
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An Al-doped SrTiO3 photocatalyst maintaining sunlight-driven overall water splitting activity for over 1000 h of constant illumination†
Hao
Lyu
a,
Takashi
Hisatomi
 b,
Yosuke
Goto‡
a,
Masaaki
Yoshida
cd,
Tomohiro
Higashi
a,
Masao
Katayama
a,
Tsuyoshi
Takata
b,
Tsutomu
Minegishi
a,
Hiroshi
Nishiyama
a,
Taro
Yamada
a,
Yoshihisa
Sakata
c,
Kiyotaka
Asakura
e and
Kazunari
Domen
*ab
b,
Yosuke
Goto‡
a,
Masaaki
Yoshida
cd,
Tomohiro
Higashi
a,
Masao
Katayama
a,
Tsuyoshi
Takata
b,
Tsutomu
Minegishi
a,
Hiroshi
Nishiyama
a,
Taro
Yamada
a,
Yoshihisa
Sakata
c,
Kiyotaka
Asakura
e and
Kazunari
Domen
*ab
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp
bCenter for Energy & Environmental Science, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, 4-17-1 Wakasato, Nagano-shi, Nagano 380-8553, Japan
cGraduate School of Sciences and Technology for Innovation, Yamaguchi University, 2-16-1 Tokiwadai, Ube-shi, Yamaguchi 755-8611, Japan
dBlue Energy Center for SGE Technology, Yamaguchi University, 2-16-1 Tokiwadai, Ube-shi, Yamaguchi 755-8611, Japan
eInstitute for Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo-shi, Hokkaido 001-0021, Japan
First published on 24th January 2019
Abstract
Photocatalytic water splitting is a viable approach to the large-scale production of renewable solar hydrogen. The apparent quantum yield for this reaction has been improved, but the lifespan of photocatalysts functioning under sunlight at ambient pressure have rarely been examined, despite the critical importance of this factor in practical applications. Herein, we show that Al-doped SrTiO3 (SrTiO3:Al) loaded with a RhCrOx (rhodium chromium oxide) cocatalyst splits water with an apparent quantum yield greater than 50% at 365 nm. Moreover, following the photodeposition of CoOOH and TiO2, this material maintains 80% of its initial activity and a solar-to-hydrogen energy conversion efficiency greater than or equal to 0.3% over a span of 1300 h under constant illumination by simulated sunlight at ambient pressure. This result is attributed to reduced dissolution of Cr in the cocatalyst following the oxidative photodeposition of CoOOH. The photodeposition of TiO2 further improves the durability of this photocatalyst. This work demonstrates a concept that could allow the design of long-term, large-scale photocatalyst systems for practical sunlight-driven water splitting.
Introduction
The development of renewable energy resources is an important issue, owing to increasing concerns regarding environmental degradation and limited fossil fuel supplies. Photocatalytic overall water splitting has attracted much attention for the past several decades owing to its potential for the large scale production of hydrogen as a renewable fuel.1–4 Certain metal oxide photocatalysts have shown apparent quantum yields (AQYs) greater than 50% during overall water splitting in response to irradiation with UV light.5–7 Photocatalytic systems exhibiting relatively high AQYs under visible light have also recently emerged.8–10 However, the practical application of photocatalytic water splitting requires systems that provide both a high level of activity and durability over prolonged operation periods.11 La-doped NaTaO3 loaded with NiO has been reported to maintain its water splitting activity over a span of 16 days without noticeable degradation at ambient pressure under UV illumination.5 A solid solution of GaN and ZnO (GaN:ZnO) loaded with rhodium chromium oxide (RhCrOx) as a hydrogen evolution cocatalyst also demonstrated constant water splitting activity over a span of three months under visible light at a reduced pressure, although this material underwent a 50% loss in activity over the following three months.12 This activity loss was attributed to oxidative dissolution of the Cr component of the RhCrOx (which is essential for suppressing backward reactions), and it was found that such dissolution could be reduced by coloading RuOx as an oxygen evolution cocatalyst. Interestingly, the durability of photocatalytic systems during overall water splitting under sunlight at ambient pressure has rarely been investigated, even though these are the conditions that would be present during the practical application of such systems.The present work examined the long-term durability of RhCrOx-loaded SrTiO3:Al (RhCrOx/SrTiO3:Al). This material was selected for study because it is one of the most active photocatalysts for overall water splitting under sunlight and has a high AQY in the near UV region (56% at 365 nm).7 The results presented herein demonstrate that the oxidative photodeposition of CoOOH on the photocatalyst stabilises the Cr in the RhCrOx cocatalyst, which would otherwise be oxidised and dissolved. The concurrent deposition of TiO2 further improves the durability and allows this photocatalyst, once fixed on a substrate, to maintain 80% of its initial activity over 1300 h of constant simulated sunlight illumination at ambient pressure.
