
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conditional disorder in proteins: functional transitions between order and disorder
Bhaswati
Devi
 a,
Niharika
Nag
b,
Vladimir N.
Uversky
*c and
Timir
Tripathi
*a
a,
Niharika
Nag
b,
Vladimir N.
Uversky
*c and
Timir
Tripathi
*a
aMolecular and Structural Biophysics Laboratory, Department of Zoology, School of Life Sciences, North-Eastern Hill University, Shillong 793022, India. E-mail: timir.tripathi@gmail.com
bMolecular and Structural Biophysics Laboratory, Department of Biochemistry, School of Life Sciences, North-Eastern Hill University, Shillong 793022, India
cDepartment of Molecular Medicine and USF Health Byrd Alzheimer's Research Institute, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA. E-mail: vuversky@usf.edu
First published on 22nd September 2025
Abstract
The classical view of protein function based on rigid, well-defined structures is being redefined by the emerging concept of intrinsic disorder. Conditionally disordered proteins (CDPs) represent a subset of cellular intrinsically disordered proteins (IDPs) that transition between ordered and disordered states in response to specific stimuli, such as redox changes, post-translational modifications, ligand binding, interaction with partners, or environmental stress. This review explores the diverse landscape of conditional disorder and encompasses cryptic or dormant disordered regions, redox-sensitive motifs, metamorphic proteins, and proteins exhibiting order–disorder–new order transitions. These dynamic transitions allow CDPs to perform specialized regulatory, signalling, and stress-responsive roles, which often act as interaction hubs in complex cellular networks. Importantly, conditional disorder is not an anomaly but a conserved and functionally relevant feature across many proteomes. We highlight mechanistic insights into disorder-to-order transitions and their implications for cellular plasticity, adaptability, and disease. We also discuss how the conformational heterogeneity of CDPs complicates structure-based drug design, while offering unique therapeutic opportunities. Future directions include the integration of advanced biophysical techniques, computational modelling, and profiling to map, characterize, and target CDPs with greater precision. Overall, understanding the molecular logics of the conditional disorder will open new frontiers in structural biology and offer a deeper appreciation of protein versatility beyond static structural paradigms.
 Bhaswati Devi | Bhaswati Devi is a PhD researcher in the Molecular and Structural Biophysics Laboratory, Department of Zoology, North-Eastern Hill University, India. She holds a Master's degree in Zoology from Gauhati University, India. Her research centers on intrinsically and conditionally disordered proteins, with a focus on their structural dynamics, interaction mechanisms, and disorder-to-order transitions. She is particularly interested in exploring the therapeutic potential of these proteins in drug discovery. |
 Niharika Nag | Niharika Nag is a PhD researcher in the Department of Biochemistry, North-Eastern Hill University, India. Her research focuses on intrinsically disordered proteins, particularly IDP–IDP interactions, their dynamics, and their role in driving liquid–liquid phase separation, with emphasis on the understanding the mechanisms underlying neurodegenerative diseases. She has published several peer-reviewed articles and book chapters and is a recipient of the DST-INSPIRE Fellowship and the BIDDS Best Research Student Award (2024). |
 Vladimir N. Uversky | Vladimir N. Uversky is a Professor at the Department of Molecular Medicine and USF Health Byrd Alzheimer's Research Institute, Morsani College of Medicine, University of South Florida, Tampa, FL, 33612, USA. He is an expert in protein physics and works on protein intrinsic disorder phenomenon and analysis of structure, function, protein folding, and misfolding, as well as liquid–liquid phase separation. He has authored over 1350 scientific publications and edited multiple books and book series related to protein science. He has been listed as Web of Science Highly Cited Researcher every year from 2014 to 2020. In 2021, he was elected as a Fellow of the Royal Society of Biology and a Fellow of the Royal Society of Chemistry. In 2024, he was elected as a Fellow of the American Institute for Medical and Biological Engineering and a Fellow of the American Association for the Advancement of Science. He collaborated with more than 12 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 colleagues from more than 2750 research organizations in 89 countries/territories.
500 colleagues from more than 2750 research organizations in 89 countries/territories. Timir Tripathi | Timir Tripathi is a Professor of Molecular Biology at North-Eastern Hill University, India. His research focuses on protein–substrate interactions, conformational dynamics, and the regulatory roles of non-catalytic protein domains in enzymatic activity. He is particularly interested in the conformational dynamics, self-assembly, stabilization, and phase separation properties of IDPs. He has published over 150 research papers, reviews, and editorials, edited seven books, and authored two textbooks. His contributions have been recognized with several awards. He currently serves as Senior Editor of Critical Insights in Biophysics, Editor of International Journal of Biological Macromolecules, Associate Editor of PLOS Neglected Tropical Diseases, Advisory Board Member of FEBS Letters, and as an editorial board member of several other leading international journals. |
1. Introduction
Intrinsically disordered proteins (IDPs) represent a distinct class of proteins that lack a stable, well-defined three-dimensional (3D) structure under physiological conditions.1 Despite this apparent structural deficiency, IDPs are abundant in nature and play indispensable roles in diverse biological processes.2 Unlike globular proteins, IDPs do not adopt a single, energetically favorable conformation but exist as dynamic ensembles of rapidly interconverting conformations (Fig. 1).3,4 The sequence characteristics of IDPs are central to their structural plasticity. IDPs are typically enriched in disorder-promoting residues (e.g., proline, serine, glutamine) and depleted in order-promoting residues (e.g., aromatic residues, leucine, isoleucine, valine), which underlie their inability to fold into a stable tertiary structure.5 This intrinsic flexibility allows IDPs to bind multiple partners with high specificity yet low affinity, often via short linear motifs (SLiMs) or molecular recognition features (MoRFs), which are disordered elements capable of disorder-to-order transition at interaction with specific partners.6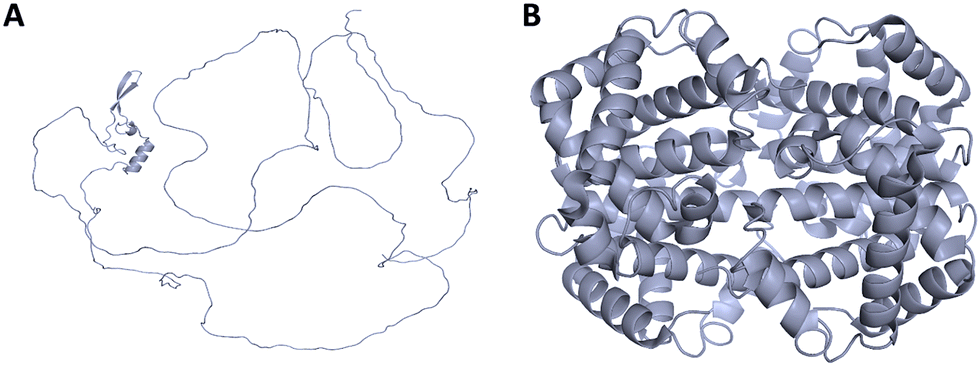 | ||
| Fig. 1 Disordered protein vs. ordered protein. (A) Structural representation of human Nup98 (disordered protein) with flexible conformation, and (B) representation of haemoglobin (ordered protein), characterized by a distinct stable folded structure. The disordered region of Nup98 (residues 1–450; UniProt ID: P52948) was modelled and visualized using PyMOL. Haemoglobin structure was obtained from the Protein Data Bank (PDB ID: 4HHB) and visualized in PyMOL. | ||
The functional versatility of IDPs arises directly from their disordered nature. They are key players in numerous cellular processes, including signal transduction, transcriptional regulation, cell cycle control, and post-translational modifications (PTMs) such as phosphorylation.5 Many IDPs undergo disorder-to-order transitions upon binding to specific targets, thereby achieving interaction specificity while maintaining adaptability.2 This conformational plasticity enables IDPs to act as central hubs in cellular networks.7 IDPs are also implicated in various pathological conditions, especially neurodegenerative diseases.8 For example, Alzheimer's disease is marked by the accumulation of neurofibrillary tangles composed of hyperphosphorylated tau and extracellular plaques of amyloid-β (Aβ), both of which involve IDPs.9 Their structural malleability renders IDPs prone to misfolding and aggregation, contributing to disease pathogenesis in disorders such as Parkinson's, Huntington's, and amyotrophic lateral sclerosis (ALS). However, their disease relevance also positions them as attractive targets for therapeutic intervention.10,11
The prevalence of intrinsically disordered regions (IDRs) in proteomes of both prokaryotes and eukaryotes is well-documented, with estimates suggesting that a significant fraction of the human proteome contains disordered segments.12 IDRs confer functional advantages, allowing proteins to engage in multiple interactions and respond dynamically to environmental cues.11,12 Transcription factors, for instance, frequently contain disordered effector domains (EDs) that enable multivalent interactions and the formation of transcriptional condensates.13 The structural plasticity of IDRs also facilitates regulation via PTMs, which can fine-tune protein function rapidly and reversibly. IDRs further act as molecular sensors, changing conformation in response to stress or other stimuli and thereby modulating cellular signalling pathways.14 Importantly, IDRs can drive liquid–liquid phase separation (LLPS), giving rise to membrane-less organelles (MLOs) such as nucleoli and stress granules. These dynamic condensates concentrate regulatory factors and enhance transcriptional efficiency among numerous other functionalities.15,16
2. Effect of environment on protein disorder
Environmental factors have a major influence on the structural plasticity of proteins, particularly in modulating transitions between ordered and disordered states. Variations in the cellular environment can markedly affect protein conformation, dynamics, and function. Changes in amino acid sequence, as well as experimental conditions such as pH, temperature, macromolecular crowding, the presence of osmolytes, and ionic strength, can impact the presence, position, and length of IDRs in proteins (Fig. 2).17 These factors affect the delicate balance of intramolecular forces governing protein conformations, thereby shifting the equilibrium between order and disorder.18 Proteins may undergo substantial structural and mechanical changes in response to alterations in pH and temperature, leading to unfolding or destabilization of their native spatial organization. Such changes influence the Gibbs free energy (ΔG) landscape, thereby modulating the stability of folded and unfolded states.19,20 Under specific environmental conditions, the conformational ensemble a protein adopts is shaped by its intrinsic properties (such as amino acid composition and PTMs) interacting with these external cues. Dysregulation of these interactions is frequently associated with pathological states.21 | ||
| Fig. 2 Effect of environmental factors on protein structure. The schematic illustrates how key external conditions, including pH, temperature, redox state, ionic strength, macromolecular crowding, and osmolarity, together impact protein conformation and stability. | ||
pH plays a particularly critical role in determining protein conformation. It alters the ionization states of amino acid side chains, thereby affecting intramolecular hydrophobic and electrostatic interactions and potentially triggering phase separation and coacervation behaviors.22 Changes in pH can alter the shape, molecular size, and adhesion properties of proteins, with extreme pH levels often decreasing structural stability.20 Even at a fixed pH, proteins with multiple ionizable residues can exist in various charge states, leading to a heterogeneous distribution of conformers. This heterogeneity is especially relevant to IDPs, whose conformational flexibility is highly sensitive to local charge environments.23 Histidine residues, which can exist in both neutral and positively charged forms, exemplify this sensitivity and can modulate IDP structure through pH-induced charge interactions.24 At acidic pH, IDPs often exhibit enhanced formation of α-helices and compactness, which can affect their aggregation propensity and functional behavior.25 By altering surface charge and electrostatic forces, pH exerts a significant impact on protein–protein interactions, which in turn affects the protein phase behavior and the tendency to aggregate.26 Electrostatic interactions are central to protein fold stability, binding specificity, and condensation processes, all of which are essential for cellular function and phase separation.27
