
Dual-template molecularly imprinted polymers for dispersive solid-phase extraction of fluoroquinolones in water samples coupled with high performance liquid chromatography†
Wenhui
Lu
ac,
Jie
Liu
bc,
Jinhua
Li
c,
Xiaoyan
Wang
c,
Min
Lv
c,
Rong
Cui
*b and
Lingxin
Chen
 *cd
*cd
aSchool of Light Industry Science and Technology, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250353, China
bSchool of Environment and Materials Engineering, Yantai University, Yantai 264005, China. E-mail: glorytre@163.com
cCAS Key Laboratory of Coastal Environmental Processes and Ecological Remediation, Research Center for Coastal Environmental Engineering and Technology, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China. E-mail: lxchen@yic.ac.cn
dCollege of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, China
First published on 4th December 2018
Abstract
Novel dual-template molecularly imprinted polymers (dt-MIPs) were prepared by simple and facile precipitation polymerization using norfloxacin (NOR) and enrofloxacin (ENR) as templates for simultaneous selective recognition and extraction of the two fluoroquinolones (FQs). The as-prepared dt-MIPs exhibited high adsorption capacity and excellent selectivity towards NOR and ENR. Several main parameters affecting the efficiency of dt-MIP based dispersive solid-phase extraction (DSPE) were systematically investigated, coupled with high performance liquid chromatography determination. Consequently, high enrichment factors of 71 and 61 were obtained for NOR and ENR respectively, and good linearity in the range of 1–200 μg L−1 was observed, with correlation coefficients (r) above 0.9977. The limits of detection and quantification for NOR were 0.22 and 0.67 μg L−1, respectively, and 0.36 and 0.98 μg L−1 for ENR. Satisfactory recoveries of the two FQs from spiked lake, sea and tap water samples at three concentration levels were attained in the range of 80.9–101.0% with relative standard deviations of 0.9–6.9%. The present study not only has great potential for applications in FQ determination, but will also enrich research into dual/multi-template imprinting.
Introduction
Fluoroquinolones (FQs) are derivates of quinolones containing a fluorine atom on the central carbon ring, which were synthesized in the late 1970s and early 1980s to resist both Gram-negative and Gram-positive bacteria.1 Over the next few decades, FQs were commonly used in human, veterinary and agricultural applications, because of their wide antimicrobial spectrum and effective antibacterial action.1 Frustratingly, the majority of FQs are discharged into the environmental system due to inadequate metabolism in human or animal bodies and limited biodegradation.2 FQ residues can promote bacterial resistance, poison plants and aquatic organisms, and have an adverse effect on human health.3 Recent research has also revealed that a multitude of FQs have been detected at levels of ng L−1 to μg L−1 in environmental water systems, and in aquatic and terrestrial organisms.4,5 Therefore, sensitive, efficient and selective analytical methods are required to determine trace levels of FQs in complicated matrices.Currently available instrumental analytical technologies for FQ determination include high performance liquid chromatography (HPLC) coupled with a fluorescence detector (FLD),6 mass spectrometry (MS),7 an ultraviolet (UV) detector8 or diode array detector (DAD),9 and capillary electrophoresis (CE).10 Given the low concentration levels of FQs in samples, a number of pretreatment technologies have been proposed to extract and enrich FQs before instrumental analysis, such as solid-phase extraction (SPE),11 dispersive solid-phase extraction (DSPE),12 solid-phase microextraction,13 matrix solid-phase dispersion,14 stir-bar sorptive extraction,15 stir-cake sorptive extraction,16 hollow fiber based liquid-phase microextraction,17 and dispersive liquid–liquid microextraction.18 Amongst them, SPE is commonly used in food,19 environmental20 and biological21 fields due to its high extraction efficiency and reusability. Recent studies have focused on the innovation of the SPE method and the preparation and application of novel adsorbents.19–24 DSPE is based on SPE technology, and sorbents are directly added into the sample without packing (into a column) and conditioning, which is simpler, more efficient and time-saving.22 He et al. used commercial magnetic graphene as a DSPE sorbent combined with HPLC for the extraction and determination of FQ residues in foods of animal origin.23 Amoli-Diva et al. synthesized magnetic multi-walled carbon nanotube nanocomposites as adsorbents for dispersive micro-solid-phase extraction coupled with surfactant-enhanced spectrofluorimetric detection for the enrichment and determination of ofloxacin and lomefloxacin in biological and environmental samples.24
Besides, molecularly imprinted polymers (MIPs) with predictable structures, specific recognition abilities and universal applications have become attractive adsorbents and have been widely applied in sample pretreatment and chromatographic separation to specifically extract the template molecules from complex matrices.25–27 MIPs have also been used for SPE of FQs in various samples. For example, Turiel et al. synthesized MIPs in one single preparation step by precipitation polymerization using ciprofloxacin (CIP) as a template, and successfully employed them both as SPE sorbents and as a stationary phase in the determination of FQs in soil samples.28 Wu and a coworker29 used levofloxacin (LEV) as a template and ionic liquids as reaction solvents to synthesize MIPs by bulk polymerization based on molecular crowding, and applied them as SPE sorbents followed by HPLC to enrich and determine FQs in milk and lake water samples. Urraca et al.30 prepared MIPs using enoxacin (ENOX) as a template by surface imprinting on silica beads, which were packed into an SPE cartridge coupled with HPLC-FLD and HPLC-MS/MS for the selective extraction and detection of six FQs in chicken muscle samples. In the above mentioned MIPs, the specific recognition sites for the template are created by a single template. However, the imprinting process does not have to be limited to a single template for practical applications.31 Although single-template MIPs have also been used to extract structural analogues of the template molecule through cross-selectivity, sometimes the prepared MIPs cannot effectively bind the structural analogues, thus diminishing their affinity toward analytes.32 Accordingly, a multi-template imprinting strategy has been proposed and increasingly used to synthesize MIPs with multiple types of recognition sites in one format, using dual/multiple targets/species as templates, which can widen the applications of MIPs for simultaneous recognition and extraction of more than one analyte.33–40 For instance, Liu et al.36 adopted a multi-template imprinting strategy using melamine and dicyandiamide as templates to prepare MIPs, which were packed into an SPE column to simultaneously extract melamine and dicyandiamide from powdered milk. Compared with single-template MIPs and non-imprinted polymers, the dual-template MIPs (dt-MIPs) showed better affinity and selectivity for the two templates. Jafari et al.38 synthesized magnetic dt-MIPs using acetaminophen and codeine as templates by combining sol–gel polymerization and surface imprinting techniques, and then used them as magnetic SPE (MSPE) adsorbents for the simultaneous enrichment of acetaminophen and codeine in urine samples. Xia et al.40 synthesized dt-MIPs using a mixture of clonazepam and 2-(trifluoromethyl)phenothiazine as templates via precipitation polymerization, and then used the dt-MIPs to prepare an SPE cartridge for the simultaneous extraction of two classes of sedatives (4 phenothiazines and 5 benzodiazepines) in swine complete formula feed samples.
Inspired by these studies, herein we chose two typical FQs as templates, namely norfloxacin (NOR) and enrofloxacin (ENR), which are often used in human and veterinary medicine, to synthesize novel dt-MIPs by simple facile precipitation polymerization for the concurrent recognition and extraction of these two FQs. Indexing of NOR and ENR can be used to evaluate the possible migration routes or spatial distribution of residual FQs in the environment, and as far as we are aware no reported studies have used them as templates for dt-MIPs. The as-prepared dt-MIPs were well characterized and assessed, and utilized as DSPE adsorbents followed by HPLC determination. To the best of our knowledge, this is the first demonstration of the extraction of FQs by dt-MIP based DSPE. The conditions for the dt-MIP based DSPE, including dt-MIP dosage, sample pH, extraction time, type and volume of desorption solvent and desorption time, were optimized. Subsequently, the dt-MIPs-DSPE-HPLC method was validated and successfully applied in real water sample analysis.