Results and discussion
SrTiO3:Al was prepared using a flux method and the RhCrOx cocatalyst was loaded by impregnation followed by calcination in air.7 Cobalt species were loaded onto the resulting RhCrOx/SrTiO3:Al by photodeposition.7 Amorphous TiO2 was also loaded on some samples of this material by photodeposition.13 Water splitting reactions were carried out using a gas flow reaction system or a panel reactor. A 300 W Xe lamp (300 nm < λ < 500 nm), a 450 W high-pressure Hg lamp (λ > 300 nm) and a solar simulator (100 mW cm−2, AM 1.5G) were used as light sources for standard, accelerated deactivation and long-term durability tests, respectively.Fig. 1a shows the time course of the water splitting activity of RhCrOx/SrTiO3:Al in response to irradiation by the high-pressure Hg lamp, and demonstrates that the water splitting rate was decreased to 14% of its original value after 168 h. Inductively-coupled plasma optical emission spectroscopy (ICP-OES) analyses determined that 37% of the Cr in the RhCrOx cocatalyst was dissolved following this trial, although there was no significant leaching of any other elements. This result is in agreement with the dissolution of Cr species that has been reported for RhCrOx/GaN:ZnO.12 The Cr in RhCrOx is thought to prevent backward reactions involving molecular oxygen,14 similar to the case for core/shell Rh/Cr2O3 cocatalysts.15 Thus, loss of the Cr species would be expected to result in exposure of the Rh species and an increase in the extent of backward reactions. Additional trials showed that the degree of deactivation was reduced under weaker irradiation when the same amounts of water were decomposed (see Fig. S1 in ESI†). These data suggest that, if the photoexcited holes generated by irradiation are not used promptly, they may oxidise Cr3+ in the RhCrOx cocatalyst to soluble Cr6+ species.
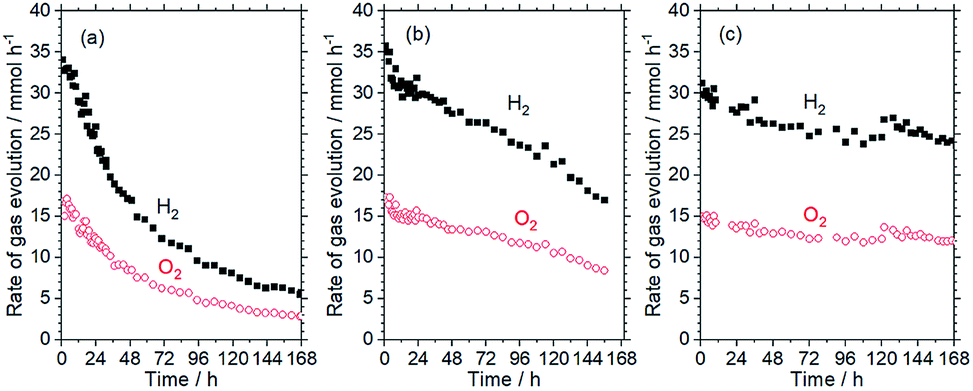 | ||
| Fig. 1 Time courses of water-splitting activity of SrTiO3:Al loaded with (a) RhCrOx, (b) RhCrOx and 0.3 wt% Co species, and (c) RhCrOx, 0.3 wt% Co species and 3 wt% TiO2. Reaction conditions: photocatalyst, 0.5 g; reaction solution, 370 mL distilled water; reaction temperature, 291 K; light source, 450 W high-pressure Hg lamp (λ > 300 nm). | ||
Cobalt oxide (CoOy) is known to extract holes from photocatalysts and to simultaneously function as an oxygen evolution cocatalyst.16 On this basis, cobalt species were loaded via photodeposition using CoCl2 in the present work. The aim was to suppress the oxidative corrosion of Cr3+ species by excess holes and thus prevent the deactivation of the RhCrOx/SrTiO3:Al photocatalyst. As shown in Fig. S2,† the RhCrOx/SrTiO3:Al maintained only 34% of its initial activity after 72 h under UV irradiation by the Xe lamp. Following the addition of Co species, an induction period was observed, during which the water splitting rate gradually increased. Notably, RhCrOx/SrTiO3:Al loaded with 0.3 wt% Co maintained stable activity under these conditions. When the Co loading was increased to 0.5 wt%, the induction period was prolonged while the water splitting rate stabilized at a lower value, suggesting excessive loading of the Co species. The water splitting reaction was also carried out using RhCrOx/SrTiO3:Al coloaded with 0.3 wt% Co species in conjunction with irradiation by the high-pressure Hg lamp (Fig. 1b). The results show that the durability of the photocatalyst was improved without sacrificing activity by loading an appropriate amount of Co species.
