
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical ADP-ribosylation: mono-ADPr-peptides and oligo-ADP-ribose
Qiang
Liu
,
Gijsbert A.
van der Marel
and
Dmitri V.
Filippov
 *
*
Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: filippov@chem.leidenuniv.nl
First published on 13th May 2019
Abstract
ADP-ribosylation is an important post-translational modification that plays a pivotal role in many cellular processes, including cell signaling, DNA repair, gene regulation and apoptosis. Although chemical synthesis of mono- or poly-ADP-ribosylated biomolecules is extremely difficult due to the challenges in regio- and stereoselective glycosylation, suitable protective group manipulations and pyrophosphate construction, synthetic procedures towards these bio-related targets have been reported in recent years. Chemically synthesized well-defined ADP-ribose derivatives serve as useful tools in biological experiments aimed to further elucidate native ADP-ribosylation. In this review, we will discuss the synthetic studies on mono-ADP-ribosylated proteins and oligo-ADP-ribose chains. Future possible synthetic targets and upcoming new methods for the synthesis of these molecules are also included.
 From left to right: Dmitri Filippov, Gijs van der Marel and Qiang Liu | Qiang Liu is a PhD student at Leiden University where he works under the supervision of Prof. Gijs van der Marel and Dmitri Filippov. He earned his B.Sc in pharmaceutics from Jining Medical University in 2012 and M.Sc in medicinal chemistry from Sichuan University in 2015 in China. Qiang focuses his research on the organic synthesis of bio-related ADP-ribosylated molecules such as linear or branched ADPr-oligomers and mono-ADP-ribosylated peptides and their derivatives. Gijs van der Marel is a Full Professor in Organic Chemistry at Leiden University. He trained with Prof. Jacques van Boom on the development of synthetic methods for oligonucleotides and obtained his PhD degree in 1981. His research has been directed at the development of synthetic chemistry to assemble all types of biopolymers: nucleic acids, peptides and carbohydrates as well as hybrids and analogues thereof to study their role in biology. He has supervised >100 PhD students and his research interests include synthetic organic chemistry methodology, biopolymer synthesis, glycobiology and chemical biology and immunology. Dmitri Filippov obtained his Ph.D. degree from Leiden University (1998) under the guidance of Jacques van Boom and Gijs van der Marel while he was investigating the application of phosphoramidite reagents to the synthesis of peptide–nucleic acid hybrid biopolymers. Dmitri has remained in Leiden ever since where he currently is an Assistant Professor. His research interests include synthetic bioorganic chemistry with the focus on the chemistry of ADP-ribosylated biomolecules and other hypermodified peptides and nucleic acids, both native and artificial. |
1. Introduction
ADP-ribosylation is a post-translation modification of proteins that occurs upon enzymatic transfer of the ADP-ribosyl moiety from NAD+ to a nucleophilic side chain of an amino acid of a protein.1–3 As a result either mono-ADP-ribose (MAR) or oligo-ADP-ribose (PAR) becomes grafted to the protein (Fig. 1). Both modifications play an important regulatory role in various physiological and pathological processes.4 The transfer of PAR to amino acids on protein substrates is catalyzed by four enzymes of the PARP family: PARP1, PARP2, and PARP5a, and PARP5b. PAR can exist as a linear or branched polymer. Other PARP family members (PARP3, 4, 6–12, and 14–16) transfer only MAR to amino acids on protein substrates. Upon ADP-ribosylation of cellular proteins, either mono- or polymers, the posttranslational modification becomes subject to further recognition and processing by proteins that are capable of removing or binding PAR or MAR (Fig. 1).1 Such variations of the ADP-ribosylation state result in a change of the intracellular signaling. Hydrolases such as PARG and enzymes from the ARH-family are responsible for the breakdown of PAR and MAR and thus for the reversal of ADP-ribosylation.5,6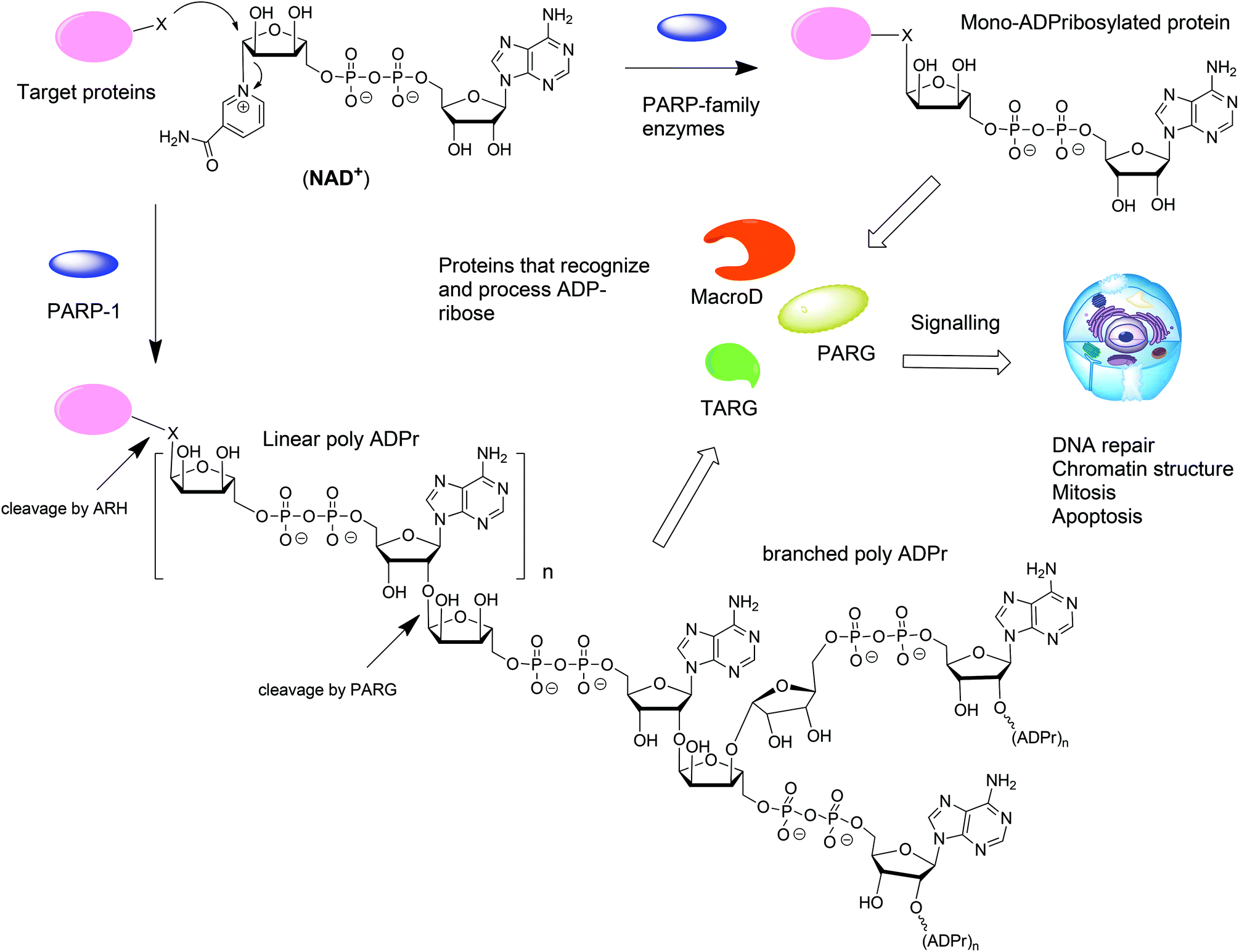 | ||
| Fig. 1 Biosynthesis and metabolism of mono- and poly-ADP-ribosylated proteins. | ||
From the point of view of a bioorganic chemist, both mono-ADP-ribosylated (MARylated) and poly-ADP-ribosylated (PARylated) biopolymers (Fig. 1) present a significant challenge. Nevertheless, synthetic well-defined ADP-ribosylated proteins or their substructures are useful for the studies that are aimed at elucidation of the biological role of ADP-ribosylation. This review describes the synthetic advances towards the synthesis of mono-ADP-ribosylated proteins and oligo-ADP-ribose chains.
2. Chemical synthesis of mono-ADPr-peptides and ADPr-oligomers
The organic synthesis of ADP-ribosylated biomolecules is challenging as the construction of these hybrid structures requires the use of elements from the synthetic chemistry of nucleic acids, oligosaccharides and oligopeptides that are sometimes incompatible. The synthetic challenge is augmented by the necessity to introduce one or even multiple pyrophosphate linkages, which are notoriously difficult to construct efficiently. In the following sections we describe synthetic approaches to the primary challenges of chemical ADP-ribosylation, that is, ribosylation of side chains of various amino acids (Fig. 2, feature A) that culminates in the synthesis of mono-ADP-ribosylated proteins, stereoselective glycosylation of the 2′-OH of adenosine (Fig. 2, feature B) and assembling the oligo-sugar pyrophosphate chain of oligo-ADPr (Fig. 2, feature C).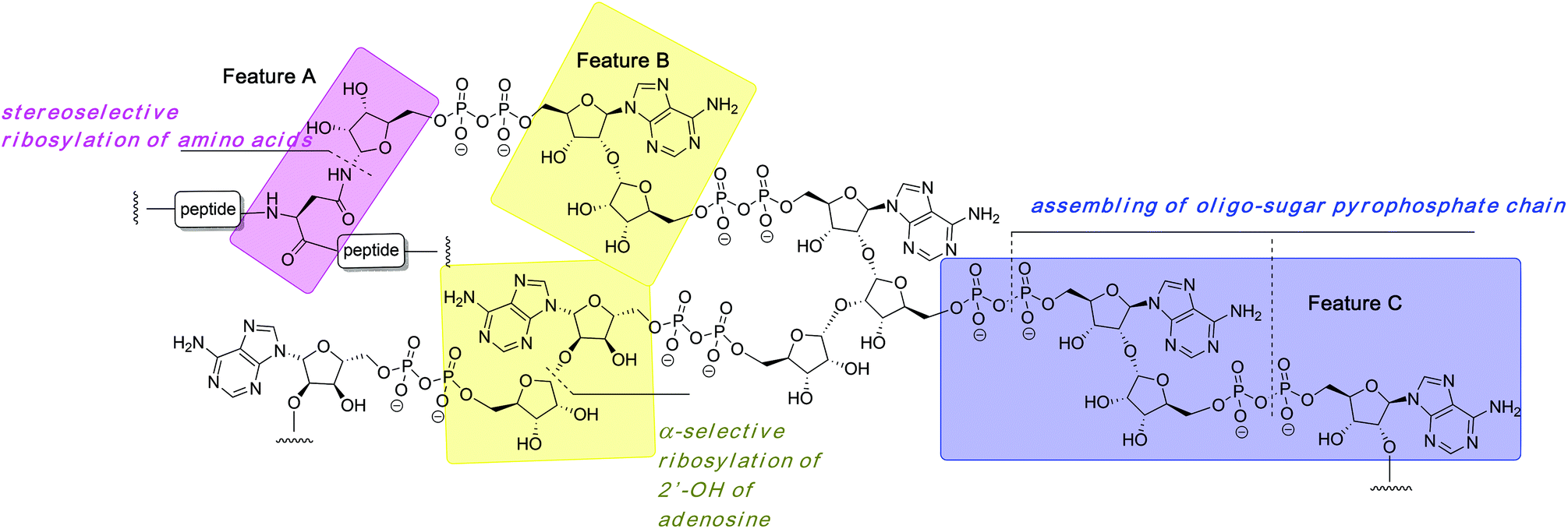 | ||
| Fig. 2 Structure of poly-ADP-ribose with its most conspicuous synthetically challenging features. | ||
2.1 Synthesis of mono-ADP-ribosylated peptides
