
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
α-Perfluoroalkyl-β-alkynylation of alkenes via radical alkynyl migration†
Xinjun Tang and
Armido Studer *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstraβe 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 4th August 2017
Abstract
Transition metal-free radical α-perfluoroalkylation with concomitant β-alkynylation of unactivated alkenes is presented. These cascades proceed via electron-catalysis and comprise a radical 1,4- or 1,5-alkynyl migration from tertiary propargylic alcoholates to secondary or tertiary C-radicals as the key step. Alkynyl migration leads to a ketyl radical anion that sustains the chain as a single electron transfer reducing reagent.
Introduction
Fluorinated compounds are highly abundant in agrochemical industry, medicinal chemistry and materials science.1 The introduction of fluorine atoms and of perfluoroalkyl groups into organic compounds increases their solubility, lipophilicity, bioavailability and metabolic stability.2,3 Therefore, it is important to develop novel synthetic methods for fluorination or perfluoroalkylation. Significant progress has been made on alkene perfluoroalkylation with concomitant β-functionalization in recent years.4 In most cases, transition-metal catalysts such as Cu(I),5 Ru(II),6 Ir(III),7 and Fe(II)8 are required to run these reactions and perfluoroalkylated products bearing alkyl, aryl, OR, Cl or CF3 groups at the β-position can be obtained (Scheme 1). However, alkene perfluoroalkylation with accompanying β-alkynylation is rare and only four papers were published along these lines. Guided by pioneering studies of the Fuchs group,9 Li10 and Yu11 recently reported radical alkene trifluoromethylation and subsequent intermolecular alkynylation using phenylethynyl p-tolyl sulfone and acetylenic triflone as the alkynylating reagents. As drawbacks of these processes, alkynyl transfer is limited to phenylalkynylation and trimethylsilylalkynylation and the methods can only be used for trifluoromethylation. During preparation of this manuscript, Zhu and co-workers reported an elegant photoredox catalyzed alkene α-trifluoromethylation-β-alkynylation comprising an intramolecular alkynyl migration.12 However, the scope of this process is limited to the trifluoromethylation and the costly Umemoto reagent has to be applied along with a photoredox catalyst. Thus, novel and efficient general methods for alkene α-perfluoroalkylation-β-alkynylation are highly demanded. Considering the economy, transition metal free processes are highly valuable.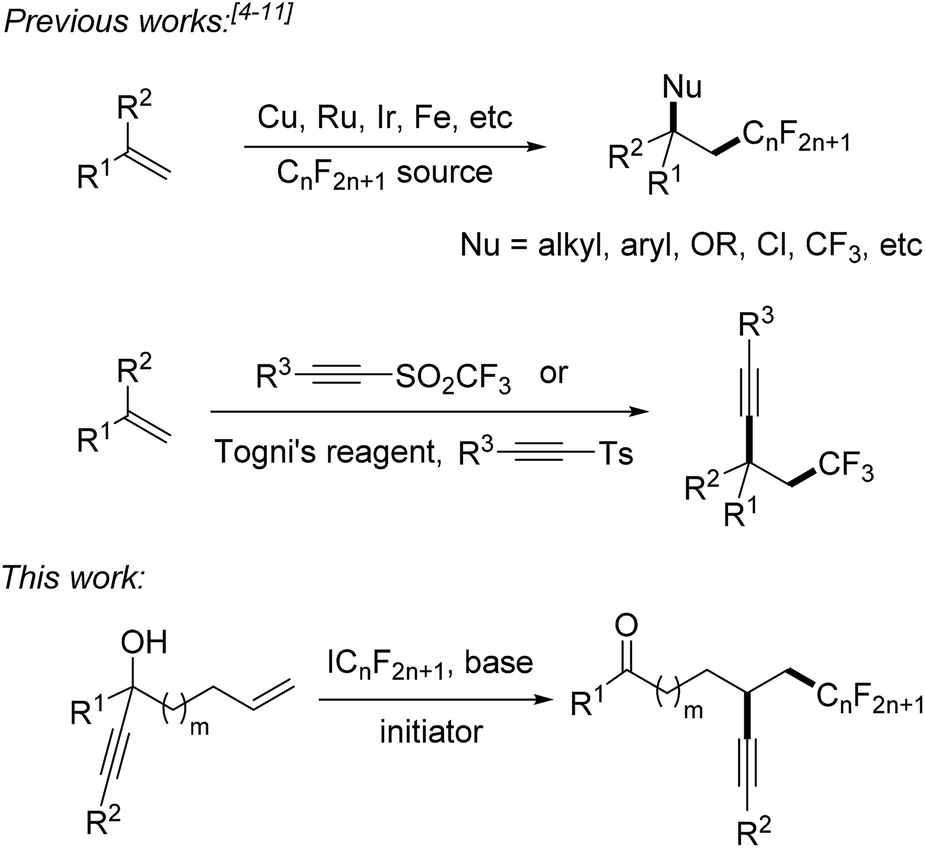 | ||
| Scheme 1 Vicinal alkene difunctionalization comprising a perfluoroalkylation. | ||