Fig. S3† and Table 1 provide Cr and Ti 2p X-ray photoelectron spectroscopy (XPS) data and the surface Cr/Ti atomic ratios for RhCrOx/SrTiO3:Al with and without the coloading of 0.3 wt% Co species, and before and after the water splitting reaction was conducted for a span of one week. In the absence of Co loading, the Cr/Ti ratio decreased from 0.12 to 0.07 during the reaction. Adding the Co species increased the Cr/Ti ratio from 0.12 to 0.21, suggesting that the Co species was photodeposited on the SrTiO3:Al photocatalyst rather than on the RhCrOx cocatalyst. Notably, the presence of the Co species decreased the reduction in the Cr/Ti ratio during the reaction, such that the ratio was reduced from 0.21 only to 0.20. Moreover, the concentration of Cr ions in the reaction solution was below the level in a blank solution (7.3 ppb, as determined by ICP-OES), indicating negligible Cr dissolution. The Co/Ti ratio was also not decreased significantly during the reaction, demonstrating the robust nature of the Co species in this material. Note that the activity of CoOOH/RhCrOx/SrTiO3:Al decreased to approximately the half although the loss of Cr content on the RhCrOx cocatalyst was marginal. The activity in overall water splitting is governed by many factors and the degree of deactivation would not be linear to the respective factors, including the loss of Cr species. Nevertheless, this observation implies that factors other than the loss of Cr species may be identified as additional causes of the deactivation.
| Cocatalyst | Reaction time/h | Cr/Ti | Co/Ti |
|---|---|---|---|
| RhCrOx | 1 | 0.12 | — |
| 168 | 0.07 | — | |
| CoOOH/RhCrOx | 0 | 0.21 | 0.27 |
| 168 | 0.20 | 0.25 | |
| TiO2/CoOOH/RhCrOx | 0 | 0.06 | 0.04 |
| 168 | 0.06 | 0.04 |
The local structure and the valence state of the Co species loaded at the optimum concentration (0.3 wt%) were investigated by X-ray absorption fine structure (XAFS) analysis in distilled water under UV illumination as well under dark conditions. Fig. 2 shows Co-K edge X-ray absorption near edge structure (XANES) data and the Fourier transformed k3-weighted extended XAFS (EXAFS) spectra for RhCrOx/SrTiO3:Al loaded with 0.3 wt% Co species. The photodeposited Co species exhibit an absorption edge at an energy greater than the value for the CoCl2·6H2O used as a precursor, and the present specimen generated a XANES spectrum similar to that of CoOOH.17,18 This result indicates that CoOOH was photodeposited on the oxidation sites (i.e., the bare surface of the SrTiO3:Al). The chemical state of the deposited Co species was also confirmed from the local structure data. The valence state and structure of the photodeposited CoOOH were unchanged during 15 h of UV illumination, in contrast to the behaviour of Co-based electrocatalysts, in which Co2+ is oxidized to Co3+ and Co4+, after which the latter species oxidise OH− to O2.19–21 The Co in the present work likely maintained a constant valence state because the quasi-Fermi level for holes in the photocatalyst was determined by the balance between the hole concentration and charge generation/consumption (by surface reactions or recombination). Conversely, the potential for electrocatalysts can be externally fixed at specific values. Additionally, the XAFS data for the Rh species in the RhCrOx cocatalyst in aqueous solution, both under darkness and following the water splitting reaction, closely matched the results expected for trivalent Rh in a corundum structure. This structure is also essentially equivalent to that of RhCrOx loaded on GaN:ZnO.22 Therefore, the bulk chemical states of the CoOOH and RhCrOx cocatalysts were both unchanged during the photocatalytic water splitting reaction.