Mono-ADP ribosylated proteins play intriguing roles in many cellular processes.7 An approach to deepen the insight into these processes to a molecular level comprises the design, synthesis and biological evaluation of well-defined synthetic mono-ADP-ribosylated derivatives. Relevant examples of such compounds are mono-ADP-ribosylated oligopeptides,8–10 as fragments of the naturally occurring proteins. The main challenges in the assembly of these ADP-ribosylated oligopeptides are an efficient procedure for the introduction of the pyrophosphate function and a method for the stereoselective α-ribosylation of the nucleophilic side chains of amino acids. In this section we will focus on the construction of the α-glycosidic bond that joins the “distal” ribose of the ADPr-moiety and an amino acid side chain in the context of the mono-ADPr-peptide synthesis. The methods that have been developed for the introduction of one pyrophosphate linkage in mono-ADP-ribosylated peptides will be discussed in this section while the introduction of multiple pyrophosphates in short fragments of poly-ADPr is the subject of discussion in section 2.2.2. Application of a solid phase approach to mono-ADP-ribosylated oligopeptides is most obvious as a solution phase synthesis would be restricted in terms of the length and composition of the oligopeptide. While the introduction of the pyrophosphate moiety is feasible on a solid support,8,9 ribosylation of partially protected and immobilized oligopeptides with a protected ribose donor is almost impossible in terms of stereoselectivity and yield. Therefore, attention has been focused on the synthesis of suitably protected ribosylated amino acid building blocks that can be applied in solid phase peptide synthesis (SPPS). The main hurdle in the synthesis of these ribosylated amino acid building blocks is the difficulty to control the 1,2-cis configuration of the ribosyl anomeric linkage at the glycosylation stage. The sensitivity of this O-glycosidic bond to an acid adds another layer of complexity.The first reported synthesis of suitably protected α-ribosylated amino acid building blocks and their application in a SPPS assembly of relevant ADP-ribosylated oligopeptides is by van der Heden van Noort et al.8 The choice for Fmoc-based peptide synthesis led to the synthesis of protected α-ribosylated asparagine (Asn) 5 and glutamine (Gln) 6 building blocks (Scheme 1). The route of synthesis started with the reduction of fully protected β-D-ribosylated azide 1 to an epimeric hemiaminal mixture 2. Subsequently, EDC-mediated coupling with Z-Glu-OBn and Z-Asp-OBn, respectively and silica gel purification gave the individual anomers 3 and 4. Protective group manipulation provided α-ribosylated Asn (5) and Gln (6) building blocks with the mutually orthogonal TBDPS and Fmoc protecting groups. Guided by the outcome of a solution phase study, SPPS was undertaken in which two procedures for the installation of the adenosine diphosphate function were explored. For that purpose, native11 model peptide 11 containing an ADP-ribosylated Asn residue and peptide 15 originating from the N-terminus of human histone H2B containing an ADP-ribosylated Gln residue were selected. In the latter case Gln was chosen as a stabilized isostere of Glu that was reported to be the natural ADP-ribosylation site.12 Hexapeptide 7 was obtained via SPPS using a BOP/HOBT Fmoc-based synthesis executed on Tentagel resin equipped with an HMBA linker. Upon removal of the TBDPS group at the 5-OH of the ribose, the immobilized peptide 7 (R = H) was phosphitylated with phosphoramidite 8 under the influence of the activator DCI, followed by oxidation using iodine in pyridine to give the activated phosphorimidazolate 9. A reaction with the protected adenosine phosphate 10 led to the formation of the protected and immobilized target ADP-ribosylated peptide. Removal of the Dmab group on Glu and subsequent treatment with ammonia methanol, to affect both the removal of the remaining protecting groups and cleavage from the resin, gave after HPLC purification ADP-ribosylated hexapeptide 11. With the aid of LC-MS analysis of the crude product the C-terminal carboxamide, the ribosyl 5-phosphomonoester and the corresponding H-phosphonate could be identified as side products. It was reasoned that the formation of H-phosphonate could be suppressed by the reversal of the procedure for pyrophosphate formation. To this end the phosphate was installed on the immobilized peptide (i.e.13), while activated phosphorimidazolate (i.e.14) was prepared in solution. The assembly of ADP-ribosylated peptide 15 started with the SPPS of heptapeptide 12 according to the same procedure as described for hexapeptide 7. Protective group manipulation led to 12 (R′ = Ac, R = H) having only base labile protecting groups. The phosphate moiety was introduced by phosphitylation with 8 under the influence of the activator DCI, oxidation of the intermediate phosphite triester with t-BuO2H and, finally, removal of the p-methoxybenzyl groups with TFA to give phosphomonoester 13. Immobilized 13 was now treated with an excess of activated phosphorimidazolate 14 to afford the immobilized and protected precursor of target 15.
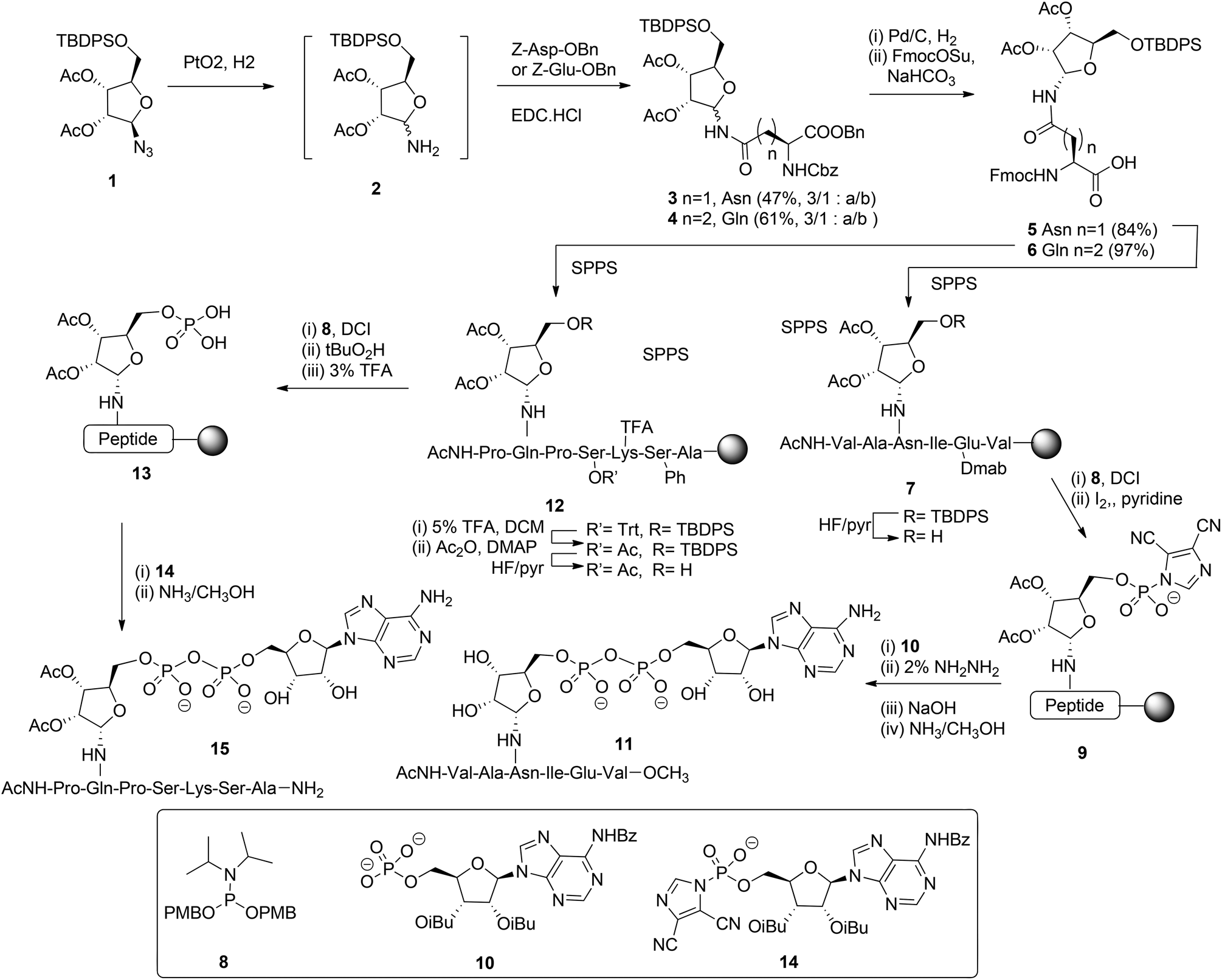 | ||
| Scheme 1 Synthesis of ADP ribosylated peptides 11 and 15. | ||
The removal of all protecting groups and concomitant cleavage from the resin gave ADP-ribosylated heptapeptide 15. However, also with this procedure the unwanted formation of the phosphate monoester (from intermediate 13) and the corresponding H-phosphonate could not be circumvented. In spite of the successful application of the α-ribosylated asparagine 5 and glutamine 6 building blocks in SPPS and the isolation of pure ADP-ribosylated peptides 11 and 15 in reasonable yields, it became apparent that the assembly of ADP-ribosylated peptides would benefit from a more efficient procedure for pyrophosphate formation.
Other authors also undertook the synthesis of protected α-ribosylated asparagine and glutamine building blocks. Thus, Bonache et al.13 have prepared such a derivative for the first time, while F. Nisic et al.14,15 developed a stereoselective synthesis of α- or β-glycofuranosyl amides with the aid of traceless Staudinger ligation of glycofuranosyl azides. Application of this approach for the synthesis of α-N-ribosyl-asparagine/glutamine building blocks is depicted in Scheme 2.16
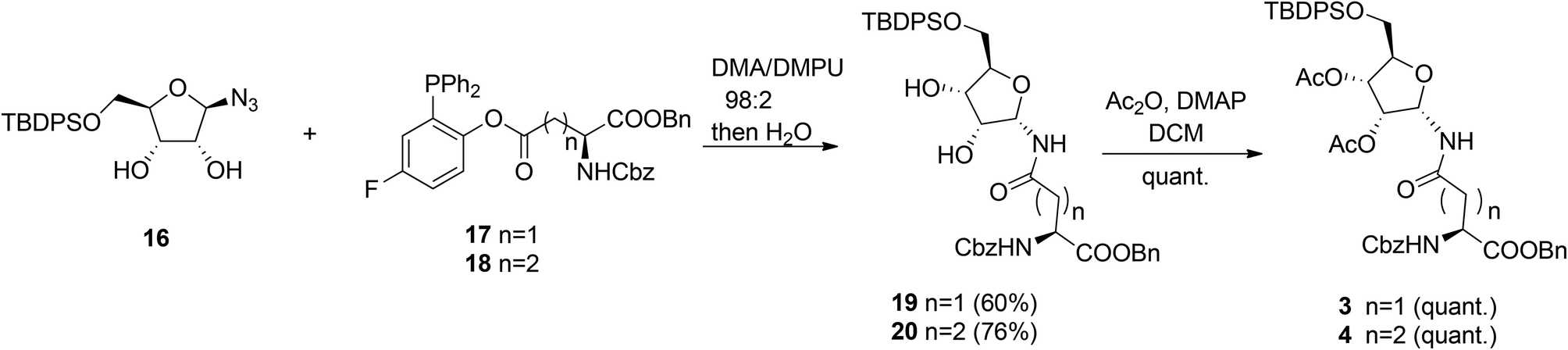 | ||
| Scheme 2 Synthesis of α-ribofuranosyl amides using fluorinated phosphines. | ||
Fluorinated triphenylphosphines functionalized with Z-Asp-OBn (17) and Z-Glu-OBn (18) were used in ligation reactions with differently protected β-D-ribofuranosyl azides. It turned out that both stereochemistry and productivity of these reactions were dependent on the protection of the hydroxyl groups in the ribose moiety. Protection of the primary 5-OH with the TBDPS group (16) produced (Asn) 19 and (Gln) 20 in good yields. Subsequent acetylation of 19 and 20 gave known8 SPPS building blocks 3 and 4.
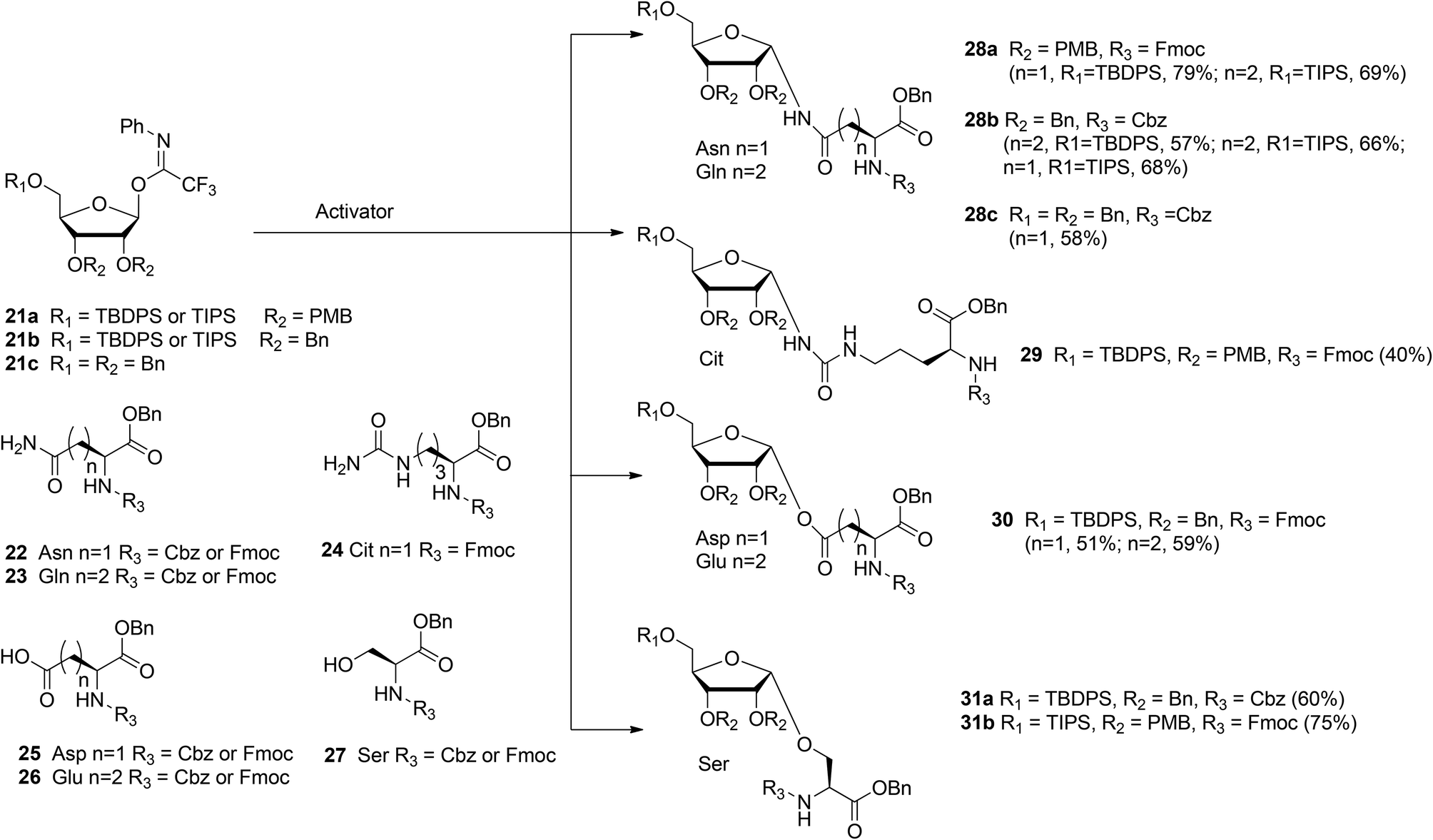
With the aim of broadening the range of the synthetically accessible ribosylated amino acids Kistemaker et al.17 developed an alternative ribosylation method that employed ribosyl donors 21a, b, and c (Scheme 3) with the N-phenyl trifluoroacetimidate leaving group and with non-participating ether protecting groups at 3- and 2-OH. The latter feature allows the formation of both O- and N-glycosidic linkages via highly α-selective acid catalyzed glycosylation. Condensation of perbenzylated donor 21c with Asn acceptor 22 (R3 = Cbz) under various conditions led to α-product 28c (n = 1). However, these conditions were not transferable to other acceptors (e.g. Glu acceptor 23 (R3 = Cbz)). It was reasoned that the selectivity of the ribosylation could be improved by replacing the benzyl group at the 5-OH in the ribose by the bulkier TBDPS or TIPS protecting groups to give donor 21b (R1 = TBDPS or TIPS). Several activator systems were tested and the results of these tests indicated that TMSOTf and HClO4-SiO2 were the most favorable activators. A reaction of donor 21b (R1 = TBDPS or TIPS) with Asn acceptor 22 (R3 = Cbz) and Gln acceptor 23 (R3 = Cbz) gave good to excellent yields of α-products 28b (n = 1, 2, respectively). Next, this glycosylation protocol was applied to Cbz- and Fmoc-protected glutamic acid (Glu, 26), aspartic acid (Asp, 25) and serine (Ser, 27). Using TMSOTf as activator protected derivatives of ribosylated Asp 30 (n = 1, α/β = 98:2, 51%), ribosylated Glu 30 (n = 2, α/β = 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 59%) and ribosylated Ser 31a (R1 = TBDPS, α/β = 1:0, 60%) were obtained.
:2, 59%) and ribosylated Ser 31a (R1 = TBDPS, α/β = 1:0, 60%) were obtained.
 | ||
| Scheme 3 Trifluoroacetimidate ribosylation of partially protected amino acids. | ||
In order to minimize protecting group manipulations towards the ribosylated amino acid building blocks suitable for SPPS, the benzyl groups at the 2-OH and 3-OH in the ribosyl donor were replaced with acid labile PMB ethers and the Cbz group in the amino acid acceptors was replaced with the Fmoc group. Condensation of imidate donor 21a (R1 = TBDPS) with Asn acceptor 22 (R3 = Fmoc) in DCM under the influence of TMSOTf furnished 28a (n = 1, α/β = 97:3, 79%). A similar condensation using the less nucleophilic citrulline (Cit) acceptor 24 proceeded in a less α-selective manner to give 29 (α/β = 78:22, 40%). The insolubility of Gln acceptor 23 (R3 = Fmoc) required a change to dioxane/DCM as the solvent system and HClO4-SiO2 as the activator to give 28a (n = 2, α/β = 93:7, 69%). Finally, condensation of Ser acceptor 27 (R3 = Fmoc) with donor 21a (R1 = TIPS) furnished ribosylated Ser 31b (α/β = 1:0, 75%).
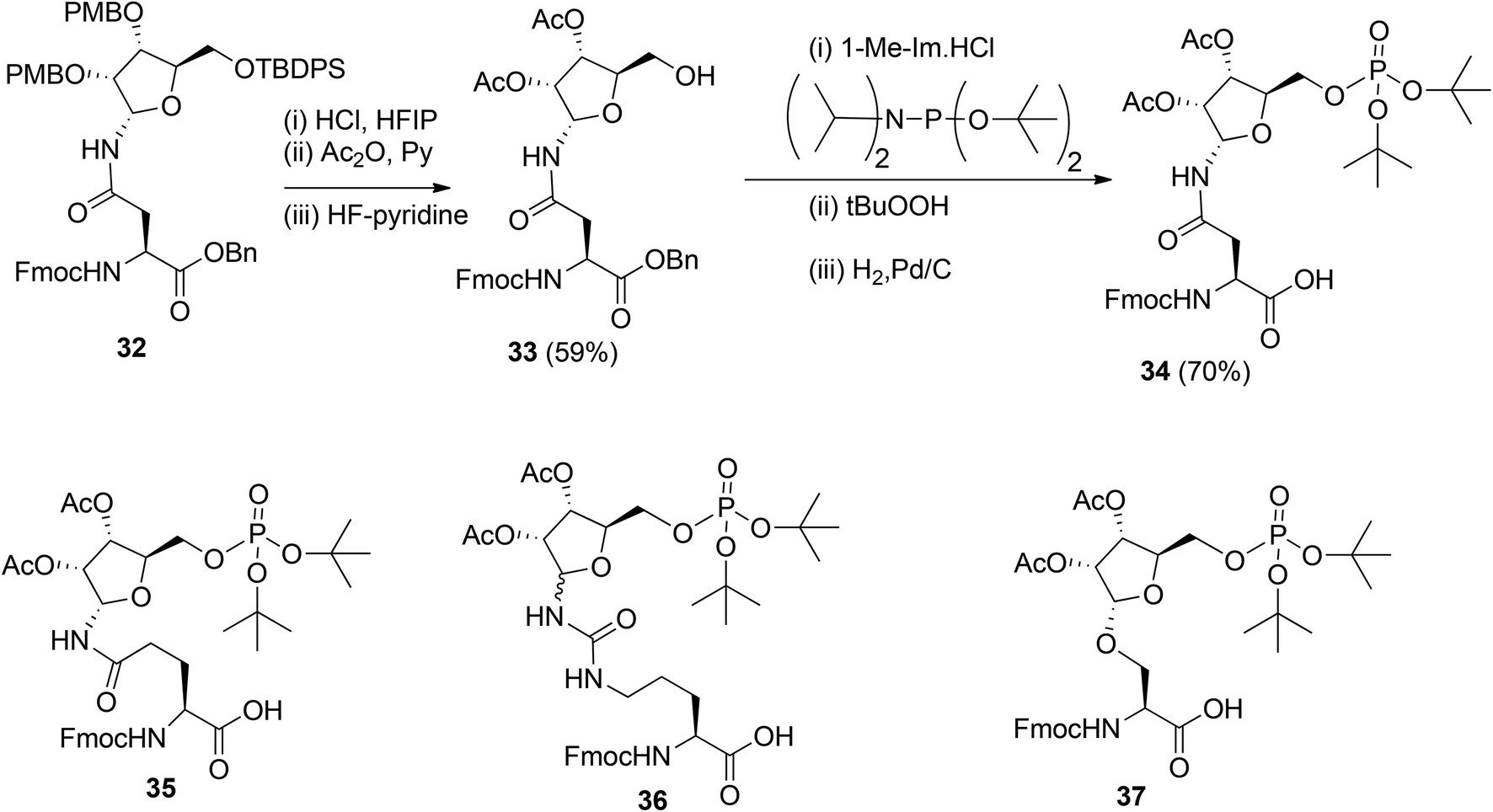
To obtain relevant ADP-ribosylated oligopeptides by SPPS, Kistemaker et al.9,18 also searched for another procedure for pyrophosphate formation. To this end, the solution phase method for the synthesis of sugar nucleotides, reported by Gold et al.,19 was adopted. This procedure9 combines phosphoramidite (PIII) with phosphate (PV) chemistry and the adaptation to a solid phase procedure required the on-resin formation of a phosphomonoester. It was reasoned that this could be circumvented by the development of the protected pre-phosphorylated amino acid building blocks 34–37 (Scheme 4). The synthesis of these phosphorylated amino acids is illustrated by the preparation of Asn building block 34. The PMB groups in fully protected ribosylated Asn 32 were replaced by acetyl groups by acidolysis, followed by acetylation while 5-OH was unmasked by desilylation to afford 33. The tert-butyl group was selected as an orthogonal phosphate protecting group.
 | ||
| Scheme 4 Ribosylated amino acids with a phosphotriester at 5-OH. | ||
The di-tert-butyl phosphate triester was installed with di-tert-butyl N,N-diisopropylphosphoramidite and subsequent oxidation of the intermediate phosphite triester. Finally, hydrogenolysis of the benzyl ester gave the α-configured Asn building block 34 suitable for SPPS. Transferring this procedure to other amino acids showed that the anomeric integrity of Gln 35 and Ser 37 remained intact while Cit 36 was obtained as an anomeric mixture, which could be separated by column chromatography.