Temperature, a fundamental thermodynamic variable, directly influences the conformational stability of proteins. Elevated temperatures tend to promote unfolding by favoring conformational entropy over enthalpic stabilization of the folded state.28 This process is often associated with the disruption of hydrophobic core interactions, decreasing protein compactness, and increasing disorder.29 For IDPs, temperature shifts can alter their functional organization and capacity to undergo phase separation. Their marginal stability makes them particularly responsive to such thermal perturbations, which is essential for their roles in dynamic cellular processes.19,20 One should also remember that, due to the temperature dependence of hydrophobic interactions, which become stronger at higher temperatures, extended IDPs (those classified as native coils and native pre-molten globules) exhibit a so-called “turned out” response to heat and may undergo partial folding when exposed to elevated temperatures.30
Redox conditions also regulate protein structure. Redox-sensitive residues such as cysteines, through reversible disulfide bond formation, can induce order–disorder transitions.31,32 A notable example is the conditionally disordered protein (CDP) CP12 from Arabidopsis thaliana, which undergoes a transition from a completely disordered state to a less disordered state upon oxidation due to the formation of disulfide bonds.33
Ionic strength of a solution profoundly affects IDP structure and interaction potential. Changes in ionic environment modulate the structure, charge distribution, and electrostatic repulsion within IDPs, thereby influencing their conformational landscape and binding affinities.34 Environmental factors such as salts have a major impact on nanopore gating and stoichiometry during cargo translocation by modulating protein behavior through the Hofmeister effect.35 The Hofmeister effect explains how specific ions impact the stability and solubility of proteins by altering hydration shells and electrostatic interactions, with kosmotropes generally stabilizing and chaotropes destabilizing protein conformations.36 Single-molecule studies have demonstrated that kosmotropic and chaotropic ions differentially regulate the conformational states and interactions of a protein by altering electrostatic forces and hydration layers, thereby regulating transmembrane channel gating and the efficiency of cargo transport.35 Ionic strength further modulates IDPs: fluctuations in charge distribution can induce dipole formation and conformational changes.37 Elevated ionic strength often promotes structural opening and reduces electrostatic repulsion, thereby enhancing the interactions of IDPs with binding partners.38 In addition, metal ions can coordinate with IDRs, stabilizing them into more ordered conformations and modifying their functional interactions. Such metal-induced folding is essential for a variety of cellular processes, including signalling, phase separation, and enzyme activation.39
LLPS is a fundamental physicochemical process underlying the formation of MLOs and biomolecular condensates.40–42 Due to their low sequence complexity and promiscuous binding potential, IDPs and proteins with large IDRs are ideally suited to drive LLPS. These condensates create specialized microenvironments that regulate biochemical reactions and cellular responses.41 IDPs play central roles in the assembly and function of MLOs, such as under stress conditions, where they contribute to the formation of stress granules that protect genetic material.43 Conditional disorder enhances the regulatory potential of proteins by allowing them to switch between functional states in response to environmental signals.19 The presence of IDPs across diverse cellular compartments further underscores their importance in facilitating complex protein interactions and dynamic communication.44
Macromolecular crowding further complicates the conformational behavior of IDPs. Crowding can alter protein–protein and protein–ligand interactions by modulating the excluded volume and available conformational space.45 IDPs, which often rely on disorder-to-order transitions upon binding, are particularly sensitive to crowding.46 Depending on their folding response, they can be classified as foldable, unfoldable, or non-foldable. Crowding can also promote LLPS, leading to the formation of MLOs that concentrate biomolecules and alter the structural behavior of IDPs.47 In such environments, amyloidogenic peptides may aggregate more readily, influencing the kinetics and morphology of pathological aggregates relevant to neurodegenerative diseases.48,49 The crowded intracellular milieu thus provides a more physiologically relevant context to study IDP function, revealing behaviors that are not observable in dilute solutions.50,51
Osmolytes, small water-soluble molecules, also influence protein disorder.52 They promote the formation of secondary structure and stabilize IDPs under osmotic stress, which enables adaptation to varying environmental conditions.30,52 Osmolytes modulate the thermodynamic balance between folded and unfolded states of a protein, thereby contributing to cellular resilience.46 Depending on their chemical nature, osmolytes can either stabilize or destabilize protein conformations by altering water structure and hydration shells.53,54
3. Binding-induced disorder-to-order transitions
3.1. Mechanisms of coupled folding
IDPs challenge the classical structure–function paradigm by functioning without a fixed 3D structure under physiological conditions.55,56 Instead, they exist as dynamic ensembles of rapidly interconverting conformations, sampling a wide range of structural possibilities.57 A hallmark feature of many IDPs is their ability to undergo binding-induced disorder-to-order transitions, also referred to as coupled folding and binding, where a conformational shift is stabilized upon interaction with a specific target.58 One common mechanism driving this process is the induced-fit model, wherein the IDP adopts an ordered conformation only after engaging with its binding partner. For instance, the phosphorylated kinase-inducible domain (pKID) of cAMP-response element binding (CREB) undergoes a transition from a disordered state to an α-helical structure upon binding to the KIX domain of CREB-binding protein (CBP) (Fig. 3).59 This process involves the formation of weak native interactions that stabilize the folded state and promote rapid compaction of the protein structure.60 An alternative mechanism is the conformational selection model, where the binding partner selects a pre-existing, native-like conformation from the IDP's ensemble.17 In this model, the protein transiently samples ordered structures even before interaction. The c-Myb activation domain, for example, binds to KIX primarily in a pre-folded state.59 Simulations of pKID binding to KIX have also demonstrated a nucleation-condensation mechanism, wherein the formation of a local native-like fragment acts as a nucleation point, driving the transition to a fully ordered complex.60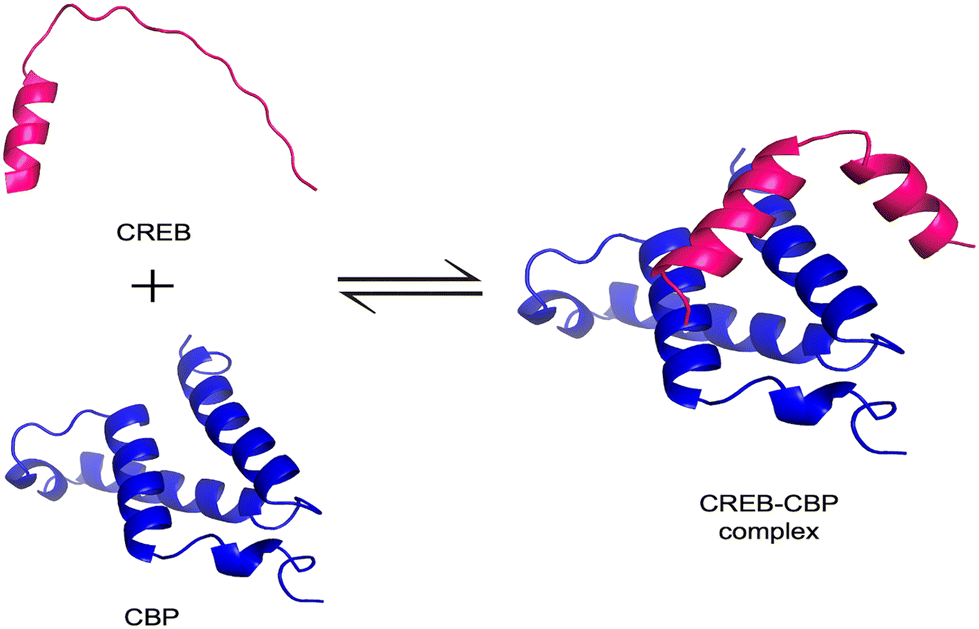 | ||
| Fig. 3 Binding-induced disorder-to-order transition in CREB. The illustration depicts the disordered region of CREB (shown in pink; residues 119–146; UniProt: P15337) in its unbound, extended state and the KIX domain of CBP (shown in blue; PDB ID: 1KDX). Upon binding, the CREB region adopts an ordered conformation as part of the complex, illustrating the disorder-to-order transition. Structures were visualized using PyMOL. | ||
IDPs achieve functionality through these intricate binding mechanisms, which often involve cooperative transitions between disordered and ordered states.61 The induced-fit and conformational selection models are not mutually exclusive but may operate concurrently or in tandem, depending on the context and partner interaction.46 For example, Zika virus protease adopts a significantly different conformation upon ligand binding, illustrating an induced-fit mechanism, whereas interaction of the dengue virus (DENV) protease (the NS2B-NS3 protease complex) with its ligands follows a conformational selection mechanism, where ligands bind to a pre-existing structural state of this protease.62
The biological relevance of binding-induced folding lies in its contribution to the functional plasticity of IDPs. This structural adaptability allows IDPs to engage in a diverse range of cellular activities and ensures context-dependent specificity.63 The kinetic and thermodynamic aspects of this transition are governed by the interplay between conformational entropy (lost during folding) and binding affinity (gained in the ordered state).64 Protein folding is fundamentally determined by a balance between enthalpic (ΔH) and entropic (TΔS) contributions, which often compensate for one another in a phenomenon known as enthalpy–entropy compensation (EEC), resulting in a narrow range of ΔG values.65 Interestingly, proteins can sometimes be stabilized despite enthalpic losses if the folded state gains configurational entropy. For example, engineered variants of acylphosphatase exhibited increased entropy in the folded state, which enhanced stability even though enzymatic activity was reduced.66 This balance allows IDPs to fine-tune their structural ensemble for optimized binding, retaining a degree of conformational heterogeneity while avoiding misfolding.67
3.2. Examples of IDPs undergoing disorder-to-order transitions
IDPs often undergo disorder-to-order transitions upon interaction with specific binding partners, stabilizing into structured conformations that are critical for their function. These transitions exemplify the functional adaptability of IDPs in diverse biological contexts. MoRFs are disordered segments that undergo structural transitions upon binding. MoRFs are categorized into α-MoRFs (that form α-helices), β-MoRFs (that form β-strands), and ι-MoRFs (that form irregular structures). Structural analysis of 258 complexes revealed 62 α-MoRFs, 20 β-MoRFs, and 176 ι-MoRFs, underscoring their role in specific molecular recognition events.68One notable example is prostate-associated gene 4 (PAGE4), an IDP implicated in prostate cancer. Phosphorylation induces distinct disorder-to-order transitions in PAGE4, modulating its structural dynamics and interactions with the AP-1 signalling axis. These changes influence cell phenotype and therapeutic response, highlighting the functional consequences of structural switching.69 Similarly, hybrid proteins combining IDPs with globular domains demonstrate how disorder-to-order transitions in the IDP component can significantly alter the overall structural and functional properties of the fusion construct.70,71 The Hahellin protein, a member of the βγ-crystallin family, is intrinsically disordered in its apo form. Upon binding of Ca2+, it adopts a well-ordered βγ-crystallin fold, exemplifying ion-induced structural stabilization.72 The N-terminal transactivation domain (TAD) of p53, intrinsically disordered in its free state, forms an α-helix upon binding to MDM2, serving as a regulatory checkpoint for the activity and degradation of p53 protein.73 The C-terminal region of this protein is able to fold differently upon interaction with various partners: it adopts an α-helical structure when binding to S100Bββ, forms a β-strand in a complex with sirtuin, takes on a β-turn when complexed with the CBP bromodomain, and exhibits an irregular structure with diverse morphologies in complexes with the histone methyltransferase Set9 and cyclin A/cyclin-dependent protein kinase 2.74–76 The linker histone H1 illustrates how binding to DNA can trigger structural rearrangement: its disordered N-terminal domain adopts a helical form upon nucleosome interaction, enhancing its affinity for DNA.77 In neurons, tau protein stabilizes microtubules through a disorder-to-order transition upon binding, which is essential for maintaining cytoskeletal integrity.78 Small molecules can also induce structural transitions in IDPs. For example, the disordered ‘lid’ region of MDM2 becomes structured upon binding to the small molecule AM-7209, significantly enhancing binding affinity and illustrating a mechanism of pharmacological stabilization.79 Likewise, α-synuclein, associated with Parkinson's disease, undergoes ordering when bound to lipid membranes, facilitating its role in synaptic vesicle trafficking.78
The transcription factor c-Myc undergoes a disorder-to-order transition upon dimerization with its partner Max, forming a stable DNA-binding complex that regulates genes involved in cell proliferation and apoptosis.2 Other transcription factors, such as antennapedia (Antp) and thyroid transcription factor-1 (TTF-1), also experience such transitions upon DNA binding. Their disordered N-terminal tails play crucial roles in DNA recognition, enhancing specificity and affinity by modulating the folding-binding coupling.80 In the high-mobility group (HMG) family, the transcription factor lymphoid enhancer-binding factor-1 (LEF-1) is only partially folded in the absence of DNA but adopts a more ordered structure upon binding to target sequences, demonstrating context-dependent structural rearrangement.81
Among viral proteins, the C-terminal domain of the measles virus nucleoprotein (NTAIL) interacts with the folded X domain of the viral phosphoprotein (XD), undergoing a folding transition essential for viral replication.82 In prion proteins (PrP), the intrinsically disordered N-terminal domain binds nucleic acids with high affinity. The conformational state of nucleic acid aptamers regulates the phase separation behavior and aggregation propensity of the Y145Stop variant of PrP, which influences transitions from liquid-like droplets to amyloid-like aggregates.83
Other examples include BtuB, a β-barrel protein involved in nutrient transport, which undergoes a calcium-induced disorder-to-order transition to facilitate function.84 In Neisseria meningitidis, the outer membrane protein OpcA exhibits structural reorganization at its receptor-binding site upon ligand binding, critical for host–cell interactions.85 Additionally, analysis of purine nucleotide-binding proteins reveals that over 22% contain segments undergoing disorder-to-order transitions, highlighting their widespread importance in nucleotide recognition and signalling pathways.86
3.3. Functional implications of binding-induced folding
When IDPs interact with their binding partners, they often undergo substantial conformational changes, transitioning from disordered to ordered states, which can significantly enhance their functional capabilities.87–89 This phenomenon exemplifies the dynamic nature of protein–protein interactions and is notably represented in the concept of mutual synergistic folding, where two disordered proteins fold together into a stable, structured complex.87 A classic example is the bacterial protein BirA, which undergoes a ligand-induced disorder-to-order transition that facilitates dimerization and stabilizes the overall structure, an essential step for its function.90 Similarly, in the case of the KIX domain binding to the transcription factor c-Myb, the binding transition state exhibits a considerable amount of native-like structure, implying that intrinsic disorder can fine-tune molecular recognition by enhancing binding specificity.91 The sequence–function relationship in binding-induced folding remains complex. Despite considerable sequence variation, many IDPs retain functional integrity, which highlights that modifications, particularly in hydrophobic side chains, can profoundly impact folding pathways and interaction dynamics without abolishing function.90 Thus, there is a delicate balance between order and disorder in protein function: while some proteins require preservation of disorder for activity, others rely on disorder-to-order transitions for their function.8Unlike globular proteins, which depend on pre-formed tertiary structures for activity, IDPs leverage their structural plasticity to achieve specificity through folding upon binding.46,63 This coupled binding and folding mechanism enables IDPs to interact with diverse partners with high specificity under context-dependent conditions, while preserving the adaptability needed for promiscuous interactions in complex signalling and regulatory networks.92 IDPs use transient conformations to fine-tune interactions, which allow them to adopt distinct structural states tailored to different targets. This functional plasticity enhances their interaction versatility and contributes to regulatory complexity in cellular pathways.93
From an evolutionary perspective, disordered regions offer significant advantages. The ability to switch between disordered and ordered states enables proteins to optimize binding affinity and specificity while remaining adaptable to novel partners. Evolutionary processes such as alternative splicing, domain shuffling, and modular recombination are enriched by the inherent flexibility of IDPs, which facilitates functional innovation without compromising protein integrity.94 The evolutionary benefit of transient disorder is clearly evident in the coupled folding-binding mechanism, where rapid conformational changes mediated by disordered segments enable swift and efficient molecular recognition.84 Such properties are particularly critical for proteins involved in signal transduction and regulatory pathways, where adaptability and responsiveness are key. By enabling a broad range of binding modes, IDPs promote evolvability, which allows organisms to develop complex multicellular functions while remaining robust to genetic variation.95,96 Cryo-EM studies reveal that the intrinsically disordered tau protein forms fibrils through a compact, structured C-terminal core that nucleates aggregation.97 Recent developments in label-free nanopore technology have further improved diagnostic potential by enabling highly sensitive detection of Alzheimer's disease-related protein aggregates and biomarkers.98
4. Proteins with cryptic disorder
4.1. Concept of cryptic disorder in ordered proteins
Moonlighting proteins, a class of multifunctional proteins, are capable of performing two or more distinct yet physiologically relevant biochemical or biophysical functions using a single polypeptide chain. A subset of these, known as cryptic enzymes, possesses hidden or dormant functions that are not evident under normal physiological conditions.99 While IDPs lack stable tertiary structures under physiological environments, many conventionally ordered proteins contain short disordered regions that remain inactive or cryptic (or dormant or latent) until triggered by specific stimuli.56 The concept of cryptic disorder challenges the classical structure–function paradigm by suggesting that intrinsic disorder can be a latent property even in folded proteins.100 This idea adds a layer of complexity to our understanding of protein dynamics, implying that ordered proteins may not exist in a single, rigid conformation but rather in an ensemble of conformational states.28 Cryptically disordered segments are often buried within the folded architecture and remain inaccessible to molecular interactions until conformational shifts, environmental changes, or binding events expose them.101Cryptic disorder confers conditional functionality, which enables proteins to undergo conformational transitions in response to cellular cues. This adaptability enhances the functional repertoire of a protein, including regulatory interactions and allosteric modulation. Such flexibility allows proteins to respond to spatial and temporal signals, contributing to dynamic cellular processes.102,103 The presence of variably ordered or disordered regions equips proteins with the ability to modulate their structures and perform context-specific functions.104 As a result, proteins with cryptic disorder often exhibit binding promiscuity and functional plasticity. Controlled unfolding of ordered regions, facilitated by cryptic disorder, allows for activation under distinct conditions. This underscores the broader functional relevance of conditional disorder in enhancing interaction versatility, even in the absence of a stable, ordered conformation.102,105 The capacity of IDPs and IDRs to adopt diverse conformations is vital to their roles in signalling and regulation.104 However, while cryptic disorder enhances biological versatility, it also increases the risk of misfolding and aggregation, which has been linked to the pathogenesis of several protein conformational diseases.106
4.2. Activation of functional sites via partial unfolding
A key mechanism for activating otherwise inaccessible functional regions involves partial unfolding events that transiently expose critical motifs embedded within the native fold (Fig. 4).107 PTMs, such as phosphorylation and acetylation, can modulate electrostatic interactions, leading to local destabilization and promoting partial unfolding of specific regions.108 Similarly, genetic mutations may alter the protein folding landscape, resulting in misfolding, gain or loss of function, or altered activity levels.109 Environmental stressors, such as changes in pH or temperature, can destabilize native structures, enhance conformational flexibility, and expose hidden functional sites. Some proteins, termed metamorphic proteins (discussed later in Section 6), illustrate the functional importance of partial unfolding by reversibly switching between multiple folded states in response to environmental stimuli, each conformation associated with distinct biological roles.110 In contrast, misfolding often exposes hydrophobic residues typically buried in the protein core, leading to aggregation and formation of non-native conformations.111Experimental evidence supports the functional relevance of such transiently unfolded states. For instance, D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), when partially unfolded in dilute guanidine hydrochloride in the presence of ATP, forms a folding intermediate that is reactivated by the chaperonin GroEL. This intermediate binds 8-anilino-1-naphthalenesulphonic acid (ANS) and exhibits enhanced fluorescence, consistent with a partially unfolded, reactivatable conformation (Fig. 4A).112 The glucocorticoid receptor (GR) is another classic example: its activation depends on the chaperone machinery of heat shock proteins (Hsps) involving Hsp90 and Hsp70. GR undergoes cycles of partial unfolding during maturation, enabling ligand binding and proper folding. These dynamic transitions within the chaperone complexes are crucial for functional activation (Fig. 4B).113 The bacterial chaperone HdeA demonstrates pH-triggered partial unfolding. In E. coli, HdeA exists as an inactive dimer at neutral pH but becomes active at acidic pH by monomerizing and partially unfolding, which allows it to prevent substrate protein aggregation under stress conditions (Fig. 4C).114 Similarly, Hsp33 relies on partial unfolding of its central linker region to transition from a self-inhibited state to a client-binding-competent form, which underscores the role of partial unfolding in molecular chaperone function (Fig. 4D).115
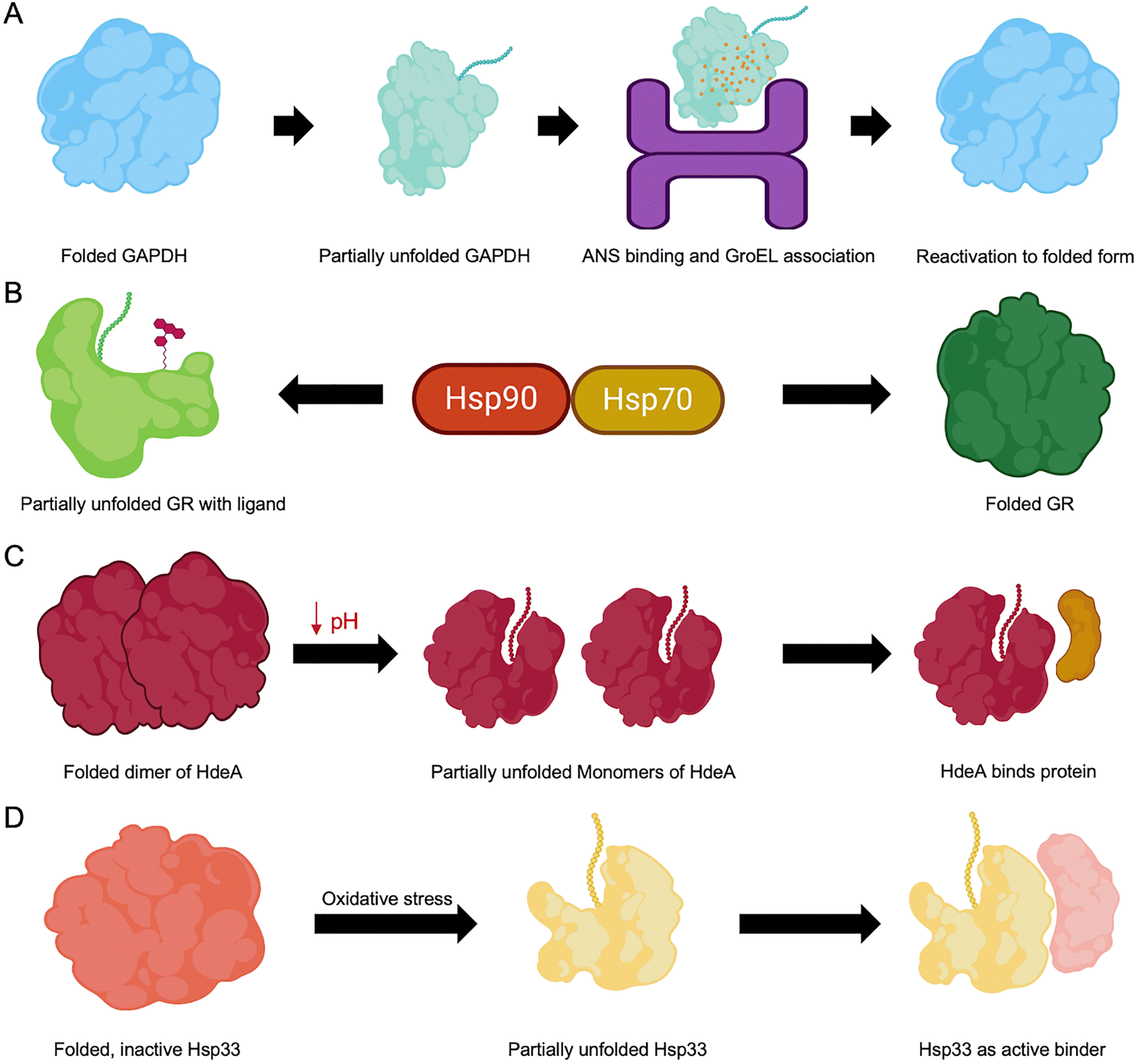 | ||
| Fig. 4 Partial unfolding and refolding of CDPs. (A) GAPDH transitions from its native folded state to a partially unfolded intermediate that binds ANS and associates with the GroEL chaperone, resulting in its reactivation. (B) The glucocorticoid receptor (GR) undergoes a cycle of partial unfolding and refolding facilitated by Hsp70 and Hsp90, enabling ligand binding. (C) HdeA exists as a folded dimer at neutral pH and, at low pH, dissociates into monomers that become partially unfolded and bind client proteins. (D) Hsp33 switches from an inactive folded form to a partially unfolded active state that interacts with client proteins. | ||
In the realm of IDPs, ligand binding can drive disorder-to-order transitions, which are often essential for allosteric regulation. These interactions are frequently “fuzzy” in nature, which retains a degree of disorder to support regulatory flexibility.116 This conformational plasticity enables IDPs to participate in complex biological processes such as signal transduction and cellular regulation – roles often inaccessible to rigid, globular proteins.117 Furthermore, as discussed earlier, disordered regions can drive LLPS, leading to the formation of biomolecular condensates that organize biochemical activities in space and time within cells.118 Molecular dynamics (MD) simulations and structural studies have revealed the existence of cryptic sites as transient surface pockets hidden in unbound states by side chains or loops. These sites can emerge during partial unfolding and enable induced-fit binding. Tools such as FTMap offer valuable insights into the structural dynamics of cryptic sites and help understand their functional significance.119
A case in point is the N-terminal oligomerization domain of nucleophosmin (Npm-N), which can transition from a folded pentameric state to a disordered monomer in response to changes in ionic strength or PTMs. This reversible switch reflects the evolution of Npm to toggle between structured and disordered conformations as a regulatory strategy.120 Moreover, the presence of cryptic amyloidogenic regions (CARs) in IDPs, which are normally masked under native conditions, has been linked to functional protein–protein interactions. These CARs appear to have evolved to enhance protein adaptability and function under varying cellular conditions.121 The ability of proteins to transition between conformational states enables precise regulation of key biological processes, including centrosome duplication, signalling, and stress response. Thus, cryptic disorder and partial unfolding emerge as central themes in protein function modulation.122
5. Redox-sensitive disordered regions
5.1. Influence of redox conditions on protein structure