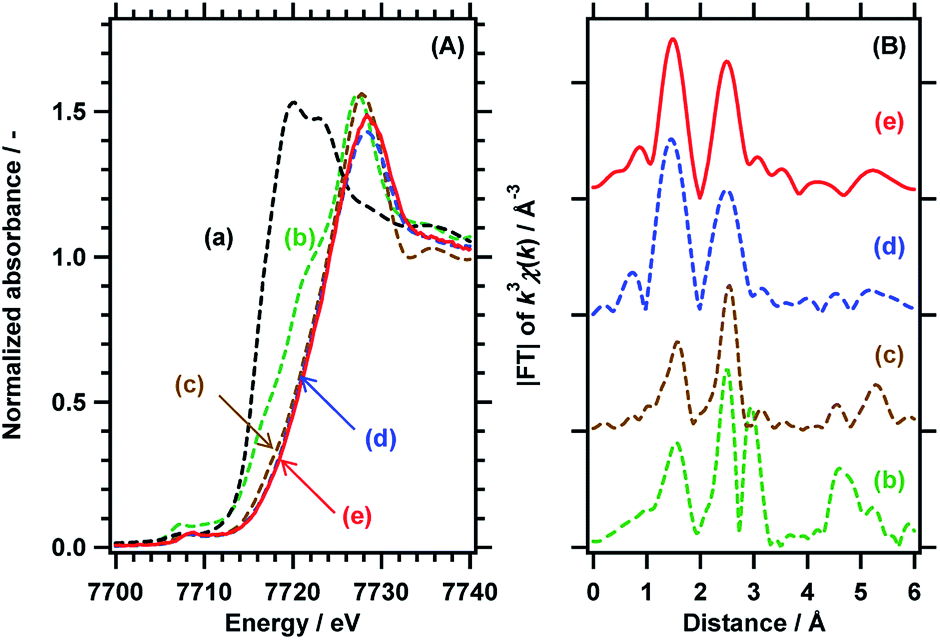 | ||
| Fig. 2 (A) Co-K edge XANES data and (B) Fourier transforms of k3-weighted Co-K EXAFS spectra for (a) CoCl2·6H2O, (b) Co3O4, (c) CoOOH, and RhCrOx/SrTiO3:Al coloaded with 0.3 wt% Co species (d) in darkness and (e) following UV illumination for 15 h by a 300 W Xe lamp. | ||
The function of CoOOH present on the RhCrOx/SrTiO3:Al surface can be interpreted as follows. In the absence of CoOOH (Fig. 3a), photoexcited holes oxidise not only water but also the RhCrOx, leading to the dissolution of Cr.12 As a result, backward reactions occur on the Cr-deficient RhCrOx cocatalyst exposed to the reaction solution. CoOOH loaded on the oxidation sites is believed to collect photoexcited holes (Fig. 3b). Thus, because holes are more likely to participate in the water oxidation reaction as opposed to reacting with the RhCrOx, oxidative corrosion of the RhCrOx cocatalyst and thereby the deactivation of the photocatalyst is suppressed. However, it is uncertain whether water oxidation occurs exclusively on CoOOH because RhCrOx/SrTiO3:Al exhibited the similar initial activities before and after the coloading of CoOOH (Fig. 1a and b). Effects of CoOOH coloading on the carrier dynamics and surface kinetics may be analysed by transient absorption spectroscopy in the presence of reactants.23
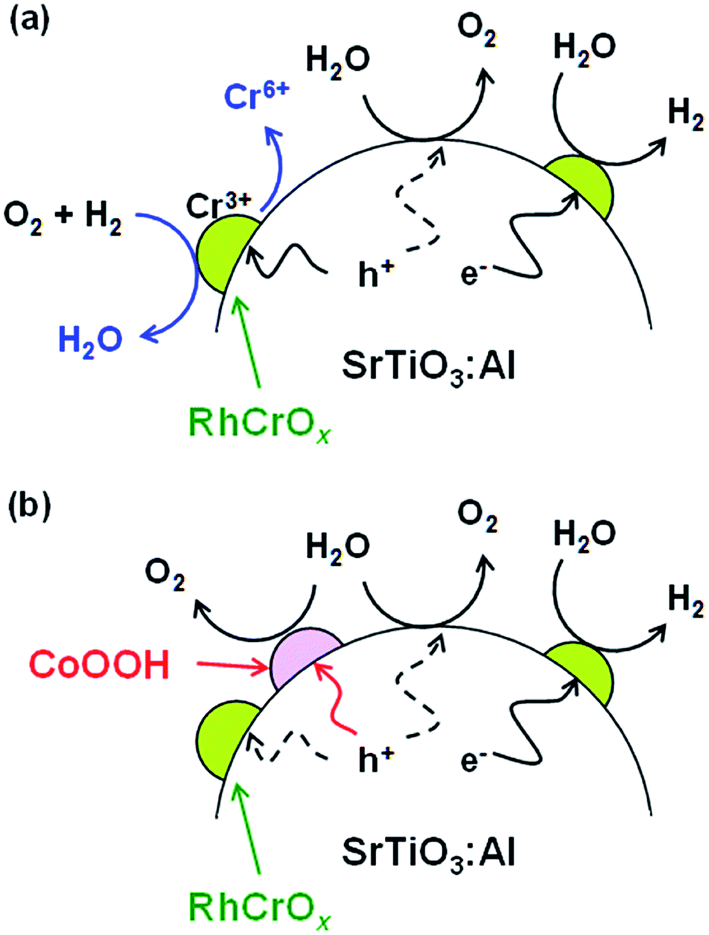 | ||
| Fig. 3 Illustrations of (a) RhCrOx/SrTiO3:Al deactivation mechanism and (b) stabilisation of RhCrOx/SrTiO3:Al by CoOOH. The stoichiometry of the reactions is ignored for simplicity. | ||
The durability of the CoOOH/RhCrOx/SrTiO3:Al photocatalyst was further enhanced by the photodeposition of amorphous TiO2 (3 wt%), as shown in Fig. 1c. This TiO2/CoOOH/RhCrOx/SrTiO3:Al photocatalyst was processed into 5 × 5 cm sheets and the durability of the material was examined in a panel-type reactor with a water depth of 1 mm under simulated sunlight at ambient pressure, to simulate a realistic operational mode.7 These sheets initially exhibited a water splitting rate of 5.7 μmol h−1 cm−2 