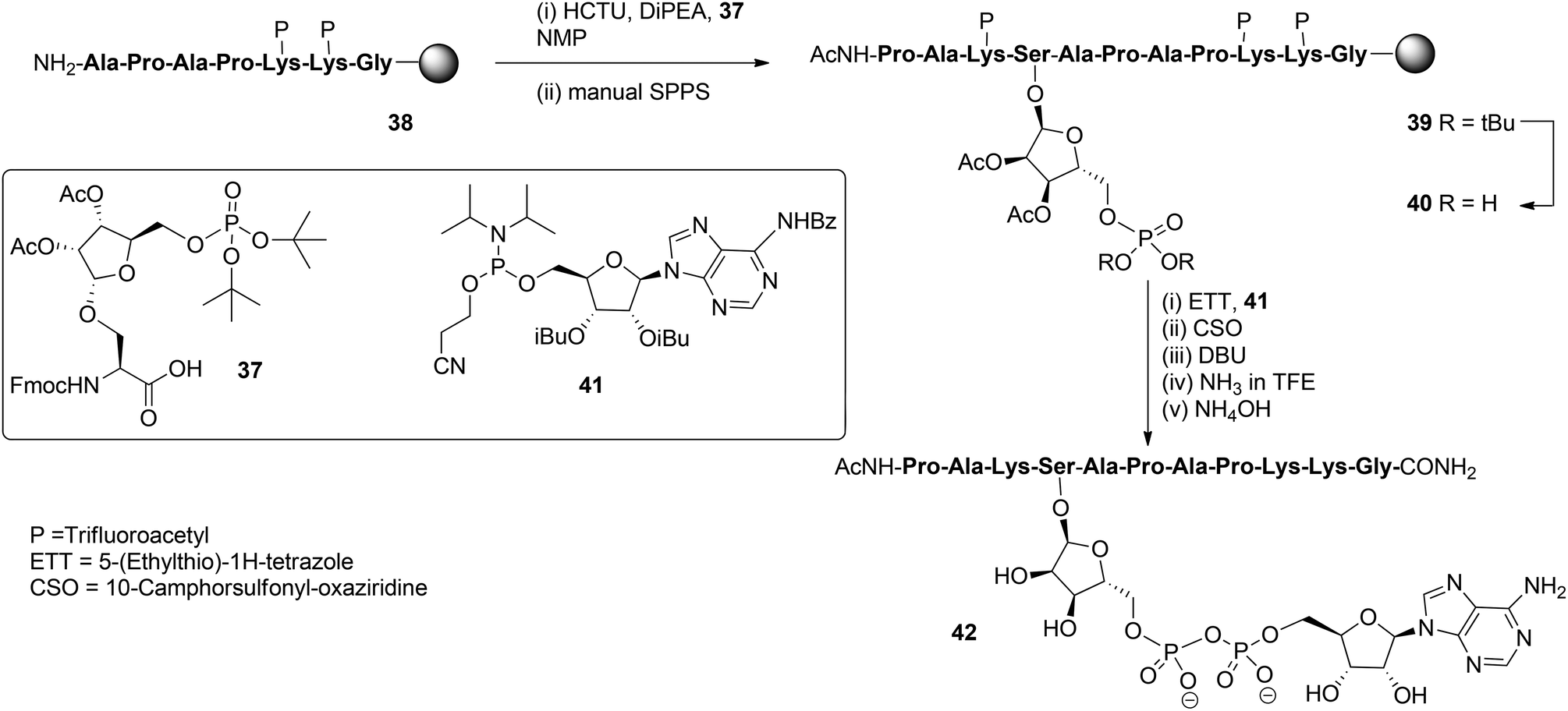
With these building blocks available SPPS could be undertaken and relevant ADP-ribosylated oligopeptide fragments from Histone H2B, RhoA protein and HNP-1 defensin were obtained.9 The synthesis of Ser-ADPr H2B peptide 42 (Scheme 5) serves as a representative example of the usefulness of this methodology for the preparation of ADP-ribosylated peptides with a native ADP-ribosylation site.10 The SPPS of hendecapeptide 42 was carried on Tentagel resin, equipped with an HMBA-linker. First, intermediate immobilized heptapeptide 38 was produced with automated SPPS utilizing Fmoc chemistry and trifluoroacetyl protected lysine residues. Subsequent elongation to phosphoribosylated peptide 39 was done manually using serine phosphotriester 37 and commercially available protected amino acids.
 | ||
| Scheme 5 SPPS of Ser-ADPr hendecapeptide originating from histone H2B. | ||
To allow pyrophosphate introduction, di-tert-butyl phosphate protective groups in the immobilized 39 were removed with TFA and DCM, followed by neutralizing with pyridine. A reaction of the obtained phosphate monoester 40 with phosphoramidite 41 under the influence of activator ETT was followed by the oxidation of the PIII–PV intermediate with CSO and, finally, the cyanoethyl group in the pyrophosphate was removed with DBU. Target pyrophosphate 42 was obtained by a two-step procedure. First, cleavage from the resin was attained by treatment with a saturated NH3 solution in trifluoroethanol. In this way the formation of a carboxylic acid at the C-terminus of the peptide is circumvented and only carboxamide is produced. Addition of NH4OH effected the removal of all remaining protective groups giving hendecapeptide 42. This is the first reported synthesis of an ADP-ribosylated peptide on serine,10 which has been found to be one of the most widespread ADPr-modification sites in histones, upon DNA damage.20–23 It is also the first real, naturally occurring ADP-ribosylated oligopeptide as all others are provided with stabilized anomeric ribosyl linkages, that is, asparagine (Asn) and glutamine (Gln)8,9 as stabilized analogues of aspartic acid (Asp) and glutamic acid (Glu), respectively.
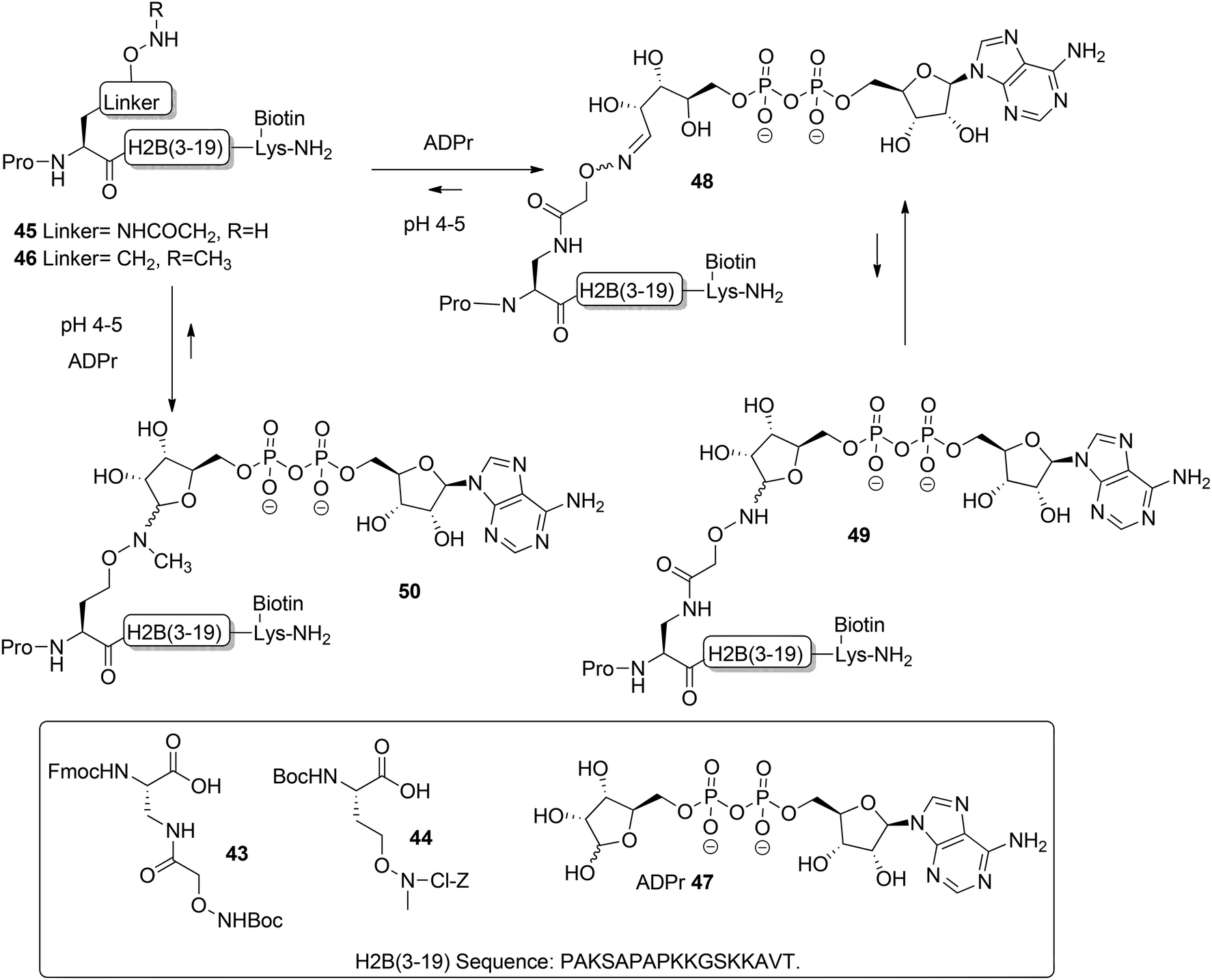
Moyle and Muir24 reported the synthesis and a biochemical evaluation of stabilized and artificial mono-ADP-ribose conjugated peptides. An N-terminal (3–19) oligopeptide24 of histone H2B protein with the glutamate residue mono-ADP-ribosylated was selected as a model (Scheme 6). With the aid of manual SPPS on MBHA resin using HBTU/DIPEA, oligopeptides 45 and 46 having either an aminooxy or N-methyl aminooxy functionality were assembled. The aminooxy-containing building block 4325 or N-methyl aminooxy containing amino acid 4426 was incorporated instead of the glutamic acid at the N-terminus. After cleavage from the resin the (N-methyl)aminooxy groups in the oligopeptides 45 and 46 were reacted with the hemiacetal of the ribose moiety in free ADP-ribose 47 producing an ADPr appendage. The aminooxy group in 45 led mainly to ring-opened ADPr peptide 48 and a small amount of the ring-closed form 49, while the N-methyl aminooxy group in 46 gave the ring-closed ADPr peptide 50 exclusively. By executing the ligation procedure at pH 4.5 the oxime formation becomes selective, leaving all natural amino acid side-chain functionalities, including those of lysine and arginine residues, intact.
