
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical dehydrogenative intramolecular C–C coupling for expedient carbazole synthesis
Mingyue Wua,
Albertus Denny Handoko a,
Kexin Zhub,
Chi-Lik Ken Leea,
Philip W. Millerb,
Mark R. Crimminb and
Balamurugan Ramalingam*ac
a,
Kexin Zhub,
Chi-Lik Ken Leea,
Philip W. Millerb,
Mark R. Crimminb and
Balamurugan Ramalingam*ac
aInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Singapore 627833, Republic of Singapore. E-mail: balamurugan_ramalingam@isce2.a-star.edu.sg
bMolecular Sciences Research Hub, Department of Chemistry, Imperial College London, London, W12 0BZ, UK
cInstitute of Materials Research and Engineering (IMRE), Agency for Science Technology and Research (A*STAR), Singapore 138634, Republic of Singapore
First published on 20th August 2025
Abstract
An electrochemical method for carbazole synthesis via dehydrogenative aryl–aryl coupling of arylamines under metal-free conditions at ambient temperature is presented. The reactivity of arylamines is rationalised by cyclic voltammetry and density functional theory (DFT) studies to provide a preliminary understanding of the observed regioselectivity.
Dehydrogenative aryl–aryl coupling is a powerful strategy for constructing aromatic molecules by directly forming C–C bonds through oxidative processes.1,2 Electrochemical dehydrogenative coupling uses the oxidation of suitable aromatic substrates to achieve the dimerisation of aryl units, providing a sustainable alternative to traditional metal-catalysed coupling reactions without chemical oxidants. When two aryl units are connected via a central nitrogen, intramolecular homo-coupling can occur, leading to the formation of carbazoles.3 Carbazole derivatives are widely used in pharmaceuticals,4 organic electronics,5,6 and functional materials.7,8 However, conventional carbazole synthesis often requires multiple reaction steps and suffers from poor atom economy. Thus, the electrochemical dehydrogenative coupling9 of diphenylamine offers an efficient alternative solution for carbazole synthesis by reducing waste generation. This method eliminates the need for pre-functionalised feedstocks or external oxidants and the use of precious transition-metal catalysts, making it atom-efficient, environmentally benign and cost-effective.
Electrochemical dehydrogenative aryl–aryl coupling has been known for more than a decade, with significant progress reported for the synthesis of biphenols,10–12 bianilides,13 meta- and para-terphenyls, as well as cross-coupled products of various heterocycles with phenols.14–16 However, the electrochemical dehydrogenative strategy has not been utilised for substrates that are pre-functionalised for carbazole synthesis. Transition metal-catalysed synthesis of carbazoles, involving oxidative pathways, has become increasingly attractive to conventional synthesis.17–19 Three major approaches (Fig. 1) have been reported for the synthesis of carbazoles adopting novel methodologies: (i) concerted C–H activation of pre-functionalised bi-aryl amines and subsequent C–N bond formation via either amination (R = H, alkyl, Bn)20 or amidation (R = acetyl)21–23 using metal catalysts or electrochemical dehydrogenative coupling (Fig. 1a);24 (ii) oxidative addition of aryl halides to metal catalysts followed by C–H activation (Fig. 1b),25–30 (iii) intramolecular oxidative C–H/C–H coupling (Fig. 1c),31,32 which can proceed in the presence of a metal catalyst or under photoredox conditions (e.g., using Fe-33 or Cu34-based catalysts) with iodine or molecular oxygen as the oxidant. While the above approaches are applicable to a broad range of substrates, the need for pre-functionalisation with either amines or halogens and the reliance on precious metals for substrate activation are disadvantageous. From a sustainability standpoint, metal-free strategies in organic synthesis are especially attractive compared to methods that depend on precious metal catalysis.
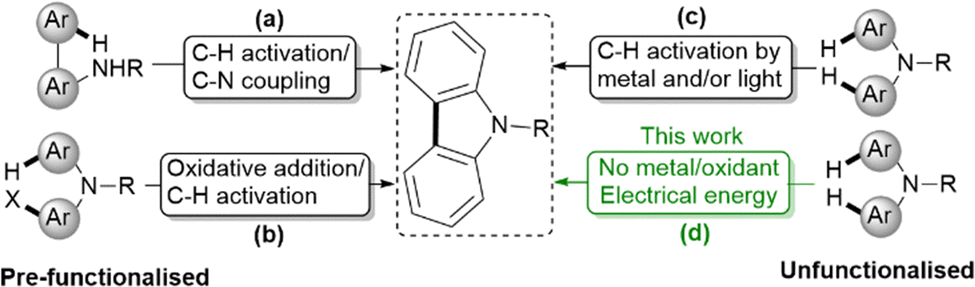 | ||
| Fig. 1 Current attractive strategies (a)–(c) for the synthesis of carbazoles compared to our sustainable approach (d). Existing strategies (a)–(c) rely on metal catalysts or oxidants and require pre-functionalized substrates. In contrast, our sustainable approach (d) uses only electricity to achieve oxidative coupling of aryl groups at room temperature, eliminating the need for oxidants or metal catalysts. | ||
To the best of our knowledge, neither simple nor substituted carbazoles have been synthesised from diaryl- or triaryl-amines via anodic oxidation without the use of oxidants, metals or photoredox catalysts. Notably, Beil et al.35 reported oxidative coupling using an active molybdenum anode under a constant current density of 7.5 mA cm−2. Under these conditions, the formation of six- or seven-membered rings was favoured, achieving isolated yields of up to 80%. It was proposed that [Mo{OCH(CF3)2}5], generated in the reaction medium, played a key role in promoting oxidative coupling. Nevertheless, the formation of carbazoles was limited to a yield of only 10%, and the synthesis of other five-membered rings remained challenging, with yields reaching up to 31%. Herein, we report an electrochemical method for carbazole synthesis carried out at room temperature, without the use of metal catalysts or external oxidants. This environmentally benign approach allows direct C–C bond formation without the use of metal catalysts. Furthermore, this study offers valuable insights into the underlying reaction pathways for the selective formation of carbazoles.
Electrochemical oxidation of tris(3,4-dimethoxyphenyl)amine (A1) was initiated in a two-electrode setup under galvanostatic control. A detailed optimisation study is presented in Table S1 (SI). The optimal conditions were then applied to various tertiary amines to explore the homo-aryl coupling reaction scope (Fig. 2). Highly electron-rich substrates like A1 can undergo oxidation readily, affording the desired carbazole C1 in 85% isolated yield. Replacing one of the OMe groups by a Me group as in C2 or the 3,4-dimethoxyphenyl moiety by phenyl (C3) decreased the yield substantially. Replacing the electron-rich aryl rings of the tertiary aryl amine with a p-halo-substituted phenyl group did not significantly impact the product yields, affording carbazoles C4–C6 in 61–89% yields. Notably, no dehalogenation or direct arylation of products was observed, which is one of the possible side reactions in metal-catalysed coupling reactions. Functional groups such as –CN (C7), –CHO (C8) and –CO2Me (C9) were unaffected under the electrooxidation conditions and provided the coupling products in good to excellent yields (61–84%). Notably, the stepwise replacement of the 3,4-dimethoxyphenyl groups with unsubstituted phenyl rings had a marked effect on the yields of C3 (63%) and C11 (0%). The optimised methodology appears to perform well with electron-rich substrates, and we believe there is significant potential to extend its applicability to electron-deficient substrates by fine-tuning the electrochemical conditions and by suppressing side reactions.
 | ||
Fig. 2 Efficiency of electrochemical dehydrogenative coupling of tertiary amines. Isolated yields are reported. Refer to the SI for the synthesis of starting materials (A1–A11) and the corresponding products (C1–C11) and analytical data. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) In the presence of 10 mol% of ferrocene as a mediator. bPossible formation of the dimerization product (C11-dimer, 38%) was observed (cf. SI, Section S6). In the presence of 10 mol% of ferrocene as a mediator. bPossible formation of the dimerization product (C11-dimer, 38%) was observed (cf. SI, Section S6). | ||
To understand the reactivity differences amongst substrates, cyclic voltammetry (CV) examinations were performed on A1, A3, and A11 under the same reaction conditions using a potentiostat (Gamry Reference 600+). Three oxidation and reduction event pairs were detected, respectively, on A1 upon the first anodic and return cathodic scans (Fig. 3a). The O1/R1 pair displayed a relatively symmetrical peak area ratio of ∼0.91 and a moderate peak-to-peak separation (ΔEp) of ∼157 mV, indicative of a (quasi)reversible single electron transfer process (Er).36 Although the ΔEp is wider than the theoretical value of 60 mV, it is comparable to that of a Fc/Fc+ reference (ΔEp ≈ 120 mV, Fig. S2a), suggesting a significant influence of iR drop or passivation that widens ΔEp. O2/R2 shows a more irreversible single electron transfer behaviour (Ei) with a larger peak area ratio of ∼1.26 and a wider ΔEp of ∼278 mV. Distinct from the rest, the O3/R3 pair is unsymmetrical, with a very large area ratio of >30 and a ΔEp of ∼257 mV, indicative of a two-step reaction consisting of irreversible electron transfer followed by an irreversible chemical reaction (Ei–Ci). As the oxidative coupling process of A1 to yield C1 is expected to involve 2 e− transfer, we hypothesise that the relevant coupling process is reflected by O1 and O2, while O3 may be linked to overoxidation that could lead to possible side-reactions decreasing the efficiency of the coupling reaction.37 Our position is supported by chronopotentiometric measurements of A1 at 10 and 15 mA (5.6 and 8.5 mA cm−2, respectively, Fig. S4), where a poor C1 yield (12%) at 10 mA applied current (Table S1, entry 5) can be rationalised to the insufficient voltage (∼0.7 V max) to reach the O2 oxidation event.
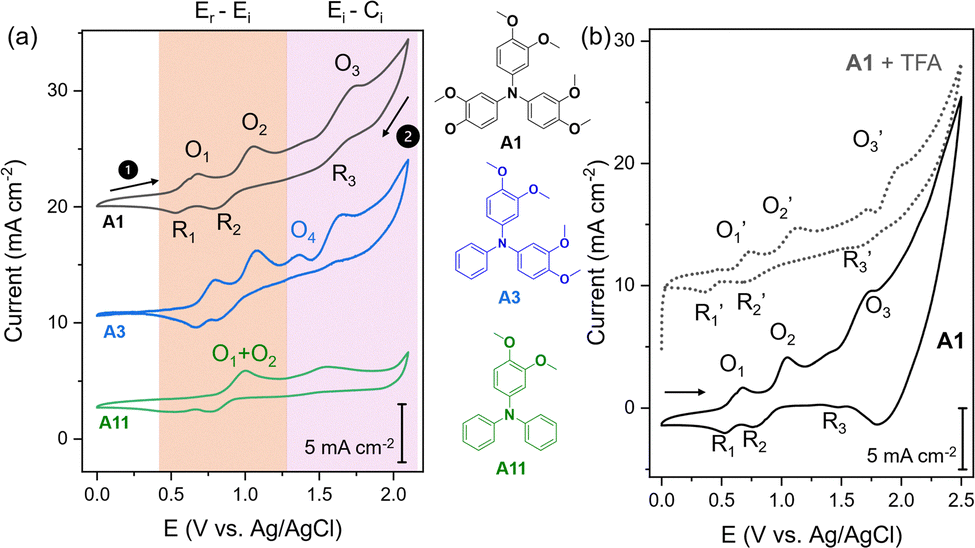 | ||
| Fig. 3 Cyclic voltammetry (CV) was performed on a mirror polished glassy carbon working electrode (∼0.071 cm2), a platinum counter electrode (∼2 cm2), and a leakless Ag/AgCl reference electrode under the optimised reaction conditions described in Fig. 2 but with a larger total volume of 10 mL. (a) Cyclic voltammetry (CV) of A1, A3 and A11 at a scan rate of 10 mV s−1 vs. Ag/AgCl. (b) CV of A1 before (solid line) and after TEA addition (dotted lines). | ||
Comparing the redox behaviour of different substrates, O1 and O2 peaks are 12–16 mV more anodic on A3 (Fig. 3a), suggesting that the oxidative coupling of the latter is more arduous. A3 also displays similar O3 potential to A1. However, a new irreversible oxidation peak, O4, was detected at 1.46 V, earlier than O3. This suggests that A3 may have additional overoxidation mechanisms that can adversely affect the C3 yield (63%). The redox behaviour of A11 (Fig. 3a) is rather different, with only two oxidation events, O5 and O3, observed in the potential range of CV. As reduction events R1 and R2 are still present around 0.50 and 0.76 V, we deduced that reduction event O5 may represent merged O1 and O2 components. The position of the O5 peak is at least 320 mV more anodic compared to O1 in A1, suggesting that the oxidative coupling process on A11 is more challenging. At the same time, the O3 peak appears 250 mV earlier compared to A1, suggesting more facile overoxidation. We believe that the combination of these factors might lead to the formation of a possible dimeric product, C11-dimer (cf. Section S6, SI), rather than the expected C11.
Next, the role of the TFA additive can be elucidated by comparing the CV of A1 with and without TFA (Fig. 3b). With TFA addition, the most notable change in the redox behaviour of A1 is the anodic shift of the O3 peak to  from 1.76 to 1.96 V, while the positions of
from 1.76 to 1.96 V, while the positions of  and
and 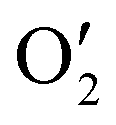 are relatively unchanged from O1 and O2, barring a 4–6 mV shift. Thus, we posit that the TFA addition does not actually make the oxidative coupling of A1 easier, but rather it makes the over-oxidation event more difficult through protonation of the central N atom. Thus, a more stable A1 (or C1) would allow the current controlled coupling process to proceed with a wider electrochemical stability window. This hypothesis is corroborated by significantly (∼5.9×) steeper yield growth compared to conversion growth with more TFA added (Fig. S4, SI). A relatively strong and electrochemically stable acid is required for this scheme, as our attempt to substitute TFA with acetic acid (Table S1, entry 10) did not improve the C1 yield. On the other hand, more acidic methanesulfonic acid (Table S1, entry 8) appears to improve the C1 yield slightly but not as good as TFA, possibly because of the better oxidation stability of TFA.38
are relatively unchanged from O1 and O2, barring a 4–6 mV shift. Thus, we posit that the TFA addition does not actually make the oxidative coupling of A1 easier, but rather it makes the over-oxidation event more difficult through protonation of the central N atom. Thus, a more stable A1 (or C1) would allow the current controlled coupling process to proceed with a wider electrochemical stability window. This hypothesis is corroborated by significantly (∼5.9×) steeper yield growth compared to conversion growth with more TFA added (Fig. S4, SI). A relatively strong and electrochemically stable acid is required for this scheme, as our attempt to substitute TFA with acetic acid (Table S1, entry 10) did not improve the C1 yield. On the other hand, more acidic methanesulfonic acid (Table S1, entry 8) appears to improve the C1 yield slightly but not as good as TFA, possibly because of the better oxidation stability of TFA.38
In the electrochemical oxidation process, we observed the favourable formation of the C6–C6’ coupling products of carbazole. A plausible reaction pathway for the formation of carbazole product C1 is proposed in Fig. S9. To gain insight into the electronic and structural properties underlying the electrochemical dehydrogenative C–C coupling reaction, we employed density functional theory (DFT) calculations. These calculations focused on the relative energies of the isomeric forms of the products and the electronic structure of potential reaction intermediates. The Gibbs free energies of formation were calculated for the three possible oxidation products obtained through coupling at the C2–C2′, C2–C6′, and C6–C6′ carbon atoms of A1 (Fig. 4a). A series of possible conformers for each product were identified based on the relative orientation of the methoxy groups with respect to their attached phenyl rings. The DFT analysis reveals that, among the various conformations, the lowest-energy structure corresponds to the C6–C6′ coupled product, which is thermodynamically more stable by 0.5 kcal mol−1 compared to the C2–C6′ product and by 6.0 kcal mol−1 compared to the C2–C2′ product. These data suggest that the selectivity of the reaction is determined by the low kinetic barrier to form the experimentally observed C6–C6′ product rather than thermodynamics.
 | ||
| Fig. 4 DFT calculations were carried out using Gaussian 16 (Revision C.02). Geometry optimisation was performed using the ωB97X-D functional including geometrical dispersion corrections described by Grimme's D2 correction. (a) Structures of three different optimised isomers of C1 (from left to right, C6–C6′, C2–C2′, and C2–C6′). (b) Spin density distribution of 2[A1-H]2+. | ||
To gain further insight into the electronic properties of potential reaction intermediates, the structures of the radical cation 2[A1]+ and its protonated analogue 2[A1-H]2+ were calculated defining each as a doublet spin state (S = 1/2). Given the experimental reaction conditions and high concentration of TFA, it is likely that the substrate exists in its protonated form in solution and 2[A1-H]2+ is a plausible intermediate following electrochemical oxidation of [A1-H]+. Examination of the spin-density plot of 2[A1-H]2+ (Fig. 4b) shows that the unpaired electron's spin is confined within just one of the aryl rings. While delocalised, there is spin-density on the C6 position, suggesting that this site should possess radical character and thereby supporting the experimentally observed site of reactivity.
In summary, this study presents an electrochemical approach for synthesising carbazoles via intramolecular dehydrogenative aryl–aryl coupling of arylamines. The methodology offers significant advantages in terms of atom economy, eliminating the need for precious metal catalysts, stoichiometric oxidants, or pre-functionalised starting materials. Trifluoroacetic acid (TFA) was identified as a critical additive, increasing the yield of C1 from 29% to 87% by expanding the electrochemical stability window and suppressing undesired overoxidation. The method afforded good to excellent yields (61–90%) across a range of electron-rich and para-halo-substituted substrates, whereas less electron-donating groups led to a marked decrease in efficiency. The reaction exhibited complete regioselectivity, consistently yielding the C6–C6' coupled product. DFT calculations supported the experimental observations, showing the observed product is the thermodynamically most stable isomer and is also likely kinetically favoured based on the electronic structure of the possible radical intermediate. Overall, this work establishes a practical and mechanistically informed method for carbazole synthesis and contributes to the advancement of methodologies in electro-organic synthesis. We are currently focusing on expanding the new methodology for the construction of oxygen- and sulphur-containing heterocycles, as well as the synthesis of carbazole-containing natural products.
This work was funded by the Horizontal Technology Coordinating Office (HTCO), Agency for Science, Technology and Research (A*STAR), Singapore, under project number C231218004. B. R. acknowledges the Materials Generative Design and Testing Framework (MAT-GDT) Program at A*STAR (Grant No. M24N4b0034) for the support.
Conflicts of interest
There are no conflicts to declare.Data availability
Detailed electrochemical studies, DFT calculations and NMR spectral data for this article have been included as part of the SI. See DOI: https://doi.org/10.1039/d5cc03841cNotes and references
- J. L. Röckl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef PubMed.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef PubMed.
- L. A. T. Allen and P. Natho, Org. Biomol. Chem., 2023, 21, 8956–8974 RSC.
- A. Caruso, J. Ceramella, D. Iacopetta, C. Saturnino, M. V. Mauro, R. Bruno, S. Aquaro and M. S. Sinicropi, Molecules, 2019, 24, 1912 CrossRef PubMed.
- Y. Liu, Y. Sun, M. Li, H. Feng, W. Ni, H. Zhang, X. Wan and Y. Chen, RSC Adv., 2018, 8, 4867–4871 RSC.
- M. Li, W. Ni, H. Feng, X. Wan, Y. Liu, Y. Zuo, B. Kan, Q. Zhang and Y. Chen, Org. Electron., 2015, 24, 89–95 CrossRef.
- X.-D. Yuan, J. Liang, Y.-C. He, Q. Li, C. Zhong, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2014, 2, 6387 RSC.
- D. Magaldi, M. Ulfa, S. Péralta, F. Goubard, T. Pauporté and T.-T. Bui, J. Mater. Sci.: Mater. Electron., 2021, 32, 12856–12861 CrossRef CAS.
- A. B. Dapkekar, J. Naveen and G. Satyanarayana, Asian J. Org. Chem., 2025, 14, e202500324 CrossRef CAS.
- B. Dahms, P. J. Kohlpaintner, A. Wiebe, R. Breinbauer, D. Schollmeyer and S. R. Waldvogel, Chem. – Eur. J., 2019, 25, 2713–2716 CrossRef CAS PubMed.
- B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2014, 53, 5210–5213 CrossRef CAS PubMed.
- B. Elsler, A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Chem. – Eur. J., 2015, 21, 12321–12325 CrossRef CAS PubMed.
- L. Schulz, M. Enders, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 4877–4881 CrossRef CAS PubMed.
- A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 14727–14731 CrossRef CAS PubMed.
- S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 13325–13329 CrossRef CAS PubMed.
- S. Lips, B. A. Frontana-Uribe, M. Dörr, D. Schollmeyer, R. Franke and S. R. Waldvogel, Chem. – Eur. J., 2018, 24, 6057–6061 CrossRef CAS PubMed.
- D.-Y. Goo and S. K. Woo, Org. Biomol. Chem., 2016, 14, 122–130 RSC.
- T. Watanabe, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 4720–4726 CrossRef CAS PubMed.
- D. J. St. Jean, S. F. Poon and J. L. Schwarzbach, Org. Lett., 2007, 9, 4893–4896 CrossRef PubMed.
- J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef CAS PubMed.
- W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560–14561 CrossRef CAS PubMed.
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed.
- W. C. P. Tsang, R. H. Munday, G. Brasche, N. Zheng and S. L. Buchwald, J. Org. Chem., 2008, 73, 7603–7610 CrossRef CAS PubMed.
- A. Kehl, N. Schupp, V. M. Breising, D. Schollmeyer and S. R. Waldvogel, Chem. – Eur. J., 2020, 26, 15847–15851 CrossRef CAS PubMed.
- R. B. Bedford and C. S. J. Cazin, Chem. Commun., 2002, 2310–2311. RSC.
- K. K. Gruner and H.-J. Knölker, Org. Biomol. Chem., 2008, 6, 3902–3904 RSC.
- D. Tsvelikhovsky and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14048–14051 CrossRef CAS PubMed.
- A. Khan, R. Karim, H. Dhimane and S. Alam, ChemistrySelect, 2019, 4, 6598–6605 CrossRef CAS.
- Y. Tanji, N. Mitsutake, T. Fujihara and Y. Tsuji, Angew. Chem., Int. Ed., 2018, 57, 10314–10317 CrossRef CAS PubMed.
- L.-C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581–590 CrossRef CAS PubMed.
- G. Brufani, S. Chen, B. Di Erasmo, A. Afanasenko, Y. Gu, C.-J. Li and L. Vaccaro, ACS Sustainable Chem. Eng., 2024, 12, 8562–8572 CrossRef CAS.
- S. Trosien, P. Böttger and S. R. Waldvogel, Org. Lett., 2014, 16, 402–405 CrossRef CAS PubMed.
- S. Parisien-Collette, A. C. Hernandez-Perez and S. K. Collins, Org. Lett., 2016, 18, 4994–4997 CrossRef CAS PubMed.
- A. C. Hernandez-Perez and S. K. Collins, Angew. Chem., Int. Ed., 2013, 52, 12696–12700 CrossRef CAS PubMed.
- S. B. Beil, T. Müller, S. B. Sillart, P. Franzmann, A. Bomm, M. Holtkamp, U. Karst, W. Schade and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 2450–2454 CrossRef CAS PubMed.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef CAS.
- C. Depecker, H. Marzouk, S. P. Trevin and J. Devynck, New J. Chem., 1999, 23, 739–742 RSC.
| This journal is © The Royal Society of Chemistry 2025 |