and a solar-to-hydrogen conversion efficiency (STH) of 0.4% during the early stage of illumination. The activity gradually decreased by 20% over 1300 h of constant illumination (8.6 kJ cm−2 day−1) but the catalyst sheet maintained a minimum 0.3% STH throughout (Fig. 4). This time span corresponds to 173 days assuming an insolation of 2.7 kJ cm−2 day−1.11 Notably, the photocatalyst sheet recovered its original water splitting activity after being kept in darkness and maintained this recovered activity for several days of additional illumination. These characteristics would be advantageous during operation under natural sunlight. In comparison, RhCrOx/SrTiO3:Al modified only with CoOOH exhibited an initial water splitting rate of 5.0 μmol h−1 cm−2 and lost 40% of its original activity after 40 days under the identical reaction conditions.7 Therefore, the photodeposition of TiO2 evidently enhanced the durability of the CoOOH/RhCrOx/SrTiO3:Al sheet while maintaining a high STH during overall water splitting. In comparison, direct deposition of TiO2 onto RhCrOx/SrTiO3:Al lowered the water splitting activity while enhanced the durability (Fig. S4 in ESI†). Amorphous TiO2 has been reported to form an insoluble, selectively permeable layer covering the entire surface of the cocatalyst/photocatalyst composite and preventing backward reactions.13 The XPS data presenting the decrease in both the Cr/Ti and Co/Ti ratios support this scenario (Table 1). It is believed that the TiO2 played similar roles in the present work, and may also have suppressed detachment of the cocatalysts by the motion of the reaction media. Excessively thick TiO2 coating would induce mass transfer resistances and lower the water splitting activity. However, it is difficult to precisely characterise the function and structure of the TiO2 layers in this work, because Ti is also a main component of the photocatalyst itself. Our group intends to perform a more detailed analysis of the degradation behaviour in future, once the appropriate experimental protocols are established.
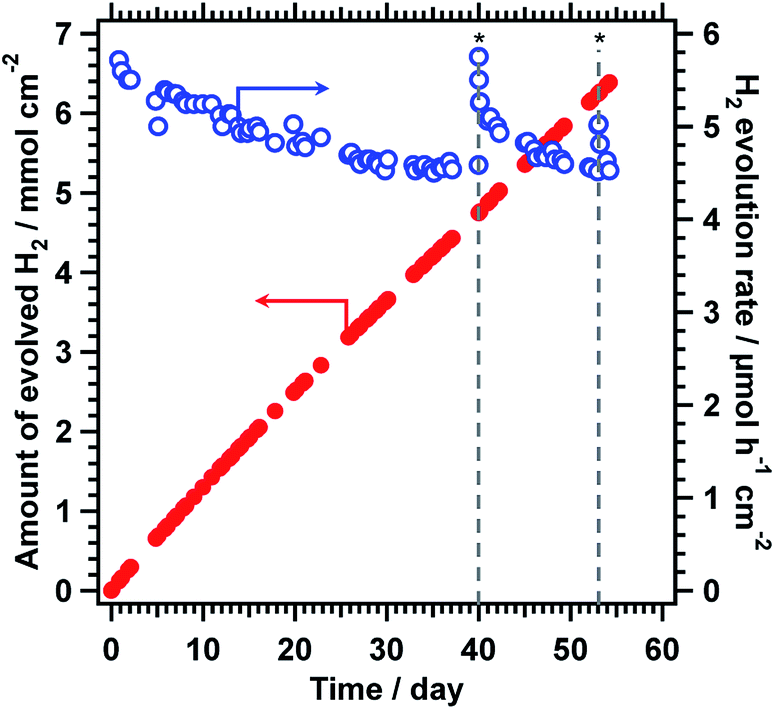 | ||
| Fig. 4 Time course of water splitting activity of 5 × 5 cm2 TiO2/CoOOH/RhCrOx/SrTiO3:Al sheet in panel reactor. Reaction conditions: photocatalyst, 20 mg; reaction solution, 1 mm deep distilled water; reaction temperature, 291 K; light source, AM 1.5G solar simulator. Asterisk symbols (*) indicate interruption of the illumination for two days. | ||
Conclusions
A RhCrOx/SrTiO3:Al photocatalyst highly active during overall water splitting was found to be stabilised via the photodeposition of CoOOH from CoCl2. The presence of CoOOH suppressed the oxidative loss of the Cr component of the RhCrOx cocatalyst. The photocatalyst could be further stabilised by photodeposition of TiO2, such that it maintained 80% of its initial activity during 1300 h of constant AM 1.5G irradiation at ambient pressure. This same material exhibited an STH of 0.3% or greater throughout the duration of the test when in the form of a photocatalyst sheet contained in a panel-type reactor. The durability of such sheets under natural sunlight could be further extended as the result of an apparent healing effect under dark conditions. The development of robust water splitting photocatalysts, as demonstrated herein, overcomes a long-standing barrier to practical applications. This technique is expected to assist in the development of sustainable large-scale solar hydrogen evolution systems.Experimental section
SrTiO3:Al was prepared using a flux method previously reported by our group.7 In this process, SrTiO3 (99.9%, Wako), Al2O3 nanopowder (Aldrich) and SrCl2 (98.0%, Kanto) were mixed at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.02:10 using an agate mortar. The mixture was then heated in an alumina crucible at 1423 K for 10 h and subsequently cooled to room temperature, after which the product was stirred in a large volume of distilled water and then retrieved by filtration to remove impurities associated with the SrCl2 flux. This rinsing process was repeated three times, after which the resulting SrTiO3:Al powder was dried at 313 K in an oven.
:0.02:10 using an agate mortar. The mixture was then heated in an alumina crucible at 1423 K for 10 h and subsequently cooled to room temperature, after which the product was stirred in a large volume of distilled water and then retrieved by filtration to remove impurities associated with the SrCl2 flux. This rinsing process was repeated three times, after which the resulting SrTiO3:Al powder was dried at 313 K in an oven.
This SrTiO3:Al powder was loaded with RhCrOx (0.1 wt% Rh and 0.1 wt% Cr) using an impregnation method,7 employing Na3RhCl6·nH2O (17.8 wt% Rh, Mitsuwa) and Cr(NO3)3·9H2O (98.0–103.0%, Kanto) as the Rh and Cr sources, respectively. In this process, SrTiO3:Al powder was dispersed in an aqueous solution containing the required amounts of the Rh and Cr precursors, and sonicated for several minutes. Following this, the solution was evaporated to dryness while being stirred, and the resulting solid was calcined in air at 623 K for 1 h. The AQY of the resulting RhCrOx/SrTiO3:Al during overall water splitting was determined to be 54% at 365 nm, which was consistent with our previous work.7
A gas flow system was used for photocatalytic water splitting reactions. The system was first purged with a mixture of gaseous Ar and N2 at ambient pressure, using the N2 as an internal standard for quantification purposes. A photocatalyst sample (0.1 g) was suspended in distilled water (100 mL) in a top-irradiation reactor, and irradiated through a Pyrex window using a 300 W xenon lamp (300 nm < λ < 500 nm) equipped with a dichroic mirror. In some experiments, a 50% transmission neutral density filter was inserted to reduce the light intensity. Trials were also performed with irradiation by a 450 W high-pressure Hg lamp (λ > 300 nm) to accelerate the water splitting reaction and thereby promote the deactivation of the photocatalyst, using an inner irradiation cell made of Pyrex glass containing a photocatalyst sample (0.5 g) suspended in distilled water (370 mL).
CoOOH was loaded onto RhCrOx/SrTiO3:Al by photodeposition at ambient pressure. The RhCrOx/SrTiO3:Al photocatalyst (0.5 g) was dispersed in distilled water (370 mL) containing CoCl2 and irradiated by a 450 W high-pressure Hg lamp (λ > 300 nm). Photodeposition was completed within 6 h. It was also found to be possible to deposit CoOOH using a top-irradiation cell under xenon lamp irradiation (see Fig. S2†), but a longer time span was required and the durability enhancement was less effective.
In preparation for the photodeposition of an amorphous TiO2 layer,13 titanium tetraisopropoxide (53 mg, 97.0%, Kanto) was added to 1 mL of an aqueous H2O2 solution (30%, Wako) and sonicated repeatedly for several minutes at a time until a pale yellow solution was obtained. An aqueous NaOH solution (1 M) was subsequently added to obtain a Na:Ti molar ratio of 2:1, which generated a transparent, light yellow solution. This solution was subsequently added to a reactant solution containing 0.5 g CoOOH/RhCrOx/SrTiO3:Al in an inner-irradiation cell so that the TiO2 concentration was 3 wt% with respect to the photocatalyst. The suspension was illuminated by a high-pressure Hg lamp (λ > 300 nm) at ambient pressure for at least 2 h. It was also possible to deposit TiO2 using a top-irradiation cell under xenon lamp irradiation. However, the durability enhancement was less effective when the resultant photocatalyst was processed into the form of sheets.
Samples were characterised by X-ray photoelectron spectroscopy (XPS; JPS-9000, JEOL) and X-ray absorption fine structure (XAFS) spectroscopy. The binding energies acquired by XPS were corrected using the Au 4f7/2 peak (84.0 eV) as a reference for each sample. Co-K edge XAFS data were acquired using the BL5S1 beamline at the Aichi Synchrotron Radiation Centre (Aichi, Japan), employing the CoOOH/RhCrOx/SrTiO3:Al photocatalyst (0.1 wt% Rh, 0.1 wt% Cr and 0.3 wt% Co). Fluorescence X-rays were detected using Si drift detectors. Rh-K edge XAFS data were obtained on the NW10A beamline (proposal no. 2017G076) at the Photon Factory of the High Energy Accelerator Research Organization (Tsukuba, Japan) using the CoOOH/RhCrOx/SrTiO3:Al photocatalyst (0.3 wt% Rh, 0.3 wt% Cr and 0.3 wt% Co). Fluorescence X-rays were detected with Ge solid-state detectors after passing through a Ru filter. XAFS analyses of the CoOOH/RhCrOx/SrTiO3:Al photocatalysts were performed using the operando technique in distilled water under UV irradiation as well as under dark conditions. The operando XAFS spectroscopy setup is pictured in Fig. S5.† CoOOH/RhCrOx/SrTiO3:Al (0.4 g) was suspended in distilled water (approximately 2 mL) in a Pyrex tube (inner diameter of 9 mm and outer diameter of 12 mm) that had a curved Kapton window along its lateral face, and was vigorously stirred using a magnetic stirrer. The photocatalyst suspension was irradiated with UV light through the glass tube, using a 300 W xenon lamp (300 nm < λ < 500 nm) equipped with a quartz optical fibre. Note that vigorous stirring was necessary because otherwise the photocatalyst powder was pushed upwards by bubbles of hydrogen and oxygen. Incident X-rays were targeted such that they irradiated the photocatalyst suspension by passing through the curved Kapton window but not through the glass tube. Fluorescence X-rays were also collected through the window. During the illumination process, the photocatalyst suspension was cooled with a fan. Reference samples were analysed in transmittance mode using pellets made of mixtures of commercial compounds and boron nitride, when available. Rh0.5Cr1.5O3 was prepared by a polymerised complex method.22 CoOOH was prepared by calcining commercial Co(OH)2 (97.0%, Kishida) in air.16 Reaction solutions were analysed by inductively-coupled plasma optical emission spectrometry (ICP-OES; ICPS-8100, Shimadzu).
Reactors of our own design were employed to assess the activity of TiO2/CoOOH/RhCrOx/SrTiO3:Al photocatalyst sheets. The RhCrOx/SrTiO3:Al photocatalyst powder was modified with CoOOH (0.3 wt% as Co) and TiO2 (3 wt% as TiO2) by photodeposition under Hg lamp irradiation for 6 and 3 h, respectively, prior to processing into sheets. The details regarding the processing of the photocatalyst into sheets and the design of the reactor have been described elsewhere.7 Distilled water was used as the reaction solution and the panel reactors were tilted at an angle of 20° (so that water bubbles could move upward as a result of the buoyant force) and were illuminated with a solar simulator (XES-40S2-CE, SAN-EI Electric) generating AM 1.5G irradiation (100 mW cm−2). The evolved gases were collected based on the upward displacement of water. The amount of hydrogen evolved was subsequently calculated based on the assumption that the molar ratio of the evolved hydrogen and oxygen was the expected stoichiometric value of 2:1.
Conflicts of interest
There are no conflicts to declare.Author contributions
K. D. led the research project. T. Hisatomi and K. D. supervised the experimental work. H. L. prepared and analysed the photocatalyst and conducted the water splitting reactions. T. Hisatomi, Y. G., M. Y., T. Higashi, M. K. and K. A conducted the XAFS analyses. T. T. conceived the TiO2 coating and conducted the water splitting reactions using photocatalyst sheets. H. N. prepared the panel-type reactors. T. Y. performed ICP-OES analyses. H. L., T. Hisatomi, Y. G., M. Y., T. Higashi, M. K., T. T., T. M, Y. S., K. A. and K. D. discussed the results. H. L., T. H. and K. D. wrote the manuscript with contributions from the other authors. All authors reviewed the manuscript.Acknowledgements
This work was financially supported by Grants-in-Aid for Scientific Research (A) (no. 16H02417) and for Young Scientists (A) (no. 15H05494) from the Japan Society for the Promotion of Science (JSPS). This work was also supported by the Artificial Photosynthesis Project of the New Energy and Industrial Technology Development Organization (NEDO). The authors thank Ms. Yasuko Kuromiya of the University of Tokyo for conducting the long-term tests of the photocatalyst activity and Dr Hideki Kato of Tohoku University for his advice regarding operand XAFS analyses. A part of this work was conducted at the Photon Factory of the High Energy Accelerator Research Organization, Japan (proposal no. 2017G076, 2017G529 and 2016G647).Notes and references
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- B. Mei, K. Han and G. Mul, ACS Catal., 2018, 8, 9154–9164 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- X. Yang and D. Wang, ACS Appl. Energy Mater., 2018, 1, 6657–6693 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- Y. Sakata, T. Hayashi, R. Yasunaga, N. Yanaga and H. Imamura, Chem. Commun., 2015, 51, 12935–12938 RSC.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2017, 2, 509–520 CrossRef.
- M. G. Kibria, F. A. Chowdhury, S. Zhao, B. AlOtaibi, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 6797 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- W. Che, W. Cheng, T. Yao, F. Tang, W. Liu, H. Su, Y. Huang, Q. Liu, J. Liu, F. Hu, Z. Pan, Z. Sun and S. Wei, J. Am. Chem. Soc., 2017, 139, 3021–3026 CrossRef CAS PubMed.
- T. Setoyama T, T. Takewaki, K. Domen and T. Tatsumi, Faraday Discuss., 2017, 198, 509–527 RSC.
- T. Ohno, L. Bai, T. Hisatomi, K. Maeda and K. Domen, J. Am. Chem. Soc., 2012, 134, 8254–8259 CrossRef CAS PubMed.
- T. Takata, C. Pan, M. Nakabayashi, N. Shibata and K. Domen, J. Am. Chem. Soc., 2015, 137, 9627–9634 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107–13112 CrossRef CAS PubMed.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- G. G. Amatucci, J. M. Tarascon, D. Larcher and L. C. Klein, Solid State Ionics, 1996, 84, 169–180 CrossRef CAS.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- E. B. Castro, C. A. Gervasi and J. R. Vilche, J. Appl. Electrochem., 1998, 28, 835–841 CrossRef CAS.
- S. Palmas, F. Ferrara, A. Vacca, M. Mascia and A. M. Polcaro, Electrochim. Acta, 2007, 53, 400–406 CrossRef CAS.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1425–1462 CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13753–13758 CrossRef CAS PubMed.
- R. Godin, T. Hisatomi, K. Domen and J. R. Durrant, Chem. Sci., 2018, 9, 7546–7555 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Time course of the water splitting activity, XPS spectra, and experimental setup for operando XAFS spectroscopy. See DOI: 10.1039/c8sc05757e |
| ‡ Department of Physics, Tokyo Metropolitan University, 1-1, Minami-Osawa, Hachioji, Tokyo 192-0397, Japan. |
| This journal is © The Royal Society of Chemistry 2019 |