
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA halogen-bonding-catalysed Nazarov cyclisation reaction†
Alexander
Dreger
,
Patrick
Wonner
,
Elric
Engelage
 ,
Sebastian M.
Walter
,
Raphael
Stoll
and
Stefan M.
Huber
*
,
Sebastian M.
Walter
,
Raphael
Stoll
and
Stefan M.
Huber
*
Department of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: stefan.m.huber@rub.de
First published on 27th June 2019
Abstract
Various neutral, mono- and dicationic halogen bond donors were screened for their ability to act as catalysts in a Nazarov cyclisation reaction. Using a highly preorganized dicationic catalyst with a noncoordinating counterion proved essential for high activity.
The Nazarov cyclisation is a versatile method to obtain highly substituted cyclopentenones.1 This carbon–carbon bond formation reaction, which involves 4π electrocyclization, is usually catalysed either by Brønsted or Lewis acids.2 In recent years, several examples of organocatalysed Nazarov reactions have also been reported.3–6 These cases include electrophilic phosphonium ions as non-metallic Lewis acids3a as well as enantioselective transformations with organoboron compounds,3b phosphoramide Brønsted acids5 or bifunctional thiourea derivatives.6 The latter represents, to the best of our knowledge, the only case of a hydrogen-bond-catalysed Nazarov reaction.
In recent years, halogen bonding7 (XB) – the attraction between electrophilic halogen substituents and Lewis bases – has emerged as a promising noncovalent interaction for various applications in solution.8 Its usage in organocatalysis is still relatively sparsely investigated.8c,d,9 Next to several examples of XB-catalysed halide abstraction reactions, there are also a few cases involving the activation of neutral electrophiles by XB.12–16 Most notably, imines/quinolines and carbonyl compounds have so far been addressed by XB donors in reduction,12 Mukaiyama-aldol addition,12c,16b Michael addition13b,14 and (aza) Diels–Alder reactions.15,16 Still, however, these cases only include three examples of carbonyl activation, and thus the Nazarov cyclization represents a perfect further test reaction to study the intricacies of XB organocatalysis.
Herein, we investigate the catalytic activity of previously reported mono- and bidentate halogen bond donors, as well as of neutral polyfluorinated variants in the reaction depicted in Scheme 1. As the chemical shifts of the starting material (divinyl ketone 1) and the product (cyclopentenone 2) are clearly separated, the reaction can easily be monitored via1H-NMR spectroscopy.
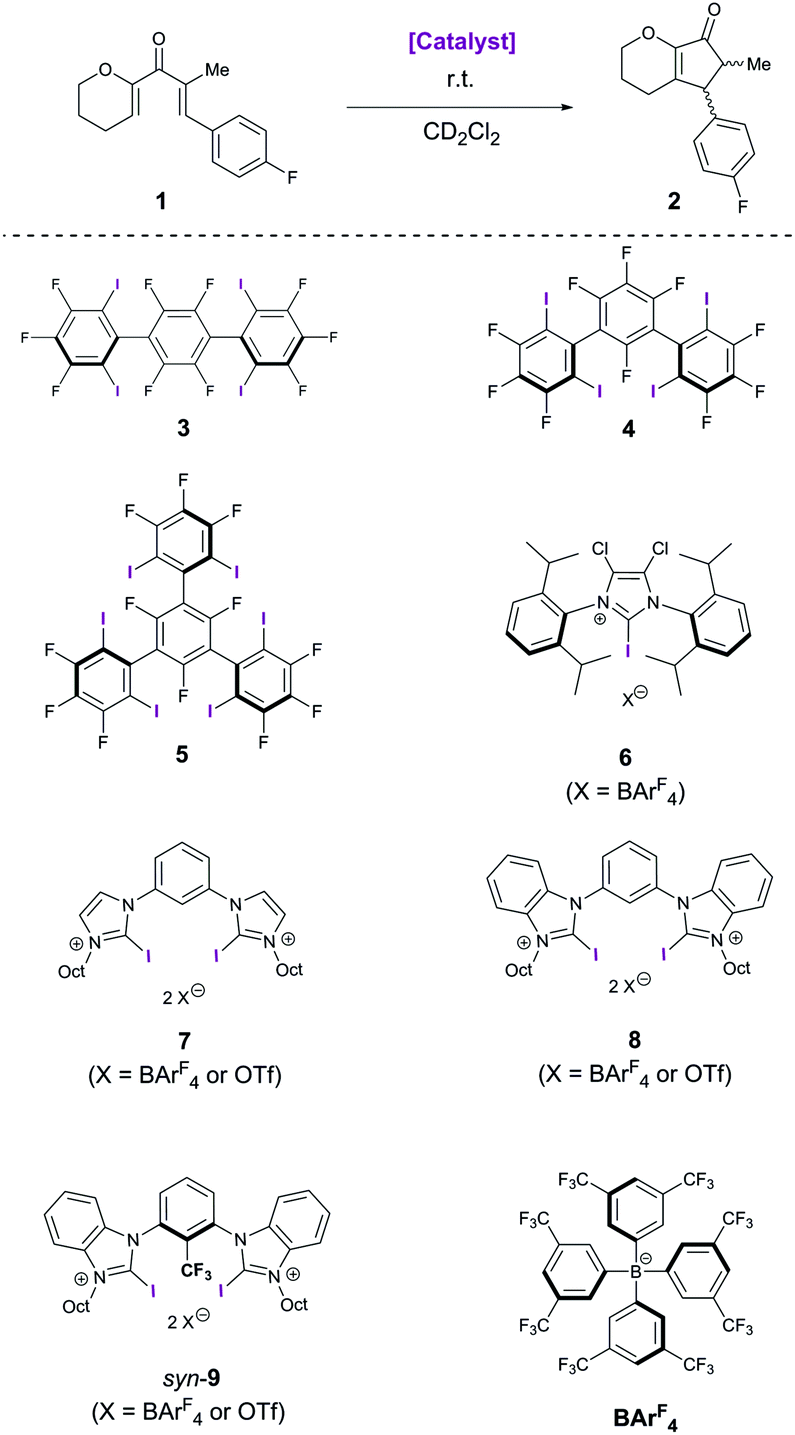 | ||
| Scheme 1 Nazarov cyclisation reaction and various XB donors as potential catalysts; Oct = n-octyl. | ||
In the absence of any catalyst, no reaction occurs at room temperature. Similarly, no catalytic activity was observed even after a few weeks for the neutral multidentate polyfluorinated XB donors 3, 4 and 5, as well as for the monodentate cationic XB donor 6. The inactivity of the former class of compounds, which are all active in halide abstraction reactions,10 illustrates the additional challenges associated with the activation of neutral substrates like divinyl ketones. As the reaction seemed to require stronger Lewis acids as catalysts, bidentate cationic XB donors were studied next. These were successfully used in a Diels–Alder and a Michael addition reaction,14,15 and in both cases a strong influence of the counterion was observed: satisfactory performance could only be achieved with noncoordinating anions like tetrakis[3,5-bis(trifluoromethyl)-phenyl]borate (BArF4) but not with counterions like triflate. Somewhat surprisingly, however, the triflate as well as the BArF4 salt of bis(imidazolium) derivative 7 showed no catalytic activity in this reaction – despite their earlier mentioned success in carbonyl activation reactions. Apparently, the Lewis acidity of these compounds is too low to allow sufficient reduction of the activation barrier, and thus the more electrophilic bis(benzimidazolium) derivatives 8 and 9 were investigated. The triflate salt of the direct benzimidazolium analogue, 8/OTf, leads to hardly noticeable formation of product 2 (≤5%) after 5 hours (Table 1).17
| Cat. (mol%) | Yield of 10 (5 h) | Yield of 2 (5 h)a | cis/trans ratio of 2a |
|---|---|---|---|
| c(1) = 15.4 mM in CD2Cl2.a Determined by 1H NMR.b After 1 h.c c0(1) = 655.4 mM in CD2Cl2 and reaction time 12 h.d Isolated yields. | |||
| None | — | — | — |
| 3–7 (5) | — | ≤5% | — |
| 8/OTf (5) | <5% | ≤5% | — |
| 8/BArF4 (5) | 71% | 26% | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| syn-9/OTf (5) | 70% | 23% | 2.3:1 |
| syn-9/BArF4 (5) | 20% | 65% | 2:1 |
| 11–13 (5) | — | — | — |
| HOTf (1) | — | ≥95% | 6.5:1 |
| I2 (5) | — | ≥95%b | 23:1b |
| syn-9/OTf (5)c | 6%c | 90%c (80%)d | 2.3:1 |
The corresponding BArF4 salt, on the other hand, showed a markedly increased performance (full consumption of 1 after 2 h with 5 mol% catalyst), which is in line with the noncoordinating nature of this counterion. In the 1H-NMR spectra of the reaction, next to the chemical shifts of starting material 1 and product 2, an additional set of signals was observed over time, which likely correspond to enol 10 (see Scheme 2 and the ESI†). An example for such spectra (with compound syn-9/OTf as a catalyst) is depicted in Fig. 1.