Redox conditions play a pivotal role in modulating protein structure, which influences conformational states and functional activities across diverse biological systems.123 The cellular redox environment, which is defined by the balance between oxidized and reduced states, critically affects proteins containing redox-sensitive residues, especially cysteines.32 Oxidation of cysteine thiol groups leads to the formation of disulfide bonds, which can drastically alter protein folding, stability, and intermolecular interactions.124 Shifts in the redox environment can trigger conformational transitions, driving proteins from ordered to disordered states or vice versa.31 This redox-responsive structural plasticity underscores the importance of integrating redox regulation into our understanding of protein function.18 Furthermore, reactive oxygen species (ROS) and reactive nitrogen species (RNS) mediate PTMs, which regulate proteolytic enzymes and contribute to cellular homeostasis.125 Dysregulation of redox balance is implicated in a wide range of pathological conditions, highlighting the need to understand the complex interplay between redox signalling, protein structure, and cellular function.126Many redox-sensitive proteins possess conditionally disordered regions that respond dynamically to oxidative stress. These regions may undergo structural transitions upon exposure to ROS, altering protein stability, activity, and interaction networks.57 For instance, the Arabidopsis thaliana protein CP12 becomes prevalently disordered upon oxidation, a conformational shift that is essential for its role in regulating photosynthesis (Fig. 5).33 Similarly, oxidation-induced structural changes can expose otherwise hidden regions, enabling processes such as ubiquitination and proteasomal degradation. For example, oxidative modification of peroxiredoxin 2 (Prx2) facilitates its breakdown via proteasomes and autophagy pathways.127 Redox-sensitive conformational switching also influences protein–protein interactions and cellular signalling. The reversible formation of disulfide bonds, as seen in proteins like the human SH3 domain (hSH3), serves as a molecular switch that fine-tunes protein function in response to oxidative cues.128
 | ||
| Fig. 5 Schematic representation of conformational forms of redox-sensitive CDP, CP12. In the reduced state, CP12 is disordered, and upon oxidation, it adopts a prevalently disordered conformation. This redox-induced change affects how CP12 binds to its binding partners GAPDH and PRK, helping assemble a fully folded complex.33 Figure adapted under open access and terms of the CC BY 4.0 licence. | ||
5.2. Mechanisms of redox-induced disorder
Redox-induced disorder in proteins primarily arises from chemical modifications to redox-sensitive amino acid residues, particularly cysteine, methionine, and tyrosine.129 Oxidation of cysteine residues can lead to disulfide bond formation, which may stabilize protein structure or, paradoxically, introduce local or global disorder depending on the structural context and redox state.101,130 These modifications can modulate protein dynamics, disrupt existing intra- or inter-molecular interactions, and promote partial or complete unfolding of structured regions. Alterations in the redox environment also influence the protonation states of amino acid side chains, affecting hydrogen bonding and electrostatic interactions essential for maintaining the protein's conformational stability. Such changes are a fundamental mechanism by which redox states regulate cellular signalling and protein function. The inherent susceptibility of proteins to oxidative PTMs offers a versatile strategy for regulating their activity in response to environmental and physiological cues.131Disulfide bonds typically confer rigidity and structural stability to proteins. Their reduction, however, can destabilize protein structure and promote conformational flexibility or disorder.132 Redox-sensitive regions often harbour cysteine residues that stabilize structure upon oxidation; their reduction can lead to disorder through unfolding or exposure of previously buried regions.57,133 Dysregulation of these redox dynamics and disulfide bond formation/breakage has been implicated in various pathological conditions, including cancer and neurodegenerative diseases such as Parkinson's disease.133 Redox-sensitive cysteine residues are particularly crucial in mediating disorder-to-order transitions. Upon oxidation, these residues can form disulfide bonds that stabilize protein conformation, driving structural ordering from a disordered state. For example, the activation of Hsp33 and COX17 involves redox-induced structural changes that confer functional regulation.31
Granulins, a family of small (∼6 kDa) multifunctional proteins derived from the proteolytic processing of their precursor progranulin, provide an illustrative example of proteins with high disulfide bond density. These proteins contain 12 conserved cysteines forming six intramolecular disulfide bonds. In their oxidized forms, granulins adopt structures dominated by irregular loops stabilized solely by disulfide bonds. Despite the absence of regular secondary structure, they exhibit remarkable thermal stability. However, reduction of the disulfide bonds renders them completely disordered.134 A recent review provides an in-depth analysis of the intricate interplay between intrinsic disorder and cysteines in proteins, highlighting how disordered sequences with interspersed cysteines can modulate protein function under stress and varying environmental conditions.135
In plant calmodulin (CaM), oxidation of methionine residues (especially Met-144 and Met-145) disrupts the structural linkage between the domains of CaM, resulting in a disordering effect that prevents activation of target proteins such as the PMCA calcium pump. Similarly, methionine oxidation contributes to helical unfolding in proteins involved in amyloidosis, exacerbating disease pathology through conformational destabilization.136 Prx2 provides another compelling example of oxidation-induced structural transitions in redox-sensitive proteins. Under oxidative stress, Prx2 undergoes a conformational change, exposing its C-terminal region, facilitating Lys191 ubiquitination, a key step in proteasomal and autophagic degradation pathways. Structural changes are further supported by elevated hydrogen/deuterium exchange rates in the GGLG and YF motifs, suggesting dynamic rearrangement upon oxidation.127
5.3. Functional significance of redox-sensitive disorder
Redox-sensitive disordered regions (RSDRs), often associated with cysteine-containing motifs and disulfide bonds, are widely present across proteomes, particularly in multicellular organisms. These regions endow proteins with the ability to dynamically alter their conformation and function in response to fluctuations in ROS, thereby serving as vital regulatory elements.57 Functioning as sensors of oxidative stress, RSDRs undergo conformational transitions that can activate signalling cascades or initiate antioxidant defense mechanisms. Notably, many signalling proteins harbour RSDRs that modulate their interactions with other molecules, enabling rapid, reversible, and finely tuned responses to shifts in cellular redox conditions.137 IDRs, due to their structural flexibility, enhance the interaction potential and functional diversity of proteins. Like structured domains, IDRs contribute to the assembly of protein complexes and higher-order structures.138 The environmental sensitivity of IDRs allows them to act as modulators of protein–protein interactions and activity. Importantly, redox dysregulation (especially involving cysteine oxidation) has been linked to protein misfolding and aggregation in several neurodegenerative diseases, such as ALS and Parkinson's disease.139,140While redox regulation is essential for normal cellular physiology, its imbalance can lead to pathological outcomes. Elevated ROS levels, characteristic of oxidative stress, can cause irreversible protein damage and contribute to conditions like cancer, cardiovascular disorders, and muscular dystrophies.141 Transcription factors such as OCT4, p53, and NRF2 are tightly regulated by redox mechanisms. For example, oxidation of cysteine residues can alter their DNA-binding capabilities and transcriptional output, thereby influencing processes like stem cell pluripotency, tumorigenesis, and apoptosis. KEAP1, which normally retains NRF2 in the cytoplasm, undergoes cysteine oxidation under oxidative stress, allowing NRF2 to translocate to the nucleus and activate the expression of antioxidant genes.142 Similarly, the activity of proto-oncogenes (e.g., c-FOS, c-JUN) and tumor suppressors (e.g., p53) is redox-dependent.
Redox-active enzymes, such as thioredoxins and NADPH oxidases, regulate ROS production and modulate protein function through reversible oxidation of cysteine residues. The thioredoxin family plays a key role in immune signalling and inflammatory responses by catalyzing thiol-disulfide exchange reactions.21
In prokaryotes, proteins such as Hsp33 serve as redox-regulated chaperones. Upon oxidative stress, Hsp33 forms disulfide bonds, undergoes partial unfolding, and binds to unfolded substrates, enhancing its chaperone activity. Analogous mechanisms exist in eukaryotic systems, where 2-Cys Prxs transition into chaperones via sulfinic acid formation.143 The pathological importance of redox-sensitive disorders is further underscored by the oxidative modification of cytoskeletal proteins (e.g., actin, tubulin) and nuclear regulators like histone deacetylases (HDACs), which influence chromatin structure and gene expression during neural differentiation.144
6. Metamorphic proteins
6.1. Definition and characteristics of metamorphic proteins
Metamorphic proteins represent a unique deviation from the traditional “one sequence, one structure” paradigm of protein folding.145 These proteins can adopt multiple distinct, stable conformations from a single amino acid sequence (Fig. 6).110 This structural plasticity equips them with functional versatility, making them the so-called “Janus proteins” of structural biology.146 Key features include: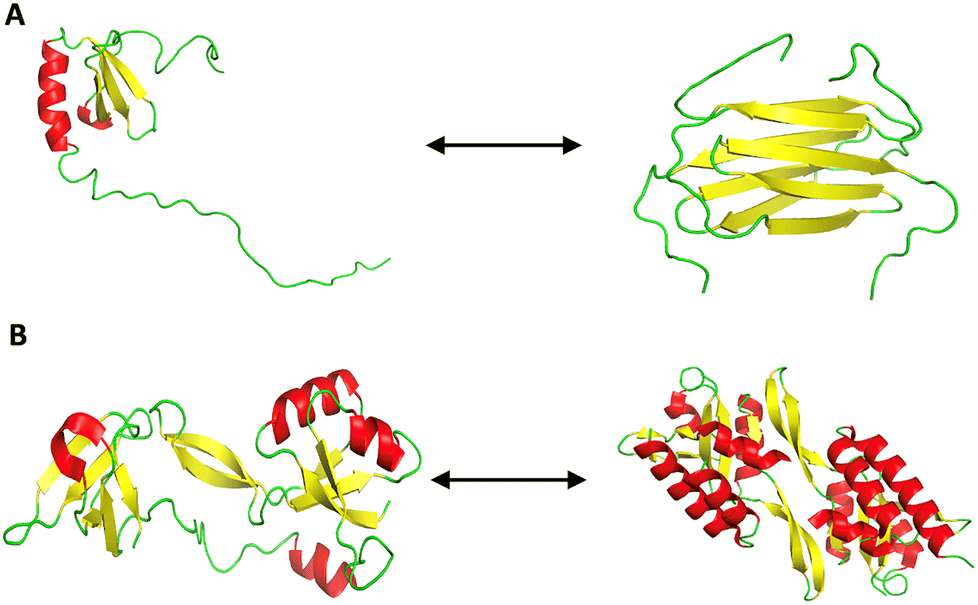 | ||
| Fig. 6 Conformational metamorphism in proteins. (A) Lymphotactin shows metamorphism by existing in two different folds. The structure on the left depicts the canonical monomeric chemokine fold (PDB ID: 1J9O) that binds to XCR1 receptor, favored at low temperature and high salt conditions. The structure on the right presents the dimeric all β-sheet fold (PDB ID: 2JP1), which facilitates glycosaminoglycan (GAG) binding, favored at high temperature and low salt conditions. (B) RfaH, a bacterial transcription factor, exhibits metamorphic folding by interconverting between two distinct conformations. The structure on the left shows the α-helical autoinhibited form (PDB ID: 5OND) and the one on the right shows the active β-fold (PDB ID: 6C6S) that engages ribosome interactions. The alpha helices are labelled in red, the beta sheets in yellow, and the coils are represented in green. | ||
(1) Multiple stable conformations: unlike canonical proteins that fold into a single native structure, metamorphic proteins exist in two or more thermodynamically stable and functional conformations.110
(2) Environmental sensitivity: external factors such as pH, temperature, and ligand binding can shift the equilibrium between different structural states.110
(3) Diverse functionality: each structural form can mediate distinct biological functions, allowing the same protein to participate in different pathways or interact with diverse molecular partners.146
(4) Conformational plasticity: a rugged, multifunnel energy landscape enables these proteins to switch between folds, often without the need for cofactors or ligands.146,147
Metamorphic proteins, such as lymphotactin (Ltn/XCL1), demonstrate how a single sequence can encode multiple structural states by modulating intramolecular contacts, oligomerization interfaces, and structural constraints (Fig. 6A).148 This contradicts classical folding theories and suggests that co-evolutionary pressures, particularly in host–pathogen dynamics, may promote metamorphism as an adaptive strategy.146 For instance, RfaH exemplifies how a metamorphic switch enables bacteria to regulate virulence and conjugation genes in response to environmental cues (Fig. 6B).149
6.2. Examples of proteins with multiple stable folds
Approximately 90 naturally occurring metamorphic proteins are currently known,150 and metamorphism has been successfully engineered into several of the most commonly occurring protein folds.151 Several well-characterized examples illustrate the phenomenon of metamorphic folding:(1) Ltn/XCL1: a chemokine that interconverts between two structurally unrelated native folds – one being a monomeric α/β chemokine-like structure and the other a dimeric β-sheet fold. This switching is rapid and reversible under physiological conditions.152
(2) Staphylococcal nuclease: exhibits at least two interconvertible native states with independent folding and unfolding pathways, as revealed by magnetization transfer NMR experiments.153
(3) ThreeFoil protein: a computationally designed, symmetric protein with high kinetic stability and long-range interactions, demonstrating the potential for engineered metamorphic behavior.154
(4) RfaH: a bacterial elongation factor that switches from an α-helical to a β-barrel structure, regulating operons associated with virulence and conjugation.149
(5) PrP: exists in a normal cellular form and a misfolded, pathogenic conformation associated with neurodegenerative disorders. Its ability to switch forms exemplifies pathological metamorphism.146
(6) Aβ peptides: demonstrate structural plasticity through fibrillogenesis, relevant to Alzheimer's disease pathology.146
(7) NusG-like transcription factors: structurally homologous to RfaH, these proteins adapt their folds to modulate gene expression in response to environmental changes.149
6.3. Biological roles and evolutionary advantages
The ability of metamorphic proteins to adopt multiple conformations expands their functional repertoire without requiring multiple genes or extensive sequence changes.110,145 Each conformation can serve a distinct biological role, which offers adaptive advantages:• Signal modulation and pathway integration: conformational switching enables dynamic regulation of signalling cascades by altering interaction profiles.131
• Environmental responsiveness: proteins like RfaH and KaiB (a clock protein) use fold-switching to modulate their activity in response to environmental or circadian changes.149,155
• Resource conservation: transitioning to an inactive conformation under stress or crowded cellular conditions may serve as an energy-saving mechanism.155
• Molecular switching: the reversible fold-switching of metamorphic proteins allows for rapid adaptation to stressors – critical for cellular survival in fluctuating environments.156
• Biotechnological potential: metamorphic proteins hold promise for the development of biosensors, responsive biomaterials, and switchable therapeutic agents.130
Despite their advantages, characterizing metamorphic proteins remains challenging due to their dynamic nature, which defies traditional structural biology techniques.157
6.4. Transitions between metamorphic states
Metamorphic proteins are distinguished by their ability to reversibly transition between distinct folded conformations, often in response to environmental cues.110,146 Their secondary structures are inherently flexible, allowing for large-scale conformational changes that can be triggered by factors such as pH, temperature, and ionic strength. These transitions occur within a polymorphic ensemble of conformations, where even minor perturbations can drastically reshape the energy landscape of the protein.108 In some cases, global unfolding is necessary to facilitate interconversion between native states, and reversible switching between folds has been observed under changes in temperature.158 This ability to morph between structures is closely linked to specific sequence features that promote flexibility and enable conformational transitions.159 Modifications in the dimer interface, along with dynamic residue contact networks and structural pliability, collectively contribute to metamorphic folding behavior.110 Ligand binding can also modulate the equilibrium between different conformational states, thereby fine-tuning the structural and functional repertoire of a protein.148For instance, Mad2 transitions between open and closed states via a denatured intermediate, although the presence of stable intermediates can hinder this switch by slowing or redirecting the transition pathway.160 Similarly, Ltn requires large-scale unfolding to switch between its native states, a process that underscores the importance of unfolding in metamorphic transitions.159 In these systems, different conformations are often thermodynamically comparable in stability, allowing reversible transitions to occur under physiological conditions.160 The kinetics of such interconversions are sensitive to environmental parameters, with temperature influencing the free energy of transition states and modulating the rates at which these transitions proceed.159
A recent comprehensive analysis of well-characterized metamorphic proteins revealed that temperature plays a crucial role in their structural transitions.161 In many cases, this temperature sensitivity is associated with partial cold denaturation, as the low-temperature state of these proteins tends to have a smaller hydrophobic core and is more disordered than their high-temperature state.161
7. Order–disorder–new order transitions
7.1. Pathways and mechanisms of structural transitions
Protein function depends on transitions between ordered, disordered, and newly ordered states, especially in IDPs. Upon interaction with physiological partners, IDPs often undergo disorder-to-order transitions. These transitions typically follow cooperative folding mechanisms, similar to those in structured proteins. However, due to the lack of a fixed folding nucleus, the folding pathways of IDPs are highly heterogeneous and flexible.46 For example, in Npm-N, phosphorylation can induce disorder under certain ionic conditions, thereby modulating its folding and assembly.162 Mutual folding occurs when disordered proteins bind each other, leading to the formation of structured complexes that are functionally active.87 The biological roles of proteins undergoing order–disorder transitions are closely linked to their enhanced conformational diversity. Upon ligand binding, more than 60% of such proteins become more structured, thereby enhancing their functional roles.163The functions of ordered proteins are dependent on the acquisition of specific structures through folding on physiological timescales. Ordered proteins may exist as partially organized intermediates under both equilibrium and non-equilibrium conditions. These intermediates typically possess some features of the fully folded state and are crucial in the folding pathway. In the presence of specific binding partners, such intermediates (commonly referred to as molten globules) can undergo disorder-to-order transitions, resulting in well-folded conformations.164–167 These reversible structural changes are essential for cellular signalling and function.168
Metamorphic proteins, due to their marginal stability, can undergo large-scale conformational shifts and respond to environmental cues such as pH, temperature, and redox conditions by switching between distinct fold topologies.108 In proteins like IscU, N-terminal order–disorder transitions play a key role in metamorphic regulation, influencing their interaction networks and contributing to structural heterogeneity.169
In Npm-N, phosphorylation and partner binding orchestrate a sequential series of disorder-mediated structural rearrangements. Phosphorylation can disrupt the coupled folding and assembly process by inducing disorder, whereas binding to physiological partners can counteract this effect by stabilizing the ordered state.162 This dynamic process involves a rapid collapse into a disordered intermediate, followed by a gradual conversion into a folded monomer, and ultimately assembly into a folded pentamer.162 The significance of binding in order–disorder transitions is also evident in mutual synergistic folding, where two disordered proteins form a stable, structured complex.87 Proteins commonly populate partially structured intermediates such as molten globules, which can transform into fully ordered conformations upon ligand binding. This transition is energetically favorable and enables proteins to reach their lowest energy structural states.168
7.2. Thermodynamic and kinetic considerations
Proteins exist along an energy continuum, from hyperstable folded states to hyperdynamic disordered conformations. This continuum is governed by both the amino acid sequence and environmental conditions, such as solvent composition, temperature, and ionic strength.170 Disordered proteins can transition into ordered structures through structural rearrangements that modulate free energy. During such transitions, the flexible peptide backbone acts as an entropic reservoir, balancing interaction enthalpy against entropy-driven conformational variability.171Statistical mechanics offers a robust framework to understand these transitions, suggesting that proteins can adopt ordered conformations under specific thermodynamic variables like temperature and interaction energy variance.172 Frequently, these order–disorder transitions are triggered by protein–protein or protein–ligand interactions. Disordered proteins, for instance, can form structured complexes through mutual synergistic folding, a process involving dynamic cooperative transitions.87 However, due to the fleeting nature of intermediate states, studying these transitions is challenging. In adenylate kinase (Adk), a unique “order–disorder–order” mechanism has been observed, where a segment within the ATP-binding subdomain locally unfolds and refolds during catalysis.173 Similarly, glucokinase exhibits glucose-induced disorder-to-order transitions that underlie its kinetic cooperativity and allosteric regulation.174
Experimental techniques like atomic force microscopy and X-ray scattering have revealed that order–disorder transitions in thin films follow linear time-dependent kinetics, typically initiated at defect sites with lower energy barriers.175 These transitions are best visualized through the free energy landscapes, which describe the thermodynamic stability of protein conformations and the energetic pathways connecting them.176 Entropic and enthalpic factors shape these landscapes and define activation barriers for transitions between states.177 A case in point is the glutamine-binding protein (GlnBP), whose conformational dynamics is reflected in its free energy landscape, with distinct basins corresponding to open and closed states.178 Transition states, representing the highest energy barriers, are critical points where folding decisions are made. By modelling the structural distribution within these states, folding mechanisms can be quantitatively predicted.179
External factors, such as temperature, further influence these landscapes by reshaping energy barriers and altering conformational minima. For example, in bovine serum albumin, thermal variation affects the balance between ordered and disordered states, reinforcing the idea that proteins often exist in dynamic equilibrium.180 This conformational plasticity is essential for functional versatility and highlights the role of protein interactions in driving order–disorder transitions, including mutual synergistic folding.87
7.3. Case studies illustrating order–disorder–new order scenarios
Several proteins exemplify the dynamic transitions between ordered, disordered, and newly ordered conformations, often in response to ligand binding, redox changes, or PTMs (Fig. 7). For instance, ligand binding near aromatic amino acid residues can induce changes in the tertiary structure of proteins.181 IDPs are uniquely characterized by their structural plasticity.182 Upon binding to their physiological partners, these proteins frequently undergo disorder-to-order transitions.46 A well-documented example is Hsp33, a heat shock protein from E. coli, which undergoes a disorder-to-order transition under reducing conditions, thereby activating its chaperone function.57 Another case is Adk, which displays an “order–disorder–order” transition involving local unfolding and refolding in its ATP-binding subdomain.173 Similarly, the C-terminal domain of troponin I exhibits order–disorder transitions, potentially impacting its role in cardiac regulation and disease.183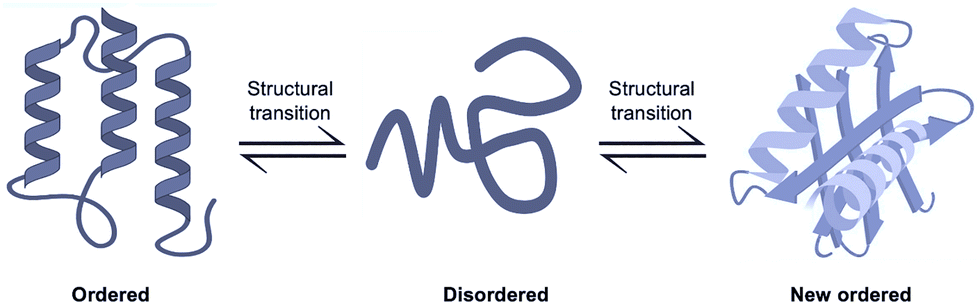 | ||
| Fig. 7 Schematic illustration of order–disorder–new order transition. Proteins can adopt a stable ordered conformation (left), switch to a disordered conformation (middle), and then refold into a new ordered conformation (right) under varying conditions. | ||
GRASP proteins display multiple disorder-to-order transitions that can be modulated by changes in the dielectric constant.184 In calcineurin, a key phosphatase involved in calcium signalling, interaction with calmodulin leads to disorder-to-order transitions in its regulatory domain. This event enables the displacement of the autoinhibitory domain from the active site, thus activating the enzyme.185 The human copper chaperone Cox17 provides another example of a CDP. It adopts a defined structured conformation upon forming a disulfide bond in the mitochondrial intermembrane space. This conformational change is essential for its role in copper transport and enzyme activation.186 Disorder-to-order transitions are also widespread among transcription factors, which allow them to remodel their structure for interactions with diverse targets. For example, phosphorylation of FoxM1 at Ser715 by Plk1 and Cdk kinases results in the release of its TAD into a disordered state. This transition permits binding by co-activators like CBP, thereby enhancing transcription during mitosis.187 Npm-N exhibits complex order–disorder transitions modulated by phosphorylation and partner interactions. These transitions influence its ability to assemble into ordered pentameric assemblies or remain disordered, depending on ionic strength and binding partners.162 A particularly striking case is RfaH, which undergoes a dramatic fold switch from an α-helical domain to a β-barrel upon DNA binding. This structural transformation shifts its role from promoting transcription elongation to facilitating translation initiation, thereby tightly coupling the two processes.188
Such adaptability underlies the function of many IDPs as dynamic switches in signalling networks.189 PAGE4, an IDP associated with prostate cancer, exemplifies this principle: it undergoes phosphorylation-dependent structural changes that alter its conformational dynamics and interaction with the AP-1 signalling axis. These transitions give rise to distinct cellular phenotypes and differential therapeutic sensitivities, highlighting the regulatory versatility encoded in the disorder of PAGE4.69 In E. coli, the biotin repressor BirA illustrates how effector binding triggers disorder-to-order transitions. Upon interaction with biotinoyl-5′-AMP, BirA dimerizes and becomes competent for DNA binding, thereby repressing transcription. This transition supports long-range allosteric regulation, demonstrating how intrinsic disorder enables precise gene control mechanisms.190
8. Functional implications of conditional disorder
Conditional disorder enables proteins to adopt specific conformations in response to specific environmental cues or PTMs.19 This structural adaptability allows proteins to reversibly fold or unfold, providing an effective mechanism for modulating function under changing physiological conditions.8 Many essential biological processes are regulated by such reversible transitions, especially in response to redox conditions, temperature fluctuations, or phosphorylation events.Proteins exhibiting conditional disorder play diverse and critical roles in biology. This phenomenon is widespread across proteomes and is particularly enriched in multicellular organisms, where it contributes to the formation of specialized functional domains. Redox-sensitive conditional disorder is a notable mechanism in many biological systems, influencing regulatory and stress response pathways.57 For instance, under reducing environments, the E. coli heat shock protein Hsp33 undergoes a transition from a disordered to an ordered state, enabling its function in protecting cells from oxidative stress.57 Similarly, some molecular chaperones exist in partially disordered forms under basal conditions and become fully active upon stress-induced unfolding, thereby enabling the recognition and binding of a wide array of aggregation-prone client proteins.101 Conditional disorder allows proteins to regulate their activity dynamically by toggling between ordered and disordered conformations. This reversible switching facilitates context-dependent molecular recognition, interaction, and signalling.8,19 IDPs enriched in signalling pathways act as dynamic hubs, enhancing the sensitivity and adaptability of cellular responses.191 These proteins often engage in interactions characterized by low affinity but high specificity, which are crucial for the precision of signalling cascades.192
Moreover, CDPs are well-suited to function as central nodes in protein–protein interaction networks. Their structural flexibility permits binding with multiple partners, enabling participation in a variety of biological processes.192,193 The presence of functional motifs within disordered regions can also increase the complexity of protein interaction networks, particularly in pathological conditions.193 Despite their functional versatility, CDPs present challenges for drug discovery, particularly when they are involved in diseases such as cancer or neurodegenerative disorders. Their lack of a stable structure complicates rational drug design and target validation.194 The dynamic structural patterns of CDPs can now be explored because of the recent advances in AI-driven protein structure prediction tools such as AlphaFold.195 By providing detailed insights into protein functions and interactions, the precisely predicted structures of AlphaFold have accelerated drug discovery. This is especially important for addressing diseases like Parkinson's and Alzheimer's that are closely associated with IDPs.196
9. Conclusions and future prospects
The study of CDPs marks a paradigm shift in our understanding of the structure–function relationship in biology. It is now evident that function does not necessarily rely on a fixed, well-folded 3D structure. Proteins exhibiting cryptic disorder, redox-sensitive disordered regions, metamorphic transitions, and intrinsic disorder challenge the classical structure-centric dogma (Fig. 8). These proteins exploit their conformational plasticity to engage in a wide range of biological processes, including signalling, regulation, stress response, and molecular recognition. The dynamic behavior of CDPs, manifested through disorder-to-order transitions upon binding or environmental cues, enables them to fine-tune their interactions in a temporally and spatially controlled manner. In particular, transitions involving “order–disorder–new order” states highlight the ability of these proteins to undergo complex structural rearrangements that expand their functional repertoire. Moving forward, it is imperative to deepen our molecular understanding of the mechanisms that govern conditional disorder.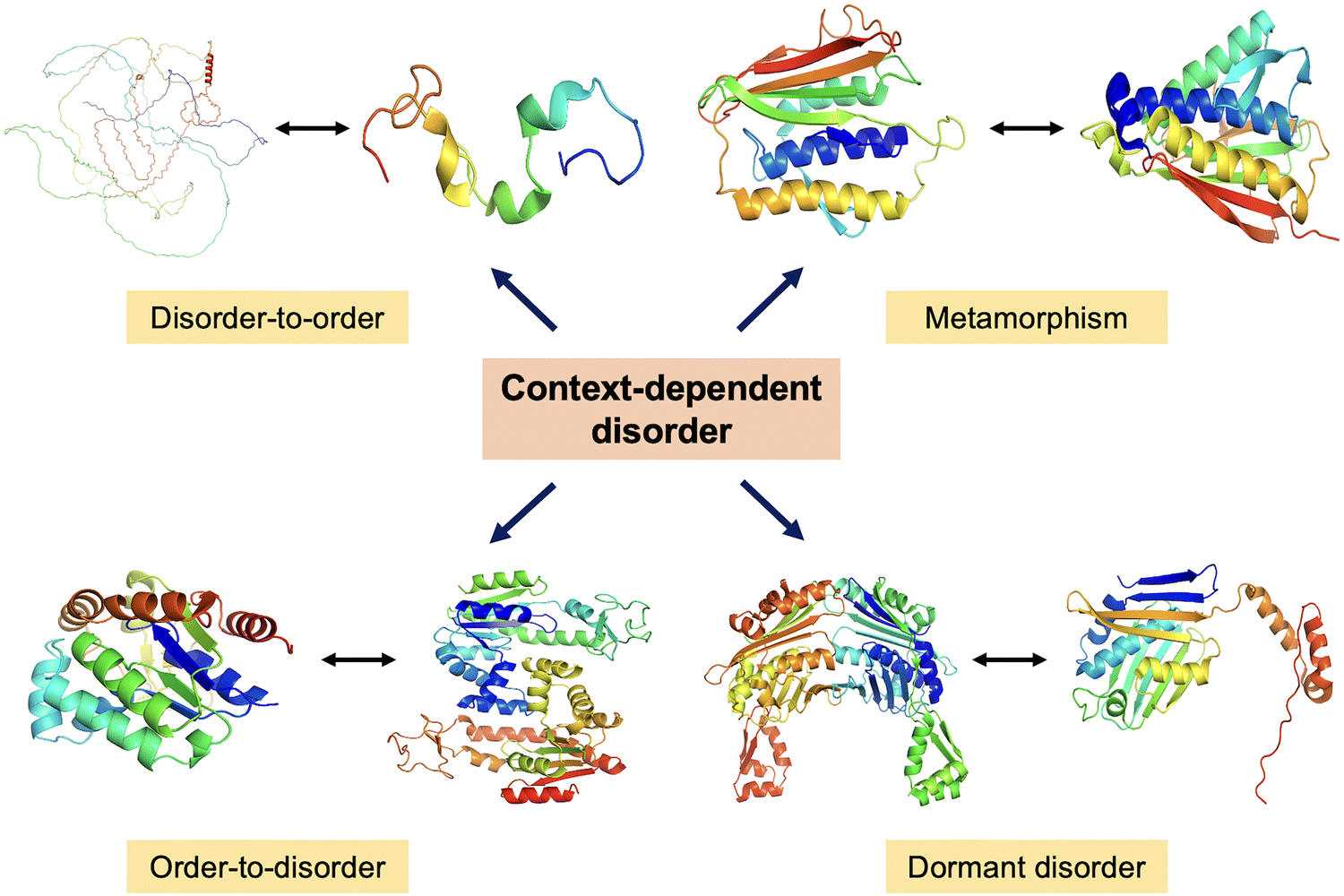 | ||
| Fig. 8 Dynamic protein structures. A schematic representation of context-dependent disorder illustrating four key transitions – disorder-to-order, metamorphism, order-to-disorder, and dormant disorder. In the disorder-to-order transition, full-length Tau protein (AlphaFold predicted structure AF-P10636-F1) adopts an ordered conformation upon binding to microtubules, shown by Tau fragment bound to microtubules (PDB ID: 2MZ7). In the metamorphism representation, MAD2 protein switches between two stable folded conformations (PDB ID: 1DUJ & 2V64). In the order-to-disorder transition, Adk undergoes partial unfolding depending on conditions (PDB ID: 1AKY & 4AKE). In dormant disorder representation, Hsp33 transitions under stress (PDB ID: 1VZY & 1HW7). | ||
Future research should aim to: (i) elucidate the conformational ensembles of CDPs using high-resolution structural techniques such as NMR spectroscopy, cryo-EM, single-molecule FRET, and hydrogen–deuterium exchange mass spectrometry, (ii) leverage computational simulations and AI-based structure prediction tools to model dynamic disorder–order transitions and predict functional states under diverse conditions, (iii) explore the role of conditional disorder in disease mechanisms, especially in cancer, neurodegeneration, and infection, where misregulation of IDPs and CDPs is often observed, (iv) identify and target transient or condition-specific structural states for therapeutic interventions—an emerging but challenging frontier in drug discovery, (v) integrate systems-level and proteomics approaches to map the distribution, dynamics, and interaction networks of CDPs across cellular and developmental contexts, (vi) ultimately, understanding how proteins leverage conditional disorder to balance structural flexibility with functional precision will offer profound insights into the fundamental principles of protein biology and provide new avenues for biomedical innovation.
Conflicts of interest
There are no conflicts to declare.Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.References
- R. B. Berlow, H. J. Dyson and P. E. Wright, FEBS Lett., 2015, 589, 2433–2440 CrossRef CAS.
- V. N. Uversky, Curr. Pharm. Des., 2013, 19, 4191–4213 CrossRef CAS.
- R. Trivedi and H. A. Nagarajaram, Int. J. Mol. Sci., 2022, 23, 14050 CrossRef CAS PubMed.
- T. Tripathi and V. N. Uversky, in The Three Functional States of Proteins, ed. T. Tripathi and V. N. Uversky, Academic Press, 2025, pp. 423–441 Search PubMed.
- P. E. Wright and H. J. Dyson, Nat. Rev. Mol. Cell Biol., 2015, 16, 18–29 CrossRef CAS.
- A. Bhattarai and I. A. Emerson, J. Biosci., 2020, 45, 29 CrossRef CAS.
- P. R. Banerjee, A. S. Holehouse, R. Kriwacki, P. Robustelli, H. Jiang, A. I. Sobolevsky, J. M. Hurley and J. T. Mendell, Trends Biochem. Sci., 2024, 49, 101–104 CrossRef CAS PubMed.
- V. N. Uversky, FEBS J., 2015, 282, 1182–1189 CrossRef CAS PubMed.
- S. Muralidar, S. V. Ambi, S. Sekaran, D. Thirumalai and B. Palaniappan, Int. J. Biol. Macromol., 2020, 163, 1599–1617 CrossRef CAS PubMed.
- S. Ayyadevara, A. Ganne, M. Balasubramaniam and R. J. Shmookler Reis, Metab. Brain Dis., 2022, 37, 147–152 CrossRef CAS PubMed.
- Z. H. Liu, M. Tsanai, O. Zhang, T. Head-Gordon and J. D. Forman-Kay, Curr. Opin. Struct. Biol., 2025, 93, 103063 CrossRef CAS.
- R. V. D. Lee, M. Buljan, B. Lang, R. J. Weatheritt, G. W. Daughdrill, A. K. Dunker, M. Fuxreiter, J. Gough, J. Gsponer, D. T. Jones, P. M. Kim, R. W. Kriwacki, C. J. Oldfield, R. V. Pappu, P. Tompa, V. N. Uversky, P. E. Wright and M. M. Babu, Chem. Rev., 2014, 114, 6589–6631 CrossRef PubMed.
- R. R. Datta, D. Akdogan, E. B. Tezcan and P. Onal, FEBS J., 2025, 292, 3014–3033 CrossRef CAS PubMed.
- D. Moses, G. M. Ginell, A. S. Holehouse and S. Sukenik, Trends Biochem. Sci., 2023, 48, 1019–1034 CrossRef CAS.
- P. Strzyz, Nat. Rev. Genet., 2018, 19, 534 CrossRef CAS.
- T. Tripathi and V. N. Uversky, The Three Functional States of Proteins: Structured, Intrinsically Disordered, and Phase Separated, Elsevier, 1st edn, 2024 Search PubMed.
- A. Mohan, V. N. Uversky and P. Radivojac, PLoS Comput. Biol., 2009, 5, e1000497 CrossRef PubMed.
- H.-C. Lu, S. S. Chung, A. Fornili and F. Fraternali, Front. Mol. Biosci., 2015, 2, 47 Search PubMed.
- A. C. Hausrath and R. L. Kingston, Cell. Mol. Life Sci., 2017, 74, 3149–3162 CrossRef CAS PubMed.
- M. Hao, Y. Ji, Y. Wang and Y. Chen, in 2019 IEEE International Conference on Manipulation, Manufacturing and Measurement on the Nanoscale (3M-NANO), 2019, pp. 333–336.
- I. Lorenzen, L. Mullen, S. Bakeschus and E.-M. Hanschmann, Oxid. Med. Cell. Longevity, 2017, 8459402 CrossRef PubMed.
- M. Nandy, K. A. Ganar, H. Ippel, I. Dijkgraaf and S. Deshpande, bioRxiv, 2025, preprint DOI:10.1101/2025.01.09.632076.
- M. J. Fossat, A. E. Posey and R. V. Pappu, Biophys. J., 2021, 120, 5438–5453 CrossRef CAS.
- R. Calinsky and Y. Levy, J. Phys. Chem. Lett., 2024, 15, 9419–9430 CrossRef CAS PubMed.
- L. Geist, M. A. Henen, S. Haiderer, T. C. Schwarz, D. Kurzbach, A. Zawadzka-Kazimierczuk, S. Saxena, S. Żerko, W. Koźmiński, D. Hinderberger and R. Konrat, Protein Sci., 2013, 22, 1196–1205 CrossRef CAS PubMed.
- A. C. Dumetz, A. M. Chockla, E. W. Kaler and A. M. Lenhoff, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 600–610 CrossRef CAS.
- H.-X. Zhou and X. Pang, Chem. Rev., 2018, 118, 1691–1741 CrossRef CAS.
- J. H. Fong, B. A. Shoemaker, S. O. Garbuzynskiy, M. Y. Lobanov, O. V. Galzitskaya and A. R. Panchenko, PLoS Comput. Biol., 2009, 5, e1000316 CrossRef PubMed.
- R. Mohana-Borges, J. Lima Silva and G. de Prat-Gay, J. Biol. Chem., 1999, 274, 7732–7740 CrossRef CAS.
- V. N. Uversky, Protein J., 2009, 28, 305–325 CrossRef CAS PubMed.
- G. Erdős, B. Mészáros, D. Reichmann and Z. Dosztányi, Proteomics, 2019, 19, 1800070 CrossRef.
- S. W. Fan, R. A. George, N. L. Haworth, L. L. Feng, J. Y. Liu and M. A. Wouters, Protein Sci., 2009, 18, 1745–1765 CrossRef CAS PubMed.
- A. Del Giudice, L. Gurrieri, L. Galantini, S. Fanti, P. Trost, F. Sparla and S. Fermani, Int. J. Mol. Sci., 2023, 24, 9308 CrossRef CAS PubMed.
- B. I. M. Wicky, S. L. Shammas and J. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9882–9887 CrossRef CAS.
- J. Li, Y. Wang, L. Wang, Y. Wang, Z. Zhang, S. Liu and L. Wang, ACS Mater. Lett., 2025, 7, 2476–2481 CrossRef CAS.
- K. D. Collins, Methods, 2004, 34, 300–311 CrossRef CAS PubMed.
- M. Phillips, M. Muthukumar and K. Ghosh, PNAS Nexus, 2024, 3, pgae367 CrossRef CAS.
- R. Vancraenenbroeck, Y. S. Harel, W. Zheng and H. Hofmann, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19506–19512 CrossRef CAS.
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014, 47, 2252–2259 CrossRef CAS PubMed.
- S. Brocca, R. Grandori, S. Longhi and V. Uversky, Int. J. Mol. Sci., 2020, 21, 9045 CrossRef CAS.
- S. Subedi, V. N. Uversky and T. Tripathi, in The Three Functional States of Proteins, ed. T. Tripathi and V. N. Uversky, Academic Press, 2025, pp. 177–195 Search PubMed.
- S. Sasidharan, N. Nag, T. Tripathi and P. Saudagar, in Droplets of Life, ed. V. N. Uversky, Academic Press, 2023, pp. 375–395 Search PubMed.
- A. S. Fefilova, I. A. Antifeeva, A. A. Gavrilova, K. K. Turoverov, I. M. Kuznetsova and A. V. Fonin, Biomolecules, 2022, 12, 1441 CrossRef CAS.
- B. Zhao, A. Katuwawala, V. N. Uversky and L. Kurgan, Cell. Mol. Life Sci., 2021, 78, 2371–2385 CrossRef CAS PubMed.
- T. Tripathi, V. N. Uversky and A. Giuliani, Cell. Mol. Life Sci., 2025, 82, 239 CrossRef CAS PubMed.
- A. Toto, F. Malagrinò, L. Visconti, F. Troilo, L. Pagano, M. Brunori, P. Jemth and S. Gianni, J. Biol. Chem., 2020, 295, 6586–6593 CrossRef CAS.
- V. N. Uversky, Curr. Opin. Struct. Biol., 2017, 44, 18–30 CrossRef CAS PubMed.
- F. Musiani and A. Giorgetti, in International Review of Cell and Molecular Biology, ed. M. Sandal, Academic Press, 2017, vol. 329, pp. 49–77 Search PubMed.
- S. P. Patel, T. Nikam, B. Sreepathi, V. S. Karankar, A. Jaiswal, S. V. Vardhan, A. Rana, V. Toga, N. Srivastava, S. A. Saraf and S. Awasthi, ACS Chem. Biol., 2024, 19, 2118–2130 CrossRef CAS PubMed.
- N. Das, T. Khan, B. Halder, S. Ghosh and P. Sen, Int. J. Biol. Macromol., 2024, 281, 136248 CrossRef CAS PubMed.
- I. M. Kuznetsova, K. K. Turoverov and V. N. Uversky, Int. J. Mol. Sci., 2014, 15, 23090–23140 CrossRef.
- S. M. Hosseiniyan Khatibi, F. Zununi Vahed, S. Sharifi, M. Ardalan, M. Mohajel Shoja and S. Zununi Vahed, Biochimie, 2019, 158, 156–164 CrossRef CAS PubMed.
- K. S. Negi, N. Das, T. Khan and P. Sen, Phys. Chem. Chem. Phys., 2023, 25, 32602–32612 RSC.
- V. Srinivasan, A. Rajendran and S. Khan, Curr. Appl. Sci. Technol., 2025, e0261080 Search PubMed.
- A. Soranno, Arch. Biochem. Biophys., 2020, 685, 108305 CrossRef CAS PubMed.
- X. Deng, J. Gumm, S. Karki, J. Eickholt and J. Cheng, Int. J. Mol. Sci., 2015, 16, 15384–15404 CrossRef PubMed.
- G. Erdős, B. Mészáros, D. Reichmann and Z. Dosztányi, Proteomics, 2019, 19, 1800070 CrossRef.
- M. Arai, Biophys. Rev., 2018, 10, 163–181 CrossRef CAS.
- M. Arai, K. Sugase, H. J. Dyson and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9614–9619 CrossRef CAS PubMed.
- M. Kurcinski, A. Kolinski and S. Kmiecik, J. Chem. Theory Comput., 2014, 10, 2224–2231 CrossRef CAS PubMed.
- A. Toto, F. Troilo, L. Visconti, F. Malagrinò, C. Bignon, S. Longhi and S. Gianni, Arch. Biochem. Biophys., 2019, 671, 255–261 CrossRef CAS PubMed.
- H. Maus, G. Hinze, S. J. Hammerschmidt, T. Schirmeister and T. Basché, Chem. – Eur. J., 2023, 1, e202300060 Search PubMed.
- P. E. Wright and H. J. Dyson, Curr. Opin. Struct. Biol., 2009, 19, 31–38 CrossRef CAS PubMed.
- C. W. Lawrence, S. Kumar, W. G. Noid and S. A. Showalter, J. Phys. Chem. Lett., 2014, 5, 833–838 CrossRef CAS PubMed.
- M. J. Benítez and J. S. Jiménez, Biophysica, 2025, 5, 2 CrossRef.
- S. Dagan, T. Hagai, Y. Gavrilov, R. Kapon, Y. Levy and Z. Reich, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10628–10633 CrossRef CAS PubMed.
- S. Hadži, A. Mernik, Č. Podlipnik, R. Loris and J. Lah, Angew. Chem., Int. Ed., 2017, 56, 14494–14497 CrossRef.
- V. Vacic, C. J. Oldfield, A. Mohan, P. Radivojac, M. S. Cortese, V. N. Uversky and A. K. Dunker, J. Proteome Res., 2007, 6, 2351–2366 CrossRef CAS PubMed.
- X. Lin, P. Kulkarni, F. Bocci, N. P. Schafer, S. Roy, M.-Y. Tsai, Y. He, Y. Chen, K. Rajagopalan, S. M. Mooney, Y. Zeng, K. Weninger, A. Grishaev, J. N. Onuchic, H. Levine, P. G. Wolynes, R. Salgia, G. Rangarajan, V. Uversky, J. Orban and M. K. Jolly, Biomolecules, 2019, 9, 77 CrossRef CAS PubMed.
- I. Sambi, P. Gatti-Lafranconi, S. Longhi and M. Lotti, FEBS J., 2010, 277, 4438–4451 CrossRef CAS PubMed.
- M. Adilović, J. Šutković, A. Hromić-Jahjefendić, T. Tripathi and V. N. Uversky, in The Three Functional States of Proteins, ed. T. Tripathi and V. N. Uversky, Academic Press, 2025, pp. 79–98 Search PubMed.
- S. Patel, B. Krishnan, R. V. Hosur and K. V. R. Chary, J. Phys. Chem. B, 2019, 123, 5086–5098 CrossRef CAS PubMed.
- P. D. Vise, B. Baral, A. J. Latos and G. W. Daughdrill, Nucleic Acids Res., 2005, 33, 2061–2077 CrossRef CAS PubMed.
- C. J. Oldfield, J. Meng, J. Y. Yang, M. Q. Yang, V. N. Uversky and A. K. Dunker, BMC Genomics, 2008, 9(Suppl 1), S1 CrossRef PubMed.
- V. N. Uversky, Int. J. Mol. Sci., 2016, 17, 1874 CrossRef PubMed.
- A. Kumar, P. Kumar, S. Kumari, V. N. Uversky and R. Giri, Arch. Biochem. Biophys., 2020, 684, 108342 CrossRef CAS PubMed.
- A. Sridhar, M. Orozco and R. Collepardo-Guevara, Nucleic Acids Res., 2020, 48, 5318–5331 CrossRef CAS PubMed.
- N. Sciolino, D. S. Burz and A. Shekhtman, Proteomics, 2019, 19, e1800055 CrossRef.
- C. Mendoza-Martinez, M. Papadourakis, S. Llabrés, A. A. Gupta, P. N. Barlow and J. Michel, Chem. Sci., 2022, 13, 5220–5229 RSC.
- Á. Tóth-Petróczy, I. Simon, M. Fuxreiter and Y. Levy, J. Am. Chem. Soc., 2009, 131, 15084–15085 CrossRef.
- J. J. Love, X. Li, J. Chung, H. J. Dyson and P. E. Wright, Biochem. J., 2004, 43, 8725–8734 CrossRef CAS.
- D. Bonetti, F. Troilo, A. Toto, M. Brunori, S. Longhi and S. Gianni, Biochemistry, 2017, 56, 3780–3786 CrossRef CAS.
- M. J. do Amaral, M. H. O. Freire, M. S. Almeida, A. S. Pinheiro and Y. Cordeiro, J. Neurochem., 2023, 166, 58–75 CrossRef CAS PubMed.
- D. P. Chimento, R. J. Kadner and M. C. Wiener, J. Mol. Biol., 2003, 332, 999–1014 CrossRef CAS PubMed.
- K. Kurup, A. K. Dunker and S. Krishnaswamy, Intrinsically Disord. Proteins, 2013, 1, e24848 CrossRef PubMed.
- A. Dan, Y. Ofran and Y. Kliger, Proteins, 2010, 78, 236–248 CrossRef CAS.
- O. O. Lebedenko, A. Sekhar and N. R. Skrynnikov, Proteins, 2024, 92, 1459–1463 CrossRef CAS PubMed.
- S. Mukhopadhyay, J. Phys. Chem. B, 2020, 124, 11541–11560 CrossRef CAS PubMed.
- F. Malagrinò, A. Diop, L. Pagano, C. Nardella, A. Toto and S. Gianni, Curr. Opin. Struct. Biol., 2022, 72, 153–160 CrossRef PubMed.
- C. Eginton, S. Naganathan and D. Beckett, Protein Sci., 2015, 24, 200–211 CrossRef CAS PubMed.
- R. Giri, A. Morrone, A. Toto, M. Brunori and S. Gianni, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14942–14947 CrossRef CAS PubMed.
- C. J. Jeffery, Front. Bioinf., 2023, 3, 1222182 CrossRef PubMed.
- C. Ghosh, S. Nagpal and V. Muñoz, Curr. Opin. Struct. Biol., 2024, 84, 102756 CrossRef CAS PubMed.
- P. Jemth, Curr. Opin. Struct. Biol., 2025, 90, 102980 CrossRef CAS PubMed.
- M. Kuravsky, C. Kelly, C. Redfield and S. L. Shammas, Nucleic Acids Res., 2024, 52, 11822–11837 CrossRef CAS PubMed.
- T. Mittag, L. E. Kay and J. D. Forman-Kay, J. Mol. Recognit., 2010, 23, 105–116 CrossRef CAS PubMed.
- R. Abskharon, M. R. Sawaya, D. R. Boyer, Q. Cao, B. A. Nguyen, D. Cascio and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119952119 CrossRef CAS PubMed.
- Q. Liu, Y. Ouyang, Y. Wang, S. Zhou, Y. Zhan and L. Wang, Adv. Healthcare Mater., 2025, 14, 2405058 CrossRef CAS PubMed.
- C. Jeffery, in Cryptic Enzymes and Moonlighting Proteins, ed. H. Irving, C. Gehring and A. Wong, Academic Press, 2025, pp. 1–10 Search PubMed.
- T. R. Alderson, I. Pritišanac, Đ. Kolarić, A. M. Moses and J. D. Forman-Kay, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2304302120 CrossRef CAS PubMed.
- J. C. A. Bardwell and U. Jakob, Trends Biochem. Sci., 2012, 37, 517–525 CrossRef CAS PubMed.
- S. DeForte and V. N. Uversky, eLS, John Wiley & Sons, Ltd, 2016, pp. 1–11 Search PubMed.
- S. DeForte and V. N. Uversky, Molecules, 2016, 21, 1090 CrossRef PubMed.
- V. N. Uversky, in Intrinsically Disordered Proteins, ed. V. N. Uversky, Springer International Publishing, Cham, 2014, pp. 1–51 Search PubMed.
- V. N. Uversky, Front. Phys., 2019, 7, 10 CrossRef.
- V. N. Uversky, Intrinsically Disord. Proteins, 2013, 1, e26782 CrossRef PubMed.
- T. P. Creamer, Intrinsically Disord. Proteins, 2013, 1, e26412 CrossRef PubMed.
- P. Kulkarni, T. L. Solomon, Y. He, Y. Chen, P. N. Bryan and J. Orban, Protein Sci., 2018, 27, 1557–1567 CrossRef CAS PubMed.
- V. Lemma, M. D’Agostino, M. G. Caporaso, M. Mallardo, G. Oliviero, M. Stornaiuolo and S. Bonatti, Sci. Rep., 2013, 3, 2659 CrossRef PubMed.
- M. Lella and R. Mahalakshmi, Biochemistry, 2017, 56, 2971–2984 CrossRef CAS PubMed.
- J. A. Johnston, C. L. Ward and R. R. Kopito, J. Cell Biol., 1998, 143, 1883–1898 CrossRef CAS PubMed.
- X.-L. Li, X.-D. Lei, H. Cai, J. Li, S.-L. Yang, C.-C. Wang and C.-L. Tsou, Biochem. J., 1998, 331, 505–511 CrossRef CAS PubMed.
- M. Castelli, A. Magni, G. Bonollo, S. Pavoni, F. Frigerio, A. S. F. Oliveira, F. Cinquini, S. A. Serapian and G. Colombo, Protein Sci., 2023, 33, e4880 CrossRef PubMed.
- L. Foit, J. S. George, B. W. Zhang, C. L. Brooks and J. C. A. Bardwell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1254–E1262 CrossRef CAS PubMed.
- B. Groitl, S. Horowitz, K. A. T. Makepeace, E. V. Petrotchenko, C. H. Borchers, D. Reichmann, J. C. A. Bardwell and U. Jakob, Nat. Commun., 2016, 7, 10357 CrossRef CAS PubMed.
- Q. Huang, L. Chen, L. Lai and Z. Liu, in Structure and Intrinsic Disorder in Enzymology, ed. M. N. Gupta and V. N. Uversky, Academic Press, 2023, pp. 327–352 Search PubMed.
- A. U. Rehman, M. U. Rahman, T. Arshad and H.-F. Chen, in Protein Allostery in Drug Discovery, ed. J. Zhang and R. Nussinov, Springer, Singapore, 2019, pp. 335–357 Search PubMed.
- S. Mukhopadhyay, Essays Biochem., 2022, 66, 817–819 CrossRef PubMed.
- D. Beglov, D. R. Hall, A. E. Wakefield, L. Luo, K. N. Allen, D. Kozakov, A. Whitty and S. Vajda, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3416–E3425 CrossRef CAS PubMed.
- D. M. Mitrea and R. W. Kriwacki, Pac. Symp. Biocomput., 2012, 152–163 CAS.
- J. Santos, I. Pallarès, V. Iglesias and S. Ventura, Comput. Struct. Biotechnol. J., 2021, 19, 4192–4206 CrossRef CAS PubMed.
- D. M. Mitrea and R. W. Kriwacki, Biocomputing 2012, World Scientific, 2011, pp. 152–163 Search PubMed.
- D. Reichmann and U. Jakob, Curr. Opin. Struct. Biol., 2013, 23, 436–442 CrossRef CAS PubMed.
- E. Mukwevho, Z. Ferreira and A. Ayeleso, Molecules, 2014, 19, 19376–19389 CrossRef PubMed.
- A. I. Petushkova and A. A. Zamyatnin, Biomolecules, 2020, 10, 650 CrossRef CAS PubMed.
- D. P. Jones and Y.-M. Go, Diabetes, Obes. Metab., 2010, 12, 116–125 CrossRef CAS PubMed.
- I.-K. Song, J.-J. Lee, J.-H. Cho, J. Jeong, D.-H. Shin and K.-J. Lee, Sci. Rep., 2016, 6, 34432 CrossRef CAS PubMed.
- J. Zimmermann, R. Kühne, M. Sylvester and C. Freund, Biochemistry, 2007, 46, 6971–6977 CrossRef CAS PubMed.
- S. Park, Nutrients, 2013, 5, 3496–3505 CrossRef PubMed.
- J.-Y. Min, K.-S. Chun and D.-H. Kim, Front. Oncol., 2023, 12, 997919 CrossRef PubMed.
- S. B. Wall, J.-Y. Oh, A. R. Diers and A. Landar, Front. Physiol., 2012, 3, 369 CAS.
- M. A. Wouters, S. W. Fan and N. L. Haworth, Antioxid. Redox Signaling, 2010, 12, 53–91 CrossRef CAS PubMed.
- T. J. Bechtel and E. Weerapana, Proteomics, 2017, 17, 1600391 CrossRef PubMed.
- G. Ghag, C. J. Holler, G. Taylor, T. L. Kukar, V. N. Uversky and V. Rangachari, Protein Sci., 2017, 26, 1759–1772 CrossRef CAS PubMed.
- A. A. Bhopatkar, V. N. Uversky and V. Rangachari, Prog. Mol. Biol. Transl. Sci., 2020, 174, 331–373 CAS.
- D. J. Bigelow and T. C. Squier, Mol. BioSyst., 2011, 7, 2101–2109 RSC.
- A. Matsuzawa and H. Ichijo, Antioxid. Redox Signaling, 2005, 7, 472–481 CrossRef CAS PubMed.
- N. S. Latysheva, T. Flock, R. J. Weatheritt, S. Chavali and M. M. Babu, Protein Sci., 2015, 24, 909–922 CrossRef CAS PubMed.
- H.-Y. Yang and T.-H. Lee, BMB Rep., 2015, 48, 200–208 CrossRef CAS PubMed.
- A. Percio, M. Cicchinelli, D. Masci, M. Summo, A. Urbani and V. Greco, Antioxidants, 2024, 13, 883 CrossRef CAS PubMed.
- M. J. Jackson, IUBMB Life, 2008, 60, 497–501 CrossRef CAS PubMed.
- G. Marsboom and J. Rehman, Oncotarget, 2016, 7, 80107–80108 CrossRef PubMed.
- C. Kumsta and U. Jakob, Biochemistry, 2009, 48, 4666–4676 CrossRef CAS PubMed.
- C. Berndt, C. Wilms, M. Thauvin and S. Vriz, in Oxidative Stress, ed. H. Sies, Academic Press, 2020, pp. 565–582 Search PubMed.
- A. F. Dishman and B. F. Volkman, ACS Chem. Biol., 2018, 13, 1438–1446 CrossRef CAS PubMed.
- K. Madhurima, B. Nandi and A. Sekhar, Open Biol., 2021, 11, 210012 CrossRef CAS PubMed.
- M. López-Pelegrín, N. Cerdà-Costa, A. Cintas-Pedrola, F. Herranz-Trillo, P. Bernadó, J. R. Peinado, J. L. Arolas and F. X. Gomis-Rüth, Angew. Chem., Int. Ed., 2014, 53, 10624–10630 CrossRef PubMed.
- A. F. Dishman, R. C. Tyler, J. C. Fox, A. B. Kleist, K. E. Prehoda, M. M. Babu, F. C. Peterson and B. F. Volkman, Science, 2021, 371, 86–90 CrossRef CAS PubMed.
- I. Artsimovitch and C. A. Ramírez-Sarmiento, Comput. Struct. Biotechnol. J., 2022, 20, 5824–5837 CrossRef CAS PubMed.
- L. L. Porter and L. L. Looger, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5968–5973 CrossRef CAS PubMed.
- B. Ruan, Y. He, Y. Chen, E. J. Choi, Y. Chen, D. Motabar, T. Solomon, R. Simmerman, T. Kauffman, D. T. Gallagher, J. Orban and P. N. Bryan, Nat. Commun., 2023, 14, 431 CrossRef CAS PubMed.
- R. L. Tuinstra, F. C. Peterson, S. Kutlesa, E. S. Elgin, M. A. Kron and B. F. Volkman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5057–5062 CrossRef CAS PubMed.
- R. O. Fox, P. A. Evans and C. M. Dobson, Nature, 1986, 320, 192–194 CrossRef CAS PubMed.
- A. Broom, S. M. Ma, K. Xia, H. Rafalia, K. Trainor, W. Colón, S. Gosavi and E. M. Meiering, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14605–14610 CrossRef CAS PubMed.
- N. Zhang, W. Guan, S. Cui and N. Ai, Commun. Chem., 2023, 6, 117 CrossRef PubMed.
- M. Das, N. Chen, A. LiWang and L.-P. Wang, Biopolymers, 2021, 112, e23473 CrossRef CAS PubMed.
- S. C. Goodchild, P. M. G. Curmi and L. J. Brown, Biophys. Rev., 2011, 3, 143–153 CrossRef CAS PubMed.
- T. L. Solomon, Y. He, N. Sari, Y. Chen, D. T. Gallagher, P. N. Bryan and J. Orban, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2215418120 CrossRef CAS PubMed.
- R. C. Tyler, N. J. Murray, F. C. Peterson and B. F. Volkman, Biochemistry, 2011, 50, 7077–7079 CrossRef CAS PubMed.
- Y. Zhao, L. Li, C. Wu, X. Jiang, B. Ge, H. Ren and F. Huang, Protein Eng., Des. Sel., 2016, 29, 23–29 CAS.
- A. LiWang and J. Orban, Proc. Natl. Acad. Sci. U. S. A., 2025, 122, e2422725122 CrossRef CAS PubMed.
- P. R. Banerjee, D. M. Mitrea, R. W. Kriwacki and A. A. Deniz, Angew. Chem., Int. Ed., 2016, 55, 1675–1679 CrossRef CAS PubMed.
- D. J. Zea, A. M. Monzon, C. Gonzalez, M. S. Fornasari, S. C. E. Tosatto and G. Parisi, Protein Sci., 2016, 25, 1138–1146 CrossRef CAS PubMed.
- O. B. Ptitsyn, V. E. Bychkova and V. N. Uversky, Philos. Trans. R. Soc. London, Ser. B, 1995, 348, 35–41 CrossRef CAS PubMed.
- O. B. Ptitsyn, Adv. Protein Chem., 1995, 47, 83–229 CrossRef CAS PubMed.
- V. E. Bychkova and O. B. Ptitsyn, Tsitologiia, 1995, 37, 1238–1250 CAS.
- V. N. Uversky and N. V. Narizhneva, Biochemistry, 1998, 63, 420–433 CAS.
- P. Mendoza-Espinosa, V. García-González, A. Moreno, R. Castillo and J. Mas-Oliva, Mol. Cell. Biochem., 2009, 330, 105–120 CrossRef CAS PubMed.
- J. Na, J. Heo, M. Jeong, S. Ji, Y. H. Ko, A. Shafiei, N. Baldir, H. DeMirci, W. Yu and J. H. Kim, Int. J. Biol. Macromol., 2025, 323, 146969 CrossRef CAS PubMed.
- M. M. Moosa, J. C. Ferreon and A. C. M. Ferreon, Semin. Cell Dev. Biol., 2020, 99, 78–85 CrossRef CAS PubMed.
- R. J. Workman, J. A. Drake and B. M. Pettitt, in Structure and Intrinsic Disorder in Enzymology, ed. M. N. Gupta and V. N. Uversky, Academic Press, 2023, pp. 97–126 Search PubMed.
- A. W. Nielsen, L. Sari, R. Fraser and M. M. Lin, Proteins: Struct., Funct., Bioinf., 2023, 91, 705–711 CrossRef CAS PubMed.
- M. Wolf-Watz and U. Olsson, Biophys. J., 2012, 102, 50a CrossRef.
- M. Larion, S. R. K. Salinas, L. Bruschweiler-Li, B. G. Miller and R. Bruschweiler, PLoS Biol., 2012, 10, e1001452 CrossRef CAS PubMed.
- S. Karmakar, M. K. Mukhopadhyay, M. K. Sanyal, A. Meinhardt and T. F. Keller, Phys. Rev. B, 2025, 111, 024105 CrossRef CAS.
- P. C. Whitford, K. Y. Sanbonmatsu and J. N. Onuchic, Rep. Prog. Phys., 2012, 75, 076601 CrossRef PubMed.
- M. Schmidt, V. Srajer, R. Henning, H. Ihee, N. Purwar, J. Tenboer and S. Tripathi, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2013, 69, 2534–2542 CrossRef CAS PubMed.
- Z.-Z. Lai, Q. Lu and J. Wang, J. Phys. Chem. B, 2011, 115, 4147–4159 CrossRef CAS PubMed.
- E. Alm and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11305–11310 CrossRef CAS PubMed.
- M. Peters, T. Zhao, S. George, V. G. Truong, S. N. Chormaic, C. Ying, R. A. Nome and R. Gordon, npj Biosensing, 2024, 1, 14 CrossRef PubMed.
- Protein Folding Dynamics and Stability: Experimental and Computational Methods, ed. P. Saudagar and T. Tripathi, Springer Nature, Singapore, 2023 Search PubMed.
- U. Jakob, R. Kriwacki and V. N. Uversky, Chem. Rev., 2014, 114, 6779–6805 CrossRef CAS PubMed.
- L. A. Metskas and E. Rhoades, J. Mol. Biol., 2016, 428, 2965–2977 CrossRef CAS PubMed.
- L. F. S. Mendes, L. G. M. Basso, P. S. Kumagai, R. Fonseca-Maldonado and A. J. Costa-Filho, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 855–865 CrossRef CAS PubMed.
- J. Rumi-Masante, F. I. Rusinga, T. E. Lester, T. B. Dunlap, T. D. Williams, A. K. Dunker, D. D. Weis and T. P. Creamer, J. Mol. Biol., 2013, 415, 307–317 CrossRef PubMed.
- H. Fraga, J. Pujols, M. Gil-Garcia, A. Roque, G. Bernardo-Seisdedos, C. Santambrogio, J.-J. Bech-Serra, F. Canals, P. Bernadó, R. Grandori, O. Millet and S. Ventura, Sci. Rep., 2017, 7, 16994 CrossRef PubMed.
- A. H. Marceau, C. M. Brison, S. Nerli, H. E. Arsenault, A. C. McShan, E. Chen, H.-W. Lee, J. A. Benanti, N. G. Sgourakis and S. M. Rubin, eLife, 2019, 8, e46131 CrossRef PubMed.
- S. H. Knauer, P. Rösch and I. Artsimovitch, RNA Biol., 2012, 9, 1418–1423 CrossRef CAS PubMed.
- V. N. Uversky, V. Davé, L. M. Iakoucheva, P. Malaney, S. J. Metallo, R. R. Pathak and A. C. Joerger, Chem. Rev., 2014, 114, 6844–6879 CrossRef CAS PubMed.
- J. Wang, G. Custer, D. Beckett and S. Matysiak, Biochemistry, 2017, 56, 4478–4488 CrossRef CAS PubMed.
- S. E. Bondos, A. K. Dunker and V. N. Uversky, Cell Commun. Signaling, 2022, 20, 20 CrossRef CAS PubMed.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, in 2007 IEEE 7th International Symposium on BioInformatics and BioEngineering, 2007, pp. 12–12.
- H. Anbo, M. Sato, A. Okoshi and S. Fukuchi, Biomolecules, 2019, 9, 88 CrossRef CAS PubMed.
- G. Zanotti, Crystallogr. Rev., 2023, 29, 48–75 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- J. Fantini, F. Azzaz, C. Di Scala, A. Aulas, H. Chahinian and N. Yahi, Pharmacol. Ther., 2025, 267, 108797 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |