Recent development of direct asymmetric functionalization of inert C–H bonds
Chao Zheng
and
Shu-Li You
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: slyou@sioc.ac.cn; Web: http://shuliyou.sioc.ac.cn Fax: +86 21-5492-5087; Tel: +86 21-5492-5085
First published on 6th December 2013
Abstract
The area of direct asymmetric functionalization of inert C–H bonds has attracted considerable attention in recent years. To realize this type of challenging but promising transformations, a lot of strategies have emerged including asymmetric C–H bond insertion by metal carbenoids or analogs, cross dehydrogenative coupling, [1,5]-hydride transfer, C–H bond functionalization involving a transient metal–carbon species and other miscellaneous methods. This review is intended to summarize and discuss the most recent developments (contributions mainly after 2009) within this area.
 Chao Zheng | Dr Chao Zheng was born in 1985 in Hubei province, China and received his BSc degree in chemistry from Shanghai Jiao Tong University in 2007. He obtained his PhD degree in Shanghai Institute of Organic Chemistry under the supervision of Prof. Shu-Li You and Prof. Yu-Xue Li in 2012. Then he joined Prof. Shu-Li You's group as an assistant professor. His current work is focused on understanding reaction mechanisms through computational methods. |
 Shu-Li You | Dr Shu-Li You received his BSc in chemistry from Nankai University in 1996. He obtained his PhD from the Shanghai Institute of Organic Chemistry (SIOC) in 2001 under the supervision of Prof. Li-Xin Dai before doing postdoctoral studies with Prof. Jeffery W. Kelly at The Scripps Research Institute. From 2004, he worked at the Genomics Institute of the Novartis Research Foundation as a Principal Investigator before returning to SIOC in 2006. His research interests include asymmetric catalysis, synthetic methodology, natural product synthesis, as well as medicinal chemistry. |
1. Introduction
The direct functionalization of inert C–H bonds is of great importance in modern synthetic organic chemistry.1 To employ the simple hydrocarbon compounds, which are abundant and cheap chemical feedstocks as the starting materials for the versatile chemical synthesis without “pre-activation”, makes the direct functionalization of inert C–H bonds meet the criteria of both atom economy2 and step economy.3 Therefore, this research area has received continuous attention and witnessed significant development in the last several decades. A large amount of catalytic systems including well-defined organometallic complexes, small molecular organocatalysts as well as enzymes have been proved effective for enabling the direct transformations of various inert C(sp2)–H and C(sp3)–H bonds. In addition, the emergence of these methodologies provides unprecedented disconnections in the retro-synthetic analyses of complex target molecules. Many elegant applications of the direct functionalization of inert C–H bonds in the total syntheses of natural products, molecules of pharmaceutical interests as well as diverse functional molecules have been reported in the literature.4 However, a principle challenge still accompanied with the high-speed development of this area is the issue of selectivity.5 Firstly, since the C–H bonds are the most fundamental and frequently encountered chemical bonds in organic molecules, to distinguish the reactivity among the numerous C–H bonds in one single molecule to achieve high level of regio- and enantioselectivity is certainly more difficult than traditional asymmetric catalysis which is in general the manipulation of functional groups. Secondly, due to the fact that the C–H bonds are among the strongest chemical bonds (BDE of C–H bonds are typically 90–110 kcal mol−1), harsh reaction conditions (high temperature, stoichiometric amount of oxidants, etc.) are usually required for the cleavage and direct functionalization of these inert bonds, which poses a problem in discriminating the diastereomeric transition states during the asymmetric catalysis. Thirdly, chiral ligands and catalysts compatible with the complex reaction conditions are limited and novel catalytic systems and strategies for the direct asymmetric functionalization of inert C–H bonds are still in great demand.Despite the aforementioned obstacles, the journey to pursuit the direct asymmetric functionalization of inert C–H bonds has already led to fruitful results in recent years. In 2009, Yu and co-workers5a published a comprehensive review discussing the diastereoselective and enantioselective transition metal-catalyzed C–H bond activation reactions. In the past five years, dozens of excellent works on the direct asymmetric functionalization of inert C–H bonds with high efficiency and largely broadened substrate scope have appeared, which definitely refreshed on our impression on this subject. Hence, a timely review collecting and discussing the most recent development in this exciting and fast developing field is highly desired. In this review, we are trying to give the readers an overview of the state-of-the-art of the methodologies (contributions mainly after 2009) for the direct catalytic asymmetric functionalization of inert C–H bonds. The reactions are classified according to the strategies and the catalytic systems employed. The suggested mechanistic scenarios for the novel transformations are also briefly described. To be noted, this review does not cover the asymmetric functionalization of the aldehyde C–H bond via hydroacylation reactions6 and N-heterocyclic carbene (NHC)-catalyzed umpolung reactions,7 as well as the asymmetric functionalization of the allylic and benzylic C–H bonds via dienamine or trienamine catalysis.8
2. C–H bond insertion by metal carbenoids or related species
2.1. C–H bond insertion by metal carbenoids
The asymmetric insertion of metal carbenoids into C–H bonds are probably the relatively more traditional type of methods to realize the direct asymmetric functionalization of inert C–H bonds compared with other ones presented in this review.9 Chiral complexes of several transition metals including Rh, Ir, Cu and Fe are typically employed in this type of transformations. α-Diazocarbonyl compounds are the most widely used carbene precursors.In 2009, Denton and Davies10 reported that the donor/acceptor carbenoids11 derived from α-aryl-α-diazoketones 1 and a chiral dirhodium complex Rh2(S-PTAD)4 (S)-C1 could insert enantioselectively into the allylic C–H bonds of 1,4-cyclohexadiene 2 in refluxing 2,2-dimethylbutane (DMB) in up to 90% yield and 89% ee (Scheme 1(a)). (In this review, we use the numbers with a prefix “C” to denote the chiral catalysts or reagent, and “L” to denote the chiral ligand.) After realizing that the destroy of the chiral catalyst by the reactive carbenoids might be the major hindrance for achieving very high catalyst turnover numbers (TONs), Davies and co-workers12 found that the highly efficient dirhodium-catalyzed asymmetric insertion reaction of methyl phenyldiazoacetate 4, a proper precursor of donor/acceptor carbenoid that has a higher stability, went smoothly at 0 °C using 2 as the solvent (Scheme 1(b)). The high concentration of 2 could probably facilitate the trapping of Rh-carbenoid species by insertion into a C–H bond before the decomposition of the catalyst. The corresponding products 5 could be afforded in 96% yield with 81% ee, and the loading of catalyst Rh2(S-DOSP)4 (S)-C2 could be reduced to 0.01 mol%.
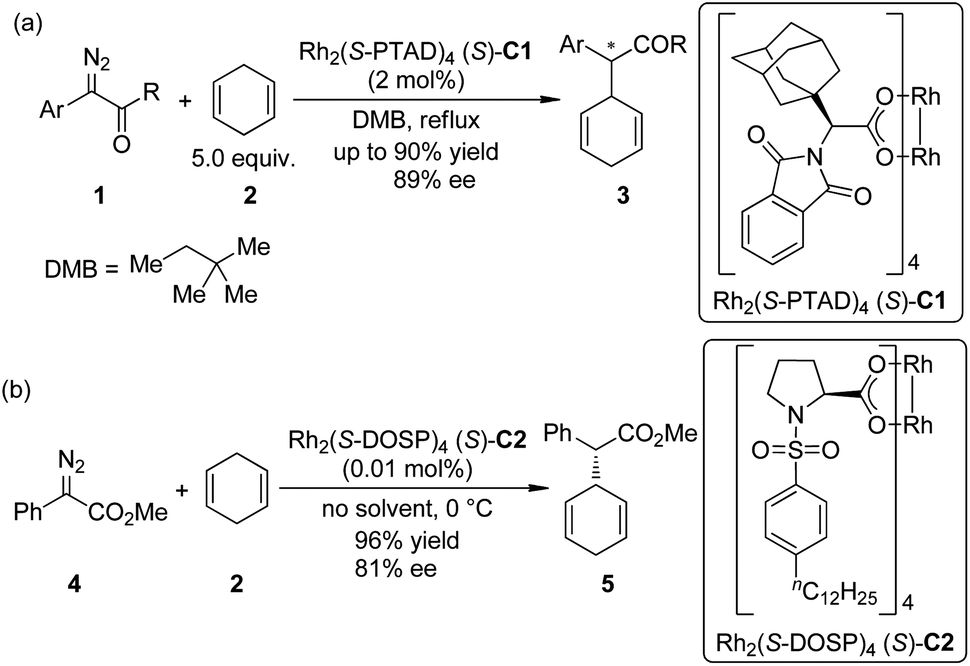 | ||
| Scheme 1 The asymmetric C–H bond insertion reactions of dirhodium carbenoids reported by Davies. | ||
Recently, Hashimoto and co-workers13 reported the Rh-catalyzed intermolecular asymmetric insertion reaction of the carbenoid derived from α-methyl-α-diazoester 6 (Scheme 2). The dirhodium complexes derived from N-tetrahalophthaloyl-(S)-tert-leucine (Rh2(S-TFPTTL)4, (S)-C3 and Rh2(S-TCPTTL)4, (S)-C4) are viable catalysts for this transformation. The insertion product 7 could be obtained in moderate yields and ee's (66% yield and 80% ee for (S)-C3, 73% yield and 61% ee for (S)-C4). Notably, when the methyl group in 6 was replaced by longer alkyl chains (ethyl or n-propyl), the Z-α,β-unsaturated esters 9 became the major or even the sole product via an [1,2]-hydride shift of the Rh-carbenoid intermediates.
 | ||
| Scheme 2 The asymmetric C–H bond insertion reactions of dirhodium carbenoids reported by Hashimoto. | ||
Indoles are the most widely distributed heterocyclic motifs in naturally occurring alkaloids. Thus, the enantioselective functionalization of indoles has been a hot research topic for a long time.14 In their seminal work on Rh-catalyzed [3 + 2] annulation of indoles, Lian and Davies15 disclosed that methyl α-phenyl-α-diazoacetate could react with 1,2-dimethylindole to afford the C3 functionalization product in 95% yield by the Rh-catalyst (S)-C2, albeit negligible asymmetric induction (<5% ee) was observed. In 2011, Fox and co-workers16 found that by using a catalytic amount of the chiral dirhodium complex Rh2(S-NTTL)4 (S)-C5, the C–H bond functionalization of indoles 10 with α-alkyl-α-diazoesters 11 could occur under mild conditions (Scheme 3(a)). Low temperature was critical to the success of the reaction. Excellent yields and ee's could be obtained for the N-aryl and N-alkyl indole derivatives with a small substituent (H or Me) at the indole C2 position. Control experiments precluded the cyclopropanation/fragmentation pathway and further density functional theory (DFT) calculations supported the mechanism that involves a Rh-ylide intermediate. The authors also suggested that the Rh-catalyst adopts the “chiral crown” conformation17 in which the four phthalimide groups are projected on the same face of the complex during the reaction. Shortly after Fox's report, the Hashimoto group18 realized the asymmetric C–H bond functionalization of N-MOM protected 2,3-unsubstituted indoles 13 with diazoester 6 by a Rh-catalyst Rh2(R-PTTEA)4 (R)-C6 (Scheme 3(b)). The corresponding product 14 could be used in the asymmetric synthesis of acremoauxin A, a potent plant-growth inhibitor.19 Markedly different from Fox's results, the 2-methylindole derivative is not a good substrate here.
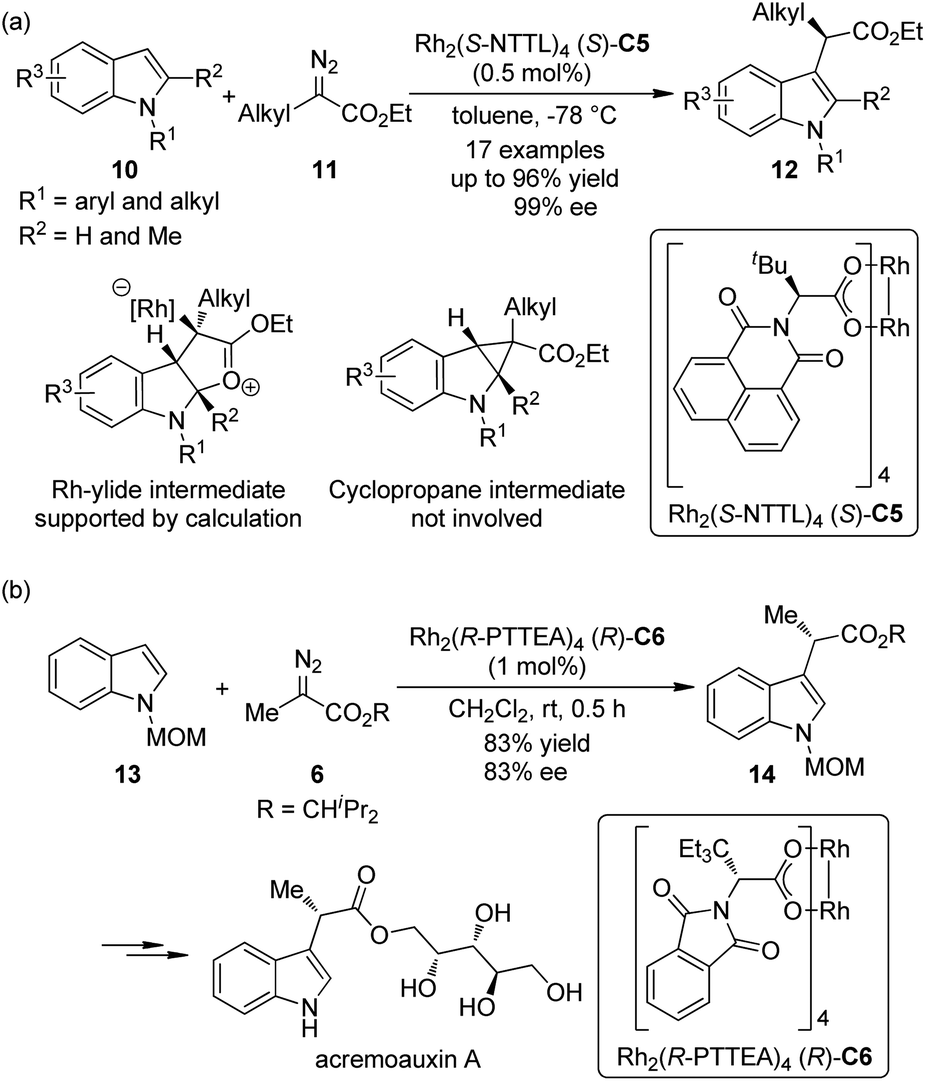 | ||
| Scheme 3 The Rh-catalyzed asymmetric C–H bond functionalization reactions of indoles reported by (a) Fox and (b) Hashimoto, respectively. | ||
The zwitterionic intermediates are proposed to exist during the C–H bond insertion reactions in which the diazo compounds participate. These intermediates usually undergo rapid proton transfer to afford the C–H bond functionalization products.20 Recently, Hu and co-workers21 utilized chiral phosphoric acid (S)-C7 activated N-aryl imines as the electrophiles to trap the zwitterionic intermediates generated from the Rh-catalyzed intra- or intermolecular carbenoid C–H bond insertion reactions (Scheme 4).22 The polyfunctionalized oxindole and indole derivatives 17 and 20 bearing two consecutive chiral centers were obtained in one single step with exceptional diastereoselectivity and enantioselectivity (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 99% ee) taking advantage of a dual catalytic system combining a transition metal salt and an organocatalyst.23
:1 dr and 99% ee) taking advantage of a dual catalytic system combining a transition metal salt and an organocatalyst.23
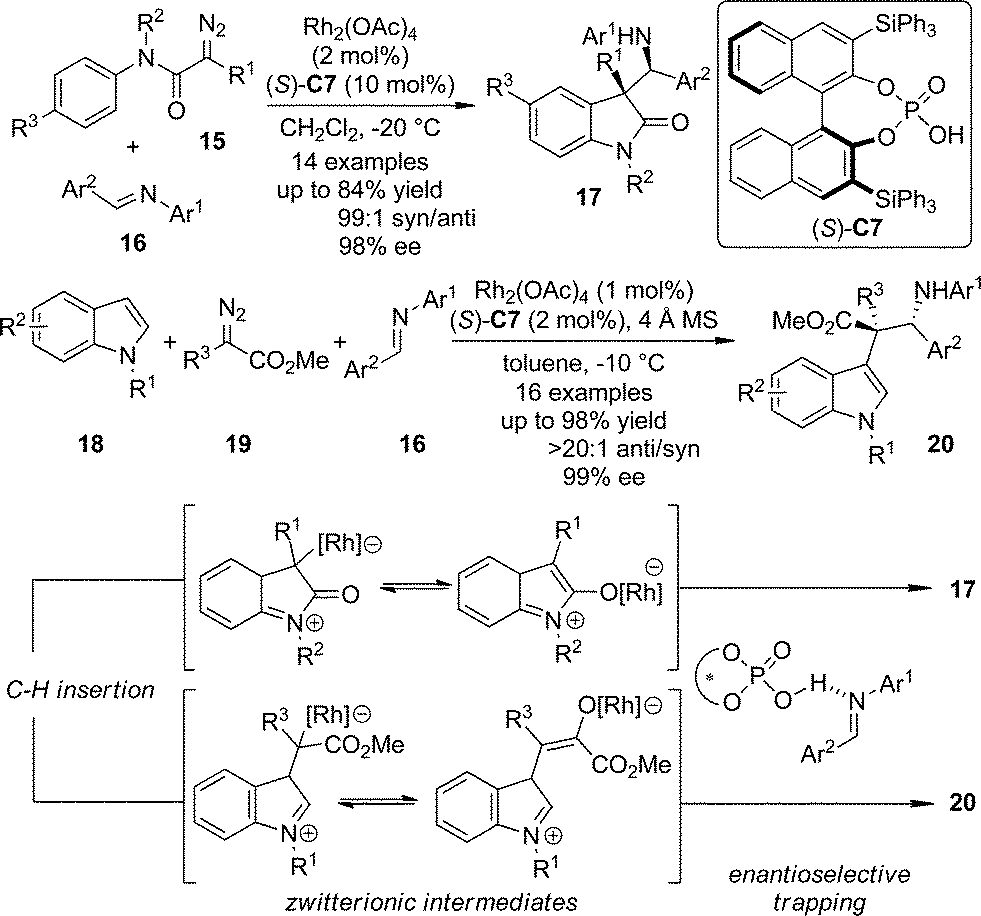 | ||
| Scheme 4 The asymmetric trapping of the zwitterionic intermediates formed via C–H bond insertion reactions of dirhodium carbenoids reported by Hu. | ||
Based on the independent studies from the Davis group24 on the Rh-catalyzed asymmetric intermolecular benzylic C–H bond insertion and the Yu group25 on the Pd-catalyzed C–H activation/C–O cyclization, a collaboration of these two laboratories26 led to the enantioselective synthesis of 2,3-dihydrobenzofurans by sequential C–H bond functionalization reactions (Scheme 5). The asymmetric insertion of the carbenoid species generated from α-aryl-α-diazoesters 21 and the dirhodium complex Rh2(R-PTTL)4 (R)-C8 into the secondary benzylic ethers 22 afforded the corresponding products 23 in good yields and excellent enantioselective control (up to 92% yield, >97:3 dr and 99% ee). After removal of the TBS group, the 2,3-dihydrobenzofurans 25 were obtained by the Pd-catalyzed C–H activation/C–O cyclization sequence employing a Pd(II)/Pd(IV) catalytic cycle. No racemization could be observed. Further diversification of the benzofuran derivative was accomplished by a third C–H bond functionalization via the oxidative Heck coupling reaction.27
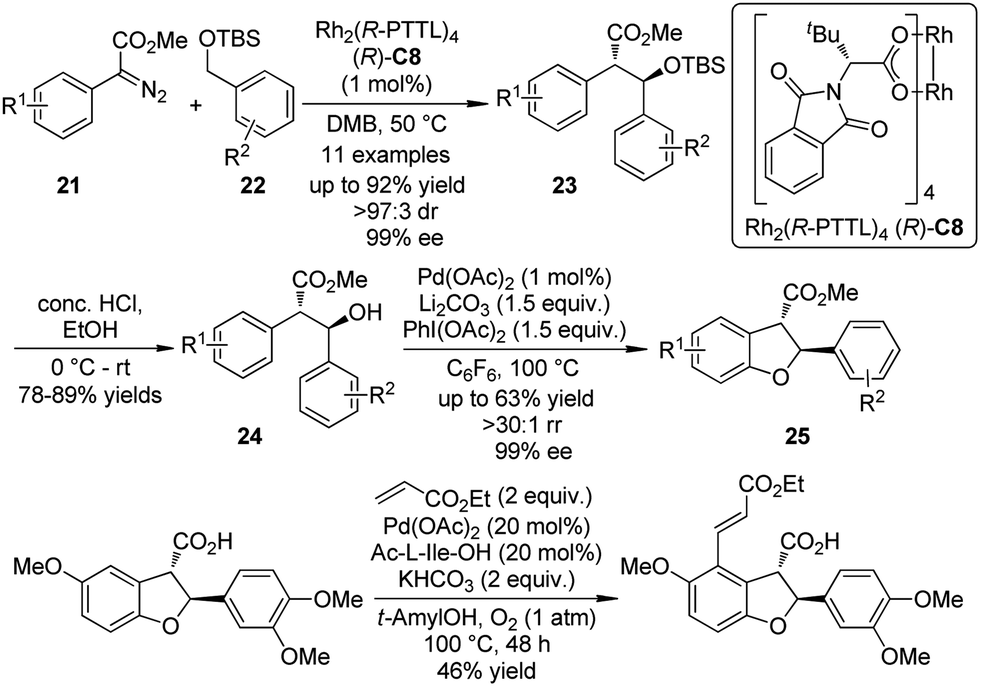 | ||
| Scheme 5 The asymmetric sequential C–H bond functionalization reactions to synthesize benzofuran derivatives reported by Yu and Davies. | ||
The α-diazo carbonyl compounds are not the only carbene precursors to realize the asymmetric C–H bond insertion reactions. It was found that the 1-sulfonyl-1,2,3-triazoles which could be easily prepared by the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction28 could serve as alternative carbene precursors for various transformations.29 In 2011, the Fokin group30 demonstrated that the azavinyl carbenoids derived from triazoles 26 and Rh-catalysts (S)-C1 or (S)-C5 underwent asymmetric C–H bond insertion reactions with a series of alkanes 27 under mild conditions (Scheme 6). The corresponding β-chiral amines 28 could be afforded after subsequent LiAlH4 reduction in up to 97% ee. The high regioselectivity (5:1) of the insertion reaction favoring tertiary C–H bonds over secondary C–H bonds was observed. Notably, the hydrolysis of the direct insertion products only gave the aldehydes in a racemic form.
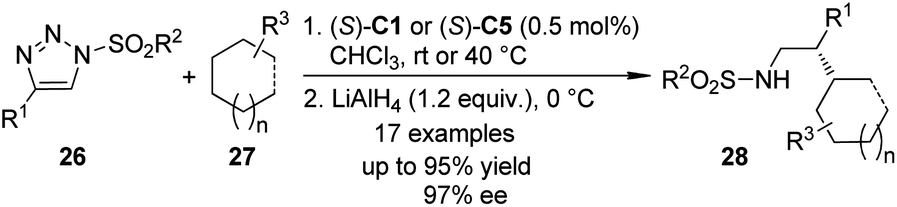 | ||
| Scheme 6 The Rh-catalyzed asymmetric C–H bond insertion reactions of azavinyl carbenoids reported by Fokin. | ||
In recent years, the Davies group31 has systematically studied the combined C–H functionalization/Cope rearrangement (CHCR), a reaction occurring between Rh-bound vinylcarbenoids and substrates containing allylic C–H bonds (Scheme 7(a)). This type of transformations offers accesses to products bearing two new stereocenters and has found broad applications in organic synthesis.32 Detailed DFT calculations have been conducted to probe the mechanism of the CHCR reactions.33 This study demonstrated that the initial C–H bond functionalization appeared to be a hydride transfer event. The charged transition state TS-HT could then bifurcate, leading to the CHCR product PA or the direct C–H bond insertion product PB (Scheme 7(b)). These results are in accord with the fact that in several cases, both products were formed and the enantioselectivity of the two products were often very similar or even identical.34 The more intriguing information gained from the computational investigation is that, in principle, the transition states with different conformation and orientation of the substrates, which will lead to different stereochemical outcomes, are all energetically accessible. Thus, the diastereoselectivity could be possibly switched by using appropriate substrates: the substrates bearing large substituents at the R2 position would prefer the s-cis/boat approach, and conversely, the substrates bearing large substituents at the R1/R4 positions would prefer the s-cis/chair approach (Scheme 7(c)). Davies and co-workers35 verified this hypothesis by employing cyclohexene 30 and cyclopentene 32 as the reaction components, respectively (Scheme 7(d)). The corresponding β-keto-α-diazoacetates 31 and 33 were generated through CHCR reaction and subsequent transformations with high stereoselectivity but in the opposite diastereomeric series, which is consistent with the working models depicted in Scheme 7(c).
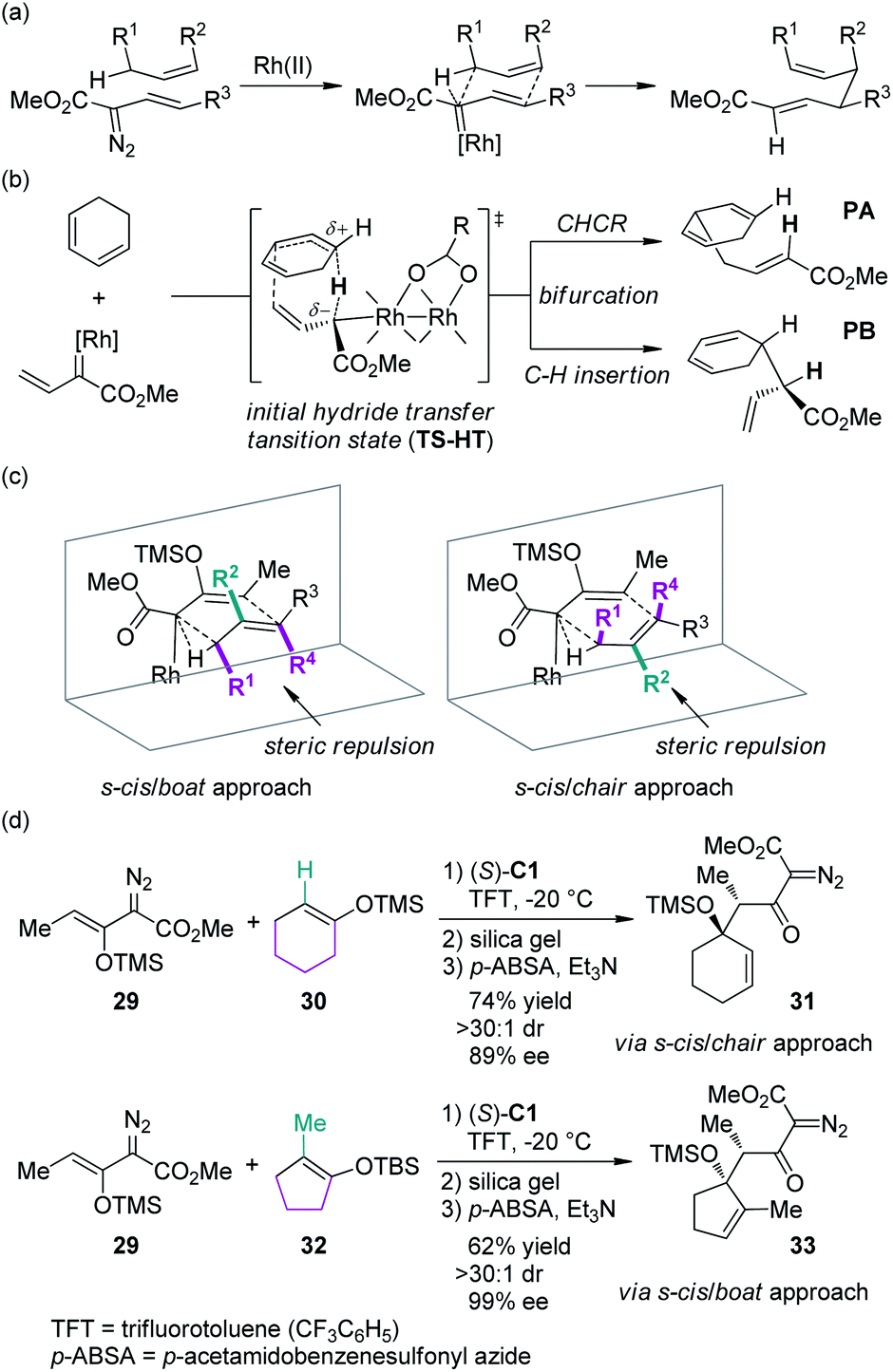 | ||
| Scheme 7 The mechanistic studies of the Rh-catalyzed CHCR reactions reported by Davies. | ||
Lian and Davies36 reported the application of the CHCR reaction of vinyl ethers as a surrogate for the vinylogous Mukaiyama aldol reactions.37 By using (S)-C2 as the catalyst, an array of Mukaiyama aldol-type products 36 were afforded in high yields with exceptional stereoselectivity (up to >30:1 dr and 99% ee) under mild conditions (Scheme 8). The absolute configuration of 36 was found to be consistent with the s-cis/boat approach. The C–H bond functionalization was highly regioselective as only the secondary C–H bond trans to the methoxyl group in E-35a was cleavaged when an 1:1 mixture of E/Z-35a was used. The kinetic resolution of the racemic vinyl ether 35b was also achieved efficiently.
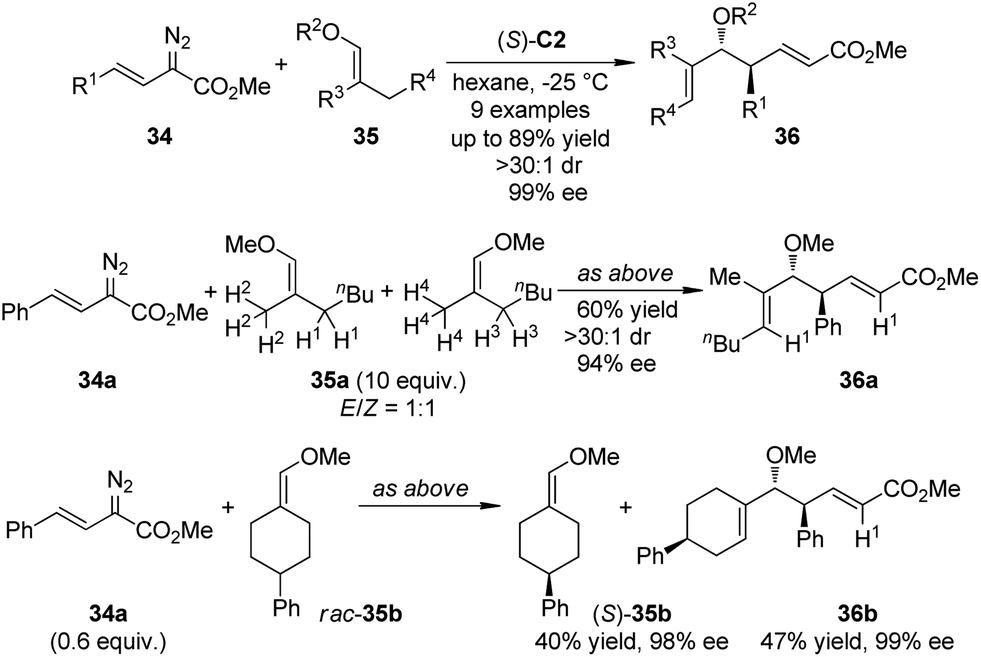 | ||
| Scheme 8 The Rh-catalyzed CHCR reactions of vinyl ethers reported by Davies. | ||
The Davies group38 also realized the asymmetric functionalization of indoles with vinylcarbenoids. It was known that the Rh-carbenoids generated from E-vinyldiazoacetates 38 could adopt s-trans or s-cis conformation. When a sterically demanding nucleophile was used, it would be possible to enhance the vinylogous reactivity of 38 by reinforcing the s-trans conformation of the carbenoid through the use of highly bulky catalysts. Indeed, when such a chiral catalyst Rh2(S-biTISP)2 (S)-C9 was employed, the desired indole functionalization products 39 were obtained in up to 86% yield and 95% ee (Scheme 9). The newly formed double bonds were in Z-configuration. The corresponding pyrrole functionalization could be also achieved at a slightly elevated temperature (−20 °C) due to the decreased reactivity of pyrroles.
 | ||
| Scheme 9 The Rh-catalyzed asymmetric C–H bond functionalization reactions of indoles reported by Davies. | ||
In addition to the extensively documented dirhodium catalysts, several chiral Ir-complexes have been used in the asymmetric C–H bond insertion reactions of metal carbenoids recently (Scheme 10). In 2009, Suematsu and Katsuki39 reported that the Ir-carbenoids derived from α-diazoesters 40 and the Ir–salen complex (Ra,R)-C10 could react with 1,4-cyclohexadiene 2 to afford the C–H bond insertion products 41 with up to 95% yield and >99% ee. Highly diastereo- and enantioselective C–H bond insertion into THF of the similar carbenoid species was also realized by using a sterically less hindered Ir–salen complex (Sa,R)-C11. The syn-42 was obtained as the major diastereo isomer with satisfactory yields and ee's. Noticeably, the α-diazopropionate (40, R1 = Me) was identified as a viable substrate for these two reactions since the product of the competing β-hydride elimination was not observed. Che and co-workers40 found that an Ir-complex (−)-C12 derived from a D4-symmetric porphyrin ligand could catalyze similar transformations. Comparable results were obtained for asymmetric C–H bond insertion between α-aryl-α-diazoesters 40 and 1,4-cyclohexadiene 2 (up to 94% yield and 98% ee). A catalyst TON of 9600 was observed in a scaled up reaction. Complementary to Katsuki's results, the asymmetric C–H bond insertion into THF catalyzed by (−)-C12 exhibited high anti-selectivity. The enantioenriched anti-42 (up to 97% ee) were the major products for a series of α-aryl-α-diazoester substrates. Very recently, Musaev, Davies, Blakey and co-workers41 developed a phebox-ligated Ir-complex (R,R)-C13 that is also capable to catalyze the asymmetric C–H bond insertion reactions between 40 and 2. The reactions could proceed at ambient conditions. Slow addition of the diazoester and low temperature were not required. Substituted 1,4-cyclohexadienes were also subjected to the C–H bond insertion reactions and the subsequent oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) led to the corresponding α,α-biarylacetates in good yields (64–98%) with excellent enantioselectivity (90–99% ee). Further DFT calculations revealed that in the active catalytic species, the carbene fragment preferred to coordinate to the Ir center in the position perpendicular to the plane of the phebox ligand. A predictive model that could explain the experimentally observed enantioselectivity was also provided.
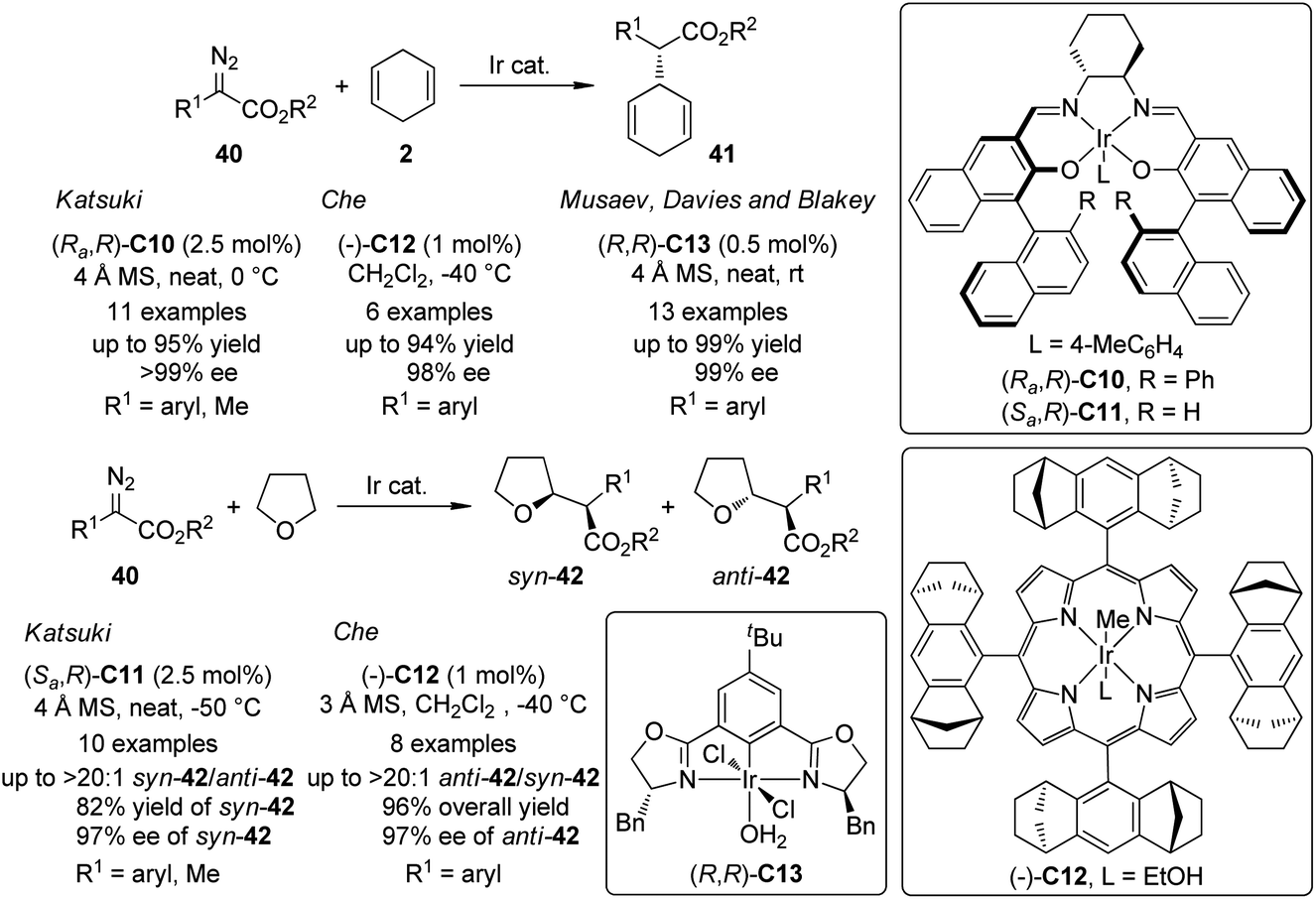 | ||
| Scheme 10 The Ir-catalyzed asymmetric intermolecular C–H bond insertion reactions reported by several groups. | ||
Che and co-workers42 expanded the scope of Ir–porphyrin complex catalyzed asymmetric C–H bond insertion reaction to intramolecular variants. By utilizing a catalytic amount of (+)-C12 (L = H2O or solvent) as the catalyst, an array of benzyl α-aryl-α-diazoesters 43 underwent intramolecular asymmetric C–H bond insertion reaction to afford cis-β-lactones in good yields (up to 87%) and enantioselectivity (up to 78% ee) (Scheme 11). Common dirhodium carboxylate catalysts such as (S)-C1 or Rh2(S-MEPY)4 (tetrakis[methyl 2-pyrrolidone-5(S)-carboxylate]dirhodium(II)) could not catalyze this reaction effectively.
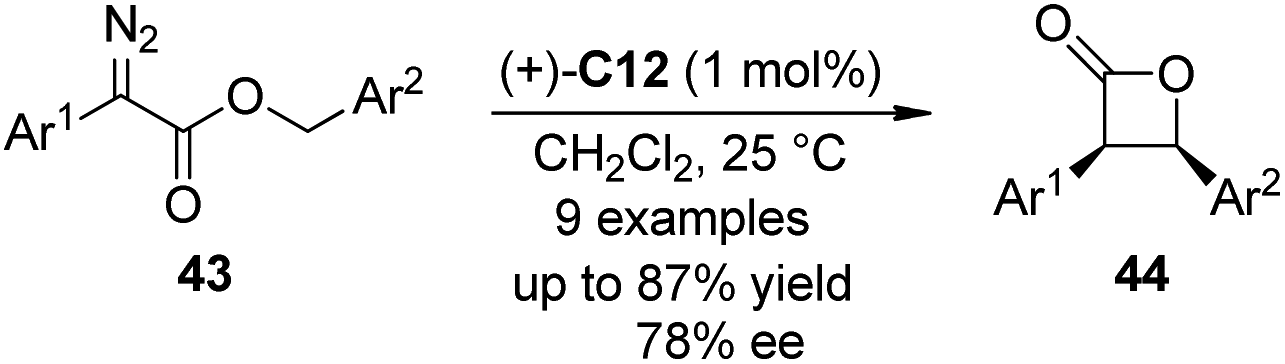 | ||
| Scheme 11 The Ir-catalyzed asymmetric intramolecular C–H bond insertion reactions reported by Che. | ||
Cu is another widely employed transition metal to catalyze the carbene transformations. However, the applications of chiral Cu catalysts to asymmetric C–H bond insertion reaction are still very limited. Fraile and co-workers43 disclosed that the homogeneous Cu complexes with bis(oxazoline) (BOX) and azabis(oxazoline) (azaBOX) ligands were able to catalyze the asymmetric insertion of α-diazo compounds 4 into the C–H bonds of cyclic ethers including THF, THP, 1,4-dioxane and 1,3-dioxolane. When the chiral complexes were immobilized by electrostatic interactions with laponite clay or supported on silica or silica-alumina, the catalytic performance was improved considerably in terms of conversion and stereoselectivity (Scheme 12). In addition, the heterogeneous catalyst could be recovered and reused for several times with similar results to those obtained in the first run. Very recently, Jiménez-Osés, Fraile and co-workers44 unambiguously determined the absolute configuration of the conformationally flexible products of the Cu-catalyzed C–H bond insertion reactions between 4 and cyclic ethers using vibrational circular dichroism (VCD) spectroscopy in combination with rigorous quantum mechanics calculations.
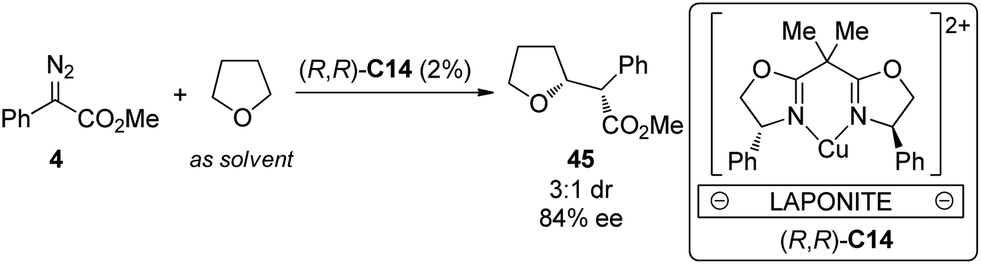 | ||
| Scheme 12 The Cu-catalyzed heterogeneous asymmetric intramolecular C–H bond insertion reactions reported by Fraile. | ||
In 2010, Maguire and co-workers45 reported the first example of Cu-catalyzed enantioselective intramolecular C–H bond insertion reactions of α-diazosulfones 46 (Scheme 13). In the presence of catalytic amount of CuCl and chiral BOX ligand (R,R)-L1, the formation of cis-thiopyrans 47 was preferred in moderate yields (up to 68%) with excellent enantioselectivity (up to 98% ee). When this option was not possible, the insertion reaction was forced to proceed to give the trans five-membered ring products 48. Notably, the dirhodium catalysts were not effective for this transformation. Shortly after these initial results, Slattery and Maguire46 realized the asymmetric intramolecular C–H bond insertion reactions of α-diazo-β-keto sulfones 49 catalyzed by similar chiral Cu-BOX complexes. It was found that using NaBARF or KBARF (BARF = tetrakis[3,5-bis(trifluromethyl)phenyl]borate) as an additive is crucial for achieving high enantioselectivity for these two reactions. Further investigations47 demonstrated that the naked alkali metal cations could alter the nature of the Cu catalyst through partial or complete chloride abstraction.
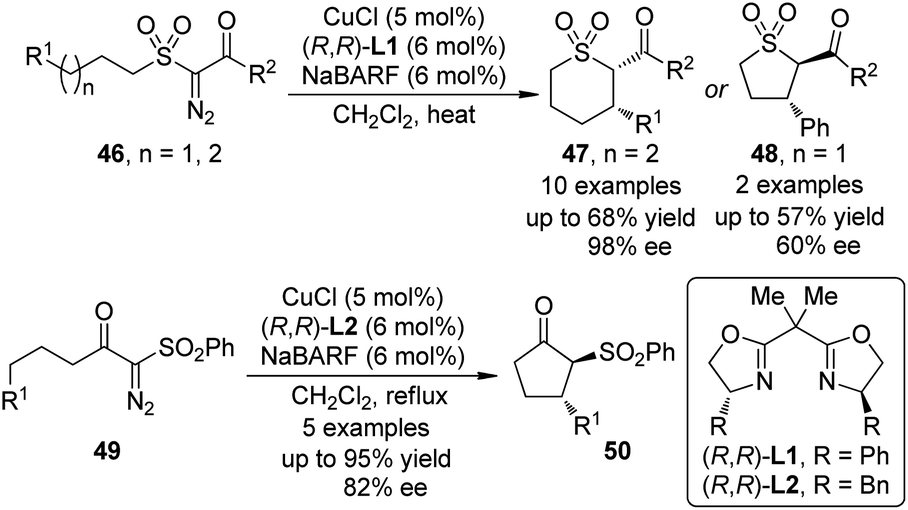 | ||
| Scheme 13 The Cu-catalyzed asymmetric intramolecular C–H bond insertion reactions of α-diazosulfones reported by Maguire. | ||
Fe-catalyzed asymmetric reactions48 have been studied intensively in recent years in that iron is cheap, non-toxic and the most abundant metal in earth crust. Zhou and co-workers49 demonstrated that the asymmetric C–H bond functionalization reactions of indoles could be accomplished by Fe-catalyzed insertion of carbenoid derived from α-aryl-α-diazoesters 51 (Scheme 14). The reaction of these substrates could not give satisfactory enantioselectivity when chiral dirhodium catalyst was utilized.15 The detailed optimization of the reaction conditions revealed that the spiro bisoxazoline (Ra,S,S)-L3 was the optimal ligand and Fe(ClO4)2 was the optimal Fe precursor. The corresponding α-aryl-α-indolylacetates 53 were constructed in up to 94% yield and 78% ee. Notably, most products are solid and can be easily purified by recrystallization. This study indicated a high potential for application of sustainable and environmentally benign catalysts in carbene transformations.
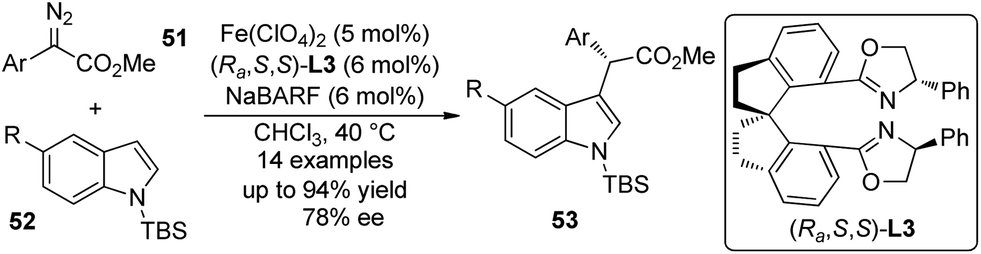 | ||
| Scheme 14 The Fe-catalyzed asymmetric C–H bond functionalization reactions of indoles reported by Zhou. | ||
Interestingly, the group of Hwang and Ryu50 reported the Lewis acid-catalyzed asymmetric insertion reactions into β-vinyl C–H bonds of cyclic enones.51 It was found that by utilizing the chiral oxazaborolidinium salts (S)-C15 or (S)-C16 as the catalysts, the β-vinyl C–H bonds of various cyclic enones 54 could be functionalized by α-alkyl-α-diazoesters 55 leading to the β-substituted cyclic enones 56 in high yields with excellent enantioselectivity under mild conditions (Scheme 15).52 In some cases, the catalyst loading could be lowered to 5 mol%. Efficient kinetic resolution was also achieved when racemic substrate 54 (n = 1, R1 = 6-Me) was employed. The synthetic utility of this transformation was further demonstrated by the formal synthesis of a natural sesquiterpene (+)-epijuvabione.53
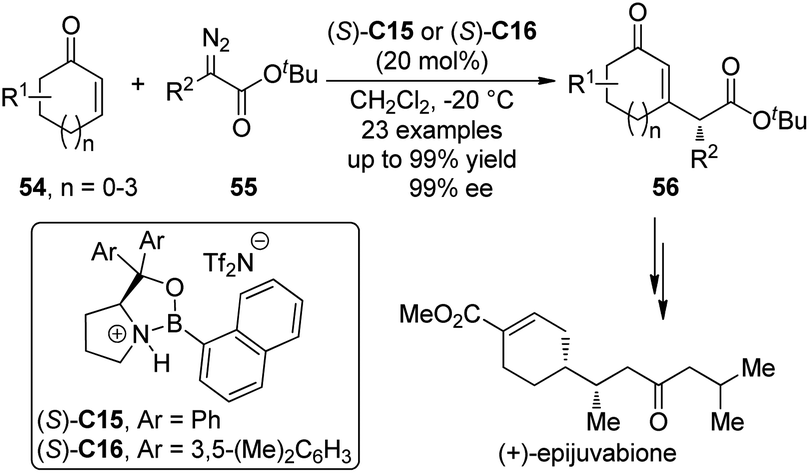 | ||
| Scheme 15 The Lewis acid-catalyzed asymmetric C–H bond functionalization reactions of enones reported by Hwang and Ryu. | ||
2.2. C–H bond insertion by metal nitrenoids
Nitrenes are the nitrogeneous isoelectronic analogs of the carbenes. The asymmetric insertion reaction of metal nitrenoids into the C–H bonds has emerged as a powerful tool for the construction of C–N bonds in recent years.54,55 Dauban and co-workers55a,b,56 systematically studied the diastereoselective nitrene C–H bond insertion reactions using a chiral sulfonimidamide (S)-C17 in combination with the chiral dirhodium catalyst Rh2(S-NTA)4 (S)-C18. The benzylic methylene units of cyclic and linear compounds, the allylic methylene units of terpenes and enol ethers as well as the tertiary C–H bonds of alkanes could be efficiently aminated with very high diastereomeric ratios under mild conditions (Scheme 16). The high regioselectivity was determined by the combination of steric, electronic and conformational factors. It is generally believed that, prior to the formation of the metal nitrenoids, an iminoiodane is generated from the reaction between the nitrogen source and the I(III) oxidant. Recent experiments57 suggested that the equilibrium for this transformation is favored for the reactants because no iminoiodane could be detected by 1H NMR. In addition, kinetic resolution could be observed when the racemic nitrene precursor (±)-C17 was employed.55b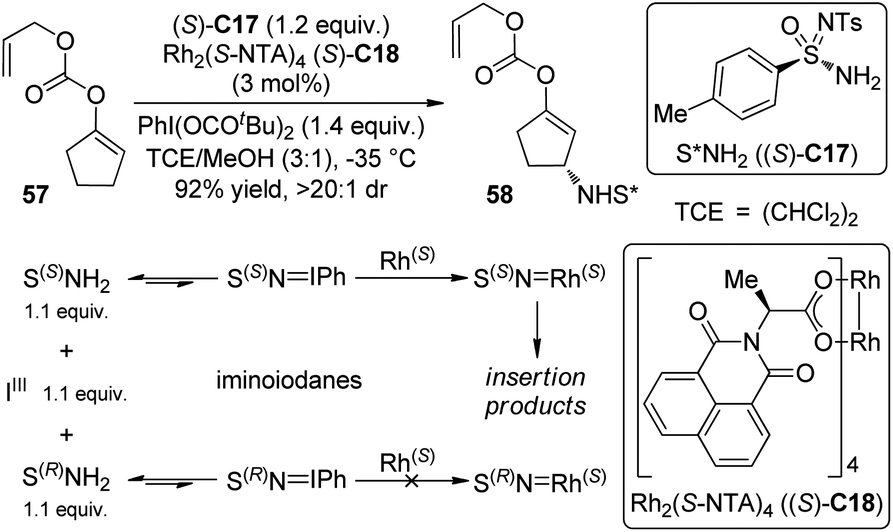 | ||
| Scheme 16 The diastereoselective C–H bond amination reactions reported by Dauban. | ||
Lebel and co-workers58 also realized Rh-catalyzed diastereoselective nitrene C–H bond insertion reactions of alkenes using a readily available chiral nitrene precursor N-tosyloxycarbamate (R)-C19. By utilizing the dirhodium complex Rh2(S-Br-NTTL)4 (S)-C20 as the catalyst, E-β-ethylstyrenes and 2-phenylcyclohexene could be converted to the corresponding allylic amination products in moderate yields and good diastereoselectivity (Scheme 17). The Ph-Troc protecting group could be easily removed using Zn/HOAc to afford the allylic amine·HCl without racemization. On the other hand, Z-β-ethylstyrenes, E- or Z-β-methylstyrenes and terminal styrenes gave aziridination products with varied levels of diastereocontrol.
 | ||
| Scheme 17 The diastereoselective C–H bond amination reactions reported by Lebel. | ||
Hashimoto and co-workers59 have documented the asymmetric synthesis of α-amino ketones by the Rh-catalyzed enantioselective amination reactions of silyl enol ethers derived from acyclic ketones or enones with (arenesulfonylimino)phenyliodanes. Interestingly, when 1-triethylsiloxy-1-cyclohexene 61 was subjected to this reaction, the expected α-amination product 64 could not be detected. Instead, β-amino ketone 63 derived from asymmetric allylic C–H bond insertion of Rh nitrenoid species was obtained. After further examination of the reaction parameters, the dirhodium complex Rh2(S-TCPTTL)4 (S)-C4 was identified as the optimal chiral catalyst, affording 63 in 79% yield with 72% ee (Scheme 18).60 By utilizing this method to construct the first chiral center, Hashimoto and co-workers accomplished the asymmetric synthesis of the key Overman's intermediate61 for the amaryllidaceae alkaloid (−)-pancracine.62
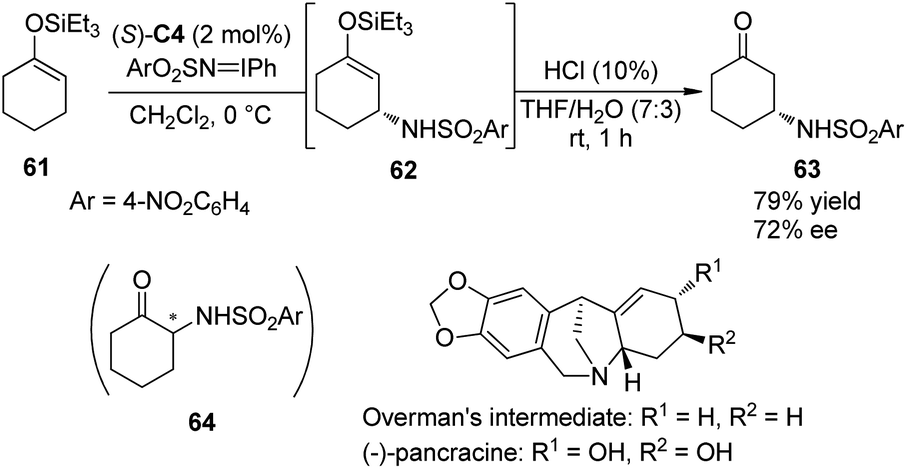 | ||
| Scheme 18 The enantioselective formal synthesis of (−)-pancracine reported by Hashimoto. | ||
(S)-(+)-Dapoxetine (Priligy) is a potent selective serotonin reuptake inhibitor (SSRI) with a short half-life and has been developed specifically for the treatment of premature ejaculation (PE).63 In 2010, Kang and Lee64 uncovered a highly efficient enantioselective synthesis of dapoxetine using the Du Bois asymmetric C–H bond amination reaction55c of the prochiral sulfamate 65 as the key transformation (Scheme 19). The (S)-(+)- and (R)-(−)-dapoxetine could be synthesized in 33% and 34% overall yields for five steps starting form 3-phenyl-1-propanol. During the course of this study, however, the absolute configuration of the major enantiomer of 66 prepared by the Du Bois asymmetric C–H bond amination reaction with Rh2(S-NAP)4 (S)-C21 as the catalyst, was determined to be (R), not (S) as was originally reported.55c
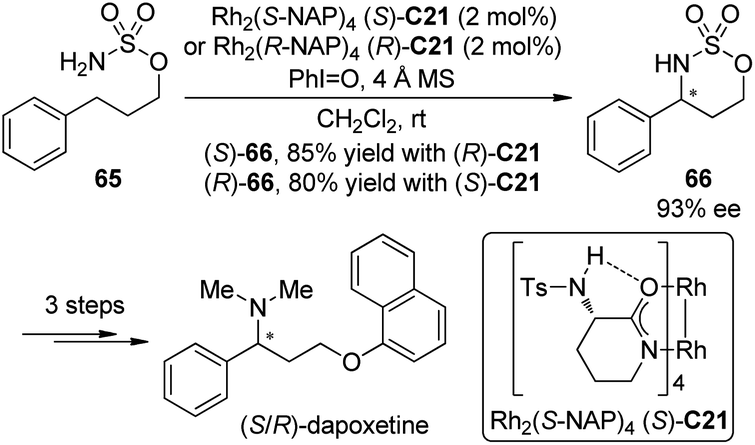 | ||
| Scheme 19 The enantioselective synthesis of dapoxetine reported by Lee. | ||
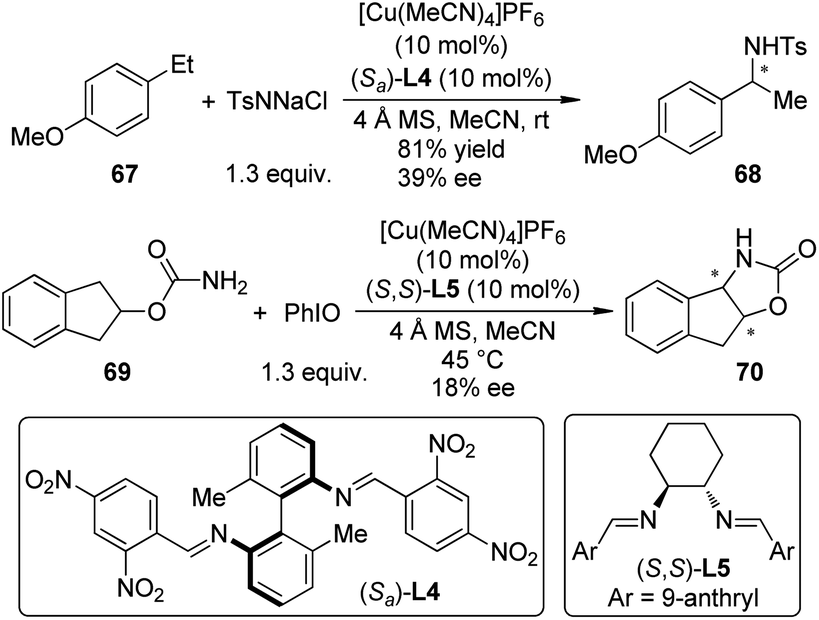
In 2010, Barman and Nicholas65 reported the Cu-catalyzed intermolecular benzylic amination reactions employing chloramine-T (TsNNaCl) as the aminating agent.66 Several classes of ligands including α-amino acids, diphosphines, diamines, diimines, etc. could enable the amination effectively. Low to moderate ee's were obtained when homochiral diimine ligand (Sa)-L4 was used for the benzylic amination of 4-ethylanisole 67 (Scheme 20). Further mechanistic probes by combining experimental and computational methods67 suggested that a ground-state triplet Cu–imido complex [(diimine)Cu![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NTs]+ was the active aminating species, which transferred the NTs unit to the benzylic substrates via a stepwise, radical process. Shortly after this work, Barman and Nicholas68 reported the synthesis of N,O-heterocycles by the intramolecular C–H bond amination reactions of carbamates and sulfamates with PhIO as the oxidant. Similar catalytic system ([Cu(MeCN)4]PF6 + (S,S)-L5), for example, was also tested, yet only 18% ee was obtained for the tricyclic product 70, probably due to the radical nature of this amination process.
NTs]+ was the active aminating species, which transferred the NTs unit to the benzylic substrates via a stepwise, radical process. Shortly after this work, Barman and Nicholas68 reported the synthesis of N,O-heterocycles by the intramolecular C–H bond amination reactions of carbamates and sulfamates with PhIO as the oxidant. Similar catalytic system ([Cu(MeCN)4]PF6 + (S,S)-L5), for example, was also tested, yet only 18% ee was obtained for the tricyclic product 70, probably due to the radical nature of this amination process.
 | ||
| Scheme 20 The Cu-catalyzed asymmetric C–H bond amination reactions reported by Nicholas. | ||
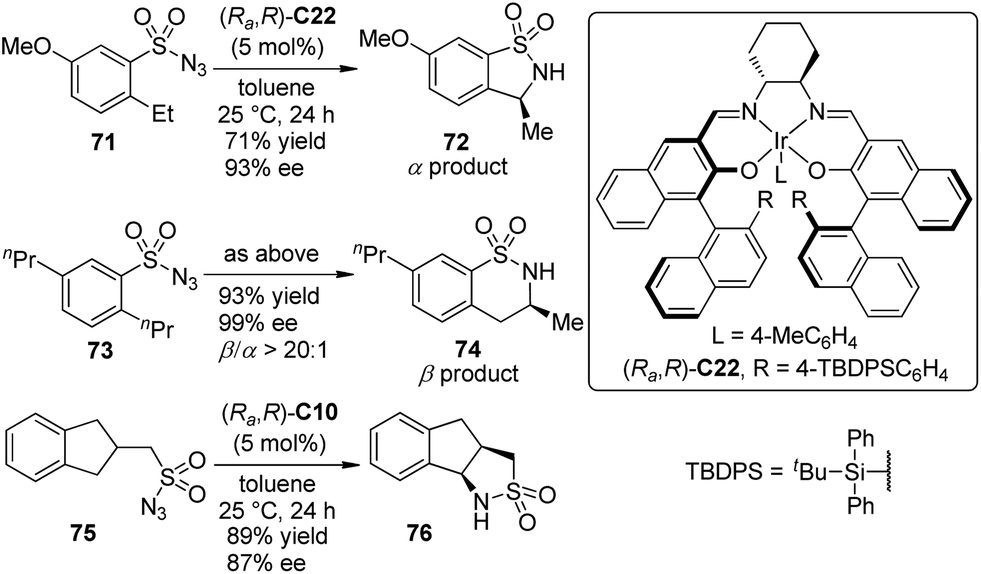
Based on their successes on Ir–salen complex catalyzed asymmetric carbenoid C–H and Si–H bond insertion reactions,39,69 Katsuki and co-workers70 demonstrated the enantioselective intramolecular benzylic C–H bond amination reaction using sulfonyl azide compounds as the atom-economical nitrene precursors. The sterically bulky Ir–salen complex (Ra,R)-C22 exhibited the superior activity, permitting the benzylic (α position) C–H bond amination reaction of 2-ethyl substituted substrates 71 with high efficiency (Scheme 21). Surprisingly, the homobenzylic (β position) C–H bond amination reaction occurred preferentially when several 2-alkyl-substituted derivatives other than 2-ethyl substituted ones 73 were employed, delivering the six-membered sultam 74 with high enantioselectivity. It was inferred that the control of the regio- and stereoselectivity largely depends on whether the substrate can adopt a conformation suitable for the orbital interaction between the C–H and Ir–nitrenoid bonds for the reaction. A higher enantioselectivity was obtained for the substrates bearing a more flexible acyclic substituent. Notably, the scope of this reaction was not limited to arylsulfonyl azides. The desymmetrization of substrate 75 with Ir–salen complex (Ra,R)-C10 as the catalyst led to tricyclic sultam 76 in 89% yield and 87% ee.
 | ||
| Scheme 21 The Ir-catalyzed asymmetric intramolecular C–H bond amination reactions reported by Katsuki. | ||
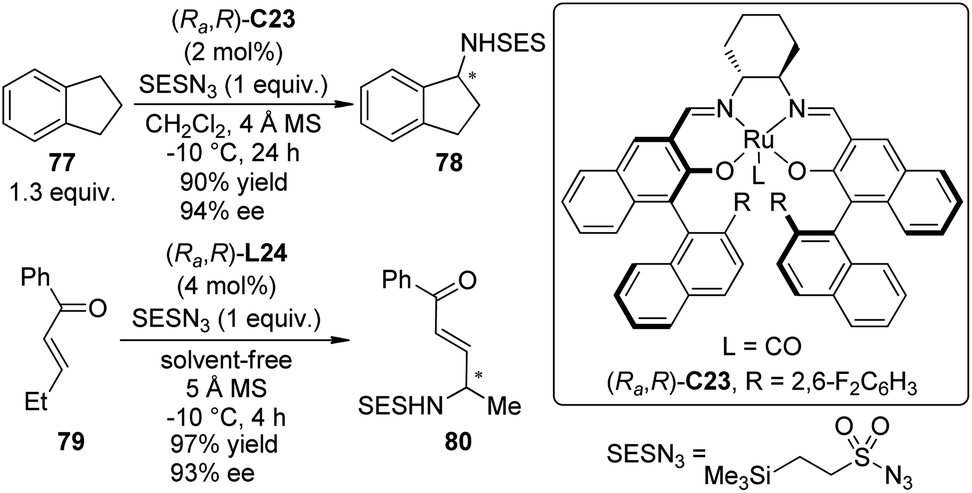
Very recently, the Katsuki group71 achieved enantioselective intermolecular C–H bond amination at the benzylic and allylic positions with 2-(trimethylsilyl)ethanesulfonyl azide (SESN3) as the nitrene precursor. A chiral Ru(CO)–salen complex (Ra,R)-C23 proved to be the best catalyst for this transformation while the Ir–salen complex (Ra,R)-C22 was not effective (Scheme 22). The amination of various alkylarenes and alkenes went smoothly in CH2Cl2, or alternatively, under solvent-free conditions. The reactions could be highly regioselective that an ethyl group was aminated selectively even in the presence of an nPr group. Preliminary mechanistic investigation supported that the C–H bond amination through a putative metal nitrenoid intermediate proceeds in a concerted manner. However, the involvement of a short-lived radical species cannot be completely ruled out.72
 | ||
| Scheme 22 The Ru-catalyzed asymmetric intermolecular C–H bond amination reactions reported by Katsuki. | ||
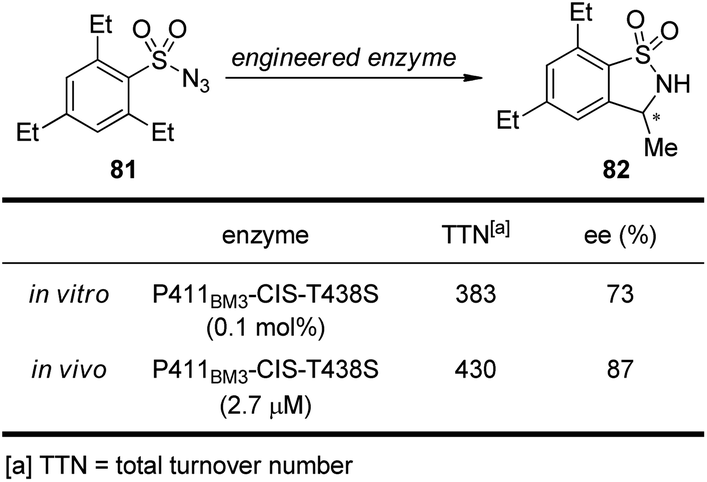
It is well known that cytochrome P450 enzymes are powerful catalysts for the C–H bond oxidation reactions in biological systems.73 However, the parallel C–H bond amination reactions via nitrenoid intermediates under natural enzyme catalysis have not been reported. In 2013, Arnold and co-workers74 realized engineered cytochrome P450 enzyme-catalyzed asymmetric intramolecular C–H bond amination reactions using sulfonylazides as the nitrenoid precursors. As shown in Scheme 23, the modified enzyme P411BM3-CIS-T438S exhibits enhanced catalytic activity and enantioselective control both in vitro and in vivo.
 | ||
| Scheme 23 The engineered cytochrome P450 enzymes catalyzed asymmetric intramolecular C–H bond amination reactions reported by Arnold. | ||
2.3. C–H bond insertion by metal oxo species
Compared with the catalytic asymmetric C–H bond insertion reactions by metal carbenoids and metal nitrenoids, the asymmetric C–H bond oxidation reactions by metal oxo species still remain challenging in organic synthesis.75 One of the most difficult issues is how to avoid or reduce the over oxidation of the newly formed C(sp3)–O bonds. In addition to the extensively studied biological catalytic systems such as cytochrome P450 enzymes,73 a few artificial catalysts based on transition metals including Fe,76 Mn77 and Ru78 have emerged as powerful tools for this transformation.A recent contribution to this field reported by Simonneaux and co-workers79 demonstrated the utility of the water-soluble Mn–porphyrin complex C24 as the chiral catalyst to enable the asymmetric benzylic hydroxylation of arylalkane 83 using H2O2 as the oxidant (Scheme 24). The reactions were conducted in the mixed MeOH–water solution. The corresponding secondary alcohols 84 were delivered as the major products with up to 57% ee. Notably, imidazole was found to play an important role in this catalytic system, working as an axial ligand to the Mn center.80
 | ||
| Scheme 24 The Mn-catalyzed asymmetric C–H bond hydroxylation reactions reported by Simonneaux. | ||
A significant advance to be highlighted here is the predictably selective aliphatic C–H bond oxidation reactions developed by the White group.81 By utilizing the highly electrophilic, sterically encumbered iron complex Fe(PDP) C25 as the catalyst, in combination with H2O2 as the oxidant, the inert tertiary C–H bonds in the complex molecules could be hydroxylated.82 Predictable selectivity was achieved on the basis of the electronic and steric properties of the C–H bonds without the need for a directing group. The products of the hydroxylation at the most electron-rich and sterically accessible C–H bonds (such as 87) could be obtained in synthetically useful yields (∼50% yields with 1 equiv. of substrates) when acetic acid, which probably worked as a ligand to the iron center, was used (Scheme 25(a)). If no suitable tertiary C–H bonds existed in the substrates, selective oxidation of methylene moieties to ketones could be achieved by the same catalytic system.83 Inspired by the importance of acetic acid, White and co-workers84 later disclosed that the carboxylic acid containing substrate 89 were more reactive than the corresponding methyl ester 88 towards the oxidation reaction. The site selectivity was controlled by the carboxyl group, even overcoming the unfavorable electronic, steric and stereoelectronic effects within the substrates to deliver the γ-lactone products 90 (Scheme 25(b)). The proposed role of carboxylic acid as a ligand was further confirmed that chiral substrate 91 exhibited matched/mismatched behavior with chiral Fe(PDP) ligand. Furthermore, when racemic 91 was subjected to the standard reaction conditions, enantiomerically pure Fe(PDP) catalyst could promote kinetic resolution of the starting material, leading to moderate level of ee's of both recovered 91 and lactone product 92 (Scheme 25(c)). A plausible mechanism was also proposed for the aliphatic C–H bond oxidation reactions.84,85 The Fe(PDP) catalyst reacts with H2O2 and carboxylic acid substrate to generate an iron oxo carboxylate as the active oxidant, which abstracts a hydrogen atom to afford a short-lived carbon-centered radical. The subsequent hydroxyl rebound step occurs very rapidly with complete stereoretention of the direct hydroxylated product as well as the lactone. Noticeably, the reaction profile of the alkyl radical diverts in a substrate-depended manner. For some carboxylic acid substrates, the second hydrogen abstraction furnishes the olefin intermediates. Further oxidation of these intermediates typically produces 10–20% yields of hydroxylactones as the “double oxidation” products (Scheme 25(d)).
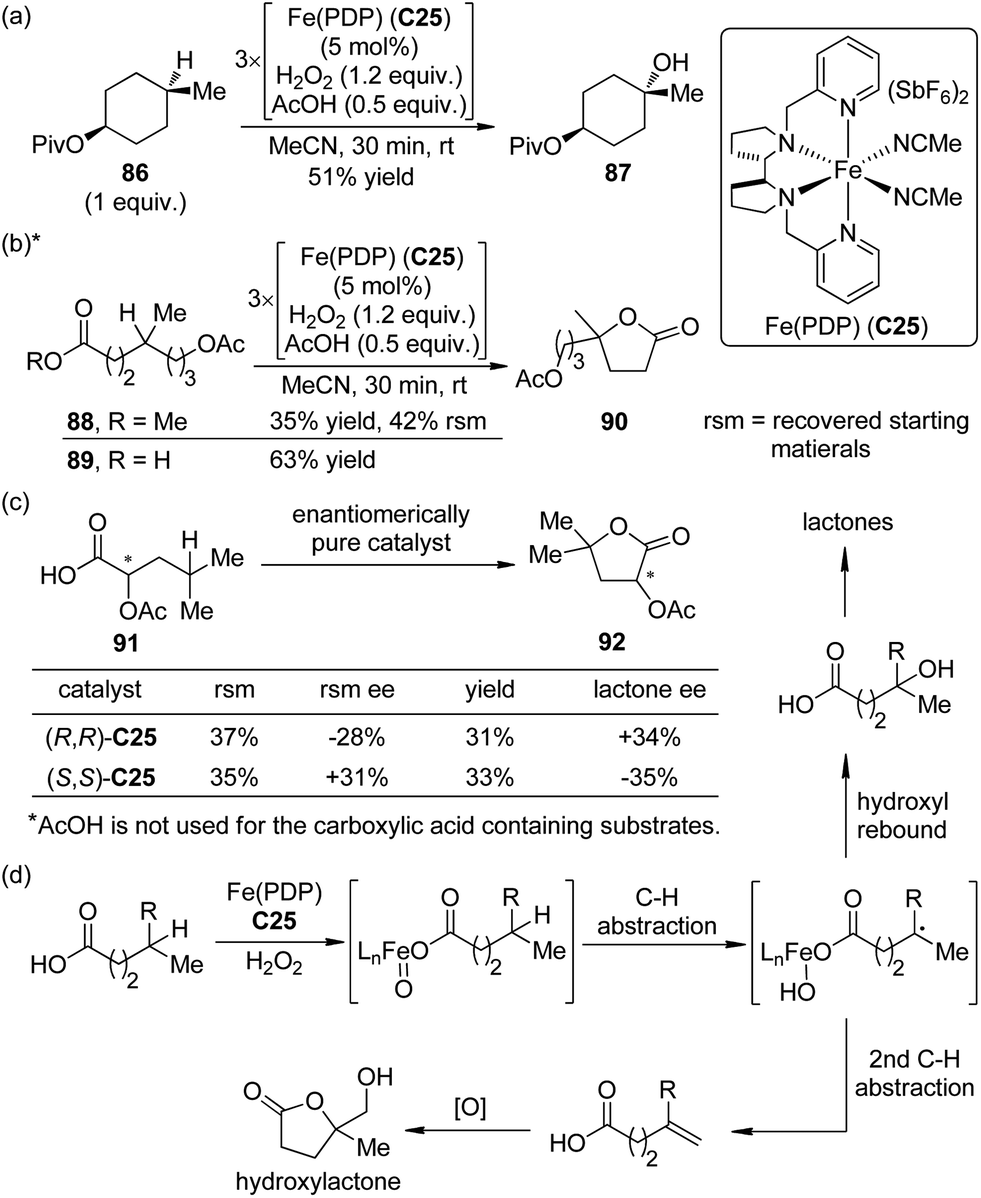 | ||
| Scheme 25 The selective aliphatic C–H oxidation reactions reported by White. | ||
3. Cross dehydrogenative coupling (CDC) reactions
The cross dehydrogenative coupling (CDC) reactions,86 pioneered by the Murahashi group87 and the Li group,88 refers to the direct coupling reactions between one C(sp3)–H bond α to a nitrogen, oxygen atom, or a carbonyl group and various C(sp)–H, C(sp2)–H or C(sp3)–H bonds under oxidative conditions without requiring preactivation. It has evolved to be an important method for the construction of C–C bonds in recent years. Asymmetric variants of this transformation have also been realized.89In 2010, Gong and co-workers90 reported the Lewis acid-catalyzed enantioselective CDC reactions between 3-indolylmethyl C–H bonds with 1,3-dicarbonyls. In the presence of a catalytic amount of Cu(OTf)2 and amino-indanol derived BOX ligand L6 as the chiral catalyst, in combination with DDQ as the stoichiometric oxidant, a variety of structurally diverse 3-arylmethylindole derivatives 93 could react with 1,3-diesters 94, affording the corresponding products 95 in up to 99% yield and 96% ee (Scheme 26(a)). Based on further experimental and computational investigations, a plausible reaction mechanism was proposed. Since no ESR signal was observed for the reaction mixture, a cationic rather than radical species was believed as the key intermediate. Lewis acid could enhance the oxidizing ability by coordinating to one oxygen atom of DDQ, facilitating the dehydrogenation of 3-arylmethylindole. The resultant Cu phenoxide deprotonated the dibenzyl malonate to generate a chiral anion, which enantioselectively attacked the vinylogous iminium cation obtained from the first deprotonation to yield the final CDC product (Scheme 26(b)).
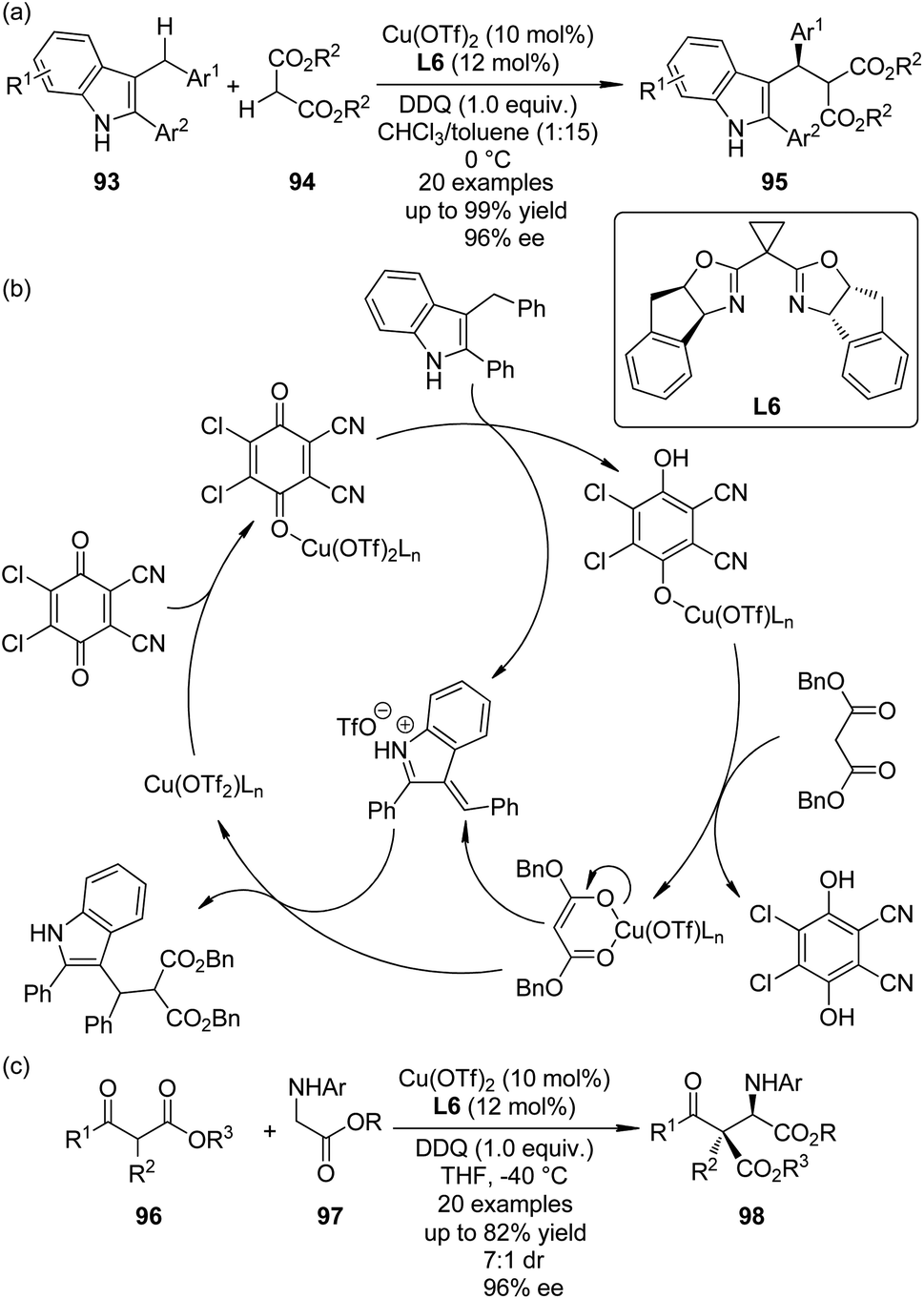 | ||
| Scheme 26 The Cu-catalyzed asymmetric CDC reactions reported by (a), (b) Gong and (c) Wang, respectively. | ||
Shortly after Gong's work, the Wang group91 reported an efficient enantioselective synthesis of α-alkyl α-amino acids 98 via the CDC reactions of glycine esters 97 with α-substituted β-ketoesters 96 enabled by the same chiral Cu catalyst and oxidant (Scheme 26(c)). Besides, this strategy could be extended to asymmetric synthesis of C1-alkylated tetrahydroisoquinolines by the similar reactions between Horner–Wadsworth–Emmons reagents and N-aryl tetrahydroisoquinolines. Different form Gong's results, preliminary mechanistic probe indicated that a radical cation intermediate might be involved in the catalytic cycle because a small amount of this species could be captured by 2,6-di-(tBu)2-4-methylphenol (BHT).
Enantioselective organo-SOMO activation92 has witnessed considerable development in last several years and become an important complementary method to the strategies of LUMO (iminium) activation and HOMO (enamine) activation of secondary amine catalysis. In 2009, Nicolaou and co-workers93 realized an asymmetric intramolecular α-arylation reactions of aldehydes by utilizing chiral secondary amine catalyst (R,R)-C26 and Ce(NH3)2(NO3)6 (CAN) as the oxidant. When 1,3,4-trisubstituted arene substrate 99 was used, the para, meta selectivity was observed, giving the corresponding arylation product 100 in moderate yield and satisfactory ee (Scheme 27(a)). The application of this methodology was further demonstrated by the total synthesis of demethyl calamenene, a potent cytotoxic agent against human adenocarcinoma A549.94
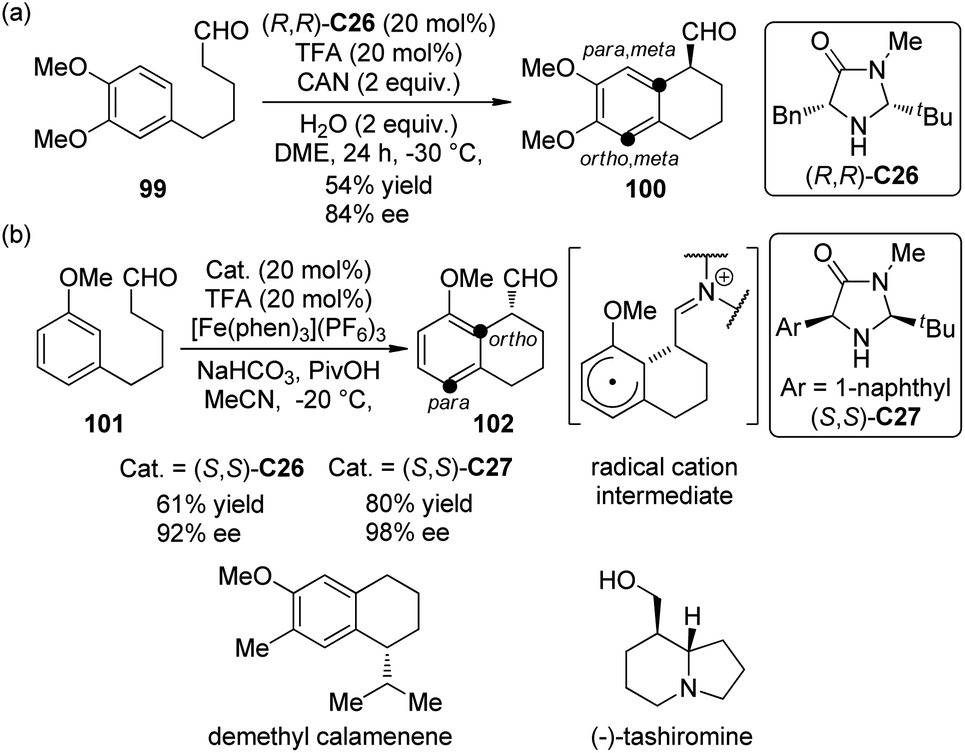 | ||
| Scheme 27 The organocatalytic asymmetric intramolecular α-arylation reactions of aldehydes reported by (a) Nicolaou and (b) MacMillan, respectively. | ||
Almost at the same time, MacMillan and co-workers95 reported a very similar asymmetric intramolecular α-arylation reactions of aldehydes catalyzed by chiral secondary amine catalysts (S,S)-C26 or (S,S)-C27. [Fe(phen)3](PF6)3 was identified as the optimal oxidant. The reaction was highly regioselective for 1,3-disubstituted arene substrate 101 and only ortho-functionalization product 102 was formed (Scheme 27(b)). This SOMO α-arylation could be combined with Hantzsch ester hydride reduction96 in a two-step organocatalytic sequence, and also be used in the total synthesis of (−)-tashiromine.97 DFT calculations by Houk, MacMillan and co-workers98 confirmed the radical cation nature of the cyclized intermediates (Scheme 27(b)). The para, meta selectivity for 99 and the ortho selectivity for 101 were also explained based on the computational results.
Impressively, Rendler and MacMillan99 accomplished the radical-mediated polyene cyclization reactions via organo-SOMO catalysis. Polyolefins that incorporate an alternating sequence of polarity-inverted CC bonds (acrylonitrile and isobutene) were the suitable substrates. The electronic match between nucleophilic olefins with electron-deficient radical intermediates (and vice versa) allowed the cascade 6-endo-trig cyclization in the presence of chiral imidazolidinone (R,R)-C27. Notably, the reactions with strong oxidants like CAN and [Fe(phen)3](PF6)3 could not afford the desired products. On the contrary, Cu(OTf)2 was found as an effective oxidant. More importantly, slow addition of the oxidant via syringe pump, which kinetically preferred for the unimolecular cascade process over the interrupted pathway involving bimolecular radical oxidation, greatly improved the yields. By applying this strategy, the hexacyclization adduct 104 could be obtained as a single diastereomer in 62% yield and as many as 6 new C–C bonds and a total of 11 contiguous chiral centers were constructed from a simple acyclic starting material 103 (Scheme 28).
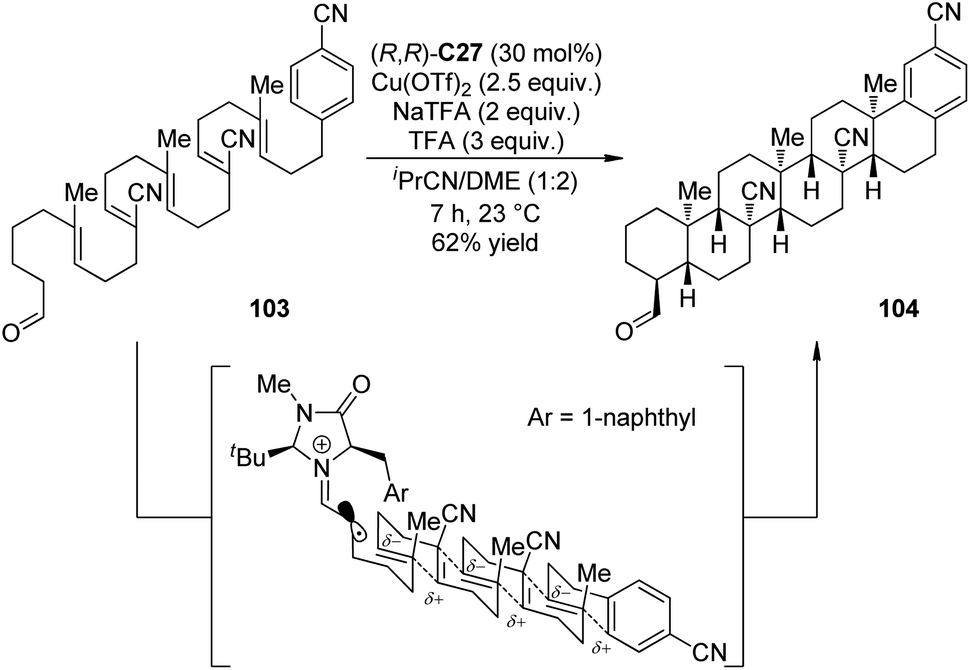 | ||
| Scheme 28 The organocatalytic asymmetric radical-mediated polyene cyclization reactions reported by MacMillan. | ||
Organocatalytic asymmetric intermolecular dehydrogenative α-functionalization of aldehydes has also been reported. Cozzi and co-workers100 disclosed that aliphatic aldehyde 105a could react with xanthene 106 smoothly to deliver the corresponding product 107a in 90% yield and 79% ee when (R,R)-C26 was used as the catalyst (Scheme 29). The SN1-type mechanism was believed to operate via the xanthene-derived benzylic carbocation generated from the oxidation by DDQ. Jang and co-workers101 found that the asymmetric α-alkylation of aldehydes with xanthene could be achieved under electro-oxidation conditions to afford the corresponding products in up to 68% yield and 68% ee with (R,R)-C26 as the catalyst. Cui, Jiao and co-workers102 developed an enantioselective CDC protocol that involved molecular O2 as the oxidant.103 By using (S)-C28 as the chiral catalyst, the α-functionalized product 107c was obtained in 71% yield and 92% ee. Although the generated water during the formation of the enamine was considered possible to lead to side reaction with carbocation, yet in this system, 10 equiv. of water was employed as an additive to enhance the enantioselectivity. It was postulated that xanthene could be oxidized to peroxides 108 and/or 108′ by a radical pathway with O2. Further investigation showed that 108′ might be the key intermediate of this transformation since independently synthesized 108′ could directly react with the aldehydes giving similar results. Xiao and co-workers104 also realized organocatalytic asymmetric CDC reactions of aldelydes with benzylic C–H bonds utilizing the chiral diarylprolinol-based silyl ether (S)-C29 as the catalyst and DDQ as the oxidant. The desired alcohol products 109 were delivered in up to 95% ee after the subsequent reduction by NaBH4.
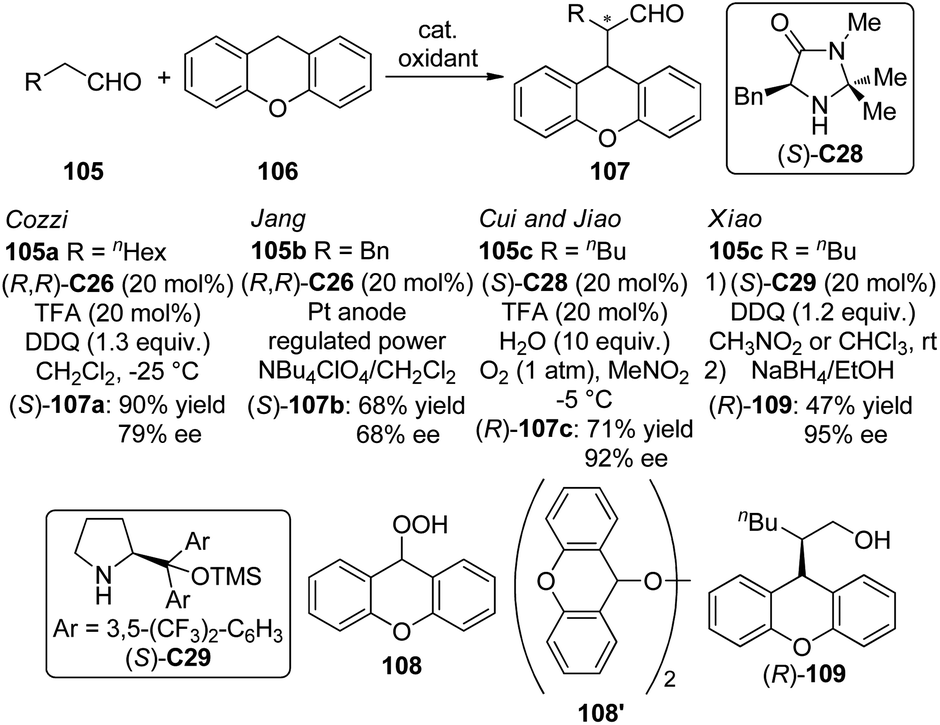 | ||
| Scheme 29 The organocatalytic asymmetric intermolecular α-functionalization reactions of aldehydes reported by several groups. | ||
Recently, Chi and co-workers105 reported the asymmetric CDC reactions between aldehydes and tertiary amines. An extensive survey revealed that (S)-C29 in combination with CuBr2 could be employed as an effective cooperative catalytic system when tBuOOH (TBHP) was used as the terminal oxidant. It was also found that using 50 mol% of AcOH could lead to improved yields. Under the optimized conditions, the reactions of various N-aryl tetrahydroisoquinolines 110 and aliphatic aldehydes 111 went smoothly. Because the direct Mannich adducts were rather unstable, the in situ NaBH4 reduction was conducted to afford their corresponding alcohols 112 with reasonable diastereoselectivity (the ratio of anti-112/syn-112 up to 72:28) and excellent enantioselectivity (up to 94% ee for anti-112 and 99% ee for syn-112) in up to 71% combined yield (Scheme 30). In consistence with the observations by the Wang group,91 the reaction pathway was suggested to involve a single-electron transfer (SET) radical mechanism. The yields of the coupling products decreased dramatically when the radical inhibitor BHT was added. Meanwhile, a racemic version of this CDC reaction was also realized via metal catalysis alone.
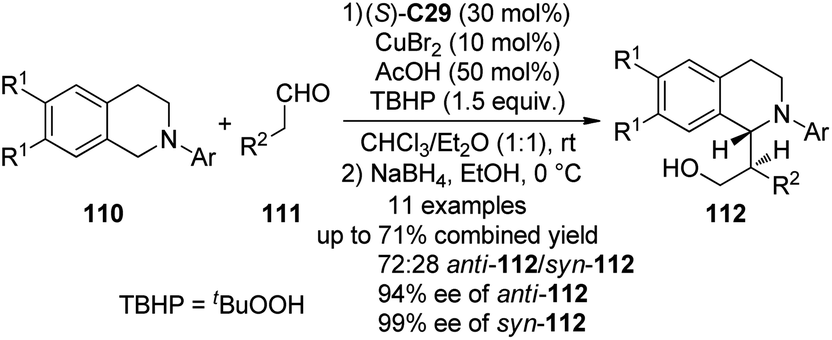 | ||
| Scheme 30 The organocatalytic asymmetric CDC reactions of aldehydes reported by Chi. | ||
The asymmetric CDC reactions between ketones and tertiary amines are much more challenging compared with their aldehyde counterparts. Several groups have been engaged in this subject. Klussmann and co-workers106 found that such reactions between tetrahydroisoquinolines 113 and acetone could be achieved by combined vanadium(IV) salt VO(acac)2 and chiral organocatalysts when TBHP was employed as the stoichiometric oxidant. However, the desired product 114a was obtained with only 17% ee in the presence of (S)-C30 (Scheme 31). Further attempts to improve the enantioselectivity by using chiral vanadium complexes were failed because the enantiopure product slowly racemized under the reaction conditions. Rueping and co-workers,107 on the other hand, realized similar transformations by utilizing the dual catalytic system containing photoredox catalyst Ru(bpy)3(PF6)2108 and L-proline (S)-C31 under the irradiation of 5 W fluorescent bulb, albeit with low enantioselectivity (8% ee for 114a). Instead of using the transition metal-based catalysts, Tan and co-workers109 utilized the organic dye Rose Bengal as the environmentally benign photoredox catalyst to promote the CDC reactions of 113 with nitroalkanes or ketones. The green LED was identified as the light source matched with Rose Bengal. With L-prolinol methyl ether (S)-C32 as the chiral secondary amine catalyst, however, the coupling product 146b could only be obtained with 15% ee.
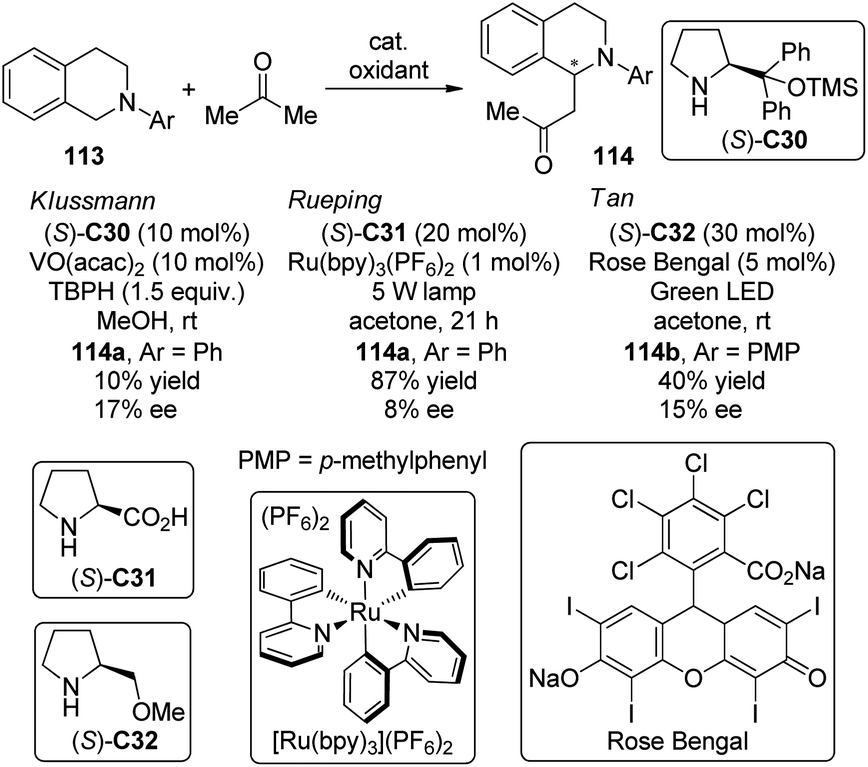 | ||
| Scheme 31 The asymmetric CDC reactions of ketones reported by several groups. | ||
Shortly after Klussmann's work, Xie and Huang110 reported the CDC reactions between N-substituted glycine esters with unmodified ketones by the cooperative catalytic system consisting of Cu(OAc)2·H2O and pyrrolidine. TBHP and DDQ were found as the two effective oxidants suitable for acetone and cyclic ketone substrates, respectively. No enantioselectivity was achieved with all the chiral ligands employed. When methyl L-proline ester (S)-C33 was used as the chiral organocatalyst for the reaction of glycine ester 115 and acetone, the product 116 was generated with 15% ee and decreased yield (47% vs. 73% with pyrrolidine) (Scheme 32).
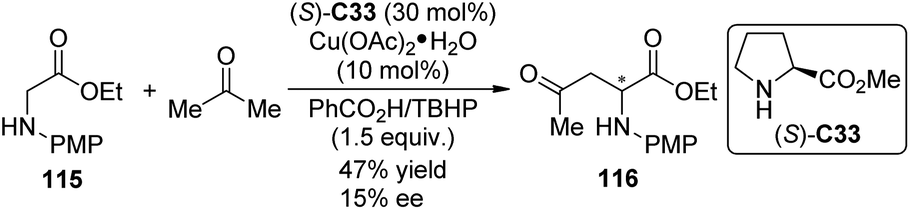 | ||
| Scheme 32 The asymmetric CDC reactions of ketones reported by Huang. | ||
Highly enantioselective CDC reactions between tertiary amines and ketones have not been realized until very recently. By utilizing L-phenylalanine (S)-C34 rather than secondary amine catalysts to activate the ketone substrates, Wang and co-workers111a accomplished the coupling reaction between N-aryl tetrahydroisoquinolines 117 and cyclic ketones 118, delivering the corresponding products 119 in up to 81% yield with good stereocontrol (up to 13:1 dr and 94% ee) when anhydrous iPrOH was added to the reaction (Scheme 33). DDQ again was identified as the optimal stoichiometric oxidant, and the copper salts were not necessary. A chiral organic contact ion-pair interaction was proposed for the reaction. The enamine intermediate was generated from the ketone and the primary amine moiety of the L-phenylalanine. The interaction between this chiral anion and the iminium cation from oxidation probably contributed to the diastereomeric discrimination, which furnished this asymmetric metal-free CDC process. In addition, Wang and co-workers111b realized intramolecular metal-free CDC reactions between tertiary amine and ketone moieties. Modest enantioselectivity (14% ee) was reached when simple chiral phosphoric acid was utilized as the catalyst.
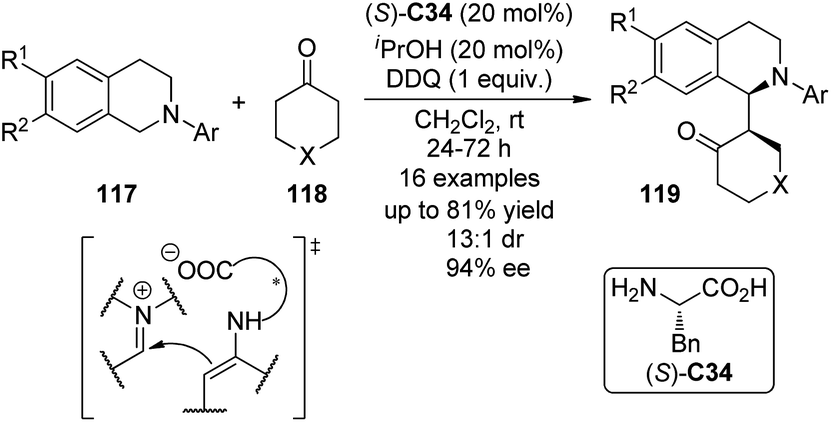 | ||
| Scheme 33 The asymmetric CDC reactions of ketones reported by Wang. | ||
Wang and co-workers112 also described an enantioselective CDC reaction of tertiary amines with the olefinic C–H bonds. In the presence of catalytic amount of quinine C35 in combination with Cu(OTf)2, N-aryl tetrahydroisoquinolines 120 reacted with the α,β-unsaturated aldehydes and ketones 121 smoothly to afford the α-functionalized products 122 (Scheme 34). Molecular O2 was found to be the sole oxidant and the addition of anhydrous Na2SO4 was beneficial to achieve higher yield and enantioselectivity for an array of products (up to 81% yield and 99% ee).
 | ||
| Scheme 34 The asymmetric CDC reactions of α,β-unsaturated aldehydes and ketones reported by Wang. | ||
DiRocco and Rovis113 realized asymmetric α-acylation reactions of tertiary amines 124 by taking advantage of a dual catalytic system of NHC and photoredox catalyst. Since the Breslow intermediate or the NHC itself can also be oxidized, a judicious choice of the oxidant and the photoredox catalyst is crucial. After careful evaluation of the reaction conditions, Ru(bpy)3Cl2 was selected as the optimal photoredox catalyst and m-dinitrobenzene (m-DNB) as the co-oxidant under irradiation with blue light. The aminoindanol-derived NHC (R,S)-C36 (in situ generated from the corresponding triazolium salt using NaH) exhibited the best asymmetric induction. An array of the desired products 125 was afforded in up to 94% yield and 92% ee (Scheme 35).
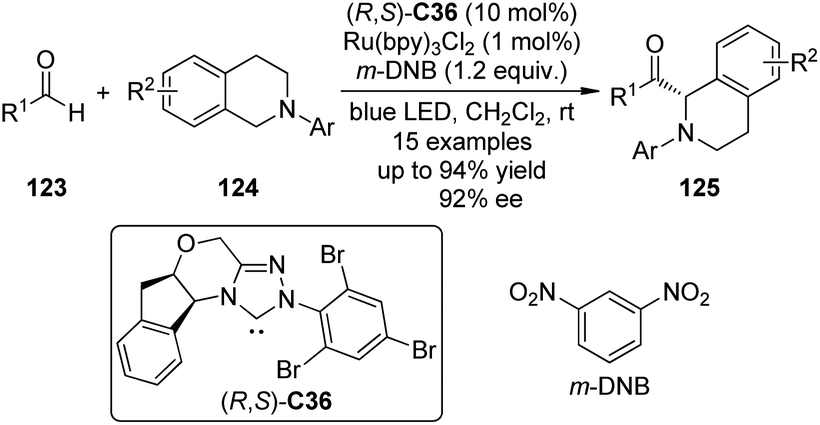 | ||
| Scheme 35 The asymmetric α-acylation reactions of tertiary amines reported by Rovis. | ||
Recently, Jacobsen, Stephenson and co-workers114 developed an enantioselective synthesis of β-amino esters by the dual catalytic system combining photoredox activation and anion binding catalysis that is related to the CDC reactions.115 In these transformations, the N-aryl tetrahydroisoquinolines 126 were firstly oxidized under photoredox conditions with CCl4 as the stoichiometric oxidant. Subsequently, the corresponding iminium intermediates activated by the chiral thiourea catalyst (S,R)-C37 via chloride binding interaction, were attacked enantioselectively by the silyl ketene acetal 127 to generate a variety of β-amino esters 128 in high yields and ee's (Scheme 36).116
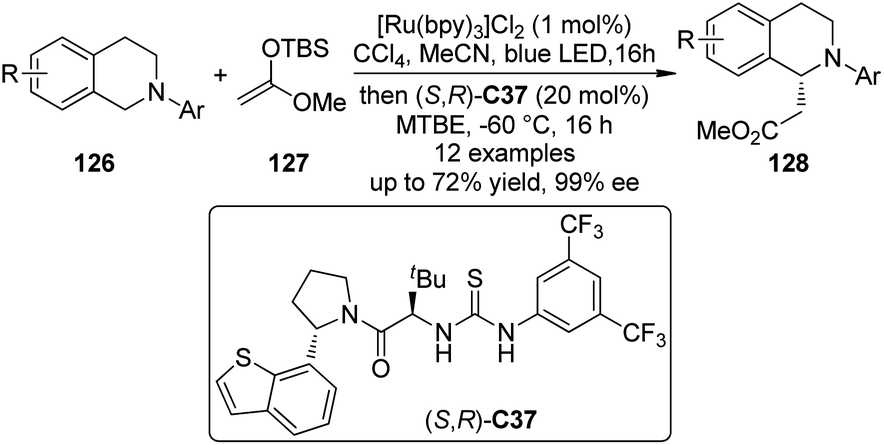 | ||
| Scheme 36 The asymmetric synthesis of β-amino esters reported by Jacobsen and Stephenson. | ||
Enzyme-catalyzed transformation has also been applied for the oxidative C–C coupling reactions. Berberine bridge enzyme (BBE) is a redox enzyme which converts (S)-reticuline as its natural substrate to the berbine derivative (S)-scoulerine at the expense of O2 (Scheme 37(a)).117 Kroutil and co-workers118 disclosed that the BBE is efficient to act as a biocatalyst to promote the coupling reactions of four unnatual 1-benzylisoquinoline substrates 129. After careful examination of the reaction parameters, it was found that the reactions worked well in a two-phase toluene–buffer mixture (7:3, pH = 9) and adding some catalase was necessary to disproportionate the formed H2O2 in order to avoid the inhibition or degradation of the enzyme. Excellent kinetic resolution behavior was observed for all the rac-129 tested, with the reactions stopped at 50% conversion. The products (S)-130 were obtained in up to 42% yield and >97% ee whereas the (R)-129 was recovered in up to 50% yield and >97% ee (Scheme 37(b)). The reactions were also highly regioselective and only minor side products 131 were formed (4–10% yield). Notably, this enzyme-catalyzed reaction could also be performed on a 500 mg scale under mild conditions.
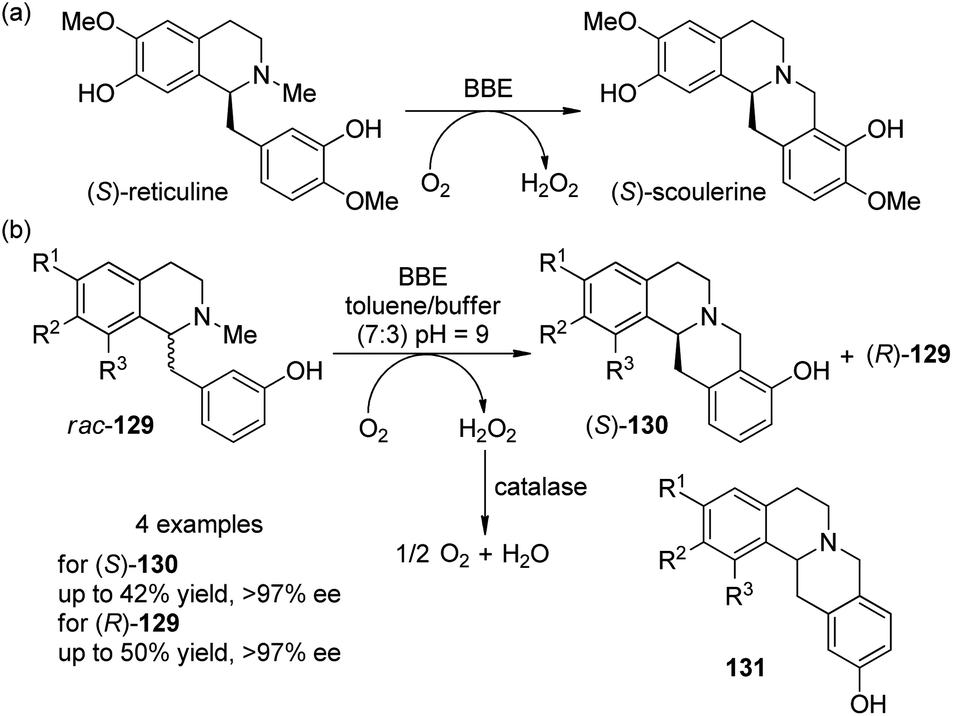 | ||
| Scheme 37 The enzyme-catalyzed oxidative C–C coupling reactions reported by Kroutil. | ||
Asymmetric CDC reactions have been found effective to construct C–N bond recently. Toste and co-workers119 envisioned that the enantioenriched cyclic aminals 133 could be delivered via intramolecular CDC reactions of substrates 132 catalyzed by a chiral counteranion with an appropriately chosen cationic oxidant. Since conventional chiral phosphate anions that only possess steric demanding groups on the skeleton of the catalyst failed to provide an effective chiral environment for the catalysis, the authors developed a series of triazole-containing chiral phosphoric acids like (S)-C38 which exhibit high level asymmetric induction and catalytic activity for the designed reactions (Scheme 38). It was hypothesized that the triazole moieties might interact with the amide-containing substrates probably via attractive hydrogen-bonding interaction, leading to additional discrimination of the diastereomeric transition states.
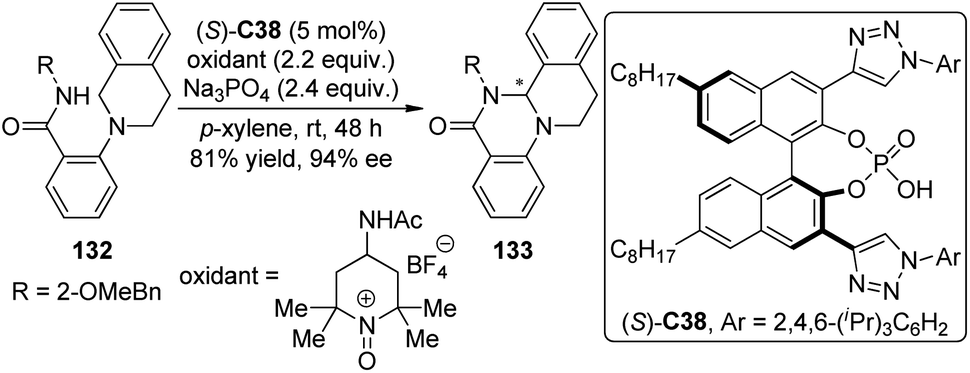 | ||
| Scheme 38 The asymmetric intramolecular CDC reactions reported by Toste. | ||
4. [1,5]-Hydride transfer reactions
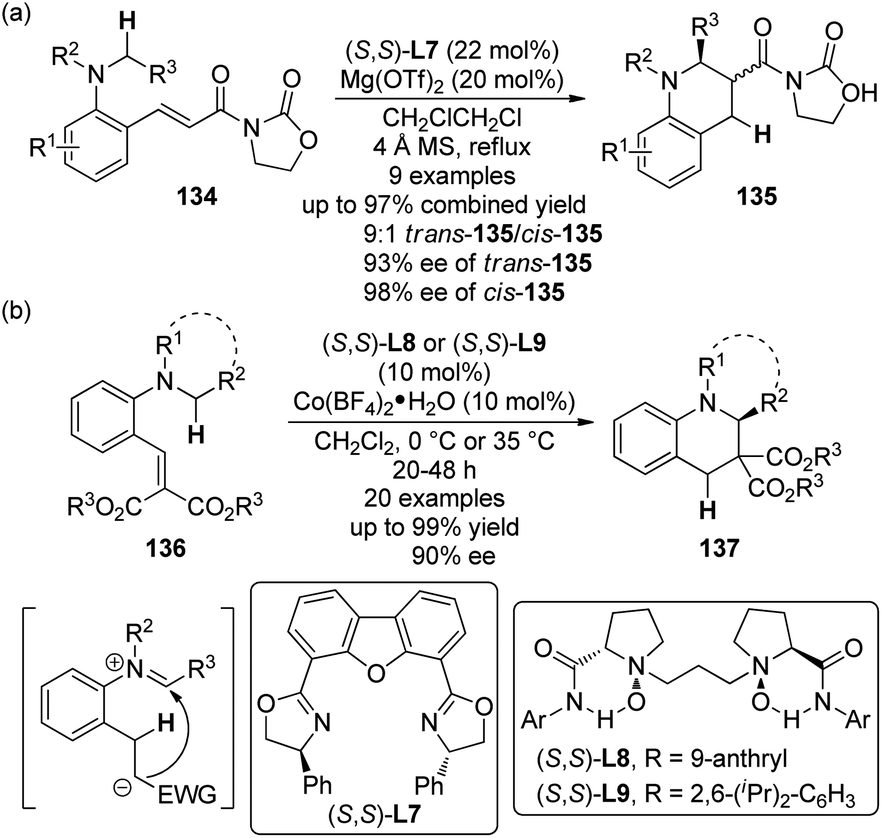
Another important strategy to furnish the C(sp3)–H bond functionalization at the α position to a tertiary amine group is the sequential intramolecular hydride transfer/ring closure reaction taking advantage of the “tert-amino effect”.120 This type of transformation features the redox-neutral process and therefore avoids the use of external oxidants. Traditionally, such reactions were typically thermal promoted.121 Recent contributions revealed that Lewis acids are also feasible to catalyze these reactions,122 thus making it possible to develop their asymmetric variants.123The first catalytic highly enantioselective [1,5]-hydride transfer reactions was reported by the Seidel group.124 The ortho-vinyl tertiary anilines 134 were selected as the substrates and the acyl oxazolidinone moiety was introduced to act as an acceptor for the chelation to a chiral metal complex. A screening of various Lewis acids and chiral ligands indicated that Mg(OTf)2 in combination with DBFox ligand (S,S)-L7 could give the best results, affording the desired tetrahydroquinoline products 135 via the [1,5]-hydride transfer and subsequent nucleophilic addition to the iminium ion generated in situ (Scheme 39(a)). Generally, good diastereoselectivity (up to 9:1 dr) and enantioselectivity (up to 93% ee for trans-135 and 98% ee for cis-135) were observed for a series of products, despite at elevated temperature (84 °C). The reaction became very sluggish at lower temperature.
 | ||
| Scheme 39 The Lewis acid-catalyzed asymmetric [1,5]-hydride transfer reactions reported by (a) Seidel and (b) Feng, respectively. | ||
On the other hand, by utilizing their chiral N,N′-dioxide ligands ((S,S)-L8 and (S,S)-L9),125 Feng and co-workers126 developed a [1,5]-hydride transfer induced asymmetric synthesis of tetrahydroquinoline derivatives 137 catalyzed by cobalt complexes (Scheme 39(b)). Notably, the reactions exhibited relatively broaden substrate scope and higher reactivity in that the reactions were typically conducted at 35 °C or 0 °C. In some cases, the catalyst loading could be lowered to 2 mol% without erosion of the ee values. Besides, a working model to explain the stereoinduction was also proposed.
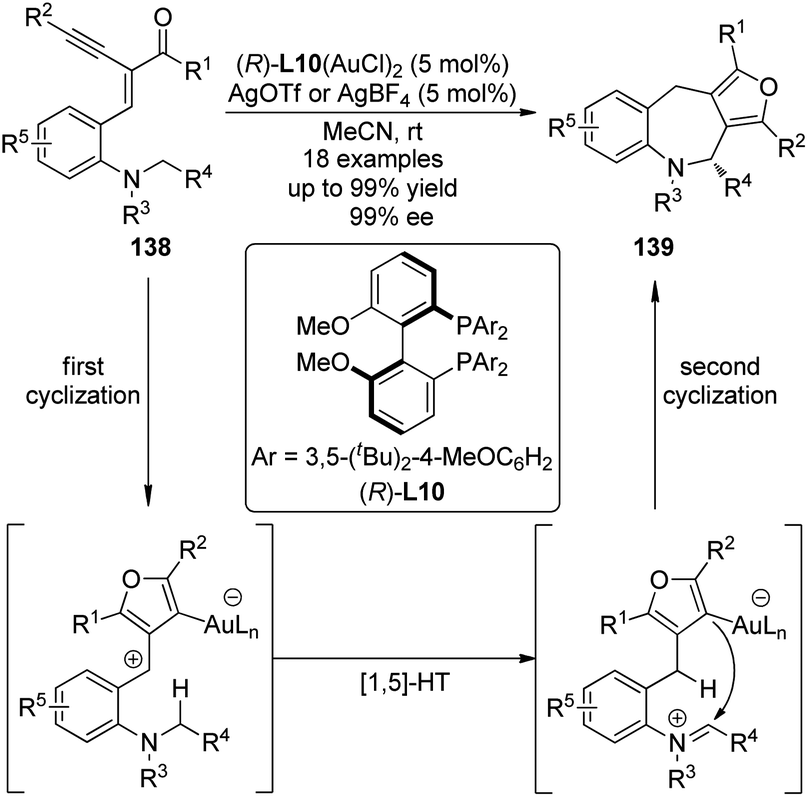
The Zhang group127 successfully integrated the [1,5]-hydride transfer reaction into the Au-catalyzed domino process128 for the synthesis of furan-fused tetrahydroazepine derivatives 139. The gold complex generated from the chiral ligand (R)-L10 and Au(SMe2)Cl enabled the first cyclization of the starting materials 138 affording the furan intermediates which underwent the subsequent [1,5]-hydride transfer and the final ring closing reaction to furnish the desired products (Scheme 40).129 The reactions could occur at room temperature with high enantioselectivity and thus offered a unique access to enantioenriched polycyclic compounds.
 | ||
| Scheme 40 The Au-catalyzed asymmetric domino reactions reported by Zhang. | ||
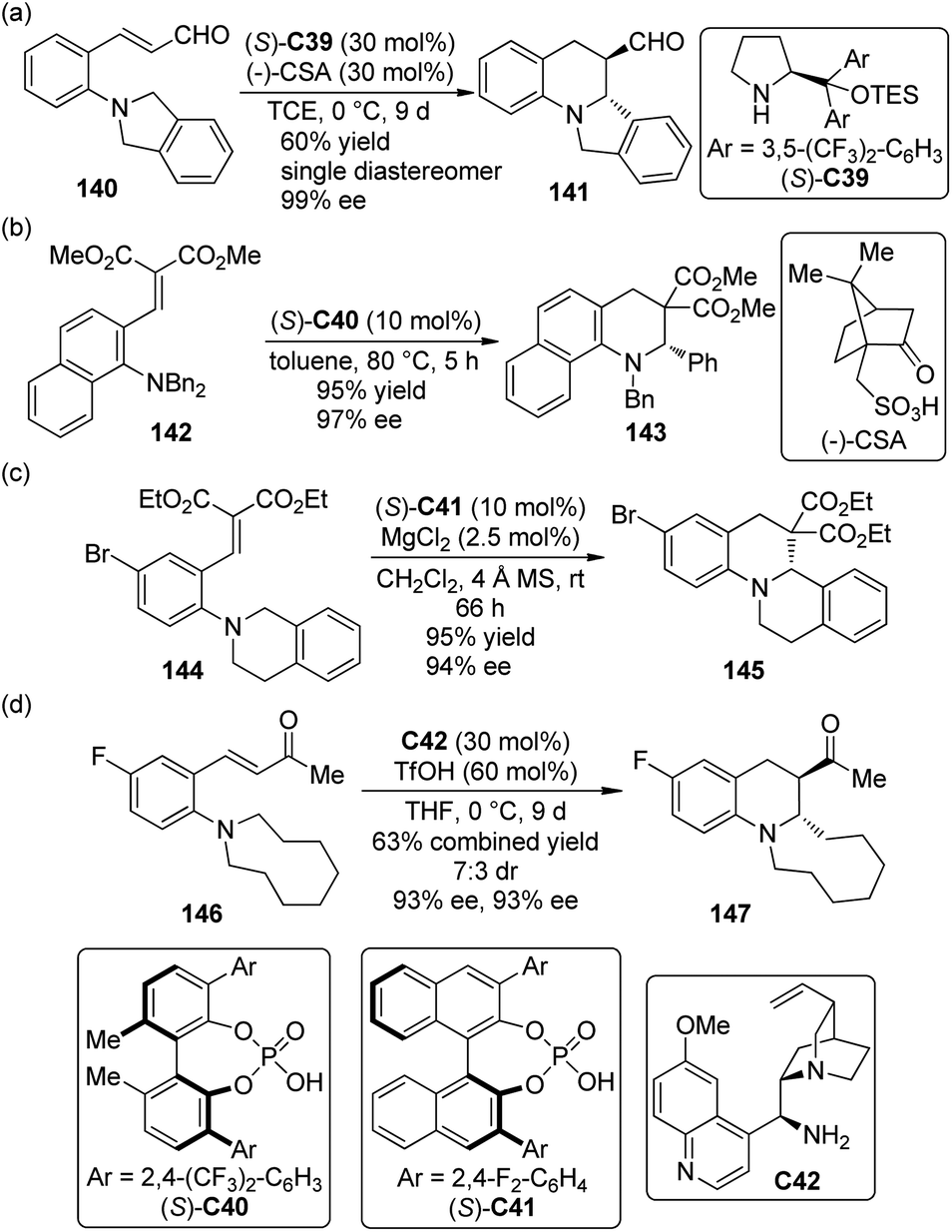
Organocatalytic strategies have also been applied to the asymmetric cascade [1,5]-hydride transfer/cyclization reactions. Kim and co-workers130 first realized this type transformations of ortho-tert-amine-substituted cinnamaldehydes 140 by utilizing chiral secondary amine catalyst (S)-C39 in combination with (−)-camphorsulfonic acid (CSA) (Scheme 41(a)). High to excellent diastereo- and enantioselectivity were obtained for a series of synthetically useful ring-fused tetrahydroquinoline derivatives 141 under mild conditions.
 | ||
| Scheme 41 The organocatalytic asymmetric [1,5]-hydride transfer reactions reported by (a) Kim, (b) Akiyama, (c) Luo and (d) Kim respectively. | ||
Akiyama and co-workers,131 on the other hand, developed similar asymmetric [1,5]-hydride transfer reactions catalyzed by the biphenyl-derived chiral phosphoric acid catalyst (S)-C40 (Scheme 41(b)).132 The reactions were highly enantioselective (up to 97% ee) even at higher temperature. It was found that a substituent ortho to the nitrogen atom could enhance the reactivity of the substrates.133 More interestingly, control experiments showed that the enantioselectivity of this catalytic system probably arose from the discrimination of the two enantiotopic α-H atoms by the chiral catalyst in the hydride transfer step instead of the enantiofacial selection of the nucleophilic attack on the iminium ion as proposed by the Seidel group124 and the Feng group.126
Luo and co-workers134 also accomplished highly enantioselective [1,5]-hydride transfer/cyclization cascade reactions catalyzed by a binary-acid system135 that comprised chiral phosphoric acid (S)-C41 as well as the Lewis acid MgCl2 or Mg(BF4)2 (Scheme 41(c)). The non-linear-effect study revealed that the active catalytic species was probably a 1:1 complex of the phosphoric acid and magnesium salt although excess amount of phosphoric acid was needed. Comprehensive theoretical calculations and experimental studies suggested that the stereospecific and suprafacial [1,5]-hydride transfer was both rate-limiting and stereocontrol step followed by a barrierless C–C bond formation.
Very recently, Kang and Kim136 expanded the substrate scope of organocatalytic asymmetric [1,5]-hydride transfer/cyclization reaction to ortho-dialkylamine substituted benzylideneacetone derivatives 146. By using chiral primary amine catalyst C42 in conjunction with TfOH, the desired tetrahydroquinoline products 147 could be afforded in moderate to high yields with high enantioselectivity (Scheme 41(d)).
In addition to the synthesis of tetrahydroisoquinolines, Gong and co-workers137 applied the chiral phosphoric acid catalyzed asymmetric [1,5]-hydride transfer/cyclization reactions for the synthesis of cyclic aminals. The bisphosphoric acid (R,R)-C43 was identified as the optimal catalyst to enable the reactions of an array of ortho-tert-amine-substituted aromatic ketoesters 148 with aromatic amines 149 (Scheme 42). Typically, the desired products 150 could be afforded in moderate to high yields (up to 81%) and diastereo- and enantioselectivity (up to 11:1 dr and 84% ee).
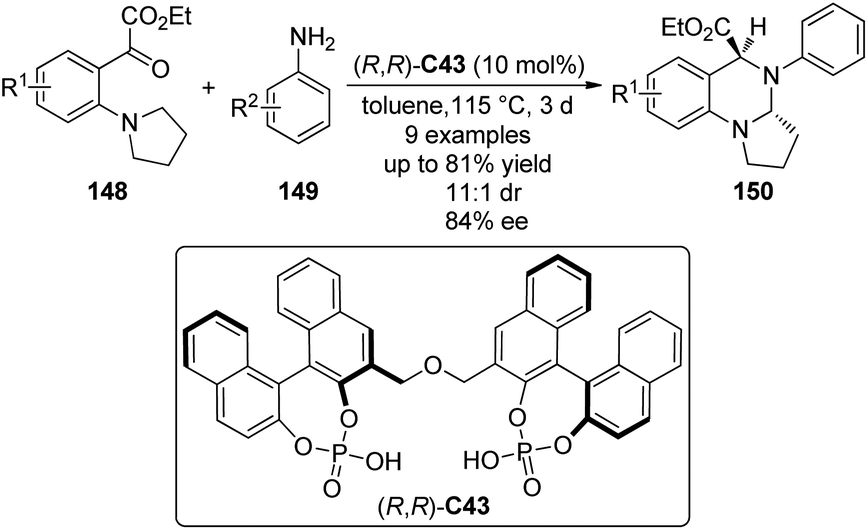 | ||
| Scheme 42 The organocatalytic asymmetric [1,5]-hydride transfer reactions reported by Gong. | ||
The catalytic functionalization of the C(sp3)–H bonds α to an oxygen atom via [1,5]-hydride transfer reactions have been well studied by Sames138 and others.133b,139 The asymmetric version of this type of transformations, however, is scarce compared to their nitrogen-containing counterparts described above. Recently, Tu and co-workers140 developed the enantioselective [1,5]-hydride transfer induced cyclization reactions of 151 by employing chiral secondary aminium salt (R,R)-C26·HCl (Scheme 43). The presence of AgSbF6 was necessary to allow the reaction to proceed by enhancing the electrophilicity of the iminium intermediates with a more weakly coordinating anion. Finally the enantioenriched spiroethers 152 could be delivered in satisfactory yields and stereoselectivity after the subsequent Wittig reaction.
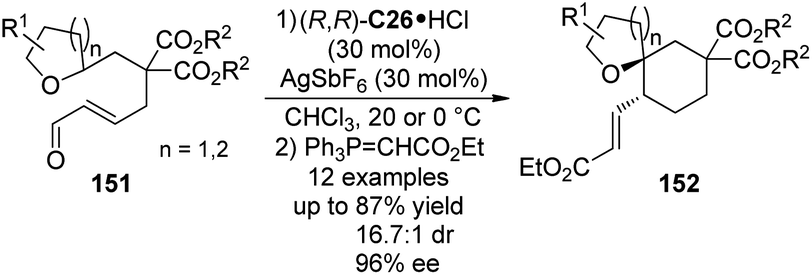 | ||
| Scheme 43 The organocatalytic asymmetric [1,5]-hydride transfer reactions reported by Tu. | ||
5. C–H bond functionalization involving a transient metal–carbon (M–C) species
5.1. Pd-Catalyzed reactions
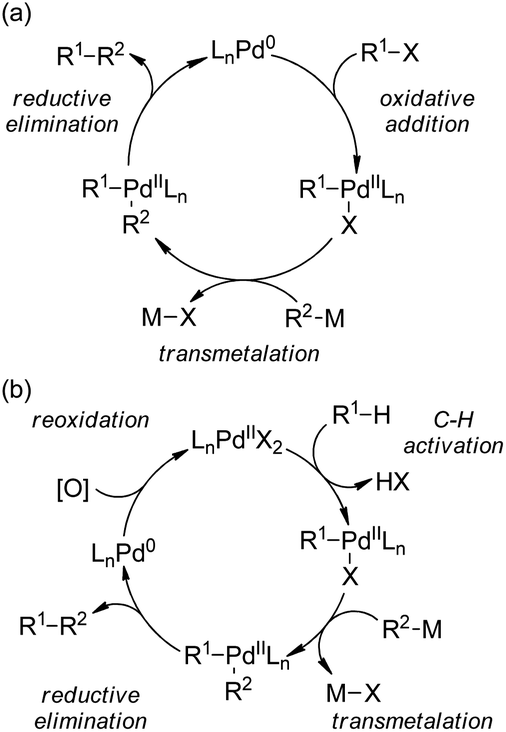 | ||
| Scheme 44 Schematic mechanistic scenarios of (a) the traditional Pd(0)-catalyzed cross-coupling reaction and (b) the Pd(II)-catalyzed C–H activation/C–C coupling reaction. | ||
A major breakthrough was the application of the mono-N-protected chiral amino acids into the Pd(II) catalyzed C–H bond functionalization reactions first reported by the Yu group143 in 2008. In order to develop a catalytic asymmetric C–H bond functionalization protocol, Yu and co-workers144 set the C–H activation/C–C coupling of the prochiral substrates 153 (R1 = 2-Me) with boronic acids 154 (R2 = nBu) as the model reaction. Inspired by the X-ray crystal structure of the dimeric Pd complex 156, they hypothesized that the asymmetric induction might be achieved by using a chiral carboxylate as the ligand (Scheme 45). After screening a series of commercially available carboxylate, it was found that mono-N-protected chiral amino acids could lead to reasonable enantioselectivity of the reaction, probably owing to the N,O-bidentate coordination manner to the Pd center. It was also suggested that the larger steric difference at the α-carbon (α-hydrogen vs. bulky side chain) can induce improved stereocontrol, and to incorporate an electron-withdrawing group to the nitrogen atom to maintain the electrophilic nature of the Pd(II) center was necessary for the C–H bond cleavage. Finally by utilizing the (−)-Men-L-Leu-OH L11 as the optimal ligand, an array of desymmetrized coupling products 155 were produced in up to 96% yield with 95% ee.
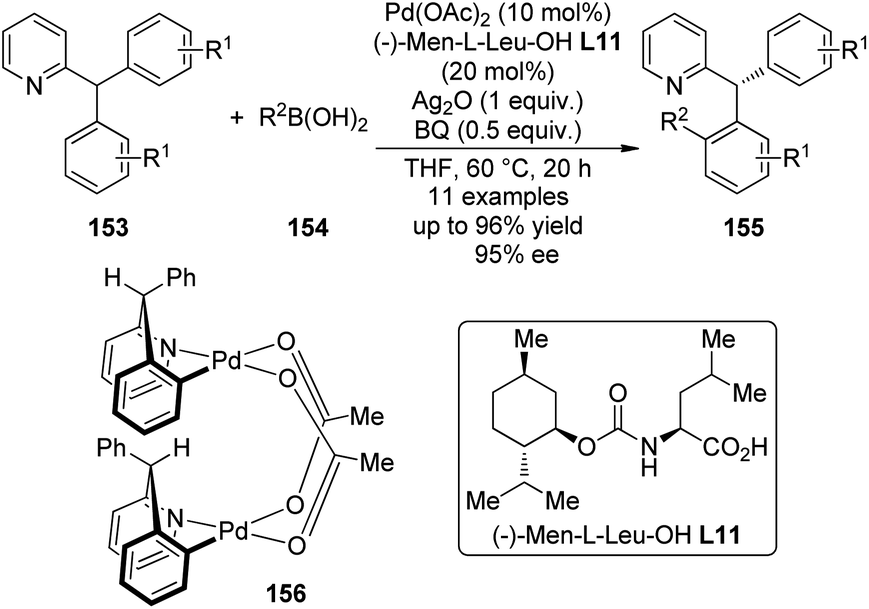 | ||
| Scheme 45 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Yu. | ||
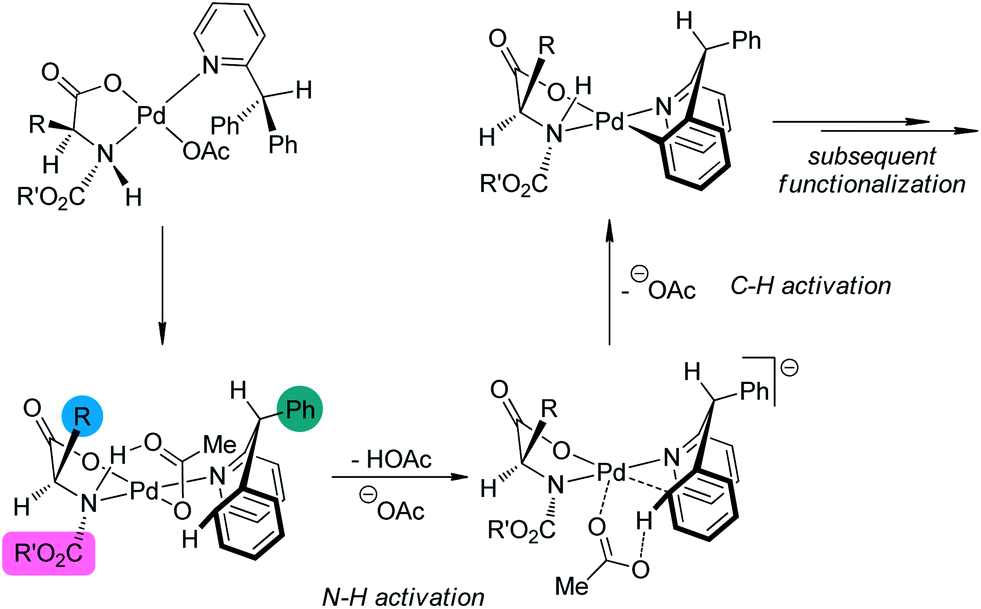
A detailed DFT calculation by Musaev, Yu and co-workers145 offered in depth understanding of the mechanism of this novel asymmetric C–H bond functionalization protocol. It was confirmed that the ligand was bound in a bidentate fashion to the Pd center. The first step of the reaction was the cleavage of the N–H moiety of the ligand by the coordinated acetate group, leaving an vacant coordination site of the Pd(II) center. At the next stage, one ortho C–H bond of the substrate was cleaved with the assistance of another acetate anion, furnishing the palladacycle with concomitant formation of a new stereocenter at the benzylic position (Scheme 46). (It is worth noting that several concepts have arisen to describe the transition metal-catalyzed C–H bond cleavage with the assistance of an external or coordinated base such as “concerted metallation deprotonation (CMD)”,146 and “ambiphilic metal–ligand activation (AMLA)”.147 The nomenclature and classification of the transition metal-catalyzed C–H bond cleavage are still topics in the literature and beyond the scope of this review. To simplify the discussion, we prefer not to use these terms here.) The chiral information at the α-carbon of the ligand could be geared to the nitrogen atom (the bulky N-protecting group is forced trans to the side chain of the amino acid ligand), which further arranges the substrate in a proper conformation for the asymmetric C–H bond functionalization. The experimentally observed preference for the formation of (R) products could also be well reproduced in the computational investigations.
 | ||
| Scheme 46 The schematic description of the mechanism of the Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Musaev and Yu. | ||
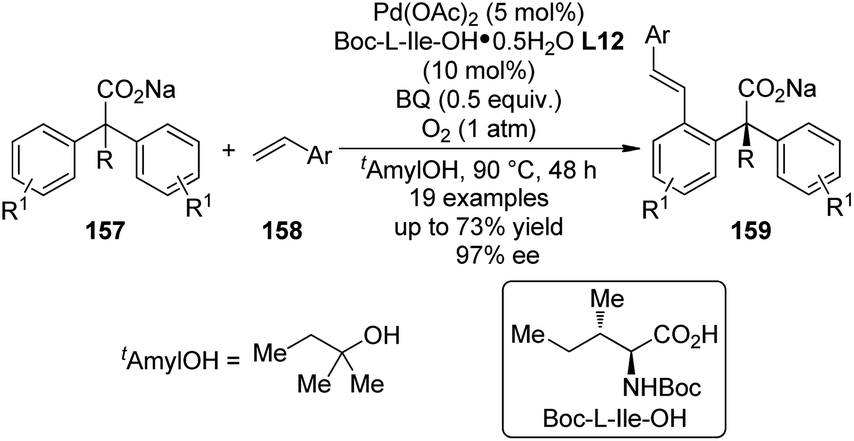
Shortly after the above success, Yu and co-workers148 were able to replace the 2-pyridyl moiety of the prochiral substrates 153 with a carboxylate group in the Pd(II)-catalyzed asymmetric C–H bond functionalization reactions. The use of a directing group that features relatively weak coordinating ability and at the same time is a useful functional group that can be readily involved in further transformation is of particular importance in developing more efficient and practical C–H functionalization methodologies.149 An extensive screening of the reaction parameters showed that Boc-L-Ile-OH·0.5H2O L12 was the best ligand for the asymmetric C–H bond olefination of the sodium diphenylcarboxylates 157 with styrene derivatives (or acrylates) 158 (Scheme 47). The loading of Pd(OAc)2 and chiral ligand could be lowered to 5 mol% and 10 mol%, respectively, and O2 (1 atm) could be used as the terminal oxidant. The desired products 159 were typically afforded in moderate yields (up to 73%) and good ee's (up to 97% ee).
 | ||
| Scheme 47 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Yu. | ||
Notably, despite enabling asymmetric C–H bond functionalization reactions, the mono-N-protected chiral amino acid ligands could also induce the Pd(II)-catalyzed C–H bond functionalization reactions in highly position-selective fashion.150 In addition, the valuable “ligand-acceleration effect” was observed for several Pd(II)-catalyzed direct C–H bond olefination150a,151 and arylation152 reactions when mono-N-protected chiral amino acid ligands were employed, possibly due to the mechanistic switch for the C–H bond breaking step.153 Fueled with these protocols, the Yu group achieved versatile direct C–H bond functionalization reactions effectively,154 even in the late stage of complex molecule synthesis.155
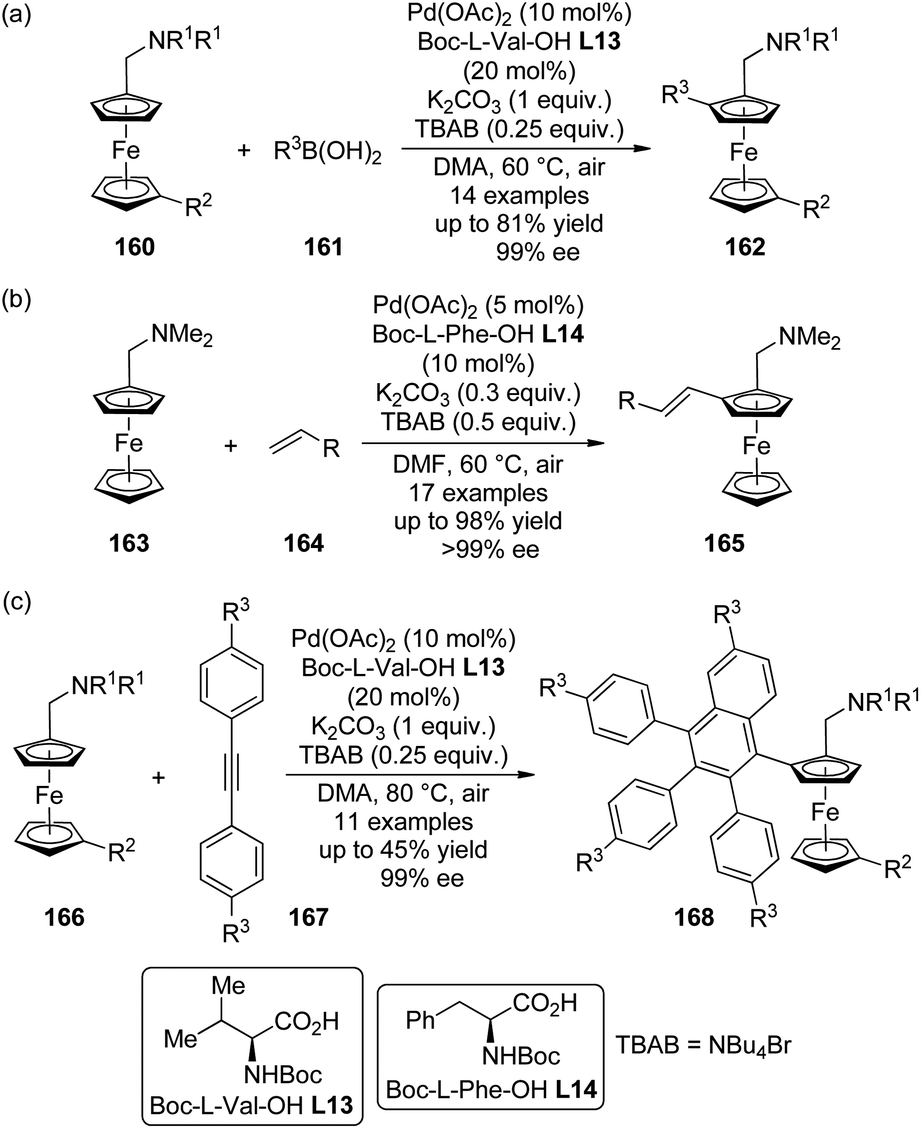
The catalytic system comprised of Pd(OAc)2 and mono-N-protected chiral amino acid ligands have recently been employed for the construction of planar-chiral ferrocene derivatives156 via Pd(II)-catalyzed direct asymmetric C–H bond functionalization reactions.157 Gu, You and co-workers158 reported such cross-coupling reactions of N,N-dialkylaminomethylferrocenes 160 with arylboronic acids 161 (Scheme 48(a)). Boc-L-Val-OH L13 was identified as the optimal ligand and adding catalytic amount of tetrabutylammonium bromide (TBAB) was critical to result in reasonable yields. A series of desired products 162 could be generated in DMA at elevated temperature (60 °C) under air in up to 81% yields with excellent enantiocontrol (up to 99% ee). Notably, a moderate kinetic resolution was observed for the formation of the bisarylation product. As the reaction time was prolonged, both the ratio of the bisarylation product and the ee of 162 increased slightly. A bromo-substituent on the other cyclopentadienyl ring was well tolerant (160, R2 = Br), which allowed further transformation of the corresponding product to a planar-chiral P,N-ligand that could enable the Pd-catalyzed asymmetric allylic alkylation reactions.
 | ||
| Scheme 48 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by (a) Gu and You, (b) Cui and Wu and (c) You, respectively. | ||
Soon after, Cui, Wu and co-workers159 communicated Pd(II)-catalyzed oxidative Heck reactions of ferrocene derivative 163 with various terminal olefins 164 including styrenes, α,β-unsaturated esters and phosphates (Scheme 48(b)). The N,N-dimethylaminomethyl moiety once again was the feasible directing group and Boc-L-Phe-OH L14 exhibited the optimal catalytic activity. The results of the controlled experiments suggested that the redox process of ferrocene scaffolds by air facilitated the regeneration of active Pd(II) catalyst from the Pd(0) species.
By applying the same strategy, You and co-workers160 realized the asymmetric synthesis of planar-chiral ferrocene derivatives 168 via Pd(II)-catalyzed direct coupling of ferrocene substrates 166 with diarylethynes 167.161 Under rather similar conditions to the previously reported asymmetric C–H bond arylation reactions,158 such transformations were achieved with excellent enantioselectivity (up to 99% ee) albeit the yields are typically moderate (Scheme 48(c)). Notably, a sterically more demanding planar-chiral P,N-ligand derived from the coupling product shows enhanced chiral induction in Pd-catalyzed asymmetric allylic substitution reactions compared to that reported previously.158
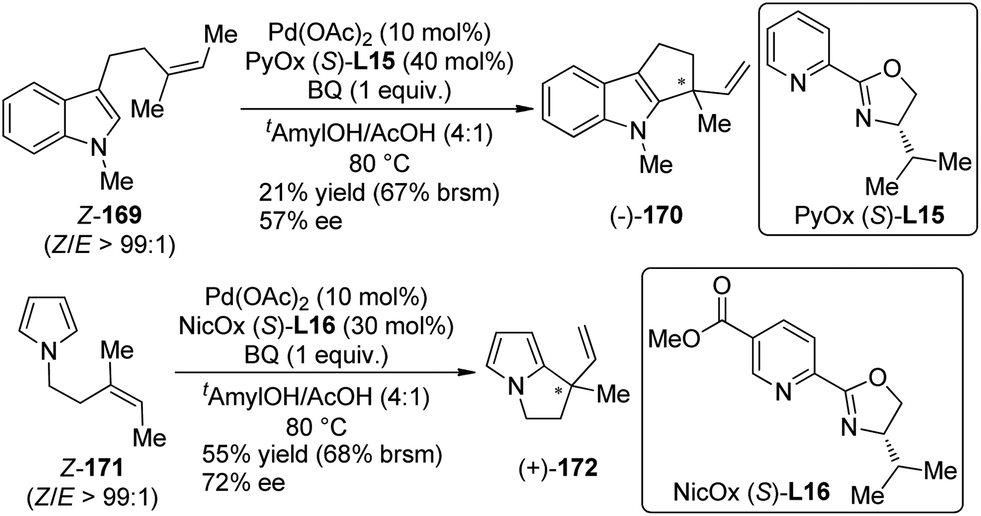
Inspired by Stoltz's work on Pd(II)-catalyzed racemic oxidation indole annulation,162 Oestreich and co-workers163 developed the enantioselective version of this intramolecular Fujiwara–Moritani reactions164 of indole and pyrrole derivatives 169 and 171. With a PyOX ligand (S)-L15 or a newly synthesized nicotine-based oxazoline ligand NicOX (S)-L16, 5-exo-trig cyclization occurred in moderate yields (up to 68% yield based on recovered starting material) and ee's (up to 72% ee), affording the desired products with a chiral quaternary carbon center (Scheme 49). Notably, the Z/E ratio of the double bond in the substrates has a strong influence of the stereochemical outcomes.
 | ||
| Scheme 49 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Oestreich. | ||
Biaryls and heterobiaryls are often important compounds of biological activity.165 The synthesis of sterically hindered (hetero)biaryls with multiple ortho-substituents can be challenging owing to the low reactivity of the hindered coupling components. Based on their previous work on the C4-selective C–H bond arylation reactions of thiophenes with arylboronic acids,166 Yamaguchi, Itami and co-workers167 disclosed that the highly C4-selective coupling reactions of the 2,3-disubstituted thiophenes 173 with the ortho-substituted arylboronic acids 174 were promoted by a catalytic amount of Pd(OAc)2 in combination with BOX ligand (S,S)-L17 and TEMPO as the oxidant (Scheme 50(a)). The yields of the desired products 175 could be significantly improved (up to 84%) by adding TFA to the reaction mixture. The heteroaromatic substrates were not limited to thiophenes in that benzofurans and indoles could also participate in such reactions. By employing a slightly modified reaction conditions, asymmetric induction was achieved with the Pd(OAc)2/(S,S)-L18 catalyst, although the yield and ee of product 175a were only moderate (Scheme 50(b)). Recently, the same group168 disclosed a dual catalytic system comprising Pd(II)-sulfoxide-oxazoline complex (Pd-sox, C44) and Fe-phthalocyanine (FePc, C45) that can enable similar asymmetric coupling under air without stoichiometric co-oxidants (Scheme 50(c)).
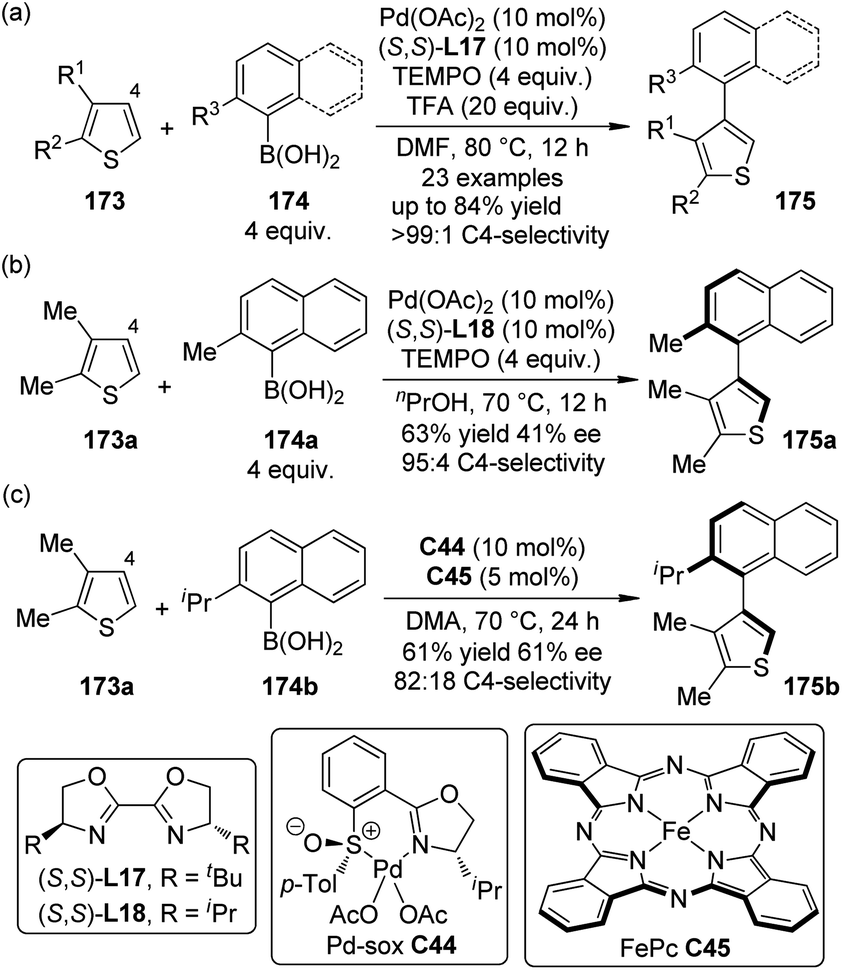 | ||
| Scheme 50 The Pd-catalyzed synthesis of heterobiaryls reported by Yamaguchi and Itami. | ||
Transition metal-catalyzed direct functionalization of C(sp3)–H bonds that involves a transient M–C species are much more difficult compared with that of C(sp2)–H bonds due to their weak coordinating ability to the metal centers.169 In their seminal report on the use of mono-N-protected amino acids as the ligands for the Pd(II)-catalyzed asymmetric aromatic C–H bond functionalization in 2008,144 Yu and co-workers described promising initial results (37% ee) for asymmetric functionalization of terminal methyl group. In 2011, the Yu group170 developed the Pd(II)-catalyzed enantioselective C–H bond functionalization reactions of cyclopropanes. The 1,1-disubstituted cyclopropanes 176 bearing an acidic N-arylamides as weakly coordinating directing groups171 were utilized as the substrates. The systematic evaluation of structurally diverse N-protecting groups as well as the amino acid backbones of the chiral ligands revealed that (S)-L19 containing a modified N-Boc group and the 2,6-difluorobenzyl as the α-substituent led to the optimal asymmetric induction of the C–H/R–BXn cross-coupling reactions (Scheme 51). The substrate scope of this transformation was general. Various alkyl and aryl groups bearing functional groups such as ethers and protected amines could be tolerated at the α-position of amide directing group of 176 (R1), affording the corresponding products 177 in up to 81% yield and 92% ee under mild conditions.
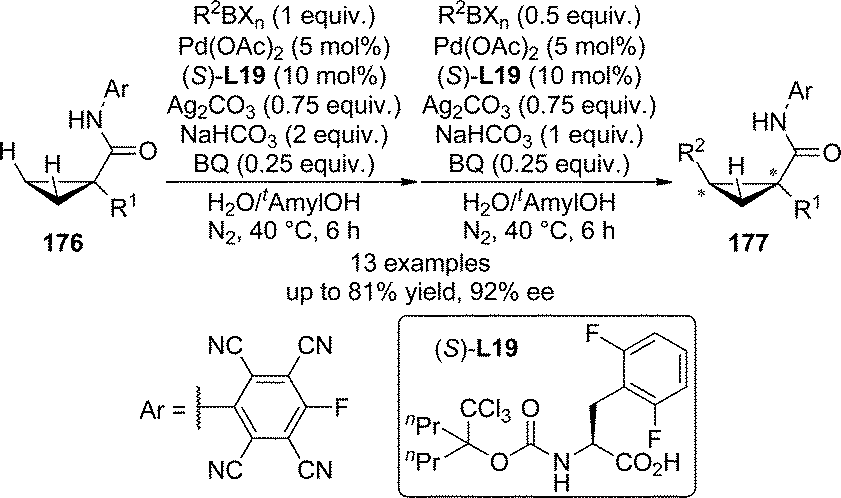 | ||
| Scheme 51 The Pd-catalyzed asymmetric C–H bond functionalization reactions of cyclopropanes reported by Yu. | ||
Another type of promising C(sp3)–H bond functionalization occurs at the allylic position172 and it can be viewed as an atom-economical alternative of the traditional Tsuji–Trost allylic substitution reactions which employs allylic halides, esters or carbonates as the substrates.173 In 2008, Shi and co-workers174 reported the formal asymmetric allylic C–H bond functionalization via Pd(0) catalyzed allylic and homoallylic diamination reactions between terminal olefins and N,N′-di-tert-butyldiaziridinone. On the other hand, Covell and White175 achieved the asymmetric branched allylic C–H bond oxidation (ABAO) of terminal olefin substrates 178 using the serial ligand catalysis strategy by Pd(II)/sulfoxide catalyst C46 in combination with a chiral Lewis acid Cr(Salen)F (R,R)-C47 to furnish the enantioenriched allylic alcohol derivatives 179 (Scheme 52). Since the enantioselectivities are modest (43–63% ee), the same authors176 were able to combined the ABAO approach with enzymatic and/or small molecule catalyst based resolutions for the construction of chiral polyoxygenated motifs with high enantiopurity.
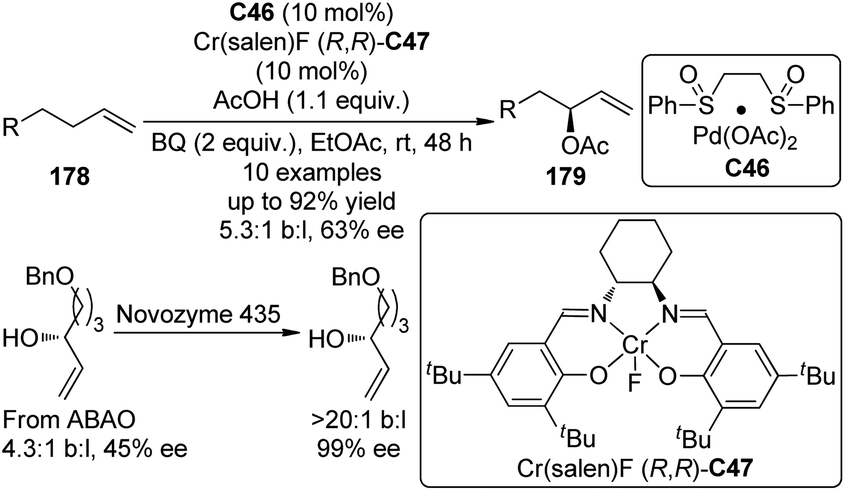 | ||
| Scheme 52 The Pd-catalyzed asymmetric allylic C–H bond oxidation reactions reported by White. | ||
In 2011, Sasai and co-workers177 reported an enantioselective cyclization of 4-alkenoic acids 180 via Pd(II)-catalyzed oxidative allylic C–H bond esterification. Among various ligands with quite different chiral backbones tested, only spiro bis(isoxazoline) ligand (M,S,S)-iPr-SPRIX L20178 exhibited satisfactory activity and asymmetric induction. Under the optimized conditions, the desired γ-lactone products 181 were afforded in up to >98% yield and 82% ee (Scheme 53). Preliminary mechanistic studies prefer the mechanism that involves a π-allyl Pd intermediate over oxypalladation/β-hydride elimination sequence.
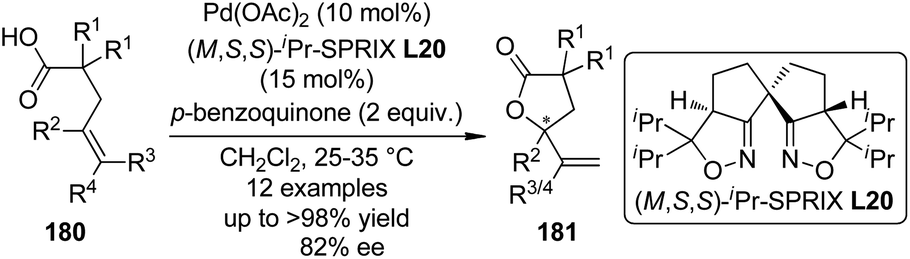 | ||
| Scheme 53 The Pd-catalyzed asymmetric allylic C–H bond esterification reactions reported by Sasai. | ||
In 2012, Chai and Rainey179 described the Pd(II)/chiral phosphoric acid catalyzed asymmetric allylic C–H bond activation/semipinacol rearrangement180 protocol for the construction of spirocyclic compounds from indene derivatives 182. Although various chiral oxazoline ligands failed to give satisfactory results, adding chiral phosphoric acids was found to have a dramatic effect on the reactivity and stereoinduction with (S)-C48 as the optimal one. The corresponding ketones 183 with an all-carbon quaternary α-chiral center were obtained in up to 78% yield (up to quantitative yield based on recovered starting material) and 98% ee (Scheme 54). A primary kinetic isotope effect (KIE) value of 2.82–3.39 confirmed that the cleavage of the allylic C–H bond is prior to or during the rate-limiting step.
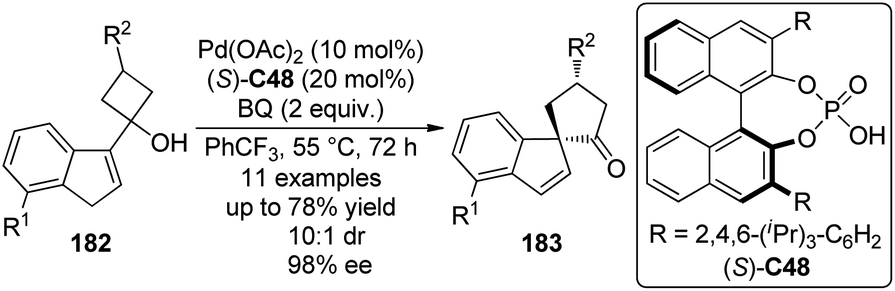 | ||
| Scheme 54 The Pd-catalyzed asymmetric allylic C–H bond functionalization reactions reported by Rainey. | ||
Recently, Trost and co-workers181 reported the Pd(II)-catalyzed asymmetric allylic alkylation reactions through C–H bond activation.182 With catalytic amount of Pd(OAc)2 and the newly synthesized chiral phosphoramidite ligand (Sa,S,S)-L21, the asymmetric allylic alkylation reactions between the cyclic α-substituted β-diketones 184 went smoothly with a series of allylarenes 185 to generate their corresponding alkylation products 186 bearing an all-carbon quaternary chiral center in good yields and ee's (Scheme 55).
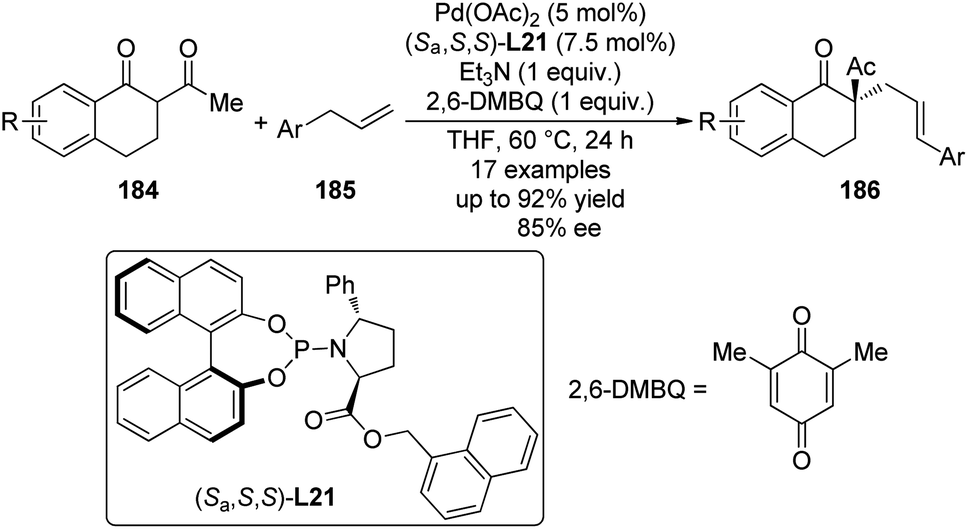 | ||
| Scheme 55 The Pd-catalyzed asymmetric allylic C–H bond functionalization reactions reported by Trost. | ||
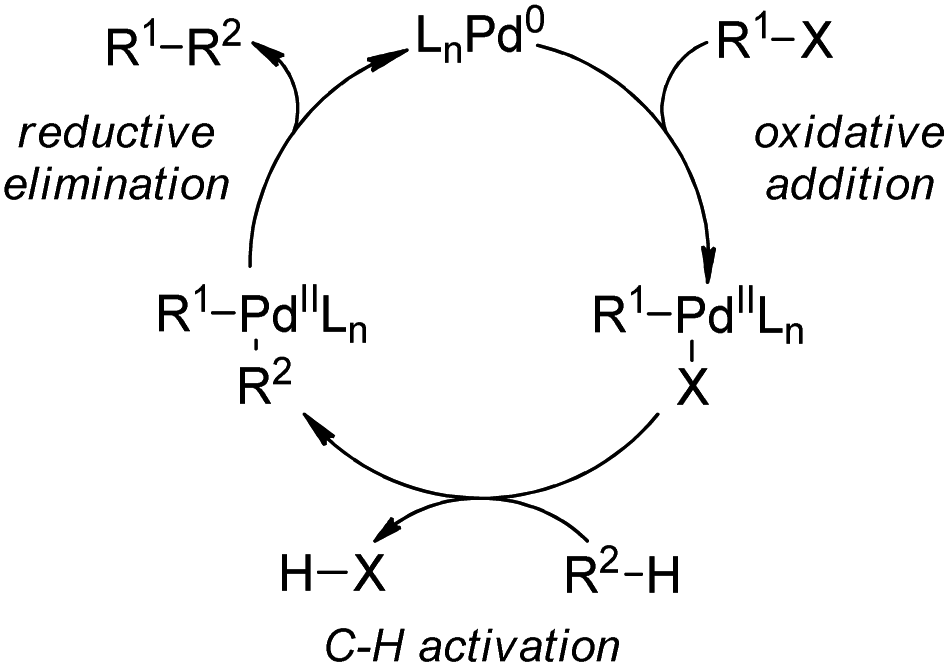 | ||
| Scheme 56 The schematic mechanistic scenarios of the Pd(0)-catalyzed C–H activation/C–C coupling reaction. | ||
The first Pd(0)-catalyzed enantioselective C(sp2)–H bond functionalization reaction was reported by Albicker and Cramer184 in 2009. The asymmetric desymmetrization of prochiral substrates 187 was expected to occur by the C–H bond functionalization via the transition state shown in Scheme 57(a). Thus only one coordination site on the Pd center was left for the external chiral ligands. In this context, a series of bulky monodentate phosphorus-based ligands were screened. While cyclohexyl-MOP ligand and Feringa-type ligands only led to moderate level of ee's, some TADDOL-based phosphoramidite ligands could result in enhanced enantioselectivity, with (R,R)-L22 as the optimal one. The desired indane derivatives 188 were obtained in up to quantitative yields and 97% ee under mild conditions. Very recently, the Cramer group185 extended this method to the enantioselective synthesis of functionalized dibenzazepinones 190 from the prochiral substrates 189. The simplest TADDOL-based phosphoramidite ligand (R,R)-L23 gave the best results and a bulky carboxylate (PivO−) was critical for the chirality relay during the C–H bond cleavage (Scheme 57(b)). Quite a lot of functional groups such as amide, chloride and heteroaromatic rings were well tolerated. Remarkably, this reaction proceeded through a challenging eight-membered palladacyclic transition state. This pathway was still preferred even when other reaction site were available on the amide protecting groups (189, R2 = Bn or iPr) for the C(sp2)–H or C(sp3)–H bond functionalization through seven- or six-membered ring transition states.
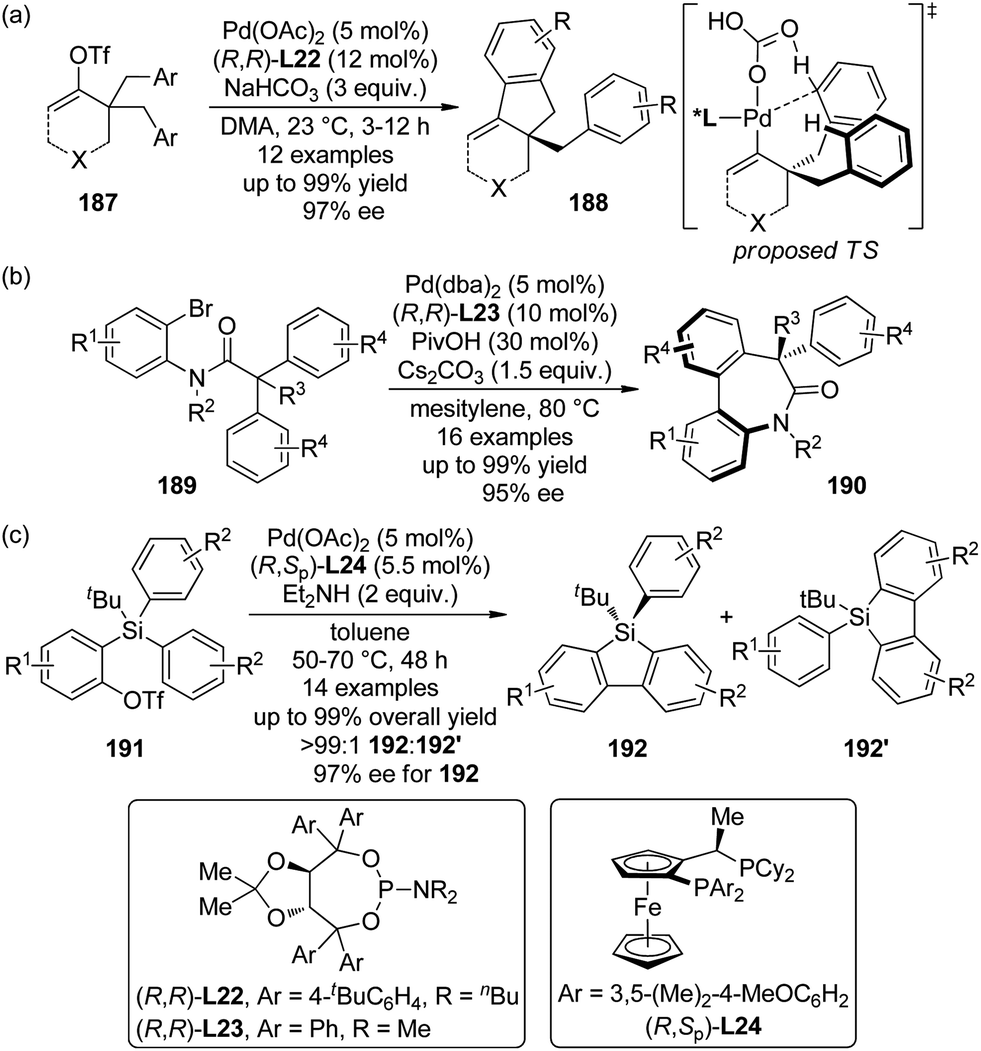 | ||
| Scheme 57 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by (a) and (b) Cramer and (c) Shintani and Hayashi. | ||
Similar strategy was applied by Shintani, Hayashi and co-workers186 to construct enantioenriched silicon-stereogenic dibenzosiloles187 via Pd(0)-catalyzed C–H bond functionalization. Different from Cramer's results, The Josiphos-type bisphosphine ligand (R,Sp)-L24 was the most effective to promote the reactions of 191 with satisfactory outcomes. The corresponding products 192 were afforded with up to 97% ee (Scheme 57(c)). Kinetic studies suggested that the initial oxidative addition of aryl triflates was likely the turnover-limiting step and the following C–H bond cleavage occurs fast. The formation of the side product 192′ involving a [1,5]-Pd migration was effectively suppressed by using the bisphosphine ligand.
Much success has been achieved in recent years in the area of Pd(0)-catalyzed asymmetric C(sp3)–H bond functionalization reactions. In 2010, Clot, Baudoin and co-workers188 realized the Pd-catalyzed β-arylation of carboxylic esters 194 with aryl halides 193. The electronegative group in the ortho position of the aryl halide was crucial to obtain the β-arylation products. Mechanistic investigations indicated that the β-arylation was not directed by the carbonyl group, but occurred via the β-hydride elimination of the initially formed palladium-C-enolate that was followed by rotation, re-insertion and the final reductive elimination (Scheme 58). Moderate level of enantioselectivity (up to 54% ee) could be achieved when BINOL-derived KenPhos (R)-L25 was utilized as the chiral ligand.
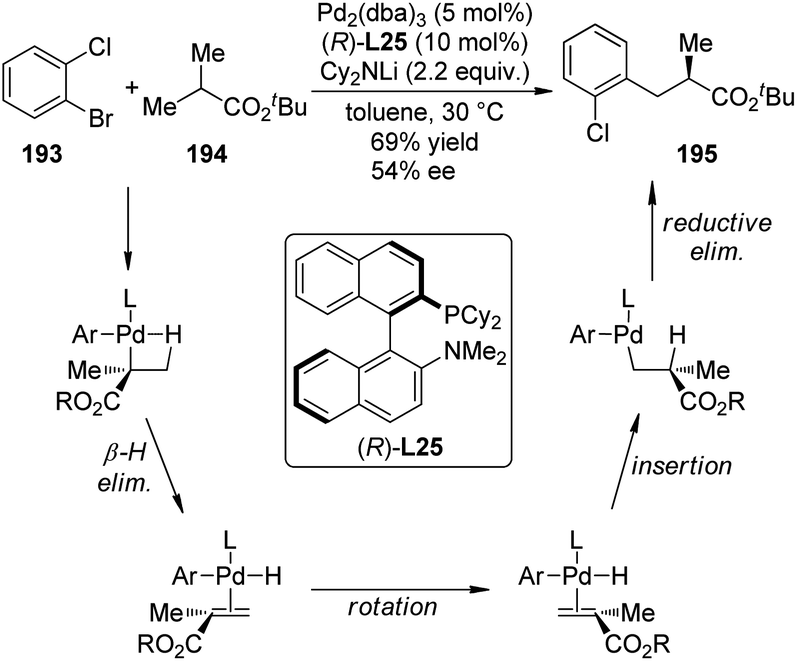 | ||
| Scheme 58 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Clot and Baudoin. | ||
Several groups have been engaged to the Pd(0)-catalyzed asymmetric C(sp3)–H bond functionalization reactions of inert methyl or methylene moiety for the synthesis of enantioenriched indoline derivatives (Scheme 59). In 2011, Kündig and co-workers189 found that the asymmetric C–H bond functionalization reactions of N-cycloalkyl substituted carbamates 196 (PG = CO2Me) could be promoted by the chiral Pd complex (either preformed or generated in situ) ligated by bulky NHC ligand (S,S)-L26. Fused indolines 197 bearing trans [3.4.0]- or [3.5.0]-bicyclic backbones could be delivered with up to 95% ee even at the elevated temperature (140 or 160 °C). Later, the Kündig group190 showed that the regiodivergent C(sp3)–H bond functionalization was possible for substrates 198 (R2 ≠ H). When NHC ligand (S,S)-L26 or (S,S)-L27 were employed, the C–H bond functionalizations both at the terminal methyl and methylene group of 198 could happen, leading to 2-substituted indoline 199 and 2,3-disubstituted indoline 200. The matched/mismatched effect exhibited between the chiral catalyst and a series of racemic substrates. The (2R)-199 were produced in higher yields but moderate level of ee's whereas the (2R,3S)-200 were the minor products but with excellent ee's. The highest selectivity was observed when R2 = Ph. In this case both (2R)-199 and (2R,3S)-200 were afforded with >95% ee when (S,S)-L26 was used. Recent mechanistic studies by Kündig and co-workers191 revealed that the ligand exchange (bromide for acetate/pivalate) appeared to be the rate-limiting step with the chiral catalysts and the C–H bond cleavage occurred fast but determined the enantioselectivity of the reaction.
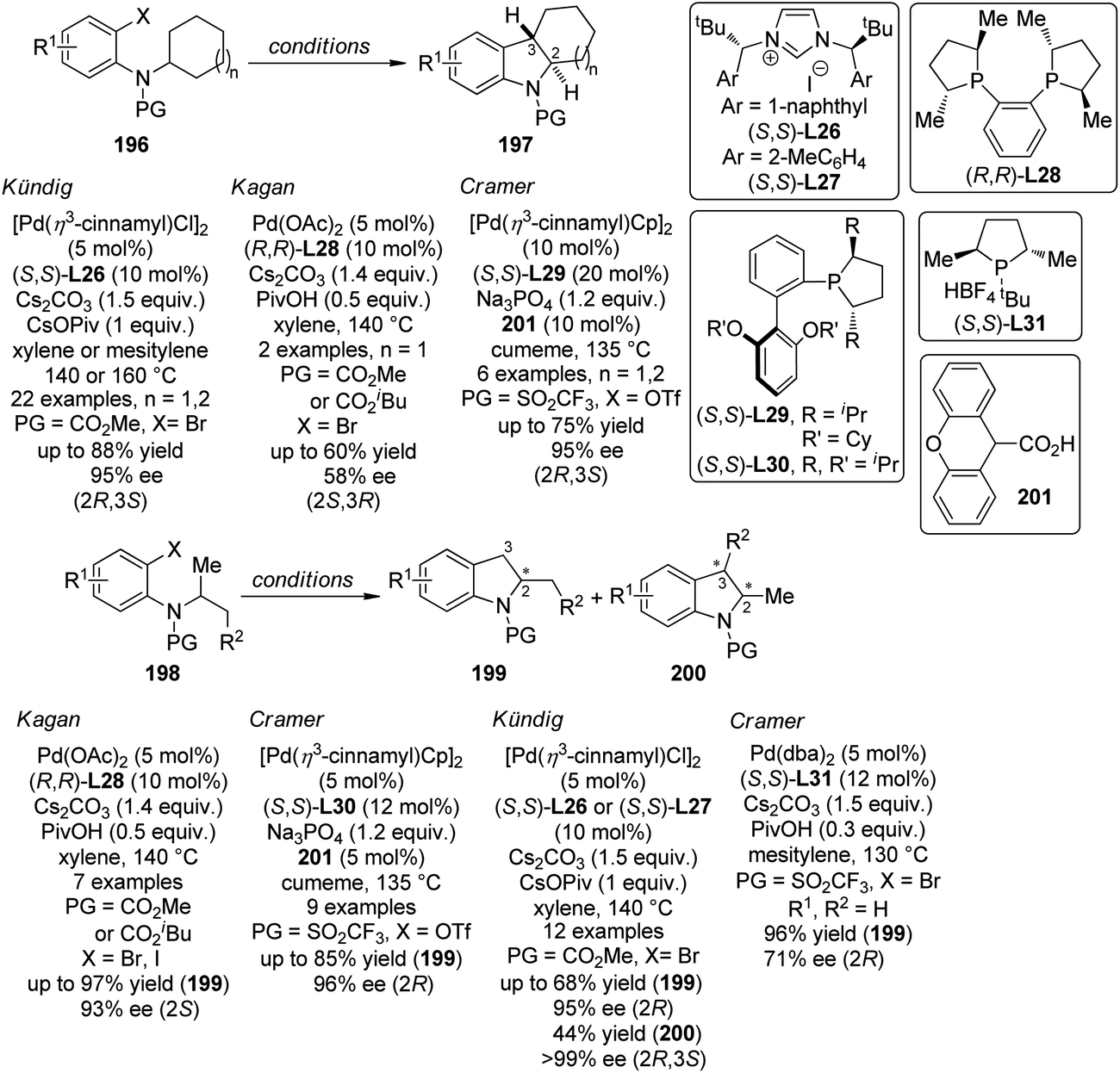 | ||
| Scheme 59 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by several groups. | ||
Almost at the same time as Kündig's first report in 2011, Kagan and co-workers192 found that the chiral bisphosphine ligand Me-DuPhos (R,R)-L28 could also be utilized in Pd-catalyzed C–H bond functionalization reactions (Scheme 59). Several 2-substituted indolines 199 could be generated with up to 93% ee from the corresponding substrates 198 (R2 = H) by the catalytic system comprised of Pd(OAc)2 and (R,R)-L28. In addition, two examples of such reactions involving substrates 196 were described, albeit the ee's of the products were only moderate.
In 2012, Cramer and co-workers193 synthesized a new class of modular monodentate phosphine ligands SagePhos by incorporating the electron-rich C2-symmetric phospholane moieties into the Buchward-type biphenyl backbone. With the novel sterically demanding ligands such as (S,S)-L29 and (S,S)-L30, the asymmetric C–H bond functionalization reaction of the substrates 196 and 198 bearing either cyclic or acyclic N-substituents were accomplished (Scheme 59). It was also disclosed that adding bulky acid like 201 to the reaction was crucial for enhancing the enantioselectivity, probably because it could help to relay the chiral information from the ligand to the reactive center. The significant cooperative effects were observed in that a pair of enantiomers of chiral acids in combination with the same chiral catalyst could result in the products in reversed absolute configuration respectively. The utilization of an enantiopure acid as the only chirality source also led to a moderate level of asymmetric induction. Recently, the Cramer group194 developed a series of chiral monodentate trialkylphosphine ligands like (S,S)-L31, and found that their Pd complexes could also catalyze the asymmetric C–H bond functionalization reaction for the synthesis of 2-substituted indolines 199 although the enantioselectivity was not high.
Besides the enantioselective synthesis of indoline derivatives via Pd catalyzed C–H bond functionalization reactions, the Cramer group195 also communicated the efficient access to functionalized tetrahydroquinolines through the direct functionalization of the cyclopropane C–H bond of substrates 202. After screening various reaction conditions, The use of catalytic amounts of Pd(dba)2, TADDOL-based phosphoramidite ligand (R,R)-L32 in conjunction with pivalic acid presented the optimal enantioselectivity (Scheme 60). Remarkably, the reaction could be performed on a gram-scale with the loading of Pd salts as low as only 1 mol%. The enantioenriched tetrahydroquinolines 203 were readily transformed to the corresponding tetrahydrobenzoazepines via the enantiospecific cyclopropane cleavage.
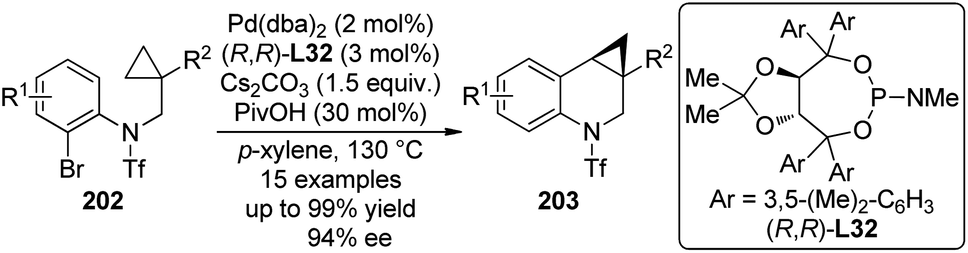 | ||
| Scheme 60 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Cramer. | ||
Baudoin and co-workers196 applied the strategy of direct functionalization of inert C–H bonds to the asymmetric synthesis of fused cyclopentane compounds. Compared with the aforementioned synthesis of indolines, the use of substrates 204 posed an additional challenge since the replacement of nitrogen atom by a quaternary benzylic carbon atom causes the existence of both diastereotopic and enantiotopic C–H bonds. An extensive survey of various Pd sources and chiral ligands indicated the combination of Pd(OAc)2 and binepine ligand (S)-L33 could give the best results. An array of desired products 205 was afforded in up to 95% yields with reasonable stereoselectivity (up to 31:1 dr and 80% ee) (Scheme 61).
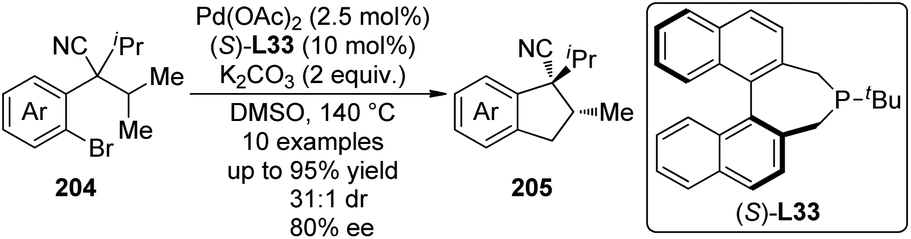 | ||
| Scheme 61 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Baudoin. | ||
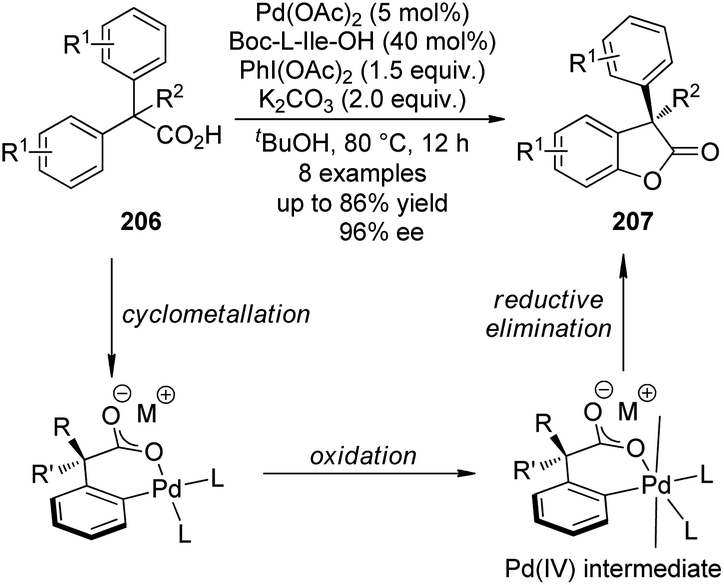 | ||
| Scheme 62 The Pd-catalyzed asymmetric C–H bond functionalization reactions reported by Wang. | ||
Very recently, Yu and co-workers200 developed a Pd(II)-catalyzed enantioselective C–H bond iodination reaction of prochiral substrates 208 with molecular I2 as the sole oxidant.201 After careful evaluation of the reaction conditions, it was finally found that Bz-L-Leu-OH L34 is the optimal ligand, and the combination of CsOAc and Na2CO3 as the base effectively promotes the reaction. Higher enantioselectivity could be obtained using a binary solvent system of tAmylOH with DMF or DMSO, probably due to DMF or DMSO could avoid racemic background reaction by sequestering the small amount of free Pd(II) species not coordinated to the chiral ligands. Thanks to this novel strategy, an array of chiral diarylmethylamines 209 could be synthesized in up to 85% yield and 99% ee under quite mild conditions (30 °C, air) (Scheme 63). However, whether a Pd(IV) intermediate is involved in this reaction is not clear at this stage.
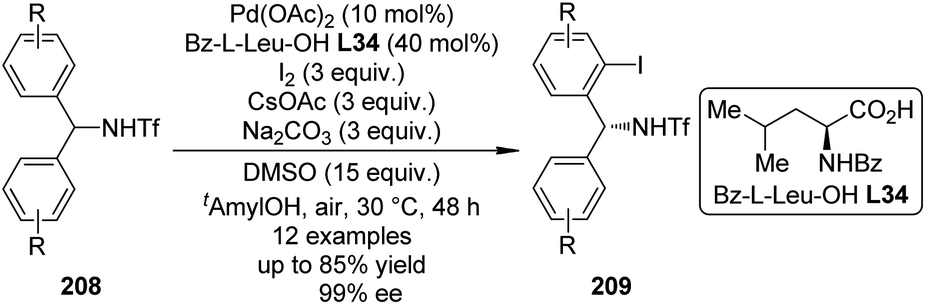 | ||
| Scheme 63 The Pd-catalyzed asymmetric C–H bond iodination reactions reported by Yu. | ||
5.2. Rh-Catalyzed reactions
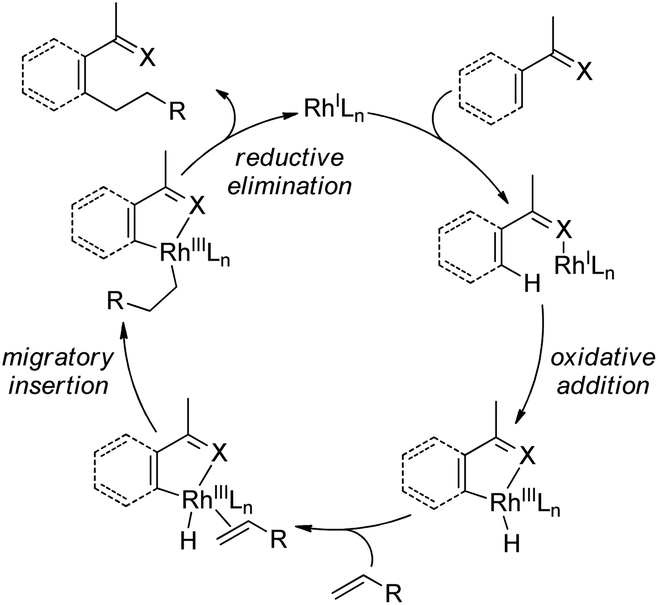 | ||
| Scheme 64 The schematic mechanistic scenarios of the Rh(I)-catalyzed C–H bond functionalization reactions. | ||
In 2009, Bergman, Ellman and co-workers206 disclosed the Rh(I)-catalyzed enantioselective cyclization of N-allylic imidazoles 210 via C–H bond functionalization reactions. Among the structurally diverse chiral ligands that were screened, only TangPhos L35 was highly active and at the same time exhibited superior asymmetric induction. Typically, with 19 mol% of L35 and 10 mol% of [Rh(coe)Cl2]2, an array of cyclized products 211 was afforded in up to 92% yield and 98% ee even at remarkably high temperature (135–175 °C) (Scheme 65). In some cases, comparable results could be achieved with prolonged reaction time where the loadings of the Rh precursor and the ligand were lowered to 2.5 mol% and 4.9 mol%, respectively. The superb performance of L35 was probably the consequence of its highly electron-rich nature and the potential to partially dissociate from the metal center, liberating a vacant coordinating site during the catalytic cycle.
 | ||
| Scheme 65 The Rh-catalyzed asymmetric C–H bond functionalization reactions reported by Bergman and Ellman. | ||
During the attempts to develop the Rh-catalyzed type II Diels–Alder reactions,207 Li and Yu208 serendipitously discovered the cyclization reactions of ene-2-diene substrates 212 to afford the cis disubstituted five-membered carbo- or heterocyclic compounds 213 via Rh-catalyzed allylic C–H bond functionalization. Later, by employing the chiral phosphoramidite ligand (S)-L36 in combination with a cationic Rh(I) catalyst, Li and Yu209 demonstrated the enantioselective variant of this reaction. The desired products bearing two consecutive chiral centers could be synthesized in up to 91% yields and excellent stereocontrol (up to >19:1 dr and 94% ee) (Scheme 66). Detailed DFT calculations210 suggested that the conjugated diene moiety is very critical for this allylic C–H bond activation reaction and the originally designed type II Diels–Alder reaction was disfavored by the bridgehead double-bond distortion. Notably, the chiral diene compounds obtained with this method could be used as efficient ligands in Rh-catalyzed asymmetric conjugated addition reactions between aryl boronic acids and α,β-unsaturated keones.211
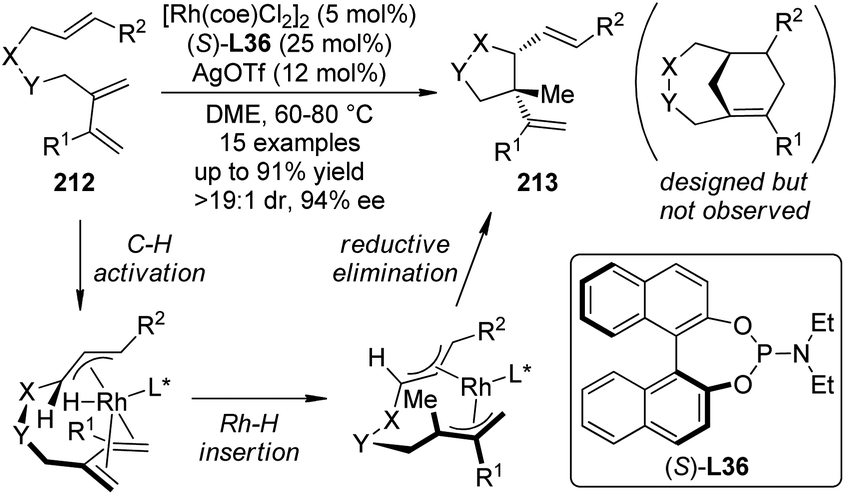 | ||
| Scheme 66 The Rh-catalyzed asymmetric C–H bond functionalization reactions reported by Yu. | ||
The group of Cramer systematically studied the versatile transformations of cyclobutanols for the construction of quaternary chiral centers induced by the β-carbon elimination.212 In 2009, Cramer and co-workers213a found that the substrates 214 could be used to synthesize multi-substituted chiral indanol derivatives 215 under Rh(I) catalysis (Scheme 67(a)). This reaction starts with the β-carbon cleavage by the initially formed Rh alkoxide species to form the primary alkyl Rh intermediate. The subsequent [1,4]-Rh migration214 leads to the aryl Rh intermediate that undergoes the addition to the formed ketone moiety. The catalytic cycle ends up with the protonation, affording the desired product. Shortly after Cramer’s work, Murakami and co-workers213b reported similar results on the asymmetric synthesis of indanol derivatives. Later, Cramer and co-workers215 envisioned that if the α-substituent of the cyclobutanol was replaced with an electron-rich aromatic ring such as 2-thienyl (216), the final step of the catalytic cycle might shift from the protonation to the β-aryl cleavage, giving rise to the corresponding indanones 217. This idea was realized with the same catalytic system (2.5 mol% of [Rh(cod)2OH] and 6 mol% of (R)-L35), in the presence of an inorganic base Cs2CO3, which probably worked as an inhibitor to the protonation (Scheme 67(b)).
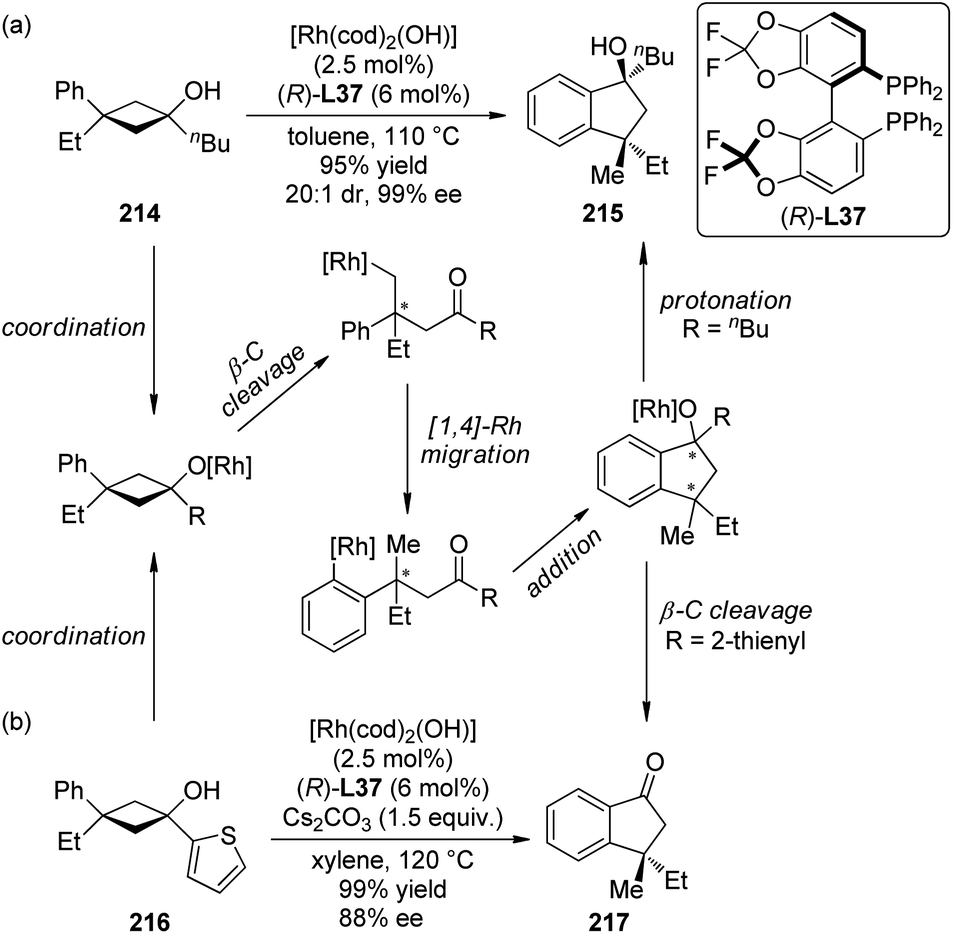 | ||
| Scheme 67 The Rh-catalyzed β-carbon cleavage induced asymmetric C–H bond functionalization reactions reported by Cramer. | ||
The C–Rh species generated from the aryl C–H bond activation/carborhodation sequence are also capable of enantioselectively adding to ketimines. This type of reactions was realized by Tran and Cramer216 who used N-unsubstituted aryl imine 218 and terminal allene 219 as the substrates with the Rh(I) catalyst. One enantioselective example was described by employing very bulky and electron-rich bisphosphine ligand (R)-L38 (Scheme 68(a)). It was suggested that the initial carborhodation occurred from the sterically less hindered face of the allene leading to the allyl-Rh intermediate that underwent the subsequent allylation via a chair-like transition state. Therefore the substituent on the allyl moiety and the free primary amine group adopted the syn configuration and the final condensation delivered the γ-lactam products 220. By replacing the terminal allenes with the internal alkynes as the reaction components, Tran and Cramer217 accomplished enantioselective Rh(I)-catalyzed annulation reactions of aromatic ketimines triggered by direct C–H bond functionalization. Another electron-rich bisphosphine ligand of increased steric bulk (S)-L10 led to the optimal results. Notably, when unsymmetrically substituted alkynes 221 was utilized, the carborhodation was highly regioselective, with the new C–C bond formed preferentially at the carbon atom proximal to the directing group (OMe). The desired product 222 was afforded in 89% yield with 93% ee and 5:1 rs (Scheme 68(b)). Very recently, Tran and Cramer218 reported the Rh(I)-catalyzed annulation reactions between racemic allenes and aryl ketimines via dynamic kinetic asymmetric transformations (DYKAT)219 in the presence of a similar catalytic system ([Rh(cod)2(OH)] + (R)-L39). In this reaction, the racemic 1,3-disubstituted allene 223 undergoes fast racemization through Rh–H insertion/β-H elimination and σ–π–σ isomerization of the allyl-Rh intermediates. The cyclometallated intermediate generated by the C–H bond activation reacts selectively with the matching allene enantiomer, leading to the desired product 224 after downstream transformations (Scheme 68(c)).
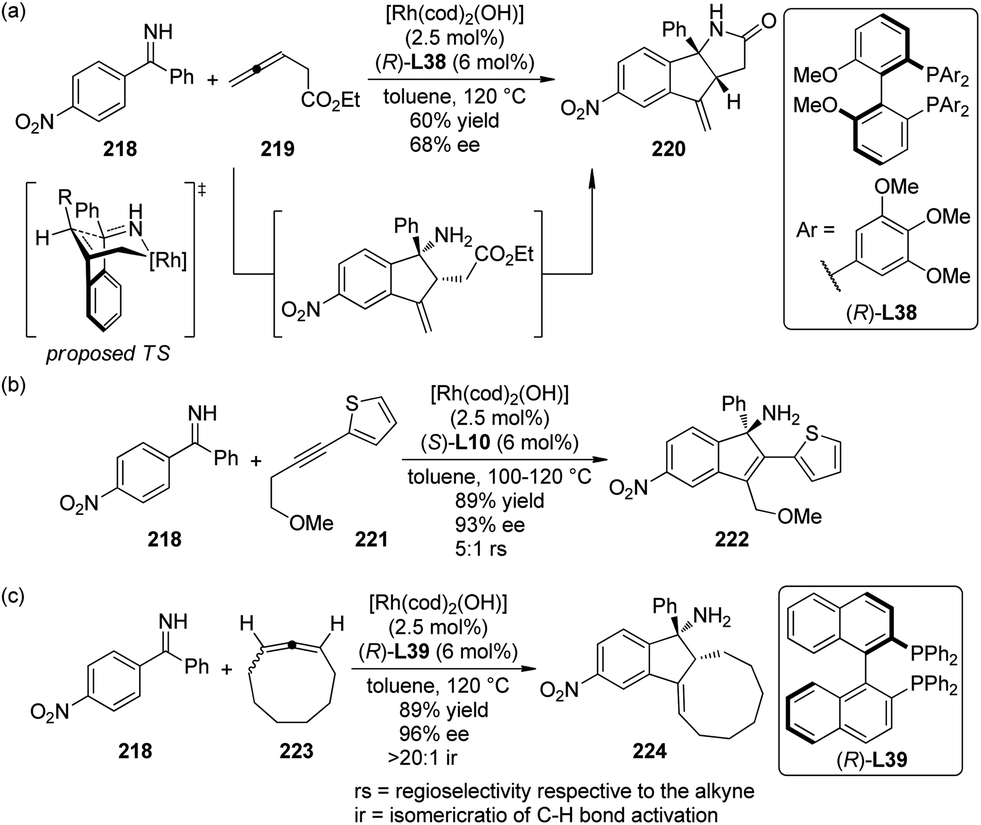 | ||
| Scheme 68 The Rh-catalyzed C–H bond functionalization/cyclization reactions reported by Cramer. | ||
Kuninobu, Takai and co-workers220 demonstrated the Rh-catalyzed asymmetric synthesis of spirosilabifluorene derivatives through double dehydrogenative intramolecular cyclization reactions. Under the optimized conditions (0.5 mol% of [Rh(cod)Cl]2 + (R)-L39), several bis(biphenyl)silanes 225 were converted to the desired silicon-stereogenic products 226 in satisfactory yields and good ee's with the concomitant extrusion of H2 (Scheme 69). It was suggested that the reaction proceeds in two consecutive cyclization steps, each of which starts with the insertion of the Rh center into a Si–H bond. The subsequent C–H bond functionalization might occur either via σ-bond metathesis or oxidative addition/reductive elimination sequence. The final reductive elimination gives rise to the desired C–Si bond.221
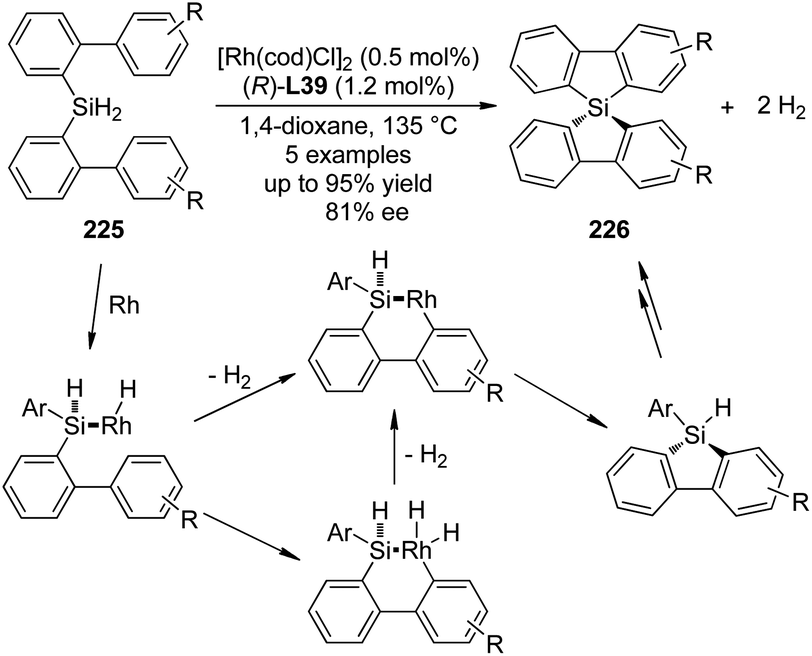 | ||
| Scheme 69 The Rh-catalyzed asymmetric synthesis of spirosilabifluorene derivatives reported by Kuninobu and Takai. | ||
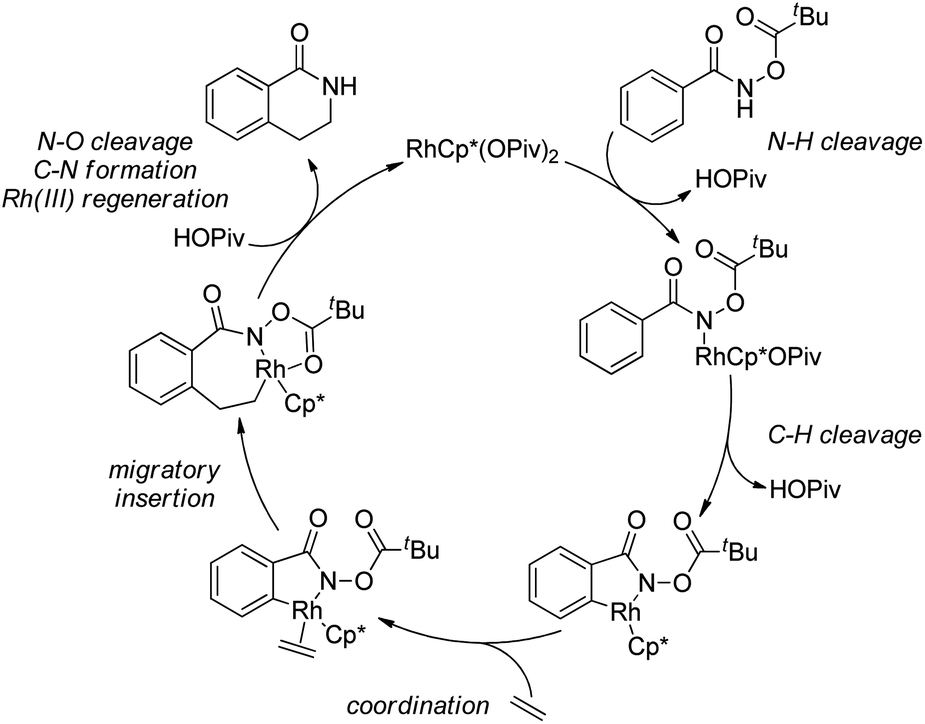 | ||
| Scheme 70 The mechanism of the Rh(III)-catalyzed C–H bond functionalization with an oxidizing directing group. | ||
However, it is pretty challenging to develop the enantioselective version of Rh(III)-catalyzed C–H bonds functionalization reactions in that the cyclopentadienyl (Cp) ligand is typically the only ligand that coordinates to the Rh center permanently throughout the catalytic cycle, and to design a chiral Cp ligand compatible with the late-transition metal-catalyzed reactions is of inherent difficulty.229 Breakthroughs in this area were achieved very recently by the group of Ward and Rovis, as well as the group of Cramer via biochemical and chemical modifications of the Cp ligand respectively.230
Ward, Rovis and co-workers231 were able to develop efficient artificial metalloenzymes232 for Rh(III)-catalyzed asymmetric C–H bond functionalization reactions by applying the Biotin–Avidin technology.233 In this study, a biotinylated Cp*Rh(III) complex named [RhCp*biotinCl2]2 was synthesized and incorporated to streptavidin (Sav) by taking advantage of the remarkable affinity between biotin and streptavidin. The ortho C–H bond functionalization reactions of benzhydroxamic acids 227 with acrylates or ethyl vinyl ketone 228 were selected as the model reactions (Scheme 71). By introducing basic residues in appropriate proximity to the metal center with the assistance of genetic engineering and computational modeling, high reactivity and enantioselectivity of the reactions were achieved. A double mutant (Ser112 to Tyr and Lys121 to Glu) of streptavidin S112Y-K121E could give the best results for a variety of substrates (up to 95% yield and 86% ee). Further mechanistic investigations showed that the C–H bond cleavage event is the turnover-limiting step and the metalloenzymes exhibit nearly 100-fold acceleration compared with the isolated Rh complex. The engineered carboxylate residue is believed crucial to the highly efficient artificial “benzannulase”.
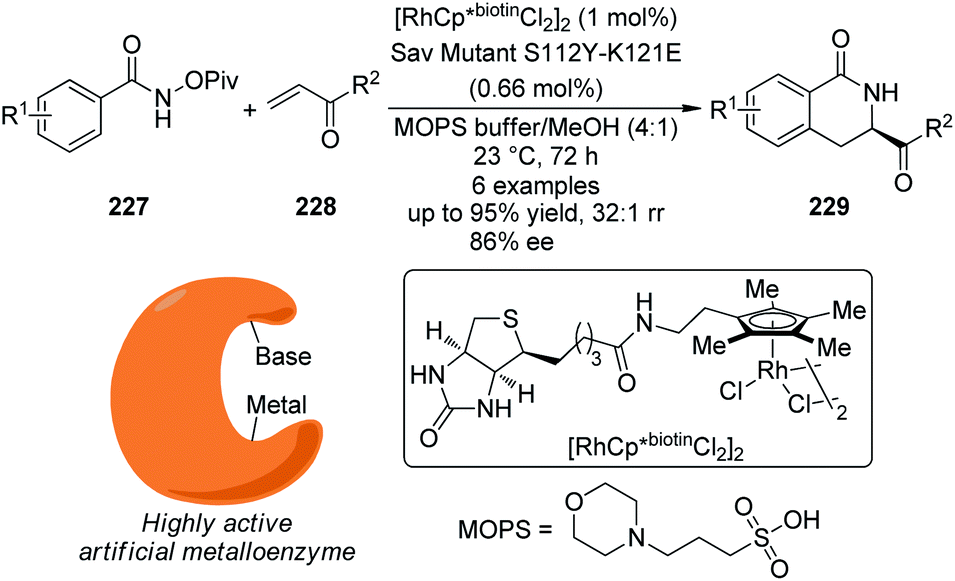 | ||
| Scheme 71 The biotinylated Rh(III) complex-catalyzed asymmetric C–H bond functionalization reactions reported by Ward and Rovis. | ||
On the other hand, Ye and Cramer234 designed and synthesized a series of C2-symmetric chiral Cp ligands. The Rh complexes of these ligands were successfully utilized in the enantioselective C–H bond functionalization reactions of hydroxamic acid derivatives 230 with various terminal and cyclic olefins 231 (Scheme 72(a)). The design of these novel chiral Cp ligands was according to the following considerations. The use of C2-symmetric Cp derivatives could avoid the formation of undesired diastereoisomer due to the coordination of the metal to the other face of the Cp ring. The introduction of two positional locks on the Cp ligand restricted the rotation of the other two ligands on the metal center and thus fixed the conformation of metal–substrate complex. A sterically bulky substituent perpendicular to the Cp plane resulted in that the incoming reactant could only approach from just one side (Scheme 72(b)). Among the synthesized chiral Rh complex, C49 led to the best results in terms of both yield and enantioselectivity, and the reaction featured a broad substrate scope. The use of stable Rh(I) precursor and DBPO as the oxidant allowed the in situ generation of a competent Rh(III) catalyst. To be noted, the catalyst loading could be lowered to 1 mol%.
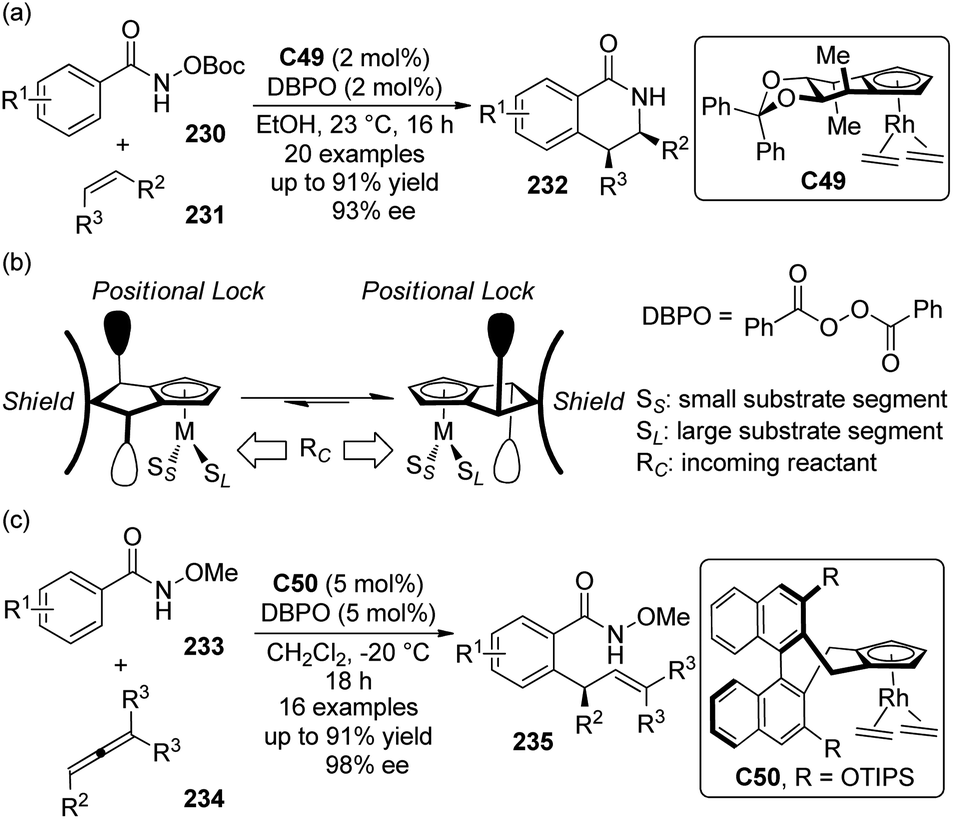 | ||
| Scheme 72 The chiral Rh(III) complex-catalyzed asymmetric C–H bond functionalization reactions reported by Cramer. | ||
Based on the same strategy, Ye and Cramer235 synthesized a tunable class of chiral Cp ligands derived from the BINOL skeleton and applied their Rh complexes to the C–H bond allylations of N–OMe protected benzamides 233 with trisubstituted allenes 234.236 In this case, the lower naphthyl portion of the ligand acts as the back wall and the ortho R group on the naphthyl ring has substantial influence to the shape of chiral pocket. After evaluation of a series of chiral Rh precursors, C50 with a bulky ortho OTIPS substituent was identified as the optimal one (Scheme 72(c)). Under very similar conditions as the previously reported annulation reaction,234 an array of allylated products 235 could be generated in up to 91% yield and 98% ee.
5.3. Ir-Catalyzed reactions
The history of iridium complexes being involved in the C–H bond activation reactions is quite long. However, the application of Ir-catalyzed functionalization of inert C–H bonds via a transient C–Ir species for the construction of C–C or C–X bonds in an enantioselective manner with high efficiency only emerges recently.237 The successful reactions fall into the area of the asymmetric alkylation of C–Ir intermediates formed by the oxidative addition of Ir(I) precursors to the C–H bonds, which mechanistically resembles the reactions under Rh(I) catalysis presented in the Section 5.2.1.In 2008, Shibata and co-workers238 reported Ir-catalyzed asymmetric C–H bond alkylation reaction with norbornene 237 as the acceptor. The 2-methylacetophenone 236 was the only viable substrate. The ortho C–H bond alkylation product 238 was obtained in 58% yield and 70% ee (Scheme 73(a)). Sevov and Hartwig239 recently expanded the substrate scope of this reaction to various heteroarenes. By using catalytic amount of [Ir(cod)Cl]2 in combination with bisphosphine ligand (S)-DTBM-Segphos (S)-L41, the C–H bond adjacent to the heteroatoms of indoles, pyrroles, thiophenes and furans could add across norbronene to afford only one single constitutional isomer in both high yield and enantioselectivity (Scheme 69(b)). Preliminary mechanistic studies indicated that the oxidative addition of Ir(I) species to the heteroarene C–H bonds occurred prior to the turnover-limiting step.
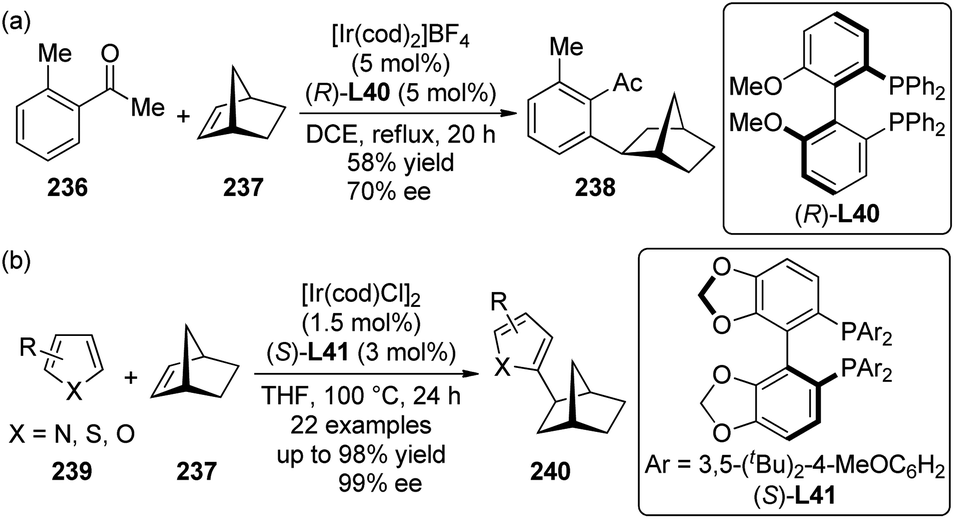 | ||
| Scheme 73 The Ir-catalyzed asymmetric C–H bond alkylation reactions reported by (a) Shibata and (b) Hartwig, respectively. | ||
The Shibata group240 also realized Ir-catalyzed asymmetric secondary C(sp3)–H bond alkylation reactions of 2-alkylaminopyridines with olefins. The cationic Ir(I) catalyst ligated by (S)-tolBINAP ligand (S)-L42 could promote the reactions of the C–H bonds adjacent to the nitrogen atom of the substrates 241 with a large array of olefins 242 including styrenes and functionalized alkenes, leading to the desired chiral amine products 230 in up to 89% yield and 99% ee (Scheme 74). The nitrogen atom in the pyridine unit is likely acting as a directing group to facilitate the C–H bond cleavage through a six-membered ring transition state.
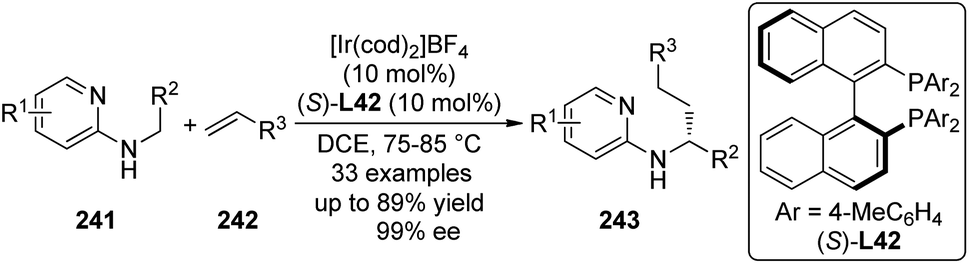 | ||
| Scheme 74 The Ir-catalyzed asymmetric C–H bond alkylation reactions reported by Shibata. | ||
Nishimura et al.241 achieved an asymmetric [3 + 2] annulation via Ir-catalyzed C–H bond functionalization to synthesize spiroaminoindane derivatives 246.242 With cationic Ir(I) catalyst generated from chiral diene ligand Me-TFB* (S,S)-L43,243 the in situ formed cyclic N-acyl ketimines from the corresponding 3-aryl-3-hydroxyisoindolin-1-ones 244 react with 1,3-dienes 245 smoothly. The desired products were obtained with high yields and exceptional enantioselectivity (Scheme 75). The reaction also features relatively broad substrate scopes. An aryl–Ir(I) species formed by ortho-C–H bond functionalization is believed as the key intermediate of the reaction.
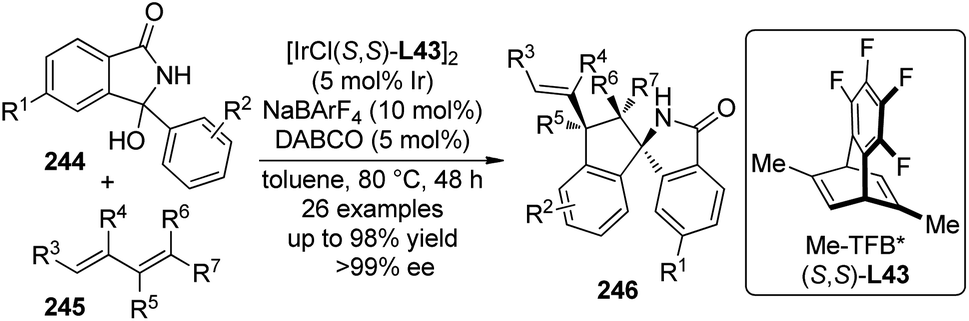 | ||
| Scheme 75 The Ir-catalyzed asymmetric [3 + 2] annulation reaction reported by Nishimura. | ||
5.4. Kharasch–Sosnovsky reactions
The Kharasch–Sosnovsky reaction that refers to the Cu-catalyzed allylic oxidation reactions of alkenes with the tert-butyl peresters as the oxidant was first reported in the late 1950s.244 The asymmetric variants of this reaction have been developed since mid-1990s.245 Various nitrogen-based chiral ligands such as C2-symmetric bis(oxazoline)s as well as proline-derived ligands were generally used. Although high level of enantioselective control could be reached, the viable substrates are mainly restricted to cyclic olefins and prolonged reaction time (up to several days at rt) is typically required. Thus, a general, highly efficient catalytic system is still in great demand. Despite different mechanistic scenarios have been proposed, it is generally postulated that an allyl–Cu(III) intermediate is involved in the Kharasch–Sosnovsky reaction (Scheme 76).246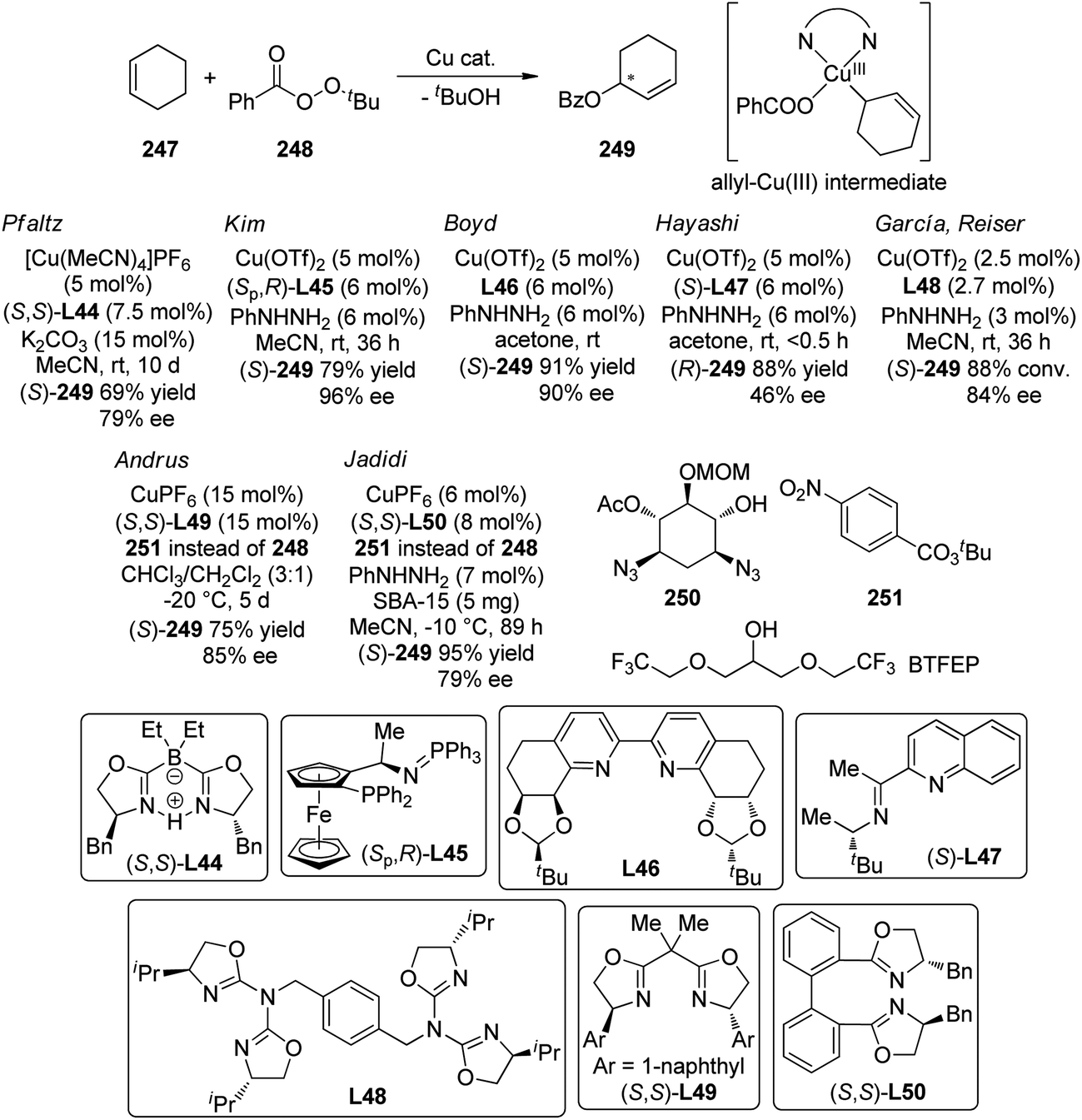 | ||
| Scheme 76 The Cu-catalyzed asymmetric Kharasch–Sosnovsky reactions reported by several groups. | ||
The Pfaltz group247 synthesized chiral boron-bridged bisoxazoline ligand (S,S)-L44 and evaluated their Cu complexes in the Kharasch–Sosnovsky reaction of cyclic olefins (Scheme 76). The enantioselectivity of the reaction was comparable with the previously reported best ones obtained using the BOX ligands.248 Kim and co-workers249 applied chiral (iminophosphoranyl)ferrocene ligands like (Sp,R)-L45 in the same reactions. By using the in situ generated Cu(I) catalyst derived from the reduction of Cu(OTf)2 with PhNHNH2, the desired product 249 of the Kharasch–Sosnovsky reaction between cyclohexene 247 and tert-butyl perbenzoate 248 could be delivered in 79% yield and 96% ee. Boyd and co-workers250 synthesized cis-dihydrodiol-derived 2,2′-bipyridine ligand L46 and used it in the Kharasch–Sosnovsky reaction of 247. The corresponding product was obtained in 91% yield and 90% ee with a similar protocol. Tan and Hayashi251 reported the Kharasch–Sosnovsky reaction catalyzed by the Cu complex generated from novel N,N-bidentate Schiff base ligand (S)-L47. This catalytic system showed high reactivity in that the reaction of 247 completed in less than 0.5 h, although the ee of the product was only moderate. This protocol was further employed in the desymmetrization of 4,5-epoxycyclohexene for the synthesis of O-protected 2-deoxystreptamine precursor 250.252 García, Reiser and co-workers253 developed an easily recoverable and reusable catalytic system based on a ditopic azabis(oxazoline) ligand L48 for the Kharasch–Sosnovsky reaction. When the reaction was complete in MeCN or BTFEP, the ligand and Cu salt were able to form coordination polymers that could precipitate after extraction with hexane. The catalyst exhibited comparable activity after several successive recycles. Zhou and Andrus254 found that the Cu complex derived from the naphthyl-substituted BOX ligand (S,S)-L49 could promote the Kharasch–Sosnovsky reaction of 247 with tert-butyl p-nitroperbenzoate 251 as the oxidant, giving rise to the desired product 249 in 75% yield and 85% ee. Jadidi and co-workers255 disclosed that in the presence of SBA-15 mesoporous silica as the additive, improved results (95% yield and 79% ee) could be observed for the Kharasch–Sosnovsky reaction catalyzed by the Cu complex derived from the atropisomeric chiral BOX ligand (S,S)-L50.
Apart from the recent development of the Kharasch–Sosnovsky reactions of cyclic olefins, the group of Zhu and Zhou256 studied this reaction of acyclic terminal olefins using the chiral spiro bisoxazoline ligand (Ra,S,S)-L51.257 A variety of the acyclic olefins 252 was investigated and the reactions of almost all the tested substrates led to the oxidation products with excellent regioselectivity (up to >20:1 b:l) favoring the branched allylic benzoates 253b in up to 64% yield and 67% ee (Scheme 77). Functional groups such as ketone, ester and silyl were tolerated. This report represented one of the best results for the Kharasch–Sosnovsky reactions of acyclic olefins.
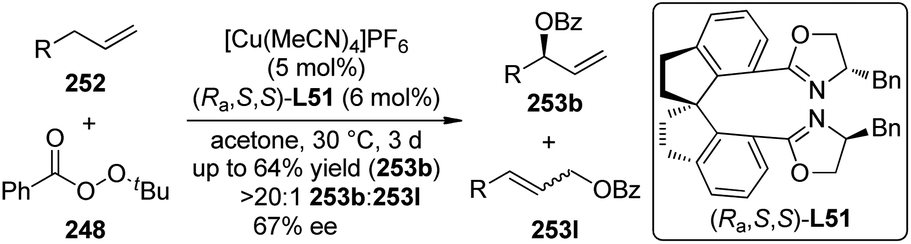 | ||
| Scheme 77 The Cu-catalyzed asymmetric Kharasch–Sosnovsky reactions reported by Zhu and Zhou. | ||
5.5. Ni-Catalyzed reactions
Very recently, Donets and Cramer258 reported a Ni-catalyzed asymmetric hydrocarbamoylation reactions of alkenes with diaminophosphine oxide ligands. It is known that secondary phosphine oxides are usually air-stable and robust preligand because of their unique tautomerization between the P(V) form and the P(III) form.259 In the presence of bases or transition metals, such equilibrium can be shifted to the trivalent phosphinous acid and thus provide the opportunity for the hetero-bimetallic catalysis where a late transition metal and an early transition metal can coordinate to the phosphorous atom and the oxygen atom, respectively (Scheme 78(a)). Bearing this idea in mind, the authors synthesized a class of chiral diaminophosphine oxide ligands260 and tested them in the Ni-catalyzed intramolecular asymmetric hydrocarbamoylation reactions of formamides 254 with AlMe3 as the other metal catalyst (Scheme 78(b)). The combination of [Ni(cod)2] and (R,R)-L52 gave the optimal catalytic activity. Substrates with various substitution patterns were converted smoothly to the desired pyrrolidinones 255 in up to 98% yield and 95% ee under mild conditions. To be noted, a catalytic amount of PPh3 was required to displace the cod from the Ni center for the generation of the competent catalytic species. In their mechanistic proposal, both AlMe3 and [Ni(cod)2] coordinate to the chiral bridging ligand. The aluminum center activates the carbonyl group of the substrate, making the nickel center approximate to the formamide C–H bonds. The C–H bond cleavage takes place via a favorable six-membered heterocycle. The corresponding product is finally released after the subsequent migratory insertion and reductive elimination. In addition, the independently prepared aluminum complex (R,R)-C51 could also be used. The loadings of this complex and the Ni precursor could be lowered to 0.25 mol% in this case. Interestingly, with (R,R)-C51 as a catalyst, neither additional AlMe3 nor PPh3 was needed for achieving excellent results (97% yield and 94% ee).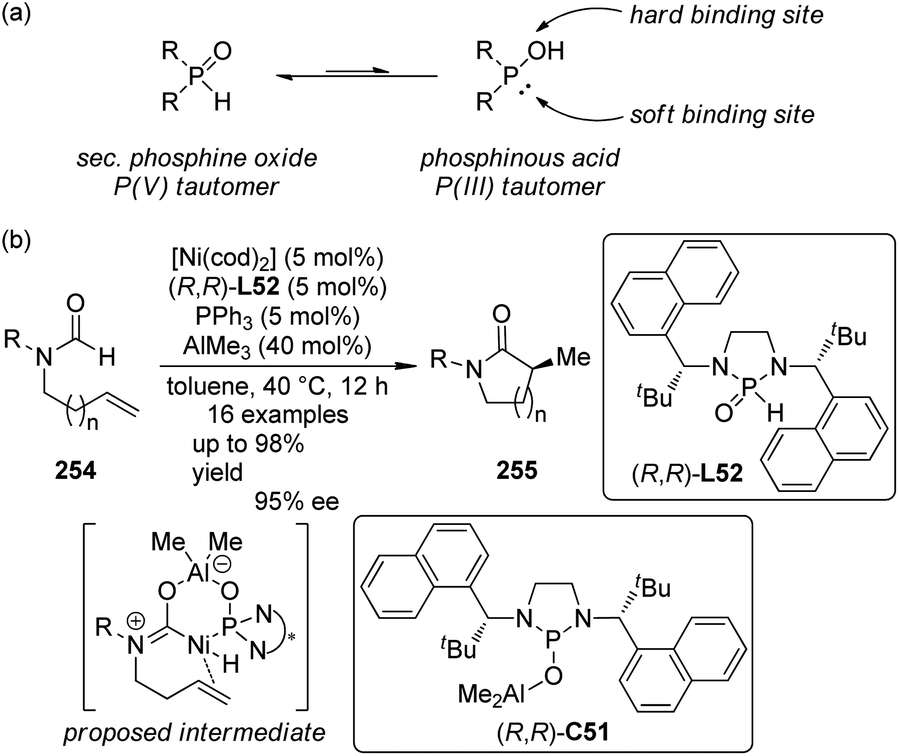 | ||
| Scheme 78 The Ni-catalyzed asymmetric hydrocarbamoylation reactions reported by Cramer. | ||
6. Miscellaneous reactions
6.1. Biaryl coupling reactions
The compounds possessing an axially chiral biaryl skeleton such as the BINOL derivatives261 are among the privileged class of molecules for the application as ligands or catalysts in asymmetric catalysis.262 Meanwhile, there are also numerous natural products that contain biaryl motifs.263 Of various methods to construct this type of molecular architecture, the asymmetric oxidative coupling of 2-naphthols is probably the most straightforward and atom-economical way.264In 2009, Egami and Katsuki265 found that by using chiral di-μ-hydroxo dimeric Fe(III)(salan) complex266 (Ra,S)-C52 or (Ra,R)-C53 as the catalyst, various 2-naphthols 256 with different substitution patterns could undergo homo-coupling via asymmetric aerobic oxidation under mild conditions to afford enantioenriched BINOL-derivatives 257 (Scheme 79(a)). Notably, high level of enantioselectivity was observed when the C3-position of the naphthyl ring was substituted (R1 ≠ H). Functional groups such as halides, alkynes are well tolerated which allows further modification of the valuable BINOL skeletons. While an electron-withdrawing ester group is incorporated (R1 = CO2Me), no oxidative coupling reaction occurs. In their follow-up study, the Katsuki group267 realized the Fe-catalyzed enantioselective synthesis of C1-symmetric BINOLs268 260 through the cross-coupling of 2-naphthols (258 and 259) having sufficiently different redox potentials (Scheme 79(b)). Mechanistic investigations suggested that the radical–anion coupling mechanism might be operative (Scheme 79(c)). The dimeric Fe catalyst first dissociated to the monomeric species and reacts with the more electron-rich 2-naphthol (Nap-EDG) giving rise to the intermediate A that was oxidized to generate the radical cationic species B. On the other hand, the less electron-rich 2-naphthol (Nap-EWG) is more acidic and thus readily coordinates to the intermediate B in the anionic alkoxide form, yielding the intermediate C. The cross-coupling product is finally produced after the subsequent oxidation. Detailed X-ray diffraction analyses and cyclic voltammetry studies269 revealed that the presence of the double hydrogen bonding in the Fe(salan) dimers, the oxidation potential of the monomeric Fe(salan) species as well as the location of the resulting radical cation have strong influence to the catalytic activity in this reaction.
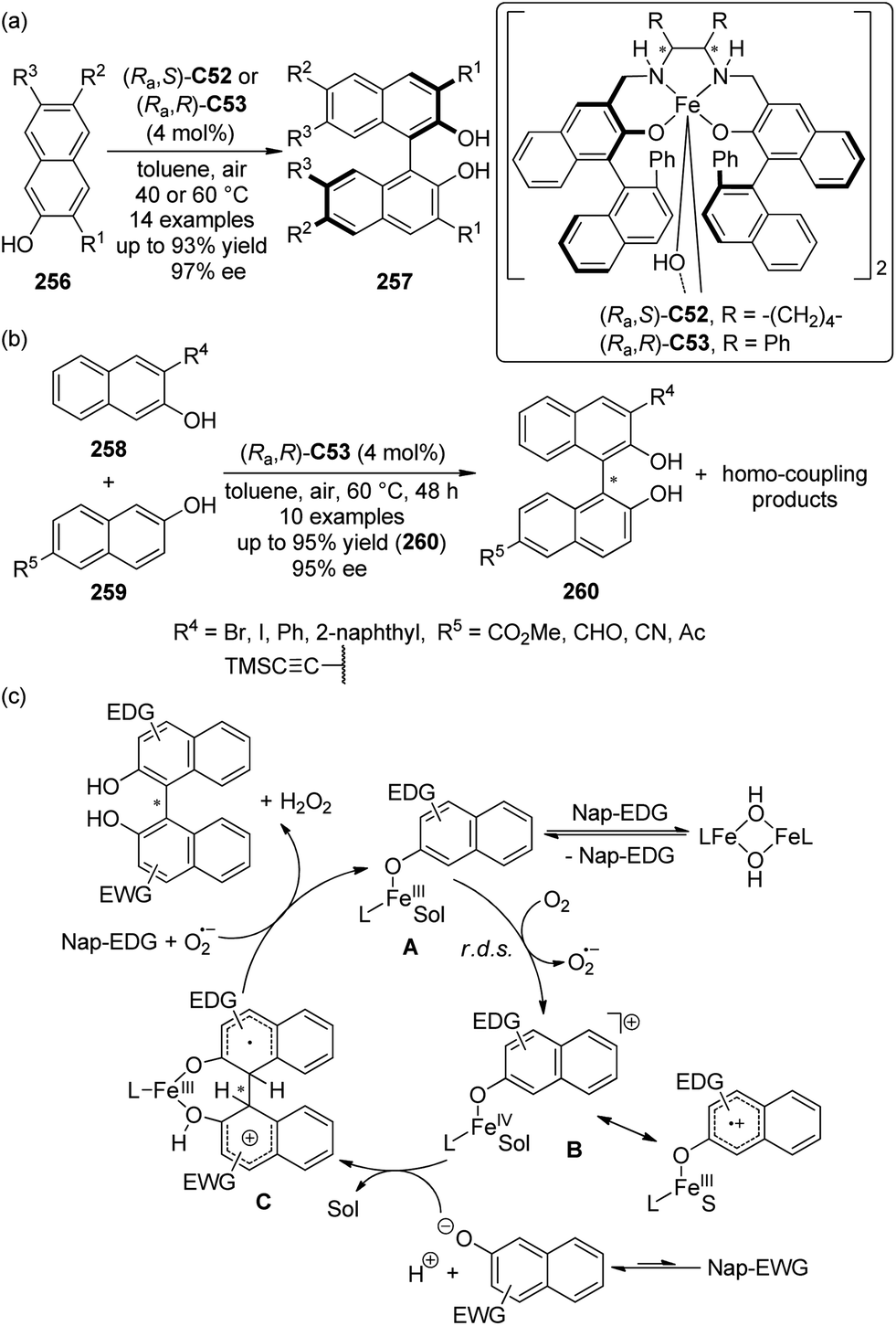 | ||
| Scheme 79 The Fe-catalyzed asymmetric biaryl coupling reactions reported by Katsuki. | ||
Prim and co-workers270 synthesized a series of chiral N-heterocyclic pyridylmethylamine ligands and found their corresponding Cu complexes could catalyze the oxidative coupling of electron-deficient 2-naphthol 261. The complex of L53 (diastereomerically pure but the absolute configuration of one chiral center not determined) showed the best asymmetric induction (61% ee) albeit the yield of the biaryl product 262 was only 50% (Scheme 80).
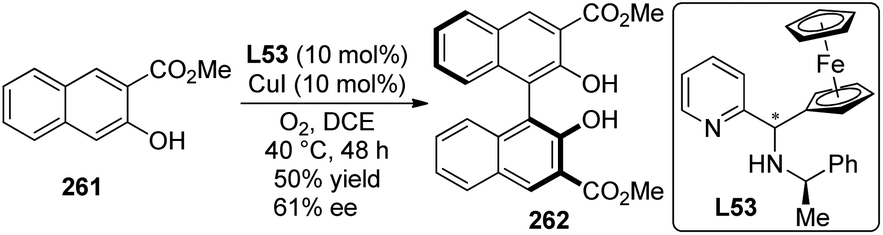 | ||
| Scheme 80 The Cu-catalyzed asymmetric biaryl coupling reactions reported by Prim. | ||
Sekar and co-workers271 utilized a Cu(I) catalytic system comprised of CuCl and (R)-BINAM ((R)-L54, BINAM = 1,1′-binaphthyl-2,2′-diamine) to realize the asymmetric oxidative coupling of 2-naphthol derivatives 263. Adding TEMPO as a co-oxidant was found to drastically increase the reaction rate as well as enantioselectivity. A series of enantioenriched BINOL products 264 bearing various ester groups at the 3,3′-positions were obtained under very mild conditions (Scheme 81). A complex of CuCl·2L54 was also isolated and characterized by single crystal X-ray diffraction analysis.
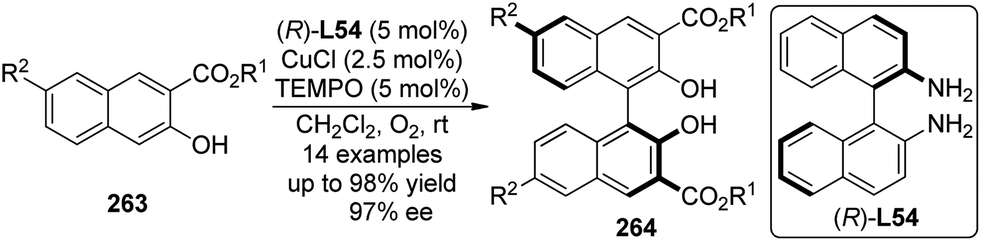 | ||
| Scheme 81 The Cu-catalyzed asymmetric biaryl coupling reactions reported by Sekar. | ||
Mikami and co-workers272 reported the Cu-catalyzed asymmetric homo-coupling reaction of 3-substituted naphthylamine 265 and its hetero-coupling reaction with 3-substituted naphthol 267. By using (−)-sparteine as the chiral ligand, the desired products 3,3′-dimethyl-2,2′-diamino-1,1′-binaphthyl 266 (DM-DABN) and 3,3′-disubstituted 2-amino-2′-hydroxy-1,1′-binaphthyl 268 (NOBIN)273 were afforded when O2 was used as the oxidant. Interestingly, it was found that the enantioselectivity of the coupling products was relatively higher with lower concentration of O2 (Ar balloon with O2 gradually introduced to the reaction mixture through the balloon) (Scheme 82).
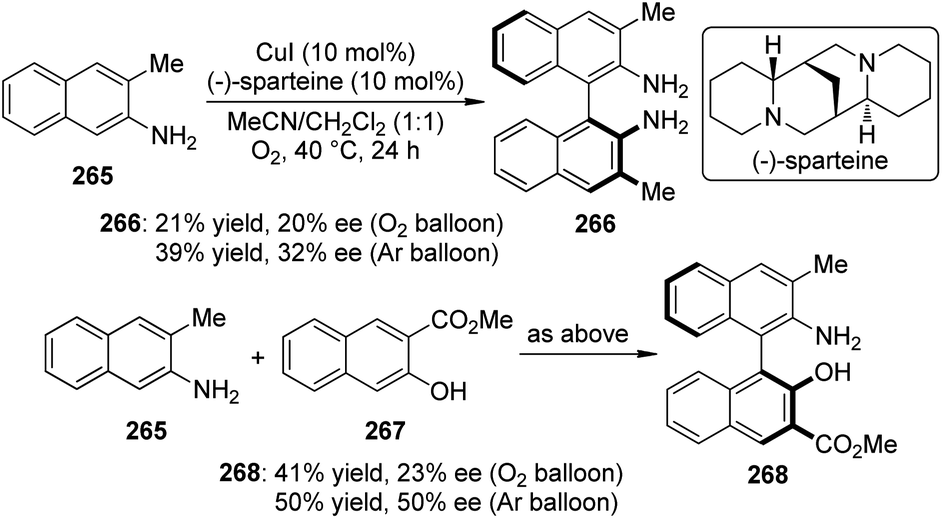 | ||
| Scheme 82 The Cu-catalyzed asymmetric biaryl coupling reactions reported by Mikami. | ||
Very recently, the Kürti group274 reported the asymmetric synthesis of BINAM derivatives via an atroposelective [3,3]-rearrangement reactions using N,N′-diaryl hydrazines 269 as the substrates. Various organocatalysts were evaluated and the chiral phosphoric acids (R)-C54 and (R)-C55 could give the optimal results in terms of both yields and ee's of the products 270 (Scheme 83). It was found that lowering the temperature and adding 4 Å molecular sieves (MS) were beneficial to improve the enantioselectivity. The reaction performed well on a 2–10 mmol scale (up to 2.8 g) and the catalysts could be recovered in nearly quantitative yield (99%) after flash chromatography. Based on DFT calculations, plausible transition states for the C–C bond forming step were postulated that the phosphate acts as a chiral counterion and creates a chiral pocket for the diastereoselective discrimination. Almost at the same time, List and co-workers275 developed a very similar strategy for the asymmetric synthesis of BINAM derivatives.276 In their cases, the chiral phosphoric acid (R)-C48 was identified as the best catalyst and the weakly acidic CG-50 resin was a viable additive, allowing lower catalyst loading with full consumption of the substrates. Notably, a significant negative nonlinear effect was observed, which is consistent with a dicationic mechanism where two catalyst anions might be involved.
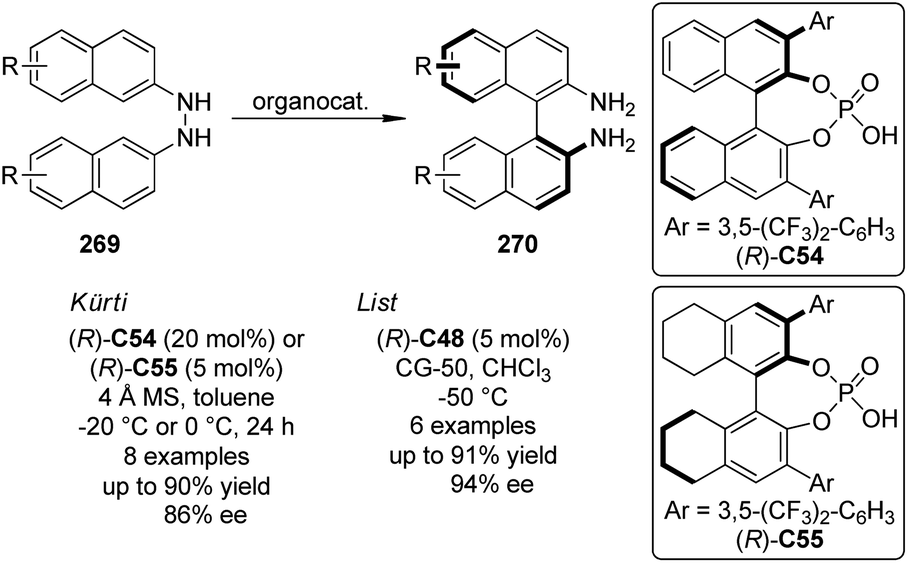 | ||
| Scheme 83 The chiral phosphoric acid-catalyzed asymmetric [3,3]-rearrangement reactions reported by Kürti and List, respectively. | ||
6.2. β-Functionalization of carbonyl compound
The asymmetric α-functionalization of carbonyl compounds has received considerable development in the past several decades and serves as prominent method for the enantioselective construction of C–C bonds in modern organic synthesis.277 However, the direct asymmetric β-functionalization of simple saturated carbonyl compounds, on the other hand, still remains elusive. Given that the strategy involving the transformation of an iminium ion to an enamine in the “iminium catalysis” provides unique opportunity to functionalize the β- and α-position of aldehydes and ketones sequentially,278 the group of Li and Wang as well as the group of Hayashi envisioned the reverse “enamine to iminium ion” process facilitated by proper oxidants could be capable to incorporate an nucleophile to the β-position of simple aldehydes and thus furnished the concept of “oxidative enamine catalysis” independently.279In 2011, Li, Wang and co-workers280 disclosed that by utilizing diphenylprolinol silyl ether (S)-C30 as the catalyst, in combination with IBX as the oxidizing agent, simple aldehydes 271 could react with fluorobis(phenylsulfonyl)methane (FBSM) 272 smoothly to afford the β-functionalized aldehydes 273 in moderate yields and high ee's (Scheme 84(a)). In addition, this methodology could be further integrated to a variety of multiple cascade reactions to deliver versatile chiral targets such as coumarin-containing pyran 276 (Scheme 84(b)). Noticeably, relatively high catalyst loading (20–30 mol%) is required since the chiral amine catalyst can be oxidized by IBX. Almost at the same time, the Hayashi group281 reported a very similar procedure employing DDQ as the oxidant and MeNO2 as the nucleophile. A number of functionalized aldehydes 278 were obtained in a one-pot manner in up to 80% yield and 95% ee (Scheme 84(c)). Very recently, Xu and co-workers282 developed an enantioselective β-functionalization reaction of aldehydes 279 with malonate diesters 280 by combining enamine/iminium ion catalysis and Pd(OAc)2 catalyzed Saegusa oxidation.283 Only O2 (1 atm) was required to serve as the oxidant (Scheme 84(d)).
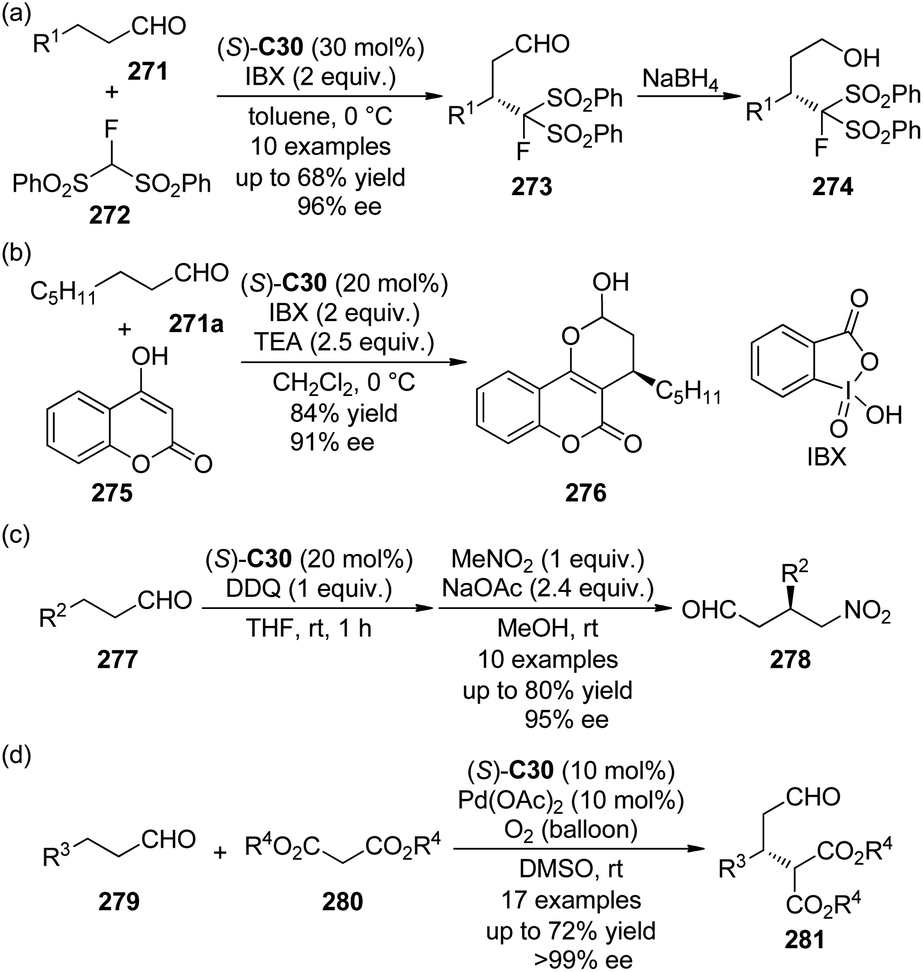 | ||
| Scheme 84 The asymmetric β-functionalization reactions of aldehydes reported by (a) and (b) Li and Wang, (c) Hayashi, and (d) Xu, respectively. | ||
Enders and co-workers284 integrated the asymmetric β-functionalization reactions of aldehydes into the organocatalytic two-component four-step “branched domino reaction” to synthesize polyfunctionalized cyclohexene derivatives. According to the proposed catalytic cycle (Scheme 85), two equivalents of β-substituted aldehydes are required. The enamine intermediate I generated from the aldehyde and secondary amine catalyst attacks a Michael acceptor leading to intermediate II. Simultaneously, I can be oxidized to the α,β-unsaturated iminium intermediate III. The following reaction between II and III gives the final product.285 In the presence of catalytic amount of (R)-C30 and 1.5 equiv. of IBX, the reactions of dihydrocinnamaldehyde 283 with nitroolefins 282 or oxindole derivatives 285 went smoothly to afford their corresponding products 284 and 286 in good yields and excellent dr's and ee's.
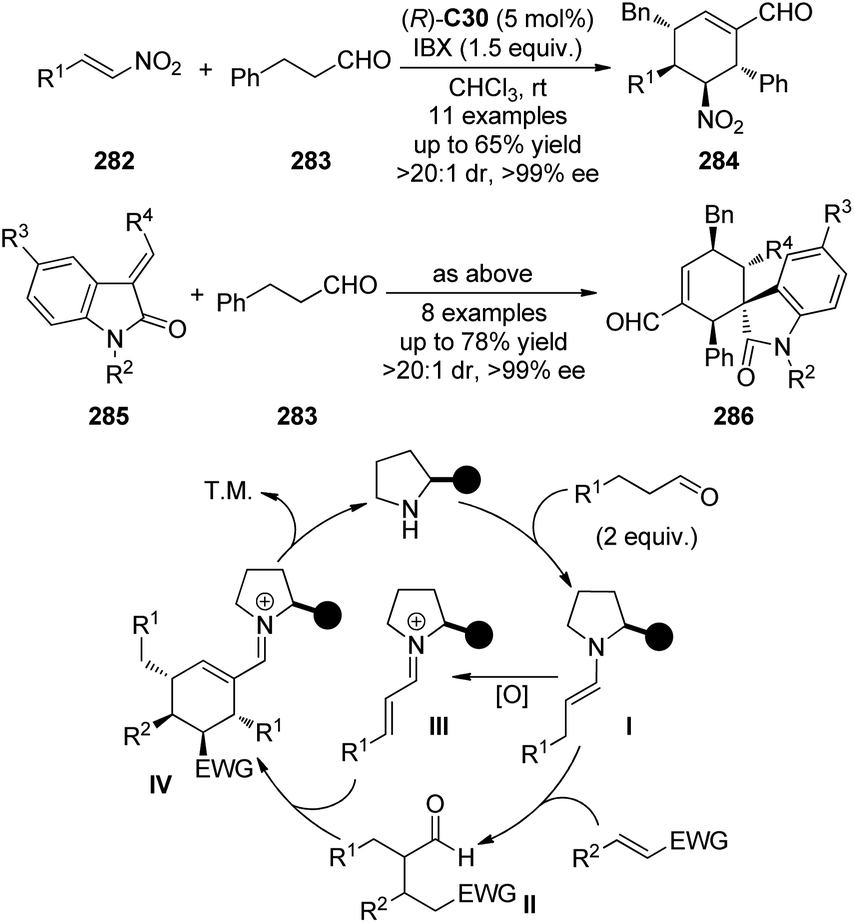 | ||
| Scheme 85 The organocatalytic domino reactions involving β-functionalization of aldehydes reported by Enders. | ||
Kang and Kim286 successfully applied the strategy of oxidative enamine catalysis into the [1,5]-hydride transfer/cyclization sequences.130 By using the secondary amine catalyst (S)-C29 in combination with IBX as the oxidant, a series of ortho-tert-amine-substituted dihydrocinnamaldehydes 287 were first oxidized to the corresponding α,β-unsaturated iminium intermediates that underwent the subsequent asymmetric [1,5]-hydride transfer/cyclization reactions to afford the desired tetrahydroquinoline derivatives 288 (Scheme 86).
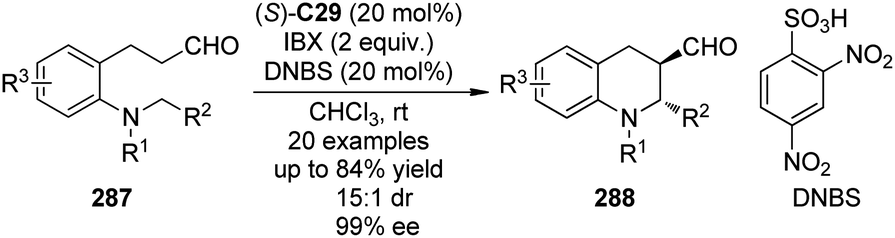 | ||
| Scheme 86 The asymmetric oxidative [1,5]-hydride transfer/cyclization reactions reported by Kim. | ||
Chi and co-workers287 realized the NHC-catalyzed asymmetric β-functionalization reactions of aldehydes via the transformation of saturated aldehydes to formal Michael acceptors by the double oxidation.288 By using the catalyst derived from chiral amino-indanol triazolium salt (R,S)-C56 in combination with quinone 291 as the oxidant, the β-aryl substituted saturated aldehydes 289 were converted to the α,β-unsaturated acyl azolium intermediates which further reacted with 1,3-dicarbonyl compounds or β-keto esters 290 to generate the corresponding δ-lactones 292 (Scheme 87). It was found the use of LiCl and 4 Å MS as additives was beneficial to improve the ee's of the products. Notably, the β-alkyl substituted saturated aldehydes were not viable substrates, probably due to the reduced acidity of the β-C–H bonds.
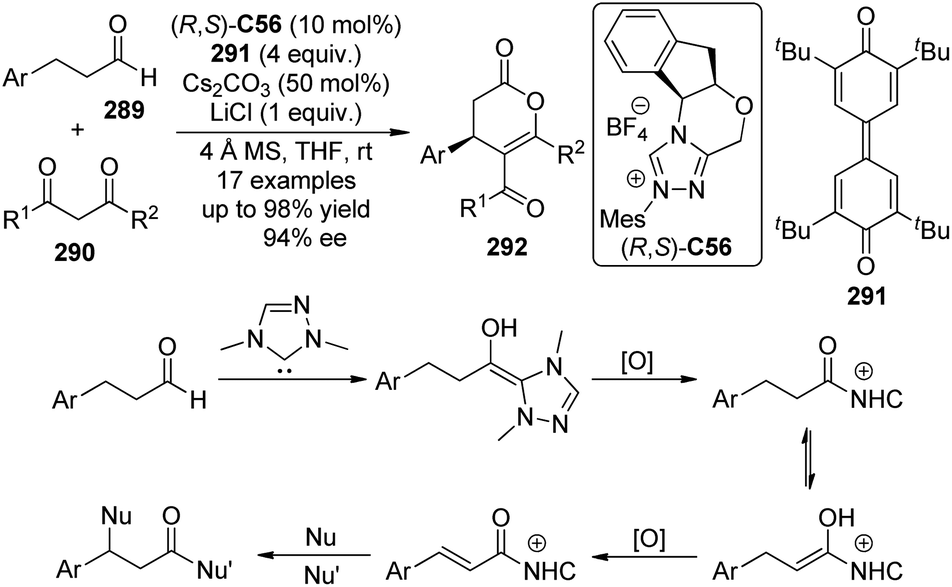 | ||
| Scheme 87 The asymmetric β-functionalization reactions of aldehydes reported by Chi. | ||
MacMillan and co-workers289 envisioned that the β-functionalization of carbonyl compounds could be realized by combining the merging visible light mediated photoredox catalysis with the amine catalysis.290 The designed dual catalytic cycle (Scheme 88(a)) starts with the formation of the excited state *Ir(III)(ppy)3 upon the visible light irradiation of the well-known photoredox catalyst Ir(III)(ppy)3. The high energy *Ir(ppy)3 is readily oxidized to Ir(IV)(ppy)3 by 1,4-dicyanobenzene via an SET process with the concomitant generation of radical anion A. Subsequently, the electron-rich enamine intermediate formed after the condensation of the carbonyl compounds and the amine catalyst is oxidized by Ir(IV)(ppy)3 to afford the corresponding radical cation B which is further transformed to the 5πe− intermediate C via the allylic proton abstraction by a proper base. The desired products will be delivered after the radical coupling between intermediates A and C and following cyanide extrusion. This elegant transformation was realized for a large array of simple aldehydes and ketones with one enantioselective example (challenging inactivated ketone 293) where cinchona-derived primary amine catalyst C57 was employed although the ee of the product 295 was only moderate (Scheme 88(b)).
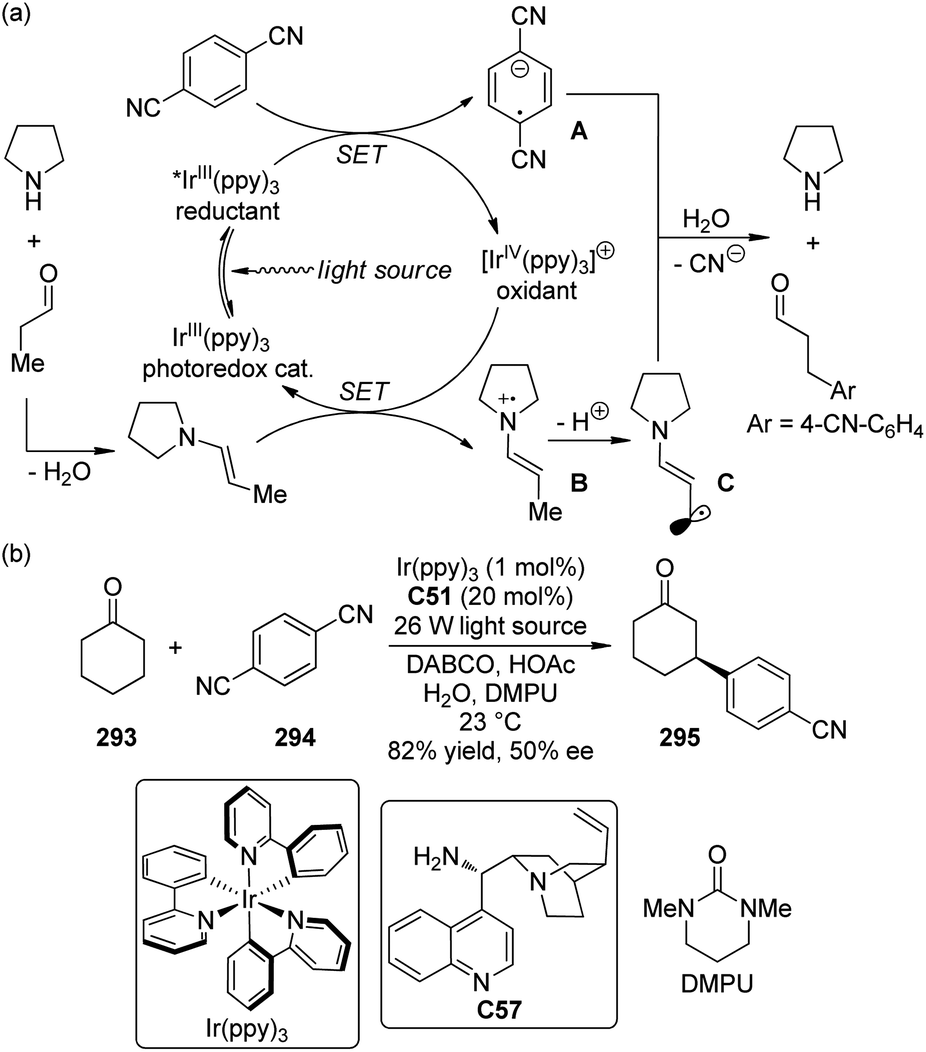 | ||
| Scheme 88 The asymmetric β-arylation reactions of aldehydes and ketones reported by MacMillan. | ||
Very recently, the Chi group291 achieved versatile enantioselective β-functionalization of esters through NHC catalysis.7 In their postulated catalytic cycle (Scheme 89), the NHC catalyst first reacts with the ester to form the acyl azolium intermediate.292 The subsequent α-C–H abstraction produced the enolate intermediate. Due to the electron-withdrawing ability of the triazolium moiety and the conjugated nature of this intermediate, the β-proton of the ester could become acidic and the deprotonation could give rise to the corresponding homoenolate intermediate that is traditionally accessed via extended umpolung of α,β-unsaturated aldehydes under NHC catalysis.293 Experimentally, the homoenolate intermediates generated from the β-activation of the esters 296 were found readily reactive to diverse electrophiles such as enones 297, trifluoromethyl ketones 299 as well as activated hydrazones 301. By utilizing chiral NHC catalyst (R)-C58, the desired highly functionalized cyclic olefins 298, γ-lactones 300 and γ-lactams 302 were all produced in moderate to high yields and high level of stereocontrol under mild conditions.
 | ||
| Scheme 89 The asymmetric β-functionalization reactions of esters reported by Chi. | ||
7. Conclusion and prospective
Summarized and discussed in this review are the recent progresses in the field of direct asymmetric functionalization of inert C–H bonds. As a result of the fast and fascinating development achieved in the last several years, a large array of inert C(sp2)–H and C(sp3)–H bonds has been found possible to be directly functionalized in a highly enantioselective fashion. Various catalytic systems including well-defined organometallic complexes, small organic molecules and enzymes are applicable to promote this type of novel transformations that provide organic chemists alternative viewpoints to execute the disconnection strategy in modern organic synthesis. However, formidable challenges still remain in the following three major aspects: (1) limited substrate scope. Most asymmetric direct C–H bond functionalization reactions presented here only occurs at some specialized positions such as in proximity to a directing group, α to a heteroatom, and benzylic or allylic positions. Therefore, it will be of considerable value to expand the scope of the asymmetric functionalization to the more inert C–H bonds. (2) Relatively poor reactivity. Due to the inherent difficulty of the direct cleavage of C–H bonds, most current methodologies to this end still require relatively high catalyst loading and elevated reaction temperature. Thus, to develop novel highly efficient catalytic systems that can facilitate asymmetric C–H bond functionalization processes will undoubtedly be the primary direction of future contributions. (3) Complex reaction conditions. Various additives, stoichiometric oxidants are needed in the procedures of many asymmetric C–H bond functionalization reactions. Some catalysts are very sensitive and not easy to handle. Accordingly, most current asymmetric C–H bond functionalization methods are limited to laboratory scale and not feasible for industrial application. Hence, it is also important to enhance the operational simplicity of these methodologies.Although there is still long way to realize the direct functionalization of arbitrarily specified C–H bonds of any chemical feedstock in high level of stereocontrol, the efforts devoted towards this ambitious goal will definitely stimulate further exciting advances of organic chemistry in the coming era.294
Acknowledgements
We thank the National Natural Science Foundation (21025209, 21121062 and 21302209) of China for generous financial supports.Notes and references
- For a book: (a) C–H Activation, in Top. Curr. Chem., ed. J.-Q. Yu and Z. Shi, Springer-Verlag Berlin Heidelberg, 2010, vol. 292 Search PubMed. For a book chapter: (b) K. M. Engle and J.-Q. Yu, Transition Metal-Catalyzed C–H Functionalization: Synthetically Enabling Reactions for Building Molecular Complexity, in Organic Chemistry – Breakthroughs and Perspectives, ed. K. Ding and L.-X. Dai, Wiley-VCH, 2012 Search PubMed. For selected reviews: (c) A. D. Ryabov, Chem. Rev., 1990, 90, 403–424 CrossRef CAS; (d) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS; (e) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed; (f) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS PubMed; (g) R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437–2450 RSC; (h) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (i) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1770 CrossRef CAS PubMed; (j) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826–834 CrossRef CAS PubMed; (k) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077–1101 CrossRef CAS; (l) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed; (m) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (n) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (o) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (p) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CAS; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS; (c) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- (a) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (b) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (c) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed; (d) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (e) D. Y.-K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452–9474 CrossRef CAS PubMed; (f) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (b) H. M. Peng, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2010, 49, 5826–5828 CrossRef CAS PubMed; (c) G. B. Shul'pin, Org. Biomol. Chem., 2010, 8, 4217–4228 RSC; (d) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed; (e) L. Yang and H. Huang, Catal. Sci. Technol., 2012, 2, 1099–1112 RSC; (f) J. Wencel-Delord and F. Colobert, Chem.–Eur. J., 2013, 19, 14010–14017 CrossRef CAS PubMed.
- For recent reviews: (a) C.-H. Jun, E.-A. Jo and J.-W. Park, Eur. J. Org. Chem., 2007, 1869–1881 CrossRef CAS; (b) M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed; (c) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS PubMed; (d) C. González-Rodríguez and M. C. Willis, Pure Appl. Chem., 2011, 83, 577–585 CrossRef; (e) J. C. Leung and M. J. Krische, Chem. Sci., 2012, 3, 2202–2209 RSC.
- For selected reviews on NHC catalysis: (a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC; (d) Z.-Q. Rong, W. Zhang, G.-Q. Yang and S.-L. You, Curr. Org. Chem., 2011, 15, 3077–3090 CrossRef CAS; (e) P.-C. Chiang and J. W. Bode, TCI MAIL, 2011, 149, 2–17 Search PubMed; (f) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (g) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS PubMed; (h) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Schiedt, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS PubMed; (i) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617–1639 CrossRef CAS PubMed; (j) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC; (k) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed.
- For recent reviews: (a) K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS PubMed; (b) J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491–1500 CrossRef CAS PubMed; (c) E. Arceo and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 5290–5292 CrossRef CAS PubMed; (d) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748–9770 CrossRef CAS PubMed; (e) D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865–887 CrossRef CAS; (f) I. Kumar, P. Ramaraju and N. A. Mir, Org. Biomol. Chem., 2013, 11, 709–716 RSC.
- For selected reviews on C–H bond insertion by metal carbenoids: (a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–936 CrossRef CAS PubMed; (b) H. M. L. Davies and E. G. Antoulinakis, J. Organomet. Chem., 2001, 617–618, 47–55 CrossRef CAS; (c) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed; (d) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (e) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (f) C. N. Slattery, A. Ford and A. R. Maguire, Tetrahedron, 2010, 66, 6681–6705 CrossRef CAS PubMed; (g) M. P. Doyle, M. Ratnikov and Y. Liu, Org. Biomol. Chem., 2011, 9, 4007–4016 RSC; (h) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918–4931 RSC.
- J. R. Denton and H. M. L. Davies, Org. Lett., 2009, 11, 787–790 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- P. Pelphrey, J. Hansen and H. M. L. Davies, Chem. Sci., 2010, 1, 254–257 RSC.
- T. Goto, T. Onozuka, Y. Kosaka, M. Anada, K. Takeda and S. Hashimoto, Heterocycles, 2012, 86, 1647–1659 CrossRef CAS PubMed.
- (a) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (b) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449–4465 RSC; (c) M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed.
- Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 440–441 CrossRef CAS PubMed.
- A. DeAngelis, V. W. Shurtleff, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 1650–1653 CrossRef CAS PubMed.
- (a) A. DeAngelis, O. Dmitrenko, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 7230–7231 CrossRef CAS PubMed; (b) V. N. G. Lindsay, W. Lin and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 16383–16385 CrossRef CAS PubMed.
- T. Goto, Y. Natori, K. Takeda, H. Nambu and S. Hashimoto, Tetrahedron: Asymmetry, 2011, 22, 907–915 CrossRef CAS PubMed.
- T. Sassa, N. Yoshida and E. Haruki, Agric. Biol. Chem., 1989, 53, 3105–3107 CrossRef CAS.
- M. P. Doyle, M. S. Shanklin, H. Q. Pho and S. N. Mahapatro, J. Org. Chem., 1988, 53, 1017–1022 CrossRef CAS.
- (a) H. Qiu, M. Li, L.-Q. Jiang, F.-P. Lv, L. Zan, C.-W. Zhai, M. P. Doyle and W.-H. Hu, Nat. Chem., 2012, 4, 733–738 CrossRef CAS PubMed. For a related Pd-catalyzed asymmetric C–H bond functionalization reaction of pyrrole derivatives: (b) D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 13356–13360 CrossRef CAS PubMed.
- For a review on the trapping of protic onium ylides with electrophiles: X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427–2440 CrossRef CAS PubMed.
- (a) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (b) M. Rueping, R. M. Koenigs and I. Atodiresei, Chem.–Eur. J., 2010, 16, 9350–9365 CrossRef CAS PubMed; (c) C. Zhong and X. Shi, Eur. J. Org. Chem., 2010, 16, 2999–3025 CrossRef; (d) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC.
- H. M. L. Davies, S. J. Hedley and B. R. Bohall, J. Org. Chem., 2005, 70, 10737–11742 CrossRef CAS PubMed.
- X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203–12205 CrossRef CAS PubMed.
- H. Wang, G. Li, K. M. Engle, J.-Q. Yu and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6774–6777 CrossRef CAS PubMed.
- D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767–5769 CrossRef CAS PubMed.
- (a) J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS PubMed. For a recent review on CuAAC reaction: (b) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- (a) T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed; (b) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS PubMed; (c) N. Grimster, L. Zhang and V. V. Fokin, J. Am. Chem. Soc., 2010, 132, 2510–2511 CrossRef CAS PubMed; (d) J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6802–6805 CrossRef CAS PubMed.
- S. Chuprakov, J. A. Malik, M. Zibinsky and V. V. Fokin, J. Am. Chem. Soc., 2011, 133, 10352–10355 CrossRef CAS PubMed.
- H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923–935 CrossRef CAS PubMed.
- (a) H. M. L. Davies and A. M. Walji, Angew. Chem., Int. Ed., 2005, 44, 1733–1735 CrossRef CAS PubMed; (b) H. M. L. Davies, X. Dai and M. S. Long, J. Am. Chem. Soc., 2006, 128, 2485–2490 CrossRef CAS PubMed; (c) H. M. L. Davies and X. Dai, Tetrahedron, 2006, 62, 10477–10484 CrossRef CAS PubMed; (d) X. Dai, Z. Wan, R. G. Kerr and H. M. L. Davies, J. Org. Chem., 2007, 72, 1895–1900 CrossRef CAS PubMed.
- J. H. Hansen, T. M. Gregg, S. R. Ovalles, Y. Lian, J. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 5076–5085 CrossRef CAS PubMed.
- H. M. L. Davies and R. E. J. Beckwith, J. Org. Chem., 2004, 69, 9241–9247 CrossRef CAS PubMed.
- Y. Lian, K. I. Hardcastle and H. M. L. Davies, Angew. Chem., Int. Ed., 2011, 50, 9370–9373 CrossRef CAS PubMed.
- Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 11940–11943 CrossRef CAS PubMed.
- For a review on the vinylogous Mukaiyama aldol reactions: G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076–3154 CrossRef CAS PubMed.
- Y. Lian and H. M. L. Davies, Org. Lett., 2012, 14, 1934–1937 CrossRef CAS PubMed.
- H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2009, 131, 14218–14219 CrossRef CAS PubMed.
- (a) J.-C. Wang, Z.-J. Xu, Z. Guo, Q.-H. Deng, C.-Y. Zhou, X.-L. Wan and C.-M. Che, Chem. Commun., 2012, 48, 4299–4301 RSC. For a Rh–porphyrin complex-catalyzed asymmetric intermolecular insertion reaction of carbenoid into primary C–H bonds: (b) H.-Y. Thu, G. S.-M. Tong, J.-S. Huang, S. L.-F. Chan, Q.-H. Deng and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9747–9751 CrossRef CAS PubMed.
- C. P. Owens, A. Varela-Álvarez, V. Boyarskikh, D. G. Musaev, H. M. L. Davies and S. B. Blakey, Chem. Sci., 2013, 4, 2590–2596 RSC.
- J.-C. Wang, Y. Zhang, Z.-J. Xu, V. K.-Y. Lo and C.-M. Che, ACS Catal., 2013, 3, 1144–1148 CrossRef CAS.
- (a) J. M. Fraile, J. I. García, J. A. Mayoral and M. Roldán, Org. Lett., 2007, 9, 731–733 CrossRef CAS PubMed; (b) J. M. Fraile, P. López-Ram-de-Viu, J. A. Mayoral, M. Roldán and J. Santafé-Valero, Org. Biomol. Chem., 2011, 9, 6075–6081 RSC; (c) J. M. Fraile, J. A. Mayoral, N. Ravasio, M. Roldán, L. Sordelli and F. Zaccheria, J. Catal., 2011, 218, 273–278 CrossRef PubMed.
- G. Jiménez-Osés, E. Vispe, M. Roldán, S. Rodríguez-Rodríguez, P. López-Ram-de-Viu, L. Salvatella, J. A. Mayoral and J. M. Fraile, J. Org. Chem., 2013, 78, 5851–5857 CrossRef PubMed.
- C. J. Flynn, C. J. Elcoate, S. E. Lawrence and A. R. Maguire, J. Am. Chem. Soc., 2010, 132, 1184–1185 CrossRef CAS PubMed.
- C. N. Slattery and A. R. Maguire, Org. Biomol. Chem., 2011, 9, 667–669 CAS.
- (a) C. N. Slattery, L.-A. Clarke, S. O'Neil, A. Ring, A. Ford and A. R. Maguire, Synlett, 2012, 23, 765–767 CrossRef CAS PubMed; (b) C. N. Slattery, L.-A. Clarke, A. Ford and A. R. Maguire, Tetrahedron, 2013, 69, 1297–1301 CrossRef CAS PubMed.
- For reviews on Fe-catalysis: (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed; (b) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (c) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed; (d) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed; (e) K. Gopalaiah, Chem. Rev., 2013, 113, 3248–3296 CrossRef CAS PubMed.
- Y. Cai, S.-F. Zhu, G.-P. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2011, 353, 2939–2944 CrossRef CAS.
- S. I. Lee, G.-S. Hwang and D. H. Ryu, J. Am. Chem. Soc., 2013, 135, 7126–7129 CrossRef CAS PubMed.
- For the racemic version of this reaction: S. I. Lee, B. C. Kang, G.-S. Hwang and D. H. Ryu, Org. Lett., 2013, 15, 1428–1431 CrossRef CAS PubMed.
- For related Lewis acid-catalyzed asymmetric functionalization reactions of aldehyde C–H bonds with diazoesters: (a) W. Li, J. Wang, X. Hu, K. Shen, W. Wang, Y. Chu, L. Lin, X. Liu and X. Feng, J. Am. Chem. Soc., 2010, 132, 8532–8533 CrossRef CAS PubMed; (b) L. Gao, B. C. Kang, G.-S. Hwang and D. H. Ryu, Angew. Chem., Int. Ed., 2012, 51, 8322–8325 CrossRef CAS PubMed. For a related Lewis acid-catalyzed asymmetric insertion of diazoesters into aryl–CHO bonds: (c) L. Gao, B. C. Kang and D. H. Ryu, J. Am. Chem. Soc., 2013, 135, 14556–14559 CrossRef CAS PubMed.
- W. S. Bowers, H. M. Fales, M. J. Thompson and E. C. Uebel, Science, 1966, 154, 1020–1021 CAS.
- For reviews: (a) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061–5074 RSC; (b) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC; (c) J. Du Bois, Org. Process Res. Dev., 2011, 15, 758–762 CrossRef CAS PubMed; (d) B. J. Stokes and T. G. Driver, Eur. J. Org. Chem., 2011, 4071–4088 CrossRef CAS; (e) J. W. W. Chang, T. M. U. Ton and P. W. H. Chan, Chem. Rec., 2011, 11, 331–357 CrossRef CAS PubMed; (f) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed; (g) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC; (h) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS PubMed; (i) R. T. Gephart, III and T. H. Warren, Organometallics, 2012, 31, 7728–7752 CrossRef.
- For selected recent examples: (a) C. Liang, F. Robert-Peillard, C. Fruit, P. Müller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641–4644 CrossRef CAS PubMed; (b) C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343–350 CrossRef CAS PubMed; (c) D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 9220–9221 CrossRef CAS PubMed; (d) E. Milczek, N. Boudet and S. Blakey, Angew. Chem., Int. Ed., 2008, 47, 6825–6828 CrossRef CAS PubMed.
- (a) F. Collet, C. Lescot, C. Liang and P. Dauban, Dalton Trans., 2010, 39, 10401–10413 RSC; (b) C. Lescot, B. Darses, F. Collet, P. Retailleau and P. Dauban, J. Org. Chem., 2012, 77, 7232–7240 CrossRef CAS PubMed.
- (a) K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562–568 CrossRef CAS PubMed; (b) K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042–3051 CrossRef CAS PubMed.
- H. Lebel, C. Spitz, O. Leogane, C. Trudel and M. Parmentier, Org. Lett., 2011, 13, 5460–5463 CrossRef CAS PubMed.
- (a) M. Anada, M. Tanaka, T. Washio, M. Yamawaki, T. Abe and S. Hashimoto, Org. Lett., 2007, 9, 4559–4562 CrossRef CAS PubMed; (b) M. Tanaka, S. Nakamura, M. Anada and S. Hashimoto, Heterocycles, 2008, 76, 1633–1645 CrossRef CAS PubMed.
- M. Anada, M. Tanaka, N. Shimada, H. Nambu, M. Yamawaki and S. Hashimoto, Tetrahedron, 2009, 65, 3069–3077 CrossRef CAS PubMed.
- (a) L. E. Overman and J. Shim, J. Org. Chem., 1991, 56, 5005–5007 CrossRef CAS; (b) L. E. Overman and J. Shim, J. Org. Chem., 1993, 58, 4662–4672 CrossRef CAS.
- W. C. Wildman and C. L. Brown, J. Am. Chem. Soc., 1968, 90, 6439–6446 CrossRef CAS.
- W. J. G. Hellstrom, Neuropsychiatr. Dis. Treat., 2009, 5, 37–46 CAS.
- S. Kang and H.-K. Lee, J. Org. Chem., 2010, 75, 237–240 CrossRef CAS PubMed.
- D. N. Barman and K. M. Nicholas, Tetrahedron Lett., 2010, 51, 1815–1818 CrossRef CAS PubMed.
- R. Bhuyan and K. M. Nicholas, Org. Lett., 2007, 9, 3957–3959 CrossRef CAS PubMed.
- (a) D. N. Barman, P. Liu, K. N. Houk and K. M. Nicholas, Organometallics, 2010, 29, 3404–3412 CrossRef CAS. For a recent mechanistic study on a related reaction: (b) M. J. B. Aguila, Y. M. Badiei and T. H. Warren, J. Am. Chem. Soc., 2013, 135, 9399–9406 CrossRef CAS PubMed.
- D. N. Barman and K. M. Nicholas, Eur. J. Org. Chem., 2011, 908–911 CrossRef CAS.
- Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510–4511 CrossRef CAS PubMed.
- M. Ichinose, H. Suematsu, Y. Yasutomi, Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2011, 50, 9884–9887 CrossRef CAS PubMed.
- Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739–1742 CrossRef CAS PubMed.
- (a) M. E. Harvey, D. G. Musaev and J. Du Bois, J. Am. Chem. Soc., 2011, 133, 17207–17216 CrossRef CAS PubMed; (b) S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036–2039 CrossRef CAS PubMed.
- For reviews: (a) M. Newcomb, P. F. Hollenberg and M. J. Coon, Arch. Biochem. Biophys., 2003, 409, 72–79 CrossRef CAS; (b) J. T. Groves, J. Inorg. Biochem., 2006, 100, 434–447 CrossRef CAS PubMed; (c) M. K. Julsing, S. Cornelissen, B. Bühler and A. Schmid, Curr. Opin. Chem. Biol., 2008, 12, 177–186 CrossRef CAS PubMed; (d) M. T. Green, Curr. Opin. Chem. Biol., 2009, 13, 84–88 CrossRef CAS PubMed; (e) J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- J. A. McIntosh, P. S. Coelho, C. C. Farwell, Z. J. Wang, J. C. Lewis, T. R. Brown and F. H. Arnold, Angew. Chem., Int. Ed., 2013, 52, 9309–9312 CrossRef CAS PubMed.
- For reviews: (a) A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 RSC; (b) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC; (c) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC.
- T. Nagataki, Y. Tachi and S. Itoh, J. Mol. Catal. A: Chem., 2005, 225, 103–109 CrossRef CAS PubMed.
- (a) J. F. Larrow and E. N. Jacobsen, J. Am. Chem. Soc., 1994, 116, 12129–12130 CrossRef CAS; (b) K. Hamachi, R. Irie and T. Katsuki, Tetrahedron Lett., 1996, 37, 4979–4982 CrossRef CAS; (c) A. Miyafuji and T. Katsuki, Synlett, 1997, 836–838 CrossRef CAS PubMed; (d) T. Hamada, R. Irie, J. Mihara, K. Hamachi and T. Katsuki, Tetrahedron, 1998, 54, 10017–10028 CrossRef CAS; (e) A. Miyafuji and T. Katsuki, Tetrahedron, 1998, 54, 10339–10348 CrossRef CAS; (f) T. Punniyamurthy, A. Miyafuji and T. Katsuki, Tetrahedron Lett., 1998, 39, 8295–8298 CrossRef CAS; (g) T. Punniyamurthy and T. Katsuki, Tetrahedron, 1999, 55, 9439–9454 CrossRef CAS; (h) S. I. Murahashi, S. Noji and N. Komiya, Adv. Synth. Catal., 2004, 346, 195–198 CrossRef CAS; (i) H. Suematsu, Y. Tamura, H. Shitama and T. Katsuki, Heterocycles, 2007, 71, 2587–2593 CrossRef CAS.
- (a) R. Zhang, W.-Y. Yu, T.-S. Lai and C.-M. Che, Chem. Commun., 1999, 1791–1792 RSC; (b) R. Zhang, W.-Y. Yu and C.-M. Che, Tetrahedron: Asymmetry, 2005, 16, 3520–3526 CrossRef CAS PubMed.
- H. Srour, P. Le Maux and G. Simonneaux, Inorg. Chem., 2012, 51, 5850–5856 CrossRef CAS PubMed.
- J.-P. Renaud, P. Battioni, J. F. Bartoli and D. Mansuy, J. Chem. Soc., Chem. Commun., 1985, 888–889 RSC.
- For a perspective: M. C. White, Science, 2012, 335, 807–809 CrossRef CAS PubMed.
- (a) M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed; (b) N. A. Vermeulen, M. S. Chen and M. C. White, Tetrahedron, 2009, 65, 3078–3084 CrossRef CAS PubMed.
- (a) M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed; (b) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef CAS PubMed.
- M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721–9726 CrossRef CAS PubMed.
- M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216–222 CrossRef CAS PubMed.
- For reviews: (a) C. J. Scheuermann, Chem.–Asian J., 2010, 5, 436–451 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (c) M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353–369 CrossRef CAS PubMed.
- S.-I. Murahashi and D. Zhang, Chem. Soc. Rev., 2008, 37, 1490–1501 RSC.
- (a) C.-J. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935–945 CrossRef CAS; (b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- (a) Z. Li and C.-J. Li, Org. Lett., 2004, 6, 4997–4999 CrossRef CAS PubMed; (b) N. Sasamoto, C. Dubs, Y. Hamashima and M. Sodeoka, J. Am. Chem. Soc., 2006, 128, 14010–14011 CrossRef CAS PubMed; (c) Z. Li, P. D. MacLeod and C.-J. Li, Tetrahedron: Asymmetry, 2006, 17, 590–597 CrossRef CAS PubMed; (d) C. Dubs, Y. Hamashima, N. Sasamoto, T. M. Seidel, S. Suzuki, D. Hashizume and M. Sodeoka, J. Org. Chem., 2008, 73, 5859–5871 CrossRef CAS PubMed.
- C. Guo, J. Song, S.-W. Luo and L.-Z. Gong, Angew. Chem., Int. Ed., 2010, 49, 5558–5562 CrossRef CAS PubMed.
- G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429–10432 CrossRef CAS PubMed.
- Seminal works: (a) T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS PubMed; (b) H.-Y. Jang, J.-B. Hong and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 7004–7005 CrossRef CAS PubMed; (c) H. Kim and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 398–399 CrossRef CAS PubMed; (d) T. H. Graham, C. M. Jones, N. T. Jui and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 16494–16495 CrossRef CAS PubMed.
- K. C. Nicolaou, R. Reingruber, D. Sarlah and S. Bräse, J. Am. Chem. Soc., 2009, 131, 2086–2087 CrossRef CAS PubMed . Addition and Correction: J. Am. Chem. Soc., 2009, 131, 6640.
- F. Bohlmann, C. Zdero, H. Robinson and R. M. King, Phytochemistry, 1979, 18, 1675–1680 CrossRef CAS.
- J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11640–11641 CrossRef CAS PubMed.
- S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32–33 CrossRef CAS PubMed.
- M. G. Banwell, D. A. S. Beck and J. A. Smith, Org. Biomol. Chem., 2004, 2, 157–159 CAS.
- J. M. Um, O. Gutierrez, F. Schoenebeck, K. N. Houk and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 6001–6005 CrossRef CAS PubMed.
- S. Rendler and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 5027–5029 CrossRef CAS PubMed.
- F. Benfatti, M. G. Capdevila, L. Zoli, E. Benedetto and P. G. Cozzi, Chem. Commun., 2009, 5919–5921 RSC.
- X.-H. Ho, S.-i. Mho, H. Kang and H.-Y. Jang, Eur. J. Org. Chem., 2010, 4436–4441 CAS.
- B. Zhang, S.-K. Xiang, L.-H. Zhang, Y. Cui and N. Jiao, Org. Lett., 2011, 13, 5212–5215 CrossRef CAS PubMed.
- For related non-enantioselective CDC coupling between benzylic C–H bonds and ketones using O2 as the sole oxidant: (a) Á. Pintér, A. Sud, D. Sureshkumar and M. Klussmann, Angew. Chem., Int. Ed., 2010, 49, 5004–5007 CrossRef PubMed; (b) Á. Pintér and M. Klussmann, Adv. Synth. Catal., 2012, 354, 701–711 CrossRef; (c) B. Schweitzer-Chaput, A. Sud, Á. Pintér, S. Dehn, P. Schulze and M. Klussmann, Angew. Chem., Int. Ed., 2013, 52, 13228–13232 CrossRef CAS PubMed.
- F. Huang, L. Xu and J. Xiao, Chin. J. Chem., 2012, 30, 2721–2725 CAS.
- J. Zhang, B. Tiwari, C. Xing, X. Chen and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 3649–3652 CrossRef CAS PubMed.
- A. Sud, D. Sureshkumar and M. Klussmann, Chem. Commun., 2009, 3169–3171 RSC.
- M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360–2362 RSC.
- For a review on C–H bond functionalization reactions by photoredox catalysis: L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682–2685 RSC.
- J. Xie and Z.-Z. Huang, Angew. Chem., Int. Ed., 2010, 49, 10181–10185 CrossRef CAS PubMed.
- (a) G. Zhang, Y. Ma, S. Wang, W. Kong and R. Wang, Chem. Sci., 2013, 4, 2645–2651 RSC; (b) G. Zhang, S. Wang, Y. Ma, W. Kong and R. Wang, Adv. Synth. Catal., 2013, 355, 874–879 CrossRef CAS.
- G. Zhang, Y. Ma, S. Wang, Y. Zhang and R. Wang, J. Am. Chem. Soc., 2012, 134, 12334–12337 CrossRef CAS PubMed.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094–8097 CrossRef CAS PubMed.
- G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112–116 RSC.
- For recent reviews on chiral anion binding catalysis: (a) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed; (c) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
- S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198–7199 CrossRef CAS PubMed.
- A. Winkler, A. Łyskowski, S. Riedl, M. Puhl, T. M. Kutchan, P. Macheroux and K. Gruber, Nat. Chem. Biol., 2008, 4, 739–741 CrossRef CAS PubMed.
- J. H. Schrittwieser, V. Resch, J. H. Sattler, W.-D. Lienhart, K. Durchschein, A. Winkler, K. Gruber, P. Macheroux and W. Kroutil, Angew. Chem., Int. Ed., 2011, 50, 1068–1071 CrossRef CAS PubMed.
- A. J. Neel, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14044–14047 CrossRef CAS PubMed.
- For reviews: (a) O. Meth-Cohn and H. Suschitzky, Adv. Heterocycl. Chem., 1972, 14, 211–278 CrossRef CAS; (b) O. Meth-Cohn, Adv. Heterocycl. Chem., 1996, 65, 1–37 CrossRef CAS.
- For reviews: (a) J. M. Quintela, Recent Res. Dev. Org. Chem., 2003, 7, 259–278 CAS; (b) P. Mátyus, O. Éliás, P. Tapolcsányi, A. Polonka-Bálint and B. Halász-Dajka, Synthesis, 2006, 2625–2639 CrossRef PubMed.
- (a) S. J. Pastine, K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2005, 127, 12180–12181 CrossRef CAS PubMed; (b) P. A. Vadola and D. Sames, J. Am. Chem. Soc., 2009, 131, 16525–16528 CrossRef CAS PubMed; (c) Y.-Y. Han, W.-Y. Han, X. Hou, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2012, 14, 4054–4057 CrossRef CAS PubMed; (d) P. A. Vadola, I. Carrera and D. Sames, J. Org. Chem., 2012, 77, 6689–6702 CrossRef CAS PubMed.
- S. Murarka, C. Zhang, M. D. Konieczynska and D. Seidel, Org. Lett., 2009, 11, 129–132 CrossRef CAS PubMed.
- S. Murarka, I. Deb, C. Zhang and D. Seidel, J. Am. Chem. Soc., 2009, 131, 13226–13227 CrossRef CAS PubMed.
- X. Liu, L. Lin and X. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed.
- W. Cao, X. Liu, W. Wang, L. Lin and X. Feng, Org. Lett., 2011, 13, 600–603 CrossRef CAS PubMed.
- G. Zhou, F. Liu and J. Zhang, Chem.–Eur. J., 2011, 17, 3101–3104 CrossRef CAS PubMed.
- For selected recent reviews on Au catalysis: (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed; (b) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed; (c) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (d) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS PubMed; (e) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (f) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448–2462 RSC.
- G. Zhou and J. Zhang, Chem. Commun., 2010, 46, 6593–6595 RSC.
- (a) Y. K. Kang, S. M. Kim and D. Y. Kim, J. Am. Chem. Soc., 2010, 132, 11847–11849 CrossRef CAS PubMed. For a related microwave-assisted organocatalytic [1,5]-hydride transfer/cyclization reaction: (b) Y. K. Kwon, Y. K. Kang and D. Y. Kim, Bull. Korean Chem. Soc., 2011, 32, 1773–1776 CrossRef CAS.
- K. Mori, K. Ehara, K. Kurihara and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 6166–6169 CrossRef CAS PubMed.
- For selected reviews on chiral phosphoric acids: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (b) M. Terada, Chem. Commun., 2008, 4097–4112 RSC; (c) M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS PubMed; (d) M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539–4549 RSC; (e) M. Rueping, B. J. Nachtsheim, W. Ieawsuwan and I. Atodiresei, Angew. Chem., Int. Ed., 2011, 50, 6706–6720 CrossRef CAS PubMed.
- (a) K. Mori, Y. Ohshima, K. Ehara and T. Akiyama, Chem. Lett., 2009, 38, 524–525 CrossRef CAS; (b) K. Mori, T. Kawasaki, S. Sueoka and T. Akiyama, Org. Lett., 2010, 12, 1732–1735 CrossRef CAS PubMed.
- (a) L. Chen, L. Zhang, J. Lv, J.-P. Cheng and S. Luo, Chem.–Eur. J., 2012, 18, 8891–8895 CrossRef CAS PubMed; (b) L. Zhang, L. Chen, J. Lv, J.-P. Cheng and S. Luo, Chem.–Asian J., 2012, 7, 2569–2576 CrossRef CAS PubMed.
- For a review: J. Lv and S. Luo, Chem. Commun., 2013, 49, 847–858 RSC.
- K. Kang and D. Y. Kim, Adv. Synth. Catal., 2013, 355, 3131–3136 CrossRef.
- Y.-P. He, Y.-L. Du, S.-W. Luo and L.-Z. Gong, Tetrahedron Lett., 2011, 52, 7064–7066 CrossRef CAS PubMed.
- (a) S. J. Pastine and D. Sames, Org. Lett., 2005, 7, 5429–5431 CrossRef CAS PubMed; (b) K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2009, 131, 402–403 CrossRef CAS PubMed; (c) K. M. McQuaid, J. Z. Long and D. Sames, Org. Lett., 2009, 11, 2972–2975 CrossRef CAS PubMed.
- (a) M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2006, 45, 1683–1684 CrossRef CAS PubMed; (b) I. D. Jurberg, Y. Odabachian and F. Gagosz, J. Am. Chem. Soc., 2010, 132, 3543–3552 CrossRef CAS PubMed; (c) M. Alajarin, B. Bonillo, M.-M. Ortin, P. Sanchez-Andrada and A. Vidal, Eur. J. Org. Chem., 2011, 1896–1913 CrossRef CAS; (d) B. Bolte and F. Gagosz, J. Am. Chem. Soc., 2011, 133, 7696–7699 CrossRef CAS PubMed.
- Z.-W. Jiao, S.-Y. Zhang, C. He, Y.-Q. Tu, S.-H. Wang, F.-M. Zhang, Y.-Q. Wang and H. Li, Angew. Chem., Int. Ed., 2012, 51, 8811–8815 CrossRef CAS PubMed.
- (a) R. F. Heck, Nobel Lecture: Palladium Reactions for Organic Syntheses, Nobelprize.org; (b) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (c) E.-i. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- For reviews: (a) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949–957 CAS; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (c) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677–685 RSC; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (e) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588–5598 RSC; (f) D.-G. Yu, B.-J. Li and Z.-J. Shi, Tetrahedron, 2012, 68, 5130–5136 CrossRef CAS PubMed.
- For a review on the ligand development: K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed.
- B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS PubMed.
- D. G. Musaev, A. Kaledin, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 1690–1698 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820–5831 RSC.
- B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460–461 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- (a) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS PubMed; (b) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14154 CrossRef CAS PubMed.
- K. M. Engle, P. S. Thuy-Boun, M. Dang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 18183–18193 CrossRef CAS PubMed.
- R. D. Baxter, D. Sale, K. M. Engle, J.-Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 4600–4606 CrossRef CAS PubMed.
- (a) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916–5921 CrossRef CAS PubMed; (b) K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169–6173 CrossRef CAS PubMed; (c) Y. Lu, D. Leow, X. Wang, K. M. Engle and J.-Q. Yu, Chem. Sci., 2011, 2, 967–971 RSC; (d) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245–1247 CrossRef CAS PubMed.
- H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef CAS PubMed.
- For a recent review on the syntheses of planar-chiral ferrocenes: D. Schaarschmidt and H. Lang, Organometallics, 2013, 32, 5668–5704 CrossRef CAS.
- Enantioenriched palladacyclic compounds derived from ferrocenes and [2.2]paracyclophanes have been synthesized using mono-N-protected chiral amino acid ligands: (a) V. I. Sokolov, L. L. Troitskaya and O. A. Reutov, J. Organomet. Chem., 1979, 182, 537–546 CrossRef CAS; (b) M. E. Gunay and C. J. Richards, Organometallics, 2009, 28, 5833–5836 CrossRef CAS; (c) N. Dendele, F. Bisaro, A.-C. Gaumont, S. Perrio and C. J. Richards, Chem. Commun., 2012, 48, 1991–1993 RSC.
- (a) D.-W. Gao, Y.-C. Shi, Q. Gu, Z.-L. Zhao and S.-L. You, J. Am. Chem. Soc., 2013, 135, 86–89 CrossRef CAS PubMed. For a synthesis of ferrocene derivatives with planar-chirality by Pd-catalyzed double C–H bond functionalization: (b) J.-B. Xia and S.-L. You, Organometallics, 2007, 26, 4869–4871 CrossRef CAS.
- C. Pi, Y. Li, X. Cui, H. Zhang, Y. Han and Y. Wu, Chem. Sci., 2013, 4, 2675–2679 RSC.
- Y.-C. Shi, R.-F. Yang, D.-W. Gao and S.-L. You, Beilstein J. Org. Chem., 2013, 9, 1891–1896 CrossRef PubMed.
- For a racemic reaction: H. Zhang, X. Cui, X. Yao, H. Wang, J. Zhang and Y. Wu, Org. Lett., 2012, 14, 3012–3015 CrossRef CAS PubMed.
- (a) E. M. Ferreira and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 9578–9579 CrossRef CAS PubMed; (b) E. M. Ferreira, H. Zhang and B. M. Stoltz, Tetrahedron, 2008, 64, 5987–6001 CrossRef CAS PubMed.
- (a) J. A. Schiffner, A. B. Machotta and M. Oestreich, Synlett, 2008, 2271–2274 CAS; (b) J. A. Schiffner, T. H. Wöste and M. Oestreich, Eur. J. Org. Chem., 2010, 174–182 CrossRef CAS.
- For the first example of the asymmetric Fujiwara–Moritani reaction: K. Mikami, M. Hatano and M. Terada, Chem. Lett., 1999, 28, 55–56 CrossRef.
- For reviews: (a) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615–624 CrossRef CAS PubMed; (b) P. Lloyd-Williams and E. Giralt, Chem. Soc. Rev., 2001, 30, 145–157 RSC.
- S. Kirchberg, S. Tani, K. Ueda, J. Yamaguchi, A. Studer and K. Itami, Angew. Chem., Int. Ed., 2011, 50, 2387–2391 CrossRef CAS PubMed.
- K. Yamaguchi, J. Yamaguchi, A. Studer and K. Itami, Chem. Sci., 2012, 3, 2165–2169 RSC.
- K. Yamaguchi, H. Kondo, J. Yamaguchi and K. Itami, Chem. Sci., 2013, 4, 3753–3757 RSC.
- For reviews: (a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (c) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654–2672 CrossRef CAS PubMed; (d) M. Wasa, K. M. Engle and J.-Q. Yu, Isr. J. Chem., 2010, 50, 605–616 CrossRef CAS; (e) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC; (f) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191–206 RSC.
- M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598–19601 CrossRef CAS PubMed.
- (a) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3680–3681 CrossRef CAS PubMed; (b) E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378–17380 CrossRef CAS PubMed.
- For reviews: (a) T. Jensen and P. Fristrup, Chem.–Eur. J., 2009, 15, 9632–9636 CrossRef CAS PubMed; (b) C. J. Engelin and P. Fristrup, Molecules, 2011, 16, 951–969 CrossRef CAS PubMed.
- For reviews: (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (c) B. M. Trost, J. Org. Chem., 2004, 69, 5813–5837 CrossRef CAS PubMed; (d) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef CAS PubMed; (e) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC.
- H. Du, B. Zhao and Y. Shi, J. Am. Chem. Soc., 2008, 130, 8590–8591 CrossRef CAS PubMed.
- (a) D. J. Covell and M. C. White, Angew. Chem., Int. Ed., 2008, 47, 6448–6451 CrossRef CAS PubMed. For a closely related non-asymmetric allylic alkylation reaction: (b) A. J. Young and M. C. White, Angew. Chem., Int. Ed., 2011, 50, 6824–6827 CrossRef CAS PubMed.
- D. J. Covell and M. C. White, Tetrahedron, 2013, 69, 7771–7778 CrossRef CAS PubMed.
- K. Takenaka, M. Akita, Y. Tanigaki, S. Takizawa and H. Sasai, Org. Lett., 2011, 13, 3506–3509 CrossRef CAS PubMed.
- For a review: G. B. Bajracharya, M. A. Arai, P. S. Koranne, T. Suzuki, S. Takizawa and H. Sasai, Bull. Chem. Soc. Jpn., 2009, 82, 285–302 CrossRef CAS.
- Z. Chai and T. J. Rainey, J. Am. Chem. Soc., 2012, 134, 3615–3618 CrossRef CAS PubMed.
- For a review on semipinacol rearrangement: B. Wang and Y. Q. Tu, Acc. Chem. Res., 2011, 44, 1207–1222 CrossRef CAS PubMed.
- B. M. Trost, D. A. Thaisrivongs and E. J. Donckele, Angew. Chem., Int. Ed., 2013, 52, 1523–1526 CrossRef CAS PubMed.
- For a highlight: G. Chen, K. Chen and Z.-J. Shi, ChemCatChem, 2013, 5, 1289–1290 CrossRef CAS.
- For reviews: (a) L.-C. Campeau and K. Fagnou, Chem. Commun., 2006, 1253–1264 RSC; (b) L.-C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35–41 CAS; (c) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269–10310 CrossRef CAS PubMed; (d) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146 CrossRef CAS PubMed.
- (a) M. R. Albicher and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 9139–9142 CrossRef PubMed. For an accounts: (b) N. Cramer, Chimia, 2012, 66, 869–872 CrossRef CAS PubMed.
- T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 7865–7868 CrossRef CAS PubMed.
- R. Shintani, H. Otomo, K. Ota and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 7305–7308 CrossRef CAS PubMed.
- For a review on silicon-stereogenic silanes: L.-W. Xu, L. Li, G.-Q. Lai and J.-X. Jiang, Chem. Soc. Rev., 2011, 40, 1777–1790 RSC.
- A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261–7265 CrossRef CAS PubMed.
- (a) M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündig, Angew. Chem., Int. Ed., 2011, 50, 7438–7441 CrossRef CAS PubMed. For accounts of Kündig's work: (b) M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündig, Chimia, 2012, 66, 241–243 CrossRef CAS PubMed; (c) E. P. Kündig, Y. Jia, D. Katayev and M. Nakanishi, Pure Appl. Chem., 2012, 84, 1741–1748 CrossRef.
- D. Katayev, M. Nakanishi, T. Bürgi and E. P. Kündig, Chem. Sci., 2012, 3, 1422–1425 RSC.
- E. Larionov, M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündug, Chem. Sci., 2013, 4, 1995–2005 RSC.
- S. Anas, A. Cordi and H. B. Kagan, Chem. Commun., 2011, 47, 11483–11485 RSC.
- T. Saget, S. J. Lemouzy and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 2238–2242 CrossRef CAS PubMed.
- P. A. Donets, T. Saget and N. Cramer, Organometallics, 2012, 31, 8040–8046 CrossRef CAS.
- T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 12842–12845 CrossRef CAS PubMed.
- N. Martin, C. Pierre, M. Davi, R. Jazzar and O. Baudoin, Chem.–Eur. J., 2012, 18, 4480–4484 CrossRef CAS PubMed.
- For reviews on Pd(II)/Pd(IV) catalytic cycle: (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712–733 RSC; (c) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478–1491 CrossRef CAS PubMed; (d) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed; (e) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- Besides the Pd(II)/Pd(IV) catalytic cycle, another type of mechanism that involves binuclear Pd(III) species was also suggested: D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840–850 CrossRef CAS PubMed.
- X.-F. Cheng, Y. Li, Y.-M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236–1239 CrossRef CAS PubMed.
- (a) L. Chu, X.-C. Wang, C. E. Moore, A. L. Rheingold and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344–16347 CrossRef CAS PubMed. For an early report on asymmetric C–H bond iodination using a chiral auxiliary: (b) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112–2115 CrossRef CAS PubMed.
- X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326–10329 CrossRef CAS PubMed.
- For reviews: (a) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (c) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed. For early reports: (d) N. Fujii, F. Kakiuchi, A. Yamada, N. Chatani and S. Murai, Chem. Lett., 1997, 26, 425–426 CrossRef; (e) F. Kakiuchi, P. Le Gendre, A. Yamada, H. Ohtaki and S. Murai, Tetrahedron: Asymmetry, 2000, 11, 2647–2651 CrossRef CAS.
- (a) R. K. Thalji, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 7192–7193 CrossRef CAS PubMed; (b) A. Watzke, R. M. Wilson, S. J. O'Malley, R. G. Bergman and J. A. Ellman, Synlett, 2007, 2383–2389 CAS; (c) H. Harada, R. K. Thalji, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2008, 73, 6772–6779 CrossRef CAS PubMed.
- For an early report: N. Fujii, F. Kakiuchi, A. Yamada, N. Chatani and S. Murai, Chem. Lett., 1997, 26, 425–426 CrossRef.
- (a) K. A. Ahrendt, R. G. Bergman and J. A. Ellman, Org. Lett., 2003, 5, 1301–1303 CrossRef CAS PubMed; (b) S. J. O'Malley, K. L. Tan, A. Watzke, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 13496–13497 CrossRef CAS PubMed; (c) R. M. Wilson, R. K. Thalji, R. G. Bergman and J. A. Ellman, Org. Lett., 2006, 8, 1745–1747 CrossRef CAS PubMed; (d) A. S. Tsai, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6316–6317 CrossRef CAS PubMed.
- A. S. Tsai, R. M. Wilson, H. Harada, R. G. Bergman and J. A. Ellman, Chem. Commun., 2009, 3910–3912 RSC.
- For a review on type II Diels–Alder reactions: B. R. Bear, S. M. Sparks and K. J. Shea, Angew. Chem., Int. Ed., 2001, 40, 820–849 CrossRef CAS.
- Q. Li and Z.-X. Yu, J. Am. Chem. Soc., 2010, 132, 4542–4543 CrossRef CAS PubMed . Addition and Correction: J. Am. Chem. Soc., 2010, 132, 6862–6862.
- Q. Li and Z.-X. Yu, Angew. Chem., Int. Ed., 2011, 50, 2144–2147 CrossRef CAS PubMed.
- Q. Li and Z.-X. Yu, Organometallics, 2012, 31, 5185–5195 CrossRef CAS.
- Q. Li, Z. Dong and Z.-X. Yu, Org. Lett., 2011, 13, 1122–1125 CrossRef CAS PubMed.
- For an account: N. Cramer and T. Seiser, Synlett, 2011, 449–460 CrossRef CAS PubMed.
- (a) T. Seiser, O. A. Roth and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 6320–6323 CrossRef CAS PubMed; (b) M. Shigeno, T. Yamamoto and M. Murakami, Chem.–Eur. J., 2009, 15, 12929–12931 CrossRef CAS PubMed.
- For a review on [1,4]-metal migration: S. Ma and Z. Gu, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS PubMed.
- T. Seiser, G. Cathomen and N. Cramer, Synlett, 2010, 1699–1703 CAS.
- (a) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 8181–8184 CrossRef CAS PubMed; (b) D. N. Tran and N. Cramer, Chimia, 2010, 65, 271–273 CrossRef.
- (a) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 11098–11102 CrossRef CAS PubMed. For a racemic reaction: (b) Z.-M. Sun, S.-P. Chen and P. Zhao, Chem.–Eur. J., 2010, 16, 2619–2627 CrossRef CAS PubMed.
- D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 10630–10634 CrossRef CAS PubMed.
- For reviews on DYKAT processes: (a) K. Faber, Chem.–Eur. J., 2001, 7, 5004–5010 CrossRef CAS; (b) B. M. Trost and D. R. Fandrick, Aldrichimica Acta, 2007, 40, 59–72 CAS; (c) J. Steinreiber, K. Faber and H. Griengl, Chem.–Eur. J., 2008, 14, 8060–8072 CrossRef CAS PubMed.
- Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520–1522 CrossRef CAS PubMed.
- T. Ureshino, T. Yoshida, Y. Kuninobu and K. Takai, J. Am. Chem. Soc., 2010, 132, 14324–14326 CrossRef CAS PubMed.
- For reviews: (a) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (b) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (c) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31–41 CAS.
- The O-methyl hydroxamic acid was first used as a directing group in Pd-catalyzed C–H bond functionalization reactions by Yu and co-workers: (a) D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190–7191 CrossRef CAS PubMed; (b) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058–14059 CrossRef CAS PubMed.
- (a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed; (b) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed.
- S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed.
- For a highlight: F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977–1979 CrossRef CAS PubMed.
- The strategy of using oxidizing directing groups has been applied in Pd-catalyzed C–H bond functionalization reactions: (a) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888–13889 CrossRef CAS PubMed; (b) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676–3677 CrossRef CAS PubMed; (c) K.-H. Ng, A. S. C. Chan and W.-Y. Yu, J. Am. Chem. Soc., 2010, 132, 12862–12864 CrossRef CAS PubMed.
- For a computational study: L. Xu, Q. Zhu, G. Huang, B. Cheng and Y. Xia, J. Org. Chem., 2012, 77, 3017–3024 CrossRef CAS PubMed.
- A. Gutnov, B. Heller, C. Fischer, H.-J. Drexler, A. Spannenberg, B. Sundermann and C. Sundermann, Angew. Chem., Int. Ed., 2004, 43, 3795–3797 CrossRef CAS PubMed.
- For highlight papers: (a) H. Wang and F. Glorius, Science, 2012, 338, 479–480 CrossRef CAS PubMed; (b) M. Hapke and C. C. Tzschucke, Angew. Chem., Int. Ed., 2013, 52, 3317–3319 CrossRef CAS PubMed; (c) J.-B. Sortais and C. Darcel, ChemCatChem, 2013, 5, 1067–1068 CrossRef CAS.
- T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed.
- For a review: M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed.
- For a review: T. R. Ward, Acc. Chem. Res., 2011, 44, 47–57 CrossRef CAS PubMed.
- B. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed.
- B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef CAS PubMed.
- (a) R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597–9600 CrossRef CAS PubMed; (b) H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318–7322 CrossRef CAS PubMed.
- For a recent review: (a) S. Pan and T. Shibata, ACS Catal., 2013, 3, 704–712 CrossRef CAS. For early reports: (b) R. Aufdenblatten, S. Diezi and A. Togni, Monatsh. Chem., 2000, 131, 1345–1350 CrossRef CAS; (c) R. Dorta and A. Togni, Chem. Commun., 2003, 760–761 RSC.
- K. Tsuchikama, M. Kasagawa, Y.-K. Hashimoto, K. Endo and T. Shibata, J. Organomet. Chem., 2008, 693, 3939–3942 CrossRef CAS PubMed.
- C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2116–2119 CrossRef CAS PubMed.
- (a) S. Pan, K. Endo and T. Shibata, Org. Lett., 2011, 13, 4692–4695 CrossRef CAS PubMed; (b) S. Pan, Y. Matsuo, K. Endo and T. Shibata, Tetrahedron, 2012, 68, 9009–9015 CrossRef CAS PubMed.
- T. Nishimura, M. Nagamoto, Y. Ebe and T. Hayashi, Chem. Sci., 2013, 4, 4499–4504 RSC.
- For a racemic reaction: T. Nashimura, Y. Ebe and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 2092–2095 CrossRef PubMed.
- For reviews on chiral olefin ligands: (a) C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS PubMed; (b) R. Shintani and T. Hayashi, Aldrichimica Acta, 2009, 42, 31–38 CAS; (c) C.-G. Feng, M.-H. Xu and G.-Q. Lin, Synlett, 2011, 1345–1356 CAS; (d) X. Feng and H. Du, Asian J. Org. Chem., 2012, 1, 204–213 CrossRef CAS.
- (a) M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS; (b) M. S. Kharasch, G. Sosnovsky and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 5819–5824 CrossRef CAS.
- For reviews: (a) D. J. Rawlinson and G. Sosnovsky, Synthesis, 1972, 1–28 CrossRef CAS; (b) J. Eames and M. Watkinson, Angew. Chem., Int. Ed., 2001, 40, 3567–3571 CrossRef CAS; (c) M. B. Andrus and J. C. Lashley, Tetrahedron, 2002, 58, 845–866 CrossRef CAS; (d) T. Punniyamurthy and L. Rout, Coord. Chem. Rev., 2008, 252, 134–154 CrossRef CAS PubMed.
- For a recent computational study: J. A. Mayoral, S. Rodríguez-Rodríguez and L. Salvatella, Chem.–Eur. J., 2008, 14, 9274–9285 CrossRef CAS PubMed.
- V. Köhler, C. Mazet, A. Toussaint, K. Kulicke, D. Häussinger, M. Neuburger, S. Schaffner, S. Kaiser and A. Pfaltz, Chem.–Eur. J., 2008, 14, 8530–8539 CrossRef PubMed.
- (a) A. S. Gokhale, A. B. E. Minidis and A. Pfaltz, Tetrahedron Lett., 1995, 36, 1831–1834 CrossRef CAS; (b) M. B. Andrus, A. B. Argade, X. Chen and M. G. Pamment, Tetrahedron Lett., 1995, 36, 2945–2948 CrossRef CAS.
- V. D. M. Hoang, P. A. N. Reddy and T.-J. Kim, Organometallics, 2008, 27, 1026–1027 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, T. Belhocine, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Chem. Commun., 2008, 5535–5537 RSC.
- Q. Tan and M. Hayashi, Adv. Synth. Catal., 2008, 350, 2639–2644 CrossRef CAS.
- Q. Tan and M. Hayashi, Org. Lett., 2009, 11, 3314–3317 CrossRef CAS PubMed.
- (a) L. Aldea, I. Delso, M. Hager, M. Glos, J. I. García, J. A. Mayoral and O. Reiser, Tetrahedron, 2012, 68, 3417–3422 CrossRef CAS PubMed; (b) L. Aldea, J. I. García and J. A. Mayoral, Dalton Trans., 2012, 41, 8285 RSC.
- Z. Zhou and M. B. Andrus, Tetrahedron Lett., 2012, 53, 4518–4521 CrossRef CAS PubMed.
- S. Samadi, K. Jadidi and B. Notash, Tetrahedron: Asymmetry, 2013, 24, 269–277 CrossRef CAS PubMed.
- B. Zhang, S.-F. Zhu and Q.-L. Zhou, Tetrahedron Lett., 2013, 54, 2665–2668 CrossRef CAS PubMed.
- For an application of this type of ligands in the Kharasch–Sosnovsky reactions of cyclic olefins: (a) B. Liu, S.-F. Zhu, L.-X. Wang and Q.-L. Zhou, Tetrahedron: Asymmetry, 2006, 17, 634–641 CrossRef CAS PubMed. For a related Cu-catalyzed non-enantioselective benzylic oxidation reaction: (b) B. Zhang, S.-F. Zhu and Q.-L. Zhou, Tetrahedron, 2013, 69, 2033–2037 CrossRef CAS PubMed.
- P. A. Donets and N. Cramer, J. Am. Chem. Soc., 2013, 135, 11772–11775 CrossRef CAS PubMed.
- For reviews: (a) N. V. Dubrovina and A. Börner, Angew. Chem., Int. Ed., 2004, 43, 5883–5886 CrossRef CAS PubMed; (b) L. Ackermann, R. Born, J. H. Spatz, A. Althammer and C. J. Gschrei, Pure Appl. Chem., 2006, 78, 209–214 CrossRef CAS; (c) L. Ackermann, Synthesis, 2006, 1557–1571 CrossRef CAS.
- Chiral diaminophosphine oxide ligands have been used in asymmetric catalysis: (a) T. Nemoto, T. Matsumoto, T. Masuda, T. Hitomi, K. Hatano and Y. Hamada, J. Am. Chem. Soc., 2004, 126, 3690–3691 CrossRef CAS PubMed; (b) T. Nemoto, T. Masuda, T. Matsumoto and Y. Hamada, J. Org. Chem., 2005, 70, 7172–7178 CrossRef CAS PubMed; (c) T. Nemoto, T. Sakamoto, T. Matsumoto and Y. Hamada, Tetrahedron Lett., 2006, 47, 8737–8740 CrossRef CAS PubMed.
- J. M. Brunel, Chem. Rev., 2005, 105, 857–897 CrossRef CAS PubMed.
- (a) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed; (b) Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011 Search PubMed.
- G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed.
- For reviews on the synthesis of chiral biaryl compounds: (a) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed; (b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC.
- H. Egami and T. Katsuki, J. Am. Chem. Soc., 2009, 131, 6082–6083 CrossRef CAS PubMed.
- K. Matsumoto, B. Saito and T. Katsuki, Chem. Commun., 2007, 3619–3627 RSC.
- H. Egami, K. Matsumoto, T. Oguma, T. Kunisu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 13633–13635 CrossRef CAS PubMed.
- (a) P. Kočovský, Š. Vyskočil and M. Smrčina, Chem. Rev., 2003, 103, 3213–3246 CrossRef PubMed; (b) M. Shibasaki and S. Matsunaga, Chem. Soc. Rev., 2006, 35, 269–279 RSC.
- K. Matsumoto, H. Egami, T. Oguma and T. Katsuki, Chem. Commun., 2012, 48, 5823–5825 RSC.
- G. Grach, G. Pieters, A. Dinut, V. Terrasson, R. Medimagh, A. Bridoux, V. Razafimahaleo, A. Gaucher, S. Marque, J. Marrot, D. Prim, R. Gil, J. G. Planas, C. Viñas, I. Thomas, J.-P. Roblin and Y. Troin, Organometallics, 2011, 30, 4074–4086 CrossRef CAS.
- S. K. Alamsetti, E. Poonguzhali, D. Ganapathy and G. Sekar, Adv. Synth. Catal., 2013, 355, 2803–2808 CrossRef CAS.
- Y. Yusa, I. Kaito, K. Akiyama and K. Mikami, Chirality, 2010, 22, 224–228 CAS.
- (a) K. Ding, X. Li, B. Ji, H. Guo and M. Kitamura, Curr. Org. Synth., 2005, 2, 499–545 CrossRef CAS; (b) K. Ding, H. Guo, X. Li, Y. Yuan and Y. Wang, Top. Catal., 2005, 35, 105–116 CrossRef CAS PubMed.
- G.-Q. Li, H. Gao, C. Keene, M. Devonas, D. H. Ess and L. Kürti, J. Am. Chem. Soc., 2013, 135, 7414–7417 CrossRef CAS PubMed.
- C. K. De, F. Pesciaioli and B. List, Angew. Chem., Int. Ed., 2013, 52, 9293–9295 CrossRef CAS PubMed.
- For closely related asymmetric Fischer indole syntheses: (a) S. Müller, M. J. Webber and B. List, J. Am. Chem. Soc., 2011, 133, 18534–18537 CrossRef PubMed; (b) A. Martínez, M. J. Webber, S. Müller and B. List, Angew. Chem., Int. Ed., 2013, 52, 9486–9490 CrossRef PubMed.
- For reviews: (a) Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000 Search PubMed; (b) J. M. Janey, Angew. Chem., Int. Ed., 2004, 44, 4292–4300 CrossRef PubMed; (c) M. Marigo and K. A. Jørgensen, Chem. Commun., 2006, 2001–2011 RSC; (d) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed.
- For reviews: (a) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570–1581 CrossRef CAS PubMed; (b) X. Yu and W. Wang, Org. Biomol. Chem., 2008, 6, 2037–2046 RSC; (c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed.
- For a highlight: J. Xiao, ChemCatChem, 2012, 4, 612–615 CrossRef CAS.
- S.-L. Zhang, H.-X. Xie, J. Zhu, H. Li, X.-S. Zhang, J. Li and W. Wang, Nat. Commun., 2011, 2, 211 CrossRef PubMed.
- Y. Hayashi, T. Itoh and H. Ishihawa, Angew. Chem., Int. Ed., 2011, 50, 3920–3924 CrossRef CAS PubMed.
- Y.-L. Zhao, Y. Wang, X.-Q. Hu and P.-F. Xu, Chem. Commun., 2013, 49, 7555–7557 RSC.
- J. Zhu, J. Liu, R. Ma, H. Xie, J. Li, H. Jiang and W. Wang, Adv. Synth. Catal., 2009, 351, 1229–1232 CrossRef CAS.
- X. Zeng, Q. Ni, G. Raabe and D. Enders, Angew. Chem., Int. Ed., 2013, 52, 2977–2980 CrossRef CAS PubMed.
- (a) D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861–863 CrossRef CAS PubMed; (b) D. Enders, M. R. M. Hüttl, J. Runsink, G. Raabe and B. Wendt, Angew. Chem., Int. Ed., 2007, 46, 467–469 CrossRef CAS PubMed; (c) D. Enders, M. R. M. Huttl, G. Raabe and J. W. Bats, Adv. Synth. Catal., 2008, 350, 267–279 CrossRef CAS.
- Y. K. Kang and D. Y. Kim, Chem. Commun., 2014, 50, 222–224 RSC.
- J. Mo, L. Shen and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 8588–8591 CrossRef CAS PubMed.
- For a review on oxidative NHC catalysis: C. E. I. Knappke, A. Imami and A. J. von Wangelin, ChemCatChem, 2012, 4, 937–941 CrossRef CAS.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nat. Chem., 2013, 5, 835–839 CrossRef CAS PubMed.
- L. Hao, Y. Du, H. Lv, X. Chen, H. Jiang, Y. Shao and Y. R. Chi, Org. Lett., 2012, 14, 2154–2157 CrossRef CAS PubMed.
- (a) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS PubMed; (b) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS PubMed.
- (a) Y. Hayashi, T. Itoh and H. Ishikawa, Adv. Synth. Catal., 2013, 355, 3661–3669 CrossRef CAS; (b) W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu, W. Yuan, J. Zhang, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201308699; (c) Z. Meng, S. Sun, H. Yuan, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201308701; (d) B. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201309207; (e) L. Souillart and N. Cramer, Chem. Sci., 2014 10.1039/c3sc52753k.
| This journal is © The Royal Society of Chemistry 2014 |