DOI:
10.1039/C4RA09914A
(Paper)
RSC Adv., 2015,
5, 3978-3998
Can elongation of the π-system in triarylamine derived sensitizers with either benzothiadiazole and/or ortho-fluorophenyl moieties enrich their light harvesting efficiency? – a theoretical study†
Received
6th September 2014
, Accepted 20th November 2014
First published on 21st November 2014
Abstract
The structural and electronic properties of five known triarylamine derived sensitizers (A1, A1-F, C218, D2 and Y123) and their associated hypothetical dyes (C218-F, D2-F, Y123-F, Y1234 and Y1234-F) have been studied using density functional theory and time-dependent density functional theory. The sensitizers primarily comprise of a triphenylamine, a 4,4′-dihexylcyclopenta[2,1-b:3,4-b]dithiophene and a cyanoacrylic acid as the electron donating, π-spacer and accepting units, respectively. The π-system is extended by incorporation of either a benzo[c][1,2,5]thiadiazol-4,7-diyl unit or an ortho-fluorophenyl unit or both. To gain insight into the effect of elongation of the π-system on the electronic properties of dye sensitized TiO2 interfaces, first-principles calculations have been carried out on sensitizer molecules co-adsorbed on the (101) surface of the anatase TiO2. The theoretical results revealed that elongating the π-system of the sensitizers with both the benzothiadiazole and ortho-fluorophenyl units increases the molecular extinction coefficient, the excited state lifetime and the light harvesting efficiency but decreases the band gap and the reorganization energy relative to the structurally comparable reference dye Y123. The calculated short circuit current density and level alignment quality showed that the π-system in the triarylamine sensitizers elongated with both benzothiadiazole and ortho-fluorophenyl units broadens their potential use in DSSCs due to the enhanced values as compared to the reference dye. The results obtained in this study will provide a valuable reference for the strategy of inserting various π-spacers in triarylamine sensitizers for dye sensitized solar cell applications.
1. Introduction
In the past decade, organic dyes have fascinated many researchers due to a wide variety of high-tech applications, such as light-emitting diodes,1 field effect transistors,2,3 and dye sensitized solar cells (DSSCs).4–6 Especially, over the last few years a number of researchers have paid increasing attention towards experimental and theoretical investigations of metal free organic dyes (MFOD) due to their possible benefits, such as relatively low cost, general ease of preparation and routinely higher molar extinction coefficients, relative to metallo-organic dyes (MOD). It is perceived that, in DSSCs the efficiency of MFOD falls beneath the MOD. For example, DSSCs based on either ruthenium7 or zinc porphyrin8 sensitizers have demonstrated overall conversion efficiencies reaching 13% under standard air mass (AM) 1.5G sunlight. On the other hand, MFODs based on coumarin,9–11 merocyanine,12,13 cyanine,14,15 indoline,16,17 hemicyanine,18–20 triarylamine,21–30 dialkylaniline,25,31,32 [bis(dimethyfluorenyl)amino]phenyl,33–36 phenothiazine,37 tetrahydroquinoline38,39 and carbazole,40,41 offer efficiencies between 4 and 10%. Among all the MFODs, the triarylamine based sensitizer (Y123) achieved respectable conversion efficiencies up to 10% with the [CoII(bpy)3](B(CN)4)2/[CoIII-(bpy)3](B(CN)4)3 redox system, owing to the promising electron-donating and steric properties of the donor group.
In DSSCs, the sunlight-to-electricity conversion efficiency (η) of solar cell devices is determined by the open-circuit photovoltage (VOC), short-circuit current density (JSC), and the fill factor (FF), as related to incident solar power (Pinc)
| |
 | (1) |
According to eqn (1), the product of VOC and JSC must be augmented to improve the efficiency of η. However, the quantity JSC directly depends on the dye properties, such as, the dye light-harvesting, kinetics of the electron injection, dye regeneration and charge transport within the cell.
A key factor that has a significant impact on the power conversion efficiency of DSSCs is the energy gap (Eg) of the sensitizer, since the rate of charge transfer (CT) is directed by the free energy of the transition that is correlated to the energy gap between the electronic potential energy levels [highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs)] of both the donors and the acceptors.42 In order to attain superior performance of DSSCs the sensitizer must have good conjugation throughout the molecule and a low energy gap between the HOMO (EH) and the LUMO (EL) to stimulate the intramolecular charge transfer and a strong absorption band in the visible and near-infrared region to maximize the light-harvesting efficiency.21,43 Briefly, for the sake of an efficient electron injection from the dye excited state onto the semiconductor surface, there must be a strong electronic coupling between the LUMO of the dye and the semiconductor conduction band (CB). The LUMOs of the triarylamine dyes are generally too low to provide perfect electron injection, and must be further improved by the optimizations. The characteristic electronic absorption spectrum of triarylamine dyes21 involves a strong transition to the first excited state (S0 → S1) at about 400–600 nm and a weak transition to the second excited state (S0 → S2) at about 300 nm. The first transition belongs to the ICT character, whereas second transition is assigned to π → π* transition. Although notable progress has been made with triarylamine dyes, the major problem is that they show very low molar extinction coefficients in the region of 500−600 nm as well as in the longer wavelength region, which greatly decreases the light harvesting abilities. Thus, the JSC of the cell decreases. In order to overcome this problem, it is essential to tune the electronic and optical properties of the sensitizers by modifying their chemical structures. As a result, the electron transfer process21 of the DSSCs will be enhanced.
A survey of the literature indicated that the photo-conversion efficiencies of DSSCs fabricated with triarylamine derived dyes was critically dependent upon the molecular structure (i.e., donor (D) and acceptor (A) groups) of the dyes and also on the amount of dye adsorbed on the TiO2 surface.21 Triphenylamine (TPA) is commonly employed as the electron donor23 and acid groups, such as cyanoacrylic acid, carboxylic acid, phosphonic acid and sulfonic acid, function as electron acceptors, which form the vital link between the dye molecules and the semiconductor surface. On the other hand, the conjugated π-system plays a vital role in the charge transfer process between the donor and the acceptor, in enhancing light harvesting, and in the suppression of the dark current. Tian et al.23 in their review article demonstrated that D–π–A structure can facilitate the electron injection due to the directional electron transfer from the donor site to the acceptor site along the D–π–A system, and the elongation of the π conjugation and loss of symmetry could also cause broadening and a red shift of the absorption bands in triphenylamine dyes. For instance, in 2010, Chen et al.44 verified that elongation of π-conjugation with the highly electronegative fluorine substituted phenyl group in dye A1, leading to dye A1-F, caused broadening and a red shift of the absorption bands and subsequently decreased the band gap and improved the photo-current response. As a consequence of the fluorinated dyes exhibiting higher hydrophobicity and offering large shielding, they moderate the direct interaction between the electrolyte and anode. Thus, water-induced desorption of the dye from the TiO2 surface is significantly reduced. Improved results were achieved with the 1,4-phenylene group with a fluorine substituent at C-3 i.e., ortho-fluorophenyl group (O-FP). On the other hand, the steric repulsion between the ortho-substituted fluorine atom and the carboxylic acid group not only influenced the D–A strength, but also increased the electronic coupling with nanostructured TiO2 electrode.21 In 2013, Gabrielsson et al.45 confirmed that higher photocurrents and solar energy conversion efficiencies can be achieved by encompassing the linker fragment, 4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT), in dye D35. Similarly, Zhang et al.43 showed that the chromophoric core comprising of CPDT and benzo[c][1,2,5]thiadiazol-4,7-diyl (BT) in conjunction with a TPA unit and a benzoic acid (BA) anchor leads to power efficiencies of over 11%. Wang et al.46 revealed that extending the CPDT π-bridge in C218 dye47 with a strong electron-withdrawing BT unit, leads to dye D2, which exhibited enhanced absorption in the long wavelength region with efficiency greater than 9%. Recently, Yella et al.48 demonstrated that the efficiency and short circuit current density of C218 and Y123 (ref. 49) dyes can be further enriched by tuning their donor fragments with a fluorine moiety. The foregoing discussion indicates that further enhancement of power conversion efficiency of DSSCs can be achieved by the rational molecular design of new triphenylamine dyes with good light harvesting ability, and also informs us that the electron-withdrawing units (O-FP and BT) increase the co-planarity of a bridged π-system without inducing intermolecular aggregation and consequently enhance the red shifted absorption spectra of triphenylamine dyes. Contrary to the report by Wang et al.46 in which improved DSSC performance was noted for incorporation of a benzothiadiazole spacer between the triphenylamine donor group and the π-system of the sensitizer, Bäuerle et al.50 have explored the incorporation of a benzothiadiazole unit placed adjacent to the cyanoacrylic acid anchoring group. In the latter study, low efficiency of the derived DSSC was noted as a consequence of enhanced back electron transfer and an increased recombination rate. The structure of our hypothetical dyes were guided by these reports and thus we have discounted hypothetical structures which incorporate the benzothiadiazole and cyanoacrylic acid grouping adjacent to one another. Therefore, for a full understanding of the dye solar cell performance and the roles of donor and π-spacer groups, the structural and electronic properties of the dyes is of crucial importance. In this concern, quantum mechanical calculations centered on DFT and TDDFT are believed to be most promising way ahead. Such state-of-the-art theoretical calculations have been successfully employed to select triarylamine derived sensitizers.51–55
The foregoing discussion clearly indicates that influence of the o-fluorphenyl unit and the thiadiazole unit have been examined independently in triarylamine derived dyes, but the examination of (i) their combined effect in triarylamine derived dyes and (ii) their combined effect in substituted triarylamine derived dyes has thus far been overlooked. Our present work redresses this oversight and aims to assess the combined effect of these individually beneficial groups. Several unique combinations of such desirable structural units are presented in the work for the first time. In order to achieve an advanced understanding of the physical properties of DSSCs, theoretical investigations of the ground and excited-state properties of five known reference dyes A1, A1-F, C218, D2 and Y123 and five hypothetical dyes C218-F, D2-F, Y123-F, Y1234 and Y1234-F are presented (see Fig. 1 for structures and their interrelationships). A1 dye contains a TPA and CPDT units with a cyanoacrylic acid acceptor unit. The substitution of either alkoxy or benzene-2,4-bis(hexyloxy) at the para-position of the phenyl rings of the TPA unit in A1 leads to C218 and Y123 dyes, respectively. The long chain alkoxy substituents on the triphenylamine unit delay contact between the donor moiety and the TiO2 surface and suppress the charge recombination to achieve high fill factor values. Such an alkoxy moiety also protects the TiO2 surface, preventing access of the electrolytes to the interface and retarding the back electron transfer. However, using cyanoacrylic acid as a terminal anchoring group increases the electron affinity (EA) in regions close to the TiO2 surface and forms a molecular dipole that is orientated away from the TiO2 surface, enabling both open-circuit voltage (VOC) and short-circuit current density to be increased simultaneously. The absorption spectra of C218 and Y123 cover only the short wavelength region of visible light. So it is possible to enhance the absorption of C218 and Y123 dyes in the long wavelength region by inserting a conjugated group BT between donor and spacer groups, forming dyes D2 and Y1234. An additional fluorine substituted phenyl group incorporated between CPDT and the cyanoacrylic acid acceptor in the series of dyes A1, C218, D2, Y123 and Y1234 leads to structures A1-F, C218-F, D2-F, Y123-F and Y1234-F, respectively, with the aim of further elongating the π-conjugation. A special emphasis is placed on the use of standard computational strategies for the study of the absorbance, the injection and recombination lifetimes at the dye/TiO2 interface. This study is organized to provide the information concerning the methodology and full computational details (Section 2), followed by the theoretical results and discussion (Section 3) and finally the conclusions.
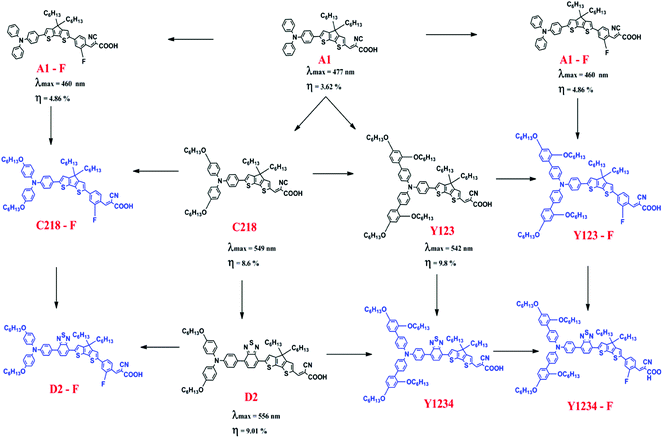 |
| | Fig. 1 Chemical structures of the triarylamine dyes investigated in the present study. Structures in black color are known (reference) dyes, whose experimental data are given below each structure and hypothetical structures are in blue color. | |
2. Theory and computational details
In DSSCs, incident photon to charge carrier efficiency (IPCE) is represented as a function of wavelength. IPCE is obtained by the following equation:24| |
 | (2) |
where JSC is the short-circuit photocurrent density for monochromatic irradiation, λ is the wavelength, and ϕsourceph(λ) = 100 mW cm−2. Here, JSC can be expressed as56| | |
JSC(λ) = q∫ηIPCEϕsourceph(λ)dλ
| (3) |
IPCE is also given by the following equation:
| ηIPCE = LHE ϕinject ηcollect |
Therefore
| |
 | (4) |
where LHE is the light-harvesting efficiency,
ϕinject is the quantum yield of electron injection, and
ηcollect is the efficiency of collecting the injected electrons at the back contact and
q is charge of electron. According to equation, if
ϕinject and
ηcollect are almost equal to unity, IPCE is determined by the LHE of the dye adsorbed on the film. Here, LHE is expressed as
56| | |
LHE = 1 − 10−A = 1 − 10−f
| (5) |
where
A and
f are the absorbance and oscillator strength of the dye associated to the
λICTmax. The oscillator strength is directly derived from the TDDFT calculations and writes
56| |
 | (6) |
where
![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) 0
0 − ICT is the dipolar transition moment associated to the electronic excitation. In order to maximize
f, both
λICTmax and
0 − ICT must be large. The amount of adsorbed dye on the surface
Γ (in mol cm
−2) can be calculated from the absorbance
A = 1000
εΓ, where
ε is the molar extinction coefficient (units M
−1 cm
−1). The Langmuir adsorption isotherm can be written as
Γ =
ΓmaxKC/(1 +
KC), with
K the binding constant for adsorption,
Γmax the maximum load on TiO
2, and
C is the dye concentration in the dye bath.
57
The molecular structure and electronic properties of a series of hypothetical triarylamine dyes and associated reference dyes were investigated using quantum chemical program Gaussian 03 (ref. 58) and GAMESS US59 in this paper. The equilibrium geometry of the molecules in neutral, cationic and anionic states were determined in the gas phase using the B3LYP/cc-PVDZ approach. Vibrational frequencies were calculated to confirm that the geometries correspond to minima on the potential energy surface. In order to reduce the computational cost, the hexyl groups have been replaced with methyl groups in each of the investigated dyes. The excited state properties have been examined using the CAM-B3LYP functional. Solvation effects, assuming acetonitrile solvent, were examined using an integral equation formalism variant Polarizable Continuum Model (IEFPCM) within the self-consistent reaction field (SCRF) theory.60,61 In order to calculate the excited state life time, firstly we optimized the excited states of dyes using the TD-B3LYP/cc-PVDZ method, then we used the excited geometries to calculate the dye relaxation in acetonitrile solvent with TD-CAM-B3LYP/DGDZVP method. To understand the nature of electronic excitations, the excited states were analysed by constructing the frontier molecular orbitals (FMOs) within the Avogadro program.62 The lambda diagnostic parameter concerning to charge transfer characteristic was calculated using GAMESS program. The reorganization energy of investigated dyes were calculated at the B3LYP/cc-PVDZ level in gas phase.
To gain insight into the electronic injection capability of dyes, the adsorption of dyes on the (TiO2)28 cluster was investigated with DFT calculations using DMol3 program.63 The (TiO2)28 configuration was fully optimised using the generalized gradient-corrected approximation (GGA) method. The Perdew–Burke–Ernzerhof (PBE) functional was used to take into account of the exchange-correlation effects with the double numerical polarization (DNP) basis set. The core electron was treated with all electrons. After optimization, the adsorption energies (Eads) of dyes on the (TiO2)28 cluster were obtained using the equation:
| | |
Eads = Edye+TiO2 − Edye − ETiO2
| (7) |
where
Edye was the total energy of isolated dye,
ETiO2 was total energy of (TiO
2)
28 cluster, and
Edye+TiO2 was the total energy of dye/(TiO
2)
28 complex. Following the above expression, a negative
Eads indicates that the molecule adsorption is exothermic and thus the adsorption system is energetically stable. It is believed that the numerical basic sets implemented in DMol
3 could minimize BSSE compared with the Gaussian basis sets.
64 Since DMol
3 uses the exact DFT spherical atomic orbitals, the molecules can be dissociated exactly to its constituent atoms. The binding energies still contain some small BSSE errors, but in the present study we have not consider the corrections.
Finally, making use of optimized geometries of Dmol3, we have calculated the excited state properties of the dye/TiO2 complexes at the CAM-B3LYP/3-21G(d) level using the Gaussian 03 program.
3. Results and discussion
3.1. Structural and electronic properties of the isolated molecules
3.1.1. Molecular geometry. The ground state optimized structural parameters of the ten triarylamine derived dyes (see Fig. 1) in neutral, cationic and anionic states computed at the B3LYP/cc-PVDZ level are presented in Table 1. The geometry optimization shows that the ten structures are non-planar. Mostly, the delocalization of the nitrogen lone pair into the phenyl rings is strongly influenced by the molecular conformation and electronic character of the three phenyl rings. Molecular structures of A1 and D2 together with some important structural parameters are presented in Fig. 2. The results showed that the structure of the central NCCC moiety (N–C–C1–C1′ in Fig. 2) in A1 and A1-F dyes is almost planar (θNCCC = 179–180°). From Table 1, we noticed that the introduction of substituents at the para-position of the phenyl rings hardly influence the nearly planar conformation of the central NCCC moiety, and the N–C bond length decreases from 1.410 Å to 1.403 Å as the electron donating strength of the donor group increases. In addition, the three phenyl rings in the propeller-shaped triphenylamine unit twist symmetrically from the central NCCC plane. Experimental results reported for the triphenylamine molecule showed a torsion angle θ1 of 41–44° in the gaseous phase,65 whereas our calculations show that θ1 is 44–45° for dyes A1 and A1-F. However, the substitution of either alkoxy or bis(hexyloxy)benzene groups at the para-position of the phenyl rings leads to an increase in torsion angle θ1 by approximately 1–6°. This slight increase in the torsion angle does not significantly influence the conjugation between the π-electrons of the phenyl groups and the central lone pair of the nitrogen atom. The steric repulsion between the inter-ring hydrogen atoms would lead to minimum energy configuration. We have also measured dihedral angles θ2 and θ3 between the donor group and the π-conjugation system, and the results were presented in Table 1. The dihedral angle θ2 is in the range ∼20.5 to 24.5° for A1, A1-F, C218 and Y123 dyes, whereas for D2, Y1234 and Y1234-F dyes, the insertion of BT between triphenylamine donor and CPDT leads to a decrease in torsion angle θ2 by 20°. This showed that the spacer and acceptor groups are almost co-planar with the central NCCC moiety. Therefore, the elongation of rigid, planar and bulky CPDT with BT and O-FP moieties, which serve as efficient linkers connect the donor and the acceptor moieties and reduce the recombination, results in the high VOC values. The rigidity constrains side paths that quench the excited states and most likely increases the lifetime of the dyes as compared with the flexible and heteroatom-containing conventional linkers.21 The calculated results clearly indicate that the dihedral angles are not only affected by the substituent (alkoxy or bis(hexyloxy)benzene) at the para-positions of the triphenylamine group, but also affected by the π-elongation of the spacer group. From Table 1, we can observe that in cationic and anionic states the C–N bond length of the investigated molecules decreases and increases, respectively, as compared with the neutral structures. However, the bond angle Φ (C1–N–C1′) of the dyes in different states increases in the order as anionic < neutral < cationic. It is worthy to mention that in the anionic and cationic states the π-conjugated spacer group in the investigated dyes is more planar with the central NCCC plane as compared with the neutral structures.
Table 1 Relevant bond lengths, angles, and dihedrals of triphenylamine derivatives at B3LYP/cc-PVDZ levela
| Compound |
State* |
Bond length (Å) |
Bond angle (°) |
Torsion angle (°) |
| N–C1 |
Φ (C1–N–C1′) |
θ1 (C1–N–C1′–C2′) |
θ2 (C–C–C–S)/(C–C–C–C) |
θ3 (C–C–C–S) |
| The bond lengths, bond angle and torsion angles are shown in Fig. 2. |
| A1 |
N |
1.410 |
120.46 |
45.07 |
23.95 |
— |
| C |
1.378 |
121.29 |
23.55 |
2.25 |
— |
| A |
1.434 |
119.01 |
34.63 |
2.66 |
— |
| A1-F |
N |
1.412 |
120.25 |
44.73 |
24.35 |
— |
| C |
1.379 |
121.37 |
22.83 |
3.16 |
— |
| A |
1.431 |
119.17 |
36.48 |
7.42 |
— |
| C218 |
N |
1.403 |
120.73 |
51.20 |
22.37 |
— |
| C |
1.389 |
120.89 |
29.29 |
4.64 |
— |
| A |
1.430 |
119.43 |
37.15 |
3.92 |
— |
| C218-F |
N |
1.405 |
120.81 |
28.76 |
23.29 |
— |
| C |
1.387 |
120.93 |
27.83 |
2.141 |
— |
| A |
1.426 |
119.77 |
39.09 |
8.44 |
— |
| D2 |
N |
1.405 |
120.73 |
48.64 |
1.146 |
0.128 |
| C |
1.388 |
120.85 |
28.80 |
0.466 |
0.044 |
| A |
1.433 |
119.31 |
34.99 |
0.00 |
0.229 |
| D2-F |
N |
1.402 |
120.84 |
27.81 |
0.281 |
0.678 |
| C |
1.386 |
120.91 |
27.16 |
0.595 |
0.269 |
| A |
1.426 |
119.85 |
40.20 |
0.454 |
0.021 |
| Y123 |
N |
1.403 |
120.50 |
46.60 |
20.54 |
— |
| C |
1.399 |
120.43 |
33.28 |
6.386 |
— |
| A |
1.433 |
119.05 |
34.03 |
0.379 |
— |
| Y123-F |
N |
1.410 |
120.40 |
46.00 |
20.98 |
— |
| C |
1.395 |
120.67 |
31.34 |
5.028 |
— |
| A |
1.431 |
119.22 |
35.15 |
0.942 |
— |
| Y1234 |
N |
1.403 |
120.74 |
49.42 |
1.681 |
0.454 |
| C |
1.398 |
120.47 |
33.00 |
0.012 |
0.591 |
| A |
1.433 |
119.29 |
34.95 |
0.477 |
0.016 |
| Y1234-F |
N |
1.404 |
120.67 |
49.07 |
1.926 |
0.666 |
| C |
1.393 |
120.94 |
29.98 |
1.423 |
0.172 |
| A |
1.430 |
119.41 |
36.67 |
0.036 |
0.148 |
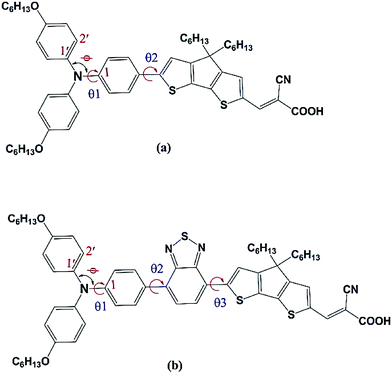 |
| | Fig. 2 Molecular structure of (a) A1 and (b) D2 dyes. For C–N bond lengths, bond, and torsion angles, see Table 1. | |
3.1.2. Electronic properties. As a general rule, a red shift in the absorption spectrum is needed to decreases the energy gap between the HOMO and the LUMO of the dye. Therefore we have calculated the potential levels of the sensitizers investigated in this work using B3LYP/cc-PVDZ level. The calculated energy levels of EH, EL and energy gap (Eg) were listed in Table 2. Results showed that the energy level obtained from the B3LYP method is close to the experimental values.
Table 2 EH (eV), EL (eV) levels, Eg (eV), ionization potential (eV), electron affinity (eV), dipole moment ρ (Debye), polarizability α (a.u), reorganization energy λ (eV) of triphenylamine based sensitizers calculated at B3LYP/cc-PVDZ levelc
| Dye |
EH (eV) |
EL (eV) |
Eg (eV) |
ρ (D) |
α (a.u) |
Hole transfer |
Electron transfer |
λint |
| IP |
λ+ |
EA |
λ− |
| See ref. 48. See ref. 46. Values in parenthesis are calculated using experimental EH and EL Values. |
| A1 |
−5.16 |
−2.69 |
2.47 |
10.21 |
596 |
6.05 |
0.25 |
1.71 |
0.42 |
0.67 |
| A1-F |
−5.08 |
−2.82 |
2.26 |
10.09 |
757 |
5.91 |
0.27 |
1.87 |
0.33 |
0.61 |
| C218 |
−4.92 (−4.99)a |
−2.61 (−2.86)a |
2.31 (2.13)a |
10.95 |
661 |
5.81 |
0.20 |
1.63 |
0.41 |
0.61 |
| C218-F |
−4.85 |
−2.74 |
2.11 |
13.53 |
825 |
5.69 |
0.21 |
1.78 |
0.33 |
0.54 |
| D2 |
−4.99 (−5.15)b |
−3.10 (−3.20)b |
1.90 (1.95)b |
11.37 |
982 |
5.73 |
0.17 |
2.21 |
0.32 |
0.50 |
| D2-F |
−4.86 |
−3.00 |
1.86 |
13.02 |
1174 |
5.63 |
0.18 |
2.24 |
0.28 |
0.46 |
| Y123 |
−4.87 (−5.08)a |
−2.59 (−2.91)a |
2.27 (2.17)a |
12.38 |
885 |
5.66 |
0.18 |
1.64 |
0.41 |
0.60 |
| Y123-F |
−4.81 |
−2.75 |
2.06 |
13.01 |
1054 |
5.57 |
0.19 |
1.80 |
0.33 |
0.52 |
| Y1234 |
−4.88 |
−3.03 |
1.85 |
12.68 |
1225 |
5.62 |
0.16 |
2.23 |
0.36 |
0.52 |
| Y1234-F |
−4.83 |
−3.00 |
1.83 |
13.03 |
1420 |
5.53 |
0.16 |
2.26 |
0.30 |
0.46 |
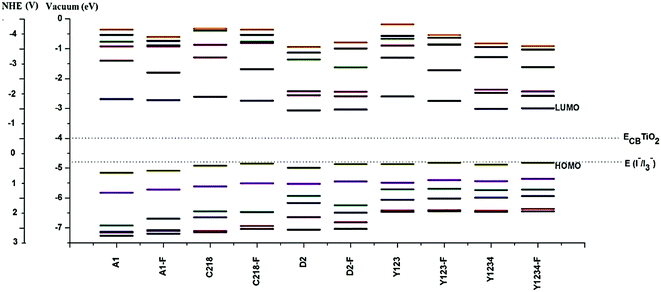
In order to design efficient triarylamine-dye sensitizers, the potential levels of the HOMO and LUMO must match to the conduction band level of the semiconductor electrode and to the I−/I3− redox potential.21 To accept electrons, i.e., for the dye regeneration process, the HOMO level must be lower than the iodine redox potential, and to inject electrons, the LUMO level must be higher than the conduction band level of the semiconductor.21 However, in dye sensitizers the low LUMO level matches with the conduction band of TiO2, which limits the further improvement of the energy conversion efficiency of DSSCs. Thus, it is very important to screen novel sensitizer candidates with a higher LUMO level. From Table 2, it can also be noted that the calculated LUMO of the dyes lies above the conduction band of the TiO2, which ensures fast and efficient electron transfer from the excited dye to the TiO2 conduction band, indicating that there will be a sufficient thermodynamic driving force for charge injection from the excited sensitizer molecule to the TiO2 conduction band. Furthermore, the calculated HOMO level of dyes lies in the gap between the conduction band and valence band, more negative than the iodide/tri-iodide redox potential (I−/I3−: −4.8 eV), indicating energetically favorable ground-state dye regeneration. Although we have compared the EH value with the experimental oxidation potential, the theoretical redox potential of the electrolyte is difficult to obtain accurately, so another reference compound was used as a benchmark in assessing the sensitizer candidates. The previously reported dye Y123, (see Fig. 1) the most effective dye from the triarylamine sensitizer family, is a good choice as the reference.49 It is predicted that sensitizer candidates with a HOMO level close to that of Y123 would be promising for dye regeneration, as Y123 can be regenerated efficiently. The calculated results in Table 2 showed that the HOMO level of hypothetical dyes are close to that of Y123, indicating that hypothetical dyes will have sufficient driving force for dye regeneration.
3.1.3. Dipole moment and polarizability. A high molecular polarizability is a prerequisite for a dye to be employed in an efficient organic solar cell.66 Such dyes with high polarizabilities exhibit strong interactions with surrounding species and induce an increase in the local concentrations of acceptor species.67 Similarly, dyes with higher dipole moment can enhance the effective charge separation between donor and acceptor units and consequently increase the open circuit voltage VOC.68 Our calculations concerning dipole moment and polarizability (see Table 2) showed that Y123-F, D2-F, Y1234 and Y1234-F, with elongated spacer groups, are highly polarized that would induce an increase in the local concentration of acceptor species at the TiO2 surface. This enhanced concentration increases the possibility of acceptor species penetrating the adsorbed dye layer leading to electron recombination within a short electron lifetime.
3.1.4. Energy gap. In DSSCs smaller energy gaps render the electrons more easily excited and thus beneficial for absorbing longer wavelength light. Hence, more photons can be absorbed at any given time, which guarantees a higher short circuit current density JSC and overall power conversion efficiency η.21 The band gap (Eg), the energy gap between the filled valance band and unfilled conduction band, of the dye can be obtained by the difference between EH and EL. It is well-known that DFT calculations often lead to overestimate the band gaps. Even though it is obtained approximately, the band gap obtained by the B3LYP method is closely related to the actual band gap (see Table 2). In order to better understand how the HOMOs and LUMOs are influenced by the π-elongation of the spacer group and substituents (alkoxy or bis(hexyloxy)benzene) at the para-position of the phenyl ring of the triphenylamine group of structure A1. We have presented the molecular energy levels of the ten dyes in Fig. 3. We can observe that the energy gap of the molecules is decreasing in the order of A1 > C218 > Y123 > A1-F > C218-F > Y123-F > D2 > D2-F > Y1234 > Y1234-F.
 |
| | Fig. 3 Energy level diagram of isolated dye molecules calculated at the B3LYP/cc-PVDZ level. | |
In general, the substituent at the para-positions of each of the phenyl rings of the triphenylamine moiety makes an important contribution to the device efficiency. The energy gap of C218 decreases by 7% when compared to A1 due to the para-alkoxy substituents of the triphenylamine phenyl rings. As a result, the power conversion efficiency of C218 increases to 8.6%. In C281, the energy levels are well separated from the HOMO and LUMO, but HOMO − 4, HOMO − 5; LUMO + 4, LUMO + 5 are energetically very close. Mostly these orbitals (HOMO − 4, HOMO − 5 and LUMO + 4, LUMO + 5) have contributions from carbon atoms of the triphenylamine group. From Fig. 3, it can also be seen that as the electron donating strength of the triphenylamine moiety increases, the energy difference between the HOMOs decreases. Similarly, introducing a bis(hexyloxy)benzene moiety at the para-position of the donor group in A1 leads to the structure Y123 in which the energy levels are well separated from each other and the HOMO–LUMO gap decreases by nearly 19% when compared to A1.
Fluorine substitution provides a significant contribution for promoting the cell performance. Elongating the π-system of the dye molecule with the O-FP group lowers both the HOMO and LUMO levels, and enriches the spatial separation between electrons and holes and reduces the dark current. When the π-system in A1 is elongated by the O-FP, led to structure A1-F, the HOMOs is shifted upward and the LUMOs downward. As a result the energy gap of the molecule A1-F decreases by 8.6% compared to A1. Here HOMO − 1 and LUMO + 1 are energetically well separated from the HOMO and LUMO levels in all molecules. It is also interesting to note that HOMO − 3 and HOMO − 4 are energetically close and well separated from HOMO, HOMO + 2 and HOMO + 5 in A1 and A1-F. This is due to the HOMOs of A1 and A1-F having a significant contribution from the nitrogen in the triphenylamine group, whereas HOMO − 3 and HOMO − 4 have hardly any contribution from nitrogen. The energy gap in C218 and Y123 can be further decreased by up to 15% compared to A1 by elongating the π-spacer with O-FP. From Fig. 3, it can be observed that in C218-F, the spacing between HOMOs is much less when compared to A1 and A1-F. Whereas the gap between LUMO and LUMO + 1 increases with the increasing electron donating strength of the triphenylamine moiety and decreases with the π-elongation of the spacer group.
Similarly, incorporation of the electron-deficient BT group between triphenylamine unit and CPDT in C218, leading to structure D2, lowers both the HOMO and LUMO energy levels, decreases the energy gap by 18% and enhances the spatial separation between electrons and holes. As a result, the efficiency of D2 (η = 9.31%) increases up to 7.62% when compared to C218 (η = 8.6%). The band gap of D2 can be further decreased by elongating the π-system with O-FP unit. The elongation of the π-system in reference dye Y123 and dye Y123-F with the BT (leads to hypothetical structures Y1234 and Y1234-F) decreases the energy gap by 18 and 11%, respectively. Therefore the hypothetical sensitizers C218-F, Y123-F, D2-F, Y1234 and Y1234-F have a smaller HOMO–LUMO gap than that of the more efficient dye Y123, showing potential for providing red-shifted electronic absorption spectra.
3.1.5. Optical properties. The absorption spectra of A1, A1-F, C218, D2 and Y123 dyes using three different LRC-DFT XC functionals (CAM-B3LYP, LC-ωPBE and ωB97XD) with DGDZVP basis set in acetonitrile solution with IEFPCM solvent model have been investigated firstly and compared with available experimental data. The calculated results were presented in Fig. 4. The results obtained with the CAM-B3LYP and ωB97XD functional are close to the experimental data (Fig. 4). Generally, the long-range corrected CAM-B3LYP offers the most stable and more rational descriptions on the relative intensities and energy spacing of energy bands, the discrepancy between the calculated and experimental excitation energies was found to be 0.2–0.3 eV. Secondly, we have adopted the CAM-B3LYP functional for calculating absorption spectra of the hypothetical dyes. The computed vertical excited singlet states, oscillator strengths of the ten dyes in acetonitrile solvent along with available experimental data were presented in the ESI (Table S1†). The simulated absorption spectra of ten dyes (Fig. 5) exhibit maxima from the localized aromatic π → π* transitions in the range 250–390 nm, while those at 400–600 nm originates from the charge transfer π → π* transition. As expected, the electronic absorption bands of the triarylamine dyes are sensitive to substituents at the para-position of the phenyl rings of the triphenylamine unit and the elongation of the π-spacer.
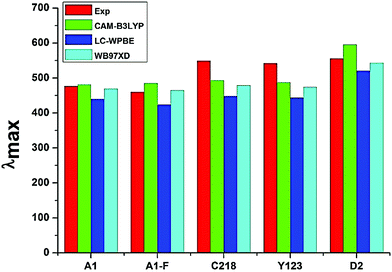 |
| | Fig. 4 Absorption maxima (in nm) of the experimentally investigated dye molecules calculated in acetonitrile solution by TD-DFT coupled with IEFPCM. | |
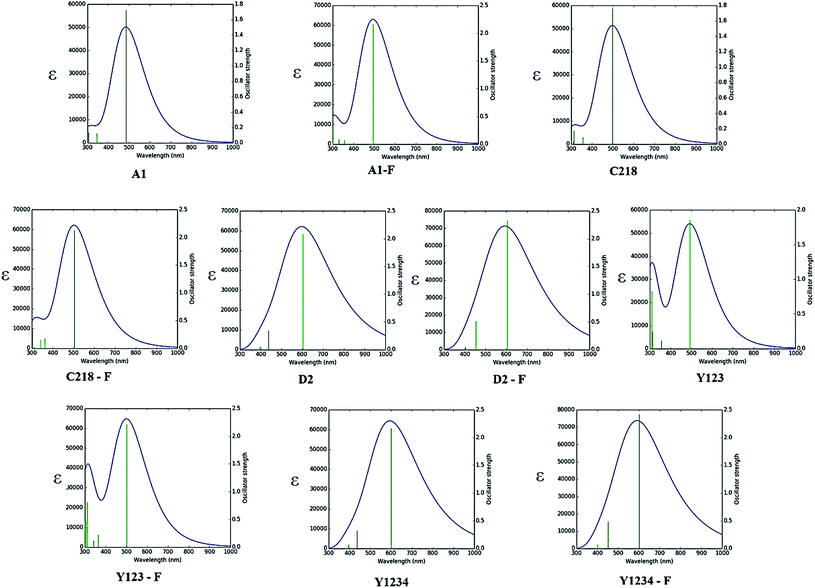 |
| | Fig. 5 Calculated absorption spectra of ten dyes in acetonitrile solution using CAM-B3LYP/DGDZPV level of theory. | |
The absorption spectra of A1 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of acetonitrile/tert-butyl alcohol exhibit an intense band at 477 nm. This band is attributed to the π → π* charge transfer transition (CT) originating from the TPA entity and part of CPDT to the electron accepting cyanoacrylic acid. Increasing the electron donating strength of the donor moiety in A1 by inserting an alkoxy group at the para-position of the TPA group, forming C218 dye, provides a ∼72 nm red shift of the absorption maximum with a similar molecular extinction coefficient to that of A1. Whereas, the calculated results show that there is a ∼10 nm red shift of the absorption spectra in acetonitrile solvent. Thus, the power conversion efficiency of C218 (η = 8.6%) increases by 135% compared to A1 (η = 3.62%). Replacement of the electron-donating alkoxy substituent at the para-position of the TPA rings in C218 with a bis(hexyloxy)benzene unit (leading to Y123) causes slight changes in the absorption properties with the absorption maximum of Y123 blue shifted by 6 nm. Generally, a dye with a low band gap can generate more electrons in the visible and near infrared regions and enhance the efficiency of DSSCs. But the probability of absorption by a molecule depends on the molecular extinction coefficient (ε). Larger values of ε indicate a high probability of absorption at a given wavelength leading to larger conversion efficiencies.69 Even though absorption bands of Y123 is blue shifted, its energy gap and molecular extinction coefficient are lower and higher than the dye C218, respectively. Therefore, the light harvesting efficiency of Y123 dye is larger than C218.
:1 solution of acetonitrile/tert-butyl alcohol exhibit an intense band at 477 nm. This band is attributed to the π → π* charge transfer transition (CT) originating from the TPA entity and part of CPDT to the electron accepting cyanoacrylic acid. Increasing the electron donating strength of the donor moiety in A1 by inserting an alkoxy group at the para-position of the TPA group, forming C218 dye, provides a ∼72 nm red shift of the absorption maximum with a similar molecular extinction coefficient to that of A1. Whereas, the calculated results show that there is a ∼10 nm red shift of the absorption spectra in acetonitrile solvent. Thus, the power conversion efficiency of C218 (η = 8.6%) increases by 135% compared to A1 (η = 3.62%). Replacement of the electron-donating alkoxy substituent at the para-position of the TPA rings in C218 with a bis(hexyloxy)benzene unit (leading to Y123) causes slight changes in the absorption properties with the absorption maximum of Y123 blue shifted by 6 nm. Generally, a dye with a low band gap can generate more electrons in the visible and near infrared regions and enhance the efficiency of DSSCs. But the probability of absorption by a molecule depends on the molecular extinction coefficient (ε). Larger values of ε indicate a high probability of absorption at a given wavelength leading to larger conversion efficiencies.69 Even though absorption bands of Y123 is blue shifted, its energy gap and molecular extinction coefficient are lower and higher than the dye C218, respectively. Therefore, the light harvesting efficiency of Y123 dye is larger than C218.
Upon inserting the O-FP group into A1, forming A1-F, the power conversion efficiency (η) increases from 3.62 % to 4.86 %. Due to the lack of planarity between CPDT and the O-FP in A2-F, the absorption maximum blue shifts by ∼20 nm with a slightly larger ε ∼ 53740 ± 130 M−1 cm−1 than that of A1. From Table 3, the calculated maximum absorption peak of dye A1-F in acetonitrile solvent is red shifted by ∼11 nm compared to A1. The π-conjugation in dyes C218 and Y123 is extended by inserting an O-FP group; forming hypothetical dyes C218-F and Y123-F. The maximum absorption band red shifts and the molecular extinction coefficient increases. As a result, the photocurrent response for these dyes is predicted to be higher than that of the C218 and Y213. Similarly, extending π-system by inserting BT group in C218, C218-F, Y123 and Y123-F, forming D2, D2-F, Y1234 and Y1234-F dyes, enhances the absorption in the long wavelength regions. The experimental results showed that the absorption maximum of D2 is red-shifted by 44 nm as compared to C218, due to better delocalization of electrons over the entire molecule. Whereas, the calculated results showed that the absorption maxima of hypothetical dyes D2-F, Y1234 and Y1234-F red shift by 110, 106 and 108 nm, respectively, with enhanced molecular extinction coefficients, as compared to the reference dye Y123.
Table 3 Absorption and emission properties of the ten dyes in acetonitrile solution calculated at the CAM-B3LYP/DGDZVP level
| Dye |
Experimental |
Theoretical |
| λabsmax (nm) |
ε (M−1 cm−1) |
Eexp0d (eV) |
λabsmax (nm) |
EA0 (eV) |
ε (M−1 cm−1) |
f |
Λ |
λemmax (nm) |
ε (M−1 cm−1) |
f |
EE0d (eV) |
EAvg00d |
| See ref. 44. See ref. 48. See ref. 46. Where average optical gap EAvg00 is calculated using sum of the optical gap obtained from absorption and emission maximum i.e., EAvg00 = (EAo + EEo)/2. |
| A1 |
477a |
51660a |
2.59 |
484 |
2.56 |
50107 |
1.724 |
0.591 |
512 |
48676 |
1.676 |
2.42 |
2.49 |
| A1-F |
460a |
53740a |
2.69 |
494 |
2.51 |
62970 |
2.168 |
0.509 |
587 |
62746 |
2.155 |
2.11 |
2.31 |
| C218 |
549b |
46000b |
2.25 |
498 |
2.49 |
51307 |
1.765 |
0.546 |
511 |
47990 |
1.627 |
2.42 |
2.46 (2.13) |
| C218-F |
— |
— |
— |
504 |
2.46 |
62112 |
2.131 |
0.467 |
521 |
61867 |
2.125 |
2.38 |
2.42 |
| D2 |
556c |
56000c |
2.23 |
596 |
2.08 |
62062 |
2.168 |
0.630 |
633 |
50594 |
1.739 |
1.96 |
2.02 (1.95) |
| D2-F |
— |
— |
— |
603 |
2.05 |
71081 |
2.329 |
0.610 |
658 |
55983 |
1.929 |
1.88 |
1.97 |
| Y123 |
542b |
50500b |
2.28 |
493 |
2.51 |
54040 |
1.857 |
0.496 |
510 |
50496 |
1.731 |
2.43 |
2.47 (2.17) |
| Y123-F |
— |
— |
— |
501 |
2.47 |
64799 |
2.219 |
0.419 |
516 |
62832 |
2.160 |
2.40 |
2.44 |
| Y1234 |
— |
— |
— |
599 |
2.06 |
64367 |
2.171 |
0.586 |
559 |
51225 |
1.633 |
2.21 |
2.14 |
| Y1234-F |
— |
— |
— |
601 |
2.06 |
73550 |
2.424 |
0.568 |
647 |
58588 |
2.019 |
1.91 |
1.99 |
The calculated results showed that the first absorption maxima of the ten dyes are derived from the HOMO → LUMO electronic transition. The HOMOs of all dyes are fairly delocalized over the TPA-donor and spacer moieties, and the LUMOs are extended over the cyanoacrylic acid unit and part of the spacer. Such a spatially well-separated orbital distribution is highly desirable for efficient intramolecular charge separation upon photoexcitation. In order to understand the charge transfer character, Tozer et al.70 introduced a spatial overlap, Λ, a quantity which measures the spatial overlap for each excitation using the formula
| |
 | (8) |
where
Oia = 〈|
ϕi‖
ϕa|〉 = ∫|
ϕi(
r)‖
ϕa(
r)|d
r is the inner product of the moduli of the two orbitals,
ϕi and
ϕa, involved in the excitation, and
κia =
Xia +
Yia is a measure of the contribution of the
i–
a orbital transition to the excitation. Because the maximum absorption of all our analogues corresponds to the HOMO → LUMO transition, the degree of CT can be approximated by simplifying
eqn (8). This is done by calculating the product of the absolute value of the molecular orbital coefficients of the HOMO and LUMO as given in
eqn (9).
| |
 | (9) |
The spatial overlap takes the value 0 ≤ ΛHL ≤ 1; 0 ← ΛHL and indicates long-range excitation, and 1 ← ΛHL signifies short range excitation. Recently, Peach et al.71 suggested that Λ values >0.4 have the greatest reliability for charge-transfer states. If Λ values <0.4, there is little overlap and significant long-range charge transfer character. Therefore, we have calculated the Λ diagnostic values for the excitation of each ground state structures using TD-CAM-B3LYP/6-31G(d) level of theory. The results were reported in Table 3 for all the dyes in the gas phase. Note that smaller values of Λ (i.e. a smaller overlap between the HOMO and LUMO orbitals) indicate a greater charge-transfer character (when Λ = 0, an electron is completely transferred).72
Fig. 6 (bottom) showed the charge density difference between the excited and ground states of ten dyes with green regions representing a depletion of density and red regions an accumulation of density upon excitation. As depicted in Fig. 6 (bottom), the density depletion zones (green) are mostly located on the conjugation spacer segments, while the regions of density enhancement (red) are mainly localized on the acceptor moiety, indicating an intra-molecular charge transfer when the transition occurs. Molecular orbital compositions of the HOMO and LUMO energy levels of the ten dyes were listed in Table 4. For a greater understanding of the molecular orbital contribution to the energy levels, the partial density of states (PDOS) of the ten molecules were presented in the ESI (Fig. S1†). The results showed that the substituent at the para-position of the phenyl rings of the triphenylamine group does not affect the contribution of the anchoring group to the LUMO of C218 and Y123 when compared to A1. Whereas, upon the insertion of the O-FP group in A1, C218 and Y123, leading to dyes A1-F, C218-F and Y123-F respectively, a 5% increase contribution of the anchoring group to LUMO was noted. The contribution of the anchoring group of our hypothetical dyes C218-F and Y123-F to LUMO is 45%, whereas the so far known best dyes C218 and Y123 contribute 40%. However the insertion of a BT group in C218, affording D2, reduces the contribution of the anchoring group LUMO to 16%, but the contribution increases up to 18% by elongating the π-system with an O-FP moiety (forming D2-F). By elongating the π-system, the contribution from the donor part in the HOMO decreases and the contribution from CPDT moiety increases. Therefore, we may anticipate that elongating the spacer group would certainly increase the electron coupling with the TiO2 surface and would be more favourable for electron injection into the TiO2 surface.
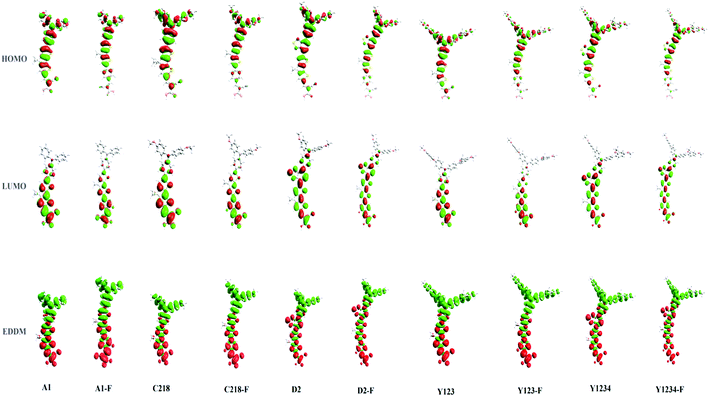 |
| | Fig. 6 HOMO, LUMO, and electron density difference maps (EDDM) between the excited and ground states of ten dyes. The contour thresholds for molecular orbitals and density differences are 0.02 and 0.0002 a.u., respectively. The green and red colors indicate a decrease and increase of charge densities, respectively. | |
Table 4 Percentage of donor, acceptor and spacer units contribution to HOMO and LUMO energy levels
| Dye |
Molecular orbital contribution (%) |
| HOMO |
LUMO |
| D |
CPDT |
OFP |
BT |
A |
D |
CPDT |
OFP |
BT |
A |
| A1 |
60 |
33 |
— |
— |
6 |
6 |
54 |
— |
— |
40 |
| A1-F |
48 |
45 |
5 |
— |
2 |
2 |
21 |
32 |
— |
45 |
| C218 |
71 |
25 |
— |
— |
4 |
6 |
54 |
— |
— |
40 |
| C218-F |
60 |
35 |
4 |
— |
1 |
2 |
21 |
32 |
— |
46 |
| D2 |
61 |
19 |
— |
17 |
3 |
9 |
32 |
— |
43 |
16 |
| D2-F |
50 |
28 |
3 |
18 |
1 |
7 |
23 |
16 |
36 |
18 |
| Y123 |
73 |
23 |
— |
— |
4 |
6 |
54 |
— |
— |
40 |
| Y123-F |
61 |
34 |
4 |
— |
4 |
2 |
21 |
32 |
— |
45 |
| Y1234 |
62 |
19 |
— |
16 |
3 |
9 |
32 |
— |
44 |
16 |
| Y1234-F |
50 |
29 |
3 |
17 |
1 |
7 |
23 |
15 |
37 |
18 |
3.1.6. Reorganization energy. In non-adiabatic Marcus electron transfer theory73 the charge transfer (Γ) between a weakly coupled electron donor and electron acceptor was defined as a function the electronic coupling, (J), the reorganization energy, (λreog), and the difference in free energy between the equilibrium states of products and reactants, ΔG0 = Gproducts − Greactants, which is written as| |
 | (10) |
where kB is the Boltzmann constant and T is the temperature. Therefore, in order to enhance the JSC one should focus on improving the LHE and ϕinject as well as decreasing the reorganization energy. Reorganization energy is the energy requisite for structural changes associated with the charge transfer of the molecules of interest (internal, inner-sphere, vibrational, or structural reorganization) and nearby molecules (external or outer-sphere reorganization). In the solid state, the external reorganization energy can be passed over because the surroundings are rigid, and the internal reorganization energy of isolated molecules can be used as an upper boundary for the charge transport energetics. Internal reorganization energy has been shown to decrease as the size of the conjugated portion of the molecule increases. So we computed the internal reorganization energy λint according to the following formula74| |
 | (11) |
where E is energy, Q is geometry, and the subscripts 0, +, and − denote neutral, cationic, and anionic states, respectively. Table 2 reported the computed internal reorganization energies, the ionization potential (IP), and the electron affinity (EA). IP and EA were calculated as E0(Q0) − E+(Q+) and E0(Q0) − E−(Q−), respectively.From the Table 2, we can observe that the total reorganization energy decreases with increasing strength of the donor moiety and elongation of the π-spacer and thus making the molecules more rigid and planar. As a result, the charge injection rate from the dye excited state to TiO2 conduction band will increase, which is a prerequisite for enhancing the short-circuit current density. When the spacer group is elongated with the O-FP, λ+ is increasing but λ− is decreasing. But when the BT group is inserted as a spacer group, both the λ+ and λ− decrease. The reorganization energy also depends on the molecular energy levels, because the LUMO energy levels generally decrease with increasing EA, HOMO energy levels decrease with increasing IP. The calculated results show that as the HOMO level increased, the hole reorganization energy is decreases. Whereas, the LUMO level decreases, the electron reorganization energy decreases (see Fig. 7). The total reorganization energy λint for the hypothetical dyes C218-F, D2-F, Y123-F, Y1234 and Y1234-F are smaller than the reference dye Y123. Therefore we can expect to obtain high electron injection rate from the excited states to the TiO2 conduction band for the hypothetical dyes due to their low reorganization energy. As a result, the JSC for these dyes is predicted to be larger than that of the reference dye Y123.
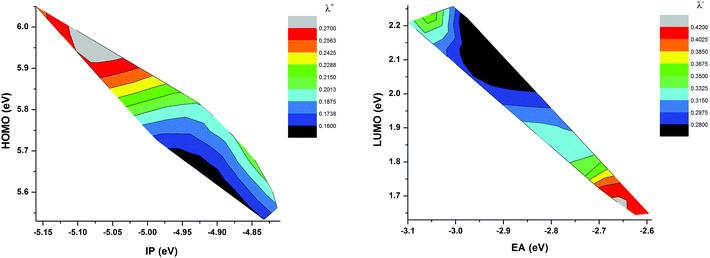 |
| | Fig. 7 Comparison graphs between (a) HOMO vs. IP and (b) LUMO vs. EA of ten dyes. The colors map in the figure indicates the reorganization energy for the corresponding energy levels, IP and EA. | |
3.1.7. Charge injection (ΔG0inj) and dye regeneration (ΔG0reg). The eqn (3) and (10) clearly indicates that the ϕinject and rate of charge transfer are related to the free energy ΔG0 of electrons injecting from the excited state of dye molecule to the TiO2 substrate. Therefore, the change in electron injection ΔG0inj affects both the electron injection rate and short circuit current density. The yield of charge collection and dye regeneration can be determined by the driving force, which is defined as the potential difference between the oxidized dyes and the electrolyte. For a fast and an efficient regeneration of the dye cation radical, the oxidation potential of the dyes must be more positive than that of electrolyte and avoiding the geminate charge recombination between oxidized dye sensitizers and the nanocrystalline TiO2 film where the photo-excited electrons are injected. According to Preat's method,54 assuming the electron injection occurs from the unrelaxed excited state of the dye, the ΔG0inj can be calculated by the following equation| |
 | (12) |
where Edye*OX is the oxidation potential of the dye in the excited state, and  is the reduction potential of the semiconductor conduction band. Though it is often difficult to accurately determine
is the reduction potential of the semiconductor conduction band. Though it is often difficult to accurately determine  , a highly sensitive datum to the conditions like the pH of the solution, thus we used the experimental value
, a highly sensitive datum to the conditions like the pH of the solution, thus we used the experimental value  (ref. 75) which corresponds to conditions where the semiconductor is in contact with the aqueous redox electrolytes of fixed pH 7.0.76,77 The excited state oxidation potential (Edye*OX) can be approximated from the redox potential of the ground state (EdyeOX),54
(ref. 75) which corresponds to conditions where the semiconductor is in contact with the aqueous redox electrolytes of fixed pH 7.0.76,77 The excited state oxidation potential (Edye*OX) can be approximated from the redox potential of the ground state (EdyeOX),54| | |
Edye*OX = EdyeOX − λICTmax
| (13) |
where λICTmax is the energy of the ICT. Note that this relation is only valid if the entropy change during the light absorption process can be neglected. Compared with dye Y123, the other dyes containing either benzothiadiazole and/or ortho-fluorophenyl moieties in the π-conjugated bridge have the slightly lower −ΔG0inj values. The calculated results indicated that the −ΔG0inj values of all the dyes are greater than 1 eV. As a result, an efficient electron injection takes place from the sensitizers into the TiO2.The maximum open circuit voltage (VOC) of the DSSC is limited by the difference between the redox potential of the redox mediator and the quasi-Fermi level of electrons in the TiO2 semiconductor. In principle, device efficiencies can be improved by choosing a redox couple with a potential closer to that of the dye, but sufficient driving force needs to be provided to ensure efficient dye regeneration by the redox mediator. Grätzel78 reports that a minimum driving force of 0.2–0.3 eV is required due to the two-electron character of the iodide oxidation reaction, which passes through I2− radicals as intermediates. Generally, at large thermodynamic driving forces for dye regeneration, energy dissipates as heat during the regeneration reaction and contributes to a major loss in energy efficiencies of DSSCs. In order to obtain the dye regeneration, we have calculated the driving force for oxidized dye regeneration as the difference between redox potential I3−/I− and redox potential of the dye ground state (ΔGreg0 = E (I−3/I−) – EdyeOX). The calculated results in Table 5 showed that the EdyeOX values of the hypothetical dyes C218-F, Y123-F, D2-F, Y1234 and Y1234-F match well with the reference dye Y123. Whereas for dyes A1, A1-F, C218 and D2, the EdyeOX values are slightly lower than the reference dye Y123. Therefore the regeneration driving forces of the hypothetical dyes C218-F, Y123-F, D2-F, Y1234 and Y1234-F are lower than the reference dye Y123. From Table 5, we can also note that the regeneration driving force of the dyes decreases as the electron donating strength of the donor group increases. This can be further decreased by π-elongation of the spacer group. Therefore, there exists a potential to obtain a higher voltage and higher power conversion efficiencies for hypothetical dyes C218-F, Y123-F, D2-F, Y1234 and Y1234-F when compared to reference dye Y123.
Table 5 Ground and excited state redox potentials (Edyeox and Edye*ox); charge injection and dye regeneration driving forces (ΔG0in and ΔG0reg) of triphenylamine based sensitizers calculated at B3LYP/cc-PVDZ level
| Dye |
Edyeox (eV) |
Edyeoxc (eV) |
ΔG0inj (eV) |
ΔG0reg (eV) |
| See ref. 48. See ref. 46. Values in parenthesis were calculated using experimental EH and EL Values. |
| A1 |
−5.16 |
−2.60 |
−1.40 |
0.36 |
| A1-F |
−5.08 |
−2.57 |
1.43 |
0.28 |
| C218 |
−4.92 (−4.99)a |
−2.43 (−2.86)a |
−1.57 (−1.26)c |
0.12 (0.19)c |
| C218-F |
−4.85 |
−2.39 |
−1.61 |
0.05 |
| D2 |
−4.99 (−5.15)b |
−2.91 (−3.20)b |
−1.09 (−1.08)c |
0.19 (0.35)c |
| D2-F |
−4.86 |
−2.81 |
−1.19 |
0.06 |
| Y123 |
−4.87 (−5.08)a |
−2.36 (−2.91)a |
−1.64 (−1.20)c |
0.07 (0.28)c |
| Y123-F |
−4.81 |
−2.34 |
−1.66 |
0.01 |
| Y1234 |
−4.88 |
−2.82 |
−1.18 |
0.08 |
| Y1234-F |
−4.83 |
−2.77 |
−1.23 |
0.03 |
3.2. Dye adsorption on (TiO2)28 substrate
The charge transfer kinetics depends largely on the configuration of the dye adsorbed on the semiconductor surface and the interface energy alignment. In order to understand the electron injection and charge recombination process, we have investigated the electronic and optical properties of the dye adsorbed on the (TiO2)28 substrate using the DFT method. Generally, the strength of the interaction between dye and the TiO2 surface depends on the adsorption energy of the dye/TiO2 system. In DSSCs, the electron-transfer rate (kET) also depends on the strength of the adsorption energy of the dye/TiO2 system, i.e., the larger the adsorption energy, the stronger the electronic coupling strength between the anchoring group of the dye and the TiO2 surface, and consequently the larger the kET value is. In order to calculate the adsorption energies, we first created the 84-atom titania nanoparticle (TiO2)28 by exposing the (101) surface of the anatase titania and then selected different initial adsorption configurations of the ten dyes onto the substrate and optimized each binding structure of them. The optimized structures of the lowest energy structures were presented in Fig. 8, in which the dyes bind onto the surface through their carboxyl group. In the initial configuration of the dye/(TiO2)28 system, the hydrogen of the –COOH anchoring group stays on the dye, but after optimization it was found to transfer to the substrate predominantly in a protonated state. As a result, the sensitizers bind almost perpendicular to the (TiO2)28 surface via bidentate bridging with the formation of two interface O–Ti bonds. Previous studies have shown that the dye links onto TiO2 surface through a bridging bidentate coordination mode which is more stable and preferable79,80 because the bidentate binding is shorter than the other binding modes and hence an increase in the rate of electron injection could be expected. The calculated bond distances between the Ti atom and the O atom of dyes are in the range 2.166–2.252 Å. The calculated adsorption energies of the most stable bidentate adsorption configurations were presented in Fig. 8. The results indicate that the adsorption energy of the known reference dyes increases with elongation of the π-system with O-FP and it decreases upon inserting the BT group between the TPA and CPDT moieties. Among all the investigated dyes, Y123-F has shown the largest adsorption energy, indicating a strong interaction between the dye and the TiO2 surface. These results clearly indicated that the binding strength between the sensitizer and TiO2 substrate can be improved by elongating the π-system with the O-FP group between the triphenylamine unit and the condensed thiophene system. As a result the electron-transfer rate will increase significantly and lead to an enhanced photovoltaic performance.
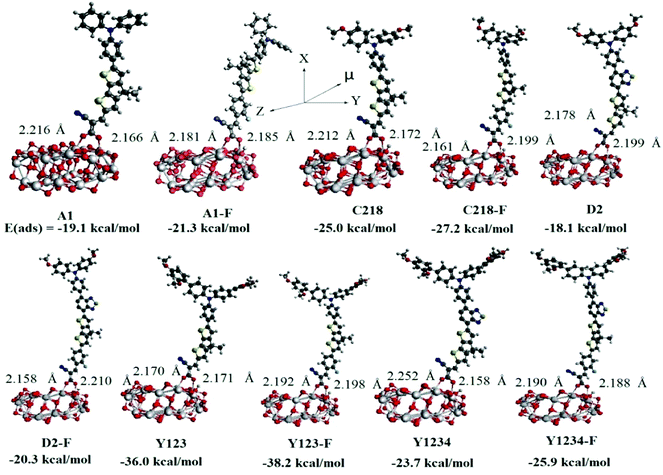 |
| | Fig. 8 Optimized bidentate bridging mode and adsorption energy of the ten triarylamine dyes on the (TiO2)28 cluster calculated by PBE/DNP on DMol3 program. | |
3.2.1. Optical properties of the dyes adsorbed on the (TiO2)28 substrate. The electronic absorption spectra of the dyes on TiO2 substrate were investigated using the CAM-B3LYP method with 3-21G* basis set to determine the relationship between the solar energy conversion efficiency and spectroscopic properties. The HOMO and LUMO energy levels of the (TiO2)28 surface in acetonitrile were −8.24 and −2.87 eV, respectively, leading to a HOMO–LUMO band gap of 5.37 eV. Even though the calculated results overestimated the energy levels and band gap, the S0 → S1 optical gap was calculated to be 3.33 eV, which is only slightly larger than the experimental band gap of ∼3.20 eV for the nano-sized (15−20 nm) TiO2,81 supporting the validity of the current theoretical approach based on the (TiO2)28 cluster. The calculated results also showed that energy gap of the TiO2 decreases upon dye adsorption. When the dye inserts the occupied orbitals within the HOMO–LUMO gap of the TiO2 substrate, the energy gap of dye/TiO2 complex decreases below the TiO2 energy gap (5.37 eV). The energy level alignment of the dyes on the TiO2 substrate was presented in Fig. 9. For dye/TiO2 complexes, the orbitals near the HOMO and LUMO are free of the combining states and have no mixing MO character. The HOMO of the complexes nearly have the characteristics of dye molecules, while the LUMO of the complexes have the same characteristics as the TiO2 substrate, which was manifested by the charge densities of the frontier MOs shown in Fig. 10.
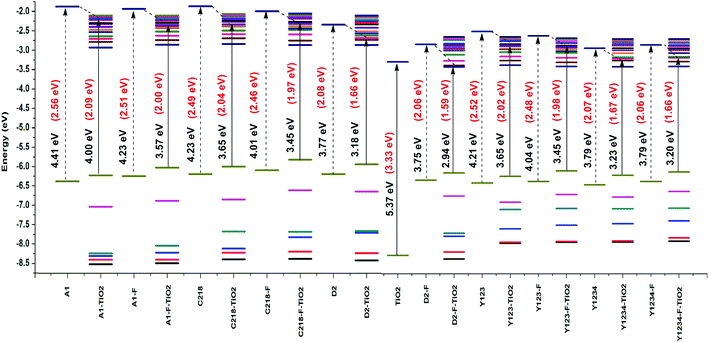 |
| | Fig. 9 Energy level alignment of isolated dyes and dye–TiO2 complexes calculated at the CAM-B3LYP/3-21G(d) level. | |
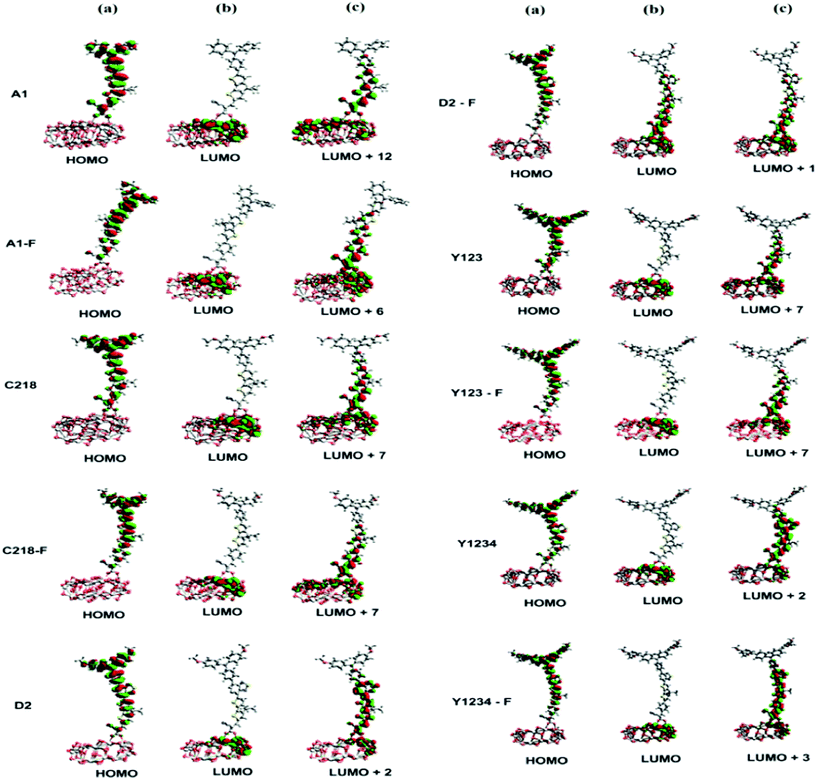 |
| | Fig. 10 (a) HOMO (b) LUMO and (c) interacting orbital isosurfaces of ten triarylamine derivatives on (TiO2)28 nanoparticle. The isovalue is 0.002 a.u. | |
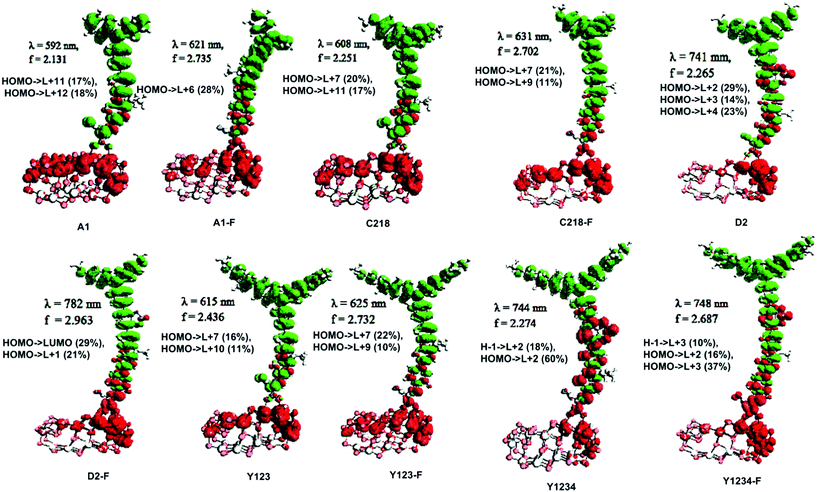
The absorption spectra of A1 and A1-F on a mesoporous TiO2 thin film show that there is a slight blue shift as well as a broadening of the absorption peak relative to the isolated dye. Fig. 11 showed the calculated absorption spectrum for the dye/TiO2 complexes in acetonitrile solution. The electronic spectrum of the dye on the (TiO2)28 surface calculated with CAM-B3LYP/3-21G(d) is overestimated in comparison with the experimental results. For example, the calculated absorption spectrum of A1-TiO2/A1-F-TiO2 complex has an absorption band extending from 400–1000 nm with the peak at 592/621 nm, whereas experimental band is at ∼460 nm. The calculated band holds several excitations with the most significant transitions (highest oscillator strengths) being listed in Table 6. All of these transitions take place from the HOMO of the complex which is typically localized on the dye and keeps the HOMO character of the isolated dye (Fig. 10). The unoccupied states involved in the transition from LUMO to LUMO + 12 in both A1-TiO2 and A1-F-TiO2. In these excitations, LUMO + 12 in A1-TiO2 and LUMO + 6 in A1-F-TiO2 show significant involvement and have the major contribution to λmax. Fig. 10 is an isosurface plot of LUMO + 12/LUMO + 6 pertaining to A1-TiO2/A1-F-TiO2, showing contributions from both the dye and the titanium dioxide. All unoccupied orbitals below LUMO + 11 in A1-TiO2 and LUMO + 6 in A1-F-TiO2 show contributions only from the surface without any dye character. On the other hand, the three highest occupied MOs (HOMO to HOMO − 2) in both the complexes show localization only on the dye. This was confirmed by visualization of these MOs with Avogadro software.
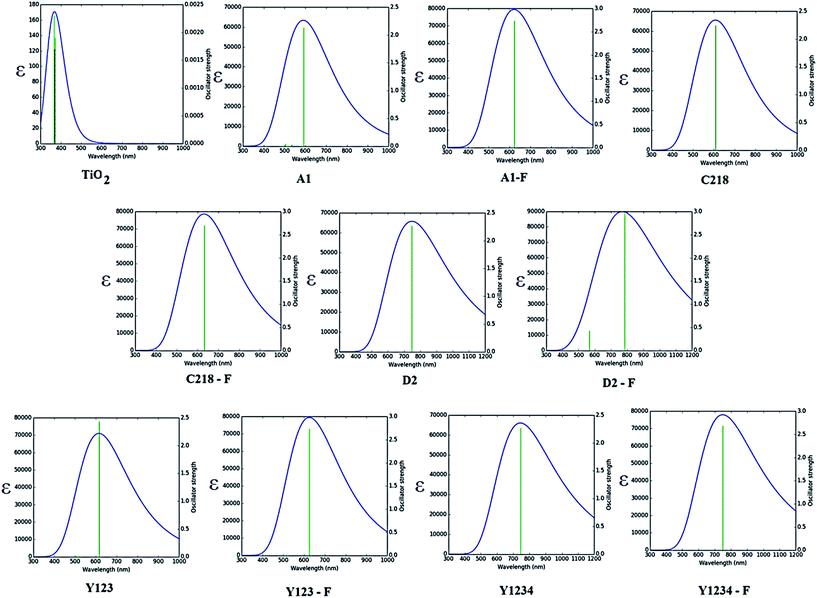 |
| | Fig. 11 Calculated absorption spectra of dye/(TiO2)28 systems. | |
Table 6 Calculated electronic properties of isolated dyes and dye–TiO2 complexes at CAM-B3LYP/3-21G(d) level
| Dye |
Isolated dye |
Dye on (TiO2)28 substrate |
| EH (eV) |
EL (eV) |
Eg (eV) |
ρ (D) |
EH (eV) |
EL (eV) |
Eg (eV) |
ρ (D) |
λabsmax (nm) |
EAo (eV) |
ε (M−1 cm−1) |
f |
LHE |
t (µm) |
JexpSC (mA cm−2) |
JcalSC (mA cm−2) |
Γℏd (eV) |
τd (fs) |
Electronic transition |
| See ref. 44. See ref. 48. See ref. 46. These results are obtained for the dye–TiO2 complexes with the B3LYP/3-21g(d) level. |
| A1 |
−6.43 |
−2.02 |
4.41 |
11.00 |
−6.28 |
−3.05 |
3.23 |
38.55 |
592 |
2.09 |
63399 |
2.13 |
0.992 |
1.6 |
6.37a |
5.26 |
0.295 |
2.23 |
H → L + 12 (18%), H → L + 11 (17%) |
| A1-F |
−6.30 |
−2.07 |
4.23 |
13.94 |
−6.08 |
−2.98 |
3.10 |
38.37 |
621 |
2.00 |
79440 |
2.73 |
0.998 |
1.6 |
7.52a |
6.82 |
0.305 |
2.15 |
H → L + 6 (28%) |
| C218 |
−6.24 |
−2.02 |
4.23 |
13.96 |
−6.05 |
−2.96 |
3.09 |
39.29 |
608 |
2.04 |
65524 |
2.25 |
0.994 |
4.0 |
14.8b |
12.77 |
0.306 |
2.15 |
H → >L + 7 (20%), H → L + 11 (17%) |
| C218-F |
−6.15 |
−2.14 |
4.01 |
15.16 |
−5.88 |
−2.99 |
2.89 |
42.45 |
631 |
1.97 |
78567 |
2.70 |
0.998 |
4.0 |
— |
15.45 |
0.310 |
2.12 |
H → L + 7 (21%), H → L + 9 (11%) |
| D2 |
−6.25 |
−2.48 |
3.77 |
13.64 |
−6.00 |
−2.99 |
3.01 |
39.90 |
747 |
1.66 |
65744 |
2.26 |
0.994 |
4.0 |
17.0c |
15.74 |
0.300 |
2.19 |
H → L + 2 (29%), H → L + 3 (14%), H → L + 4 (23%) |
| D2-F |
−6.16 |
−2.38 |
3.77 |
15.01 |
−5.95 |
−3.01 |
2.94 |
48.89 |
782 |
1.59 |
89751 |
2.96 |
0.998 |
4.0 |
— |
21.35 |
0.296 |
2.22 |
H → L (29%), H → L + 1 (21%) |
| Y123 |
−6.23 |
−2.03 |
4.21 |
14.27 |
−6.04 |
−2.96 |
3.08 |
42.56 |
615 |
2.02 |
71001 |
2.43 |
0.996 |
4.0 |
14.1b |
13.83 |
0.303 |
2.17 |
H → L + 7 (16%), H → L + 10 (11%) |
| Y123-F |
−6.19 |
−2.15 |
4.04 |
15.34 |
−5.90 |
−3.00 |
2.90 |
48.98 |
625 |
1.98 |
79400 |
2.73 |
0.998 |
4.0 |
— |
15.44 |
0.309 |
2.12 |
H → L + 7 (22%), H → L + 9 (10%) |
| Y1234 |
−6.28 |
−2.49 |
3.79 |
14.54 |
−6.02 |
−3.01 |
3.01 |
43.84 |
744 |
1.67 |
66042 |
2.27 |
0.994 |
4.0 |
— |
15.73 |
0.303 |
2.17 |
H − 1 → L + 2 (18%), H → >L + 2 (60%) |
| Y1234-F |
−6.19 |
−2.40 |
3.79 |
15.16 |
−5.92 |
−3.00 |
2.92 |
49.15 |
748 |
1.66 |
77931 |
2.68 |
0.997 |
4.0 |
— |
18.19 |
0.289 |
2.27 |
H − 1 → L + 3 (10%), H → L + 2 (16%), H → >L + 3 (37%) |
An experimental study performed by Yella et al.48 for C218 and Y123 on a TiO2 film showed a blue shift as well as broadening of the absorption peak compared to the isolated dye. The calculated absorption spectra of C218-TiO2 and Y123-TiO2 have shown only a major excitation for λmax at 608 and 615 nm, respectively. This excitation corresponds to the S0 → S1 transition and shows the contribution from HOMO → LUMO + 7 and HOMO → LUMO + 11 for C218-TiO2 and HOMO → LUMO + 7 and HOMO → LUMO + 10 for Y123-TiO2 (Table 6). However, LUMO + 7 in both C218-TiO2 and Y123-TiO2 show largest contribution in the excitation and delocalizes over the linker and acceptor moieties of the dye as well as on the TiO2 (Fig. 10). The unoccupied orbitals below LUMO + 7 in both C218-TiO2 and Y123-TiO2 localize at the TiO2 nanoparticle only. Similar to the A1-TiO2 complex, HOMO to HOMO – 2 in both the complexes shows only dye character. This is also indicated in Fig. 10. In the case of A1-TiO2, the optical gap (Eopt) between HOMO and LUMO + 12 is 2.09 eV (Fig. 9). For C218-TiO2 and Y123-TiO2 complexes, the optical gap between the MOs involved in the highest intensity transition is 2.04 eV and 2.02 eV, respectively. On the other hand, the hypothetical C218-F-TiO2 and Y123-F-TiO2 complexes have shown maximum absorption at 631 and 635 nm, respectively. This excitation corresponds to S0 → S1 and has a maximum contribution from HOMO → LUMO + 7 for each of the complexes. The MOs show that the HOMO exhibits the HOMO character of the dye and that LUMO + 7 shows considerable involvement of both dye and TiO2. The absorbance spectrum of C218-TiO2/Y123-TiO2 shows a blue shift compared to C218-F-TiO2/Y123-F-TiO2.
Recently, Wang et al.46 reported that the absorption peak for D2 adsorbed onto 4.8 µm thick TiO2 film is blue-shifted to 536 nm. In fact, D2 had shown a hypsochromic shift by 20 nm in the absorption spectrum. The computed absorption band of the D2-TiO2 complex expands from ∼400 to ∼1200 nm with a maximum at 741 nm. This excitation corresponds to S0 → S2 transition with high oscillator strength and shows a contribution from HOMO → LUMO + 7. The LUMO + 7 is an admixture of π* orbitals of the dye and the empty states derived mainly from d-orbitals of TiO2. Here, LUMO + 7 is delocalized over the entire BT, CPDT, cyanoacrylic acid anchor and continues over to the TiO2 substrate attached to the anchor. The delocalization of the LUMO demonstrates a strong coupling between the dye and the oxide surface. Fig. 9 showed that the optical gap between the HOMO and LUMO + 7 is 1.66 eV. All unoccupied states below the LUMO + 7 are localized on the TiO2 substrate. Whereas, hypothetical D2-F-TiO2 complex showed a maximum absorption band at 782 nm red-shifted by 20 nm as compared to that of the D2-TiO2 complex. In this case, the S0 → S2 transition has the highest contribution from HOMO → LUMO. The LUMO of the complex shows resemblance to the unoccupied orbital of the isolated dye with sufficient surface contribution from TiO2 (Fig. 10). The top four occupied MOs in the D2-F-TiO2 complex are derived mostly from the dye. HOMO and LUMO are located at −5.95 eV and −3.01 eV respectively with Eg (Eopt) of 2.96 eV (1.59 eV). Fig. 11 shows another low intensity absorption peak at 565 nm for D2-F-TiO2. This transition originates from the three highest occupied MOs (HOMO to HOMO − 2). Several unoccupied MOs contribute significantly to this transition. With the exception of the LUMO, the other unoccupied orbitals involved in the transition are localized on the TiO2 substrate (Fig. 10).
The computed Y1234-TiO2 and Y1234-F-TiO2 complexes have shown maximum absorption around 744 and 748 nm, respectively. The main contribution to this excitation for Y1234-TiO2 (Y1234-F-TiO2) is HOMO → LUMO + 2 (HOMO → LUMO + 3). The isosurface plot of LUMO + 2 (LUMO + 3) shows the contribution is from both the dye and the TiO2. For the Y1234-TiO2 (Y1234-F-TiO2) complex, the Eopt gap between the MOs involved in the highest intensity transition is 1.67 eV (1.66 eV). The excited state calculations for Y1234-TiO2 and Y1234-F-TiO2 show a red shift in the absorption spectrum as compared with the Y123-TiO2 complex.
In 2008, Jose et al.82 indicated that the short circuit current density (JSC) is directly related to the probability density at the anchor moiety. This observation was reinforced by Angelis et al.83 who exposed an improved photocurrent with an increase in electron coupling between dye and the TiO2 surface. Furthermore, the placement of the excited states in the conduction band of the complexes as well as their isosurface plots can throw light on the mechanism involved in the electron injection from excited dye to TiO2. Therefore, in order to understand the charge transfer character of the complexes we have presented the electron density difference maps of the main excitation transition in Fig. 12. From Fig. 12, we can observe that upon insertion of the fluorophenylene and benzothiadazole groups in the triarylamine derivatives, the negative charge transfer from dye to TiO2 substrate increases and consequently enhances the JSC.
 |
| | Fig. 12 Electron density difference maps of investigated dyes on (TiO2)28 nanoparticle. The contour threshold for isosurfaces is 0.0002 a.u. The green and red colors indicate a decrease and increase of charge density, respectively. | |
3.2.2. Factors influencing the photovoltaic performance. Eqn (2) clearly indicates that ηIPCE is directly proportional to JSC, so in order to enhance the JSC, the LHE must be as large as possible. Therefore we calculated LHE of ten dye-titanium complexes at the CAM-B3LYP/3-21G(d) level of theory and the results were presented in Table 6. The results showed that the LHE of the dye is enhanced by increasing the strength of the donor moiety and elongating the conjugated system. The LHE of the hypothetical dyes are much better than Y123-TiO2. As a result, the photocurrent response for the model dyes is predicted to be larger than that of the reference dye. In order to check this, we calculated the JSC value using eqn (4) by considering approximate parameters K ≈ 104 M−1, C ≈ 10−6 M and Γmax ≈ 5 × 10−8 mol cm−2 for a 6 µm thick TiO2 film.56 The JSC value obtained with DFT method are well comparable with the experimental results. From Table 6, we can observe that JSC values for dyes C218-TiO2, D2-TiO2 and Y123-TiO2 are larger than the complex A1-TiO2, whereas for hypothetical systems C218-F-TiO2, D2-F-TiO2, Y123-F-TiO2, Y1234-TiO2 and Y1234-F-TiO2, JSC values are larger than the reference dye Y123. As a result, the IPCE of the model dyes is greater than the reference system Y123-TiO2.In DSSCs, the VOC is another important factor which effect the efficiency of the cell and is defined as the potential difference between the electrolyte redox potential and the quasi-Fermi level of electrons in the semiconductor TiO2; the VOC can be calculated with the empirical formula given below84
| |
 | (14) |
where
NCB is the density of accessible states in the conduction band;
nc is the number of injected electrons in TiO
2 due to dye adsorption;
q is the charge of electron and ΔCB is the shift of
ECB when dyes are adsorbed on TiO
2 and can be expressed as
84| |
 | (15) |
In this expression, µnormal denotes the dipole moment of individual dye molecules perpendicular to the surface of the TiO2, q is the charge of electron and γ is the surface concentration of the dye. ε0 and ε represent the vacuum permittivity and dielectric permittivity, respectively.
From the above equations it is clear that VOC is strongly influenced by the structural parameters of the dye. Because the dye adsorbed on the TiO2 substrate induces the change in dipole moment and charge distribution, as a result, the conduction band edge of the TiO2 substrate shifts upwards. Therefore we compared the important structural parameters of the dyes before and after the adsorption on the TiO2 surface calculated at the same level of theory (GGA-PBE/DNP). The important structural parameters of the dyes were presented in the ESI (Table S2†). From Table S2,† it can be observed that there are appreciable changes in the structural parameters of the dyes before and after adsorption on TiO2 substrate, which leads to large changes in the electronic properties of dye–TiO2 complexes. Normally, the dyes with a dipole pointing toward the TiO2 surface plane cause a decrease of the VOC, whereas the opposite is true for a dipole pointing away from the surface. In order to obtain higher VOC, the dipole moment of the dye–TiO2 complexes must be as large as possible and point away from the surface. The calculated results indicated that the dyes on the TiO2 surface adsorb almost perpendicularly with their total dipole moments forming an angle of roughly 45° to the TiO2 surface. The dipole moment of dye–TiO2 complexes increases by up to 3 times (Table 6) when compared with an isolated dye, which is a prerequisite for enhancing charge separation between the dye and the TiO2 substrate. The dipole moment of complexes A1, A1-F, C218 and D2 all fall beneath that of Y123, whereas for hypothetical complexes (C218-F, D2-F, Y123-F, Y1234 and Y1234-F) the dipole moments are larger than the reference dye complex. This indicates that the CB shift of the hypothetical dye complexes will be larger than the Y123 complex. However, from Fig. 9 we noticed that the CB shift of the hypothetical dye–complexes (except C218-F) is comparable to Y123 complex, whereas, for A1, A1-F, C218, and D2 the CB shifts are larger than Y123. Consequently, the VOC of A1, A1-F, C218, and D2 complexes will be larger than Y123. But it is in contradiction with the experimentally observed VOC.21 Therefore, on the basis of dipole moment we predict that the VOC of C218-F, D2-F, Y123-F, Y1234 and Y1234-F will be larger and A1, A1-F, C218, and D2 complexes are lower than Y123 complex. These results indicate that hypothetical dyes (C218-F, D2-F, Y123-F, Y1234 and Y1234-F) offer potential for DSSCs applications.
Another factor that influences the photovoltaic performance is the excited state lifetime of the dyes. The electron lifetime must be as large as possible,85 i.e. to realize long-term stability, the dyes must remain stable in the cationic form for a long time. Jiang et al.86 reported that the remarkable performance of the triarylamine dye D35 is due to its stable excited state. In contrast, the longest electron lifetime with triarylamine dyes was ascribed to the blocking effect by steric hindrance of the nonplanar structure of the donor. Generally, in DSSCs, the excited state decay of the dye is dominated by the nonradiative process.87 Therefore to calculate the nonradiative electron injection time (τ) between the sensitizer and the semiconductor surface, we employed the Newns–Anderson model88,89 in the formulation used by Persson et al.90,91 The complete procedure for obtaining nonradiative electron lifetimes of the sensitizers were given in ESI1.† The calculated results were presented in Table 6. The calculated results highlighted that the nonradiative electron life times of the C218, D2 and Y123 dyes will decrease and increase, respectively with the inclusion of O-FP and BT groups. The calculated results showed that the electron lifetime for hypothetical dyes containing both BT and O-FP moieties (D2-F, Y1234, and Y1234-F) is much larger than that of the reference dye Y123. As a result, the hypothetical dyes will retard the charge recombination process and enhance the efficiency of the DSSCs.
3.2.3. Level alignment quality (η). A no-loss DSSC level alignment quality is another important factor which can be used to screen the organic sensitizers for DSSC applications. Recently, Ørnsø et al.92 investigated the efficiency of a functionalized zinc porphyrin using level alignment quality. Level alignment quality is defined as| |
 | (16) |
where
here Ec − EH is the distance from the HOMO level to the conduction band, E1 is the optical gap of the dye, Θ(E − E1) is a step function representing the absorption of the dye molecules, JSC(E) is the short-circuit current density and  . In order to calculate the level alignment quality, we adopted two different models for open circuit voltage i.e., VOC = Ec − EH and VOC = 1 V.92 The calculated results presented in Table 7 showed that the η values of A1 and C218 dyes are lower than the reference dye Y123. This indicates that the η value can be improved with increasing the strength of the donor group. It is also interesting to observe that the level alignment quality of A1 is enhanced by elongating the π-system with O-FP, leads to structure A1-F. However, when the spacer group in C218 is elongated with the BT group, producing dye D2, the η value decreases. The calculated results showed that η values for dyes elongated with O-FP (C218-F, Y123-F) or with both O-FP and BT (Y1234-F) are larger than the reference dye Y123. Whereas for the remaining dyes (A1, A1-F, C218, D2 and D2-F) the η values are smaller than the Y123. Based on the above results, we concluded that elongating the π-system in triarylamine dyes with both O-FP and BT offers more potential for DSSC applications.
. In order to calculate the level alignment quality, we adopted two different models for open circuit voltage i.e., VOC = Ec − EH and VOC = 1 V.92 The calculated results presented in Table 7 showed that the η values of A1 and C218 dyes are lower than the reference dye Y123. This indicates that the η value can be improved with increasing the strength of the donor group. It is also interesting to observe that the level alignment quality of A1 is enhanced by elongating the π-system with O-FP, leads to structure A1-F. However, when the spacer group in C218 is elongated with the BT group, producing dye D2, the η value decreases. The calculated results showed that η values for dyes elongated with O-FP (C218-F, Y123-F) or with both O-FP and BT (Y1234-F) are larger than the reference dye Y123. Whereas for the remaining dyes (A1, A1-F, C218, D2 and D2-F) the η values are smaller than the Y123. Based on the above results, we concluded that elongating the π-system in triarylamine dyes with both O-FP and BT offers more potential for DSSC applications.
Table 7 Calculated Ec − EH, E1 and η values for ten triarylamine dyes
| Dye |
Ec − EH (eV) |
E1a (eV) |
ηconb |
ηdync |
ηexp |
| Optical band gap calculate by E1 = 1240 eV/λ, where λ absorption maximum of dye–TiO2 complex calculated at the CAM-B3LYP/3-21G(d) level. VOC = 1 V. VOC = Ec − EH. See ref. 44. See ref. 48. See ref. 46. |
| A1 |
4.00 |
2.09 |
0.11 |
0.21 |
0.036d |
| A1-F |
3.57 |
2.00 |
0.14 |
0.24 |
0.048d |
| C218 |
3.65 |
2.04 |
0.26 |
0.47 |
0.086e |
| C218-F |
3.45 |
1.97 |
0.30 |
0.53 |
— |
| D2 |
3.18 |
1.66 |
0.26 |
0.50 |
0.090f |
| D2-F |
2.94 |
1.59 |
0.34 |
0.63 |
— |
| Y123 |
3.65 |
2.02 |
0.28 |
0.50 |
0.098e |
| Y123-F |
3.45 |
1.98 |
0.31 |
0.53 |
— |
| Y1234 |
3.23 |
1.67 |
0.26 |
0.51 |
— |
| Y1234-F |
3.20 |
1.66 |
0.30 |
0.58 |
— |
4. Conclusions
A first-principles study has been carried out to gain insight into the effect of π-elongation on the structural and electrical properties of ten triarylamine dye molecules adsorbed on the (TiO2)28 anatase (101) surface. The geometry optimization of the isolated molecules in the gas phase showed that the central NCCC moiety is significantly influenced by the elongation of the π-system. As a result, the three phenyl rings of the triarylamine unit are twisted symmetrically from the central NCCC plane by 34–49°. The dihedral angles are affected not only by the π-elongation, but also by the substituent (alkoxy or bis(hexyloxy)benzene) at the para-position of the phenyl rings of the triphenylamine group. The energy gap between HOMO and LUMO energy levels decreases further by elongating the π-system with either BT or O-FP units or both. The absorption spectra of the isolated molecules containing either BT or O-FP units or both in the π-system show pronounced red shifts of the maximum absorption band with enhanced molecular extinction coefficient compared to reference dye Y123. The simulated absorption spectra of the ten dyes showed intense bands in the range 250–390 nm and 400–600 nm, which are ascribed to the localized aromatic π → π* transitions and the charge transfer π → π* transition, respectively. The calculated results also showed that the excited state life-times and reorganization energies of the investigated dyes are larger and lower, respectively, than the reference dye. The dye/TiO2 results indicate that the adsorption energy of the hypothetical dyes increases with elongating the π-system with O-FP group but decreases with inserting BT group between TPA and CPDT moieties. However, the absorption spectra of dye/TiO2 complexes show pronounced red shifts of the absorption maxima with large molecular extinction coefficients after the elongation of π-system. Due to elongation of the π-system, the optical gap and light harvesting efficiency of the dye/TiO2 complexes decreases and increases, respectively, as compared to reference Y123. The short circuit current density and level alignment quality factors of the hypothetical systems are larger than those of Y123. Finally, we concluded that elongating the π-conjugated spacer group in triarylamine dyes with BT and O-FP groups offers a potential approach for optimizing DSSC performance. The elongation of the π-system using carefully placed spacer groups guarantees the dyes with larger molecular excitation coefficient, electron lifetime, LHE, JSC, level alignment quality and smaller electron reorganization energy and energy gap than the reference dye. Thus, to summarize, the elongation of the π-system in a series of hypothetical triphenylamine dyes by the judicious placement either a fluorophenyl or benzothiadiazole group, or a combination of both groups, results in an improved light harvesting efficiency in DSSC devices.
Acknowledgements
The authors thank the National Science Foundation of China for supporting this work under Grant no. 21372116.
References
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- M. A. Muccini, Nat. Mater., 2006, 5, 605–613 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J. L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- J. Gong, J. Liang and K. Sumathy, Renewable Sustainable Energy Rev., 2012, 16, 5848–5860 CrossRef CAS PubMed.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed.
- C. Y. Chen, M. Wang, J. Y. Li, N. Pootrakulchote, L. Alibabaei, C. H. Ngoc-le, J. D. Decoppet, J. H. Tsai, C. Grätzel, C. G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS PubMed.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, J. Phys. Chem. B, 2003, 107, 597–606 CrossRef CAS.
- Z. S. Wang, Y. Cui, K. Hara, Y. Dan-oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138–1141 CrossRef CAS.
- Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224–7230 CAS.
- K. Sayama, K. Hara, N. Mori, M. Satsuki, S. Suga, S. Tsukagoshi, Y. Abe, H. Sugihara and H. Arakawa, Chem. Commun., 2000, 1173–1174 RSC.
- K. Sayama, S. Tsukagoshi, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, J. Phys. Chem. B, 2002, 106, 1363–1371 CrossRef CAS.
- X. Ma, J. Hua, W. Wu, Y. Jin, F. Meng, W. Zhan and H. Tian, Tetrahedron, 2008, 64, 345–350 CrossRef CAS PubMed.
- W.-H. Zhan, W.-J. Wu, J.-L. Hua, Y.-H. Jing, F.-S. Meng and H. Tian, Tetrahedron Lett., 2007, 48, 2461–2465 CrossRef CAS PubMed.
- T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218–12219 CrossRef CAS PubMed.
- L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Gratzel, Adv. Mater., 2005, 17, 813–815 CrossRef CAS.
- Z.-S. Wang, F.-Y. Li and C.-H. Huang, J. Phys. Chem. B, 2001, 105, 9210–9217 CrossRef CAS.
- Z.-S. Wang, F.-Y. Li and C.-H. Huang, Chem. Commun., 2000, 2063–2064 RSC.
- Y.-S. Chen, C. Li, Z.-H. Zeng, W.-B. Wang, X.-S. Wang and B.-W. Zhang, J. Mater. Chem., 2005, 15, 1654–1661 RSC.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- S. Hwang, J. H. Lee, C. Park, H. Lee, C. Kim, C. Park, M.-H. Lee, W. Lee, J. Park, K. Kim, N.-G. Park and C. Kim, Chem. Commun., 2007, 4887–4889 RSC.
- Z. Ninga and H. Tian, Chem. Commun., 2009, 5483–5495 RSC.
- M. Liang, W. Xu, F. Cai, P. Chen, B. Peng, J. Chen and Z. Li, J. Phys. Chem. C, 2007, 111, 4465–4472 CAS.
- W. Xu, B. Peng, J. Chen, M. Liang and F. Cai, J. Phys. Chem. C, 2008, 112, 874–880 CAS.
- M.-S. Tsai, Y.-C. Hsu, J. T. Lin, H.-C. Chen and C.-P. Hsu, J. Phys. Chem. C, 2007, 111, 18785–18793 CAS.
- T. Kitamura, M. Ikeda, K. Shigaki, T. Inoue, N. A. Anderson, X. Ai, T. Lian and S. Yanagida, Chem. Mater., 2004, 16, 1806–1812 CrossRef CAS.
- Z. Ning, Q. Zhang, W. Wu, H. Pei, B. Liu and H. Tian, J. Org. Chem., 2008, 73, 3791–3797 CrossRef CAS PubMed.
- M. Velusamy, K. R. Justin Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Org. Lett., 2005, 7, 1899–1902 CrossRef CAS PubMed.
- C. Teng, X. Yang, C. Yang, S. Li, M. Cheng, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 9101–9110 CAS.
- S.-L. Li, K.-J. Jiang, K.-F. Shao and L.-M. Yang, Chem. Commun., 2006, 2792–2794 RSC.
- K. Hara, T. Sato, R. Katoh, A. Furube, T. Yoshihara, M. Murai, M. Kurashige, S. Ito, A. Shinpo, S. Suga and H. Arakawa, Adv. Funct. Mater., 2005, 15, 246–252 CrossRef CAS.
- S. Kim, J. K. Lee, S. O. Kang, J. Ko, J. H. Yum, S. Fantacci, F. De Angelis, D. Di Censo, M. K. Nazeeruddin and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 16701–16707 CrossRef CAS PubMed.
- I. Jung, J. K. Lee, K. H. Song, K. Song, S. O. Kang and J. Ko, J. Org. Chem., 2007, 72, 3652–3658 CrossRef CAS PubMed.
- H. Choi, J. K. Lee, K. Song, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 3115–3121 CrossRef CAS PubMed.
- H. Choi, C. Baik, S. O. Kang, J. Ko, M.-S. Kang, M. K. Nazeeruddin and M. Gratzel, Angew. Chem., Int. Ed., 2008, 47, 327–330 CrossRef CAS PubMed.
- H. Tian, X. Yang, R. Chen, Y. Pan, L. Li, A. Hagfeldt and L. Sun, Chem. Commun., 2007, 3741–3743 RSC.
- R. Chen, X. Yang, H. Tian, X. Wang, A. Hagfeldt and L. Sun, Chem. Mater., 2007, 19, 4007–4015 CrossRef CAS.
- R. Chen, X. Yang, H. Tian and L. Sun, J. Photochem. Photobiol., A, 2007, 189, 295–300 CrossRef CAS PubMed.
- N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256–14257 CrossRef CAS PubMed.
- D. Kim, J. K. Lee, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 1913–1922 CrossRef CAS PubMed.
- Y. Zhao and W. Z. Liang, Chem. Soc. Rev., 2012, 41, 1075–1087 RSC.
- M. Zhang, Y. Wang, M. Xu, W. Ma, R. Li and P. Wang, Energy Environ. Sci., 2013, 6, 2944 CAS.
- D. Y. Chen, Y. Y. Hsu, H. C. Hsu, B. S. Chen, Y. T. Lee, H. Fu, M. W. Chung, S. H. Liu, H. C. Chen, Y. Chi and P.-T. Chou, Chem. Commun., 2010, 46, 5256–5258 RSC.
- E. Gabrielsson, H. Ellis, S. Feldt, H. Tian, G. Boschloo, A. Hagfeldt and L. Sun, Adv. Energy Mater., 2013, 3, 1647–1656 CrossRef CAS.
- X. Wang, J. Yang, H. Yu, F. Li, L. Fan, W. Sun, Y. Liu, Z. Y. Koh, J. Pan, W. L. Yim, L. Yan and Q. Wang, Chem. Commun., 2014, 50, 3965–3968 RSC.
- J. Liu, J. Zhang, M. Xu, D. Zhou, X. Jing and P. Wang, Energy Environ. Sci., 2011, 4, 3021–3029 CAS.
- A. Yella, R. H. Baker, B. F. E. Curchod, N. A. Astani, J. Teuscher, L. E. Polander, S. Mathew, J. E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, M. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 2733–2739 CrossRef CAS.
- H. N. Tsao, C. Yi, T. Moehl, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Gratzel, ChemSusChem, 2011, 4, 59–594 CrossRef PubMed.
- S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J.-E. Moser, R. H. -Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291–1302 CrossRef CAS.
- J. Preat, D. Jacquemin and E. A. Perpète, Environ. Sci. Technol., 2010, 44, 5666–5671 CrossRef CAS PubMed.
- W. Fan, D. Tanab and W. Deng, Phys. Chem. Chem. Phys., 2011, 13, 16159–16167 RSC.
- H. B. Li, J. Zhang, Y. Wu, J. L. Jin, Y. A. Duan, Z.-M. Su and Y. Geng, Dyes Pigm., 2014, 108, 106–114 CrossRef CAS PubMed.
- J. Preat, C. Michaux, D. Jacquemin and E. A. Perpète, J. Phys. Chem. C, 2009, 113, 16821–16833 CAS.
- W. Fan, D. Tan and W. Q. Deng, ChemPhysChem, 2012, 13, 2051–2060 CrossRef CAS PubMed.
- J. Feng, Y. Jiao, W. Ma, M. K. Nazeeruddin, M. Gratzel and S. Meng, J. Phys. Chem. C, 2013, 117, 3772–3778 CAS.
- M. Pazoki, P. W. Lohse, N. Taghavinia, A. Hagfeldt and G. Boschloo, Phys. Chem. Chem. Phys., 2014, 16, 8503–8508 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- Y. Wang and H. Li, J. Chem. Phys., 2010, 133, 034108 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS PubMed.
- Y. Inada and H. Orita, J. Comput. Chem., 2008, 29, 225 CrossRef CAS PubMed.
- I. Reva, L. Lapinsky, N. Chattopadhyay and R. Fausto, Phys. Chem. Chem. Phys., 2003, 5, 3844–3850 RSC.
- I. Bruder, A. Ojala, C. Lennartz, S. Sundarraj, J. Schoneboom, R. Sens, J. Hwanga, P. Erk and J. Weis, Sol. Energy Mater. Sol. Cells, 2010, 94, 310–316 CrossRef CAS PubMed.
- J. I. Nishida, T. Masuko, Y. Cui, K. Hara, H. Shibuya, M. Ihara, T. Hosoyama, R. Goto, S. Mori and Y. Yamashita, J. Phys. Chem. C, 2010, 114, 17920–17925 CAS.
- B. Liu, B. Wang, R. Wang, L. Gao, S. Huo, Q. Liu, X. Lia and W. Zhu, J. Mater. Chem. A, 2014, 2, 804–812 CAS.
- M. G. Neumann, W. G. M. C. C. Schmitt, F. A. Rueggeberg and I. C. Correa, J. Dent., 2005, 33, 525–532 CrossRef CAS PubMed.
- A. D. Dwyer and D. J. Tozer, Phys. Chem. Chem. Phys., 2010, 12, 2816–2818 RSC.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- N. Kuritz, T. Stein, R. Baer and L. Kronik, J. Chem. Theory Comput., 2011, 7, 2408–2415 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- V. Vaissier, P. Barnes, J. Kirkpatrick and J. Nelson, Phys. Chem. Chem. Phys., 2013, 15, 4804–4814 RSC.
- J. B. Asbury, Y. Q. Wang, E. Hao, H. Ghosh and T. Lian, Res. Chem. Intermed., 2001, 27, 393–406 CrossRef CAS PubMed.
- R. Katoh, A. Furube, T. Yoshihara, K. Hara, G. Fujihashi, S. Takano, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 4818–4822 CrossRef CAS.
- A. Hagfeldt and M. Gratzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1789 CrossRef PubMed.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Grätzel, J. Phys. Chem. B, 2000, 104, 1300–1306 CrossRef CAS.
- M. K. Nazeeruddin, R. Humphry-Baker, P. Liska and M. Grätzel, J. Phys. Chem. B, 2003, 107, 8981–8987 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- R. Jose, A. Kumar, V. Thavasi, K. Fujihara, S. Uchida and S. Ramakrishna, Appl. Phys. Lett., 2008, 93, 23125–23133 CrossRef PubMed.
- F. De Angelis, S. Fantacci, A. Selloni, M. Gratzel and M. K. Nazeeruddin, Nano Lett., 2007, 7, 3189–3195 CrossRef CAS PubMed.
- W. L. Ding, D. M. Wang, Z. Y. Geng, X. L. Zhao and Y. F. Yan, J. Phys. Chem. C, 2013, 117, 17382–17398 CAS.
- T. N. Murakami, N. Koumura, M. Kimura and S. Mori, Langmuir, 2014, 30, 2274–2279 CrossRef CAS PubMed.
- X. Jiang, K. M. Karlsson, E. Gabrielsson, E. M. J. Johansson, M. Quintana, M. Karlsson, L. Sun, G. Boschloo and A. Hagfeldt, Adv. Funct. Mater., 2011, 21, 2944–2952 CrossRef CAS.
- B. E. Hardin, A. Sellinger, T. Moehl, R. H. Baker, J. E. Moser, P. Wang, S. M. Zakeeruddin, M. Gratzel and M. D. McGehee, J. Am. Chem. Soc., 2011, 133, 10662–10667 CrossRef CAS PubMed.
- J. P. Muscat and D. M. Newns, Prog. Surf. Sci., 1978, 9, 1–43 CrossRef CAS.
- C. Cohen-Tannoudji, B. Diu and F. Laloe, Quantum Mechanics, J.Wiley & Sons, Paris, 1977, vol. 2 Search PubMed.
- P. Persson, M. J. Lundqvist, R. Ernstorfer, W. A. Goddard and F. Willig, J. Chem. Theory Comput., 2006, 2, 441–451 CrossRef CAS.
- M. J. Lundqvist, M. Nilsing, S. Lunell, B. Åkermark and P. Persson, J. Phys. Chem. B, 2006, 110, 20513–20525 CrossRef CAS PubMed.
- K. B. Ørnsø, J. M. G. Lastraa and K. S. Thygesena, Phys. Chem. Chem. Phys., 2013, 15, 19478–19486 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Contains the computed vertical excited singlet states, oscillator strengths of ten dyes in acetonitrile solvent and PDOS of dyes calculated at the CAM-B3LYP/DGDZVP level of theory. The important structural parameters of the dyes before and after the adsorption on TiO2 surface calculated at the GGA-PBE/DNP level of theory using Dmol3. See DOI: 10.1039/c4ra09914a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 

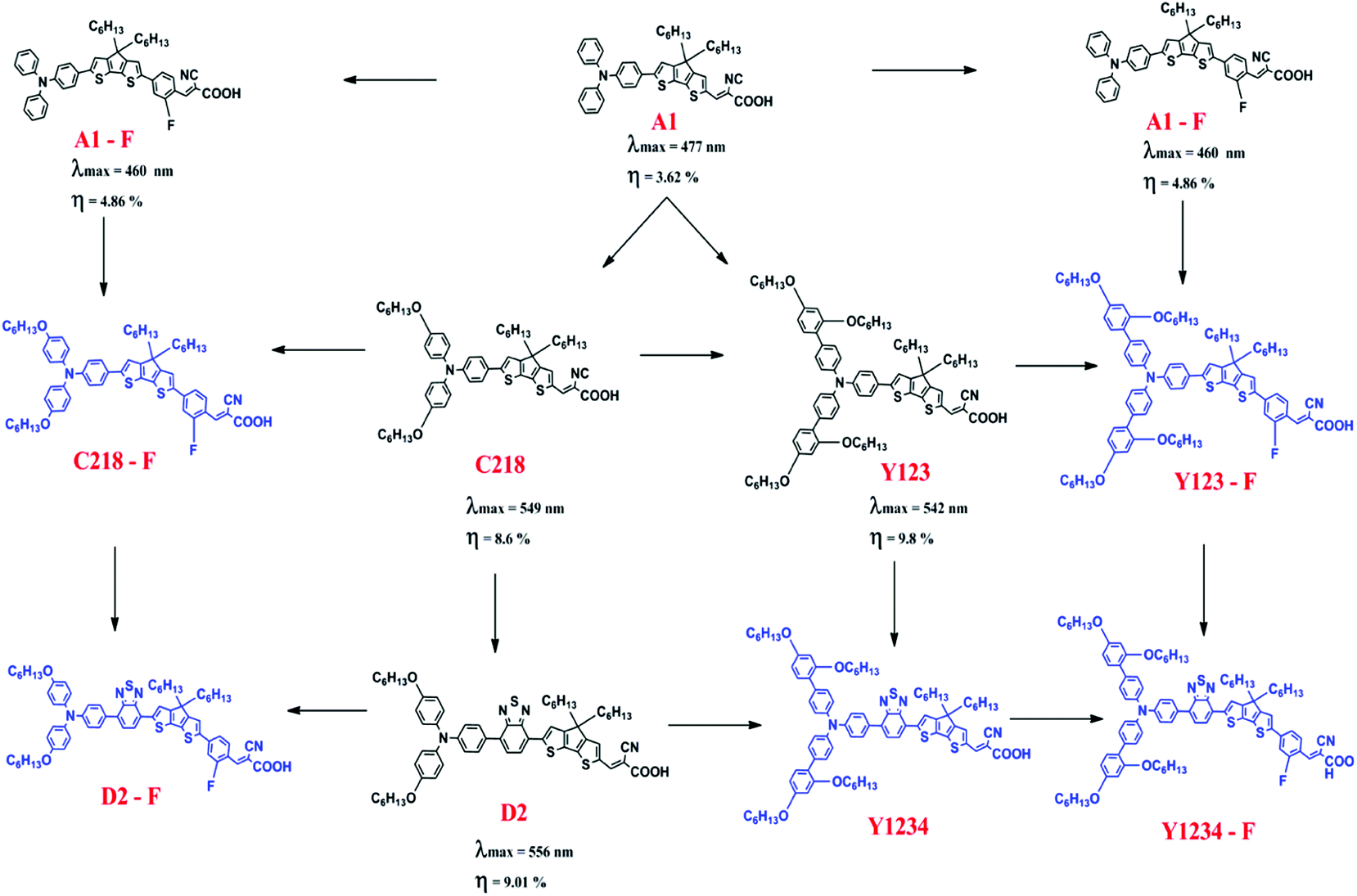




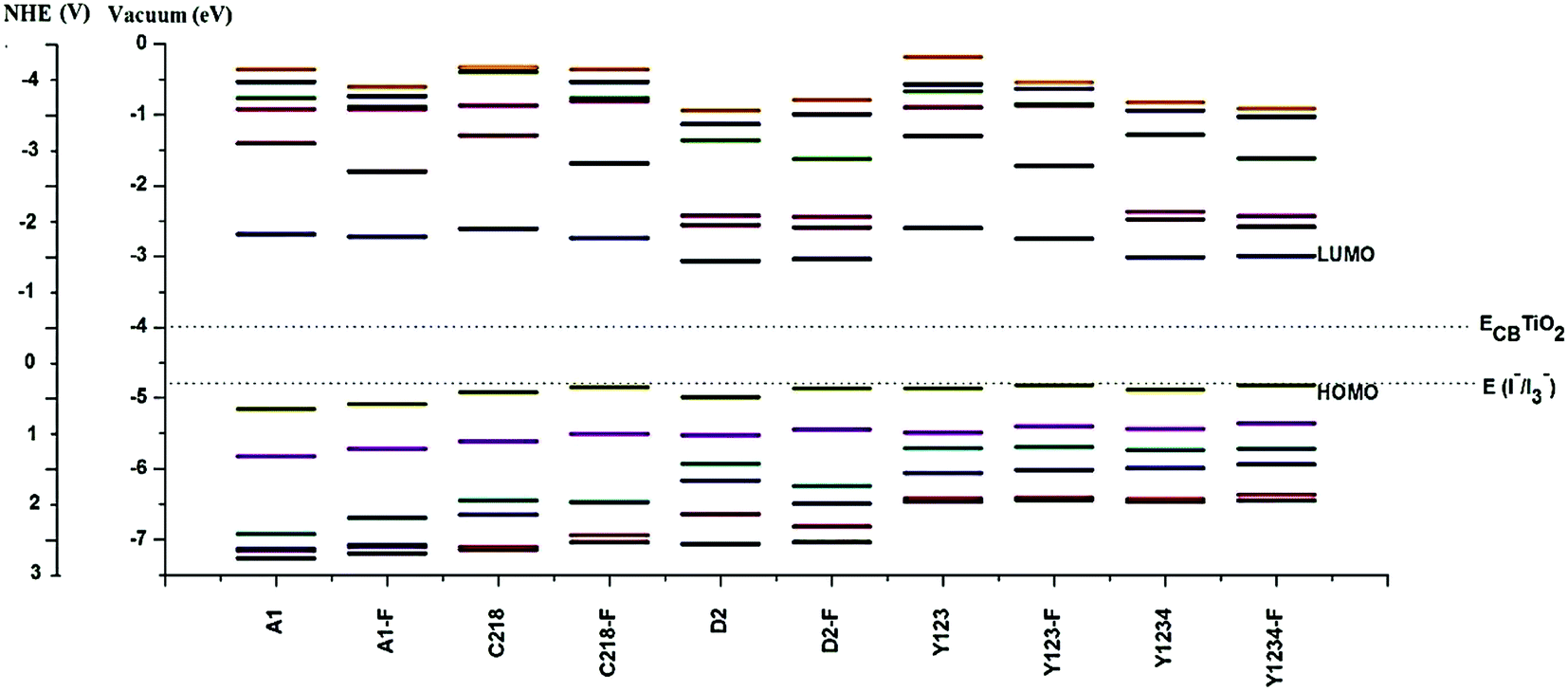
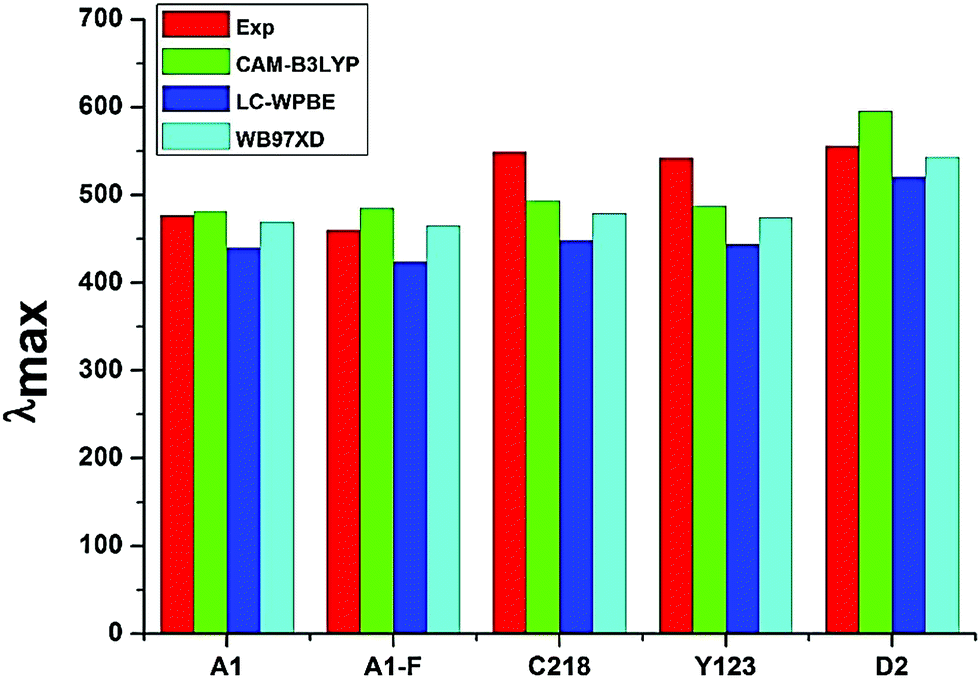
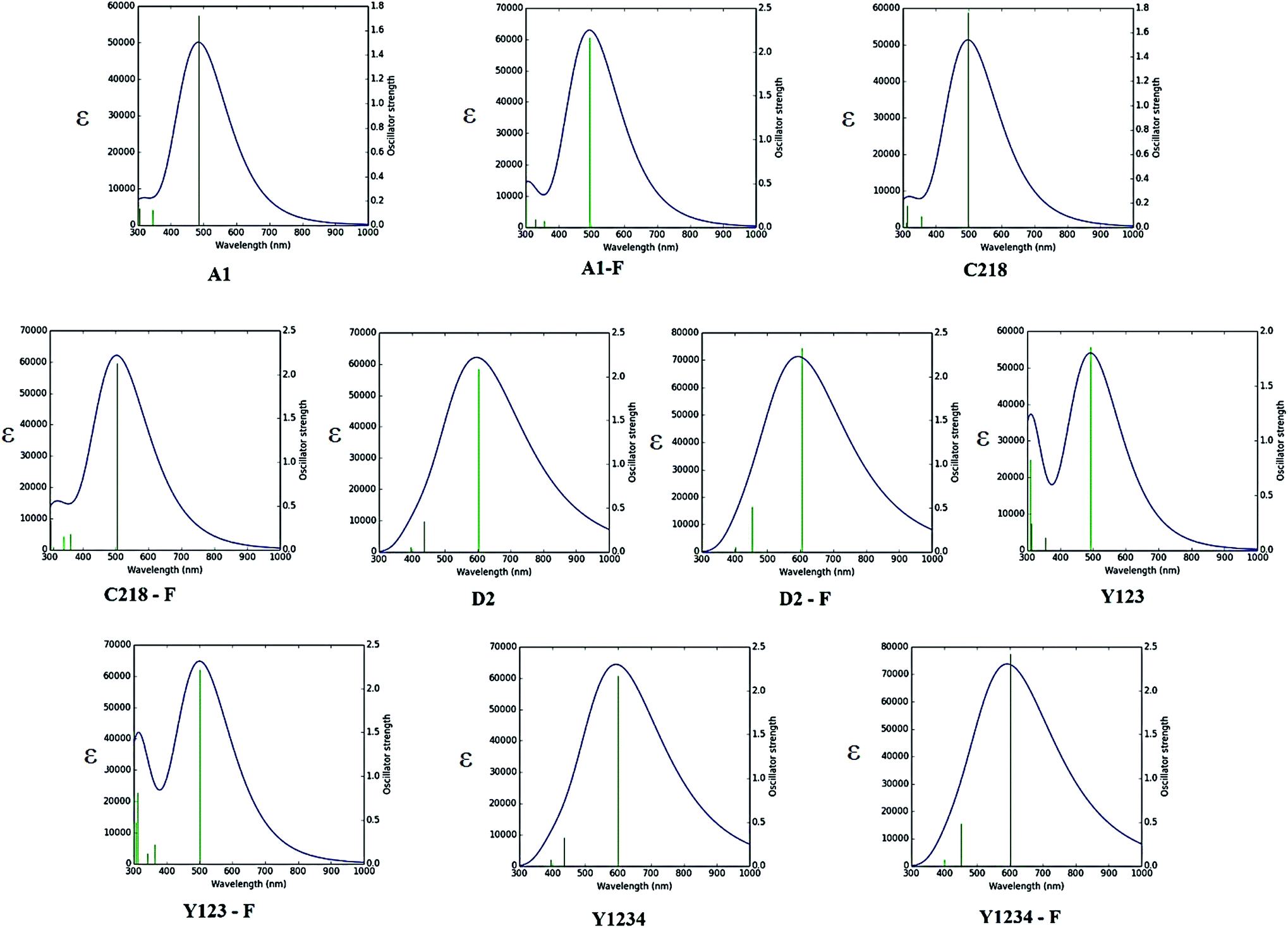


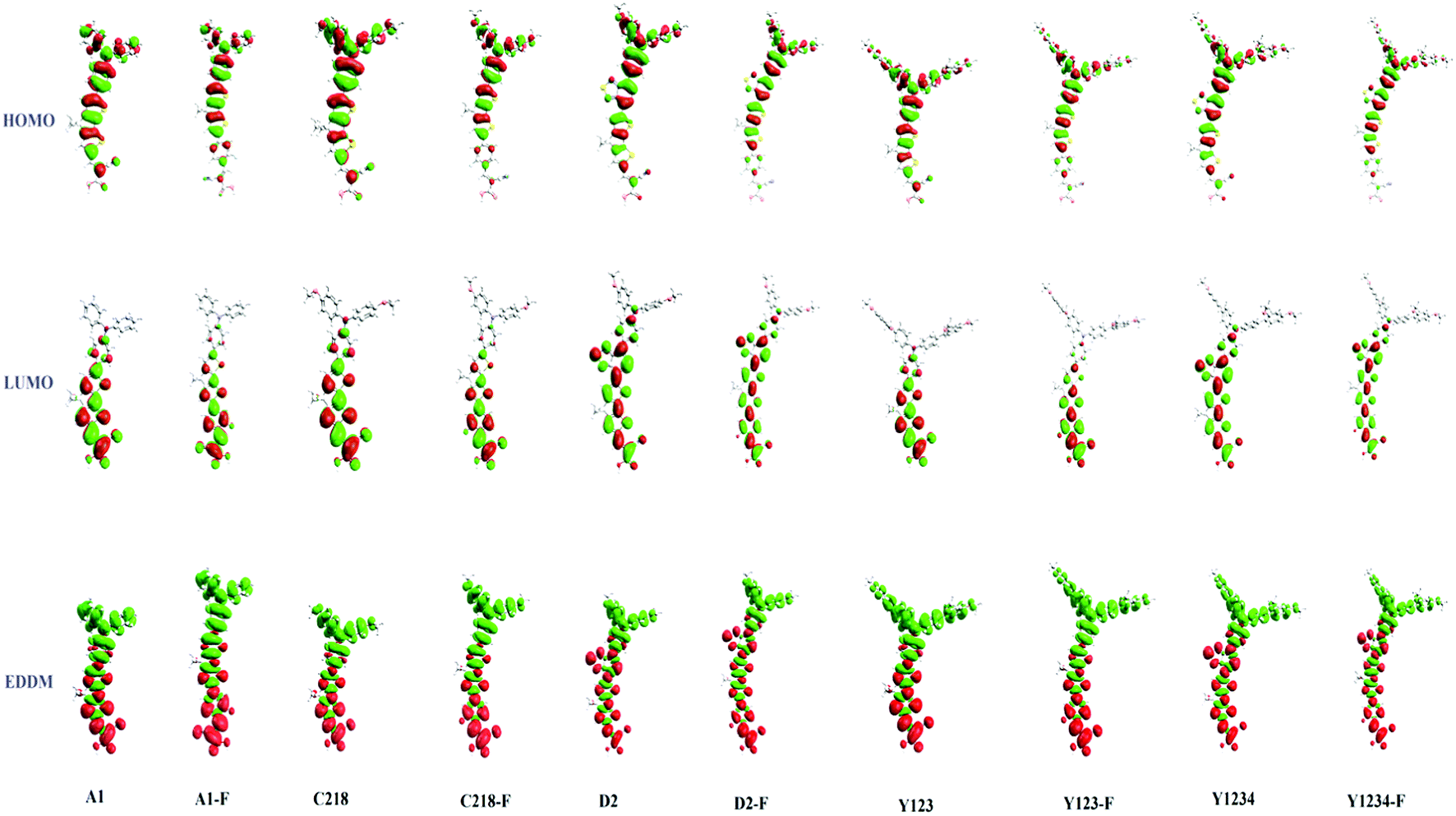


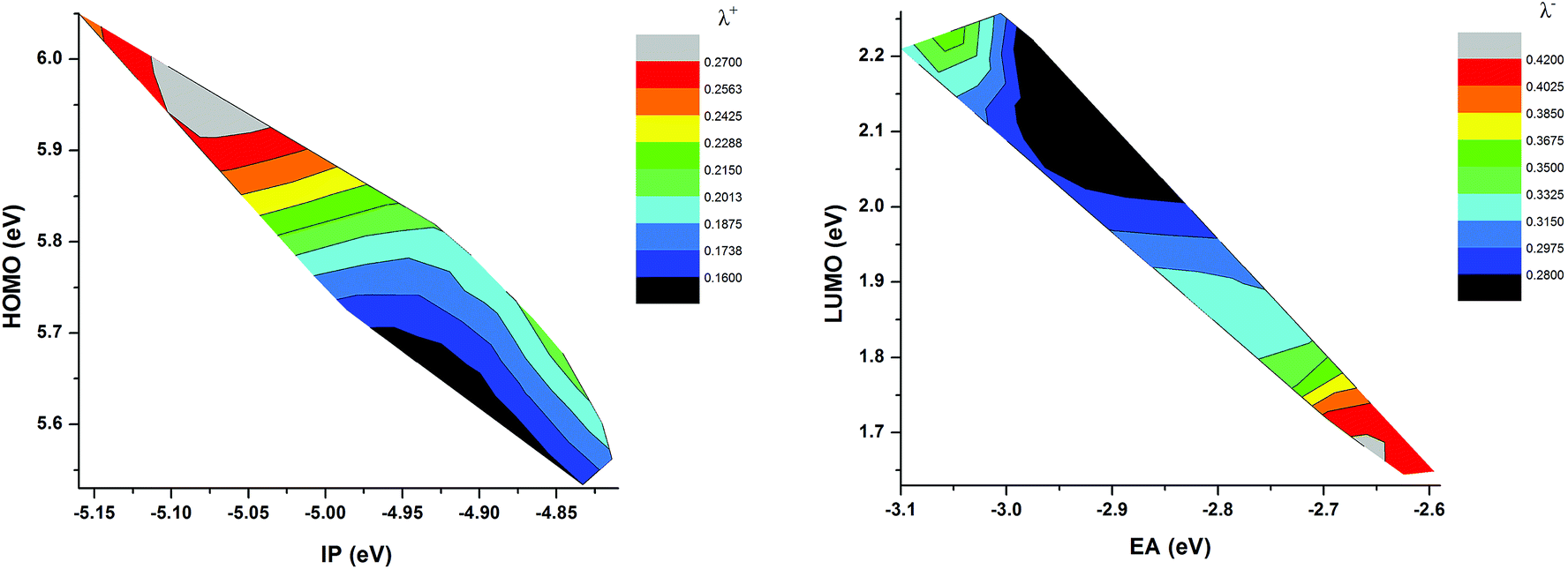

 is the reduction potential of the semiconductor conduction band. Though it is often difficult to accurately determine
is the reduction potential of the semiconductor conduction band. Though it is often difficult to accurately determine  , a highly sensitive datum to the conditions like the pH of the solution, thus we used the experimental value
, a highly sensitive datum to the conditions like the pH of the solution, thus we used the experimental value  (ref. 75) which corresponds to conditions where the semiconductor is in contact with the aqueous redox electrolytes of fixed pH 7.0.76,77 The excited state oxidation potential (Edye*OX) can be approximated from the redox potential of the ground state (EdyeOX),54
(ref. 75) which corresponds to conditions where the semiconductor is in contact with the aqueous redox electrolytes of fixed pH 7.0.76,77 The excited state oxidation potential (Edye*OX) can be approximated from the redox potential of the ground state (EdyeOX),54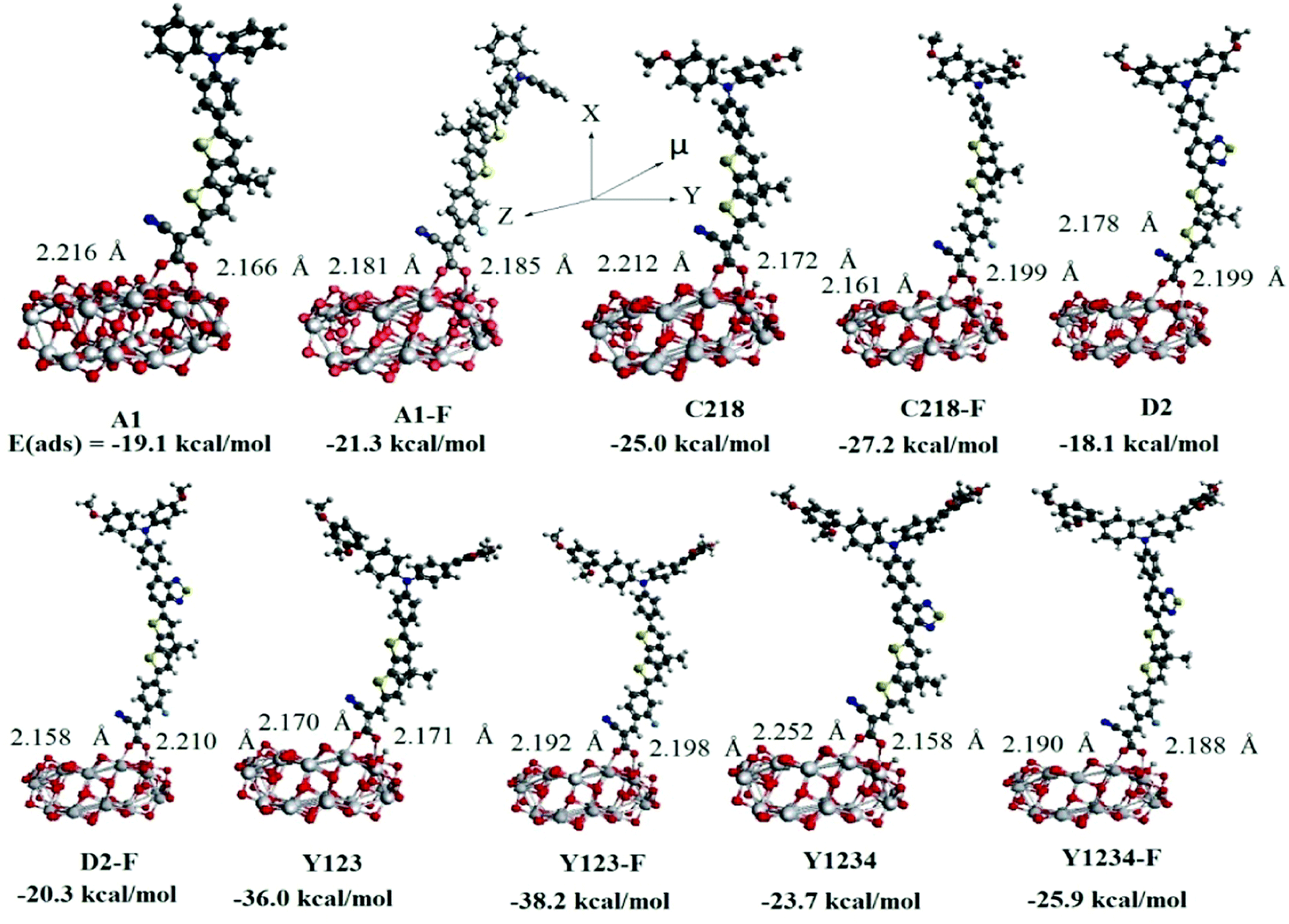
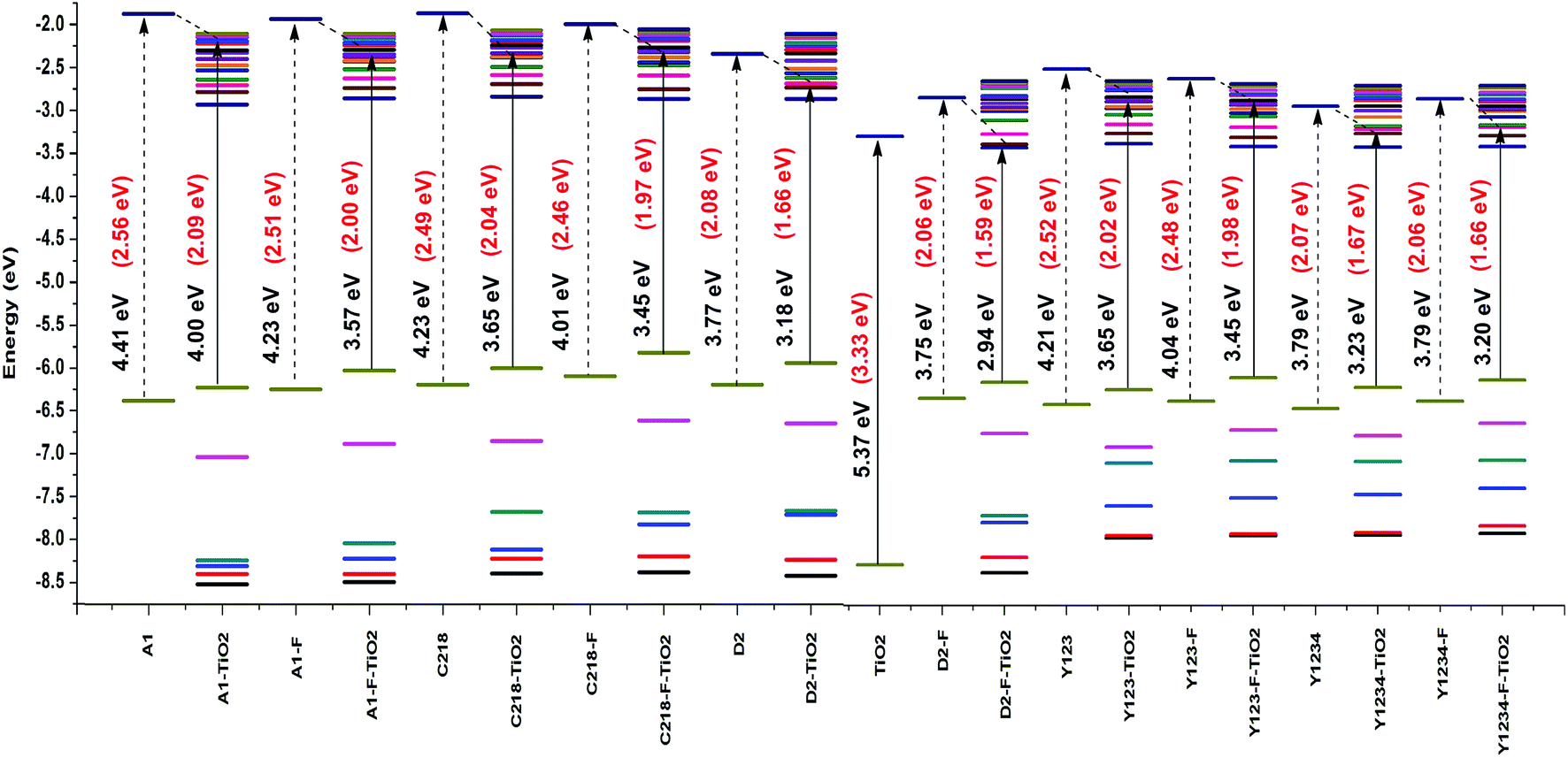
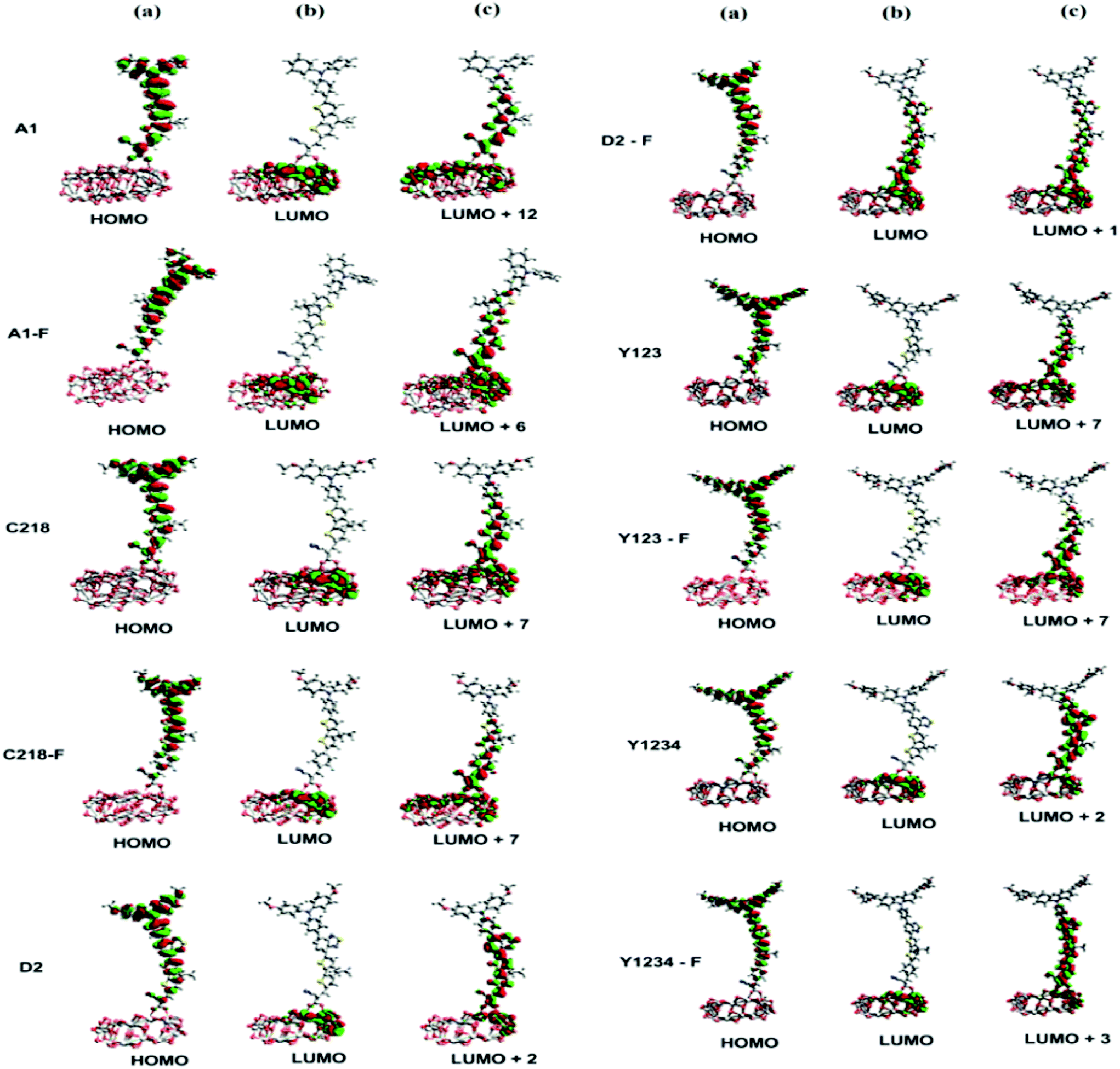
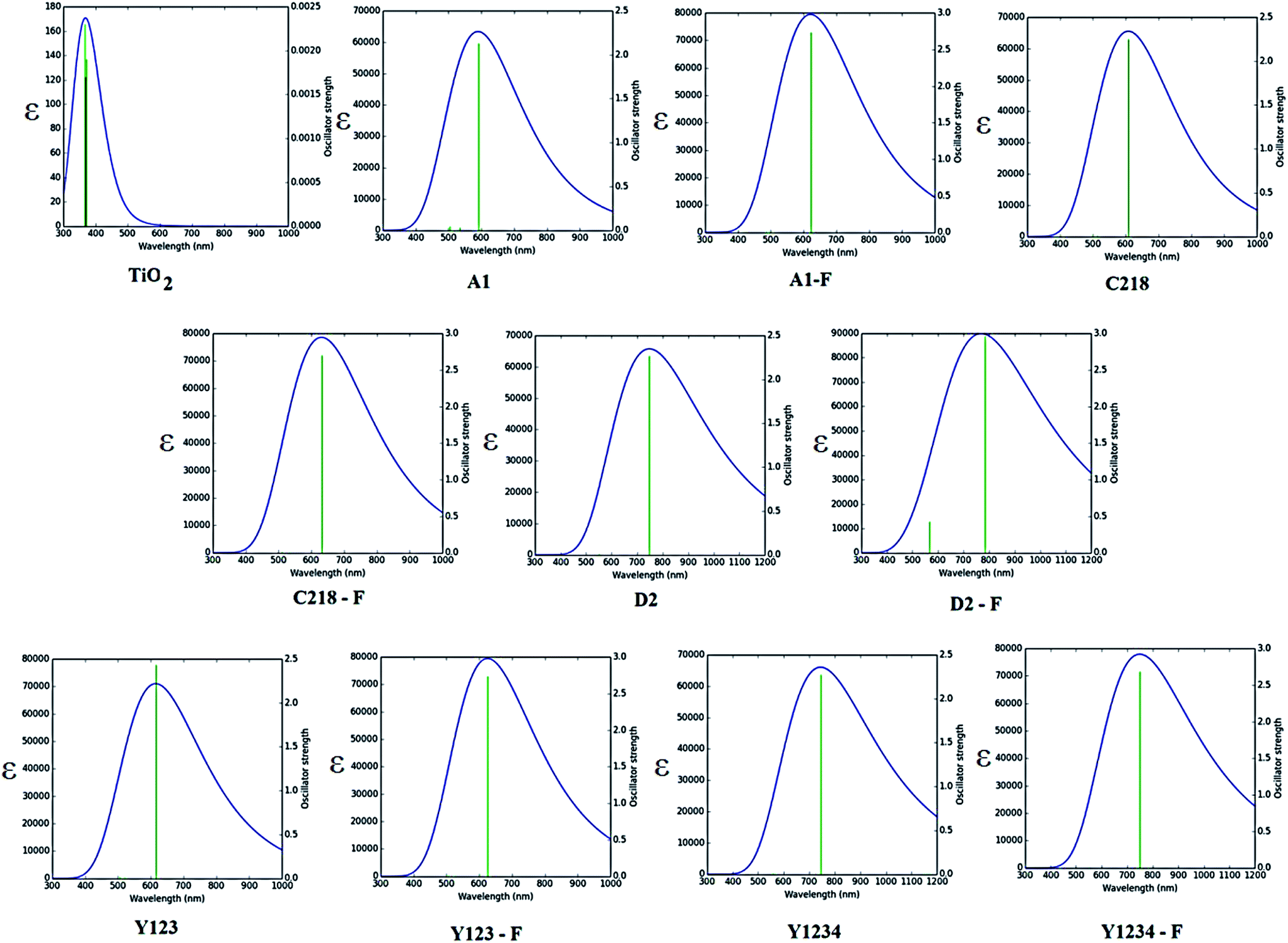





 . In order to calculate the level alignment quality, we adopted two different models for open circuit voltage i.e., VOC = Ec − EH and VOC = 1 V.92 The calculated results presented in Table 7 showed that the η values of A1 and C218 dyes are lower than the reference dye Y123. This indicates that the η value can be improved with increasing the strength of the donor group. It is also interesting to observe that the level alignment quality of A1 is enhanced by elongating the π-system with O-FP, leads to structure A1-F. However, when the spacer group in C218 is elongated with the BT group, producing dye D2, the η value decreases. The calculated results showed that η values for dyes elongated with O-FP (C218-F, Y123-F) or with both O-FP and BT (Y1234-F) are larger than the reference dye Y123. Whereas for the remaining dyes (A1, A1-F, C218, D2 and D2-F) the η values are smaller than the Y123. Based on the above results, we concluded that elongating the π-system in triarylamine dyes with both O-FP and BT offers more potential for DSSC applications.
. In order to calculate the level alignment quality, we adopted two different models for open circuit voltage i.e., VOC = Ec − EH and VOC = 1 V.92 The calculated results presented in Table 7 showed that the η values of A1 and C218 dyes are lower than the reference dye Y123. This indicates that the η value can be improved with increasing the strength of the donor group. It is also interesting to observe that the level alignment quality of A1 is enhanced by elongating the π-system with O-FP, leads to structure A1-F. However, when the spacer group in C218 is elongated with the BT group, producing dye D2, the η value decreases. The calculated results showed that η values for dyes elongated with O-FP (C218-F, Y123-F) or with both O-FP and BT (Y1234-F) are larger than the reference dye Y123. Whereas for the remaining dyes (A1, A1-F, C218, D2 and D2-F) the η values are smaller than the Y123. Based on the above results, we concluded that elongating the π-system in triarylamine dyes with both O-FP and BT offers more potential for DSSC applications.