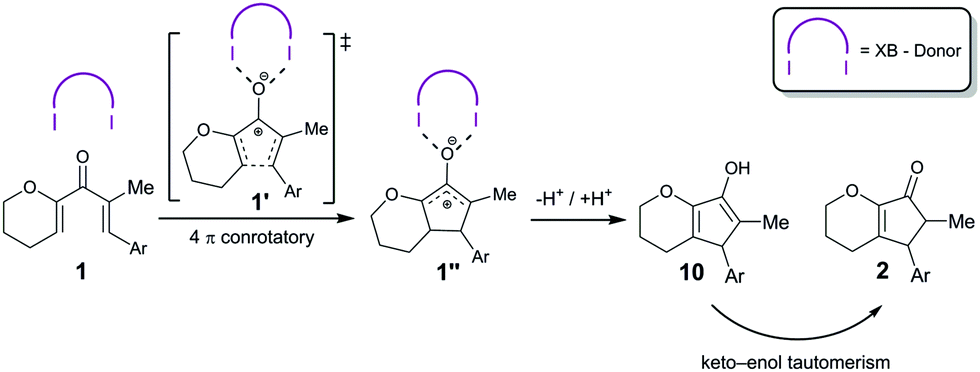 | ||
| Scheme 2 Postulated mechanism of the halogen bond catalysed Nazarov reaction. | ||
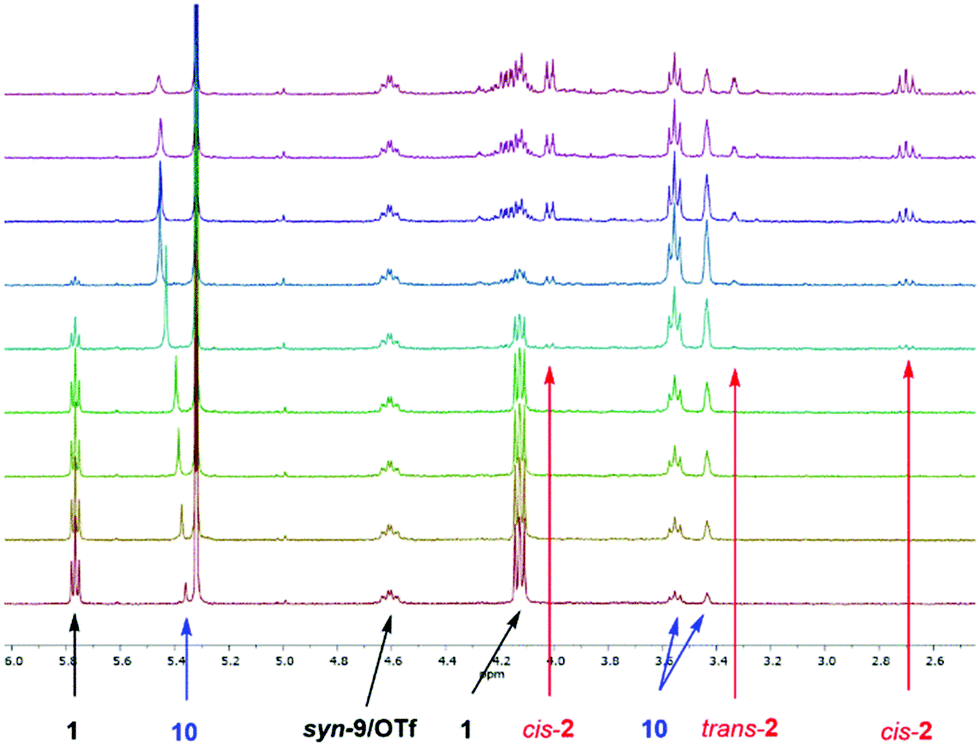 | ||
| Fig. 1 Selected section of the 1H-NMR kinetics in CD2Cl2 of the Nazarov reaction (c0(1) = 15.4 mM) catalysed by XB Donor syn-9/OTf (10 mol%). The 1H-NMR signals of the corresponding compounds 1, 2, syn-9/OTf and enol 10 are marked with arrows. | ||
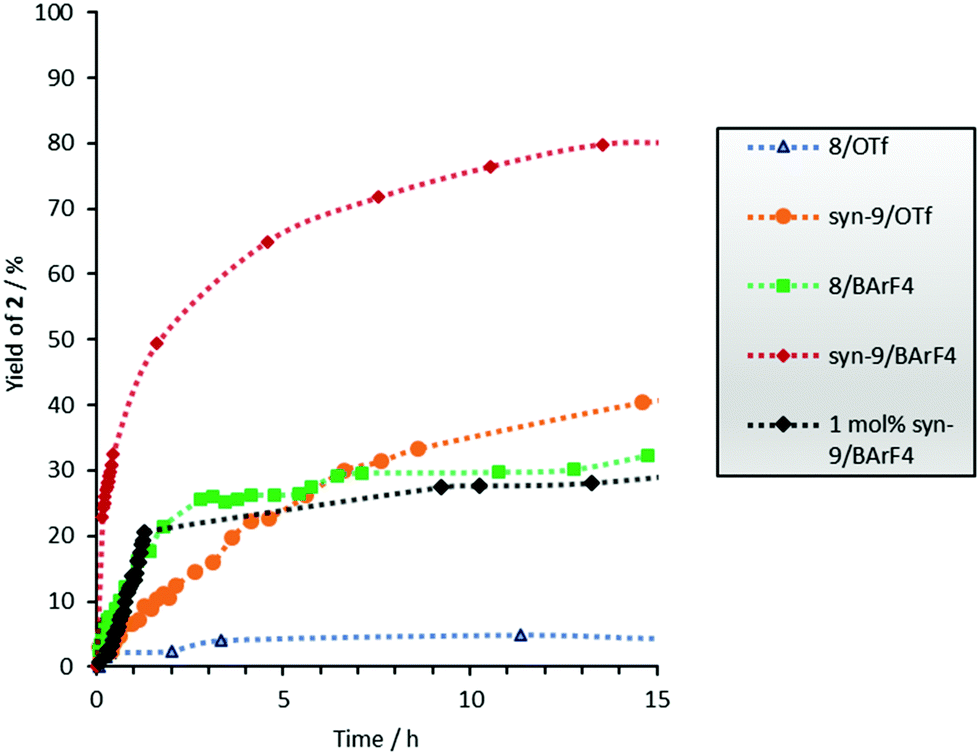
Since we postulate that halogen bonding will only influence the initial step (electrocyclization) of the overall reaction, but not the following keto–enol tautomerization (see Scheme 2), the analysis will first focus on the rate acceleration of the electrocyclization (which can be conveniently monitored via the consumption of starting material 1). The kinetic profile of this step in the presence of catalysts 8 or 9 is shown in Fig. 2.
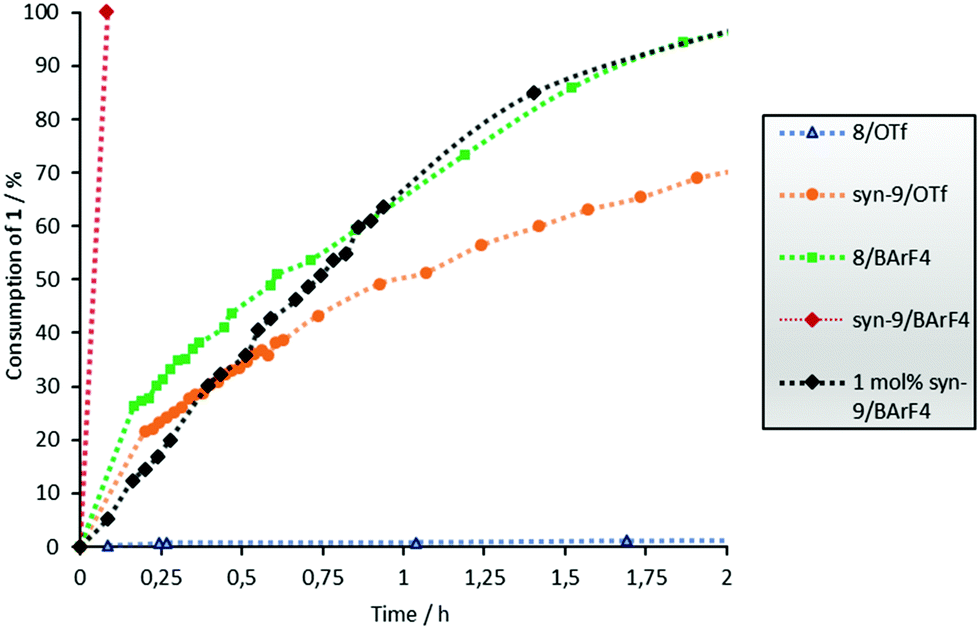 | ||
| Fig. 2 Consumption of 1versus the time profile of the Nazarov cyclisation with 5 mol% catalyst (unless noted otherwise) and c0(1) = 15.4 mM. No reaction without a catalyst and with compounds 3–7 and 11–13. The error is approximately 5%. | ||
It becomes immediately apparent that the better performance of 8/BArF4vs.8/OTf in overall product formation is also observed in the electrocyclization step. The Lewis acidity of these bidentate XB donors can be further increased by the introduction of a trifluoromethyl group in the central benzene core,11 which prevents rotation of the XB-donating moieties and allows the isolation of preorganized syn-atropisomer 9. The superior performance of 9vs.7 in a halide abstraction case11 and a Michael addition reaction has been demonstrated before.14
A strong effect of this preorganization was also found in the Nazarov reaction: triflate salt syn-9/OTf showed a markedly better performance compared to its almost inactive analogue 8/OTf (70% vs. 2% consumption of 1 after 2 h, see Fig. 2). As expected, the highest catalytic activity was achieved with the BArF4 salt syn-9/BArF4 – in less than 5 min, and the complete conversion of starting material 1 to enol 10 was observed with only 5 mol% of the catalyst. A reasonable kinetic profile of the electrocyclization step could only be obtained after reducing the amount of catalyst to 1 mol% (Fig. 2). In all cases mentioned so far, the cis/trans ratio of product 2 was in the range between 2:1 and 3:1 (Table 1).
Thus, the two key parameters to increase the performance of 8/OTf are the change of the counterion to BArF4 and the introduction of preorganization. Closer inspection of Fig. 2 indicates that the former has a larger impact: exchanging the counterion provides a more active catalyst (8/BArF4, green line) compared to the more preorganized one with the same counterion (syn-9/OTf, orange line).
In order to establish whether the observed activity was in indeed due to halogen bonding, non-iodinated compounds 11, 12 and 13 were also tested as catalysts (Fig. 3). Even though they share all structural features except the iodine substituents with catalysts 7–9, none of them showed any activity. Consequently, activation by hydrogen bonding or electrostatic interactions can be ruled out.
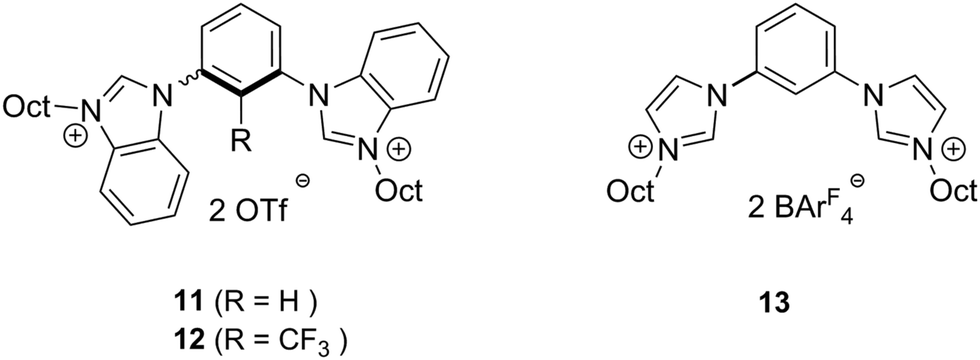 | ||
| Fig. 3 Structures of reference compounds. | ||
Two possible catalytically active decomposition products of the XB donors are elemental iodine and acid traces. A comparison experiment with 5 mol% of elemental iodine showed that it indeed induces quantitative product formation after 30 minutes.18 The exact origin of this activity is not entirely clear, as elemental iodine has several pathways (XB, hidden acid catalysis19 or iodonium formation) to activate organic molecules.20 It is, however, very unlikely that the observed activity of XB donors 8 and syn-9 is due to the decomposition to elemental iodine, for various reasons: (a) no spectroscopic evidence of decomposition was obtained (only one 19F signal was observed in the case of syn-9 after the reaction); (b) the consumption of the starting material follows sigmoidal kinetics with 0.1 mol% of iodine (see Fig. S8 in the ESI† and compare Fig. 2 for 8 and 9); and (c) a drastically different cis/trans ratio of product 2 (23:1) was found with elemental iodine.
Likewise, addition of 1 mol-% of HOTf triggered full conversion of compound 1 to product 2 within 1 hour. The kinetic profile of the acid-catalysed reaction, however, is entirely different from the ones observed for catalysts 8 and syn-9, in which the enol is accumulated (see the ESI†). In addition, the cis/trans ratio of product 2 is once again markedly different (6.5:1). As a consequence, catalysis by elemental iodine or acid traces – either as impurities or decomposition products – appears very unlikely and the mode of activation is most probably halogen bonding.
Even though the focus of this study was on the electrocyclization step, it is noteworthy that the rate of the keto–enol tautomerization was apparently not significantly influenced by the catalysts. Also, in some cases the rate of formation of cyclopentenone 2 decreases significantly after full consumption of starting material 1 (see Fig. 4).
 | ||
| Fig. 4 1H-NMR yield vs. time profile for the formation of cyclopentenone 2. The error is approximately 5%. c0(1) = 15.4 mM. | ||
Finally, the nature of XB catalysis in this reaction was also investigated by DFT calculations using the M06-2X functional,21 Grimme D3 dispersion corrections22 and the def2-TZVP(D) basis set.23 The corresponding transition state of the Nazarov cyclisation of substrate 1 with a truncated version24 of XB donor syn-9 is shown in Fig. 5.
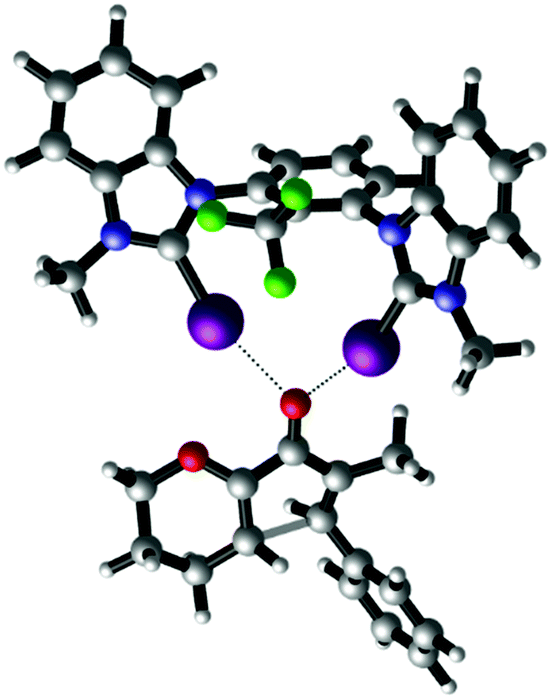 | ||
| Fig. 5 DFT-calculated transition state structure of the Nazarov cyclisation catalysed by halogen bond-donor syn-9 (M06-2X D3 def2-TZVP(D)).25 Selected distances [Å] and angles [°]: I–O 2.68 and 2.69; C–I–O 162 and 164. Graphic generated using CYLview.26 | ||
The optimized geometry features a clear bidentate binding to the carbonyl oxygen with iodine–oxygen bond distances of 2.68 Å and 2.69 Å. The corresponding barrier of activation (corrected with the SMD1825 intrinsic solvent model for dichloromethane) is 26.4 kcal mol−1. This result is in good agreement with the experimental findings. The barrier for the uncatalysed Nazarov cyclisation (33.1 kcal mol−1) is significantly higher.
In conclusion, the first halogen-bonding-catalysed Nazarov cyclisation reaction was reported, which is also just the 4th example of carbonyl activation by this interaction. The catalytic activity could be clearly traced back to halogen bonding via comparison experiments. The effect of counterion exchange and preorganization was investigated and compared, and strong performance could only be achieved by a combination of both. In fact, it seems that this reaction is the most challenging one activated by halogen bonding so far (as 7/BArF4 did not show any effect in contrast to all examples reported earlier).
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (Grant Agreement No. 638337) and from the Fonds der Chemischen Industrie (Dozentenstipendium to S. M. H.). We thank Martin Gartmann for assistance with NMR spectroscopic measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) I. N. Nazarov, I. B. Torgov and L. N. Terekhova, Izv. Akad. Nauk SSSR Otd. Khim. Nauk, 1942, 200 CAS ; For the most recent reviews, see: ; (b) A. J. Frontier and C. Collison, Tetrahedron, 2005, 61, 7577 CrossRef CAS; (c) M. A. Tius, Eur. J. Org. Chem., 2005, 2193 CrossRef CAS; (d) T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531 CrossRef CAS; (e) D. R. Wenz and J. Read de Alaniz, Eur. J. Org. Chem., 2015, 23 CrossRef CAS; (f) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2017, 15, 8245 RSC.
- (a) G. Liang, S. N. Gradl and D. Trauner, Org. Lett., 2003, 26, 4931 CrossRef PubMed; (b) V. K. Aggarwal and A. J. Belfield, Org. Lett., 2003, 26, 5075 CrossRef PubMed; (c) W. He, X. Sun and A. J. Frontier, J. Am. Chem. Soc., 2003, 125, 14278 CrossRef CAS PubMed; (d) G. Liang and D. Trauner, J. Am. Chem. Soc., 2004, 126, 9544 CrossRef CAS PubMed; (e) K. Murugan, S. Srimurugan and C. Chen, Chem. Commun., 2010, 46, 1127 RSC; (f) G. E. Hutson, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 4988 CrossRef CAS; (g) J. Davies and D. Leonori, Chem. Commun., 2014, 50, 15171 RSC; (h) T. Mietke, T. Cruchter, V. A. Larionov, T. Faber, K. Harms and E. Meggers, Adv. Synth. Catal., 2018, 360, 2093 CrossRef CAS.
- (a) M. Vogler, L. Süsse, J. H. W. LaFortune, D. W. Stephan and M. Oestreich, Organometallics, 2018, 37, 3303 CrossRef CAS; (b) L. Süsse, M. Vogler, M. Mewald, B. Kemper, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 11441 CrossRef PubMed.
- G. Pouss, A. Devineau, V. Dalla, L. Humphreys, M. Lasne, J. Rouden and J. Blanchet, Tetrahedron, 2009, 65, 10617 CrossRef.
- (a) M. Rueping, W. Ieawsuwan, A. P. Antonchick and B. J. Nachtsheim, Angew. Chem., Int. Ed., 2007, 46, 2097 CrossRef CAS PubMed; (b) A. Jolit, P. M. Walleser, G. P. A. Yap and M. A. Tius, Angew. Chem., Int. Ed., 2014, 53, 6180 CrossRef CAS PubMed.
- (a) A. K. Basak, N. Shimada, W. F. Bow, D. A. Vicic and M. A. Tius, J. Am. Chem. Soc., 2010, 132, 8266 CrossRef CAS PubMed; (b) A. H. Asari, Y. Lam, M. A. Tius and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 13199 CrossRef PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CAS.
- For the most recent reviews, see: (a) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed; (b) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed; (c) D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434 CrossRef CAS PubMed; (d) R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004 CrossRef CAS PubMed.
- (a) S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187 CrossRef CAS PubMed; (b) F. Kniep, L. Rout, S. M. Walter, H. K. V. Bensch, S. H. Jungbauer, E. Herdtweck and S. M. Huber, Chem. Commun., 2012, 48, 9299 RSC; (c) A. Dreger, E. Engelage, B. Mallick, P. D. Beer and S. M. Huber, Chem. Commun., 2018, 54, 4013 RSC.
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028 CrossRef CAS PubMed.
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110 CrossRef CAS PubMed.
- (a) A. Bruckmann, M. A. Pena and C. Bolm, Synlett, 2008, 900 CAS; (b) W. He, Y. Ge and C. Tan, Org. Lett., 2014, 16, 3244 CrossRef CAS PubMed; (c) K. Matsuzaki, H. Uno, E. Tokunaga and N. Shibata, ACS Catal., 2018, 8, 6601 CrossRef CAS.
- (a) D. von der Heiden, S. Bozkus, M. Klussmann and M. Breugst, J. Org. Chem., 2017, 82, 4037 CrossRef CAS PubMed; (b) D. von der Heiden, E. Detmar, R. Kuchta and M. Breugst, Synlett, 2018, 1307 CAS.
- J. Gliese, S. H. Jungbauer and S. M. Huber, Chem. Commun., 2017, 53, 12052 RSC.
- S. H. Jungbauer, S. M. Walter, S. Schindler, L. Rout, F. Kniep and S. M. Huber, Chem. Commun., 2014, 50, 6281 RSC.
- (a) Y. Takeda, D. Hisakuni, C. Lin and S. Minakata, Org. Lett., 2015, 17, 318 CrossRef CAS PubMed; (b) R. Haraguchi, S. Hoshino, M. Sakai, S. Tanazawa, Y. Morita, T. Komatsu and S. Fukuzawa, Chem. Commun., 2018, 54, 10320 RSC; (c) F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3892 CrossRef.
- After longer reaction times, formation of product 2 is observed with sigmoidal kinetics (Fig. S4, ESI†). Currently, we cannot explain their origin.
- Lower amounts of iodine lead to sigmoidal kinetics, see Fig. S8 in the ESI†.
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353 CrossRef CAS PubMed.
- M. Breugst and D. von der Heiden, Chem. – Eur. J., 2018, 24, 9187 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- This basis set consists of def2-TZVP (F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297) for all main group elements up to Kr and def2-TZVPD (D. Rappoport, F. Furche, J. Chem. Phys. 2010, 133, 134105) for all other elements.
- The counterion (BArF4) and the para-fluorine substituent of the aryl group of the substrate were omitted and the N-octyl-chains were exchanged to N-methyl for reasons of computational costs.
- E. Engelage, N. Schulz, F. Heinen, S. M. Huber, D. G. Truhlar and C. J. Cramer, Chem. – Eur. J., 2018, 24, 15983 CrossRef CAS PubMed.
- Legault, C. Y. CYLview, 1.0b; Université de Sherbrooke: Sherbrooke, 2009, http://www.cylview.org.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1898636. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc02816a |
| This journal is © The Royal Society of Chemistry 2019 |