
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpin polarized current in chiral organic radical monolayers†
Niccolò
Giaconi
 a,
Michela
Lupi
a,
Tapan Kumar
Das
b,
Anil
Kumar
b,
Lorenzo
Poggini
c,
Caterina
Viglianisi
a,
Lorenzo
Sorace
a,
Stefano
Menichetti
a,
Ron
Naaman
b,
Roberta
Sessoli
a and
Matteo
Mannini
*a
a,
Michela
Lupi
a,
Tapan Kumar
Das
b,
Anil
Kumar
b,
Lorenzo
Poggini
c,
Caterina
Viglianisi
a,
Lorenzo
Sorace
a,
Stefano
Menichetti
a,
Ron
Naaman
b,
Roberta
Sessoli
a and
Matteo
Mannini
*a
aDepartment of Chemistry “Ugo Schiff” (DICUS) & INSTM Research Unit, University of Florence, Sesto Fiorentino 50019, Italy. E-mail: matteo.mannini@unifi.it
bDepartment of Chemical and Biological Physics, Weizmann Institute of Science, Rehovot 76100, Israel
cIstituto di Chimica dei Composti Organo-Metallici (ICCOM-CNR), Sesto Fiorentino 50019, Italy
First published on 3rd June 2024
Abstract
The chirality-induced spin selectivity (CISS) effect is the capability of chiral molecules to act as spin filters, i.e. to selectively sort flowing electrons based on their spin states. The application of this captivating phenomenon holds great promise in the realm of molecular spintronics, where the primary focus lies in advancing technologies based on chiral molecules to regulate the injection and coherence of spin-polarized currents. In this context, we conducted a study to explore the spin filtering capabilities of a monolayer of the thia-bridged triarylamine hetero[4]helicene radical cation chemisorbed on a metallic surface. Magnetic–conductive atomic force microscopy revealed efficient electron spin filtering at exceptionally low potentials. Furthermore, we constructed a spintronic device by incorporating a monolayer of these molecules in between two electrodes, obtaining an asymmetric magnetoresistance trend with signal inversion in accordance with the handedness of the enantiomer involved, indicative of the presence of the CISS effect. Our findings underscore the significance of thia[4]azahelicene organic radicals as promising candidates for the development of quantum information operations based on the CISS effect as a tool to control the molecular spin states.
Introduction
Spintronics seeks to use the spin as an additional degree of freedom,1,2 in contrast to molecular and traditional electronics that rely solely on the electron's charge. The majority of materials employed nowadays as spin injectors or detectors are inorganic.3 Organic materials were previously used only as spin transport channels due to their comparatively low spin injection efficiency induced by the weak spin–orbit coupling.4 The discovery of efficient spin filtering of electrons transmitted through chiral organic molecules has changed this scenario. The chirality induced spin selectivity (CISS) effect5 has been observed using many techniques adopting different molecular–inorganic hybrid architectures. Most experiments are based on the measurement of magnetoresistance in spin valve-like devices.6,7 Very recently, CISS has also been observed in solution on isolated molecules constituted by donor–acceptor dyads linked by a chiral bridge.8 The electron transfer has been promoted by photoexcitation and the spin polarization of the transferred electron monitored by time-resolved electron paramagnetic resonance (EPR) spectroscopy.9–11 This seminal result opens the perspective of using CISS for spin-qubit initialization and readout with small error even at moderate temperatures.12Dealing with hybrid devices, many types of molecules have been adopted as chiral units to promote the CISS effect, ranging from molecules with stereogenic centres, such as DNA and oligopeptides, to supramolecular or sterically hindered structures.11,13–19 Among the latter, helicenes are very efficient building blocks for the development of chiral molecule-based spintronic devices, reaching high spin polarization values.20 Furthermore, additional functions can be achieved by inserting heteroatoms in the helicene scaffold. For instance, some of us studied molecules belonging to the class of thia[4]azahelicenes. Interestingly, they have a high energy barrier for racemization (131.9–132.6 kJ mol−1)21 similar to that of carbo[5]helicene, that prevents racemization in solution and on the surface.22 A molecular monolayer of neutral thia[4]azahelicenes provided spin polarization at room temperature above 60%, also detectable at unusually low voltages.23 Additionally, these molecules undergo a reversible one-electron oxidation providing a stable radical cation where the spin density is localized on the nitrogen atom.24
Paramagnetic chiral molecules were previously employed in the detection of CISS, resulting in a small but sizeable increase in the spin filtering.25 However, the paramagnetic centre was not directly involved in the chiral electron transmission. The possibility of exploiting the CISS effect to control and initialize the spin state of an unpaired electron would open significant perspectives in terms of manipulating the spin state of magnetic molecules for quantum information operations.12 Therefore, given the interest in harnessing CISS effects to control the spin of species that can act as molecular qubits,12,26 we investigated here the spin filtering properties of the thia[4]azahelicene radical cation species (3,7,11-trimethylbenzo[5,6][1,4]thiazino[2,3,4-kl]phenothiazin-2-yl 16-(acetylthio) hexadecanoate hexafluoroantimonate radical cation, from here on RadESAc), whose structure is reported in Scheme 1. For this purpose, we functionalized the two enantiomers with an aliphatic thioacetyl tether and deposited a monolayer on a gold surface. X-Ray photoelectron spectroscopy (XPS) and electron paramagnetic resonance (EPR) confirmed the retention of the chemical and paramagnetic properties after the deposition process. Finally, the CISS effect was investigated and quantified by collecting I/V curves using magnetic conductive atomic force microscopy (mc-AFM) and by assembling micrometric devices comprising an enantiopure monolayer for magnetoresistance measurements. Spin selectivity induced by the presence of chiral radical cationic molecules was detected by comparing the response of the two enantiomers when the magnetic field is reversed. The local probe of mc-AFM showed a spin polarization up to 60% at room temperature, whereas MR measurements on large area devices evidenced the occurrence of the CISS effect with an asymmetric conductance between 1% and 2% with opposite sign according to the handedness of the helicene. These values are comparable to those observed for the parent neutral species.23 As the spin density of the radical mainly resides on the tensioned nitrogen atom, thia[4]azahelicenes appear as a promising platform to investigate CISS-based manipulation of radical spins.
 | ||
| Scheme 1 Scheme of the oxidation synthetic step to obtain enantiomer (P)-RadESAc. | ||
Experimental
Instruments
XPS measurements were performed using a microfocused monochromatic Al Kα radiation source (1486.6 eV, model SPECS XR-MS Focus 600) and a multichannel detector electron analyser (model SPECS Phoibos 150 1DLD) with a pass energy of 40 eV to ensure an appropriate resolution. Spectra were acquired in normal emission geometry with the X-ray source mounted at 54.44° with respect to the analyser. Spectra were calibrated by rescaling the binding energy value to the Au 4f7/2 peak at 84 eV.27 Fitting analysis was performed using CasaXPS software introducing mixed Gaussian and Lorentzian contributions for each component. The background was fitted using the Shirley or the linear method. The bulk reference sample was prepared by dropcasting a 2 mM solution of molecules. The semiquantitative elemental analysis was performed by employing the cross-section values extracted from the literature.28 Continuous wave X-band (ν ≈ 9.40 GHz) EPR spectra of the sample were recorded on a Bruker Elexsys E500 spectrometer equipped with an SHQ cavity (see the ESI† for further EPR experimental details). Spectra were acquired at 30 K, which was found to be the best temperature in terms of maximization of the intensity of the signal. Low temperatures were achieved by using a ESR900 (Oxford Instruments) 4He continuous flow cryostat.The mc-AFM experiment was carried out using a multimode magnetic scanning probe microscopy (SPM) system equipped with a Beetle-type Ambient AFM setup and an electromagnet with R9 electronic controller (RHK Technology). I–V measurements were performed under a ±0.5 T magnetic field perpendicular to the sample surface at room temperature, applying voltage ramps between ±0.3 V with a Pt-coated tip (DPE-XSC11, μmasch) in contact mode (applied force ca. 8–10 nN). At least 150 curves were scanned for each point, and several points were investigated all over the surface for a proper statistical analysis.
Devices were fabricated by optical lithography, followed by e-beam evaporation. On a precleaned Si wafer, a 50 μm wide bottom electrode consisting of a Ti adlayer (8 nm) and Au (60 nm) were deposited by evaporation. The substrate was then cleaned by immersing it in boiling acetone for 10 minutes and in boiling ethanol for an additional 10 minutes. Finally, the substrate was kept under a UV/ozone atmosphere for 15 minutes. On top of the gold layer, a self-assembled monolayer of RadESAc was deposited following the procedure described in the following section. Finally, as the top electrode, the insulating buffer layer of MgO (2 nm), Ni (40 nm), and Au (20 nm) layers were evaporated using a shadow mask with a line width of 20 μm. The device was subsequently attached to a cryogenic chip carrier and electrically connected by a wire bonder (Au wires). All electrical measurements were performed within the cryogenics system made by Cryogenics, Ltd. A magnetic field of up to 1 T was applied perpendicular to the sample plane, and the resistance of the device was measured using the standard four-probe method. A constant current of 0.5 mA was applied using a Keithley current source (model 2400), and the voltage across the junction was measured using a Keithley nanovoltmeter (model 2182A).
Synthesis of RadESAc
The diamagnetic precursor NeuSAc was obtained in the enantiopure form by using a synthetic procedure previously reported in the literature followed by HPLC.23 An oxidation reaction was carried out on each enantiomer by using AgSbF6 as the oxidizing agent to obtain the corresponding radical cation (Scheme 1); further details about the synthesis procedure can be found in the ESI†.Monolayer deposition
A polycrystalline gold film deposited on different substrates and cleaned with specific protocols depending on the substrate (see the ESI† for more details), was incubated overnight in a diluted solution of RadESAc to induce the formation of a well-ordered layer. At the end of the incubation time the substrate was vigorously rinsed with pure solvent and dried under a nitrogen atmosphere to ensure the removal of the molecules physisorbed on the surface.Results and discussion
Deposition and characterization of the RadESAc monolayer
The procedure adopted to deposit a monolayer of RadESAc on the surface (RadESAc@Au) provided the formation of covalent bonds between the deprotected terminal sulfur atoms and the gold substrate. To confirm the correct assembly of the monolayer on the surface we performed an XPS characterization using as a reference for the starting bulk radical a thick film (ca. 100 nm) prepared via drop casting on a gold surface. S2p, C1s, F1s and N1s regions have been investigated to obtain qualitative information as well as a semi-quantitative analysis of the chemical composition of the molecular deposit.The S2p region is fundamental for confirming the formation of a covalent bond between the sulfur atoms of the thioacetyl group of RadESAc and the gold surface. As expected, in the bulk sample just one component (163.2 eV) and its relative spin–orbit coupled one are observed (Fig. 1) as the sulfur atoms of the helicene structure and that of the free thioacetyl group are characterized by comparable binding energy. In contrast, in RadESAc@Au an additional contribution is detected at a lower binding energy (161.8 eV), demonstrating S–Au bond formation (Fig. 1).29 Furthermore, the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio observed between these two components is consistent with the stoichiometric ratio between the two sulfur atoms in the helicene structure and the thioacetyl one, bound to gold. The C1s region of the RadESAc bulk sample features four different components, as reported in Fig. S2 (ESI†): a main component at 283.9 eV, a second one at 285.1 eV, and two additional minor components at higher binding energies. The main signal can be attributed to C–C/C
:1 ratio observed between these two components is consistent with the stoichiometric ratio between the two sulfur atoms in the helicene structure and the thioacetyl one, bound to gold. The C1s region of the RadESAc bulk sample features four different components, as reported in Fig. S2 (ESI†): a main component at 283.9 eV, a second one at 285.1 eV, and two additional minor components at higher binding energies. The main signal can be attributed to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C atoms of the helicene,30 whereas the second one is assigned to carbons of C–N/C–S groups.31 The minor components are due to the presence of C–O/CO functional groups that might come from environmental contamination,32 since samples were exposed to the air during the preparation. The spectrum acquired on the monolayer is also characterized by four components that are comparable to those observed in the bulk phase. Indeed, there is a dominant component at 284.1 eV, plus three additional signals at 285.1 eV, 286.2 eV, and 288.3 eV, which are attributable to the same species described above. The F1s spectrum of the bulk sample shows one single component at 685 eV attributable to fluorine atoms of SbF6− anions. The same signal is observed in the spectrum acquired on the monolayer sample, which thus confirms the presence of the counterion after the deposition process (Fig. S3, ESI†). The N1s XPS region is the most sensitive to the radical moiety since the spin density of the unpaired electron lies mainly on the nitrogen atom of the helicene.24 Unfortunately, these spectra do not allow assessing the preservation of the radical functionality because of the radiation damage occurring when organic radicals are exposed to X-rays. Indeed, under X-rays exposure molecules undergo a photoinduced reduction process (Fig. S4 and S5, ESI†), a phenomenon already observed in other monolayer composed of molecules with the spin density located on nitrogen atoms.22,33,34 Finally, we carried out a semi-quantitative analysis on the investigated spectral regions and obtained an estimation of the stoichiometry of the molecules in the bulk sample and in the one assembled on the surface (see Table S1, ESI†). Considering the experimental error of the XPS technique, this semiquantitative analysis supported that intact molecules are deposited and that a monolayer of chemically bound molecules is formed. An EPR characterization was performed on the bulk sample as well as on the monolayer of RadESAc to assess if the radical nature is retained after the deposition process and the formation of covalent bonds between molecules and the surface. In Fig. 2, the EPR spectrum acquired at room temperature on a powder sample of RadESAc is reported. It features a single, partially structured absorption signal attributable to a S = 1/2 paramagnetic system with incomplete anisotropic averaging. Despite the signal being close to the sensitivity limit of the technique (ca. 1013 spins/G), hundreds of acquisitions at 30 K on RadESAc@Au revealed a signal whose intensity is consistent with a monolayer sample. In particular, the Landé g factor of the observed resonance (g = 2.0071) is comparable to that of the powder and of the previous reports (g = 2.0072). We stress here that the corresponding solution spectrum (CH2Cl2/toluene, Fig. S6, ESI†) exhibits a completely different pattern. Indeed, the hyperfine structure due to the interaction between the unpaired electron and nuclear spins of nitrogen and hydrogen atoms is clearly visible, and could be simulated with the parameters reported in Table S2 (ESI†) using the software EasySpin.35 These values are consistent with those already reported in the literature for the pristine helicene radical cation.24 In this respect, the absence in the RadESAc@Au spectrum of the hyperfine structure observed in solution and of the residual anisotropy observed in the powder suggests a highly packed structure promoting relevant intermolecular interactions. As a whole, EPR confirms the persistence of paramagnetism at the monolayer level.
C atoms of the helicene,30 whereas the second one is assigned to carbons of C–N/C–S groups.31 The minor components are due to the presence of C–O/CO functional groups that might come from environmental contamination,32 since samples were exposed to the air during the preparation. The spectrum acquired on the monolayer is also characterized by four components that are comparable to those observed in the bulk phase. Indeed, there is a dominant component at 284.1 eV, plus three additional signals at 285.1 eV, 286.2 eV, and 288.3 eV, which are attributable to the same species described above. The F1s spectrum of the bulk sample shows one single component at 685 eV attributable to fluorine atoms of SbF6− anions. The same signal is observed in the spectrum acquired on the monolayer sample, which thus confirms the presence of the counterion after the deposition process (Fig. S3, ESI†). The N1s XPS region is the most sensitive to the radical moiety since the spin density of the unpaired electron lies mainly on the nitrogen atom of the helicene.24 Unfortunately, these spectra do not allow assessing the preservation of the radical functionality because of the radiation damage occurring when organic radicals are exposed to X-rays. Indeed, under X-rays exposure molecules undergo a photoinduced reduction process (Fig. S4 and S5, ESI†), a phenomenon already observed in other monolayer composed of molecules with the spin density located on nitrogen atoms.22,33,34 Finally, we carried out a semi-quantitative analysis on the investigated spectral regions and obtained an estimation of the stoichiometry of the molecules in the bulk sample and in the one assembled on the surface (see Table S1, ESI†). Considering the experimental error of the XPS technique, this semiquantitative analysis supported that intact molecules are deposited and that a monolayer of chemically bound molecules is formed. An EPR characterization was performed on the bulk sample as well as on the monolayer of RadESAc to assess if the radical nature is retained after the deposition process and the formation of covalent bonds between molecules and the surface. In Fig. 2, the EPR spectrum acquired at room temperature on a powder sample of RadESAc is reported. It features a single, partially structured absorption signal attributable to a S = 1/2 paramagnetic system with incomplete anisotropic averaging. Despite the signal being close to the sensitivity limit of the technique (ca. 1013 spins/G), hundreds of acquisitions at 30 K on RadESAc@Au revealed a signal whose intensity is consistent with a monolayer sample. In particular, the Landé g factor of the observed resonance (g = 2.0071) is comparable to that of the powder and of the previous reports (g = 2.0072). We stress here that the corresponding solution spectrum (CH2Cl2/toluene, Fig. S6, ESI†) exhibits a completely different pattern. Indeed, the hyperfine structure due to the interaction between the unpaired electron and nuclear spins of nitrogen and hydrogen atoms is clearly visible, and could be simulated with the parameters reported in Table S2 (ESI†) using the software EasySpin.35 These values are consistent with those already reported in the literature for the pristine helicene radical cation.24 In this respect, the absence in the RadESAc@Au spectrum of the hyperfine structure observed in solution and of the residual anisotropy observed in the powder suggests a highly packed structure promoting relevant intermolecular interactions. As a whole, EPR confirms the persistence of paramagnetism at the monolayer level.
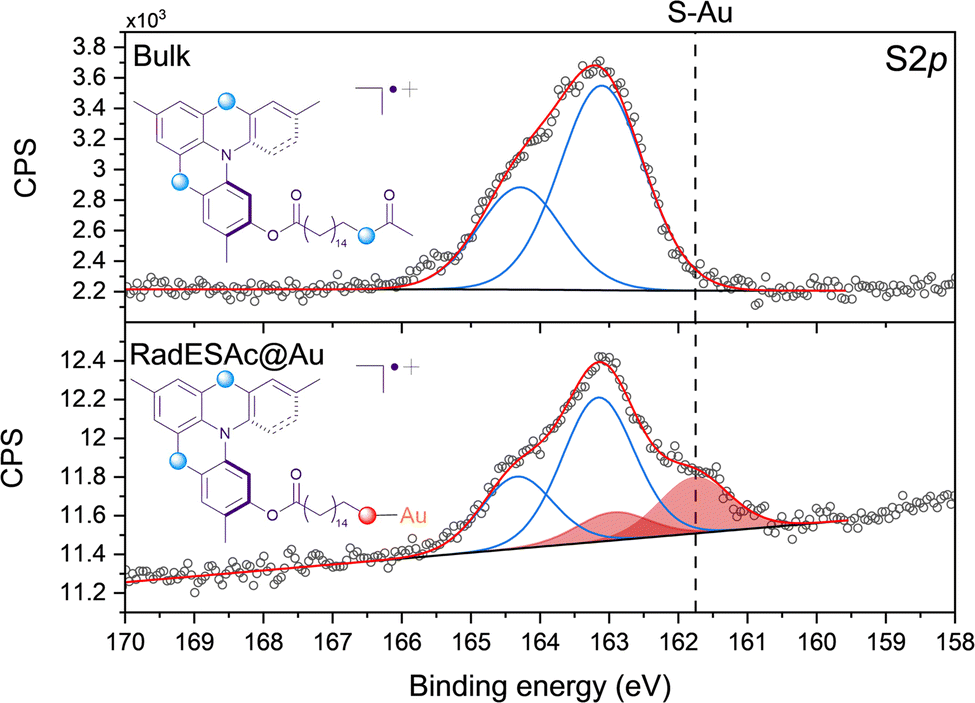 | ||
| Fig. 1 S2p XPS region acquired on the RadESAc bulk sample (top) and on the RadESAc@Au monolayer deposited on Au(111). The dashed line highlights the position of the component attributed to the formation of S–Au bond. | ||
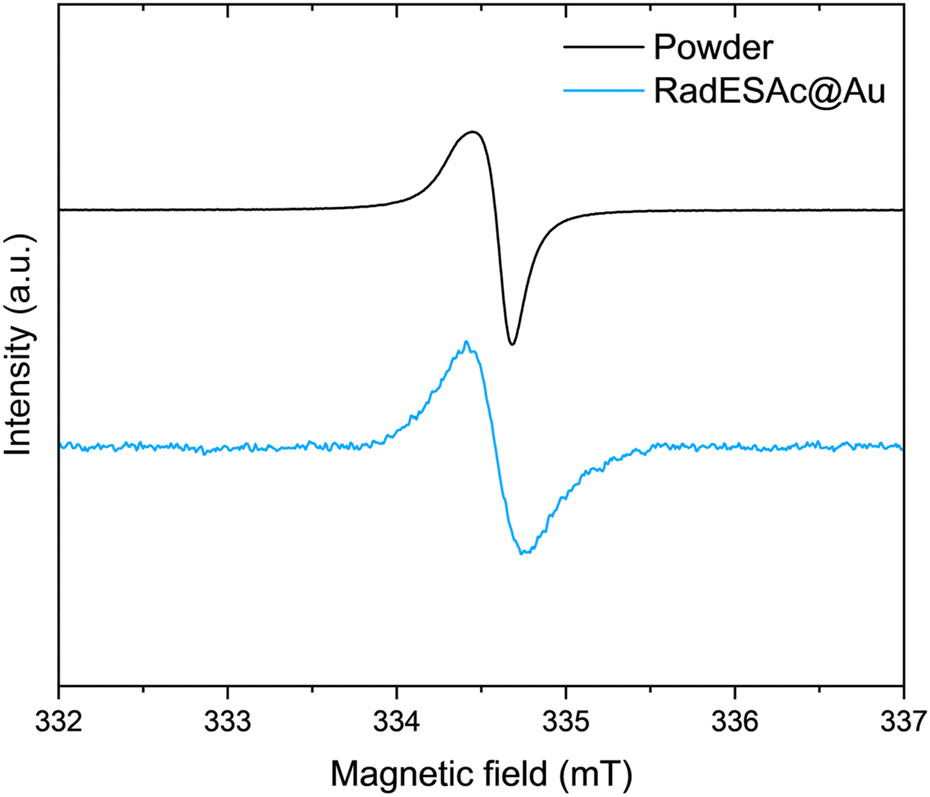 | ||
| Fig. 2 X-band (ν = 9.399030 GHz) EPR spectra acquired on RadESAc powder and on the RadESAc@Au monolayer assembled on gold at 30 K. | ||
mc-AFM measurements
The spin filtering properties of the molecular deposit were investigated by performing mc-AFM, i.e. by measuring the current between the tip and molecules anchored on a ferromagnetic substrate. For this purpose, an enantiopure monolayer of RadESAc was assembled on a Ni/Au surface following the procedure described above. Measurements were carried out by applying the voltage to the surface kept in contact with the tip, which is grounded. In Fig. 3a and b, the average of hundreds of I/V curves acquired on (P)-RadESAc@Au as well as on (M)-RadESAc@Au are reported applying either positive (up) or negative (down) magnetic field perpendicular to the ferromagnetic surface. The intensity of the current varies according to the direction of the magnetization of the Ni substrate, exhibiting the opposite behaviour for the two handednesses of the helicene molecule. This demonstrates that the flowing of electrons injected in a specific spin state is favoured for one handedness over the other one, and thus the occurrence of a spin-filtering process induced by the chirality of the molecules of the monolayer. The CISS effect is detectable for very weak applied voltages, similarly to what was observed in the corresponding neutral species.23 It is indeed possible to appreciate a spin selective process already within a ±0.3 V range, paralleling recent observations in supramolecular assemblies,36 while reported small molecules exhibit CISS for applied voltages above at least ±1.5 V.19,37,38 The “zero current” values are shifted slightly by about 40 mV to a negative potential due to the dipole moment of the adsorbed molecules. As already observed, the deposition of organic molecular layer induces a modification in the contact potential difference39 that can be appreciated, among all, by using a local probe such as an AFM tip, confirming the molecular nature of the junction. The spin polarization percentage was calculated for the whole applied voltage range using the equation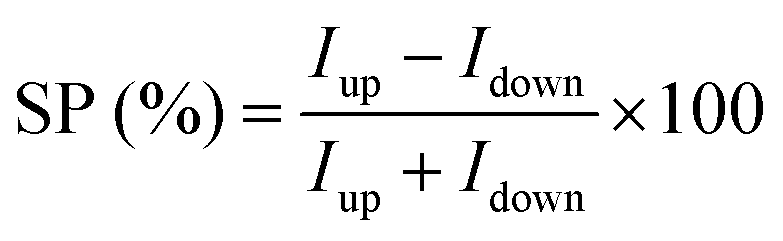 , where Iup and Idown are the intensities of the current measured applying a positive and a negative magnetic field, respectively. Both enantiomers reach a spin polarization of up to 60% at room temperature (Fig. 3c) but with the opposite sign, which is comparable to that of the previously investigated neutral species.23
, where Iup and Idown are the intensities of the current measured applying a positive and a negative magnetic field, respectively. Both enantiomers reach a spin polarization of up to 60% at room temperature (Fig. 3c) but with the opposite sign, which is comparable to that of the previously investigated neutral species.23
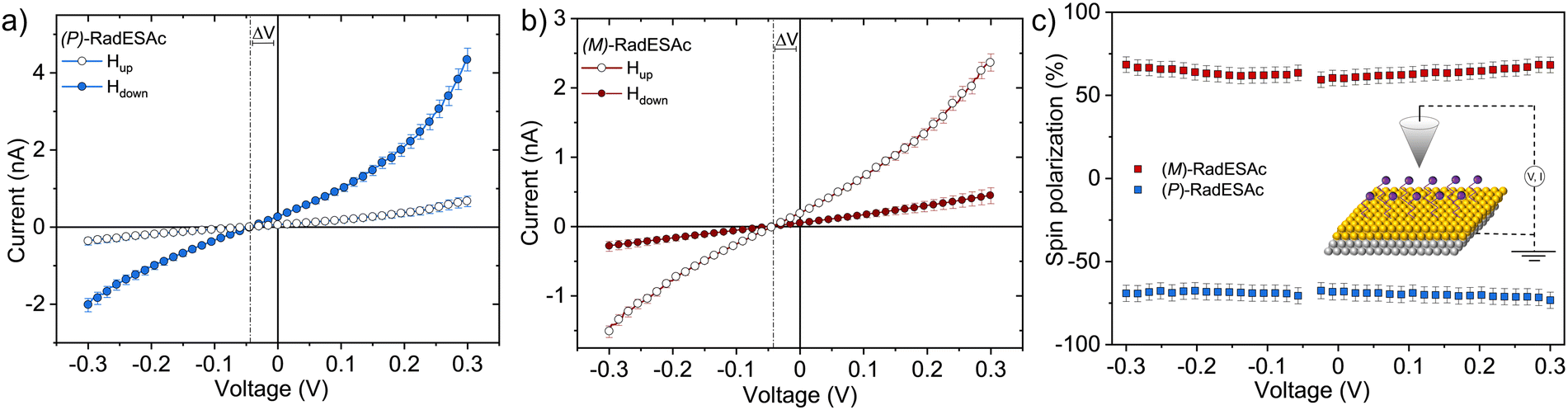 | ||
| Fig. 3 I/V curves acquired on (a) (P)-RadESAc@Au and on (b) (M)-RadESAc@Au applying either positive or negative magnetic field equal to ±0.5 T. Vertical dashed lines indicate the zero-current bias voltage. (c) Spin polarization percentage extracted from the corresponding I/V curves for both the enantiomers at room temperature. Error bars are presented for each point. Inset: Scheme of the experimental setup of mc-AFM. | ||
Magnetoresistance device
To gain further insight into the spin selectivity properties of RadESAc, a molecular-based spintronic device was assembled, embedding a monolayer deposit of enantiopure helicenes. A scheme of the micrometric device is reported in Fig. 4a. A monolayer of RadESAc was self-assembled on a gold electrode following the procedure described in the Experimental section. A thin insulating MgO layer was deposited between the monolayer and the ferromagnetic top electrode to avoid pinholes that result in Ni atoms penetrating the layer. Finally, an Au layer was added to prevent Ni oxidation. The magnetoresistance (MR) of the device was measured by a standard four-probe setup, by applying a constant current of 0.5 mA and sweeping the magnetic field in the range ±1 T, perpendicular to the vertical device. The field dependence of the resistance was measured at different temperatures, between 5 and 250 K. The values of magnetoresistance (Fig. 4b and c) were calculated as , where RB and R0 are the resistance measured in the presence of a magnetic field and at zero magnetic field, respectively (see Fig. S7, ESI†). In contrast to the classical organic vertical spin valve, where a symmetric signal with respect to the magnetic field is expected;40 in this case, an asymmetric trend of the MR occurred with an opposite behaviour for the two handednesses of the embedded molecules (Fig. 4b and c). This feature clearly indicates the occurrence of a spin selectivity process induced by the molecular layer. Finally, the temperature dependence of the MR was monitored detecting the typical behaviour of the CISS effect already confirmed in several systems,41i.e. an enhancement of the MR percentage with increasing temperature. Fig. 4d reports |ΔMR (%)| calculated as |MR (%)1T + MR (%)−1T|, where MR (%)1T and MR (%)−1T are the absolute values of the magnetoresistance percentage at +1 T and −1 T, respectively. An almost linear temperature dependence is observed for both enantiomers. To better compare the MR results with the spin-dependent conductivity measured by mc-AFM, we extracted the conductance asymmetry between the two different spin states following Julliere's model, as recently suggested by Weiss et al.42,43 In Fig. 5 we report
, where RB and R0 are the resistance measured in the presence of a magnetic field and at zero magnetic field, respectively (see Fig. S7, ESI†). In contrast to the classical organic vertical spin valve, where a symmetric signal with respect to the magnetic field is expected;40 in this case, an asymmetric trend of the MR occurred with an opposite behaviour for the two handednesses of the embedded molecules (Fig. 4b and c). This feature clearly indicates the occurrence of a spin selectivity process induced by the molecular layer. Finally, the temperature dependence of the MR was monitored detecting the typical behaviour of the CISS effect already confirmed in several systems,41i.e. an enhancement of the MR percentage with increasing temperature. Fig. 4d reports |ΔMR (%)| calculated as |MR (%)1T + MR (%)−1T|, where MR (%)1T and MR (%)−1T are the absolute values of the magnetoresistance percentage at +1 T and −1 T, respectively. An almost linear temperature dependence is observed for both enantiomers. To better compare the MR results with the spin-dependent conductivity measured by mc-AFM, we extracted the conductance asymmetry between the two different spin states following Julliere's model, as recently suggested by Weiss et al.42,43 In Fig. 5 we report 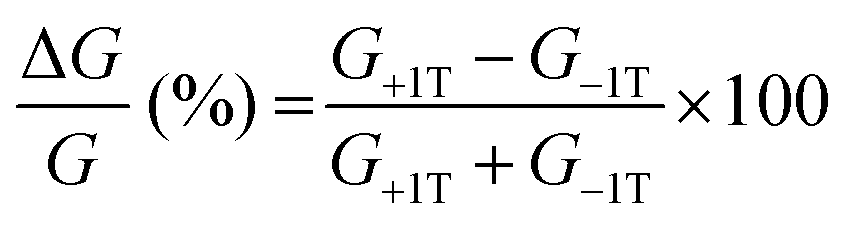 , where G+1T and G−1T is the conductance at a ±1 T applied magnetic field. The two enantiomers show the opposite sign of
, where G+1T and G−1T is the conductance at a ±1 T applied magnetic field. The two enantiomers show the opposite sign of 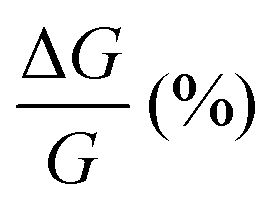 and similar temperature dependence. Despite the unambiguous occurrence of the CISS effect, the conductance asymmetry obtained in these devices is in the range of a few %, thus significantly lower than the spin polarization percentage measured with the mc-AFM setup. This difference can be attributed to the intrinsic nature of the measurements. Indeed, in the MR device both the electrons passing through the molecules and those flowing through SAM defects as pinholes or bare gold are collected. The latter are detrimental to the total yield of the MR percentage.23,44 In contrast, the local nature of mc-AFM measurements allows for evaluating the effective spin polarization percentage more accurately, avoiding part of the surface that presents bad-quality molecular deposits.
and similar temperature dependence. Despite the unambiguous occurrence of the CISS effect, the conductance asymmetry obtained in these devices is in the range of a few %, thus significantly lower than the spin polarization percentage measured with the mc-AFM setup. This difference can be attributed to the intrinsic nature of the measurements. Indeed, in the MR device both the electrons passing through the molecules and those flowing through SAM defects as pinholes or bare gold are collected. The latter are detrimental to the total yield of the MR percentage.23,44 In contrast, the local nature of mc-AFM measurements allows for evaluating the effective spin polarization percentage more accurately, avoiding part of the surface that presents bad-quality molecular deposits.
 | ||
| Fig. 4 (a) Scheme of the magnetoresistance device with magnification on the cross junction. Magnetoresistance percentage in the range ±1 T of (b) (P)-RadESAc@Au and of (c) (M)-RadESAc@Au monolayers. (d) |ΔMR (%)| values as a function of the temperature for both the enantiomers. | ||
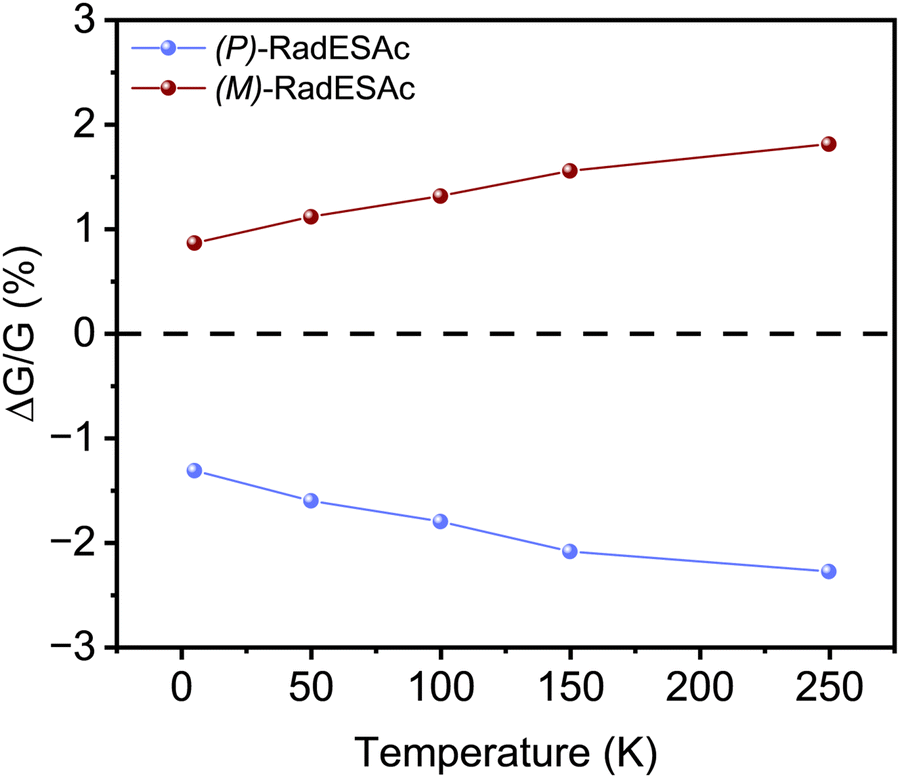 | ||
| Fig. 5 Conductance asymmetry values as a function of the temperature. | ||
We notice that (P)-RadESAc@Au shows slightly larger absolute values of both ΔMR (%) and  than (M)-RadESAc@Au. Although symmetry between the two enantiomers is expected in ideal devices, the observed difference might be due to the unequal quality of the molecular deposits. Finally, in Fig. S8 (ESI†) a comparison between the
than (M)-RadESAc@Au. Although symmetry between the two enantiomers is expected in ideal devices, the observed difference might be due to the unequal quality of the molecular deposits. Finally, in Fig. S8 (ESI†) a comparison between the  of RadESAc and the corresponding neutral species23 is reported. A slight enhancement of the conductance can be appreciated in both radical samples compared to the neutral ones. It is however not possible to unambiguously ascribe this increase to the presence of paramagnetic species on the surface. Indeed, we might also hypothesize that the higher percentage observed in the radical samples could be due to a different arrangement of the molecular deposit embedded in the device.
of RadESAc and the corresponding neutral species23 is reported. A slight enhancement of the conductance can be appreciated in both radical samples compared to the neutral ones. It is however not possible to unambiguously ascribe this increase to the presence of paramagnetic species on the surface. Indeed, we might also hypothesize that the higher percentage observed in the radical samples could be due to a different arrangement of the molecular deposit embedded in the device.
Conclusions
In this work, we successfully deposited a monolayer of enantiopure radicals of the hetero helicene family on a gold surface, as probed by using XPS and EPR spectroscopy. By using two complementary techniques, mc-AFM and MR, we proved the spin selective properties of a monolayer of enantiopure organic radicals assembled on a gold surface, evidencing a noticeable (60%) efficiency at room temperature and under a low voltage bias. The spin-selectivity is comparable, even higher at low temperatures, to that of the previously investigated neutral diamagnetic parent compound.23 A few examples of paramagnetic species exhibiting the CISS effect have already been reported in the literature25,45 and, to the best of our knowledge, this is the first observation of the CISS effect in a monolayer of an organic radical.The combination of the geometrical stability of the chiral skeleton imparted by the heteroatoms, the magnetic properties, and the good conductivity at a low voltage in a unique molecular system makes thia[4]azahelicenes an exceptional platform to be exploited for the development of molecule-based spintronic devices. Moreover, the spin density of the radical resides on the chiral scaffold without any detrimental impact on the spin polarization of the transmitted electrons, thus giving the possibility for exploiting the CISS effect as a control tool on the molecular spin state. The recent observation of the CISS effect in unsupported donor–acceptor dyads connected by a chiral linker suggests further exploring the chemistry of this class of molecules.8 Given the easy and reversible oxidation of the neutral form, thia[4]azahelicenes could act as a chiral donor to generate a spin-polarized chiral radical if properly connected to an acceptor. In addition, CISS is expected to affect the spin polarization in both charge transfer and recombination processes,9 thus showing great potential for quantum applications.12 Our observation of similar CISS efficiency in both neutral and radical species of thia[4]azahelicenes suggests them to be promising building blocks for exploring CISS at the molecular level.
Author contributions
N. G. – formal analysis, investigation (synthesis, XPS, EPR, mc-AFM, device), conceptualization, writing – original draft; M. L. investigation (synthesis); T. K. D. – investigation (device); A. K. – investigation (mc-AFM); L. P. – investigation (XPS), data discussion, writing – review & editing; C. V. – investigation (synthesis); L. S. – investigation (EPR), writing – review & editing; S. M. – supervision; R. N. – supervision; R. S. – conceptualization, funding acquisition, project administration, supervision, validation, writing – review & editing; M. M. – conceptualization, funding acquisition, project administration, supervision, validation, writing – review & editing.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
Funded by the European Union ERC-SyG CASTLe (No. 101071533). This work has also received funding under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 - Call for tender No. 341 funded by the European Union – NextGenerationEU, award number PE0000023, adopted by the Italian Ministry of University and Research (MUR), CUP D93C22000940001, project title “National Quantum Science and Technology Institute” (NQSTI). Finally, this work was funded also by MUR through PRIN Project 2017CR5WCH Q-chiSS “Quantum detection of chiral-induced spin selectivity at the molecular level” and through Dipartimenti di Eccellenza 2023–2027 (DICUS 2.0, CUP B97G22000740001) to the Department of Chemistry “Ugo Schiff” of the University of Florence as well as from Fondazione Cassa di Risparmio di Firenze for SPIN-E2 project (ref. 2020.1634). All authors acknowledge MatchLab Interdepartmental Research Unit staff of the Università degli Studi di Firenze.Notes and references
- I. Žutić, J. Fabian and S. Das Sarma, Rev. Mod. Phys., 2004, 76, 323–410 CrossRef.
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336 RSC.
- J. Camarero and E. Coronado, J. Mater. Chem., 2009, 19, 1678 RSC.
- D. Sun, E. Ehrenfreund and Z. Valy Vardeny, Chem. Commun., 2014, 50, 1781–1793 RSC.
- K. Ray, S. P. Ananthavel, D. H. Waldeck and R. Naaman, Science, 1999, 283, 814–816 CrossRef CAS PubMed.
- S.-H. Yang, R. Naaman, Y. Paltiel and S. S. P. Parkin, Nat. Rev. Phys., 2021, 3, 328–343 CrossRef.
- Z. Shang, T. Liu, Q. Yang, S. Cui, K. Xu, Y. Zhang, J. Deng, T. Zhai and X. Wang, Small, 2022, 18, 2203015 CrossRef CAS PubMed.
- H. J. Eckvahl, N. A. Tcyrulnikov, A. Chiesa, J. M. Bradley, R. M. Young, S. Carretta, M. D. Krzyaniak and M. R. Wasielewski, Science, 2023, 382, 197–201 CrossRef CAS PubMed.
- J. Luo and P. J. Hore, New J. Phys., 2021, 23, 043032 CrossRef CAS.
- A. Chiesa, M. Chizzini, E. Garlatti, E. Salvadori, F. Tacchino, P. Santini, I. Tavernelli, R. Bittl, M. Chiesa, R. Sessoli and S. Carretta, J. Phys. Chem. Lett., 2021, 12, 6341–6347 CrossRef CAS PubMed.
- A. Privitera, E. Macaluso, A. Chiesa, A. Gabbani, D. Faccio, D. Giuri, M. Briganti, N. Giaconi, F. Santanni, N. Jarmouni, L. Poggini, M. Mannini, M. Chiesa, C. Tomasini, F. Pineider, E. Salvadori, S. Carretta and R. Sessoli, Chem. Sci., 2022, 13, 12208–12218 RSC.
- A. Chiesa, A. Privitera, E. Macaluso, M. Mannini, R. Bittl, R. Naaman, M. R. Wasielewski, R. Sessoli and S. Carretta, Adv. Mater., 2023, 35, 2300472 CrossRef CAS PubMed.
- G.-F. Du, H.-H. Fu and R. Wu, Phys. Rev. B, 2020, 102, 035431 CrossRef CAS.
- P.-J. Hu, S.-X. Wang, X.-H. Gao, Y.-Y. Zhang, T.-F. Fang, A.-M. Guo and Q.-F. Sun, Phys. Rev. B, 2020, 102, 195406 CrossRef CAS.
- Q. Zhu, Y. Kapon, A. M. Fleming, S. Mishra, K. Santra, F. Tassinari, S. R. Cohen, T. K. Das, Y. Sang, D. K. Bhowmick, C. J. Burrows, Y. Paltiel and R. Naaman, Cell Rep. Phys. Sci., 2022, 3, 101157 CrossRef CAS.
- S. Mishra, A. K. Mondal, S. Pal, T. K. Das, E. Z. B. Smolinsky, G. Siligardi and R. Naaman, J. Phys. Chem. C, 2020, 124, 10776–10782 CrossRef CAS.
- V. Kiran, S. R. Cohen and R. Naaman, J. Chem. Phys., 2017, 146, 092302 CrossRef.
- H. Aizawa, T. Sato, S. Maki-Yonekura, K. Yonekura, K. Takaba, T. Hamaguchi, T. Minato and H. M. Yamamoto, Nat. Commun., 2023, 14, 4530 CrossRef CAS PubMed.
- R. Rodríguez, C. Naranjo, A. Kumar, K. Dhbaibi, P. Matozzo, F. Camerel, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, Chem. – Eur. J., 2023, 29, e202302254 CrossRef PubMed.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. Hernández Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957–1962 CrossRef CAS PubMed.
- G. Lamanna, C. Faggi, F. Gasparrini, A. Ciogli, C. Villani, P. J. Stephens, F. J. Devlin and S. Menichetti, Chem. – Eur. J., 2008, 14, 5747–5750 CrossRef CAS PubMed.
- N. Giaconi, A. L. Sorrentino, L. Poggini, M. Lupi, V. Polewczyk, G. Vinai, P. Torelli, A. Magnani, R. Sessoli, S. Menichetti, L. Sorace, C. Viglianisi and M. Mannini, Angew. Chem., Int. Ed., 2021, 60, 15276–15280 CrossRef CAS PubMed.
- N. Giaconi, L. Poggini, M. Lupi, M. Briganti, A. Kumar, T. K. Das, A. L. Sorrentino, C. Viglianisi, S. Menichetti, R. Naaman, R. Sessoli and M. Mannini, ACS Nano, 2023, 17, 15189–15198 CrossRef CAS PubMed.
- S. Menichetti, S. Cecchi, P. Procacci, M. Innocenti, L. Becucci, L. Franco and C. Viglianisi, Chem. Commun., 2015, 51, 11452–11454 RSC.
- R. Torres-Cavanillas, G. Escorcia-Ariza, I. Brotons-Alcázar, R. Sanchis-Gual, P. C. Mondal, L. E. Rosaleny, S. Giménez-Santamarina, M. Sessolo, M. Galbiati, S. Tatay, A. Gaita-Ariño, A. Forment-Aliaga and S. Cardona-Serra, J. Am. Chem. Soc., 2020, 142, 17572–17580 CrossRef CAS PubMed.
- C. D. Aiello, J. M. Abendroth, M. Abbas, A. Afanasev, S. Agarwal, A. S. Banerjee, D. N. Beratan, J. N. Belling, B. Berche, A. Botana, J. R. Caram, G. L. Celardo, G. Cuniberti, A. Garcia-Etxarri, A. Dianat, I. Diez-Perez, Y. Guo, R. Gutierrez, C. Herrmann, J. Hihath, S. Kale, P. Kurian, Y.-C. Lai, T. Liu, A. Lopez, E. Medina, V. Mujica, R. Naaman, M. Noormandipour, J. L. Palma, Y. Paltiel, W. Petuskey, J. C. Ribeiro-Silva, J. J. Saenz, E. J. G. Santos, M. Solyanik-Gorgone, V. J. Sorger, D. M. Stemer, J. M. Ugalde, A. Valdes-Curiel, S. Varela, D. H. Waldeck, M. R. Wasielewski, P. S. Weiss, H. Zacharias and Q. H. Wang, ACS Nano, 2022, 16, 4989–5035 CrossRef CAS PubMed.
- M. T. Anthony and M. P. Seah, Surf. Interface Anal., 1984, 6, 95–106 CrossRef CAS.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- M. S. Deleuze, J. Chem. Phys., 2002, 116, 7012–7026 CrossRef CAS.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- J. Rieß, M. Lublow, S. Anders, M. Tasbihi, A. Acharjya, K. Kailasam, A. Thomas, M. Schwarze and R. Schomäcker, Photochem. Photobiol. Sci., 2019, 18, 1833–1839 CrossRef PubMed.
- L. Poggini, A. Lunghi, A. Collauto, A. Barbon, L. Armelao, A. Magnani, A. Caneschi, F. Totti, L. Sorace and M. Mannini, Nanoscale, 2021, 13, 7613–7621 RSC.
- M. Mannini, L. Sorace, L. Gorini, F. M. Piras, A. Caneschi, A. Magnani, S. Menichetti and D. Gatteschi, Langmuir, 2007, 23, 2389–2397 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- D.-Y. Zhang, Y. Sang, T. K. Das, Z. Guan, N. Zhong, C.-G. Duan, W. Wang, J. Fransson, R. Naaman and H.-B. Yang, J. Am. Chem. Soc., 2023, 145, 26791–26798 CrossRef CAS PubMed.
- T. Kumar Das, A. K. Mondal, O. S. Tiwari, P. Makam, G. Leitus, E. Gazit, F. Claudio and R. Naaman, Phys. Chem. Chem. Phys., 2023, 25, 22124–22129 RSC.
- J. Labella, D. K. Bhowmick, A. Kumar, R. Naaman and T. Torres, Chem. Sci., 2023, 14, 4273–4277 RSC.
- F. Tassinari, D. R. Jayarathna, N. Kantor-Uriel, K. L. Davis, V. Varade, C. Achim and R. Naaman, Adv. Mater., 2018, 30, 1706423 CrossRef PubMed.
- Z. H. Xiong, D. Wu, Z. Valy Vardeny and J. Shi, Nature, 2004, 427, 821–824 CrossRef CAS PubMed.
- S. Alwan, S. Sarkar, A. Sharoni and Y. Dubi, J. Chem. Phys., 2023, 159, 3257–3264 CrossRef PubMed.
- M. Julliere, Phys. Lett. A, 1975, 54, 225–226 CrossRef.
- T. Liu and P. S. Weiss, ACS Nano, 2023, 17, 19502–19507 CrossRef CAS PubMed.
- R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709–7719 CrossRef PubMed.
- R. Nakajima, D. Hirobe, G. Kawaguchi, Y. Nabei, T. Sato, T. Narushima, H. Okamoto and H. M. Yamamoto, Nature, 2023, 613, 479–484 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00944d |
| This journal is © The Royal Society of Chemistry 2024 |