 | ||
| Scheme 6 The aminooxy and N-methyl aminooxy functionalized peptides 48–50 of the group of Muir. | ||
Liu et al.27 reported another type of ADPr analogue that can be easily introduced in oligopeptides with the aid of a copper-catalyzed azide–alkyne cycloaddition (CuAAC) to give triazole-linked ADP-ribosylated peptides (Scheme 7). With the assistance of SPPS and standard amino acid building blocks, either azido-alanine or azido-homoalanine was incorporated at specific positions of peptides, the sequences of which were based on those of biologically relevant ADP-ribosylated proteins. Both azido functionalized oligopeptides (54a–d) and ubiquitin protein 54e in which Arg42 was replaced by azido-homoalanine were assembled by standard SPPS. After cleavage from the resin and concomitant removal of all protecting groups, followed by purification with RP-HPLC, peptides 54a–e were subjected to the critical CuAAC reaction with alkyne 53. This ADPr building block was efficiently prepared by the reaction of imidate donor 21a with propargyl alcohol. Isolation of the pure α-anomer, followed by protective group manipulation gave glycoside 51. Next, phosphitylation, oxidation to the phosphodiester and removal of the tBu protecting groups gave phosphomonoester 52 that was used in combination with phosphoramidite 41 to provide pyrophosphate 53via the same PIII–PV procedure19 as described for the synthesis of the Ser-ADPr hendecapeptide in Scheme 5. The CuAAC reaction of 53 with 54a–e in Tris buffer at pH 7.6 under the influence of CuSO4, sodium ascorbate and a tris-triazole ligand proceeded efficiently to give triazole-linked adenosine diphosphate ribosylated peptides 55. The CuAAC reaction of 53 proved to be successful for small proteins as illustrated by the total chemical synthesis of a biologically active ADP-ribosylated ubiquitine derivative, demonstrating that triazole linked ADPr can be employed as bio-isosteres of ADPr-Arg in peptides or proteins (Scheme 7).27 A different CuAAC mediated approach to the synthesis of triazole linked peptides has been reported by Li et al.28
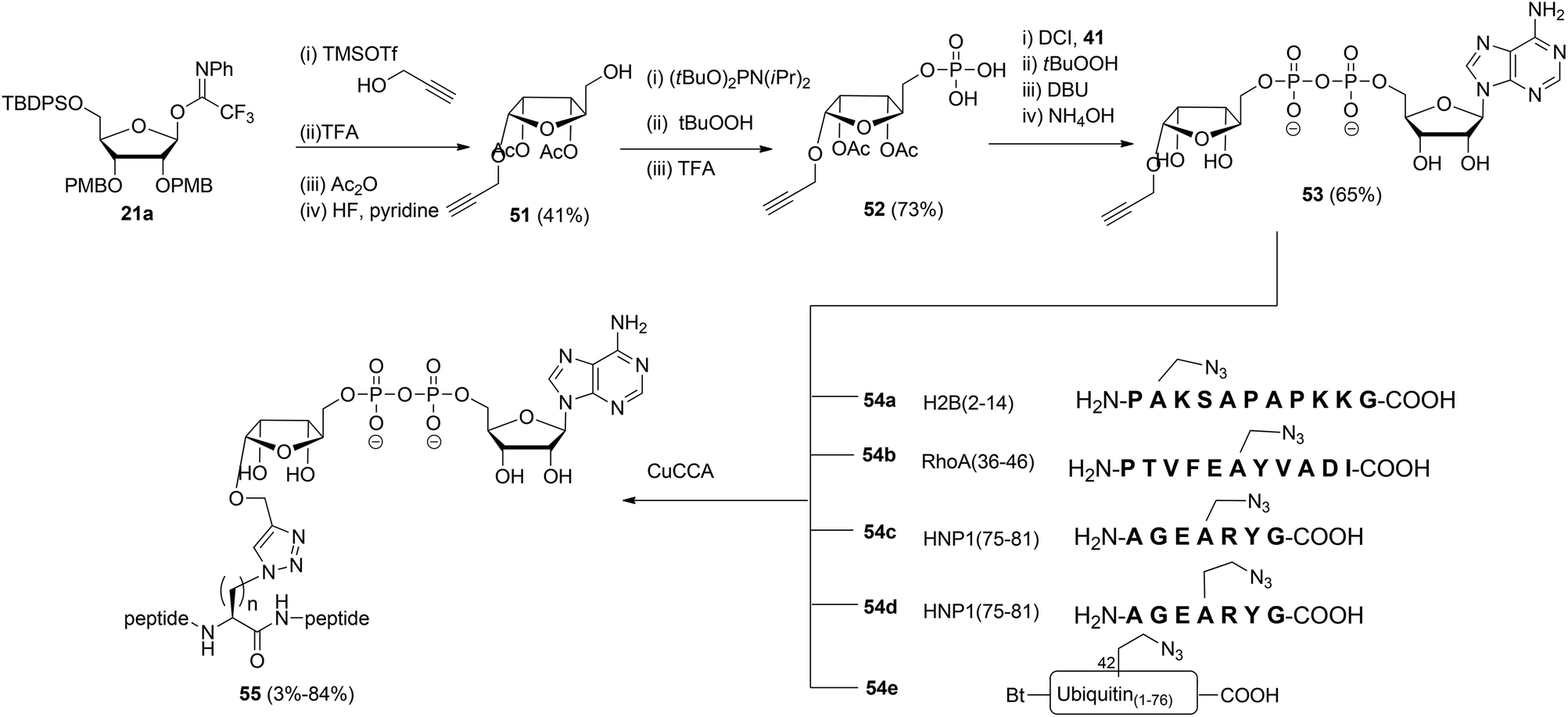 | ||
| Scheme 7 Synthesis of triazole linked ADPr-peptides by CuAAC chemistry. | ||
2.2 Synthesis of poly-ADPr chain
The organic synthesis of fragments of poly-ADP-ribose (PAR, Fig. 1) comprises a repetitive introduction of both an α-glycosidic bond between the ribose and the 2′-OH of adenosine and a pyrophosphate linkage between the primary OHs of the adenosine and the ribose moiety. To acquire ADPr oligomers of a certain length both solution and solid phase approaches require the design and synthesis of suitably protected and functionalized building blocks. Monomeric building blocks could be envisaged, in which a pyrophosphate moiety is incorporated but these must then act as ADP-ribofuranosyl donors that are suitable for repetitive α-ribosylation of the 2′-OH of the terminal adenosine moiety of the growing PAR-chain. Although, such a method would resemble the biosynthesis of PAR, in which NAD+ fulfills the role of the ADP-ribofuranosyl donor this approach should be rejected because the repetitive introduction of multiple α-ribosidic bonds in the presence of (anionic) pyrophosphates is almost impossible. Therefore the α-ribosidic bond should be preinstalled in the building block while the pyrophosphate moiety is then repetitively introduced during the assembly of the oligo-ADP-ribose chain. Both syntheses of ADP-ribose oligomers that are reported to date use the latter strategy.18,29In the following sections, we first describe the methods that have been developed for the synthesis of 2′-O-ribosylated adenosine building blocks (section 2.2.1), and next, the methods of pyrophosphate formation in the framework of the assembly of fragments of poly-ADPr-ribose will be discussed (section 2.2.2).
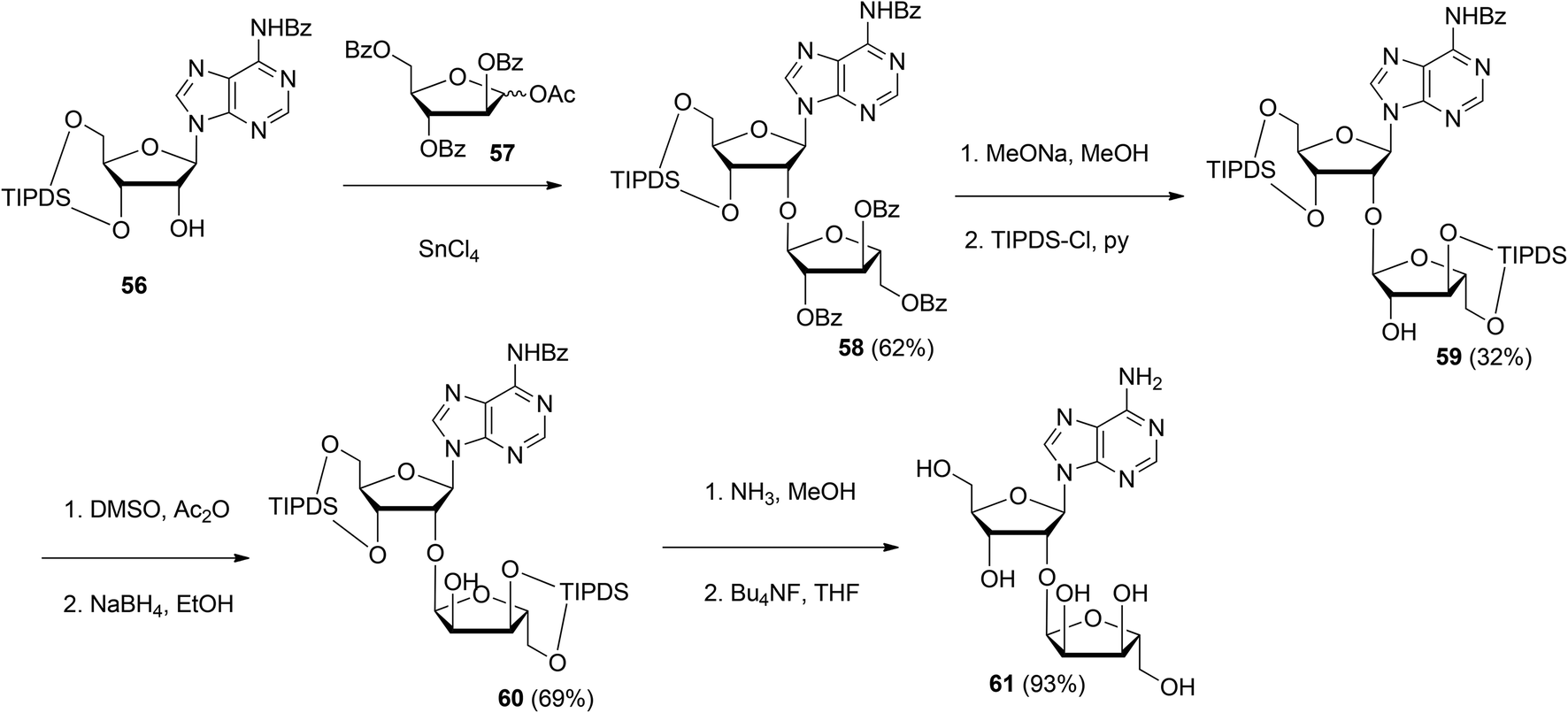 | ||
| Scheme 8 Synthesis of 2′-O-α-ribosylated adenosine 61 using 1-O-acetyl-2,3,5-tri-O-benzoyl-D-arabinofuranose 57 as the donor. | ||
Although the method of Mikhailov et al.30 is robust the route of synthesis to a monomeric building block suitable for the assembly of oligo-ADP-ribose is rather lengthy due to the necessity to invert the 2′-OH position of ribose and the subsequent introduction of orthogonal protective groups. For the synthesis of oligo-ADP-ribose, more direct approaches to attain α-selective glycosylation of adenosine were developed. Van der Heden van Noort et al.33 reported the synthesis of 2′-O-α-D-ribosylated adenosine (64, Scheme 9A) with TBDPS and DMT as orthogonal protecting groups on the primary hydroxyl functions of the ribose moieties. The key step is the TMSOTf mediated condensation of (N-phenyl)-2,2,2-trifluoroacetimidate donor 21c and adenosine acceptor 62 to furnish fully protected ribofuranosyladenosine 63 in an α-selective manner. Subsequent protecting group manipulation yielded 64, amenable for the assembly of oligo-ADP-ribose. Recently, Shirinfar et al.32 reported the synthesis of a protected phosphorylated ribofuranosyladenosine building block, using the same glycosylation procedure.
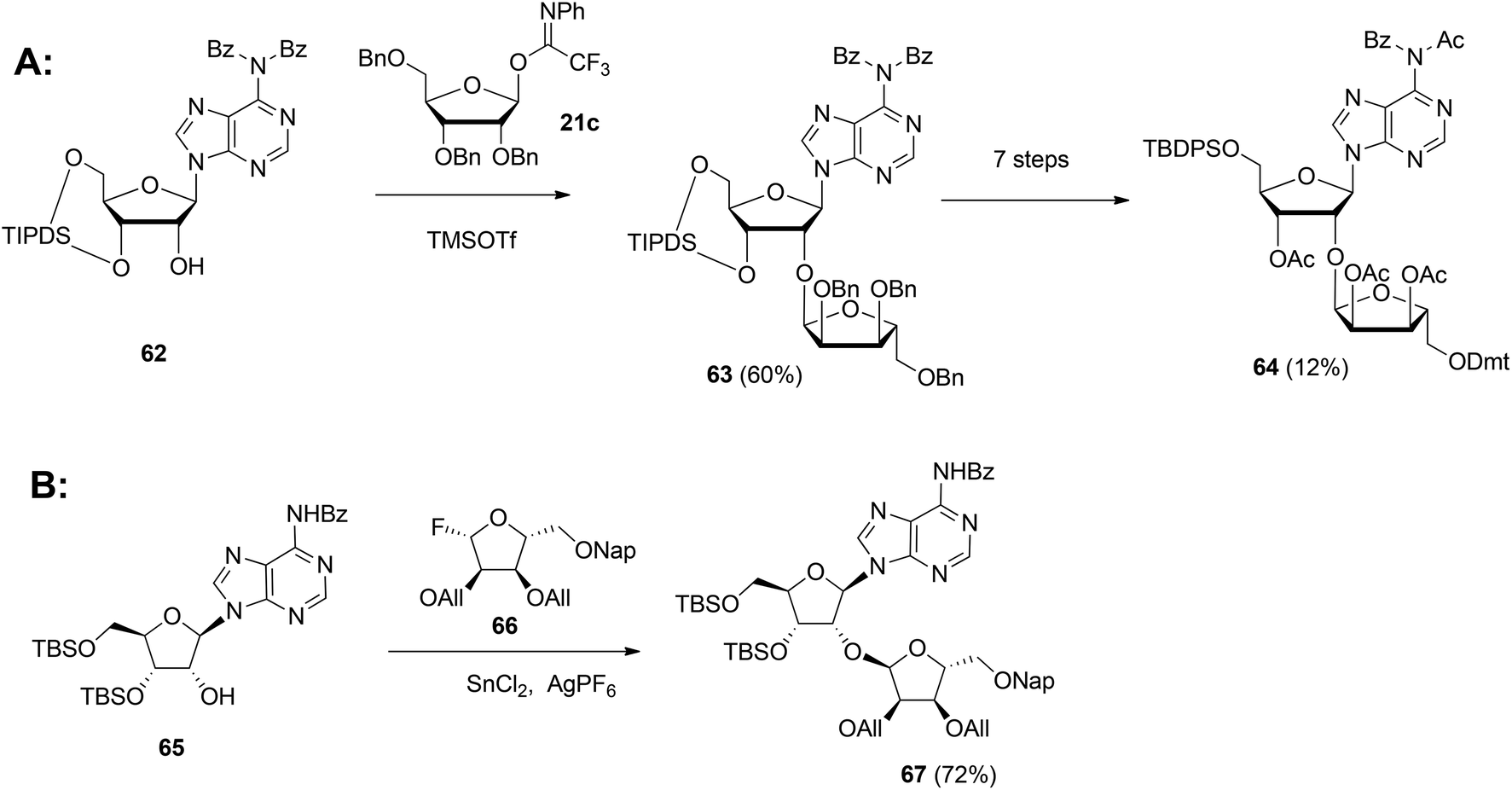 | ||
| Scheme 9 Synthesis of orthogonally protected ribosylated adenosine with (A): 1-O-(N-phenyl)-2,2,2-trifluoroacetimido-2,3,5-tri-O-benzyl-D-ribofuranose 21c and (B): glycosyl fluoride 66 as the donors. | ||
In 2015 Lambrecht et al.29 reported the synthesis of orthogonally protected ribosyl adenosine 67 (Scheme 9B) by the α-selective condensation of β-fluoride donor 66, obtained in six steps from ribose with adenosine acceptor 65. Crucial for the productivity of this reaction was the use of AgPF6/SbCl2 as an activator combination.
Guided by the need to scale up the process and to acquire sufficient quantities of a suitable ribosyl-adenosine building block Kistemaker et al.18 developed a new method. Side reactions on the nucleobase often accompany the glycosylation of protected nucleosides limiting the scalability of such synthetic strategies.33 Therefore it was decided to install the adenine base by a Vorbrüggen reaction after the ribosylation event.34,35 As depicted in Scheme 10 a reaction of benzylated (N-phenyl)-2,2,2-trifluoroacetimidate donor 21b and commercially available acceptor 1,3,5-tri-O-benzoylribose 68 led to the isolation of α-configured disaccharide 69. After hydrogenolysis and acetylation, 70 was obtained. Vorbrüggen coupling of 70 and Bz-adenine under the influence of the immobilized acid (HClO4-SiO2), introduced the adenine base both regio- and β-stereoselectively. It is noteworthy that this approach gives access to a large amount of 71, the precursor of a suitable 2-O-ribosylated adenosine building block.
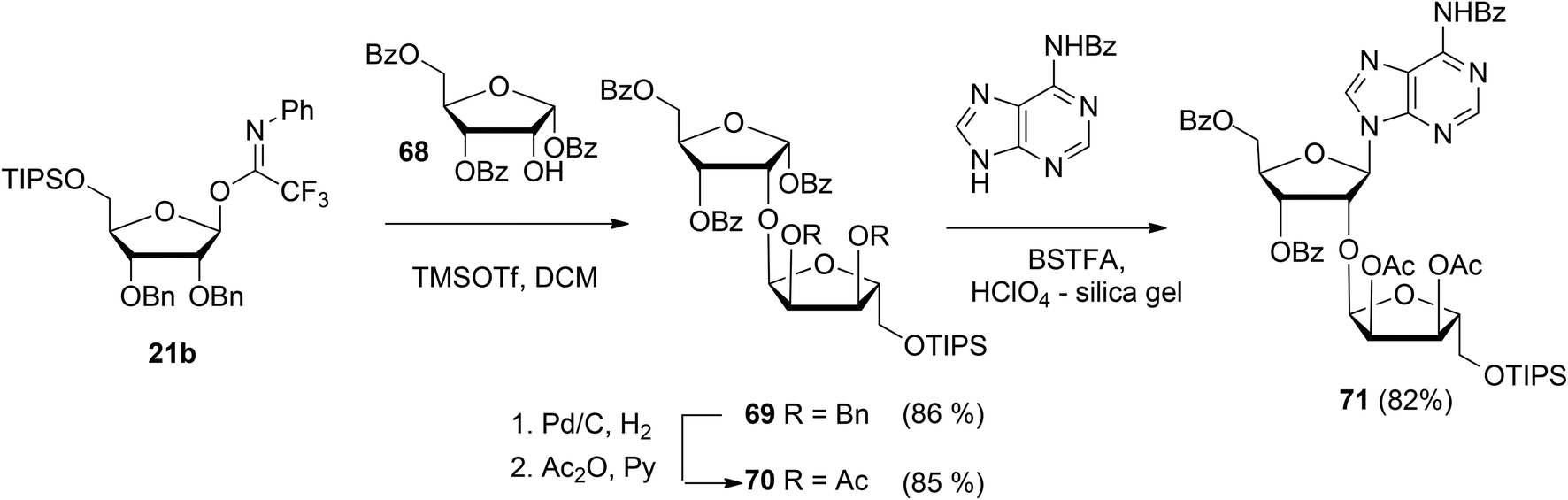 | ||
| Scheme 10 Synthesis of ribosylated adenosine via Vorbrüggen type glycosylation. | ||
This glycosylation strategy was adopted towards the synthesis of the branching point of ADPr-chains, culminating in the construction of both unphosphorylated36 and triphosphorylated37 branched ADPr fragments. The assembly of O-α-D-ribofuranosyl-(1′′′ → 2′′)-O-α-D-ribofuranosyl-(1′′ → 2′)-adenosine-5′,5′′,5′′′-tris(phosphate) 77 is depicted in Scheme 11. Protected disaccharide 69, termed parobiose,37 was converted into 72, in which the non-reducing ribose is provided with a free 2-OH group. Subsequent TMSOTf mediated condensation of 72 with benzylated (N-phenyl)-2,2,2-trifluoroacetimidate donor 21b led to all alpha configured tri-riboside 73 (protected parotriose).37 After protective group manipulation the adenine base was introduced through a similar Vorbrüggen type glycosylation as described above to afford protected O-α-D-ribofuranosyl-(1′′′ → 2′′)-O-α-D-ribofuranosyl-(1′′ → 2′)-adenosine 75. Transformation of 75 to 76 allowed the three-fold introduction of di-tert-butyl-phosphotriesters with the aid of phosphoramidite chemistry. Treatment of 76 with 10 equivalents of di-tert-butyl-N,N-diisopropylphosphoramidite under the influence of 1-methylimidazole and 1-methylimidazolium chloride as the activator, followed by oxidation of the intermediate phosphite triesters and global deprotection yielded target triphosphate 77 which proved to be identical to the native structure38,39 as ascertained by NMR spectroscopy.
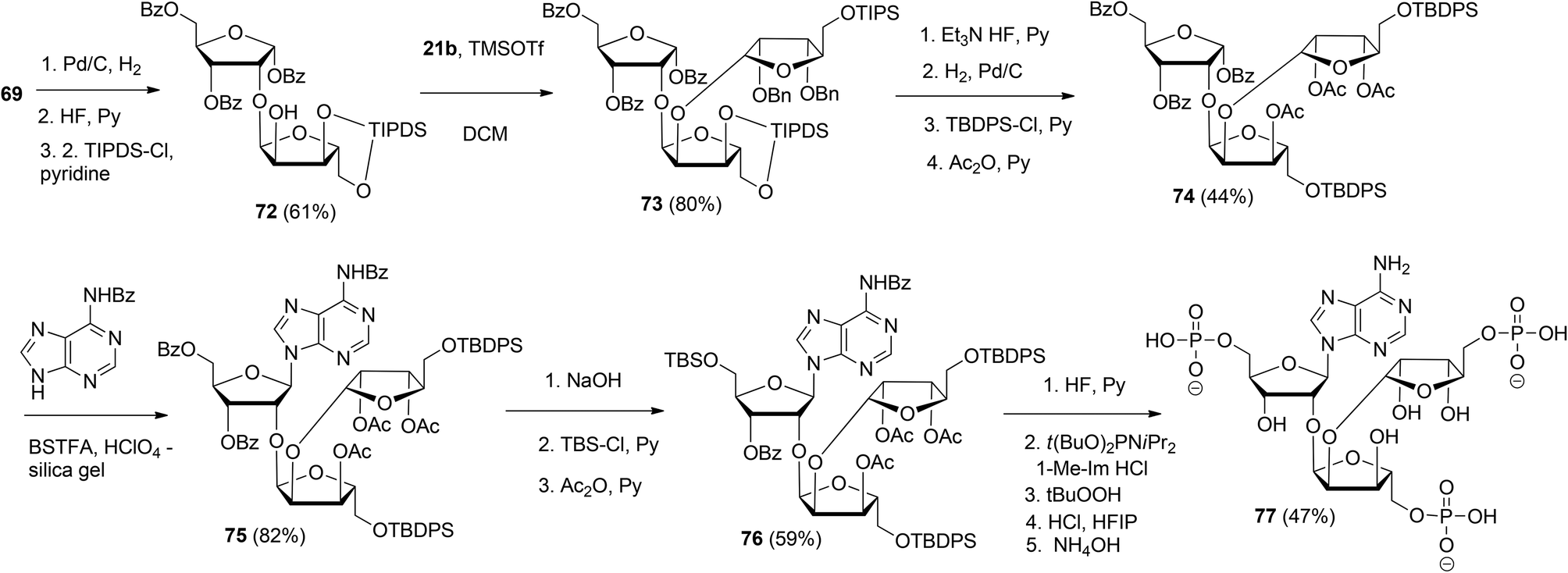 | ||
| Scheme 11 Synthesis of O-α-D-ribofuranosyl-(1′′′ → 2′′)-O-α-D-ribofuranosyl-(1′′ → 2′)-adenosine-5′,5′′,5′′′-tris(phosphate): the branching point of ADPr-chain. | ||
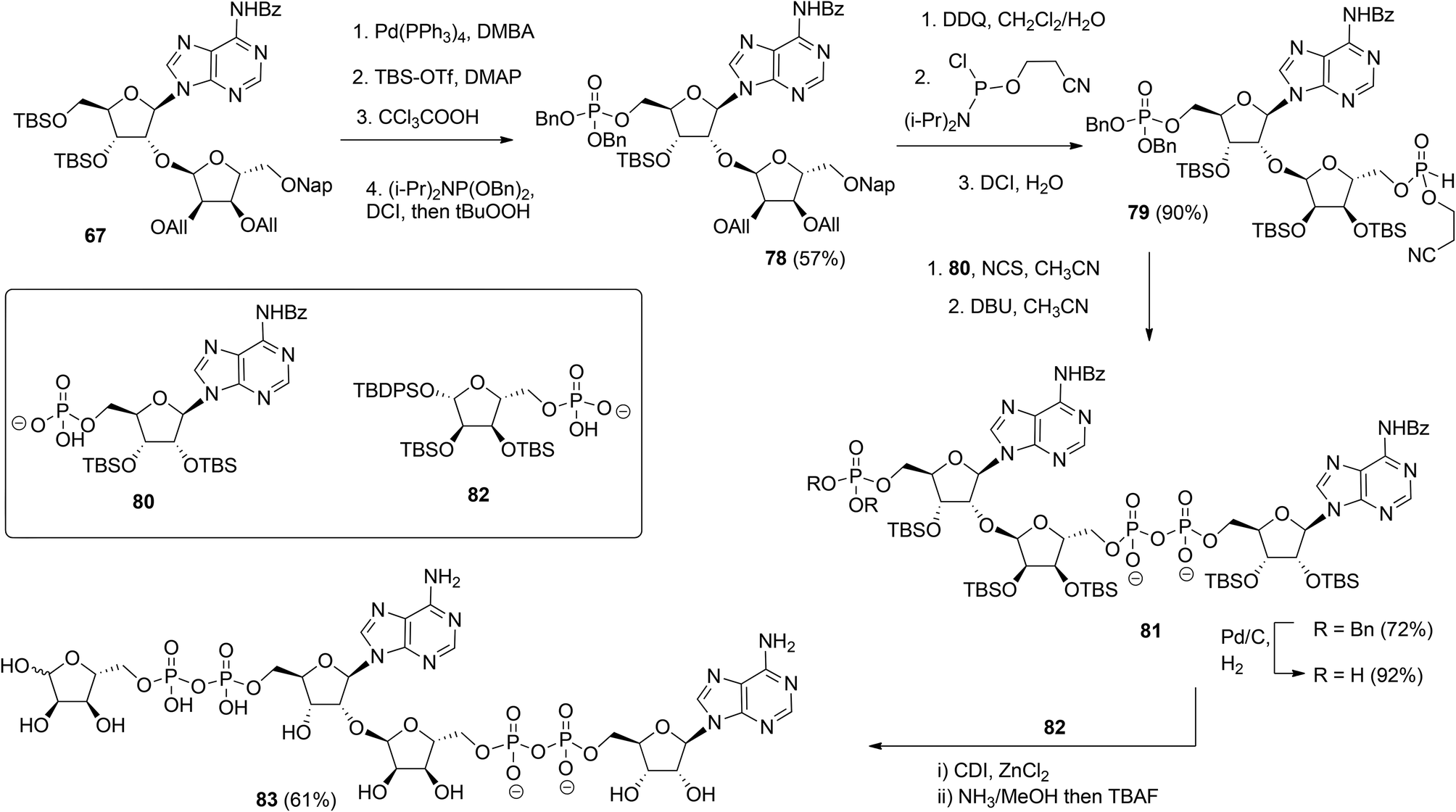 | ||
| Scheme 12 Synthesis of ADPr dimer 83 by Lambrecht et al. | ||
Kistemaker et al. reported a solid phase synthesis of both an ADPr dimer and trimer (Scheme 14).18 To be able to introduce multiple pyrophosphates a method to access sugar-nucleotides which was based on the combination of PIII–PV chemistry was investigated.19 This methodology proved to be convenient and expedient not only for the synthesis of mono-ADP-ribosylated peptides (Scheme 5) but also for the synthesis of various bioorganic pyrophosphate derivatives both in solution47,49,50 and on the solid phase.51,52 It was expected that this PIII–PV method would be uniquely suitable for the repeated pyrophosphorylation on the solid phase, not least due to its mild nature and fast kinetics. To be able to introduce multiple pyrophosphate functions building block 86, provided with di-tert-butyl phosphotriester and 2-cyanoethyl N,N-diisopropylphosphoramidite, was designed. The synthesis of building block 86 (Scheme 13) started with 2-O-ribosyladenosine 71 which was obtained in sufficient quantities and good yield as described above (Scheme 10). Protective group manipulation of dimer 71 gave 84 in which the free primary OH in the ribose moiety was provided with a di-tert-butyl phosphotriester using standard phosphoramidite chemistry, followed by oxidation of the intermediate phosphite triester. Finally, removal of the DMT group to give 85 and a reaction with 2-cyanoethyl N,N-diisopropylchlorophosphoramidite resulted in the isolation of building block 86 that contains both a phosphoramidite and a protected precursor of the phosphate monoester.