Experimental
Reagents and materials
Norfloxacin (NOR, 99%), enrofloxacin (ENR, 98.5%), ciprofloxacin (CIP, 99%), pefloxacin (PEF, 99%) and danofloxacin (DAN, 98%) were purchased from Dalian Meilun Biological Technology Co., Ltd (Dalian, China), and enoxacin (ENO, 98%) was obtained from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). Standard solutions of ENR in methanol with concentration of 100 mg L−1 and NOR in ethanol with concentration of 100 mg L−1 were purchased from Agro-Environmental Protection Institute, Ministry of Agriculture (Tianjin, China) and stored at 4 °C in a refrigerator before use. HPLC grade acetonitrile and formic acid were obtained from Sigma-Aldrich (Shanghai, China). Methacrylic acid (MAA), 4-vinylpyridine (4-VP) and ethylene glycol dimethacrylate (EGDMA) were purchased from Sigma-Aldrich and purified prior to use in order to remove stabilizers. Acrylamide (AAm) was purchased from Aladdin (Shanghai, China) and recrystallized in water prior to use. 2,2′-Azo-bis-isobutyronitrile (AIBN) was obtained from Aladdin (Shanghai, China), and was recrystallized in ethanol prior to use. Methanol, acetonitrile and acetic acid were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).Ultrapure water with a specific resistance of 18.2 MΩ cm was produced by a Pall Cascada™ lab water purification system (Pall Corp., USA) for aqueous solution preparation throughout the study. All other reagents were analytical reagent grade and used without further purification steps.
Instrumentation
Analyses were performed using an Agilent 1260 Infinity HPLC system (Agilent Technologies, Palo Alto, CA, USA) equipped with a diode array detector (DAD). Optimized fluoroquinolone separation was achieved in a reversed-phase column (Agilent ZORBAX SB-C18, 150 × 4.6 mm i.d., particle size: 5 μm) by isocratic elution using a mobile phase consisting of water (containing 0.05% formic acid)–acetonitrile (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15, v/v) at a flow rate of 1 mL min−1. The injection volume, column temperature and detection wavelength were individually set as 20 μL, 30 °C and 280 nm.
:15, v/v) at a flow rate of 1 mL min−1. The injection volume, column temperature and detection wavelength were individually set as 20 μL, 30 °C and 280 nm.
The morphologies of the polymer particles were examined by scanning electron microscopy (SEM, Hitachi S-4800, Japan). All samples were sputter-coated with thin gold film before observation. The size distributions of the MIPs and non-imprinted polymers (NIPs) were tested using a laser particle analyzer (Malvern Mastersizer 2000F, UK). Infrared spectra of samples were obtained using a Fourier transform infrared (FT-IR) spectrometer (Thermo Nicolet Corporation, USA) by using a pressed KBr tablet. The thermal stability of the MIPs was measured by thermogravimetric analysis (TGA) using a Mettler 5MP/PF7548/MET/400 W thermal analyzer (Mettler Toledo, Switzerland) in a nitrogen atmosphere between 40 and 750 °C with an nitrogen flow rate of 50 mL min−1 and a heating rate of 20 °C min−1, in order to obtain TGA and derivative thermogravimetry (DTG) data. Specific surface areas were obtained using the Brunauer–Emmett–Teller (BET) method and pore diameters were determined using the Barrett–Joyner–Halenda (BJH) method, which were performed on a Full-automatic Specific Surface Instrument (3H-2000PS4, Beishide Instrument Technology, China).
Preparation of dt-MIPs
dt-MIPs were prepared according to a non-covalent approach by a simple facile precipitation polymerization using NOR and ENR as the templates, and the schematic procedure is shown in Fig. 1A. Briefly, NOR (0.1 mmol), ENR (0.1 mmol), and MAA (0.8 mmol) were dissolved in acetonitrile (29 mL) and methanol (1 mL) in a 100 mL glass flask, and were pre-polymerized at 4 °C in the dark for 12 h. The cross-linker (EGDMA, 4 mmol) and the initiator (AIBN, 20 mg) were then successively added to the solution. The solution was sonicated for 5 min and deoxygenated with nitrogen for 15 min to remove dissolved oxygen. Then, the flask was sealed under a nitrogen atmosphere and placed in a water bath. The temperature of the water bath was increased from room temperature to 60 °C over approximately 2 h and then kept at 60 °C for 24 h for the polymerization. After the polymerization, the polymers were washed with acetonitrile to remove the unreacted reagents and then washed with methanol/acetic acid (90:10, v/v) to remove the template molecules until no template was detected by HPLC. Finally, the polymer particles were washed with methanol to remove acetic acid and dried under vacuum at 40 °C before use. As a control, the corresponding NIPs were synthesized using the same conditions in the absence of the template molecules.
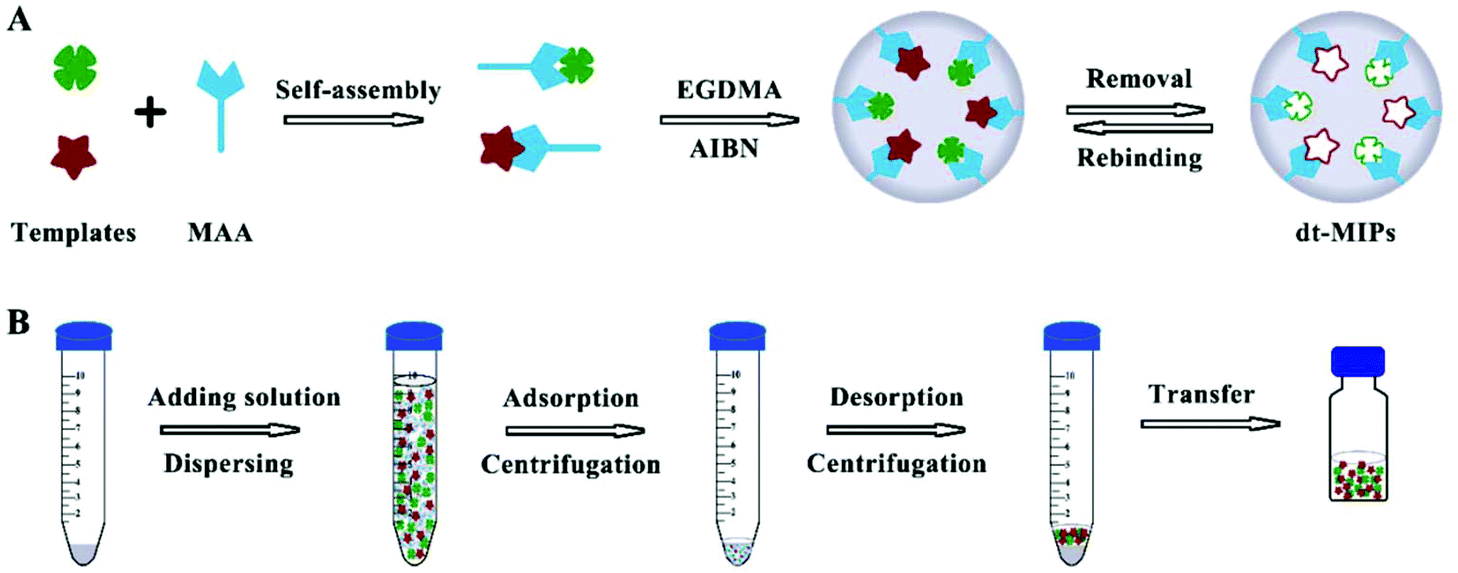 | ||
| Fig. 1 Schematic procedures for the preparation of dt-MIPs by precipitation polymerization (A) and dt-MIPs-DSPE (B). | ||
Adsorption experiments
Static adsorption experiments were carried out to investigate the adsorption capacity of the synthesized polymers. In short, 5.0 mg dt-MIPs or NIPs were dispersed in 10 mL plastic centrifuge tubes containing 4 mL ENR and NOR solutions of various concentrations from 5 to 100 mg L−1. After ultrasonic dispersion, the mixtures were shaken for 24 h at room temperature. After centrifugation and filtration through 0.22 μm cellulose acetate membrane filters, the supernatant was collected and tested by HPLC. The adsorption capacity (Q) was calculated according to the following equation:where C0 (mg L−1) and Ce (mg L−1) are the initial and equilibrium concentrations of the template compounds, respectively. V (mL) is the volume of the solution, and m (g) is the mass of dt-MIPs or NIPs.
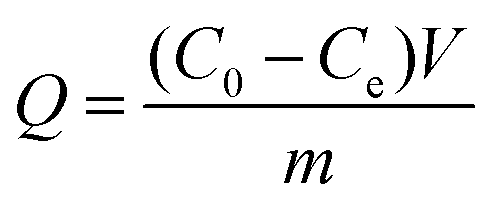
According to the static adsorption procedure, the specific adsorption performances of the dt-MIPs were investigated using a mixed solution of NOR, ENR and other four analogues (PEF, DAN, ENO and CIP) at individual concentration levels of 80 mg L−1. The imprinting factor (α), distribution coefficient (Kd), selectivity coefficient (K) and relative selectivity coefficient (K′) were used to evaluate the selectivity of the dt-MIPs. The related descriptions and equations are given in Experimental S1.†
DSPE procedure and real sample preparation
The dt-MIPs-DSPE procedure is schematically shown in Fig. 1B. Briefly, 10 mg dt-MIPs were added into a centrifugation tube containing 10 mL NOR and ENR solutions at concentrations of 50 μg L−1. The dt-MIPs were dispersed by ultrasonication for 2 min, and then the tube was continuously shaken for 3 h at room temperature. After centrifugation at 9000 rpm for 10 min and discarding the supernatant, the polymers and adsorbed target molecules at the bottom of the tube were dried in a vacuum drying oven. Then, 150 μL methanol/acetic acid (90:10, v/v) were added and the analytes were desorbed by ultrasound for 5 min, followed by centrifugation at 9000 rpm for 10 min and collection of the supernatant. Finally, 20 μL of the desorption solvent was injected into the HPLC system for analysis after filtration through 0.22 μm organic nylon membrane filters.
Tap water was collected in the laboratory after it had flowed for about 5 min. Surface seawater was obtained from Haichang Fisherman's Wharf at the Yellow Sea located in the coastal zone area of Yantai City, and lake water was collected from an artificial lake named Sanyuan Lake located in the school yard of Yantai University. All water samples were stored at 4 °C and filtered through a 0.22 μm hydrophilic polypropylene membrane for the removal of possible suspended impurities prior to use.
Results and discussion
Preparation of dt-MIPs using NOR and ENR
The dt-MIPs were synthesized by precipitation polymerization using NOR and ENR as templates, which are typical FQs commonly used in human and veterinary applications. In order to synthesize dt-MIPs with homogeneous morphology and good adsorption and recognition properties, typical parameters should be optimized, such as type and concentration of porogen, functional monomer and cross-linker. Generally, porogens act as both solvents and pore forming agents in the polymerization process. Especially in non-covalent interaction systems, porogens can also influence the bonding strength between the monomer and the template, and the properties and morphology of the imprinted polymers.41 In this study, methanol, acetonitrile and chloroform were used as porogens for the preparation of dt-MIPs. It was found that soft bulk polymers were obtained when using methanol and chloroform as porogens. Because the solubility of NOR in methanol was better than that in acetonitrile, a suitable volume of methanol was added when using acetonitrile as the porogen, and consequently imprinted polymers with uniform morphology were obtained. Moreover, the effect of volume of porogen (acetonitrile containing methanol) was also investigated and the results indicated that the obtained dt-MIPs had comparatively higher adsorption capacity and more uniform morphology when the total volume of porogen was 30 mL (acetonitrile/methanol = 29:1, v/v). The functional monomer plays an important role in providing functional groups which can form a pre-polymerization complex with the template by covalent or non-covalent interactions.41,42 MAA, AAm and 4-VP were chosen as monomers and the results showed that it was easier to obtain more uniform spherical polymers with higher adsorption capacity when using MAA as monomer than when using AAm or 4-VP as monomer. Also, the molar ratio of template and monomer can affect the affinity and imprinting effect of dt-MIPs toward the templates.43 Molar ratios of template and monomer of 1:2 and 1:4 were investigated, while the molar ratio of template to cross-linker (EGDMA) was set at 1:20. The dt-MIPs prepared using a molar ratio of template/monomer/cross-linker of 1:4:20 showed excellent adsorption affinity for NOR and ENR. Therefore, under the optimized preparation conditions, new dt-MIPs were prepared by simple and facile precipitation polymerization.
Characterization of dt-MIPs
The as-prepared dt-MIPs were characterized by SEM, BET, FT-IR and TGA as follows. Fig. 2A shows the SEM images of dt-MIPs (a) and NIPs (b), demonstrating that uniform and spherical particles, relatively rough surfaces and smaller average diameters were observed for the MIPs. As seen from Fig. 2B, the dt-MIPs and NIPs exhibited good size distribution, and the dominant distribution peak was at about 190 nm for dt-MIPs (a) and 350 nm for NIPs (b), possibly resulting from the influence of the template on particle growth during the precipitation polymerization.44 This phenomenon might be attributed to different forms of MAA in the dt-MIP and NIP reaction systems. During the preparation of NIPs, MAA could form hydrogen-bonded dimers in the absence of template, and both free MAA and MAA dimers were present in the pre-polymerization solution. In the dt-MIP reaction system, additional molecular interactions between MAA and the template were formed, which might affect the growth of the cross-linked polymer nuclei and result in smaller polymer particles.45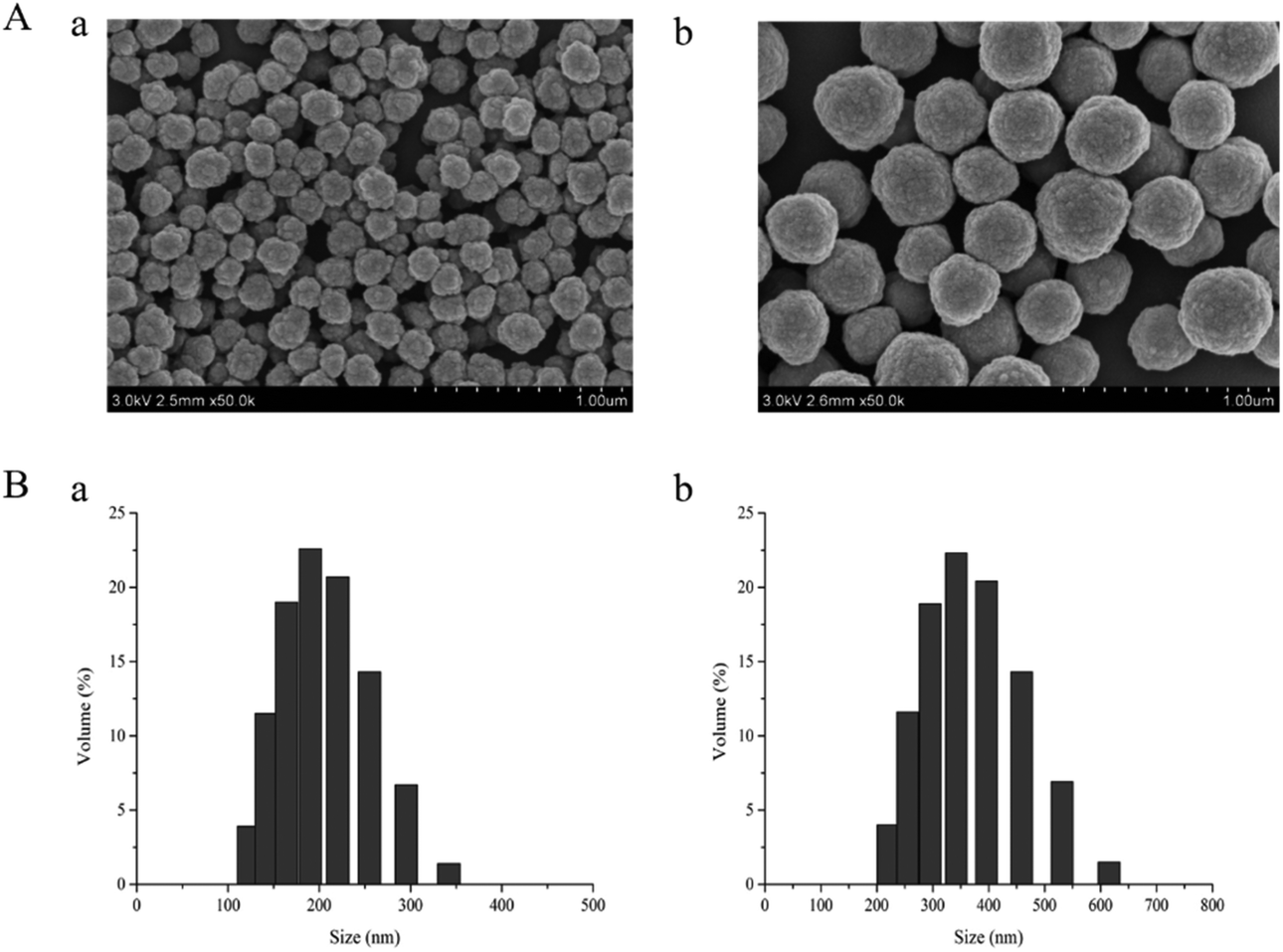 | ||
| Fig. 2 (A) SEM images of dt-MIPs (a) and NIPs (b) at the same scale (1 μm). (B) Particle size distribution profiles of dt-MIPs (a) and NIPs (b). | ||
Surface area and pore volume are important parameters that affect the adsorption capacity of imprinted polymers. By BET analysis, the specific surface area of the dt-MIPs was 48.73 m2 g−1, much larger than that of the corresponding NIPs (14.40 m2 g−1), probably leading to a higher adsorption capacity for the dt-MIPs. Likewise, the dt-MIPs possessed much larger pore volume (0.43 mL g−1) than the NIPs (0.18 mL g−1), which might be attributed to the formation of binding cavities on the surface of the dt-MIPs. Moreover, the dt-MIPs and NIPs exhibited average pore sizes of 24.84 and 21.77 nm, respectively. According to the commonly used classification of the International Union of Pure and Applied Chemistry (IUPAC), the pores should be classified as mesopores (between 2 nm and 50 nm). The mesopores can also affect the polymers’ adsorption properties to some degree.
FT-IR spectra of the dt-MIPs before and after template removal are shown in Fig. 3A. It can be seen from curves a and b that strong absorption bands at around 3428 cm−1 and 1731 cm−1 were observed, which could be ascribed to the O–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the carboxylic acid group of MAA. The significant absorption bands at around 1257 cm−1 and 1157 cm−1 could be assigned to the symmetric and asymmetric C–O stretching vibrations of EGDMA.29 The peaks at 2985 cm−1 and 2954 cm−1 were attributed to the C–H stretching vibrations of –CH3 and –CH2 groups. Compared with the IR spectra of NOR (curve c) and ENR (curve d), the C–F stretching vibration at 960 cm−1 disappeared and the CC vibration of the benzene ring at around 1465 cm−1 became weak after template removal (curve b), which could indicate that the dt-MIPs were successfully prepared.
O stretching vibrations of the carboxylic acid group of MAA. The significant absorption bands at around 1257 cm−1 and 1157 cm−1 could be assigned to the symmetric and asymmetric C–O stretching vibrations of EGDMA.29 The peaks at 2985 cm−1 and 2954 cm−1 were attributed to the C–H stretching vibrations of –CH3 and –CH2 groups. Compared with the IR spectra of NOR (curve c) and ENR (curve d), the C–F stretching vibration at 960 cm−1 disappeared and the CC vibration of the benzene ring at around 1465 cm−1 became weak after template removal (curve b), which could indicate that the dt-MIPs were successfully prepared.
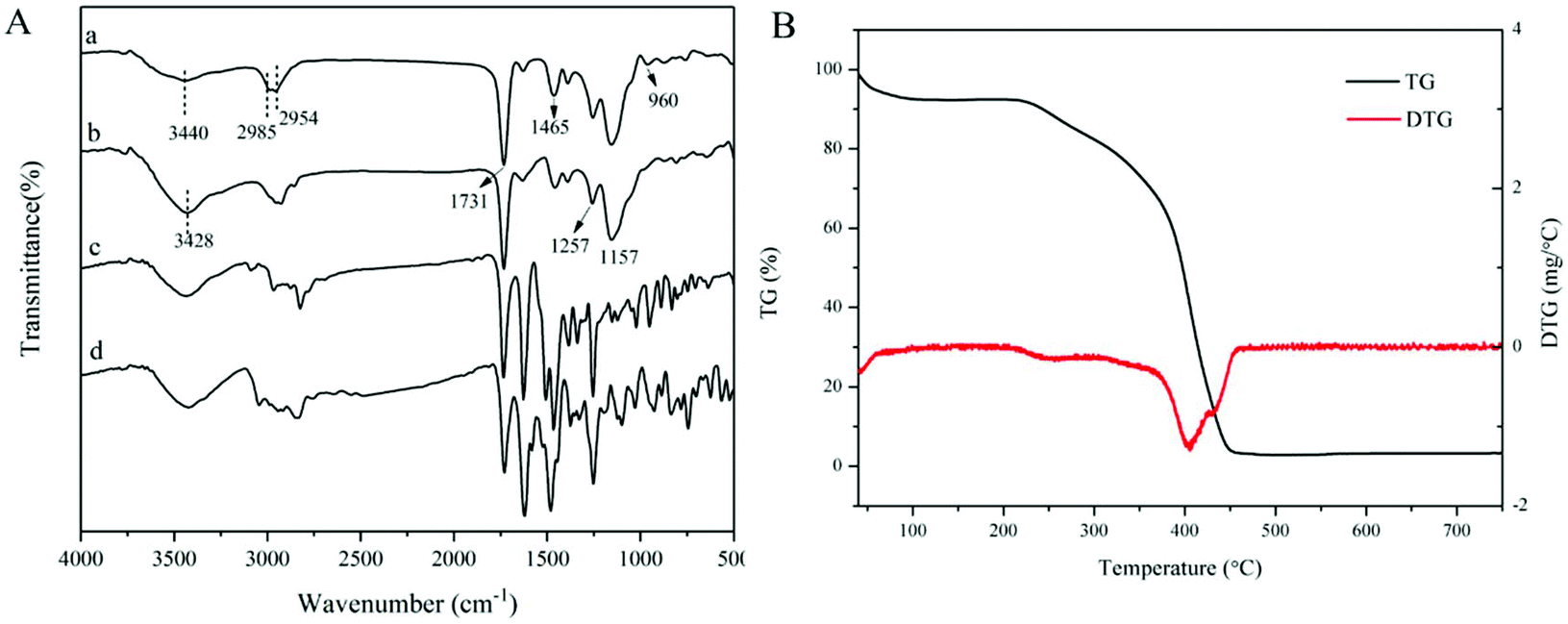 | ||
| Fig. 3 (A) FT-IR spectra of dt-MIPs before (a) and after (b) template removal, and spectra of NOR (c) and ENR (d). (B) TG/DTG curves of dt-MIPs. | ||
TGA was used to evaluate the thermal stability of the dt-MIPs. As shown in Fig. 3B, the dt-MIPs exhibited a slight weight loss with an increase in temperature from 40 °C to 220 °C, which might be attributed to water evaporation.46 At temperatures ranging from 220 °C to 450 °C, a rapid weight loss occurred, indicating decomposition of the material. Then, the weight of the dt-MIPs tended to stabilize at higher temperature, with the full release of a volatile compound. Therefore, the synthesized dt-MIPs showed good thermal stability at temperatures lower than 220 °C. This phenomenon can also be clearly observed from the corresponding DTG curve (Fig. 3B).
Adsorption analysis
The adsorption capacities of the dt-MIPs and NIPs for NOR and ENR were evaluated by static adsorption isotherms, as shown in Fig. S1.† As seen from the figure, for the dt-MIPs, the adsorption capacities for NOR and ENR increased continuously with an increase in initial concentration and then became stable above the equilibrium concentration of 80 mg L−1, while the NIPs exhibited a similar trend but lower binding capacities. The maximum adsorption capacities of the dt-MIPs for NOR and ENR were 32.0 and 21.8 mg g−1, respectively. Whereas, the maximum adsorption capacities of the NIPs were lower, 14.0 mg g−1 and 13.1 mg g−1 for NOR and ENR, respectively. Based on the principle of adsorption–desorption equilibrium between polymers and templates, the templates can only occupy a small portion of the recognition sites of the dt-MIPs at low initial concentration, resulting in a low adsorption capacity. With an increase in concentration, the recognition sites of the dt-MIPs were gradually occupied by templates, so that the adsorption capacity increased sharply and reached saturation. Because of the absence of specific recognition sites in the NIPs, a comparatively lower adsorption capacity was observed due to the dominating physical adsorption. On the other hand, the maximum adsorption capacity of the dt-MIPs was higher for NOR than for ENR, which might be ascribed to the different interaction strength between each template and the functional monomer. As a result, the synthesized dt-MIPs could selectively recognize and adsorb the two template compounds simultaneously. And the dt-MIPs with high adsorption capacity would achieve high extraction efficiency for the trace analysis of NOR and ENR.To estimate the selectivity of the dt-MIPs for NOR and ENR, four other structurally related FQs (PEF, DAN, ENO and CIP) were chosen and investigated. The chemical structures of the FQs used in this study and the adsorption capacities of the dt-MIPs and NIPs for them are shown in Fig. S2 and S3,† respectively. As seen from Fig. S3,† similar low adsorption capacities of dt-MIPs and NIPs for PEF, DAN, ENO and CIP were attained. The p values from the Student’s t-test were all more than 0.05, which illustrated that there were no statistically significant differences. The above mentioned data confirmed that non-specific adsorption possibly occurred between the structural analogues and the dt-MIPs/NIPs. In contrast, the adsorption capacities of the dt-MIPs were much higher for the two templates, and the p values were lower than 0.01, which was considered as a highly significant difference, possibly resulting from the fact that specific recognition sites for NOR and ENR in terms of shape, size and functionality were generated on the dt-MIPs. Also, the imprinting factors (α), distribution coefficients (Kd), selectivity coefficients (K) and relative selectivity coefficients (K′) of the templates and analogues on dt-MIPs and NIPs were calculated for selectivity evaluation, as summarized in Table S1.† As shown in the table, good imprinting effects were indicated by the higher imprinting factors for NOR and ENR than for the analogues. The Kd values of the dt-MIPs for NOR and ENR were greater than those of the NIPs, which demonstrated that the distribution of imprinted cavities/recognition sites in the dt-MIPs was ordered according to a predetermined orientation. The K values of the dt-MIPs for NOR and ENR were significantly larger than those of the NIPs, implying that the dt-MIPs had higher recognition selectivity for the two templates. The K′ values indicated an enhancement in adsorption affinity and selectivity for the template molecules on the dt-MIPs with respect to the NIPs. The difference in binding ability between the two templates and the other analogues on the dt-MIPs revealed that the geometric affinity played an essential role in the selectivity of the dt-MIPs. The above results confirmed that the dt-MIPs had higher recognition and binding affinity for NOR and ENR than the NIPs, and that the dt-MIPs could highly selectively extract these two templates simultaneously. Moreover, it was found that the adsorption capacities of the NIPs for the two templates, NOR and ENR, were much larger than for the four structural analogues (Fig. S3†). For the selective adsorption experiments, the adsorption capacities of the NIPs for the templates and the structural analogues are mainly influenced by the adsorption performance of the NIPs, the adsorption conditions and the physicochemical properties of the analytes.47–50 In our study, considering that the adsorption experiments were carried out under the same conditions, the adsorption capacities of the NIPs for the FQs should be related to the pore structure and specific surface area of the NIPs, and to the steric hindrance and water solubility of the FQs.
Optimization of dt-MIPs-DSPE conditions
The proposed dt-MIPs with spherical morphology and high adsorption capacity were used as adsorbents for DSPE, coupled with HPLC-DAD, for simultaneous extraction and determination of NOR and ENR in aqueous solutions. Several main parameters affecting the extraction efficiency of dt-MIPs-DSPE were investigated in detail and optimized, including dt-MIP dosage, sample pH, type and volume of desorption solvent, and extraction and desorption time. All experiments were performed in triplicate using a standard aqueous solution with individual NOR and ENR concentrations of 50 μg L−1.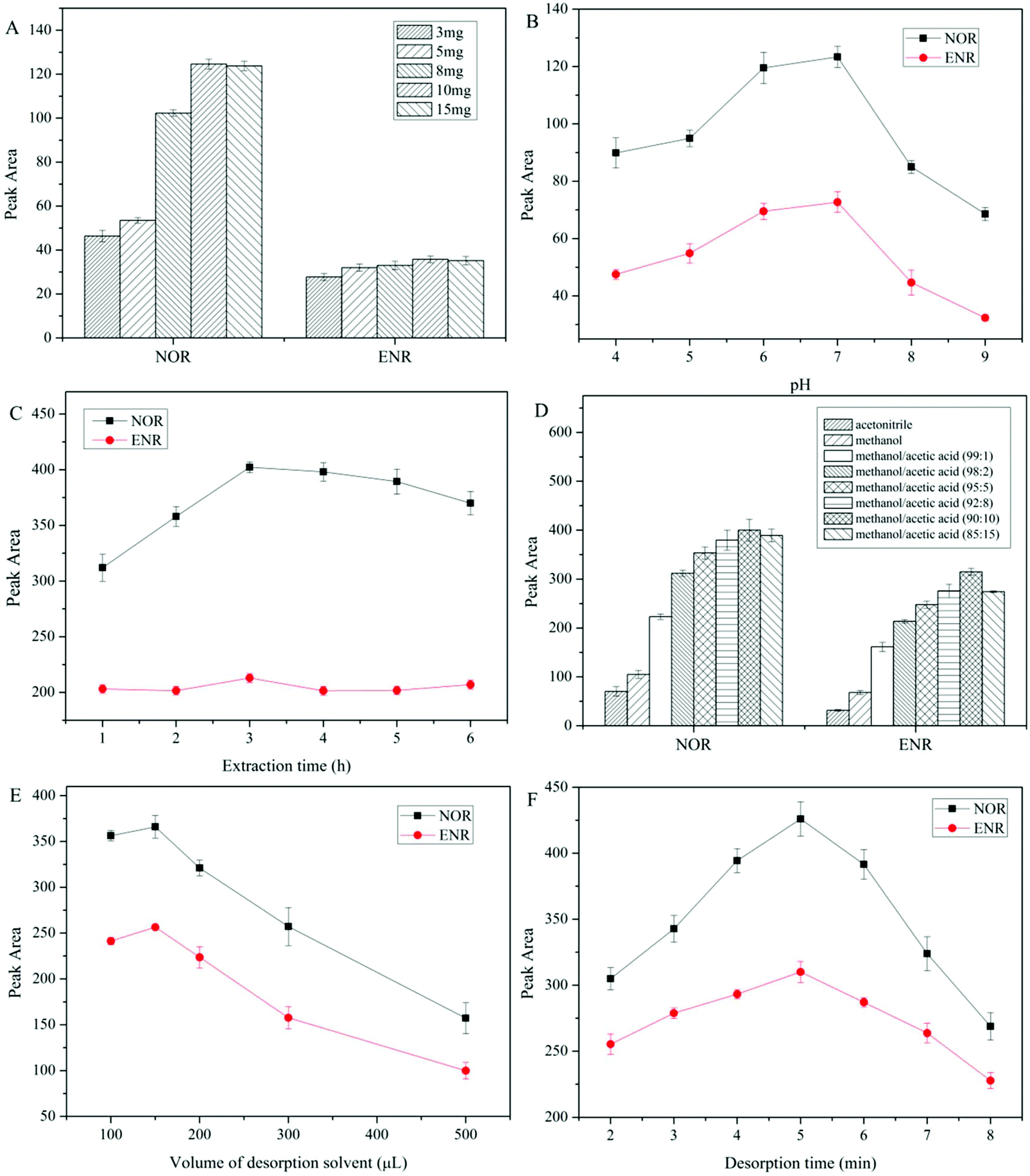 | ||
| Fig. 4 Effect of dt-MIP dosage (A), sample pH (B), extraction time (C), type of desorption solvent (D), volume of desorption solvent (E) and desorption time (F) on the extraction performance of dt-MIPs-DSPE. Extraction conditions: sample volume, 10.0 mL; (A) sample pH, 7.0; extraction time, 3 h; desorption solvent and volume, 150 μL methanol; desorption time, 5 min; (B) dt-MIP dosage, 10 mg; extraction time, 3 h; desorption solvent and volume, 150 μL methanol; desorption time, 5 min; (C) dt-MIP dosage, 10 mg; sample pH, 7.0; desorption solvent and volume, 150 μL methanol containing 10% acetic acid (v/v); desorption time, 5 min; (D) dt-MIP dosage, 10 mg; sample pH, 7.0; extraction time, 3 h; desorption volume, 150 μL; desorption time, 5 min; (E) dt-MIP dosage, 10 mg; sample pH, 7.0; extraction time, 3 h; desorption solvent, methanol containing 10% acetic acid (v/v); desorption time, 5 min; (F) dt-MIP dosage, 10 mg; sample pH, 7.0; extraction time, 3 h; desorption solvent and volume, 150 μL methanol containing 10% acetic acid (v/v). | ||
Volumes of desorption solvent ranging from 100 to 500 μL were also investigated, and the maximum extraction efficiency was attained using 150 μL, as obviously seen from Fig. 4E. The FQs bound in the dt-MIPs could not be desorbed sufficiently using a lower volume of elution solvent, while larger volumes reduced the extraction efficiency. Thus, 150 μL methanol/acetic acid (90:10, v/v) was employed for desorption.
The effect of desorption time on the extraction efficiency of FQs was studied for a range of 2–8 min. As observed in Fig. 4F, with an extension of desorption time, the extraction efficiency increased gradually and reached a maximum at 5 min, and then decreased. This indicated that 5 min was enough time to desorb the retained FQs from the dt-MIPs, whereas longer ultrasonic desorption times would make a small volume of the desorption solvent splash on the centrifuge tube wall, resulting in a decline in extraction efficiency. Accordingly, the optimal conditions for desorption were set as using 150 μL methanol/acetic acid (90:10, v/v) and a desorption time of 5 min.
Method validation and practical application of the dt-MIPs-DSPE
The selective dt-MIPs-DSPE coupled with HPLC was validated by investigating linearity, limit of detection (LOD), limit of quantification (LOQ), intraday/interday precision, and extraction recovery (ER), as well as the enrichment factor (EF). The equations for EF and ER are shown in Experimental S2† and all the results are presented in Table 1. As seen, good linearity was obtained in the range of 1–200 μg L−1, with correlation coefficients (r) of 0.9977 and 0.9989 for NOR and ENR, respectively. The LOD and LOQ were 0.22 and 0.67 μg L−1 for NOR, respectively, and 0.36 and 0.98 μg L−1 for ENR, obtained based on the signal-to-noise ratio (S/N = 3 and 10, respectively). The intraday and interday precision are expressed as the relative standard deviations (RSDs), which were calculated via five replicate analyses at 50 μg L−1 on the same day and five different days, respectively. Values of 3.0 and 7.3% respectively were obtained for NOR, and values of 3.3 and 4.3% respectively were obtained for ENR. The RSD values suggest that the method is highly accurate and reliable. The high EF and extraction recovery values (Table 1) fully demonstrate the highly selective pretreatment ability of the dt-MIPs-DSPE, which enabled the highly sensitive and concurrent determination of the two FQs.| FQ | Calibration curvea | Correlation coefficient (r) | Linear range (μg L−1) | LOD (μg L−1) | LOQ (μg L−1) | Intraday precisionb (RSD, %) | Interday precisionb (RSD, %) | EFb | ERb (%) |
|---|---|---|---|---|---|---|---|---|---|
| a y and x stand for the peak area and the concentration (μg L-1) of the analytes, respectively. b Intraday precision (n = 5), interday precision (n = 5), EF and extraction recovery (ER) were calculated at individual NOR and ENR concentrations of 50 μg L-1. | |||||||||
| NOR | y = (2.48 ± 0.07)x + (89.9 ± 6.85) | 0.9977 | 1–200 | 0.22 | 0.67 | 3.0 | 7.3 | 71 | 106.4 |
| ENR | y = (2.44 ± 0.05)x + (44.3 ± 4.74) | 0.9989 | 1–200 | 0.36 | 0.98 | 3.3 | 4.3 | 61 | 91.2 |
Furthermore, the validated DSPE method was applied to real water sample analysis including lake water, sea water and tap water. Endogenous FQs were not detected in the three water samples, owing to their quite low content in complicated matrices. Whereas, for water samples spiked with NOR and ENR at three concentration levels of 5, 20 and 50 μg L−1, the two FQs were easily found with complete base separation in less than 7 min, which could be attributed to the selective recognition ability and high adsorption capacity of the as-prepared dt-MIPs. Taking the samples spiked at 50 μg L−1 as examples, as shown in Fig. S4,† there are no obvious matrix interferences in the extraction and analysis of all three real water samples. Moreover, as listed in Table 2, satisfactory recoveries of the above three concentrations of FQs in the three kinds of water samples were attained, i.e., 80.9–101.0% with RSDs of 0.9–6.9%. For instance, in the sea water samples, the recoveries ranged from 83.7 to 99.1% with RSDs of 1.2–5.6% for NOR, and from 80.9 to 97.1% with RSDs of 1.1–6.7% for ENR. Consequently, the dt-MIP based DSPE method was shown to be practically feasible for effective enrichment, separation and determination of NOR and ENR simultaneously in complicated water samples. And therefore, a convenient and cost-effective method was successfully proposed and developed for the simultaneous selective analysis and abatement of more than one target analyte at trace levels in complicated matrices.
| FQ | Spiked concentration (μg L−1) | Lake water | Sea water | Tap water | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Average recovered concentration ± SDa (μg L−1) | Recovery (%) | RSD (%) | Average recovered concentration ± SD (μg L−1) | Recovery (%) | RSD (%) | Average recovered concentration ± SD (μg L−1) | Recovery (%) | RSD (%) | ||
| a SD means the standard deviation. | ||||||||||
| NOR | 5.0 | 4.8 ± 0.3 | 97.0 | 5.6 | 5.0 ± 0.1 | 99.1 | 1.2 | 5.0 ± 0.1 | 99.3 | 2.3 |
| 20.0 | 16.2 ± 0.1 | 81.2 | 0.9 | 16.7 ± 0.6 | 83.7 | 3.5 | 19.4 ± 0.7 | 96.8 | 3.4 | |
| 50.0 | 43.8 ± 0.9 | 87.6 | 2.0 | 46.7 ± 2.6 | 93.3 | 5.6 | 43.0 ± 1.6 | 86.1 | 3.6 | |
| ENR | 5.0 | 5.0 ± 0.1 | 100.8 | 1.8 | 4.8 ± 0.2 | 97.1 | 3.7 | 5.0 ± 0.2 | 101.0 | 3.4 |
| 20.0 | 17.1 ± 1.2 | 85.5 | 6.9 | 16.2 ± 0.2 | 80.9 | 1.1 | 19.9 ± 1.1 | 99.4 | 5.4 | |
| 50.0 | 47.9 ± 1.6 | 95.9 | 3.4 | 46.8 ± 3.1 | 93.6 | 6.7 | 48.8 ± 2.6 | 97.5 | 5.4 | |
Method performance comparison
The analytical performance of this developed method was compared with that of reported MIP based SPE-HPLC methods for the extraction of FQs, as listed in Table S2.†28,29,47,56,57 As seen from the table, our dt-MIPs were prepared by a simpler polymerization method than other reported MIPs.29,47,56,57 Although a one step preparation process was used to synthesize the polymers for both bulk polymerization and precipitation polymerization, the subsequent treatment procedure was complicated for bulk polymerization. The obtained bulk polymers needed to be crushed, ground and sieved before use in SPE,28 even though precipitation polymerization used more porogen. For surface imprinting,56,57 two or more preparation steps were required to synthesize the MIPs, which needed a longer preparation time and expended more organic solvents. For instance, magnetic MIPs exhibited high adsorption capacity and thereby achieved fast and selective magnetic extraction of FQs from biological fluids, however the sensitivity was lower than that of our MIPs, and relatively complex surface imprinting was required.47 Using the same precipitation polymerization for the MIPs,28 a lower amount of adsorbent was needed for our DSPE procedure, which achieved a high extraction efficiency comparable to that of traditional SPE. On the whole, our developed dt-MIPs-DSPE-HPLC method demonstrated high selectivity and sensitivity, simple and convenient operation, rapid concurrent determination of the FQs and good practical applicability.
Conclusions
To conclude, novel dt-MIPs were prepared by a precipitation polymerization and multi-template imprinting strategy using NOR and ENR as templates, and employed as DSPE adsorbents coupled with HPLC-DAD for simultaneous selective extraction and determination of two FQs in environmental water samples. The developed simple, rapid and cost-effective dt-MIPs-DSPE-HPLC method proved to be highly sensitive and practically applicable for the simultaneous selective determination of trace FQ antibiotics in complex aqueous samples.On the other hand, based on some published studies regarding dt-MIPs in sample pretreatment techniques, the feasibility of the dual/multi-template imprinting strategy has also been fully confirmed. By summarizing these reports on dt-MIPs, it can be found that the innovation and research content mainly lie in the synthesis methods for the dt-MIPs (including the choice of template, functional monomer, cross-linker, initiators and preparation methods), the sample pretreatment techniques (mode choice and condition optimization), the target analytes, and the applications in real samples. However, dual/multi-template imprinting strategy related studies still have great challenges and promising opportunities. Especially, the synthesis of functionalized dt-MIPs (e.g. magnetic dt-MIPs37,38) and the utilization of dt-MIPs for more than two target analytes39,40 are highly appreciated and should be strongly promoted. Furthermore, we can foresee a rapid advance in the dual/multi-template imprinting strategy and wider applications of the resulting dt/mt-MIPs in preparative technologies, high-throughput contaminant analysis and abatement.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21876199, 21477160, 41601525, 21804010, 41776110), the Natural Science Foundation of Shandong Province of China (ZR2014BL031, ZR2016BL25, ZR2016DB07), and the Department of Science and Technology of Shandong Province of China (GG201709290055).Notes and references
- N. Janecko, L. Pokludova, J. Blahova, Z. Svobodova and I. Literak, Environ. Toxicol. Chem., 2016, 35, 2647–2656 CrossRef PubMed.
- Q. T. Dinh, E. Moreau-Guigon, P. Labadie, F. Alliot, M. J. Teil, M. Blanchard and M. Chevreuil, Chemosphere, 2017, 168, 483–490 CrossRef PubMed.
- M. Rusch, A. Spielmeyer, J. Meissner, M. Kietzmann, H. Zorn and G. Hamscher, J. Agric. Food Chem., 2017, 65, 3118–3126 CrossRef PubMed.
- X. Van Doorslaer, J. Dewulf, H. Van Langenhove and K. Demeestere, Sci. Total Environ., 2014, 500–501, 250–269 CrossRef PubMed.
- X. Liu, J. C. Steele and X. Z. Meng, Environ. Pollut., 2017, 223, 161–169 CrossRef.
- M. M. Zheng, R. Gong, X. Zhao and Y. Q. Feng, J. Chromatogr. A, 2010, 1217, 2075–2081 CrossRef PubMed.
- M. Denadai and Q. B. Cass, J. Chromatogr. A, 2015, 1418, 177–184 CrossRef PubMed.
- S. Qu, X. Wang, C. Tong and J. Wu, J. Chromatogr. A, 2010, 1217, 8205–8211 CrossRef.
- X. Liu, X. Wang, F. Tan, H. Zhao, X. Quan, J. Chen and L. Li, Anal. Chim. Acta, 2012, 727, 26–33 CrossRef PubMed.
- V. H. Springer and A. G. Lista, Electrophoresis, 2015, 36, 1572–1579 CrossRef PubMed.
- A. Speltini, F. Maraschi, R. Govoni, C. Milanese, A. Profumo, L. Malavasi and M. Sturini, J. Chromatogr. A, 2017, 1489, 9–17 CrossRef.
- X. He, G. N. Wang, K. Yang, H. Z. Liu, X. J. Wu and J. P. Wang, Food Chem., 2017, 221, 1226–1231 CrossRef.
- Y. J. Tang, J. Q. Xu, C. Le, J. L. Qiu, Y. Liu and G. F. Ouyang, Talanta, 2017, 175, 550–556 CrossRef.
- H. Yan, F. Qiao and K. H. Row, Anal. Chem., 2007, 79, 8242–8248 CrossRef.
- W. Y. Fan, M. He, X. R. Wu, B. B. Chen and B. Hu, J. Chromatogr. A, 2015, 1418, 36–44 CrossRef.
- M. Mei and X. J. Huang, J. Sep. Sci., 2016, 39, 1908–1918 CrossRef.
- M. R. Payan, M. A. B. Lopez, R. Fernandez-Torres, J. A. O. Gonzalez and M. C. Mochon, J. Pharm. Biomed. Anal., 2011, 55, 332–341 CrossRef.
- I. Timofeeva, S. Timofeev, L. Moskvin and A. Bulatov, Anal. Chim. Acta, 2017, 949, 35–42 CrossRef.
- Y. Zhao, L. Xu, M. Liu, Z. Duan and H. Wang, Food Chem., 2018, 239, 40–47 CrossRef.
- Z. Duan, M. Yin, C. Zhang, G. Song, S. Zhao, F. Yang, L. Feng, C. Fan, S. Zhu and H. Wang, Analyst, 2018, 143, 392–395 RSC.
- X. Zhu, Y. Cui, X. Chang and H. Wang, Talanta, 2016, 146, 358–363 CrossRef.
- W. H. Lu, W. N. Ming, X. S. Zhang and L. X. Chen, Electrophoresis, 2016, 37, 2487–2495 CrossRef.
- X. He, G. N. Wang, K. Yang, H. Z. Liu, X. J. Wu and J. P. Wang, Food Chem., 2017, 221, 1226–1231 CrossRef.
- M. Amoli-Diva, K. Pourghazi and S. Hajjaran, Mater. Sci. Eng., C, 2016, 60, 30–36 CrossRef.
- L. X. Chen, X. Y. Wang, W. H. Lu, X. Q. Wu and J. H. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- S. Farooq, J. Nie, Y. Cheng, Z. Yan, J. Li, S. A. S. Bacha, A. Mushtaq and H. Zhang, Analyst, 2018, 143, 3971–3989 RSC.
- S. Xu, H. Lu and L. Chen, J. Chromatogr. A, 2014, 1350, 23–29 CrossRef.
- E. Turiel, A. Martin-Esteban and J. L. Tadeo, J. Chromatogr. A, 2007, 1172, 97–104 CrossRef.
- X. Wu and L. T. Wu, J. Sep. Sci., 2015, 38, 3615–3621 CrossRef CAS.
- J. L. Urraca, M. Castellari, C. A. Barrios and M. C. Moreno-Bondi, J. Chromatogr. A, 2014, 1343, 1–9 CrossRef CAS.
- A. C. Meng, J. LeJeune and D. A. Spivak, J. Mol. Recognit., 2009, 22, 121–128 CrossRef CAS.
- S. A. Zaidi, Electrophoresis, 2013, 34, 1375–1382 CrossRef CAS.
- R. X. Gao, Y. Hao, S. Q. Zhao, L. L. Zhang, X. H. Cui, D. C. Liu, Y. H. Tang and Y. S. Zheng, RSC Adv., 2014, 4, 56798–56808 RSC.
- W. H. Lu, X. Y. Wang, X. Q. Wu, D. Y. Liu, J. H. Li, L. X. Chen and X. S. Zhang, J. Chromatogr. A, 2017, 1483, 30–39 CrossRef CAS.
- X. L. Song, J. H. Li, S. F. Xu, R. J. Ying, J. P. Ma, C. Y. Liao, D. Y. Liu, J. B. Yu and L. X. Chen, Talanta, 2012, 99, 75–82 CrossRef CAS.
- J. Liu, H. Song, J. Liu, Y. Liu, L. Li, H. Tang and Y. Li, Talanta, 2015, 134, 761–767 CrossRef CAS PubMed.
- M. Liu, X. Li, J. Li, Z. Wu, F. Wang, L. Liu, X. Tan and F. Lei, J. Colloid Interface Sci., 2017, 504, 124–133 CrossRef CAS.
- M. T. Jafari, B. Rezaei and H. Bahrami, Anal. Sci., 2018, 34, 297–303 CrossRef CAS.
- K. Yang, G. N. Wang, H. Z. Liu, J. Liu and J. P. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1046, 65–72 CrossRef CAS.
- W. Q. Xia, P. L. Cui, G. N. Wang, J. Li and J. P. Wang, Anal. Methods, 2018, 10, 3001–3010 RSC.
- X. L. Song, J. H. Li, J. T. Wang and L. X. Chen, Talanta, 2009, 80, 694–702 CrossRef CAS PubMed.
- L. X. Chen, S. F. Xu and J. H. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- J. Qiao, H. Yan, H. Wang and Y. Lv, J. Sep. Sci., 2011, 34, 2668–2673 CrossRef CAS PubMed.
- P. Xiao, Y. Dudal, P. F. X. Corvini, P. Spahr and P. Shahgaldian, React. Funct. Polym., 2012, 72, 287–293 CrossRef CAS.
- F. Tan, D. M. Sun, J. S. Gao, Q. Zhao, X. C. Wang, F. Teng, X. Quan and J. W. Chen, J. Hazard. Mater., 2013, 244–245, 750–757 CrossRef CAS.
- H. L. de Oliveira, S. d. S. Anacleto, A. T. Maria da Silva, A. C. Pereira, W. d. S. Borges, E. C. Figueiredo and K. B. Borges, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1033–1034, 27–39 CrossRef.
- D. Xiao, P. Dramou, N. Xiong, H. He, H. Li, D. Yuan and H. Dai, J. Chromatogr. A, 2013, 1274, 44–53 CrossRef CAS.
- T. Zhao, X. Guan, W. Tang, Y. Ma and H. Zhang, Anal. Chim. Acta, 2015, 853, 668–675 CrossRef CAS.
- Y. Tang, J. Lan, X. Gao, X. Liu, D. Zhang, L. Wei, Z. Gao and J. Li, Food Chem., 2016, 190, 952–959 CrossRef CAS.
- P. Guo, X. Yuan, J. Zhang, B. Wang, X. Sun, X. Chen and L. Zhao, Anal. Bioanal. Chem., 2018, 410, 373–389 CrossRef CAS.
- S. Babić, A. J. M. Horvat, D. Mutavdžić Pavlović and M. Kaštelan-Macan, TrAC, Trends Anal. Chem., 2007, 26, 1043–1061 CrossRef.
- M. S. Burkhead, H. Wang, M. Fallet and E. M. Gross, Anal. Chim. Acta, 2008, 613, 152–162 CrossRef CAS PubMed.
- D. A. Volmer, B. Mansoori and S. J. Locke, Anal. Chem., 1997, 69, 4143–4155 CrossRef CAS.
- The Merck Index: An Encyclopedia of Chemicals, Drugs and Biologicals, 2006 Search PubMed.
- H. B. He, X. X. Lv, Q. W. Yu and Y. Q. Feng, Talanta, 2010, 82, 1562–1570 CrossRef CAS PubMed.
- X. L. Sun, X. M. Tian, Y. Z. Zhang and Y. J. Tang, Food Anal. Methods, 2013, 6, 1361–1369 CrossRef.
- Y. K. Lv, Y. Ma, X. B. Zhao, C. L. Jia and H. W. Sun, Talanta, 2012, 89, 270–275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an02133c |
| This journal is © The Royal Society of Chemistry 2019 |