Herein we disclose a simple and efficient novel method for the α-perfluoroalkylation with concomitant β-alkynylation of unactivated alkenes proceeding via radical 1,4- or 1,5-alkynyl migration using perfluoroalkyl iodides13,14 as cheap and commercially available perfluoroalkyl radical sources in combination with readily prepared tertiary propargylic alcohols as substrates (Scheme 1). Notably, the potential of radical alkene perfluoroalkylation with concomitant β-functionalization via intramolecular formyl,15a aryl,15b and heteroaryl4e migration has recently been documented.15c,d
Results and discussion
Initial investigations were performed with 3-methyl-1-phenylhept-6-en-1-yn-3-ol 1a and perfluorobutyl iodide using Li-hexamethyldisilazide (LiHMDS) (1.2 equiv.) as a base in DME. The alcohol was first deprotonated with LiHMDS at room temperature (0.5 h). After complete deprotonation, DABCO (1.5 equiv., to mediate chain initiation)14 and perfluorobutyl iodide 2a (1.8 equiv.) were added sequentially and the mixture was stirred under visible-light irradiation (using a Philips Master HPI-T Plus (400 W) bulb) at 50 °C for 18 hours. To our delight, the target ketone 3a was obtained in 53% yield (Table 1, entry 1). The base heavily affects the reaction outcome and the yield dropped to 42% upon using LiOH (Table 1, entry 2) but a better result was achieved with NaOH (63%) (Table 1, entry 3). Surprisingly, perfluoroalkylation alkynyl migration failed using KOH as a base (Table 1, entry 4).|
|
||||
|---|---|---|---|---|
| Entry | Base | Additive | Solvent | Yield of 3ab [%] |
| a The reaction was conducted with 1a (0.1 mmol), 2a (1.8 equiv.), DABCO (1.5 equiv.), base (1.2 equiv.), and additive (0.42 equiv.) in 1.25 mL of DME under visible-light irradiation (using a Philips Master HPI-T Plus (400 W) bulb) at 50 °C for 18 h. b Determined by 1H NMR analysis using 1-fluoro-4-methylbenzene as the internal standard. c Isolated yield in parenthesis. d Bn2NH was used instead of DABCO. e Et3N was used instead of DABCO. f The reaction was conducted without DABCO. g The reaction was conducted without visible-light irradiation. h No base and additive were used. | ||||
| 1 | LiHMDS | — | DME | 53 |
| 2 | LiOH | — | DME | 42 |
| 3 | NaOH | — | DME | 63 |
| 4 | KOH | — | DME | — |
| 5 | LiHMDS | NaOH | DME | 58 |
| 6 | LiHMDS | LiOH | DME | 79 (79) |
| 7 | LiHMDS | KOH | DME | 6 |
| 8 | LiHMDS | LiOH | THF | 45 |
| 9 | LiHMDS | LiOH | 1,4-Dioxane | 28 |
| 10d | LiHMDS | LiOH | DME | 39 |
| 11e | LiHMDS | LiOH | DME | 8 |
| 12f | LiHMDS | LiOH | DME | 27 |
| 13g | LiHMDS | LiOH | DME | — |
| 14h | — | — | DME | 6 |

Having noted the importance of base, we started to screen mixtures of two bases. The addition of NaOH (0.42 equiv.) to LiHMDS did not affect the reaction outcome to a large extent (Table 1, entry 5) but with additional LiOH (0.42 equiv.) the yield significantly improved (79%) (Table 1, entry 6). However, with KOH as an additional base the yield dropped to 6% (Table 1, entry 7). The reaction proceeded less efficiently using THF and 1,4-dioxane as solvents (Table 1, entries 8 and 9). Replacing DABCO with Bn2NH or Et3N, or without DABCO, lower yields were obtained (Table 1, entries 10–12) and the cascade did not work without any visible-light irradiation (Table 1, entry 13). Base was found to be important and the target 3a was formed in only 6% in the absence of any base (Table 1, entry 14). Thus, 1a (0.1 mmol), 2a (1.8 equiv.), LiHMDS (1.2 equiv.), LiOH (0.42 equiv.), and DABCO (1.5 equiv.) in 1.25 mL of DME with stirring under visible-light irradiation (using a Philips Master HPI-T Plus (400 W) bulb) at 50 °C for 18 h were identified as the optimal reaction conditions for this sequence.
With the optimized reaction conditions in hand, we then turned to examine the scope with respect to the alkene component, keeping perfluorobutyl iodide 2a as the C-radical precursor (Table 2). The aryl group in these propargylic alcohols was first varied by replacing the phenyl substituent in 1a with differently substituted aryl groups. Electronic effects at the para-position do not play an important role as good results were achieved for both electron-rich and also electron-poor systems (3b–f, Table 2, entries 1–5). A similar result was noted for the meta-methyl substituted alcohol 1g to provide 3g, and also its ortho-congener 1g gave the target ketone 3h in a good yield (Table 2, entries 6 and 7). Thus, steric effects at the aryl moiety are not important, a fact which is further supported by the successful transformations of ortho, ortho′-disubstituted aryl alkynes 1i and 1j (see 3i, j, Table 2, entries 8 and 9). Notably, the 1-naphthyl and 2-pyridyl groups are both tolerated as R-substituents, and the corresponding ketones 3k and 3l were obtained in good yields (Table 2, entries 10 and 11).
|
|
|||
|---|---|---|---|
| Entry | R | Product | Yield [%]b |
| a The reaction was conducted with 1 (0.1 mmol), 2a (1.8 equiv.), DABCO (1.5 equiv.), LiHMDS (1.2 equiv.), and LiOH (0.42 equiv.) in 1.25 mL of DME under visible-light irradiation (using a Philips Master HPI-T Plus (400 W) bulb) at 50 °C for 18 h. b Isolated yield. | |||
| 1 | 1b, 4-MeC6H4 | 3b | 76 |
| 2 | 1c, 4-tBuC6H4 | 3c | 56 |
| 3 | 1d, 4-MeOC6H4 | 3d | 67 |
| 4 | 1e, 4-FC6H4 | 3e | 68 |
| 5 | 1f, 4-ClC6H4 | 3f | 83 |
| 6 | 1g, 3-MeC6H4 | 3g | 68 |
| 7 | 1h, 2-MeC6H4 | 3h | 78 |
| 8 | 1i, 2,4,6-Me3C6H2 | 3i | 61 |
| 9 | 1j, 3,5-(MeO)2C6H3 | 3j | 57 |
| 10 | 1k, 1-naphthyl | 3k | 71 |
| 11 | 1l, 2-pyridyl | 3l | 60 |
| 12 | 1m, 1-cyclohexenyl | 3m | 44 |
| 13 | 1n, nC4H9 | 3n | 40 |
| 14 | 1o, iPr3Si | 3o | 82 |
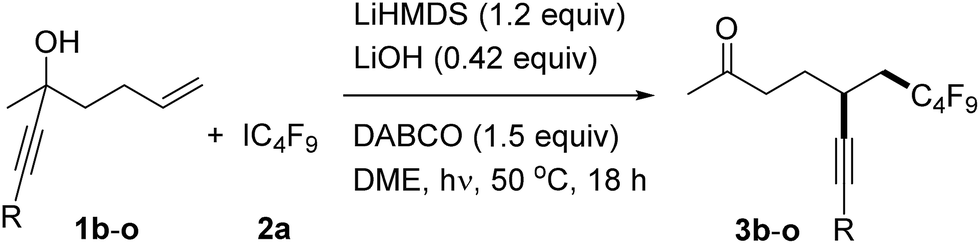
However, replacing the phenyl group in 1a by a 1-cyclohexenyl (1m) or n-butyl substituent (1n) led to significantly reduced yields of the corresponding alkynylated ketones 3m and 3n (Table 2, entries 12 and 13). Likely the rate constant for the radical cyclization is smaller for these less activated alkynes. A good result was also achieved in the transformation of the silylated alkyne 1o to provide 3o in 82% isolated yield.
We continued the studies by replacing the methyl group in 1a with other alkyl and aryl substituents in combination with 2a as the C-radical precursor (Scheme 2). The 1-butyl and 1-i-isopropyl propargylic alcohols 1p and 1q worked well and the dialkyl ketones 3p and 3q were isolated in 64% and 62% yield, respectively. An excellent yield was also obtained for the reaction with the tertiary benzylic alcohol 1r to give 3r (88%). Pleasingly, quaternary centres can be built up via the alkynyl migration as documented by the successful preparation of 3s (73%). The trisubstituted alkene 1t also acted as an acceptor and surprisingly an acceptable yield of the alkynyl migration product 3t derived from initial addition at the more hindered alkene position was obtained. The geminally dimethylated alcohol 1u provided the ketone 3u in 57% yield. Notably, the perfluoroalkyl group in these product ketones is readily varied by switching the radical precursor (see 3v–y). ICF2CF2Cl reacted chemoselectively at the C–I bond to give 3z and the corresponding product derived from C–Cl cleavage was not identified. We also tested other alkyl iodides such as cyclohexyl iodide and adamantyl iodide that are typical substrates in radical I-atom transfer reactions. As expected, alkylation/alkynyl migration did not work due to the fact that such non-fluorinated alkyl iodides cannot be readily SET-reduced. In addition, we tested α-bromo acetophenone but could not identify the target product, and large amounts of starting material remained unreacted.16 We are currently not sure whether the problem lies in the initiation step or in the intermolecular SET transfer from the ketyl radical anion to the bromo ketone (see mechanistic discussion below). Importantly, the novel sequence is not restricted to 1,4-alkynyl migration and reaction of alcohol 1v with perfluorobutyl iodide gave the ketone 3va resulting from a 1,5-alkynyl migration in 66% yield.
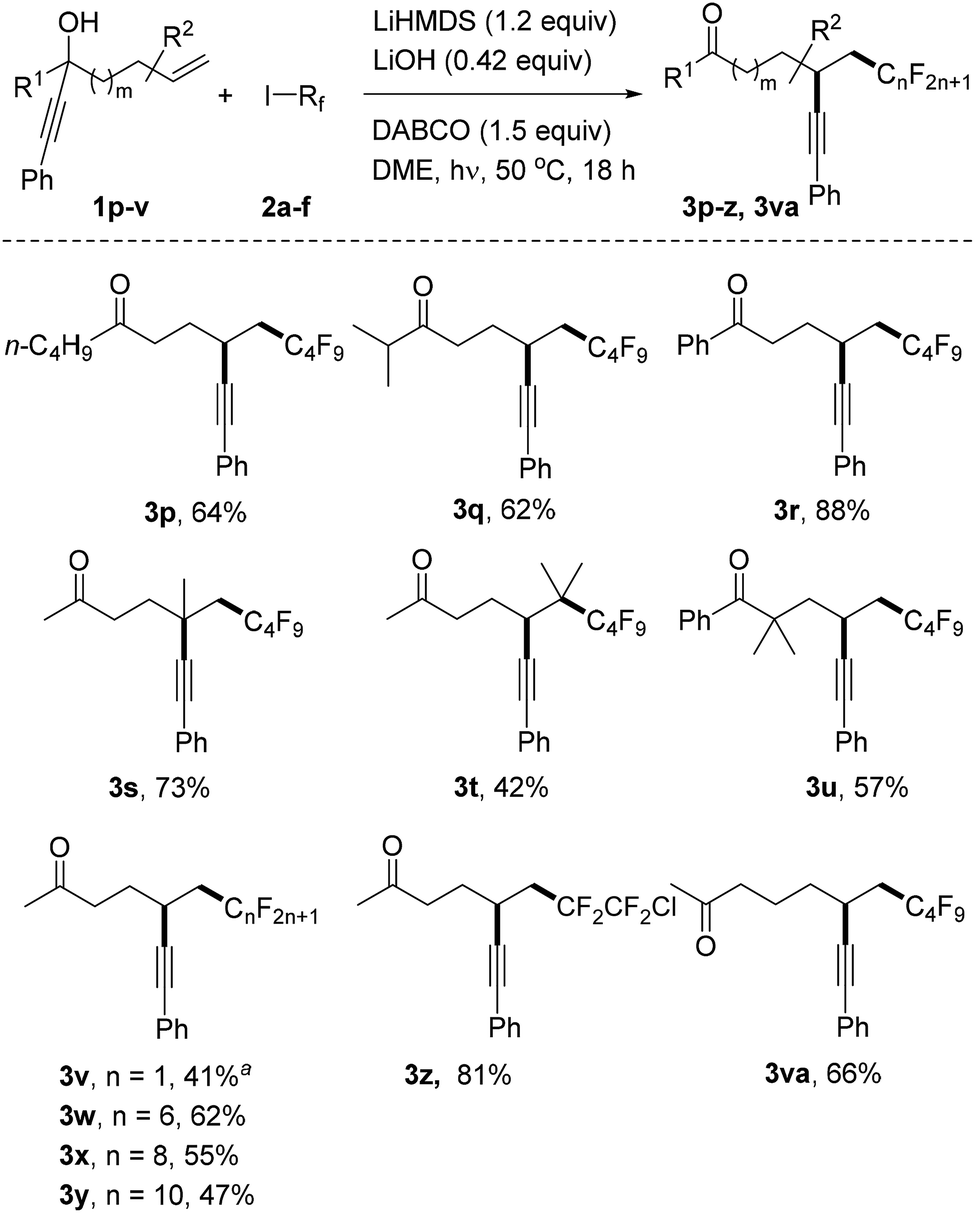 | ||
| Scheme 2 Variation of the radical acceptor and the perfluoroalkyl iodides. a3.6 equivalents of CF3I were added in two parts and the reaction time was 24 hours. | ||
To test whether alkynyl migration proceeds stereospecifically, we repeated the reaction with enantioenriched propargylic alcohol 1r (25% ee) as the starting material in combination with perfluorobutyl iodide and obtained the desired product 3r in 82% yield and 7% ee showing that only very low stereospecificity is achieved for this sequence (for details, see the ESI†).
The proposed mechanism is depicted in Scheme 3. Initiation occurs by irradiation of the perfluoroalkyl iodide in the presence of DABCO11 to give the corresponding perfluoroalkyl radical. This C-radical then adds to the terminal position of the alkene in the deprotonated alcoholate 1–Li to give the alkyl radical A. 5- or 6-exo radical cyclization leads to vinyl radical B, which further reacts by regioselective β-C–C bond cleavage to generate the ketyl radical anion C. As recently shown by us, such ketyl radical anions are good SET reducing reagents.17 Hence, electron transfer from C to the starting perfluoroalkyl iodide gives the product ketone 3 along with the corresponding perfluoroalkyl radical thereby sustaining the chain, and the overall cascade therefore belongs to an electron-catalyzed process.18
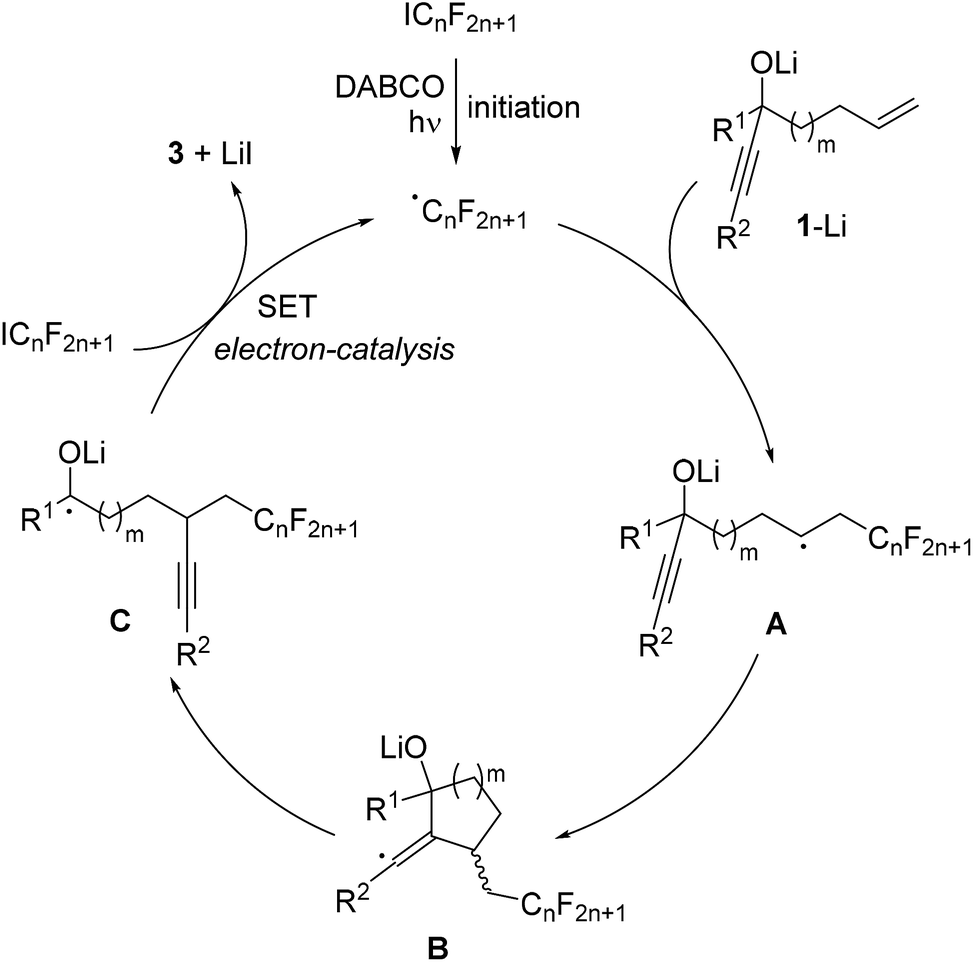 | ||
| Scheme 3 Proposed mechanism. | ||
We also investigated the follow-up chemistry using 3a as the starting material (Scheme 4). Chemoselective reduction of the carbonyl group in 3a was achieved using LiAlH4 at 80 °C to provide alcohol 4 in 76% yield as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoisomeric mixture. The triple bond in 3a was chemoselectively hydrogenated with Pd/C and H2 to form ketone 5 in 80% yield. TfOH-catalyzed cyclization of 3a afforded the α,β-unsaturated ketone 6 in 76% isolated yield.19 The same product could also be obtained using Au-catalysis, albeit in a slightly lower yield.20
:1 diastereoisomeric mixture. The triple bond in 3a was chemoselectively hydrogenated with Pd/C and H2 to form ketone 5 in 80% yield. TfOH-catalyzed cyclization of 3a afforded the α,β-unsaturated ketone 6 in 76% isolated yield.19 The same product could also be obtained using Au-catalysis, albeit in a slightly lower yield.20
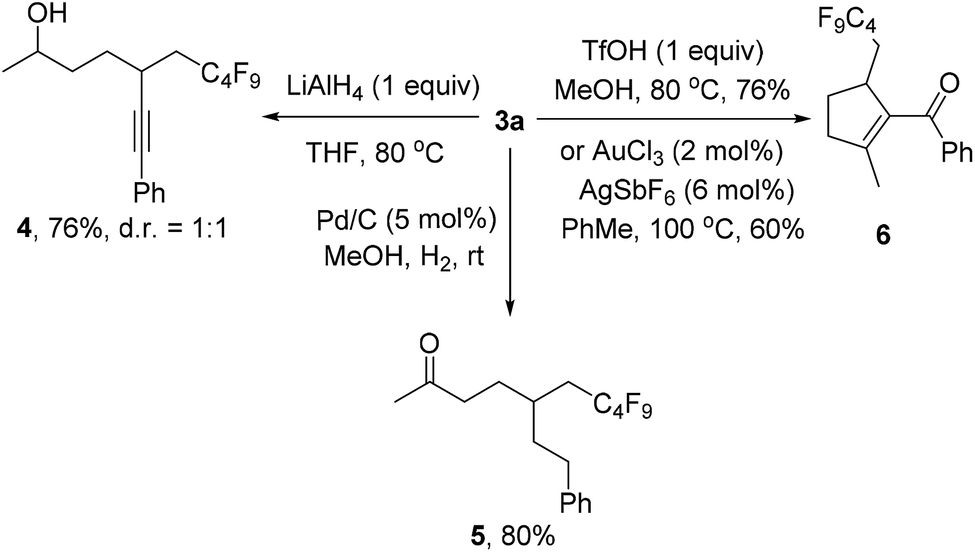 | ||
| Scheme 4 Follow-up chemistry. | ||
Conclusions
In summary, we have developed a novel and efficient method for the transition metal-free α-perfluoroalkylation β-alkynylation of unactivated alkenes via radical 1,4- or 1,5-alkynyl migration. Reaction conditions are mild and the substrate scope is broad. The starting propargylic alcohols are readily prepared and commercially available perfluoroalkyl iodides are used as C-radical precursors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the WWU Münster and the European Research Council (ERC Advanced Grant agreement no. 692640) for their financial support.Notes and references
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
- (a) M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- (a) Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221–8224 CrossRef CAS PubMed; (b) M. Hartmann, Y. Li and A. Studer, J. Am. Chem. Soc., 2012, 134, 16516–16519 CrossRef CAS PubMed; (c) L. Zhang, Z. Li and Z.-Q. Liu, Org. Lett., 2014, 16, 3688–3691 CrossRef CAS PubMed; (d) N.-Y. Yang, Z.-L. Li, B. Tan and X.-Y. Liu, Chem. Commun., 2016, 52, 9052–9055 RSC; (e) Z. Wu, D. Wang, Y. Liu, L. Hua and C. Zhu, J. Am. Chem. Soc., 2017, 139, 1388–1391 CrossRef CAS PubMed.
- (a) X. Mu, T. Wu, H.-Y. Wang, Y.-L. Guo and G. Liu, J. Am. Chem. Soc., 2012, 134, 878–881 CrossRef CAS PubMed; (b) H. Egami, R. Shimizu and M. Sodeoka, J. Fluorine Chem., 2013, 152, 51–55 CrossRef CAS; (c) X. Dong, R. Sang, Q. Wang, X.-Y. Tang and M. Shi, Chem.–Eur. J., 2013, 19, 16910–16915 CrossRef CAS PubMed; (d) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962–6966 CrossRef CAS PubMed; (e) H. Egami, R. Shimizu, S. Kawamura and M. Sodeoka, Angew. Chem., Int. Ed., 2013, 52, 4000–4003 CrossRef CAS PubMed; (f) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480–14483 CrossRef CAS PubMed; (g) Z.-M. Chen, W. Bai, S.-H. Wang, B.-M. Yang, Y.-Q. Tu and F.-M. Zhang, Angew. Chem., Int. Ed., 2013, 52, 9781–9785 CrossRef CAS PubMed; (h) G. Han, Q. Wang, Y. Liu and Q. Wang, Org. Lett., 2014, 16, 5914–5917 CrossRef CAS PubMed; (i) F. Wang, D. Wang, X. Mu, P. Chen and G. Liu, J. Am. Chem. Soc., 2014, 136, 10202–10205 CrossRef CAS PubMed; (j) Y.-T. He, L.-H. Li, Z.-Z. Zhou, H.-L. Hua, Y.-F. Qiu, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2014, 16, 3896–3899 CrossRef CAS PubMed; (k) B. Yang, X.-H. Xu and F.-L. Qing, Org. Lett., 2015, 17, 1906–1909 CrossRef CAS PubMed; (l) J. Zheng, Z. Deng, Y. Zhang and S. Cui, Adv. Synth. Catal., 2016, 358, 746–751 CrossRef CAS; (m) J.-Y. Guo, R.-X. Wu, J.-K. Jin and S.-K. Tian, Org. Lett., 2016, 18, 3850–3853 CrossRef CAS PubMed.
- (a) P. Xu, J. Xie, Q. Xue, C. Pan, Y. Cheng and C. Zhu, Chem.–Eur. J., 2013, 19, 14039–14042 CrossRef CAS PubMed; (b) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Chem. Commun., 2014, 50, 14197–14200 RSC; (c) B. Sahoo, J.-L. Li and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 11577–11580 CrossRef CAS PubMed; (d) L. Zheng, C. Yang, Z. Xu, F. Gao and W. Xia, J. Org. Chem., 2015, 80, 5730–5736 CrossRef CAS PubMed.
- P. Xu, K. Hu, Z. Gu, Y. Cheng and C. Zhu, Chem. Commun., 2015, 51, 7222–7225 RSC.
- H. Egami, R. Shimizu, Y. Usui and M. Sodeoka, Chem. Commun., 2013, 49, 7346–7348 RSC.
- J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486–4487 CrossRef CAS.
- S. Zhou, T. Song, H. Chen, Z. Liu, H. Shen and C. Li, Org. Lett., 2017, 19, 698–701 CrossRef CAS PubMed.
- H. Jiang, Y. He, Y. Cheng and S. Yu, Org. Lett., 2017, 19, 1240–1243 CrossRef CAS PubMed.
- Y. Xu, Z. Wu, J. Jiang, Z. Ke and C. Zhu, Angew. Chem., Int. Ed., 2017, 56, 4545–4548 CrossRef CAS PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed.
- (a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed; (b) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed; (c) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC; (d) A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC; (e) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (f) X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638–4641 CrossRef CAS PubMed.
- (a) Z.-L. Li, X.-H. Li, N. Wang, N.-Y. Yang and X.-Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 15100–15104 CrossRef CAS PubMed; (b) L. Li, Z.-Z. Li, F.-L. Wang, Z. Guo, Y.-F. Cheng, N. Wang, X.-W. Dong, C. Fang, J. Liu, C. Hou, B. Tan and X.-Y. Liu, Nat. Commun., 2016, 7, 13852 CrossRef PubMed; for related cyano migrations, see: (c) Z. Wu, R. Ren and C. Zhu, Angew. Chem., Int. Ed., 2016, 128, 10979–10982 CrossRef; (d) M. Ji, Z. Wu, J. Yu, X. Wan and C. Zhu, Adv. Synth. Catal., 2017, 359, 1959–1962 CrossRef CAS.
- T. Xu and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 1307–1311 CrossRef CAS PubMed.
- A. Dewanji, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2016, 55, 6749–6752 CrossRef CAS PubMed.
- (a) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- T. Jin, F. Yang, C. Liu and Y. Yamamoto, Chem. Commun., 2009, 3533–3535 RSC.
- T. Jin and Y. Yamamoto, Org. Lett., 2007, 9, 5259–5262 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02175e |
| This journal is © The Royal Society of Chemistry 2017 |