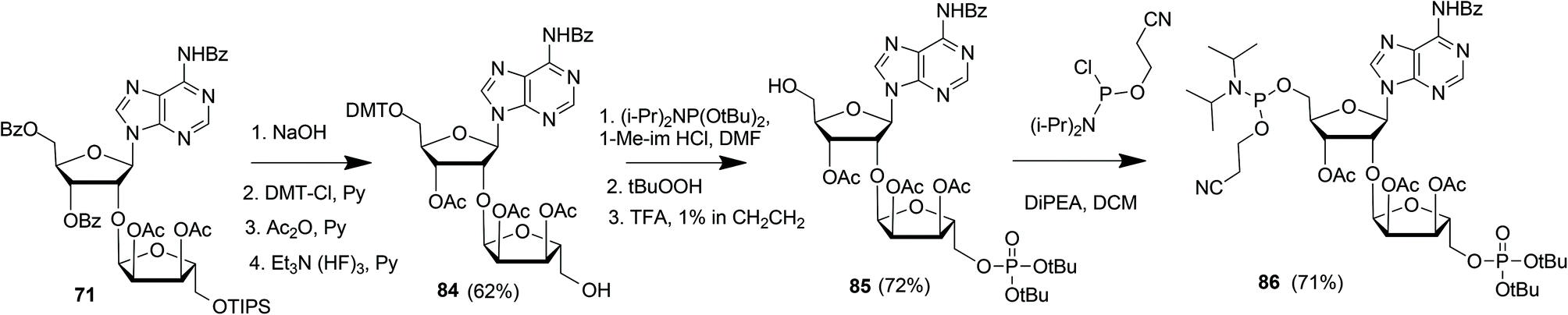 | ||
| Scheme 13 Synthesis of ribosylated adenosine building block 86 suitable for solid-phase preparation of oligo-ADPr fragments. | ||
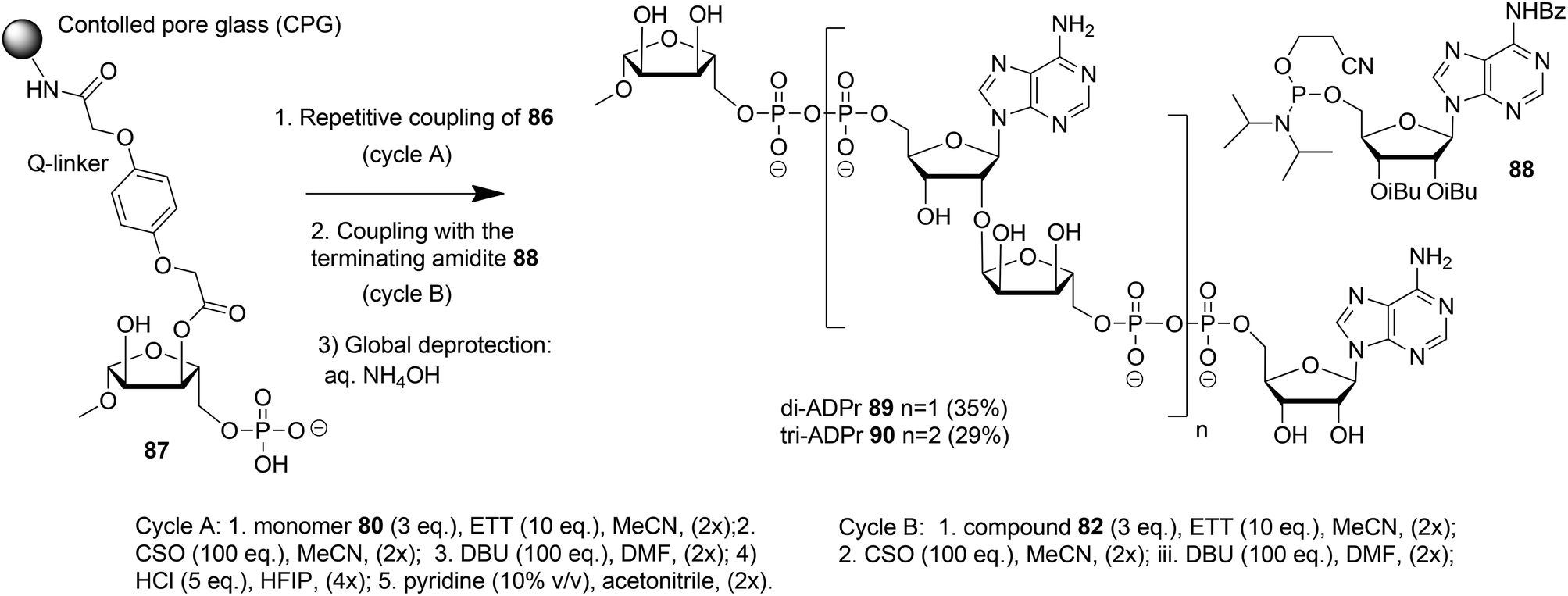 | ||
| Scheme 14 Solid-phase synthesis of ADPr dimer 89 and trimer 90. | ||
The solid phase synthesis of an ADPr dimer 89 and trimer 90 using building block 86 and terminating building block 88 is shown in Scheme 14.
Guided by the state of the art in automated DNA synthesis, controlled pore glass (CPG)18 with long alkyl amine chains was used as the solid support while hydroquinone-O,O′-diacetic acid (Q-linker) was selected as a linker for its improved resistance to DBU that was used to cleave 2-cyanoethyl protection from the pyrophosphate. Functionalization of this solid support with protected ribose and introduction of the phosphate monoester at the primary position gave “initiator” 87. At this stage, up to two coupling cycles with building block 86 were undertaken. The coupling cycle took one hour and involved 5-ethylthiotetrazole (ETT) mediated condensation of 80 with the immobilized monophosphate, oxidation of the obtained labile phosphite–phosphate (PIII–PV) intermediate by (1S)-(+)-(10-camphorsulfonyl)oxaziridine (CSO), and removal of the 2-cyanoethyl group in the partially protected pyrophosphate intermediate with DBU. The final unmasking of the di-tert-butyl phosphotriester with HCl/HFIP followed by neutralization with pyridine allowed the next elongation with either 86 or terminating building block 88. The immobilized and partially protected ADPr dimer and trimer were cleaved from the resin and completely deprotected by treatment with aqueous ammonia and purified to give milligram quantities of ADPr dimer 89 and trimer 90.
3. Conclusions and outlook
It can be concluded that significant progress has been achieved in the last ten years towards the synthesis of both mono-ADPr peptides and oligo-ADPr.For the synthesis of mono-ADPr-peptides most successes have been achieved in the preparation of the constructs, in which the ADPr-moiety is attached to the peptide backbone via isosteres of the natural amino acids either close, such as Gln9 instead of native Glu, or remote,22 for example, a triazole linkage27 as a substitute for the arginine side chain. Native ADPr-Ser containing peptides10 have been synthesized nevertheless. A fully synthetic ADPr-protein has been prepared via a convergent approach based on copper-catalyzed click chemistry and has been shown to be biologically active.27 The synthetic ADPr-peptides have been extensively applied in biochemical9,53 and proteomics studies.54–56 Thus, ADPr-peptides have been used as the essential calibration standard to enable quantification of ADP-ribosylation in the proteome.54,55 Another application of the synthetic ADPr-peptides is to validate the pull-down steps in the ELTA-methodology that has been developed for selective labeling of ADP-ribose and ADP-ribosylated proteins.57 Concerning the synthetic ADPr-oligomers, the dimeric ADPr was cocrystallized with PARG to investigate the details of the mechanism of the catalytic action of this enzyme. The synthetic dimeric and trimeric ADPr fragments were essential for determining the minimum length of PAR necessary to bind ALC1, which is a chromatin remodeler involved in oncogenesis.53 Looking towards the future applications, one can envisage the use of synthetic ADPR-oligomers in experiments for which less well-defined and, arguably, less homogeneous PAR-fragments of a biological origin have been applied up to now.58–61 Future synthetic studies will probably focus on the native ADP-ribosylated peptides, containing ADPr-Arg, ADPr-Cys, ADPr-Tyr and ADPr-His as the ADP-ribosylation sites. To make the synthesis of ADPr-peptides more practical, it is imperative to develop a synthetic methodology that does not require significant alterations in the standard SPPS protocols such as the use of alkali labile side-chain protection and linkers. Another important avenue of research is the development of more chemically robust derivatives, for example, mono-ADPr-peptides that contain carba-ribose62 residues instead of native ribofuranose and methylene bisphosphonate63 as pyrophosphate isosteres. Such stabilized derivatives of ADP-ribosylated biomolecules may prove invaluable for structural studies on the native enzymes because one can then forego the use of catalytically inactive mutants.64 Methylene bisphosphonates have been used in the past as substitutes of the native pyrophosphate in studies aimed to generate antibodies for mono-ADP-ribosylated proteins.65 It has been found that rendering the native pyrophosphate nuclease resistant by converting it into methylene bisphosphonate was essential to raise antibodies against ADP-ribose.
Both solid-phase18 and solution29 methodologies have been published for the synthesis of ADPr-dimers. It seems, however, that the solid-phase synthesis is more practical as evident from the fact that tri-ADPr was prepared using a solid-phase approach. Various methods toward the introduction of the key α-glycosidic bond that connects the monomers in the ADPr-chain have been explored. It appears that the methods that involved direct glycosylation of the 2′-OH of ribose, whether in the absence of the adenine nucleobase or having the nucleobase preinstalled, are the most useful ones for the monomer synthesis. In particular, glycosylation of the 2-OH of ribose followed by the introduction of adenine via Vorbrüggen type condensation allowed the preparation of the branching point of the ADPr-chain.36,37 Up to now, the P(III)–P(V) approach to pyrophosphate formation19 has been proven to be suitable for the synthesis of the ADPr-chains up to trimers on a solid phase, while the classic method of pyrophosphate formation allowed the synthesis of di-ADPr in solution. Such short fragments already proved to be useful in the biochemical53 and structural studies.29
Further developments in the methodology of chemical ADP-ribosylation can be envisaged. Improved temporal protective groups for phosphate monoesters should enable the synthesis of longer ADPr-chains, perhaps up to 15–25 ADPr units long. Various resins, such as polystyrene-based resins, could be explored for scale up and the preparation of oligo-ADPr fragments grafted to peptides. The synthesis of branched oligo-ADPr and ADPr-chains attached to peptides should also be possible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the support Q. L. received from China Scholarship Council (CSC).References
- B. Luscher, M. Butepage, L. Eckei, S. Krieg, P. Verheugd and B. H. Shilton, Chem. Rev., 2018, 118, 1092–1136 CrossRef PubMed.
- P. O. Hassa, S. S. Haenni, M. Elser and M. O. Hottiger, Microbiol. Mol. Biol. Rev., 2006, 70, 789–829 CrossRef CAS PubMed.
- W. L. Kraus, Mol. Cell, 2015, 58, 902–910 CrossRef CAS PubMed.
- M. S. Cohen and P. Chang, Nat. Chem. Biol., 2018, 14, 236–243 CrossRef CAS PubMed.
- D. Slade, M. S. Dunstan, E. Barkauskaite, R. Weston, P. Lafite, N. Dixon, M. Ahel, D. Leys and I. Ahel, Nature, 2011, 477, 616–620 CrossRef CAS PubMed.
- P. Fontana, J. J. Bonfiglio, L. Palazzo, E. Bartlett, I. Matic and I. Ahel, eLife, 2017, 6, e28533 CrossRef PubMed.
- M. Butepage, L. Eckei, P. Verheugd and B. Luscher, Cells, 2015, 4, 569–595 CrossRef PubMed.
- G. J. van der Heden van Noort, M. G. van der Horst, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, J. Am. Chem. Soc., 2010, 132, 5236–5240 CrossRef CAS PubMed.
- H. A. Kistemaker, A. P. Nardozza, H. S. Overkleeft, G. A. van der Marel, A. G. Ladurner and D. V. Filippov, Angew. Chem., Int. Ed., 2016, 55, 10634–10638 CrossRef CAS PubMed.
- J. Voorneveld, J. G. M. Rack, I. Ahel, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Org. Lett., 2018, 20, 4140–4143 CrossRef CAS PubMed.
- A. Sekine, M. Fujiwara and S. Narumiya, J. Biol. Chem., 1989, 264, 8602–8605 CAS.
- N. Ogata, K. Ueda and O. Hayaishi, J. Biol. Chem., 1980, 255, 7610–7615 CAS.
- M. A. Bonache, F. Nuti, A. Le Chevalier Isaad, F. Real-Fernández, M. Chelli, P. Rovero and A. M. Papini, Tetrahedron Lett., 2009, 50, 4151–4153 CrossRef CAS.
- F. Nisic, G. Speciale and A. Bernardi, Chem. – Eur. J., 2012, 18, 6895–6906 CrossRef CAS PubMed.
- F. Nisic and A. Bernardi, Carbohydr. Res., 2011, 346, 465–471 CrossRef CAS PubMed.
- G. Speciale, A. Bernardi and F. Nisic, Molecules, 2013, 18, 8779–8785 CrossRef CAS PubMed.
- H. A. Kistemaker, G. J. van Noort, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Org. Lett., 2013, 15, 2306–2309 CrossRef CAS PubMed.
- H. A. Kistemaker, L. N. Lameijer, N. J. Meeuwenoord, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Angew. Chem., Int. Ed., 2015, 54, 4915–4918 CrossRef CAS PubMed.
- H. Gold, P. van Delft, N. Meeuwenoord, J. D. C. Codée, D. V. Filippov, G. Eggink, H. S. Overkleeft and G. A. van der Marel, J. Org. Chem., 2008, 73, 9458–9460 CrossRef CAS PubMed.
- O. Leidecker, J. J. Bonfiglio, T. Colby, Q. Zhang, I. Atanassov, R. Zaja, L. Palazzo, A. Stockum, I. Ahel and I. Matic, Nat. Chem. Biol., 2016, 12, 998–1000 CrossRef CAS PubMed.
- J. J. Bonfiglio, P. Fontana, Q. Zhang, T. Colby, I. Gibbs-Seymour, I. Atanassov, E. Bartlett, R. Zaja, I. Ahel and I. Matic, Mol. Cell, 2017, 65, 932–940 CrossRef CAS PubMed.
- L. Palazzo, O. Leidecker, E. Prokhorova, H. Dauben, I. Matic and I. Ahel, eLife, 2018, 7, e34334 CrossRef PubMed.
- Q. Liu, B. I. Florea and D. V. Filippov, Cell Chem. Biol., 2017, 24, 431–432 CrossRef CAS PubMed.
- P. M. Moyle and T. W. Muir, J. Am. Chem. Soc., 2010, 132, 15878–15880 CrossRef CAS PubMed.
- F. Wahl and M. Mutter, Tetrahedron Lett., 1996, 37, 6861–6864 CrossRef CAS.
- M. R. Carrasco and R. T. Brown, J. Org. Chem., 2003, 68, 8853–8858 CrossRef CAS PubMed.
- Q. Liu, H. A. V. Kistemaker, S. Bhogaraju, I. Dikic, H. S. Overkleeft, G. A. van der Marel, H. Ovaa, G. J. van der Heden van Noort and D. V. Filippov, Angew. Chem., Int. Ed., 2018, 57, 1659–1662 CrossRef CAS PubMed.
- L. Li, Q. Li, S. Ding, P. Xin, Y. Zhang, S. Huang and G. Zhang, Molecules, 2017, 22, 1346 CrossRef PubMed.
- M. J. Lambrecht, M. Brichacek, E. Barkauskaite, A. Ariza, I. Ahel and P. J. Hergenrother, J. Am. Chem. Soc., 2015, 137, 3558–3564 CrossRef CAS PubMed.
- S. N. Mikhailov, I. V. Kulikova, K. Nauwelaerts and P. Herdewijn, Tetrahedron, 2008, 64, 2871–2876 CrossRef CAS.
- M. Zheng, M. Mex, K. H. Gotz and A. Marx, Org. Biomol. Chem., 2018, 16, 8904–8907 RSC.
- B. Shirinfar and N. Ahmed, Helv. Chim. Acta, 2018, 101, e1700226 CrossRef.
- G. J. van der Heden van Noort, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Org. Lett., 2011, 13, 2920–2923 CrossRef CAS PubMed.
- H. Vorbrüggen and K. Krolikiewicz, Angew. Chem., Int. Ed. Engl., 1975, 14, 421–422 CrossRef.
- H. Vorbrüggen, K. Krolikiewicz and B. Bennua, Chem. Ber., 1981, 114, 1234–1255 CrossRef.
- H. A. Kistemaker, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Org. Lett., 2015, 17, 4328–4331 CrossRef CAS.
- Q. Liu, H. A. V. Kistemaker, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Chem. Commun., 2017, 53, 10255–10258 RSC.
- M. Miwa, M. Ishihara, S. Takishima, N. Takasuka, M. Maeda, Z. Yamaizumi, T. Sugimura, S. Yokoyama and T. Miyazawa, J. Biol. Chem., 1981, 256, 2916–2921 CAS.
- M. Miwa, N. Saikawa, Z. Yamaizumi, S. Nishimura and T. Sugimura, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 595–599 CrossRef CAS PubMed.
- M. Dong, T. Kirchberger, X. Huang, Z. J. Yang, L. R. Zhang, A. H. Guse and L. H. Zhang, Org. Biomol. Chem., 2010, 8, 4705–4715 RSC.
- X. Xue, R. B. Zheng, A. Koizumi, L. Han, J. S. Klassen and T. L. Lowary, Org. Biomol. Chem., 2018, 16, 1939–1957 RSC.
- F. Ravalico, I. Messina, M. V. Berberian, S. L. James, M. E. Migaud and J. S. Vyle, Org. Biomol. Chem., 2011, 9, 6496–6497 RSC.
- A. R. Kore and G. Parmar, Synth. Commun., 2006, 36, 3393–3399 CrossRef CAS.
- I. B. Yanachkov, E. J. Dix, M. I. Yanachkova and G. E. Wright, Org. Biomol. Chem., 2011, 9, 730–738 RSC.
- S. Wendicke, S. Warnecke and C. Meier, Angew. Chem., Int. Ed., 2008, 47, 1500–1502 CrossRef CAS PubMed.
- A. Hofer, G. S. Cremosnik, A. C. Muller, R. Giambruno, C. Trefzer, G. Superti-Furga, K. L. Bennett and H. J. Jessen, Chem. – Eur. J., 2015, 21, 10116–10122 CrossRef CAS PubMed.
- Q. Zhang, P. L. Howell, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel and J. D. C. Codee, Carbohydr. Res., 2017, 450, 12–18 CrossRef CAS PubMed.
- N. Qi, K. Jung, M. Wang, L. X. Na, Z. J. Yang, L. R. Zhang, A. H. Guse and L. H. Zhang, Chem. Commun., 2011, 47, 9462–9464 RSC.
- I. Pavlovic, D. T. Thakor, J. R. Vargas, C. J. McKinlay, S. Hauke, P. Anstaett, R. C. Camuna, L. Bigler, G. Gasser, C. Schultz, P. A. Wender and H. J. Jessen, Nat. Commun., 2016, 7, 10622 CrossRef CAS PubMed.
- J. G. M. Rack, A. Ariza, B. S. Drown, C. Henfrey, E. Bartlett, T. Shirai, P. J. Hergenrother and I. Ahel, Cell Chem. Biol., 2018, 25, 1533–1546 CrossRef CAS PubMed.
- G. S. Cremosnik, A. Hofer and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 286–289 CrossRef CAS PubMed.
- B. A. Anderson and R. Krishnamurthy, Chem. – Eur. J., 2018, 24, 6837–6842 CrossRef CAS PubMed.
- H. R. Singh, A. P. Nardozza, I. R. Moller, G. Knobloch, H. A. V. Kistemaker, M. Hassler, N. Harrer, C. Blessing, S. Eustermann, C. Kotthoff, S. Huet, F. Mueller-Planitz, D. V. Filippov, G. Timinszky, K. D. Rand and A. G. Ladurner, Mol. Cell, 2017, 68, 860–871 CrossRef CAS PubMed.
- V. Bilan, N. Selevsek, H. A. V. Kistemaker, J. Abplanalp, R. Feurer, D. V. Filippov and M. O. Hottiger, Mol. Cell. Proteomics, 2017, 16, 949–958 CrossRef CAS PubMed.
- J. Abplanalp, M. Leutert, E. Frugier, K. Nowak, R. Feurer, J. Kato, H. V. A. Kistemaker, D. V. Filippov, J. Moss, A. Caflisch and M. O. Hottiger, Nat. Commun., 2017, 8 CAS.
- R. L. McPherson, R. Abraham, E. Sreekumar, S.-E. Ong, S.-J. Cheng, V. K. Baxter, H. A. V. Kistemaker, D. V. Filippov, D. E. Griffin and A. K. L. Leung, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1666–1671 CrossRef CAS PubMed.
- Y. Ando, E. Elkayam, R. L. McPherson, M. Dasovich, S. J. Cheng, J. Voorneveld, D. V. Filippov, S. E. Ong, L. Joshua-Tor and A. K. L. Leung, Mol. Cell, 2019, 73, 845–856 CrossRef CAS PubMed.
- J. Fahrer, R. Kranaster, M. Altmeyer, A. Marx and A. Burkle, Nucleic Acids Res., 2007, 35, e143 CrossRef PubMed.
- Q. Chen, M. A. Kassab, F. Dantzer and X. Yu, Nat. Commun., 2018, 9, 3233 CrossRef PubMed.
- T. I. Kam, X. Mao, H. Park, S. C. Chou, S. S. Karuppagounder, G. E. Umanah, S. P. Yun, S. Brahmachari, N. Panicker, R. Chen, S. A. Andrabi, C. Qi, G. G. Poirier, O. Pletnikova, J. C. Troncoso, L. M. Bekris, J. B. Leverenz, A. Pantelyat, H. S. Ko, L. S. Rosenthal, T. M. Dawson and V. L. Dawson, Science, 2018, 362 Search PubMed.
- K. A. Krukenberg, S. Kim, E. S. Tan, Z. Maliga and T. J. Mitchison, Chem. Biol., 2015, 22, 446–452 CrossRef CAS PubMed.
- S. Shuto, M. Fukuoka, A. Manikowsky, Y. Ueno, T. Nakano, R. Kuroda, H. Kuroda and A. Matsuda, J. Am. Chem. Soc., 2001, 123, 8750–8759 CrossRef CAS PubMed.
- S. B. Engelsma, N. J. Meeuwenoord, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Angew. Chem. Int. Ed., 2017, 56, 2955–2959 CrossRef CAS PubMed.
- M. F. Langelier, L. Zandarashvili, P. M. Aguiar, B. E. Black and J. M. Pascal, Nat. Commun., 2018, 9, 844 CrossRef PubMed.
- T. Meyer and H. Hilz, Eur. J. Biochem., 1986, 155, 157–165 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |