 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective production of mono-aromatics from lignocellulose over Pd/C catalyst: the influence of acid co-catalysts
Xiaoming
Huang†
a,
Xianhong
Ouyang†
a,
Bart M. S.
Hendriks†
a,
O. M. Morales
Gonzalez
a,
Jiadong
Zhu
a,
Tamás I.
Korányi
a,
Michael D.
Boot
b and
Emiel J. M.
Hensen
 *a
*a
aSchuit Institute of Catalysis, Inorganic Materials Chemistry, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
bCombustion Technology, Department of Mechanical Engineering, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 20th February 2017
Abstract
The ‘lignin-first’ approach has recently gained attention as an alternative whole biomass pretreatment technology with improved yield and selectivity of aromatics compared with traditional upgrading processes using technical lignins. Metal triflates are effective co-catalysts that considerably speed up the removal of lignin fragments from the whole biomass. As their cost is too high in a scaled-up process, we explored here the use of HCl, H2SO4, H3PO4 and CH3COOH as alternative acid co-catalysts for the tandem reductive fractionation process. HCl and H2SO4 were found to show superior catalytic performance over H3PO4 and CH3COOH in model compound studies that simulate lignin–carbohydrate linkages (phenyl glycoside, glyceryl trioleate) and lignin intralinkages (guaiacylglycerol-β-guaiacyl ether). HCl is a promising alternative to the metal triflates as a co-catalyst in the reductive fraction of woody biomass. Al(OTf)3 and HCl, respectively, afforded 46 wt% and 44 wt% lignin monomers from oak wood sawdust in tandem catalytic systems with Pd/C at 180 °C in 2 h. The retention of cellulose in the solid residue was similar.
Introduction
Lignocellulosic biomass is the most abundant source of renewable carbon with significant potential for the production of sustainable chemicals and fuels.1,2 Together with cellulose and hemi-cellulose, lignin is a main component of land-based biomass. Lignin makes up about 20–35% of the mass of dry biomass.3 It is typically obtained as a by-product in the paper and pulping industry and 2nd generation biorefineries. Such technical lignin is usually contaminated by sulphur, ash, proteins and carbohydrates residue.3,4 During biomass separation, the lignin structure is altered, rendering technical lignins much less reactive than the native lignin contained in the biomass.5–7 Abundant weak ether linkages, such as β-O-4 ones, are replaced by more recalcitrant C–C linkages.8 The resulting highly recondensed structure of lignin poses significant challenges for its efficient conversion to chemicals and fuels. Such processes require harsh conditions and typically yield a wide range of aromatic compounds in a low overall yield.9–14Recent studies have demonstrated that mild depolymerization strategies can also be effective when reactive lignin feedstock rich in β-O-4 ether linkages is used. For instance, a two-step strategy has been developed for aspen lignin, consisting of selective oxidation of the secondary alcohol of β-O-4 linkages followed by a redox-neutral bond cleavage at 110 °C.5,15 A similar strategy was also applied to the depolymerization of a reactive dioxasolv birch lignin (an organosolv lignin extracted by 1,4-dioxane) at 80 °C.16 The use of a hydrosilane reductant and a Lewis acidic (B(C6F5)3) catalyst facilitated the conversion of extracted wood lignin into mono-aromatics with a yield of 7–24 wt% at room-temperature.17 A combination of triflic acid and a heterogeneous Ru or homogeneous Ir catalyst was also effective in depolymerizing dioxasolv lignin in 1,4-dioxane at 140 °C.18 A similar catalytic system involving the use of a metal triflate and a homogeneous Rh catalyst was employed for the depolymerization of dioxansolv lignins in a 1,4-dioxane/water solvent mixture at 175 °C.19 Two-step approaches consisting of a mild pretreatment of woody biomass followed by catalytic depolymerization have also been reported.20,21 A suitable pretreatment method for the preparation of reactive lignin that preserves high ether linkage content is essential to these mild lignin depolymerization approaches.
An alternative approach is to directly convert the lignin in its native form in the lignocellulosic biomass matrix.22–25 For instance, Kou et al. reported that lignin in birch wood can be selectively depolymerized to alkylmethoxyphenols in 46 wt% yield in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) dioxane/water solvent mixture over carbon-supported noble metal (Pt, Ru, Pd and Rh) catalysts at 200 °C for 4 h.26 Li et al. reported the direct catalytic conversion of carbohydrate and lignin fragments from the whole woody biomass into diols and alkylmethoxyphenols in water over a carbon-supported Ni–W2C catalyst at 235 °C for 4 h.27 Song et al. were able to convert about half of the lignin in birch wood into alkylmethoxyphenols in alcohols over a Ni/C catalyst.22 Ferrini, Rinaldi and others reported a catalytic upstream biorefining process that converts lignin from woody biomass into low molecular weight lignin oil and a (hemi-)cellulose-rich pulp over a RANEY® Ni catalyst in a mixture of H2O/2-propanol.23,28 The group of Abu-Omar used a bimetallic Zn/Pd/C catalyst to convert lignocellulosic biomass into two alkylmethoxyphenols and carbohydrate pulp residue in methanol. A yield of 52 wt% of lignin monomers was obtained from birch hardwood after a 12 h reaction at 225 °C.24,29 Sels and co-workers reported a lignin-first process that allows simultaneous delignification and reductive depolymerization in the presence of Ru/C or Pd/C in methanol under a H2 atmosphere at elevated temperature, resulting in a carbohydrate pulp and a lignin oil rich in alkylmethoxyphenol monomers in ∼50% yield.6,30 The addition of acids, such as H3PO4, improved the overall efficiency of the process due to enhanced delignification.31 A similar catalytic lignocellulose fractionation process without the addition of external hydrogen or co-catalysts in a 1:1 (v/v) ethanol/water solvent mixture was also reported recently.32 Related to these lignin-first or catalytic upstream biorefining processes, we have also demonstrated a tandem catalytic system that couples delignification and reductive depolymerization of wood lignin in one pot using methanol as a solvent by a combination of Lewis acid metal triflates (e.g. Al(OTf)3) and Pd/C.33,34 An important function of the acidic co-catalyst is the efficient extraction of lignin from the biomass, which is a relatively slow step. Conventional Lewis acids, such as AlCl3 and ZnCl2, were found to be less effective in deconstructing lignocellulose compared with metal triflates.33,34 Moreover, these conventional Lewis acids are moisture sensitive and tend to decompose in the presence of water generated in etherification and hydrogenolysis. Another drawback is that their recovery and reuse are difficult.
:1 (v/v) dioxane/water solvent mixture over carbon-supported noble metal (Pt, Ru, Pd and Rh) catalysts at 200 °C for 4 h.26 Li et al. reported the direct catalytic conversion of carbohydrate and lignin fragments from the whole woody biomass into diols and alkylmethoxyphenols in water over a carbon-supported Ni–W2C catalyst at 235 °C for 4 h.27 Song et al. were able to convert about half of the lignin in birch wood into alkylmethoxyphenols in alcohols over a Ni/C catalyst.22 Ferrini, Rinaldi and others reported a catalytic upstream biorefining process that converts lignin from woody biomass into low molecular weight lignin oil and a (hemi-)cellulose-rich pulp over a RANEY® Ni catalyst in a mixture of H2O/2-propanol.23,28 The group of Abu-Omar used a bimetallic Zn/Pd/C catalyst to convert lignocellulosic biomass into two alkylmethoxyphenols and carbohydrate pulp residue in methanol. A yield of 52 wt% of lignin monomers was obtained from birch hardwood after a 12 h reaction at 225 °C.24,29 Sels and co-workers reported a lignin-first process that allows simultaneous delignification and reductive depolymerization in the presence of Ru/C or Pd/C in methanol under a H2 atmosphere at elevated temperature, resulting in a carbohydrate pulp and a lignin oil rich in alkylmethoxyphenol monomers in ∼50% yield.6,30 The addition of acids, such as H3PO4, improved the overall efficiency of the process due to enhanced delignification.31 A similar catalytic lignocellulose fractionation process without the addition of external hydrogen or co-catalysts in a 1:1 (v/v) ethanol/water solvent mixture was also reported recently.32 Related to these lignin-first or catalytic upstream biorefining processes, we have also demonstrated a tandem catalytic system that couples delignification and reductive depolymerization of wood lignin in one pot using methanol as a solvent by a combination of Lewis acid metal triflates (e.g. Al(OTf)3) and Pd/C.33,34 An important function of the acidic co-catalyst is the efficient extraction of lignin from the biomass, which is a relatively slow step. Conventional Lewis acids, such as AlCl3 and ZnCl2, were found to be less effective in deconstructing lignocellulose compared with metal triflates.33,34 Moreover, these conventional Lewis acids are moisture sensitive and tend to decompose in the presence of water generated in etherification and hydrogenolysis. Another drawback is that their recovery and reuse are difficult.
In this work, we explore a range of Brønsted acid co-catalysts, such as HCl, H2SO4, H3PO4 and CH3COOH, as alternatives to the benchmark Al(OTf)3 catalyst. The main goal was to identify a suitable and cheaper acid co-catalyst that helps in the delignification of the biomass but leaves the (hemi)cellulose part as much as possible unconverted. The strategy of this work is to evaluate the performance of these acids as co-catalysts in the cleavage of model compounds relevant to carbohydrate–lignin interlinkages and lignin intralinkages. Finally, oak wood sawdust is used to verify the trends and establish the potential of these cheap acid co-catalysts to replace Al(OTf)3.
Results and discussion
The catalytic process
Fig. 1 illustrates the overall concept of the tandem catalytic system for the valorization of lignin from woody biomass. Metal triflates are effective in the extraction of lignin from the lignocellulosic matrix by cleaving phenyl glycoside, phenyl ether and ester lignin–carbohydrate intralinkages.31,32 These lignin fragments are then disassembled by Pd-catalyzed hydrogenolysis reactions involving molecular hydrogen. Metal triflates strongly promote the reductive fractionation of lignocellulose, thus shortening the time needed to obtain a high lignin monomer yield.31,32 The metal triflates are also active for the depolymerization of some of the ether bonds in these lignin fragments and they also modify the performance of the Pd/C catalyst used to depolymerize these fragments. In a typical experiment (2000 mg of birch wood sawdust, 200 mg of Pd/C, 15 mg of aluminum triflate (Al(OTf)3), 40 ml of methanol, 180 °C, 2 h, 30 bar H2), the yield of C9 lignin monomers was 45 wt% (based on 23.6 wt% lignin content of birch wood). The main products are 4-n-propylsyringol (PS-H), 4-n-propanolsyringol (PS-OH) and 4-n-methoxy propyl syringol (PS-OCH3), and their corresponding guaiacyl counterparts, namely PG-H, PG-OH and PG-OCH3. The formation of PS-OCH3 and PG-OCH3 is caused by the etherification of the propanol side-group with methanol catalyzed by the Al(TOf)3 Lewis acid catalyst. The lignin monomers distribution can be controlled by varying the ratio of Pd/C to Al(TOf)3. A higher Pd/Al ratio results in more PS/G-OH and PS/G-H, and less PS/G-OCH3 products.34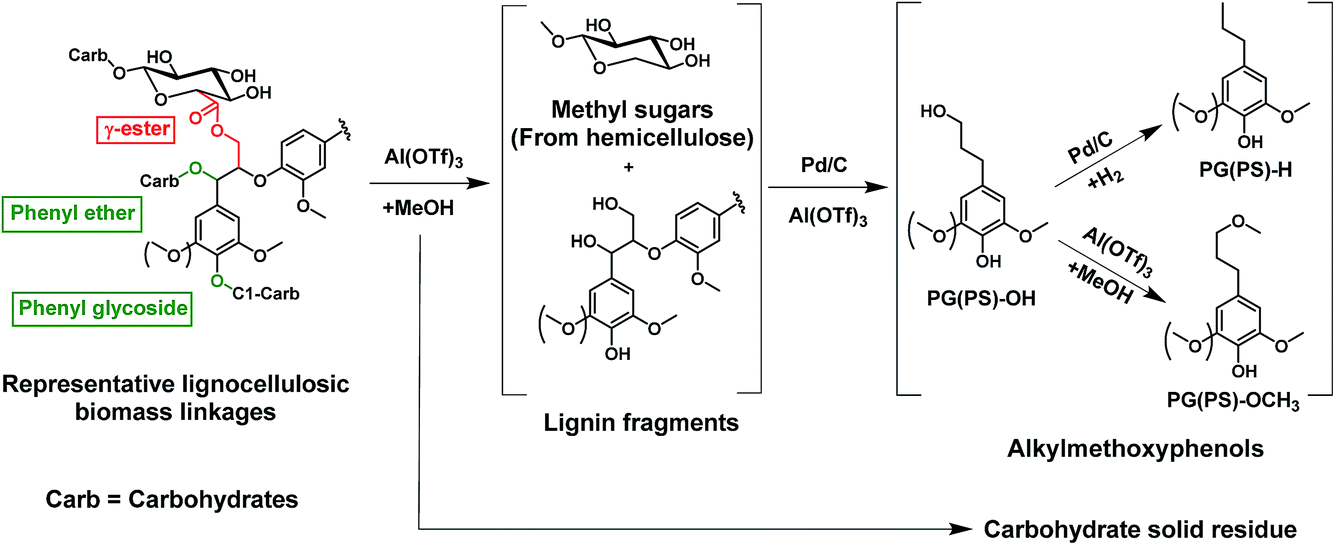 | ||
| Fig. 1 The cleavage of representative linkages between carbohydrates and lignin (phenyl glycoside, benzyl ether and γ-ester) by a tandem metal triflate and Pd/C catalyst system. | ||
Fig. 2 shows a detailed analysis of the composition of birch wood, the solid residue and the product oil after upgrading by a combination of Pd/C and Al(OTf)3. The solid residue is comprised mainly of cellulose, hemicellulose and a small amount of lignin together with the Pd/C catalyst. About 56 wt% of the hemicellulose and 81 wt% of the lignin were released from the raw birch wood and, respectively, converted into predominantly methyl xylose and mono-aromatics. The cellulose fraction was nearly all unconverted as evident from the unchanged cellulose content.
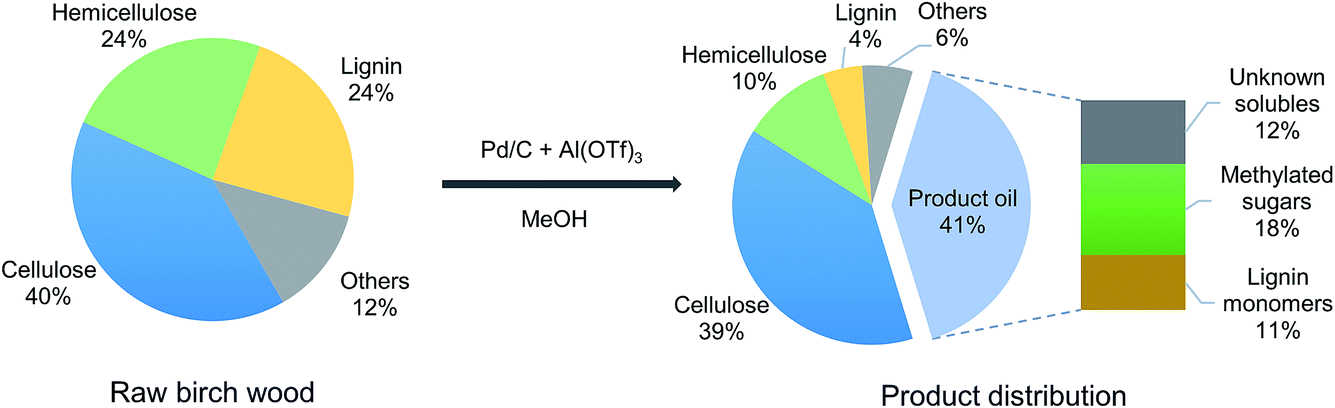 | ||
| Fig. 2 Composition of birch wood, the carbohydrate residue and product oil mixture after reductive fractionation over a tandem Al(OTf)3 and Pd/C catalyst system. (Reaction conditions: 2000 mg of extracted birch wood sawdust (125–300 μm), 200 mg of 5 wt% Pd/C, 15 mg of Al(OTf)3, 40 ml of methanol, 30 bar H2, stirring speed: 500 rpm, 2 h.) | ||
For the comparison of this optimized catalyst system to common Brønsted acid catalysts, we used another hardwood oak sawdust (125–300 μm) as the starting material. The amount of Pd/C was reduced from 200 mg to 100 mg. The amount of acid was equivalent to the amount of Al(OTf)3 (0.0316 mmol, 15 mg). The effect of the acid concentration was also investigated by increasing the amount of acid to 4, 16 and 32 times the standard concentration. For the model compound studies, the same concentrations of acids were used.
Influence of acid co-catalysts: model compound studies
The selective cleavage of lignin–carbohydrate bonds and lignin–lignin ether bonds is essential for the one-pot reductive fraction process. Typical linkages between lignin and carbohydrates in woody biomass are phenyl glycoside, benzyl ether and γ-ester bonds.35 With the purpose of understanding the role of acid in the delignification of woody biomass, we selected phenyl glycoside (PG) and glyceryl trioleate (GT) as model compounds, representing phenyl glycosidic and γ-ester type linkages, respectively. The reactions were conducted at 160 °C for 2 h in a small autoclave of 12 ml. When Pd/C was used without an acid co-catalyst, the conversions of PG and GT were 22% and 5%, respectively (Fig. 3). Methanol is nearly inactive in converting PG.34 The conversion of PG is mainly due to hydrogenation and hydrogenolysis reactions catalyzed by the Pd/C catalyst (Fig. 3a). Hydrogenation of ester bonds typically requires high reaction temperature and hydrogen pressure.36 GT can be converted in methanol by trans-esterification catalyzed by the weak acidity of methanol under sub-critical conditions.16 Trans-esterification can be significantly enhanced by an acid. The use of Al(OTf)3 is very effective in cleaving the phenyl glycosidic bond of PG. The overall product yield of phenol-derivatives is 75% after a 2 h reaction at 160 °C. Al(OTf)3 also shows a high activity towards ester bond cleavage in GT with a 80% yield of methyl octadecanoate, the product of ester bond cleavage.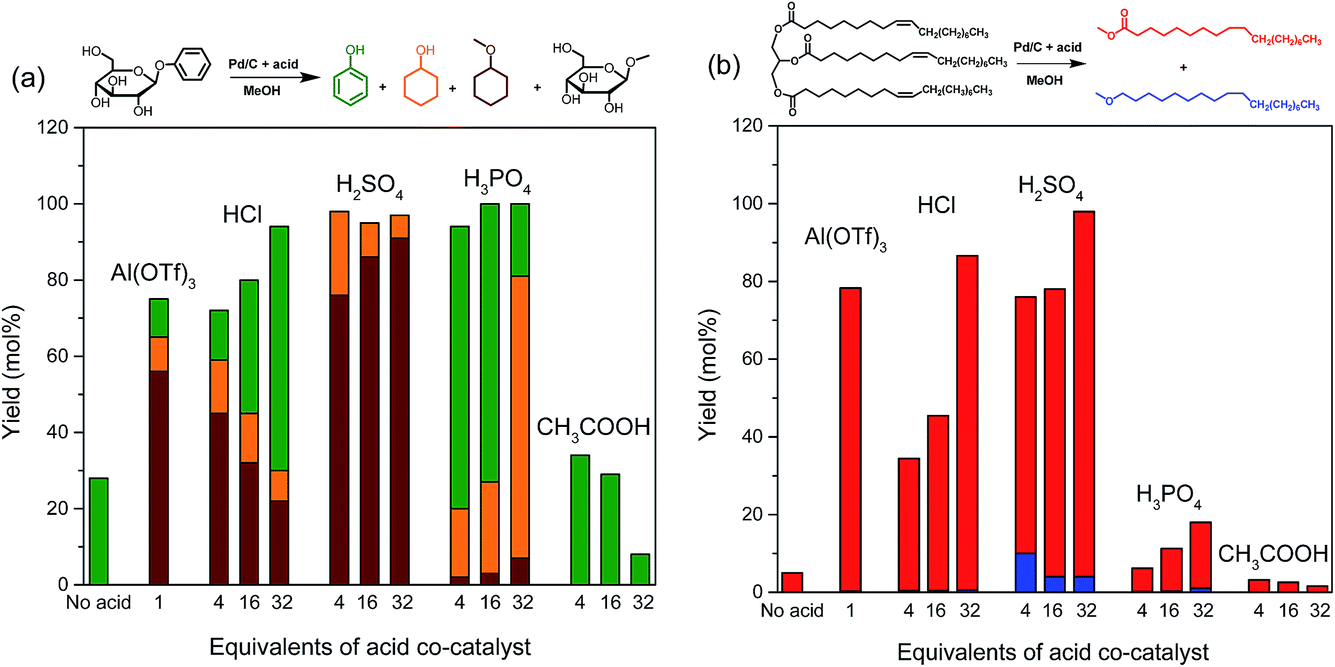 | ||
| Fig. 3 Reactions of (a) phenyl glycoside (PG) and (b) glyceryl trioleate (GT) in methanol over Pd/C and/or different acid co-catalysts at 160 °C for 2 h in 30 bar H2. | ||
The use of acids resulted in similar or higher conversions of PG (Fig. 3). Increasing the HCl concentration led to higher conversions and suppressed aromatic ring hydrogenation. Nearly all the PG was converted when H2SO4 was the co-catalyst. In this case, all the aromatic products were hydrogenated. H3PO4 is very effective in catalyzing the cleavage of the phenol–glycoside bond in PG and it also strongly suppresses aromatic ring hydrogenation. Acetic acid was the least effective acid and phenol was the only product observed from the phenyl part of the reactant.
The conversion of GT was less efficient with HCl and larger amounts were needed to obtain a similar conversion as that obtained for PG. H2SO4 was nearly as active as the metal triflate in cleaving the GT model reactant. H3PO4 and CH3COOH were much less active in cleaving the ester bond in GT. Overall, these data show that cleaving the phenyl glycosidic bond is more facile than cleaving the ester bond. The substantial difference (∼70%) between PG and GT conversion observed for H3PO4 cannot be explained by the small activity difference (∼20%) observed when only the Pd/C catalyst was used. Accordingly, we conclude that strong acids, such as HCl and H2SO4, are required to catalyze trans-esterification, while a weak acid (H3PO4) is sufficient to catalyze trans-etherification of the phenyl glycosidic bond. The use of CH3COOH as a co-catalyst is not efficient. It appears that acetic acid suppresses the cleavage of the bonds compared to the reference case. A tentative explanation could be that esterification between CH3COOH and methanol decreases the acidity. In line with this, we observed the formation of methyl acetate by GC-FID. Based on these results, we conclude that HCl and H2SO4 acids are potential co-catalysts for the lignin-first approach.
With the purpose of gaining insight into the performance of the acid co-catalyst in the depolymerization of lignin fragments, guaiacylglycerol-β-guaiacyl ether (GG) was used as a model substrate. GG is the most suitable model for the most abundant (50–65%) β-O-4 ether linkage in native lignin.14 For this reason and also because it is readily available, GG is frequently used in mechanistic studies on lignin deconstruction.5,16,18,37–39 The reactions were performed under the same conditions as the previous model compound reactions (160 °C, 2 h). Without acid, Pd/C is already active in cleaving the β-O-4 ether linkage in GG. The product of the ether bond cleavage, guaiacol, is obtained in 40% yield (Fig. 4). A combination of Pd/C and Al(OTf)3 resulted in a higher conversion (∼60%) of the β-O-4 ether bond. Interestingly, at the lowest HCl concentration, the yield (∼20%) was lower than in the Pd-only case. Although a higher HCl concentration slightly improved the GG conversion, the overall product yields remain lower than for Pd/C. In contrast, the use of H2SO4 is already effective in low concentrations. At a higher concentration, the yield of GG conversion products decreased. Similar trends were found when H3PO4 or CH3COOH were the co-catalyst (Fig. 4). Notably, the combination of 4 or 16 equivalents of CH3COOH and Pd/C shows superior performance in cleaving the ether bond of GG (∼65%).
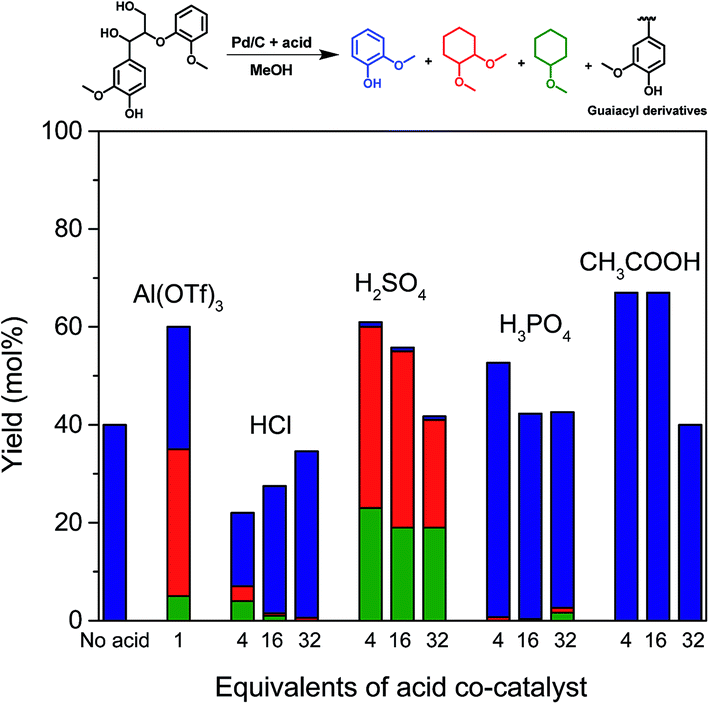 | ||
| Fig. 4 Reactions of guaiacylglycerol-β-guaiacyl ether (GG) in methanol over Pd/C and/or different acid co-catalysts at 160 °C for 2 h in 30 bar H2. | ||
In line with the results of PG conversion (Fig. 3a), the addition of acid promoted the hydrogenation activity of Pd/C. With Al(OTf)3, the guaiacol selectivity was about 50% – the remainder being methylated and ring-saturated products such as 1,2-dimethoxycyclohexane and a small amount of methoxycyclohexane (Fig. 4). The promoting effect for hydrogenation is stronger with H2SO4: nearly all of the guaiacol-derived products are ring saturated. On the other hand, such a promoting effect was not apparent with HCl, H3PO4 and CH3COOH as co-catalysts, different from the results obtained during PG conversion (Fig. 3a). During PG conversion, the addition of HCl or H3PO4 yielded more ring-saturated phenol-derivatives (Fig. 3a). The difference is the higher stability of the aromatic ring in guaiacol compared with phenol.40 This was confirmed by carrying out two separate hydrogenation reactions using guaiacol and phenol as the reactant under identical conditions (Pd/C, 160 °C, 30 bar H2, 2 h). With guaiacol as the reactant, the use of Pd/C resulted in a conversion to ring-saturated products of 61%, while all the phenol was converted to methoxycyclohexane (Fig. 5a).
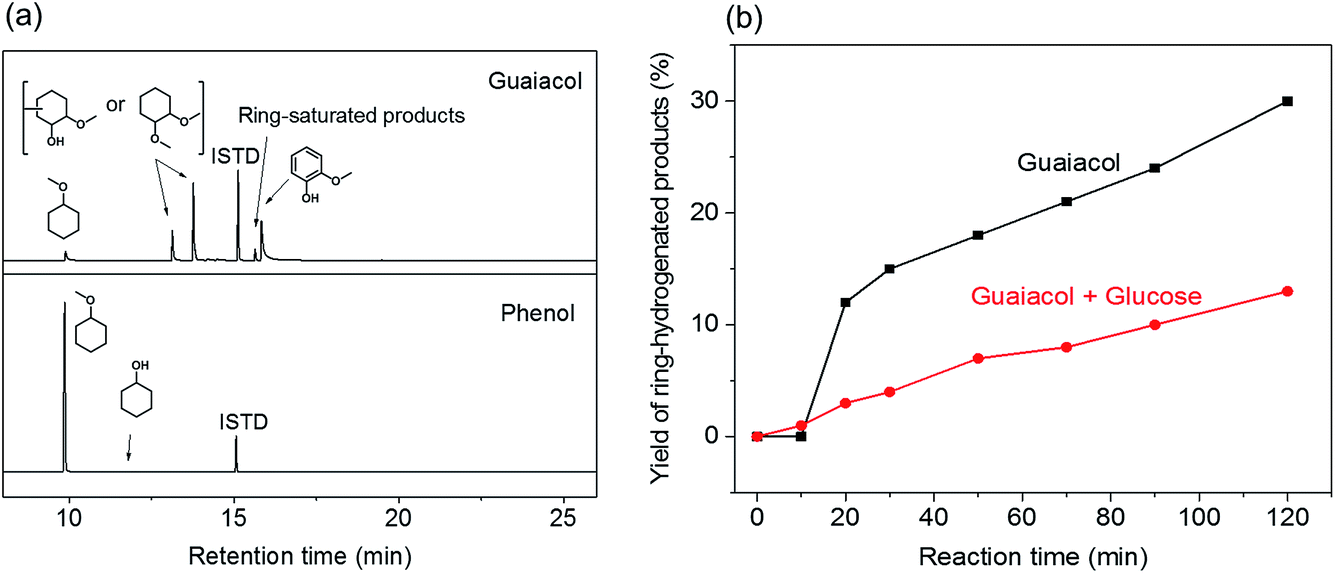 | ||
| Fig. 5 (a) Gas chromatograms of the reactions in methanol using phenol and guaiacol as model substrates; (b) yield of ring-hydrogenated products obtained from the hydrogenation reaction of guaiacol (black) and guaiacol + glucose (red) in methanol (conditions: 50 mg of substrate, 30 ml of methanol, 10 mg of Pd/C, 160 °C, 2 h, 30 bar H2). | ||
A tentative explanation for the promoting effect of acid co-catalysts on the aromatic ring hydrogenation by Pd/C is the removal of hydroxyl groups by their methylation catalyzed by acids. We have shown earlier that hydroxyl groups (e.g. as in propanol and sugars) suppress hydrogenation of aromatic rings.33 This is further supported by two additional experiments in which guaiacol was converted in the presence and absence of an equivalent mass of glucose. Fig. 5b shows the yield of the ring-hydrogenated products as a function of reaction time. The results attest to the lower rate of aromatic ring hydrogenation in the presence of sugars. This result confirms that hydroxyl-rich substrates hinder the hydrogenation activity of Pd/C, presumably by hindering specific adsorption models of the aromatic rings on the Pd surface that would lead to complete ring saturation.33 In the presence of an acid, the hydroxyl groups are readily converted into methoxy groups in the methanol solvent. This is also supported by the formation of substantial amounts of methylated cyclic products when PG and GG were converted with Pd/C and Al(OTf)3 or H2SO4 as co-catalysts (Fig. 3a and 4).
Based on these results, we conclude that combinations of Pd/C and H2SO4 or HCl are effective in cleaving two different types of lignin–carbohydrate (phenyl glycosidic and ester) as well as lignin–lignin (β-O-4 ether) linkages. The combination of Pd/C and weak H3PO4 acid is only active in cleaving the phenyl glycosidic bond and β-O-4 ether bonds, and not in the cleavage of ester type lignin–carbohydrate linkages. CH3COOH, the weakest among the tested acids, is not effective in cleaving any of the lignin–carbohydrate linkages.
Oak sawdust upgrading
With these model compound experiment results in hand, we evaluated the performance of the acids as co-catalysts in the upgrading of oak wood. In a typical experiment, 2000 mg of extracted oak wood sawdust, 100 mg of 5 wt% Pd/C and different equivalents of acid (1 equivalent = 0.0316 mmol) were added to a 100 ml autoclave together with 40 ml of methanol (180 °C, 2 h, 30 bar H2). The acid concentration was the same as in the model compound experiments. An aliquot of the reaction mixture was analyzed by GC-MS and GC-FID to quantify the lignin monomers and methylated sugars. Some of the reaction mixtures were also analyzed by gel permeation chromatography (GPC). Table 1 summarizes the overall results of the tandem-catalyzed reductive fractionation of oak sawdust using different acid co-catalysts under different conditions.| Entry | Catalyst | Acid co-catalyst | Solid residue mass (mg) | Lignin monomers yielde wt% (mg) | Methyl C5 sugars (mg) | Total yield (wt%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | Amount (mmol) | pHa | Amountb (mg) | Delignificationc (wt%) | 0 hd | 1 h | 2 h | ||||
| a pH values were measured at room temperature using an electronic pH meter after replacing 20 vol% of the acidic methanol solution with water. b Mass of the residue excluding Pd/C catalyst. c Delignification was determined by analyzing the Klason lignin content of the solid residue, assuming the Pd/C is homogenously mixed with the biomass residue. d Heating time (30 min). e Lignin yield is based on the acid-insoluble (Klason) lignin content (22.4 wt%) of the pre-extracted oak sawdust. | |||||||||||
| 1 | — | — | — | 6.6 | 1655 | 35 | 1 | 5 | 6 (26) | 4 | 84 |
| 2 | Pd/C | — | — | 6.6 | 1619 | 57 | 4 | 14 | 19 (81) | 4 | 85 |
| 3 | Pd/C | Al(OTf)3 | 0.0316 (1×) | 3.4 | 1175 | 82 | 18 | 40 | 46 (206) | 274 | 83 |
| 4 | Pd/C | HCl | 0.0316 (1×) | 3.0 | 1546 | N/A | 4 | 11 | 14 (62) | 5 | 81 |
| 5 | Pd/C | 0.126 (4×) | 2.2 | 1422 | N/A | 6 | 15 | 18 (78) | 20 | 76 | |
| 6 | Pd/C | 0.506 (16×) | 1.7 | 1211 | N/A | 12 | 21 | 23 (100) | 133 | 72 | |
| 7 | Pd/C | 1.011 (32×) | 1.4 | 1148 | 75 | 21 | 41 | 44 (194) | 249 | 80 | |
| 8 | Pd/C | H2SO4 | 0.0316 (1×) | 2.9 | 1562 | N/A | 4 | 9 | 10 (44) | 5 | 81 |
| 9 | Pd/C | 0.126 (4×) | 2.2 | 1017 | 76 | 23 | 31 | 35 (153) | 257 | 71 | |
| 10 | Pd/C | 0.506 (16×) | 1.7 | 328 | N/A | 28 | 35 | 40 (176) | 46 | 28 | |
| 11 | Pd/C | H3PO4 | 0.0316 (1×) | 3.6 | 1761 | N/A | 1 | 6 | 13 (55) | 20 | 92 |
| 12 | Pd/C | 0.126 (4×) | 3.1 | 1600 | N/A | 3 | 9 | 13 (55) | 19 | 84 | |
| 13 | Pd/C | 0.506 (16×) | 2.8 | 1448 | N/A | 3 | 13 | 17 (75) | 30 | 78 | |
| 14 | Pd/C | 1.011 (32×) | 2.6 | 1358 | 69 | 5 | 14 | 26 (117) | 31 | 77 | |
| 15 | Pd/C | CH3COOH | 0.0316 (1×) | 5.0 | 1598 | N/A | 4 | 10 | 15 (65) | 5 | 83 |
| 16 | Pd/C | 0.126 (4×) | 4.1 | 1577 | N/A | 3 | 8 | 10 (41) | 6 | 81 | |
| 17 | Pd/C | 0.506 (16×) | 3.7 | 1639 | N/A | 1 | 5 | 8 (35) | 7 | 84 | |
| 18 | Pd/C | 1.011 (32×) | 3.5 | 1649 | 40 | 1 | 4 | 6 (26) | 3 | 84 | |
Without a catalyst, the reaction mixture contains only 6 wt% C9 lignin monomers (entry 1). A large amount (1655 mg) of solid biomass residue was obtained (83 wt% based on the mass of the starting biomass). This solid residue was subjected to further analysis to determine the Klason lignin content. The results show that delignification is about 35 wt%, suggesting that methanol is not effective in extracting lignin from woody biomass at 180 °C. The low monomer yield and high solid residue retention obtained in the blank experiment are caused by the low degree of delignification and lignin depolymerization. By adding the Pd/C catalyst, the lignin monomer yield increased to 19 wt%, with PS-H, PS-OH and PG-OH as the main products (Table 1 entry 2, Fig. 6a). The improved yield is mainly due to the enhanced lignin depolymerization. Besides, the extent of delignification was also found to be slightly increased (57 wt%) compared with the blank reaction (entry 1, 35 wt%). This could be due to the fact that Pd/C is also effective in cleaving the phenyl glycosidic linkage. However, the delignification is limited by the poor accessibility of the solid biomass for the solid Pd/C catalyst.
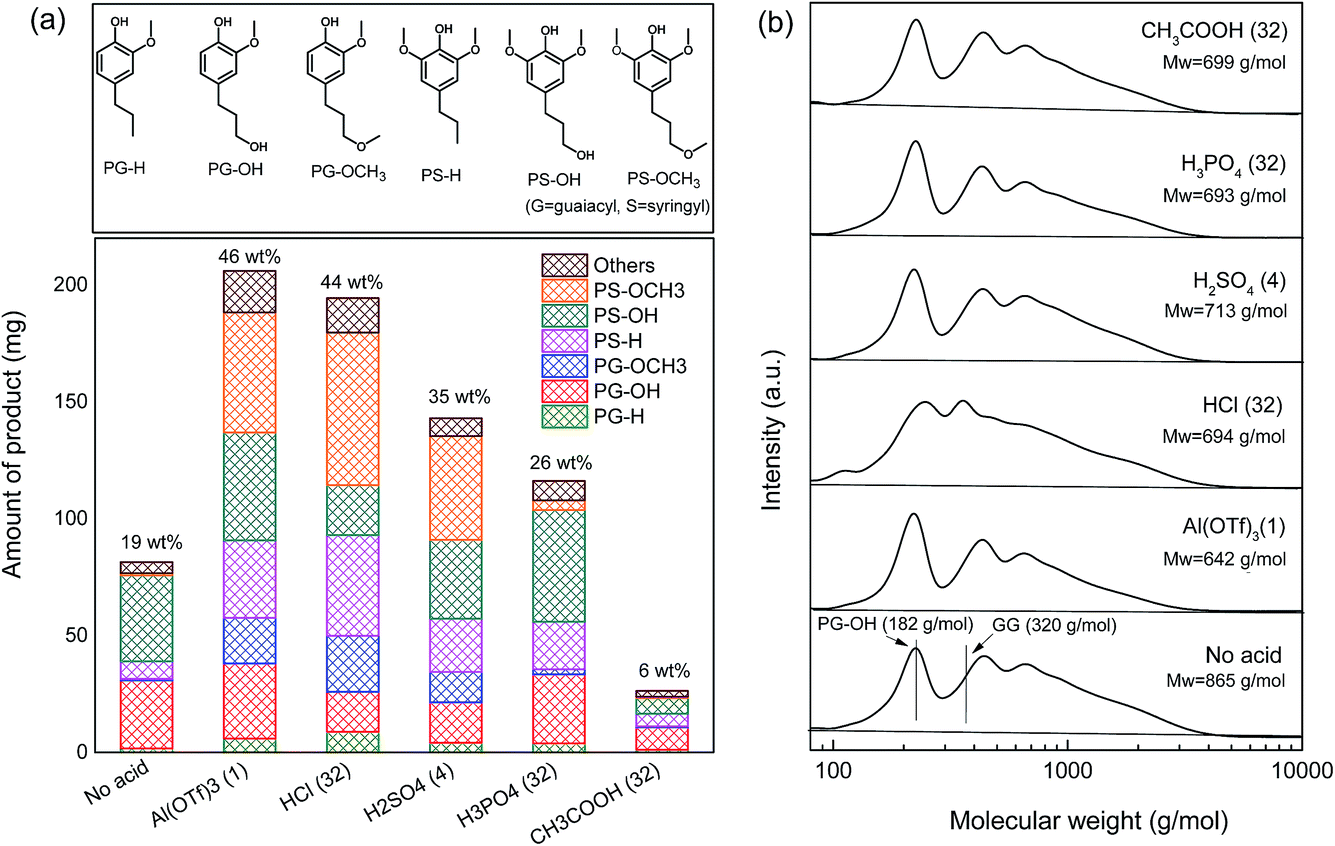 | ||
| Fig. 6 (a) Lignin monomeric product distributions and (b) GPC chromatograms of the reaction mixtures obtained from the reactions over Pd/C in combination with different acid co-catalysts at 180 °C for 2 h. (Conditions: 2000 mg of pre-extracted oak sawdust, 100 mg of Pd/C, 30 bar H2, 40 ml of methanol; 1 eqv. = 0.0316 mmol of acid; the numbers on top of the bars in (a) are the lignin monomer yields; GPC results of two reference compounds (GG: guaiacylglycerol-β-guaiacyl ether and PG-OH: 4-n-propanolguaiacol) are included.) | ||
In the benchmark case using 1 equivalent of Al(TOf)3 as the co-catalyst, the yield of lignin monomers increased substantially. A yield of 40 wt% C9 lignin monomer was obtained after a 1 h reaction at 180 °C and the yield increased to 46 wt% after 2 h (entry 3). The delignification degree is 82 wt%. The C9 lignin monomers are mainly PS-H, PS-OH and PS-OCH3, and small amounts of PG-H, PG-OH and PG-OCH3 (Fig. 6a). Apart from these mono-aromatics, an amount of 274 mg of methylated C5 sugars was formed. No cellulose-derived C6 sugars nor sugar degradation products were observed. Sugar degradation was only observed when a larger amount of metal triflate was used, consistent with our earlier work.34 For instance, some aliphatic ketones, such as 2,5-hexanedione, and esters, such as the methyl ester of levulinic acid, were observed when 3 equivalents of Al(OTf)3 were used in combination with 200 mg of Pd/C for the reductive fractionation of birch wood sawdust at 180 °C for 2 h.34 The incomplete mass balance is due to the presence of lignin dimers and oligomers, which were not identified. Their formation was confirmed by GPC analysis (vide infra).
The influence of other acid co-catalysts was evaluated under identical conditions. The use of small amounts of HCl did not improve the lignin monomer yield compared with the Pd/C-only case (entries 4–5). However, larger amounts improved the yields of lignin monomers and also increased the amount of methylated C5 sugars (entries 6–7). With the largest amount of HCl, the lignin monomer yield was 44 wt% (entry 7). The amount of solid residue and the monomeric lignin product distribution (Fig. 5a) are very similar to the results obtained with Al(TOf)3.
At the lowest concentration, H2SO4 hardly affected the conversion as with HCl (entry 8). The lignin monomer yield increased drastically to 35 wt% when 4 equivalents of H2SO4 were used (entries 9–10). The lignin monomeric product distribution and the amount of residue (1017 mg) are very similar to the best result obtained with HCl (1148 mg, 32 eqv., entry 7) and the Al(TOf)3 reference (1175 mg, entry 3). The delignification degree in this case is 76 wt%. Although increasing the dosage of H2SO4 acid resulted in a slightly higher yield of lignin monomers (40 wt%), much less solid residue was left, suggesting that a significant fraction of cellulose was converted. Moreover, darkening of the solution and the very low mass balance (28 wt%) point to humin formation from the released C6 sugars. These findings limit the use of high concentrations of H2SO4.
The influence of H3PO4 was minor with lignin monomer yields below 20 wt% (entries 11–13). Moreover, it is seen that the solid carbohydrate residue is decreasing with increasing H3PO4 content. The lignin monomer yields are even lower than the reference case without H3PO4 (19 wt%, entry 2). A maximum yield of 26 wt% lignin monomers was achieved after a 2 h reaction at 180 °C at the highest H3PO4 concentration (entry 14). The yield is slightly lower than the one reported in the literature.31 It was reported that the addition of 2.5 g L−1 (equal to 27.8 equivalents) H3PO4 in combination with Pd/C resulted in a lignin monomer yield of approximately 40 mol%, compared with the yield obtained from the neutral methanol solvent (26 mol%) at 200 °C using pre-extracted poplar sawdust feedstock. This difference could be caused by the lower reaction temperature (180 °C vs. 200 °C), the different feedstock (oak vs. poplar sawdust) and the different calculation methods (wt% vs. mol%). Nevertheless, it can be said that the addition of an appropriate amount of H3PO4 improves the overall efficiency of the reductive fractionation process. Yet, compared with Al(TOf)3 and the other stronger Brønsted acids (HCl and H2SO4), the promoting effect of H3PO4 is less pronounced. Our model compound experiments show that the combination of H3PO4 and Pd/C is active in cleaving the phenyl glycosidic lignin–carbohydrate and β-O-4 ether types of lignin–lignin linkages, but less active in cleaving the ester type lignin–carbohydrate linkages. We surmise that part of the lignin (linked by ester bonds) might not be effectively extracted from the biomass matrix. This is further confirmed by the relatively low degree of delignification (68 wt%), revealed by the compositional analysis of the solid residue obtained at the highest H3PO4 concentration. Furthermore, we observed that the lignin monomers are mostly non-etherified products, such as PS-H, PS-OH and PG-OH (Fig. 6a). Also, a small amount of methylated C5 sugars was formed. These results indicate that methylation/etherification is not occurring due to the low acidity of H3PO4 (entries 11–14). It is known that sugars and their derivatives (e.g. 5-HMF, furfural) can condense with each other and lignin-derived products under acidic conditions into humins.41,42 Methylation stabilizes sugars.43,44 In our earlier work, it was also confirmed that methylation of the lignin phenolic intermediates plays a key role in hindering repolymerization of lignin fragments and avoiding char formation.12 Accordingly, we argue that the low rate of etherification might be another reason for the lower efficiency of H3PO4.
With CH3COOH as the co-catalyst, a lignin monomer yield of 15 wt% was obtained at the lowest concentration (entry 15), which is close to the yield obtained in the case of the Pd/C catalyst. When the concentration was increased, the lignin monomer yield decreased significantly (entries 15–18). The lignin products were mostly non-etherified PS-OH, PS-H and PG-OH (Fig. 5). The amount of solid residue was nearly unchanged, suggesting that the delignification could not be improved by increasing the amount of CH3COOH. Analysis of the Klason lignin content of the solid residue showed that 40 wt% lignin was released from the woody biomass at the highest CH3COOH concentration. Based on the decreasing lignin monomer yield and unchanged solid residue yield, we infer that the addition of CH3COOH negatively affects the lignin conversion, consistent with the model compound results. The lack of delignification and etherification is likely the cause of the low lignin monomer yield.
Based on the lignin monomer yields and the solid carbohydrate residue, lignin oil samples obtained from the following experiments were subjected to GPC analysis: no acid, Al(OTf)3 (1 eqv.), HCl (32 eqv.), H2SO4 (4 eqv.), H3PO4 (32 eqv.) and CH3COOH (32 eqv.). Fig. 6b shows the results of the GPC analysis. Surprisingly, all of the selected lignin oils gave very similar results. Three broad peaks were observed related to lignin monomers, dimers and oligomers. For all the acid-catalyzed lignin oil samples, the molecular weights (Mw) were between 642 g mol−1 (Al(OTf)3) and 713 g mol−1 (H2SO4 (4 eqv.)), which are slightly lower than the molecular weight for the Pd/C-only case (865 g mol−1). This confirms that the addition of acid contributes to lignin depolymerization.34 The dimers and oligomers are likely to be linked by strong C–C bonds. The possible formation of dimers and oligomers is shown in Fig. 7. These C–C linkages either originate from the native structure (e.g. β-1, 5–5′) or they derive from the cleavage of some ether linkages (e.g. β–β′, β-5/cyclic α-O-4). The cleavage of C–C linkages requires a higher temperature. Accordingly, the fraction of ether linkages (typically 50–65% β-O-4 ether linkages in hardwood) puts an upper limit on the lignin monomers yield.
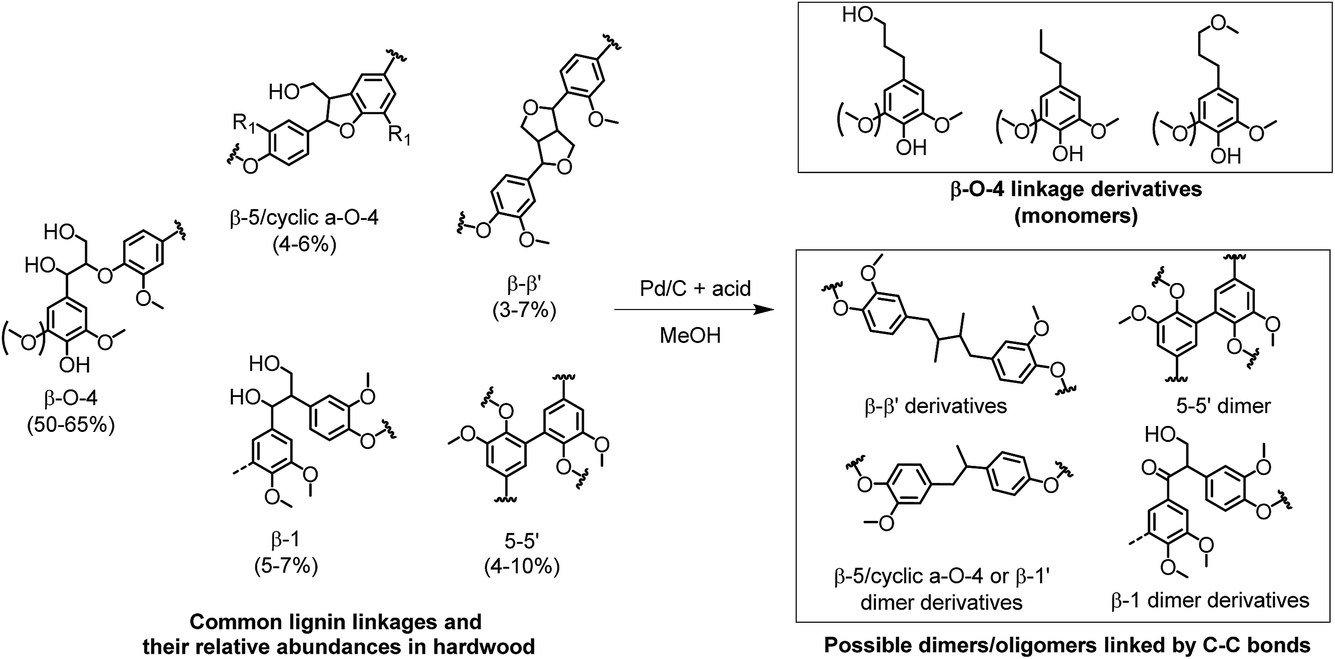 | ||
| Fig. 7 Formation of lignin monomers and proposed dimers and oligomers in the process of reductive fractionation of lignin from woody biomass. (The proposed structure and relative abundances are based on the literature.14,22,25) | ||
In brief, the most promising replacement of Al(OTf)3 is HCl, which is able to obtain a 44 wt% yield of lignin monomers from oak wood sawdust in reductive fractionation using Pd/C at 180 °C in 2 h. A drawback of the use of HCl is corrosion of the process equipment, which will add to the overall cost of biomass upgrading. The alkylmethoxyphenols resulting from this approach can be used in the fragrance industry (e.g. dihydroeugenol) or upgraded to other valuable base chemicals, such as (alkyl)phenols. One challenge in this lignin-first approach is that the metal catalyst ends up in the solid residue. A solution is to separate the delignification step and hydrogenolysis steps. This approach has been demonstrated in a recent study.33 The resulting carbohydrate solid residue is an ideal starting material for the paper industry and can also be used in the production of biobased chemicals, such as ethanol, levulinic acid, and γ-valerolactone.
Conclusions
We explored Brønsted acids as alternatives to Al(OTf)3 co-catalysts in the tandem reductive delignification of biomass to aromatics using Pd/C as the hydrogenolysis catalyst. The main role of the acid catalyst is to delignify lignocellulosic biomass, while Pd/C cleaves the ether bonds in the released lignin fragments. The solid residue in this process is a cellulose-rich fraction that also contains some hemicellulose and lignin. We systematically compared HCl, H2SO4, H3PO4 and CH3COOH as co-catalysts for the tandem reductive fractionation of woody biomass in methanol using Pd/C at 180 °C for 2 h. Model compound experiments (PG, GT and GG) show that the combinations of Pd/C and strong Brønsted acids (H2SO4 and HCl) are suitable to cleave phenyl glycosidic and ester type lignin–carbohydrate linkages as well as lignin–lignin (β-O-4 ether) linkages. The combination of Pd/C and the weaker acid H3PO4 was only able to cleave phenyl glycosidic bonds and β-O-4 ether bonds but not ester type lignin–carbohydrate linkages. CH3COOH is not effective in cleaving the model lignin–carbohydrate linkages. These model compound results explain the better catalytic performance of Al(OTf)3, HCl and H2SO4 compared to H3PO4 and CH3COOH for the upgrading of oak wood biomass. The poor performance of the latter two weak acids is due to their low activity in delignification. The most promising replacement of Al(OTf)3 is HCl. Under optimum conditions, a 44 wt% yield of lignin monomers was obtained from oak wood sawdust, similar to the result obtained with the metal triflate.Experimental section
Chemicals and materials
Oak wood sawdust was obtained from Houtzagerij Mennen (http://www.houtzagerijmennen.nl). Birch wood was obtained from the Energy Research Center of Netherlands (ECN). 5 wt% Pd/C, Al(OTf)3 (≥99%), CH3COOH (≥99%) and H3PO4 (85 wt% in water) were purchased from Sigma Aldrich. HCl (37 wt% in water) was purchased from Merck Emsure. H2SO4 (95–97%, 99.99% trace metals basis) was purchased from Merck Emsure. 4-n-Propanolguaiacol (PG-OH) was purchased from TCI. 4-n-Propylsyringol (PS-H) and 4-n-methoxy propyl syringol (PS-OCH3) were recovered from the reaction mixture; the detailed procedure can be found in our previous work.33 These phenolic compounds were used to determine the weight response factors relative to n-dodecane. Glyceryl trioleate (≥97.0%), phenyl β-D-glucopyranoside (≥97%) and benzyl phenyl ether were purchased from Sigma Aldrich. Guaiacylglycerol-β-guaiacyl ether (GG, ≥97%) was purchased from TCI. Extra-dry absolute methanol was purchased from Biosolve. All the other commercial chemicals were analytical grade and were used without further purification.Feedstock pretreatment
Oak wood sawdust was obtained in the sawdust form and sieved to obtain particles with sizes between 150 μm and 300 μm. The wood sawdust was subjected to a Soxhlet extraction with water (∼4 h) followed by ethanol (∼4 h) to remove extractives. After extraction, the feedstock particles were dried at 105 °C overnight for the catalytic reaction.Catalytic activity measurements
In a typical reaction experiment, the autoclave was charged with a suspension of 2000 mg of feedstock, 100 mg of Pd/C (5 wt% Pd loading), a certain amount of acid (e.g. 0.0316 mmol of Al(OTf)3) and 30 μl of n-dodecane internal standard in 40 ml of methanol. The reactor was sealed and purged with nitrogen and hydrogen several times sequentially. After leak testing, the pressure was brought to 30 bar with hydrogen and the reaction mixture was heated to the reaction temperature under continuous stirring at 500 rpm within 0.5 h. A sampling valve was installed, which allowed the removal of liquid samples during the reaction. About 0.4 ml solutions were taken out from the autoclave during the reaction and analysed directly by GC-MS. After the reaction, the heating oven was removed and the reactor was cooled to room temperature in an ice-water bath. After releasing the pressure, the autoclave was opened and the mixture was collected and combined with the solution obtained from washing the autoclave with methanol. The collected reaction mixture was filtered by using a filter crucible (porosity 4) under vacuum. The carbohydrate and Pd/C solid residue was left in air to dry for one day to determine the amount of all-solid residue. In some cases, the solid residue was further subjected to a two-stage acid hydrolysis to determine the Klason lignin content following the protocol described earlier.33An autoclave with a volume of 12 ml was used for the reactions with the model compounds. In a typical run, 25 mg (PG, GT and GG) of model compound, 10 mg of Pd/C and 10 μl of n-dodecane internal standard were added to 5 ml of methanol, which contained a certain amount of acid (the same concentrations as the corresponding woody biomass reactions described above). After sealing, purging and checking for leaks, the autoclave was heated to 160 °C and maintained at that temperature for 2 h. After the reaction, an aliquot of ∼1 ml was taken from the reaction mixture and directly analysed by GC-MS without dilution following filtration with a 0.45 μm syringe filter. In the experiments involving guaiacol and phenol model compounds, a 100 ml Parr autoclave was used as the reactor, which allows sampling during the reaction. In a typical run, 50 mg of reactant (guaiacol, phenol and glucose), 10 mg of Pd/C, and 30 μl of n-dodecane internal standard were added to the reactor together with 30 ml of methanol. The reaction was operated at 160 °C for 2 h.
Product analysis and characterization
The liquid phase products were analysed by a Shimadzu 2000 GC-MS system equipped with a RTX-1701 column (60 m × 0.25 mm × 0.25 μm) and a flame ionization detector (FID) together with a mass spectrometer (MS) detector. For the lignin monomers, the product peaks were identified using MS and by authentication with reference compounds. All the quantitative analyses of the liquid phase products were based on GC-FID. Experimentally determined weight response factors (RF) were used for calculation relative to n-dodecane, which was the internal standard. For some of the commercially unavailable compounds (side-products obtained in small amounts), the RF of PS-OCH3 was used for quantification. The yield of lignin monomers was calculated using following equation:| Yield of monomers (wt%) = (weight of monomers)/(weight of starting feedstock × Klason lignin weight percentage in oak wood) × 100% |
For the model lignin compound reactions (PG, GT and GG), the FID response factors were calculated using the Effective Carbon Number (ECN) method45 to determine the relative response factors corrected by the molecular weight of the compounds relative to n-dodecane, which served as the internal standard.
Gel permeation chromatography (GPC)
GPC analyses were performed using a Shimadzu apparatus equipped with two columns in series (Mixed-C and Mixed-D, polymer laboratories) and a UV-Vis detector at 254 nm. The column was calibrated with polystyrene standards. Analyses were carried out at 25 °C using THF as the eluent at a flow rate of 1 ml min−1. For the lignin residue analysis, the sample was prepared at a concentration of 2 mg ml−1. All the samples were filtered using a 0.45 μm filter membrane prior to injection.References
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- Y. C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- F. P. Bouxin, A. McVeigh, F. Tran, N. J. Westwood, M. C. Jarvis and S. D. Jackson, Green Chem., 2015, 17, 1235–1242 RSC.
- J. L. Wen, B. L. Xue, S. L. Sun and R. C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 1663–1671 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2015, 17, 4941–4950 RSC.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Chem., 2014, 6, 1035–1036 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- E. Feghali, G. Carrot, P. Thuery, C. Genre and T. Cantat, Energy Environ. Sci., 2015, 8, 2734–2743 CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2016, 9, 2074–2079 CrossRef CAS PubMed.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Green Chem., 2017, 19, 202–214 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. D. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Y. Cai, Y. H. Wang, J. J. Zhang, W. Q. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS PubMed.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed.
- C. Z. Li, M. Y. Zheng, A. Q. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 CAS.
- P. Ferrini, C. A. Rezende and R. Rinaldi, ChemSusChem, 2016, 9, 3171–3180 CrossRef CAS PubMed.
- I. Klein, C. Marcum, H. Kenttämaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- T. Renders, W. Schutyser, S. Van den Bosch, S. F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS PubMed.
- X. Huang, J. Zhu, T. I. Koranyi, M. D. Boot and E. J. Hensen, ChemSusChem, 2016, 9, 3262–3267 CrossRef CAS PubMed.
- X. Huang, O. M. Morales Gonzalez, J. Zhu, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2017, 19, 175–187 RSC.
- T. T. You, L. M. Zhang, S. K. Zhou and F. Xu, Ind. Crops Prod., 2015, 71, 65–74 CrossRef CAS.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- J. Tsujino, H. Kawamoto and S. Saka, Wood Sci. Technol., 2003, 37, 299–307 CrossRef CAS.
- H. L. Wang, L. B. Zhang, T. S. Deng, H. Ruan, X. L. Hou, J. R. Cort and B. Yang, Green Chem., 2016, 18, 2802–2810 RSC.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- H. Shafaghat, P. S. Rezaei and W. M. A. W. Daud, RSC Adv., 2015, 5, 33990–33998 RSC.
- X. M. Zhang, P. Murria, Y. Jiang, W. H. Xiao, H. I. Kenttamaa, M. M. Abu-Omar and N. S. Mosier, Green Chem., 2016, 18, 5219–5229 RSC.
- P. Sannigrahi, A. J. Ragauskas and S. J. Miller, BioEnergy Res., 2008, 1, 205–214 CrossRef.
- G. J. M. Gruter and F. Dautzenberg, EP1834950 A1, 2007.
- G. J. M. Gruter and F. Dautzenberg, EP1834951 A1, 2007.
- R. L. Grob and E. F. Barry, Modern practice of gas chromatography, Wiley, Hoboken, 2004, pp. 302–303 Search PubMed.
Footnote |
| † Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |