, we report the colloidal SERS performance of AuNS substrates using a handheld Raman spectrometer. In-solution SERS measurements were carried out with AuNS synthesized using HEPES and EPPS buffer at numerous ratios with and without using an aggregating agent (NaCl). This is to determine the optimal buffer and buffer to gold ratio for colloidal SERS analysis. The AuNS are characterized based on their LSPR, shape and size, and Raman intensity enhancement with and without NaCl. Following optimization with a chemisorbed Raman probe, the AuNS are used as a dispersible substrate to detect and quantify methimazole (MTZ) in synthetic urine. All Raman analyses are performed using a handheld Raman device to show the field deployability aspect of a colloidal AuNS substrate and to demonstrate rapid measurement performance.
Experimental
Reagents
Gold(III) chloride trihydrate (99.995%, HAuCl4), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer solution (1 M in H2O, HEPES), 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (99.5%, EPPS), sodium chloride (≥99.5%, NaCl), malachite green oxalate salt technical grade (MG), methimazole (analytical standard, MTZ), ciprofloxacin (≥98%, Cipro) and Surine™ negative urine control were purchased from Sigma-Aldrich Canada (Oakville, Ontario). 4-Mercaptobenzonitrile (MBN) was purchased from Combi-Blocks, Inc. (San Diego, California, USA). Sodium hydroxide (NaOH) was purchased from Fisher Scientific Canada. Transmission electron microscopy (TEM) grids (400-mesh carbon) were purchased from Electron Microscopy Sciences. Fisherbrand™ Class A clear glass threaded vials (1 dram) were purchased from Fisher Scientific Canada. The gold nanoparticles (30 nm) were purchased from Ted Pella, Inc. Deionized (DI) water (18.2 MΩ cm) was used for all syntheses and measurements in this work.Preparation of gold nanostars
AuNS were synthesized according to previously developed methods with slight modifications.32 The pH of the 1 M HEPES buffer solution was adjusted to 7.20 ± 0.01 using a solution of 1 M NaOH and a Fisher Scientific accumet research AR15 pH meter. Similarly, a solution of 0.5 M EPPS buffer was prepared in DI water and the pH was also adjusted to 7.20 ± 0.01. A stock solution of HAuCl4 (26 mM) was prepared in DI water. The synthesis of AuNS is governed by the ratio of the precursors in solution and is defined as R = [Buffer]/[HAuCl4], where [Buffer] is the concentration of HEPES or EPPS, and [HAuCl4] is held constant at 0.2 mM. The ratios assessed for each buffer in this work are 100, 300, 500, 700 and 1000 (buffer concentration = 20, 60, 100, 140 and 200 mM, respectively). The Good's buffer is initially mixed with DI water at 400 rpm. HAuCl4 solution is then added to the solution and mixed for 5 minutes. The solution is then removed and stored in a dark environment for 24 hours. The AuNS concentration at HR100 and ER100 is estimated to be ∼2.2 and 1.2 nM, respectively, as calculated using a previously published method.39 Throughout this manuscript, a codename is used to describe the AuNS such as HR100. The first letter (in this case H) is the buffer used, where H stands for HEPES and E stands for EPPS, the R represents ratio, and the numbers represent the ratio being examined.Characterization
UV-Vis spectroscopy experiments were completed using a PerkinElmer Lambda 35 spectrometer. All experiments used a slit width of 1 nm and a scan rate of 960 nm min−1. The samples were analyzed in 1.5 mL BRAND® polystyrene disposable cuvettes (unless stated otherwise) and monitored from 400 to 1000 nm.Transmission electron microscopy (TEM) imaging was performed using a JEOL JEM-ARM200CF S/TEM. Samples underwent a washing step to remove excess buffer by centrifuging three times: 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes, 9000 rpm for 10 minutes and 6000 rpm for 10 minutes (Eppendorf Centrifuge 5417 R). The samples were re-dispersed in water and sonicated for 2 minutes between centrifugation steps. After the final centrifugation step the sample was concentrated by a factor of 10 for imaging. The sample (10 μL) was drop-casted onto a TEM grid for 10 minutes and the excess solution was wicked away. Samples were imaged at 200 kV. Image processing and analysis was completed using Gatan Digital Micrograph software.
000 rpm for 10 minutes, 9000 rpm for 10 minutes and 6000 rpm for 10 minutes (Eppendorf Centrifuge 5417 R). The samples were re-dispersed in water and sonicated for 2 minutes between centrifugation steps. After the final centrifugation step the sample was concentrated by a factor of 10 for imaging. The sample (10 μL) was drop-casted onto a TEM grid for 10 minutes and the excess solution was wicked away. Samples were imaged at 200 kV. Image processing and analysis was completed using Gatan Digital Micrograph software.
Dynamic light scattering (DLS) and zeta potential measurements of AuNS were determined using a Malvern Zetasizer Nano-ZS. The instrument is equipped with a 4 mW HeNe laser (633 nm) and 173° backscattering angle. The 1 mL aliquot samples were measured in 2.5 mL BRAND® polystyrene disposable cuvettes (pathlength = 1 cm). Each measurement was performed with an equilibrium time of 120 seconds and at a temperature of 25 °C. For the DLS experiment, the standard deviation is representative of n = 5 with each measurement consisting of 13 sub-runs. For the zeta potential experiment, the standard deviation is representative of n = 3 with each measurement consisting of 20 sub-runs.
Raman analysis
Raman analysis using the AuNS was completed with a B&W Tek TacticID handheld Raman device with 785 nm laser (spot size 100 μm). The handheld was standardized using a polystyrene standard and all samples were analyzed using a liquid cell adapter at 100% power (300 mW). All measurements were performed in 1 dram vials. The analytes were mixed with the AuNS for 2 minutes using a vortex before analysis. In some experiments, NaCl was used as an aggregating agent to produce additional enhancement. Studies using salt were mixed for an additional 2 minutes after the addition of NaCl (total of 4 minutes) before taking the measurement. All samples contained the same concentration of AuNS and a total sample volume of 1 mL. Care was taken to choose an analyte concentration range that would not aggregate the AuNS before the addition of salt. This range was investigated by assessing low concentrations to concentrations that visibly aggregate the AuNS. This ensured partial control over the aggregation throughout the analyses. The acquisition time from the handheld cannot be user controlled. All Raman spectra were normalized to the device acquisition time. The spectral analysis was done using Spectragryph open-source software.40Results and discussion
A combination of easy-to-use, solution based SERS substrates and handheld Raman instrumentation has the potential to be a powerful portable measurement platform. Anisotropic metal nanoparticles, such as AuNS, have shown a great deal of promise as in-solution SERS substrates. The influence of the type of buffer and the [buffer]/[HAuCl4] ratio (R) on the structure and plasmonic behavior of AuNS has been explored by Odom and coworkers.32 Although their work provides a clear set of design rules for achieving a certain shape and optical properties, the effect of buffer type and R on the performance of colloidal AuNS as a water dispersible SERS substrate has not been fully explored to the best of our knowledge. In this work, AuNS synthesized in two different Good's buffers (HEPES and EPPS) with R ranging from 100 to 1000 were investigated (Fig. 1). AuNS used in SERS applications are usually prepared in this buffer ratio range as shown in Table 1.25–28,34,41,42 In the work described here, we characterize the structure of AuNS from HEPES and EPPS at various ratios. The in-solution SERS performance of the AuNS is then investigated using a probe molecule and a handheld Raman device. Finally, we present results for the measurement of the anti-thyroid pharmaceutical drug, methimazole (MTZ) in urine. | ||
| Fig. 1 Schematic of AuNS synthesis using Good's buffers (HEPES and EPPS). | ||
| Type of synthesis | Buffer type and ratio | Platform | Application | Ref. |
|---|---|---|---|---|
| a Seedless/seeded – AuNS are initially synthesized using a seedless method. Then AuNS are used as seeds to produce nanostars with longer branches via further addition of HAuCl4. | ||||
| Seedless/seededa | EPPS | Cellular | SERS imaging of breast cancer cells | 34 |
| R = ∼300 | ||||
| Seedless/seededa | EPPS | Colloidal | Detection of uranyl | 27 |
| R = 400 | ||||
| Seedless/seededa | EPPS | Surface | Detection of uranyl | 28 |
| R = 200 | ||||
| Seedless | HEPES | Surface | Lateral flow assay | 25 |
| R = ∼310 | ||||
| Seedless | HEPES | Surface | Lateral flow assay | 26 |
| R = ∼625 | ||||
| Seedless | HEPES | Surface | Development of SERS graphene-nanostar composite nanoprobe | 41 |
| R = 200 | ||||
| Seedless | HEPES | Surface | Synthesis of graphene nanostar nanocomposite | 42 |
| R = 83–500 | ||||
| Seedless | HEPES/EPPS | Colloidal | SERS optimization and detection | This work |
| R = 100–1000 | ||||
Synthesis and characterization of AuNS using HEPES and EPPS buffers
The synthesis of AuNS in Good's buffers was performed using previously reported methods.16,32 The size and shape of the AuNS were manipulated based on R.32Fig. 2 shows the extinction spectra and TEM images of AuNS synthesized using HEPES and EPPS. The AuNS extinction spectra is at ratios of 100, 300, 500, 700 and 1000, while only the AuNS extremities were imaged. The inset photographs are a visualization of the LSPR red-shift through a solution colour change from purple/blue to grey as the ratio is increased. Fig. 2A are the results for HEPES. It is known that low R values for HEPES result in more spherical particles with short branches and higher R mixtures yield longer branches.32 It has also been reported that HEPES induces branch growth in the [111] crystal plane direction.32 The extinction spectrum of HR100 in Fig. 2A exhibits a single LSPR mode centered at 566 nm. This mode is attributed to the near-spherical core of the AuNS (see Fig. SI1†).16,32,43 The TEM image of the HR100 shows a near-spherical structure with a size of 39 ± 11 nm (tip-to-tip) and branches of 7 ± 2 nm in length (size distribution found in Fig. SI2†). The lattice spacing within the branches measured from high-resolution TEM images (Fig. SI2†) is 0.235 ± 0.003 nm and is consistent with branch growth along the [111] direction.16,32,44 The extinction spectra of AuNS prepared with R 300 to 1000 all exhibit a primary band that is associated with the branches that red shifts as R increases. The red-shift has previously been attributed to the change in branch length and the branch tip sharpness.16 As shown in the TEM image of the HR1000, a high R produces anisotropic AuNS containing branches up to 100 nm in length with sharper tips. A low intensity band between 520 and 550 nm is also observed for R > 100 that is attributed to spherical by-products that are minimized with stirring.32 The results in Fig. 2A agree with those reported by Odom and co-workers for AuNS synthesized from HEPES.32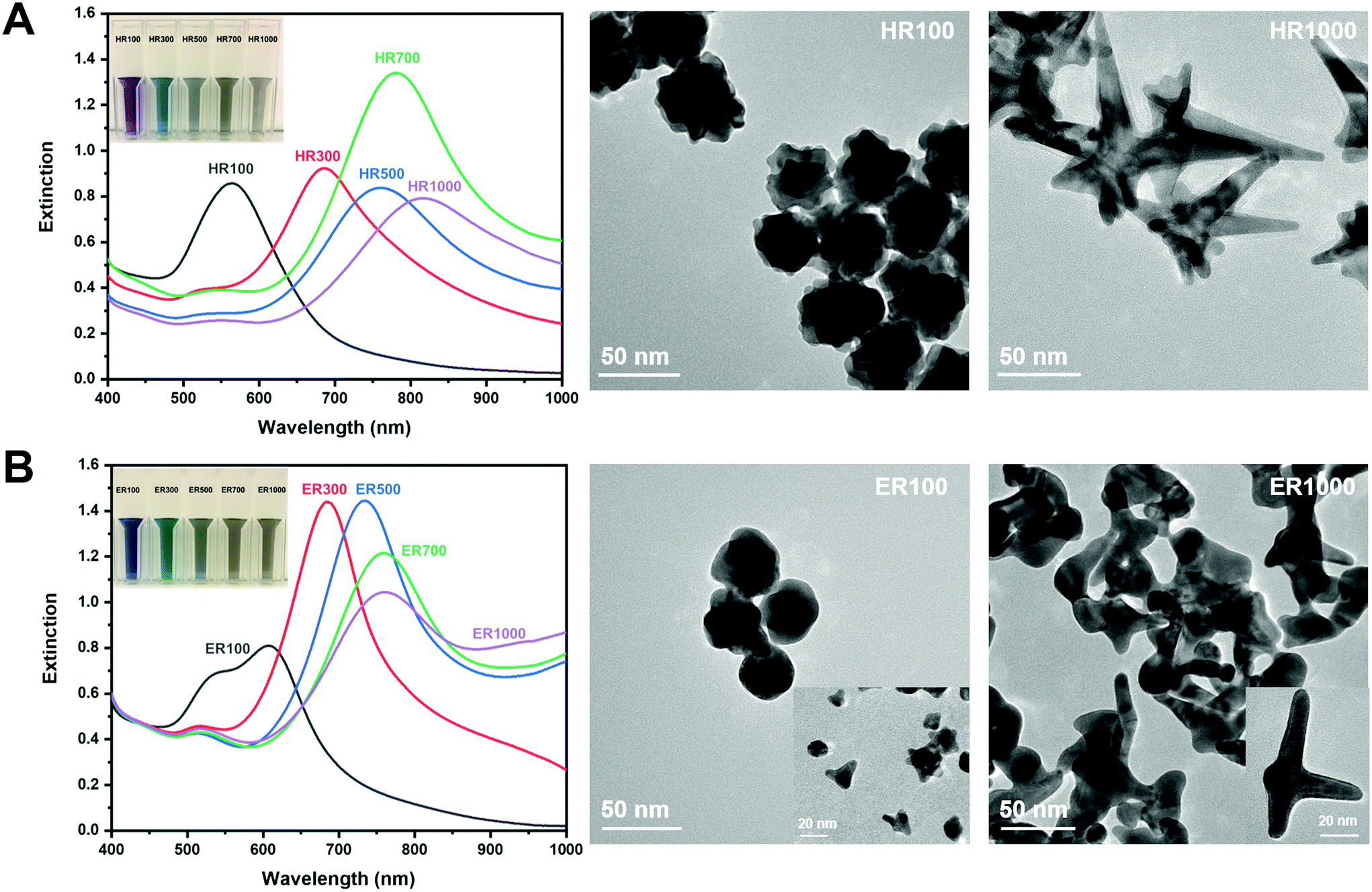 | ||
| Fig. 2 Extinction spectra of (A) HEPES and (B) EPPS buffers. The inset images show the colour of the AuNS solutions at the given ratios. TEM images correspond to AuNS at R 100 and 1000. | ||
The optical properties of AuNS synthesized using EPPS buffer are qualitatively similar to those from HEPES as shown in Fig. 2B. The primary mode is red-shifted as R is increased from 100 to 1000. One notable difference between the HEPES and EPPS is the presence of two plasmon modes for ER100. This suggests a spherical core (26 ± 9 nm, tip-to-tip, or furthest tip points) with smaller branch lengths (6 ± 2 nm) in comparison to nanostars synthesized at higher ratios. Odom and co-workers have thoroughly examined the extinction spectra of AuNS synthesized using EPPS, including subtle changes at higher ratios.32 Our results are consistent with their observations. The TEM images for ER100 and ER1000 indicate a shape evolution from more spherical NPs with small branches to NPs dominated by long branches. In general, the λmax for NPs synthesized in EPPS is lower than those in HEPES for a given R. It has been reported that the branches grow in the [110] direction in EPPS32 and the difference in λmax is likely due to the variations in crystal structure of the branches. The results in Fig. 2 are qualitatively similar to those reported in the literature32 and describe the evolution of both the shape and optical properties of AuNS with buffer type and concentration.
In-solution SERS performance of AuNS
In-solution SERS using colloidal NPs has proven to be a challenge. Spherical NPs do not yield substantial SERS intensities without inducing aggregation.45,46 Nanoparticles with shapes such as rods47 and dog-bones48,49 have demonstrated promise as colloidal SERS substrates. As shown in Table 1, most of the SERS applications of AuNS have been in the solid-state with the exception of work by Haes and co-workers in the measurement of uranyl with functionalized AuNS.27 In the work presented here, we examine the SERS performance of AuNS synthesized using HEPES and EPPS buffers with different R and using a handheld Raman instrument. Mercaptobenzonitrile (MBN) was chosen as a Raman probe due to its ability to specifically adsorb to the AuNS surface. We chose to conduct our measurements without any centrifugation or cleaning steps following synthesis. This represents a facile approach of performing SERS measurements directly after NP synthesis. Fig. 3 shows the SERS spectra of 5 μM MBN mixed with AuNS prepared from R100 solutions for both HEPES and EPPS buffers. The spectra were collected with a handheld instrument following 2 min of stirring (vortex). The major Raman bands are identified in Table SI1† according to previously published papers.50–52 Note that the only evidence of the buffer itself is the shoulder at 1044 cm−1 (see Fig. SI3†). Both buffers yield AuNS that provide in-solution SERS spectra with reasonable signal-to-noise without the need to form aggregate generated hot-spots.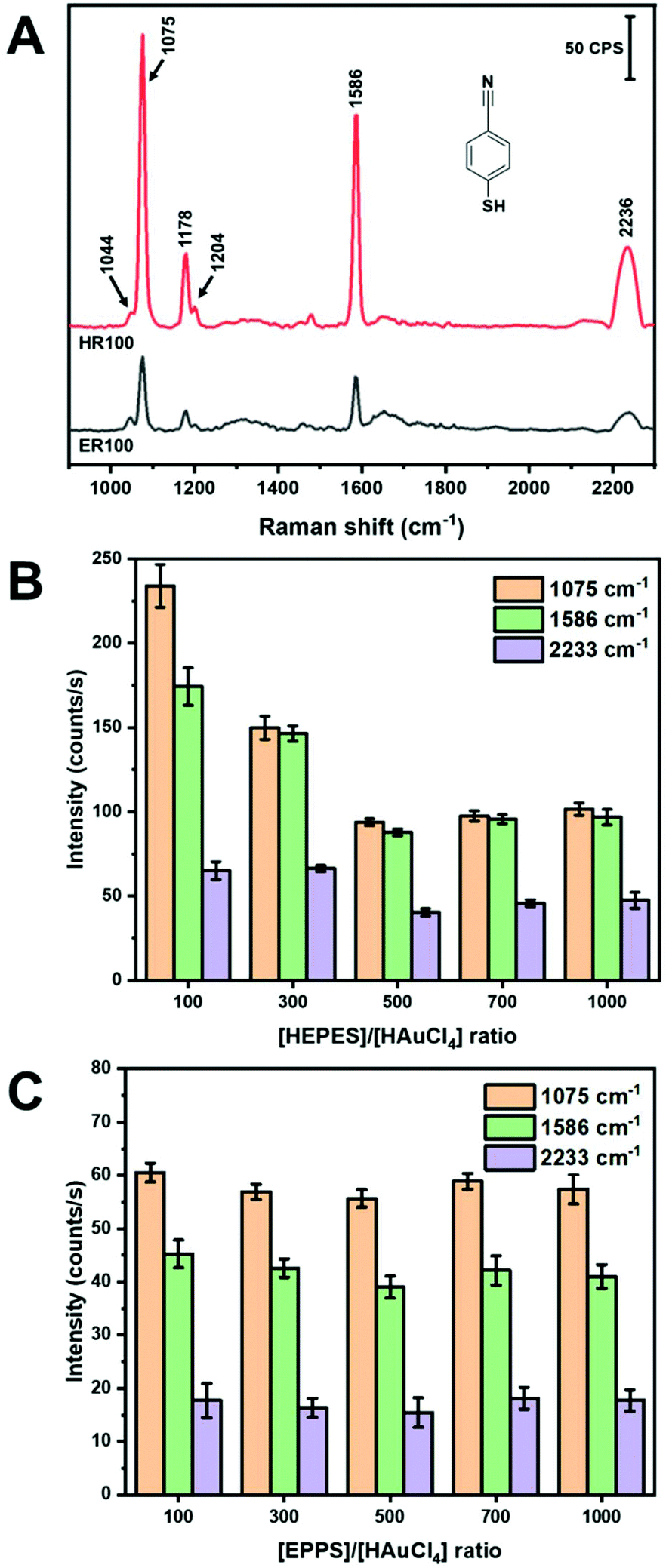 | ||
| Fig. 3 (A) SERS spectra of 5 μM MBN comparing AuNS HR100 ([AuNS] = 2.2 nM) and ER100 ([AuNS] = 1.2 nM). Column graph of the peak height of different vibrational modes of MBN at different ratios using (B) HEPES and (C) EPPS. | ||
The band intensities observed in Fig. 3A for the AuNS from HEPES are approximately 4× more intense than those in EPPS. A summary of the intensities of the three major bands at 1075 cm−1 (C–S stretch, νCS, and C–C stretch, νCC), 1586 cm−1 (C–C stretch, νCC), and 2233 cm−1(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch, νCN) of MBN for the different buffers and concentration ratios are shown in parts B and C of Fig. 3.50–52 The results for HEPES show a dependence on R, while the intensity is insensitive to R for EPPS. In Fig. 3B, the intensity of all bands decrease as R increases with the highest intensities observed at HR100 and are 2× that of HR1000. This is surprising considering the shapes of the AuNS, where the longer and sharper branches formed from higher R may be expected to yield higher SERS intensity. However, the variations of the SERS intensities observed are, at most, a factor of 4, and as described below, we believe these differences are primarily due to surface coverage of the probe MBN rather than any large electromagnetic differences.
N stretch, νCN) of MBN for the different buffers and concentration ratios are shown in parts B and C of Fig. 3.50–52 The results for HEPES show a dependence on R, while the intensity is insensitive to R for EPPS. In Fig. 3B, the intensity of all bands decrease as R increases with the highest intensities observed at HR100 and are 2× that of HR1000. This is surprising considering the shapes of the AuNS, where the longer and sharper branches formed from higher R may be expected to yield higher SERS intensity. However, the variations of the SERS intensities observed are, at most, a factor of 4, and as described below, we believe these differences are primarily due to surface coverage of the probe MBN rather than any large electromagnetic differences.
HEPES and EPPS both act as reducing, shape directing and stabilizing agent in the synthesis of AuNS.32,37 The piperazine moiety contains two cationic free radical tertiary amines that can reduce Au3+ to Au0.32,36 Additionally, the terminal alkanesulfonate group interacts with the AuNS surface and acts a shape directing agent, while the terminal hydroxyl group acts as a stabilizing agent and facilitates bilayer formation of the buffer ligand.32,33,37 MBN will have to compete with the HEPES or EPPS for surface sites. Previous studies suggest that at lower HEPES concentration the formation of a bilayer is incomplete or collapsed.33,37 Conversely, at high HEPES concentration (>100 mM) the formation of a bilayer inhibits the adsorption of MBN. Thus, MBN can compete more favourably for AuNS surface sites when R = 100.
Fig. 3 plots the intensities of the MBN bands for ER100 to ER1000. A comparison of the intensities between parts B and C of Fig. 3 shows that the AuNS prepared from HEPES provide more intense SERS signals for all R values. This difference is a factor of 4 for R100 and a factor of 2 for higher buffer concentrations. Again, we believe these modest differences in SERS intensities are due to the amount of probe adsorbed in each case. The additional methylene group in EPPS may promote a more densely packed monolayer on the AuNS through hydrophobic interactions similar to alkanethiols monolayers leading to less available surface sites for Raman active molecules.53 Another factor involves the growth direction of the AuNS branches in each buffer. As previously noted, it has been shown that HEPES induces branch growth in the [111] direction while EPPS results in branches in the [110] direction.32 It is known that the surface coverage of thiol-derived monolayers depends on the gold crystal face.54 Thus, surface coverage of MBN binding to the AuNS will be influenced by these different crystal faces. Another observation from Fig. 3C is that the peak intensities of MBN are relatively constant regardless of the EPPS buffer to gold ratio. One possible explanation involves the energetics of bilayer formation for HEPES–HEPES vs. EPPS–EPPS. A computational study using the polarizable continuum model determined that HEPES–HEPES and EPPS–EPPS dimers have a binding energy of 25.7 and −1.0 kJ mol−1, respectively.33 Thus, while HEPES can form stable bilayers at higher concentrations, bilayer formation with EPPS is less favorable. The AuNS formed in EPPS are thus coated with a more densely packed monolayer of stabilizing agent that does not vary with concentration of EPPS. In summary, colloidal AuNS prepared from HR100 solutions provide the highest in-solution SERS intensity for the MBN probe and overall greater enhancement than EPPS. The experiments described in the next section are all carried out with HR100 AuNS.
Reproducibility and additional enhancement
One of the potential benefits of using a colloidal SERS substrate and in-solution measurements is high reproducibility provided by sample homogeneity. Fig. 4A shows SERS spectra of MBN from AuNS within the same batch on different days. The peak intensity from the C–S stretching vibration on different days is plotted in Fig. 4B. The AuNS HR100 were exposed to 5 μM MBN and measured on day 0. The samples were then measured after being stored at 4 °C for the given period of time. Slight deviations in intensity may be the result of several factors. The initial spectra (day 0) is obtained after 2 minutes of mixing and the thiolate layer may be incomplete. Additionally, over 78 days adsorption/desorption of the thiolate, changes in molecular orientation and gold surface reconstruction could result in slight variations in intensity. However, any significant destabilization of the AuNS would result in substantial changes in intensity. Over a period of 78 days, the AuNS showed a percent relative standard deviation (%RSD) range (1–5%) within the same day and %RSD of 8% over the entire time period suggesting an excellent colloidal stability and a long-term storage capability at 4 °C. This demonstrates the applicability to store functionalized AuNS long-term.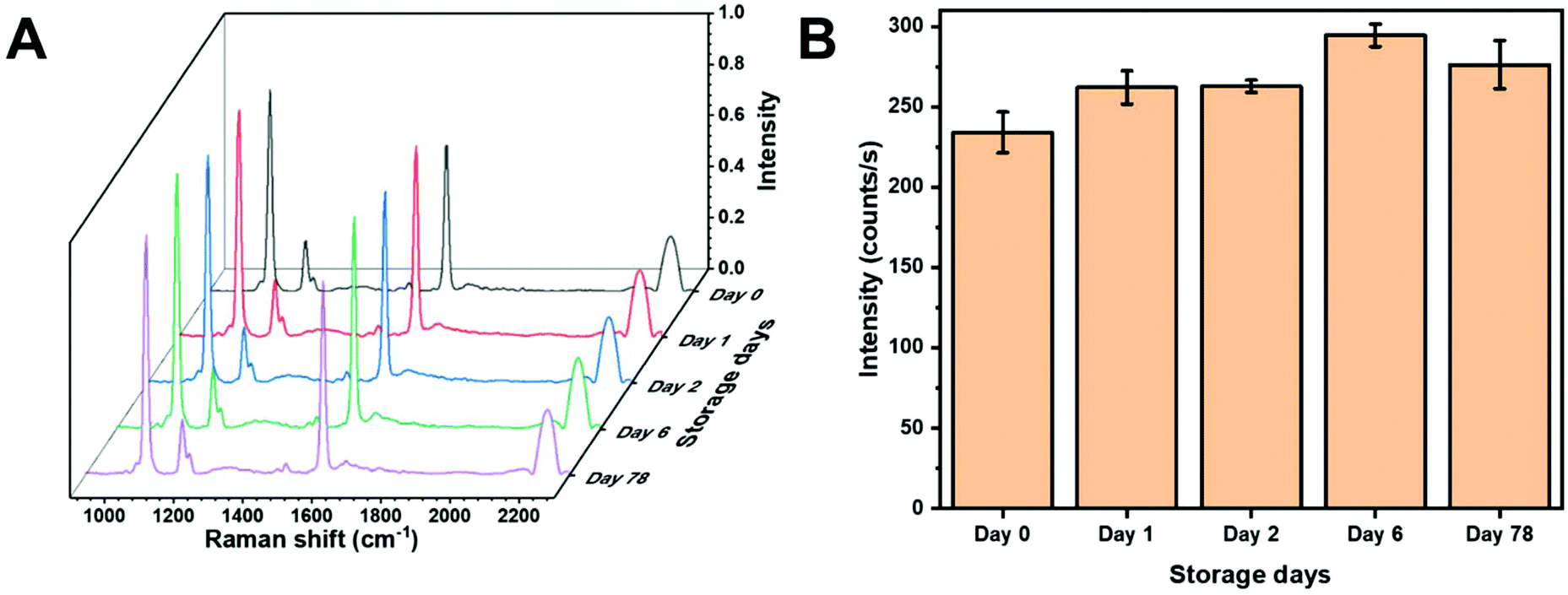 | ||
| Fig. 4 (A) SERS spectra of MBN showing signal reproducibility from AuNS substrates. (B) Bar chart of MBN C–S stretch peak intensity showing stability of AuNS at 4 °C. | ||
Aggregating agents are commonly used to enhance the SERS signals in solution by entrapping molecules between interstitial hot spots.55,56 We explored the possibility of increasing the observed signals by adding NaCl (200 mM) to produce dispersible AuNS nano-aggregates. Fig. 5A shows in-solution SERS spectra of MBN in AuNS HR100 before and after the addition of NaCl. The addition of NaCl results in a significant increase in peak intensity; a factor of about 10 for the C–S stretch at 1075 cm−1. The extinction spectra in Fig. 5B provides evidence for the formation of salt induced nano-aggregates. The AuNS HR100 exhibits a LSPR λmax at 566 nm. The addition of MBN indicates a slight decrease in the extinction and the appearance of a tail in the 600–775 nm range of the spectrum. There is also a slight colour change from light purple to dark purple. This is attributed to MBN adsorbing to the AuNS surface and changing the local dielectric constant. After the addition of NaCl, there is a large red-shift and broadening of the LSPR peak indicating the formation of AuNS aggregates. The colour of the AuNS changes from a dark purple to a light blue/grey. The UV-Vis extinction results are consistent with the formation of AuNS nano-aggregates with the addition of NaCl, and this explains the large enhancement observed in the SERS data.
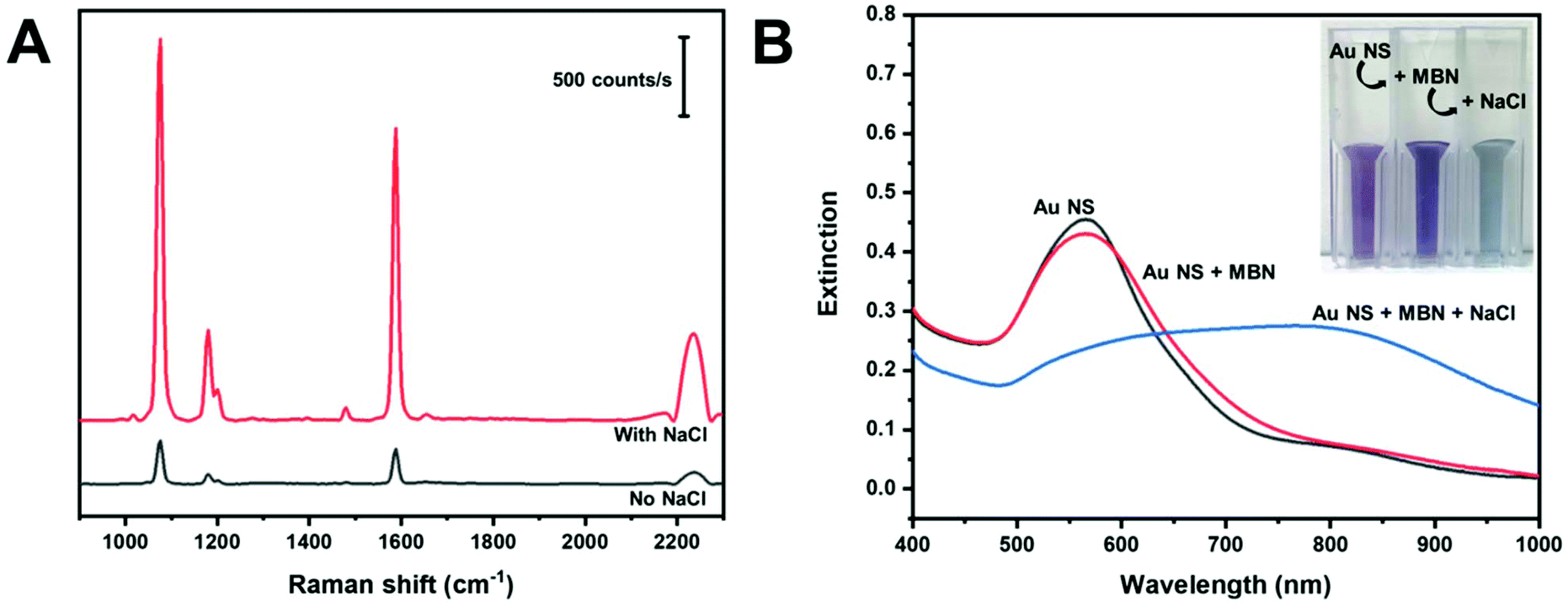 | ||
| Fig. 5 (A) SERS spectra of MBN using AuNS HR100 before and after salt addition. (B) UV-Vis spectra showing the extinction of AuNS HR100, after the addition of MBN and after the addition of MBN and NaCl. | ||
The size and colloidal stability of the AuNS nano-aggregates were studied using DLS and zeta potential measurements, and the results are shown in Table 2. The AuNS HR100 have a z-average diameter of 49.4 ± 0.9 nm. With the addition of MBN, we observe an increase in the z-average diameter due to the adsorption of MBN onto the AuNS surface through a gold–thiol linkage. Addition of NaCl causes the z-average diameter to increase to 379 nm indicating the formation of AuNS nano-aggregates. The polydispersity index (PDI) decreases with the addition of MBN and NaCl indicating the AuNS become more uniformly dispersed. The zeta potential remains relatively constant with the addition of MBN and NaCl.
| AuNS HR100 | z-Average diametera (nm) | PDIa | Zeta potentialb (mV) |
|---|---|---|---|
| a n = 5 with each measurement consisting of 13 sub-measurements. Analysis is based on a measurement time of 8 minutes. b n = 5 with each measurement consisting of 20 sub-measurements. Analysis is based on a measurement time of 5 minutes. pH values of the AuNS solutions were at 6.5–7 during Zeta potential measurement. | |||
| Unmodified | 49.4 ± 0.9 | 0.38 ± 0.04 | −35 ± 1 |
| +MBN | 54.7 ± 0.4 | 0.29 ± 0.01 | −36.9 ± 0.9 |
| +NaCl | 379 ± 54 | 0.23 ± 0.03 | −34 ± 2 |
Applications of AuNS for in-solution SERS
In the absence of any programmed molecular recognition, analytes will have to adsorb onto the AuNS in order to be detected. In this work, different adsorption mechanisms were investigated. Fig. 6 contains the in-solution SERS spectra of four different analytes, before and after nano-aggregate formation, that are expected to interact with the AuNS via different mechanisms. All analytes were detected in solution within five minutes. Thiabendazole (TBZ) (Fig. 6A) and methimazole (MTZ) (Fig. 6B) are examples of sulfur containing molecules which adsorb on AuNS by chemisorption through a S–Au interaction. Malachite green (MG) (Fig. 6C) and ciprofloxacin (Cipro) (Fig. 6D) are examples of non-sulfur containing molecules that will likely physisorb through the aromatic rings or N groups. The significance of these analytes and their band assignments are listed in the ESI.† All of these analytes can be rapidly measured (within 5 minutes) in the low μM range using colloidal AuNS via either chemisorption or physisorption mechanisms using the handheld Raman device. This points to the ability of a range of molecular structures to compete with HEPES for surface sites on the AuNS surface. As shown in Fig. 6, the analytes are detectable with un-aggregated AuNS. However, with the addition of NaCl the analyte signal is significantly enhanced due to aggregation and the subsequent formation of SERS hot spots.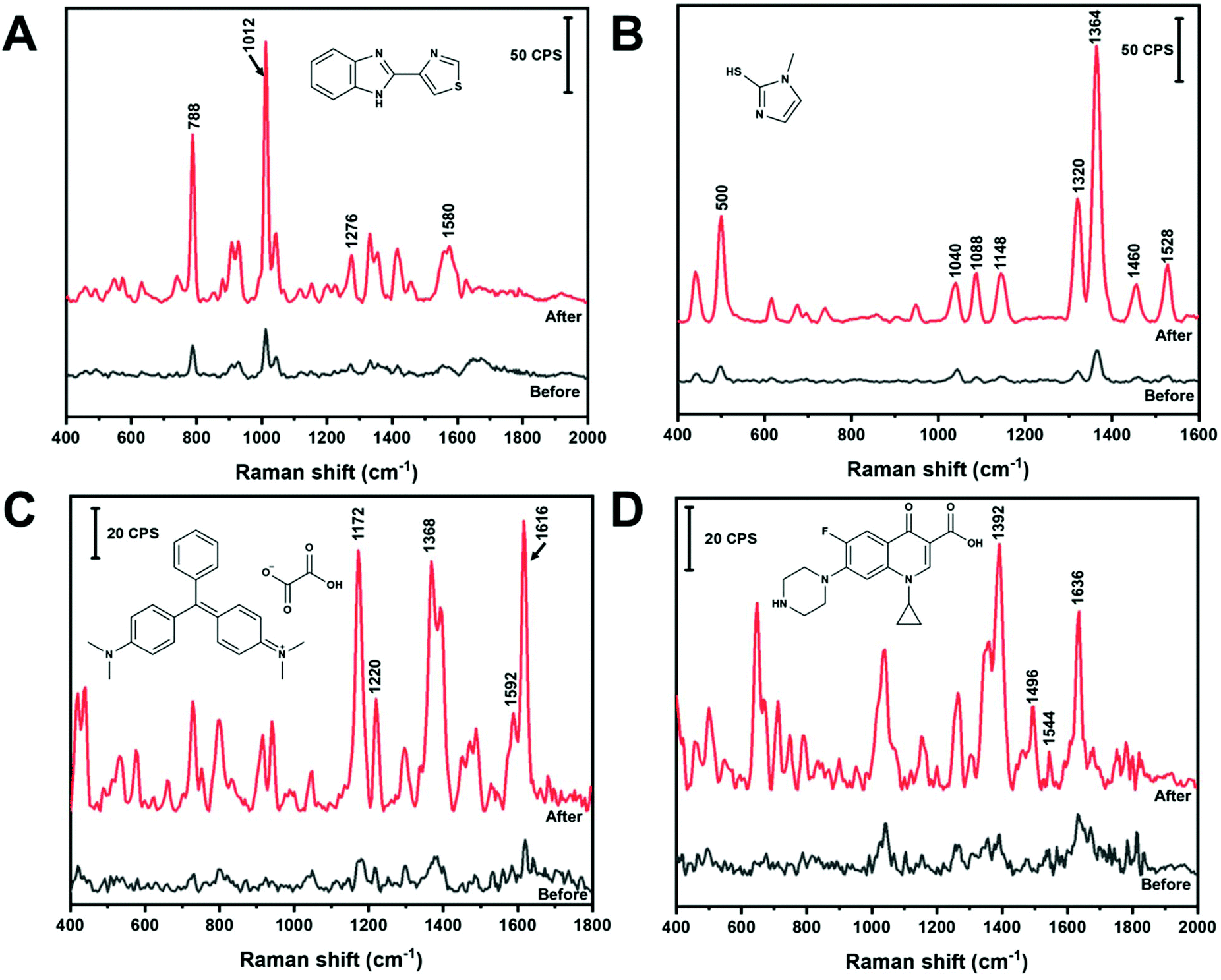 | ||
| Fig. 6 (A) 5 μM thiabendazole in 0.1 M HCl, (B) 1 μM methimazole in water, (C) 0.5 μM malachite green in water and (D) 5 μM ciprofloxacin in 0.1 M HCl using a colloidal AuNS HR100 substrate before and after the addition of NaCl (200 mM). | ||
Further development of a quantitative method and measurement in a complex matrix was carried out for MTZ. MTZ is commonly used as an anti-hormone pharmaceutical to treat hyperthyroidism by regulating the production of thyroxine and triiodothyronine.57,58 Moreover, MTZ is often illegally applied to animal feed to promote animal weight gain by increasing water retention in tissues.57,59 Monitoring and limiting human exposure is important as MTZ is known to have multiple side effects. These side effects include skin irritation, allergic reactions, impaired taste, pharyngitis, nephritis and liver cirrhosis.57–59 In addition, metabolization of MTZ leads to further MTZ intermediates known to have cytotoxic effects in the body.59 Chromatography is commonly used to detect and quantify MTZ in urine.58 However, the chromatographic method requires instrumental expertise, sample extraction, pre-concentration, or derivatization treatment before analysis.57,58 The limit of detection of MTZ by liquid chromatography methods can range from a concentration of nM to μM.57,58 A simple and less labour intensive method that can competitively quantify MTZ in urine would be useful. Fig. 6B shows the SERS spectrum of 1 μM MTZ before and after the addition of NaCl adsorbed on AuNS. This demonstrates the promise of in-solution SERS with a handheld device to measure this analyte.
Fig. 7 contains the results for the method development and quantitation of MTZ using in-solution SERS. First, we estimated the in-solution SERS enhancement factor of the MTZ AuNS system with the handheld device. Fig. 7A contains the SERS spectra of MTZ in water in the absence and presence of AuNS. The band intensity at 1364 cm−1 is low and only slightly visible above the background for 16 mM MTZ in water. The MTZ (0.917 μM)-AuNS nano-aggregate mixture exhibit a large enhancement at 1364 cm−1. The enhancement factor for AuNS HR100 was determined using the analytical enhancement factor formula,60 as shown in eqn (1):
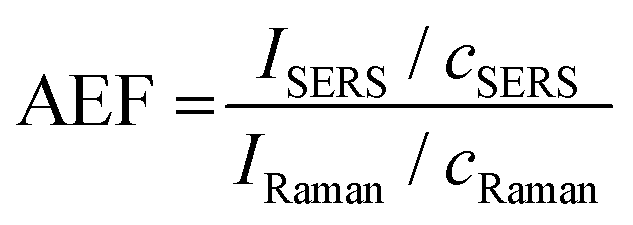 | (1) |
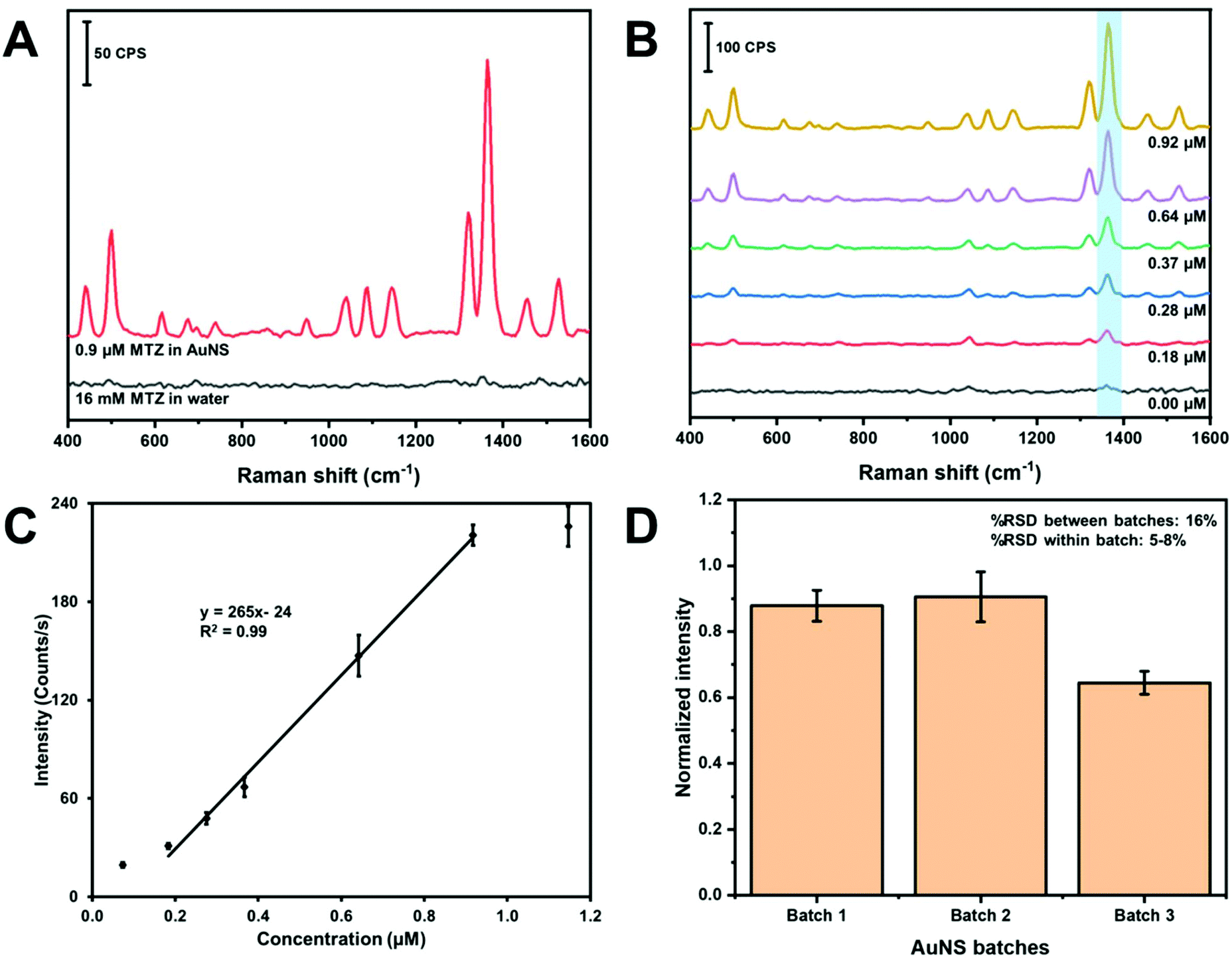 | ||
| Fig. 7 (A) SERS spectra showing the enhancement of MTZ in the presence of AuNS HR100. (B) SERS spectra of MTZ at various concentrations using AuNS HR100 and NaCl. (C) Calibration curve of MTZ using colloidal AuNS HR100. (D) Bar graph of the batch to batch reproducibility of AuNS using MTZ. | ||
Parts B and C of Fig. 7 demonstrate quantitation of MTZ using colloidal AuNS nano-aggregates as a water dispersible SERS substrate. The SERS spectra of MTZ at different concentrations in water is shown in Fig. 7B. Each concentration is represented by an average spectrum composed of three samples measured three times at different spots in glass vial (n = 9). The N–C stretching band at 1364 cm−1 used for quantitation is highlighted in light blue and band intensities increase with concentration.
Fig. 7C is a plot of the intensity of the 1364 cm−1 band versus concentration. The plot exhibits a linear portion and curves at low and high concentration. The curvature at lower concentration indicates the limit of the measurement. The “levelling-off” observed at high concentrations likely indicates that the adsorption sites on the AuNS nano-aggregates are saturated. A linear least squares fit of a portion of the plot can be used as a calibration curve. From this linear portion, the limit of detection and limit of quantification were calculated to be 0.1 and 0.3 μM. The dynamic range is determined to be between 0.3 and 0.9 μM.
The batch-to-batch reproducibility of the AuNS was monitored using the peak intensity at 1364 cm−1 of MTZ. Fig. 7D shows a bar chart of the peak intensity using three different AuNS batches. Each batch consists of three samples analyzed three times at different spots on the vial (n = 9). The %RSD between samples within the same batch was between 5–8%, while the %RSD between different batches was 16%. The %RSD for SERS substrates is generally reported up to about 15–20%.63
Human urine is a complex matrix consisting of non-protein nitrogenous compounds (e.g. urea), inorganic ions and salts, water soluble toxins and haemoglobin by-products.64 Individuals that use MTZ to treat hyperthyroidism will excrete excess amounts in their urine.57,58 The amount of MTZ retained in tissues can be harmful to the individuals health.57 The amount of MTZ retained in tissues can be quantified by monitoring the amount of MTZ excreted in urine. In this study, we spiked synthetic urine with MTZ and used our colloidal AuNS SERS substrate to quantify the amount of MTZ. Fig. 8 shows the Raman spectra of MTZ found in urine along with the corresponding control experiments. A band at 1008 cm−1 is prominent in the spectrum of pure urine that likely corresponds to the symmetrical CN stretch of urea.64
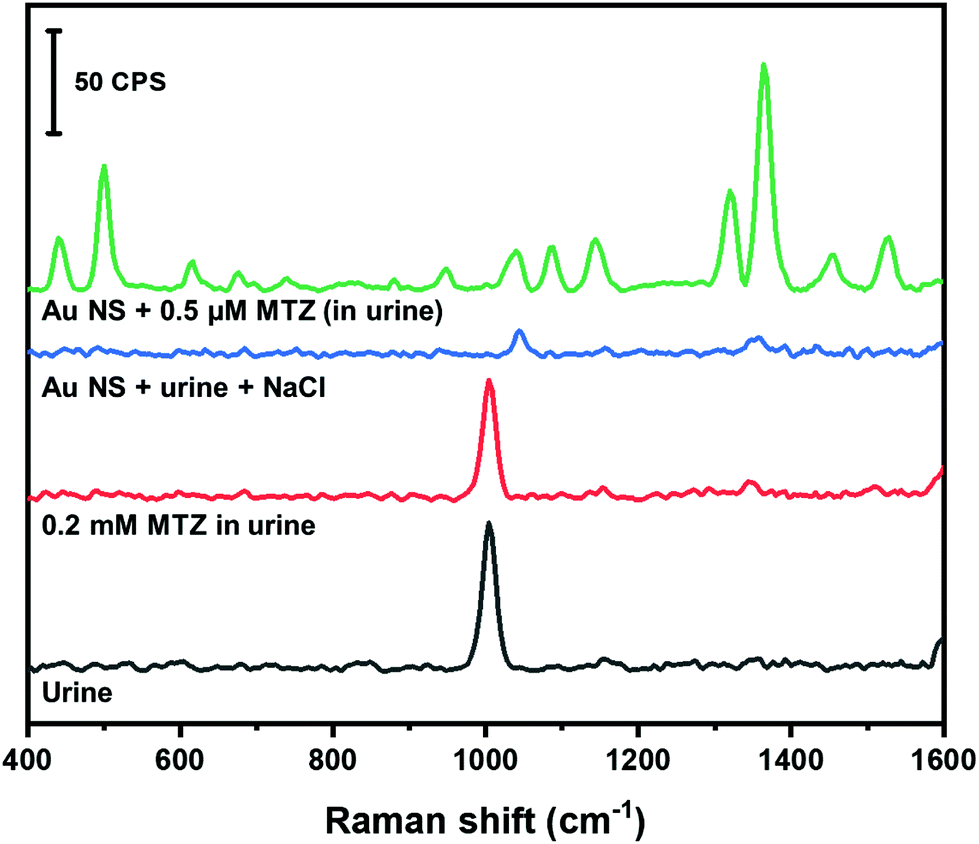 | ||
| Fig. 8 In-solution SERS spectrum of MTZ in urine and associated control spectra. | ||
When urine is spiked with MTZ (0.2 mM as final concentration), no bands corresponding to MTZ are observed. When urine is mixed with the AuNS nanoaggregate substrate, only a band from the HEPES buffer is visible. The disappearance of the 1008 cm−1 is due to the 200× dilution of the urine into the AuNS. The urine sample spiked with MTZ (0.5 μM final concentration) mixed with the AuNS nanoaggregates shows a large enhancement of MTZ Raman bands. Using the calibration curve obtained in water (Fig. 7C), MTZ in urine was examined at 0.5 and 0.8 μM and our method provided recoveries of nearly 100% (Table 3).
| Spiked amount of MTZ (μM) | Calc. amount of MTZ (μM) | % Recovery | % RSD |
|---|---|---|---|
| 0.5 | 0.495 ± 0.009 | 99 ± 2 | 2% |
| 0.8 | 0.83 ± 0.05 | 104 ± 6 | 5% |
Conclusions
The synthesis of AuNS using HEPES and EPPS buffer were systematically explored to determine the optimal buffer and buffer-to-gold ratio for in-solution SERS analysis. Differences in SERS intensities between HEPES and EPPS at different R values are primarily due to surface coverage of MBN on the AuNS instead of enhanced electromagnetic effects. AuNS synthesized using HEPES at R 100 produced the highest signal intensities. The AuNS showed a %RSD of 8% in signal intensity over 78 days at 4 °C and the ability to detect a variety of analytes in under 5 minutes through chemisorption and physisorption mechanisms. The addition of NaCl to the AuNS increases the SERS intensity by a factor of about 10 through the formation of nanoaggregates. A SERS assay was developed for the detection and quantitation of MTZ. The assay showed a limit of detection of 0.1 μM and a recovery of nearly 100% when MTZ was spiked in urine. The merging of in-solution substrates and handheld Raman spectrometers open new avenues for rapid and reproducible on-site SERS analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada Discovery Grant. We would like to acknowledge Dr Nikola Pekas from National Research Council of Canada – Nanotechnology Research Centre for providing use of the handheld Raman spectrometer. This work made use of the NanoFAB facility at the University of Alberta.Notes and references
- R. A. Crocombe, Appl. Spectrosc., 2018, 72, 1701–1751 CrossRef CAS PubMed.
- R. Deidda, P.-Y. Sacre, M. Clavaud, L. Coïc, H. Avohou, P. Hubert and E. Ziemons, TrAC, Trends Anal. Chem., 2019, 114, 251–259 CrossRef CAS.
- R. Pilot, J. Raman Spectrosc., 2018, 49, 954–981 CrossRef CAS.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- Y. Xia and N. J. Halas, MRS Bull., 2011, 30, 338–348 CrossRef.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. V. Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- A. Gole and C. J. Murphy, Chem. Mater., 2004, 16, 3633–3640 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- X. Kou, W. Ni, C. K. Tsung, K. Chan, H. Q. Lin, G. D. Stucky and J. Wang, Small, 2007, 3, 2103–2113 CrossRef CAS PubMed.
- S. E. Skrabalak, L. Au, X. Li and Y. Xia, Nat. Protoc., 2007, 2, 2182–2190 CrossRef CAS PubMed.
- B. Pelaz, V. Grazu, A. Ibarra, C. Magen, P. del Pino and J. M. de la Fuente, Langmuir, 2012, 28, 8965–8970 CrossRef CAS PubMed.
- H. Ahsan, T. Masaaki and W. Y. Guang, Bull. Chem. Soc. Jpn., 2005, 78, 262–269 CrossRef.
- C. L. Nehl and J. H. Hafner, J. Mater. Chem., 2008, 18, 2415–2419 RSC.
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS.
- M. J. Banholzer, J. E. Millstone, L. Qin and C. A. Mirkin, Chem. Soc. Rev., 2008, 37, 885–897 RSC.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS PubMed.
- C. J. Murphy, A. M. Gole, S. E. Hunyadi, J. W. Stone, P. N. Sisco, A. Alkilany, B. E. Kinard and P. Hankins, Chem. Commun., 2008, 544–557 RSC.
- M. Rycenga, M. R. Langille, M. L. Personick, T. Ozel and C. A. Mirkin, Nano Lett., 2012, 12, 6218–6222 CrossRef CAS PubMed.
- J. I. Gersten, J. Chem. Phys., 1980, 72, 5779–5780 CrossRef CAS.
- F. Tian, F. Bonnier, A. Casey, A. E. Shanahan and H. J. Byrne, Anal. Methods, 2014, 6, 9116–9123 RSC.
- L. Rodríguez-Lorenzo, R. A. Álvarez-Puebla, I. Pastoriza-Santos, S. Mazzucco, O. Stéphan, M. Kociak, L. M. Liz-Marzán and F. J. García de Abajo, J. Am. Chem. Soc., 2009, 131, 4616–4618 CrossRef PubMed.
- A. S. D. S. Indrasekara, S. Meyers, S. Shubeita, L. C. Feldman, T. Gustafsson and L. Fabris, Nanoscale, 2014, 6, 8891–8899 RSC.
- M. Sánchez-Purrà, M. Carré-Camps, H. de Puig, I. Bosch, L. Gehrke and K. Hamad-Schifferli, ACS Infect. Dis., 2017, 3, 767–776 CrossRef PubMed.
- M. Sánchez-Purrà, B. Roig-Solvas, C. Rodriguez-Quijada, B. M. Leonardo and K. Hamad-Schifferli, ACS Omega, 2018, 3, 10733–10742 CrossRef PubMed.
- G. Lu, T. Z. Forbes and A. J. Haes, Analyst, 2016, 141, 5137–5143 RSC.
- G. Lu, A. J. Johns, B. Neupane, H. T. Phan, D. M. Cwiertny, T. Z. Forbes and A. J. Haes, Anal. Chem., 2018, 90, 6766–6772 CrossRef CAS PubMed.
- E. Nalbant Esenturk and A. R. Hight Walker, J. Raman Spectrosc., 2009, 40, 86–91 CrossRef.
- P. Senthil Kumar, I. Pastoriza-Santos, B. Rodríguez-González, F. J. García de Abajo and L. M. Liz-Marzán, Nanotechnology, 2007, 19, 015606 CrossRef PubMed.
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 CrossRef PubMed.
- K. Chandra, K. S. B. Culver, S. E. Werner, R. C. Lee and T. W. Odom, Chem. Mater., 2016, 28, 6763–6769 CrossRef CAS.
- H. Liu, Y. Xu, Y. Qin, W. Sanderson, D. Crowley, C. H. Turner and Y. Bao, J. Phys. Chem. C, 2013, 117, 17143–17150 CrossRef CAS.
- J. Cai, V. Raghavan, Y. J. Bai, M. H. Zhou, X. L. Liu, C. Y. Liao, P. Ma, L. Shi, P. Dockery, I. Keogh, H. M. Fan and M. Olivo, J. Mater. Chem. B, 2015, 3, 7377–7385 RSC.
- R. Chen, J. Wu, H. Li, G. Cheng, Z. Lu and C.-M. Che, Rare Met., 2010, 29, 180–186 CrossRef CAS.
- J. K. Grady, N. D. Chasteen and D. C. Harris, Anal. Biochem., 1988, 173, 111–115 CrossRef CAS PubMed.
- W. Xi and A. J. Haes, J. Am. Chem. Soc., 2019, 141, 4034–4042 CrossRef CAS PubMed.
- D. H. M. Dam, J. H. Lee, P. N. Sisco, D. T. Co, M. Zhang, M. R. Wasielewski and T. W. Odom, ACS Nano, 2012, 6, 3318–3326 CrossRef CAS PubMed.
- H. de Puig, J. O. Tam, C.-W. Yen, L. Gehrke and K. Hamad-Schifferli, J. Phys. Chem. C, 2015, 119, 17408–17415 CrossRef CAS PubMed.
- F. Menges, Spectragryph - optical spectroscopy software, http://www.effemm2.de/spectragryph/, (accessed November 28, 2019) Search PubMed.
- G. Jalani and M. Cerruti, Nanoscale, 2015, 7, 9990–9997 RSC.
- S. Z. Nergiz, N. Gandra and S. Singamaneni, Carbon, 2014, 66, 585–591 CrossRef CAS.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS PubMed.
- J. A. Webb, W. R. Erwin, H. F. Zarick, J. Aufrecht, H. W. Manning, M. J. Lang, C. L. Pint and R. Bardhan, J. Phys. Chem. C, 2014, 118, 3696–3707 CrossRef CAS.
- K. Faulds, R. E. Littleford, D. Graham, G. Dent and W. E. Smith, Anal. Chem., 2004, 76, 592–598 CrossRef CAS PubMed.
- D. Graham, D. G. Thompson, W. E. Smith and K. Faulds, Nat. Nanotechnol., 2008, 3, 548–551 CrossRef CAS PubMed.
- B. Saute, R. Premasiri, L. Ziegler and R. Narayanan, Analyst, 2012, 137, 5082–5087 RSC.
- B. Saute and R. Narayanan, Analyst, 2011, 136, 527–532 RSC.
- B. Saute and R. Narayanan, J. Raman Spectrosc., 2013, 44, 1518–1522 CrossRef CAS.
- D. Gkogkou, B. Schreiber, T. Shaykhutdinov, H. K. Ly, U. Kuhlmann, U. Gernert, S. Facsko, P. Hildebrandt, N. Esser, K. Hinrichs, I. M. Weidinger and T. W. H. Oates, ACS Sens., 2016, 1, 318–323 CrossRef CAS.
- R. Holze, J. Solid State Electrochem., 2013, 17, 1869–1879 CrossRef CAS.
- E. Villarreal, G. G. Li, Q. Zhang, X. Fu and H. Wang, Nano Lett., 2017, 17, 4443–4452 CrossRef CAS PubMed.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559–3568 CrossRef CAS.
- N. C. III, C. E. D. Chidsey, G. Y. Liu and G. Scoles, J. Chem. Phys., 1993, 98, 4234–4245 CrossRef.
- S. E. J. Bell and N. M. S. Sirimuthu, J. Am. Chem. Soc., 2006, 128, 15580–15581 CrossRef CAS PubMed.
- N. R. Yaffe, A. Ingram, D. Graham and E. W. Blanch, J. Raman Spectrosc., 2010, 41, 618–623 CrossRef CAS.
- H. Ebrahimzadeh, A. A. Asgharinezhad, L. Adlnasab and N. Shekari, J. Sep. Sci., 2012, 35, 2040–2047 CrossRef CAS PubMed.
- K. Kuśmierek and E. Bald, Talanta, 2007, 71, 2121–2125 CrossRef PubMed.
- M. B. Genter, J. Biochem. Mol. Toxicol., 1998, 12, 305–314 CrossRef CAS PubMed.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS.
- L. Osinkina, T. Lohmüller, F. Jäckel and J. Feldmann, J. Phys. Chem. C, 2013, 117, 22198–22202 CrossRef CAS.
- K. Kneipp, H. Kneipp, R. Manoharan, E. B. Hanlon, I. Itzkan, R. R. Dasari and M. S. Feld, Appl. Spectrosc., 1998, 52, 1493–1497 CrossRef CAS.
- R. Panneerselvam, G.-K. Liu, Y.-H. Wang, J.-Y. Liu, S.-Y. Ding, J.-F. Li, D.-Y. Wu and Z.-Q. Tian, Chem. Commun., 2018, 54, 10–25 RSC.
- L. P. Moreira, L. Silveira, M. T. T. Pacheco, A. G. da Silva and D. D. F. M. Rocco, J. Photochem. Photobiol., B, 2018, 185, 223–234 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: UV-Vis spectrum, particle size distribution histograms, TEM images, Raman spectra, tabulated Raman band assignments and analyte descriptions. See DOI: 10.1039/c9an02439e |
| ‡ Both authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |