
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriplet formation inhibits amplified spontaneous emission in perylene-based polycyclic aromatic hydrocarbons†
Sergio
Moles Quintero
 a,
Jose C.
Mira-Martínez
b,
Ya
Zou
c,
Marcos
Díaz-Fernández
a,
Pedro G.
Boj
d,
Jishan
Wu
*c,
María A.
Díaz-García
*b,
Jose M.
Marín-Beloqui
*a and
Juan
Casado
*a
a,
Jose C.
Mira-Martínez
b,
Ya
Zou
c,
Marcos
Díaz-Fernández
a,
Pedro G.
Boj
d,
Jishan
Wu
*c,
María A.
Díaz-García
*b,
Jose M.
Marín-Beloqui
*a and
Juan
Casado
*a
aDepartment of Physical Chemistry, University of Malaga, Campus de Teatinos s/n, Malaga 29071, Spain. E-mail: jm.marinbeloqui@uma.es; casado@uma.es
bDepartamento Física Aplicada and Instituto Universitario de Materiales de Alicante, Universidad de Alicante, Alicante 03080, Spain. E-mail: maria.diaz@ua.es
cDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmwuj@nus.edu.sg
dDepartamento Óptica, Farmacología y Anatomía, and Instituto Universitario de Materiales de Alicante, Universidad de Alicante, Alicante 03080, Spain
First published on 26th February 2024
Abstract
Polycyclic aromatic hydrocarbons (PAHs) have demonstrated potential as active laser materials, showing good amplified stimulated emission (ASE) properties. However, the molecular origin of their ASE properties is still unclear and depends on each particular compound. In this work we study the ASE properties of polystyrene films hosting three different perylene-based PAHs YZ–1, YZ–2 and YZ–3, where only YZ–3 has displayed ASE. Their molecular structure has been systematically changed to establish the connection between their molecular structure and their ASE properties. A complete spectroscopic study, with ground state and time-resolved techniques, show that, even at low yields, triplets play a critical role as a major loss mechanism. Triplet slow relaxation to the ground state completely hinders the required imbalance of the S1 and S0 states for successful ASE. Quantum chemical calculations suggest that a lower number of available triplet states for YZ–3 are responsible for the blockage in triplet formation and, therefore, do not restrict ASE. This work not only presents a new PAH showing ASE, but also unequivocally proves the massive importance of triplet states in the development of organic lasers.
Introduction
Organic electronics has attracted the attention of scientists around the world due to their suitability for low-cost fabrication and manufacturing. An advantage of organic materials in opposition to their inorganic counterparts is the ability to fine-tune their properties with small changes in their molecular structure. This makes organic compounds (OCs) suitable for an ample range of applications from organic photovoltaics to transistors and sensors.1–3One of the applications of OCs is to serve as active units in lasing devices.4,5 Particularly interesting are those in which the OCs are dispersed in thermoplastic polymer films because they can be easily processed as low-loss waveguide films, leading to the creation of the so-called thin film organic lasers (TFOLs). A way to assess the suitability of a given OC for a TFOL is to identify the presence of amplified spontaneous emission (ASE). This phenomenon is caused by the amplification of the spontaneous emission, conventionally fluorescence, due to the presence of stimulated emission if the pumping excitation is strong enough. TFOLs open the door to new applications which need large area emitters or applications where it is important to have a smaller size than conventional inorganic laser devices.
In this work, we study the photophysical and ASE properties of three perylene-based polycyclic aromatic hydrocarbons (PAHs denoted as YZ1–3; Fig. 1), whose synthesis was recently reported.6 PAHs have several advantages due to their unique structure,7,8 mainly based on their great planarity provided by the extended π-conjugation. Besides, PAHs have structural tunability since they can be synthesised in a wide range of shapes which endow them with an ample range of properties.9–11 In terms of ASE, these molecules have the great advantage that their planarity usually provides a great fluorescence quantum yield (ΦF).12–14 Moreover, the possibility to make chemical modifications to modify their bandgap could in principle allow obtaining organic lasers operating at a wide range of wavelengths.
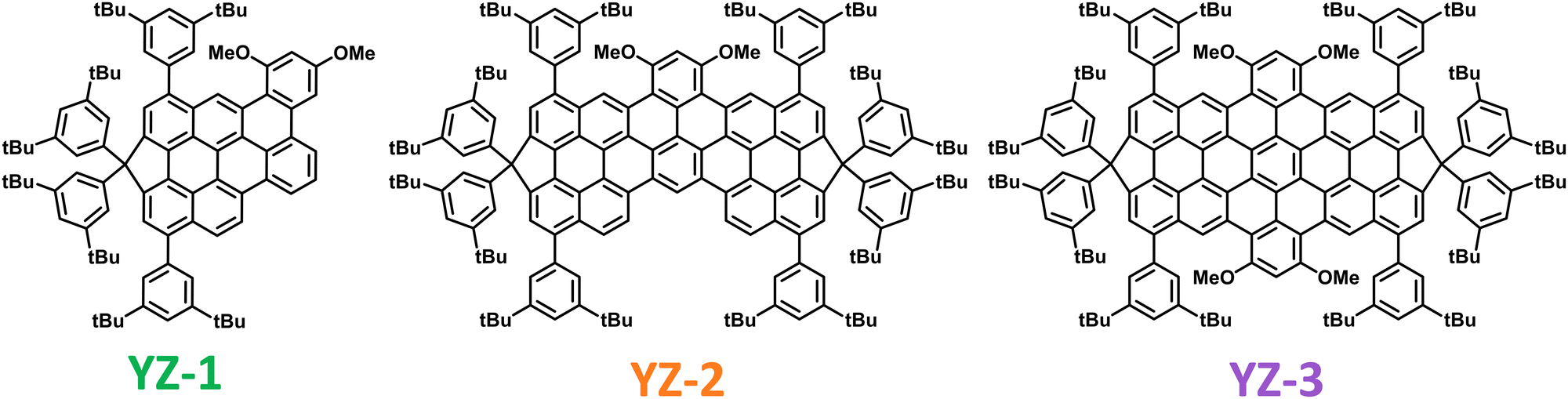 | ||
| Fig. 1 Structures of the molecules used for this work. | ||
There are many studies aiming to establish structure–property relationships in different PAH families,8,15,16 while less work has been done with π-extended PAHs considering the relatively recent demonstration of ASE and lasing.13,17–19 The establishment of suitable structure–property relationships for these compounds will lead to the diversified and optimal design of ASE molecules with larger efficiency.
YZ1–3 were deposited as films over quartz plates, using a small concentration of YZ molecules (0.5–6 wt%) dispersed in polystyrene (PS), used as a hosting passive matrix. Surprisingly, only YZ–3 showed ASE despite the three molecules share similar ΦF values; indeed YZ–3 shows the lowest value for ΦF. To ascertain the origin of this phenomenon, their excited state behaviour was studied by transient absorption spectroscopy (TAS). TAS is a very powerful pump–probe technique that allows direct probing of photoexcited species, as well as following their kinetics. TAS measures the difference in optical density (ΔOD) before and after the laser pulse. This makes the ΔOD intensity proportional to the number of photogenerated excited states.20,21 The thorough analysis of the TA data showed the formation of triplets in YZ–1 and YZ–2 though their TAS signals were almost obscured by the large absorbance of the co-existing long-lived singlet excited states. Despite the low triplet yield displayed in these molecules, it was enough to completely hinder the possibility of displaying ASE. Furthermore, with the help of DFT quantum calculations, it has been possible to link the YZ–3 chemical structure to the lack of triplet formation in this sample. This establishes a very important property–structure relationship which will help to synthesise further compounds with increased ASE efficiency.
Results
Optical properties
Fig. 2 shows the absorbance/emission properties in 2-MeTHF solution of the studied compounds. This solvent was used due to its ability to create an invisible solid matrix when the temperature is close to 80 K. As seen, the absorbance spectra at room temperature (300 K) for the three compounds show many narrow bands with the lowest energy band maximum at 483, 560 and 632 nm, for YZ–1, YZ–2 and YZ–3, respectively. The red-shift seen from YZ–1 to YZ–3 in the absorption spectra is in accordance with a larger effective conjugation length on YZ–1 → YZ–3.22,23 The PL spectra for the three molecules show a large 0–0 emission transition with a vibronic band deeper in the red region of the spectrum which can be likely associated with the 0–1 transition. The considerably narrow absorption and emission bands are attributed to a largely constrained molecular structure. This is further confirmed with the observed smaller Stokes shift which is directly correlated with the vibrational relaxation from the as-generated hot excited state upon vertical excitation and the lowest energy first excited state. In the studied compounds the Stoke shifts observed are 0.04, 0.03 and 0.02 eV, for YZ–1, YZ–2 and YZ–3 at 80 K (vide infra), respectively. This indicates a larger stiffness of the molecular structure. Moreover, the ΦF, i.e. the number of photons emitted per photon absorbed, was measured in 2-MeTHF solution. The obtained ΦF values were 0.70, 0.55 and 0.52, for YZ–1, YZ–2 and YZ–3, respectively. These results follow the same trend as the results obtained for CH2Cl2 solution obtained from ref. 6, where there is no larger ΦF for YZ–3. To eliminate any equipment/functional error from lab to lab, we also have repeated the experiments using CH2Cl2 as solvent using the same fluorimeter and integrating sphere (Fig. S1 and Table S1, ESI†). Indeed, no major differences were seen in terms of optical properties when the solvent was changed, besides a larger ΦF when CH2Cl2 is used as solvent.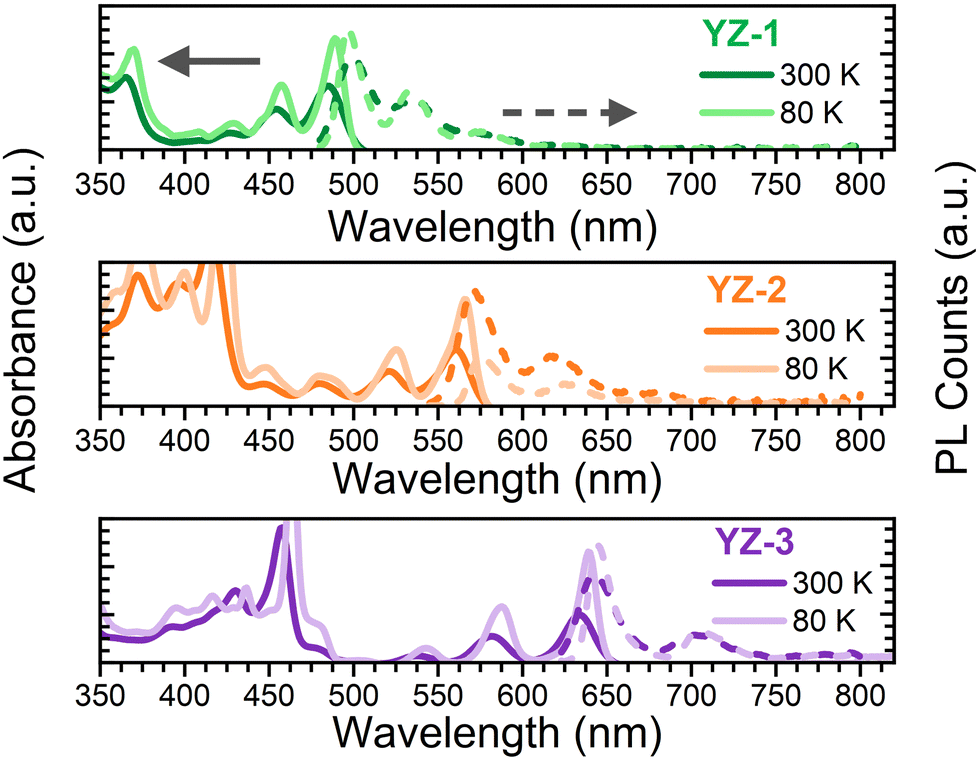 | ||
| Fig. 2 Absorbance (solid line) and PL (dashed line) obtained at room temperature (darker colour) and 80 K (lighter colour) for (top) YZ–1, (middle) YZ–2 and (bottom) YZ–3 in 2-MeTHF solutions. | ||
Fluorescence lifetimes have also been characterised (Table S1, ESI†). There is an increase in the fluorescence lifetime when increasing the size of the molecule (3.4, 8.0 and 9.3 ns for YZ–1, YZ–2 and YZ–3, respectively). These lifetimes are in line with the obtained ΦF values, as a shorter fluorescence lifetime is usually associated with a larger efficiency in the fluorescence process, and, therefore, a larger ΦF.
The absorbance and fluorescence were also measured at lower temperature (80 K) (Fig. 2). The data show a clearer resolution of all vibronic transition bands due to the decrease of spectral broadening with cooling.
ASE characterisation
For ASE characterisation, the compounds were embedded in PS, forming thin films deposited over quartz plates, and excited under ns optical pulses (see details in the Experimental section in the ESI†). Film thickness (values in Table S1, in the ESI†) was properly adjusted to ensure well confined transversal waveguide modes propagating through the waveguide, which is important to ensure an optimized ASE performance.24–26 The existence of ASE is revealed through the observation of narrowing of the PL spectrum, at a given excitation energy intensity (the so-called ASE threshold), together with a drastic increase of the emission intensity (Fig. 3). ASE was observed in the YZ–3-based films, but surprisingly not in the films containing YZ–1 and YZ–2, although the latter were excited under very severe excitation density conditions (up to 20 mJ cm−2) and various dye contents were tried (see Table S1, ESI†). In particular, Fig. 3(c) shows the clear presence of two different fluorescence linear regimes, in terms of intensity and bandwidth (blue squares and red circles, respectively) for YZ–3. The crossing energy point where one regime is transformed into the other one is the ASE threshold, which was 14.5 mJ cm−2 for YZ–3. This ASE threshold is rather large compared to the most efficient PAHs reported, which have shown values ranging from 30 μJ cm−2 to 5.3 mJ cm−2.13,27–29 Interestingly, the ASE spectral shape for the YZ–3 film has the maximum intensity matching the 0–0 transition of the PL spectrum. Generally, the most intense ASE band is associated with the 0–1 PL transition (and sometimes with the 0–2 PL one) as the close proximity between the 0–0 absorption and emission bands diminishes the gain by reabsorption of the emitted photons.30–33 Our hypothesis to explain why ASE occurs close to the 0–0 emission will be discussed in the next sections.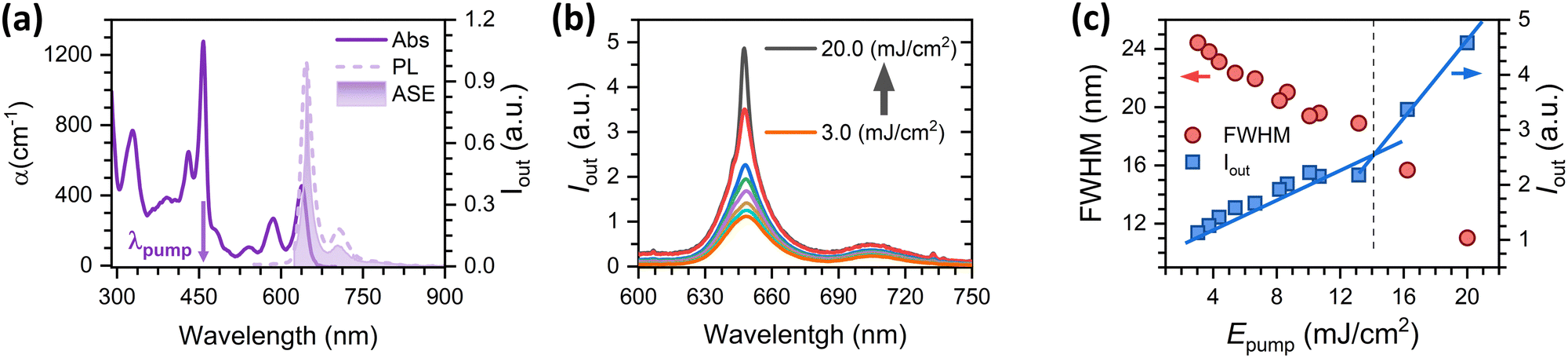 | ||
| Fig. 3 Results of ASE characterisation of a 1 wt% YZ–3 doped PS film. (a) Ground state absorbance (solid line), fluorescence (dashed line) and ASE emission (filled curve). (b) Emission spectra obtained at different pump energies of the same film. (c) Output intensity (right axis, blue squares) and emission linewidth defined as full with at half maximum, FWHM (left axis, red circles) plotted against different pump energies. Blue lines are a guide to the eye. PL excitation wavelength was set at 447 nm and ASE measurements were performed under pumping at the maximum of the absorption (λp = 458 nm). | ||
Transient absorption spectroscopy
To elucidate the origin for the lack of ASE in YZ–1 and YZ–2, despite their structures being similar to that of YZ–3, transient absorption spectroscopy characterisation was performed (Fig. 4). A similar analysis can be done for the three compounds. First, it is possible to see a large negative signal in a similar position to the largest ground state absorbance band at 483, 560 and 632 nm for YZ–1, YZ–2 and YZ–3, respectively. These bands are associated with ground state bleach (GSB). Also, with negative ΔOD, we find the stimulated emission (SE) of the molecules, matching their PL spectra, with maxima at 500, 570 and 640 nm for YZ–1, YZ–2 and YZ–3, respectively. The SE is also the origin for the local minima found in the positive signal at 535, 625 and 710 nm for YZ–1, YZ–2 and YZ–3, respectively, associated with the 0–1 band of their PL spectrum. Interestingly, the position of the 0–0 GSB band is red-shifted a few nm in comparison with the ground state absorbance. This is the result of the GSB and SE 0–0 bands merge due to their minimal Stokes shift. This merge hinders proper characterisation of GSB or SE lifetimes in their 0–0 band, and, instead, the 0–1 band has been used for the study of both processes (Fig. S2, ESI†). Despite SE being a common feature in the fs-characterisation of organic molecules for organic electronics,34,35 the large SE lifetime of these compounds is remarkable, reaching the nanosecond timescale. These large SE lifetimes have been seen previously in the literature in organic molecules displaying ASE, indicating that this could be an important fingerprint for this kind of molecules. On the other hand, the TA data shows in all cases a large positive band in the 600–800 nm region (with maxima at 630, 695 and 745 nm for YZ–1, YZ–2 and YZ–3, respectively). These bands start to decay in a few nanoseconds with little spectral modification along the technique timescale. These bands were associated with excited state absorption (ESA) of the excited singlet species given their lifetime similarity to the obtained PL lifetime (Table S1, ESI†). | ||
| Fig. 4 Femtosecond transient absorption spectroscopy results of (a) YZ–1, (b) YZ–2 and (c) YZ–3 in 2-MeTHF solution. Normalised species associated spectra obtained by global analysis for (d) YZ–1, (e) YZ–2, and (f) YZ–3 fs-TAS data. Fs-TAS was obtained by exciting at 490, 560 and 640 nm for YZ–1, YZ–2 and YZ–3, respectively with a power of 0.25 mW. | ||
As seen, the singlet absorbance spectra for the three molecules are in the same region as the bands associated with the SE. This spectral overlap between the SE and the ESA is associated with a lack of gain in other compounds and restrains their abilities to obtain net optical gain, and therefore hinders the possibilities of ASE.36 The spectral overlap of the SE and the ESA means that the ASE photons could be re-absorbed by the excited species decreasing the stimulated emission efficiency. YZ–3 shows a narrower ESA in comparison with the two other molecules. However, the differences are not enough to justify YZ–3 being the only one displaying ASE.
This overlap between the ESA and the SE also serves as the reason for the observation of the ASE spectrum showing the 0–0 PL transition. This is quite unusual since in most situations the ASE matches the 0–1 or 0–2 transitions, particularly in compounds with small Stokes shifts (see above).30–32 In YZ–3, the 0–1 and 0–2 SE transitions are coincident with the ESA, which has a similar extinction coefficient to the ground state (given the similar amplitude of ESA and GSB at short time delays, where only singlet states are present). Hence, given this similarity in the absorption coefficient, YZ–3 emitted photons along the whole spectrum are going to be reabsorbed to a similar extent. The photons corresponding to the 0–1 and 0–2 transitions are reabsorbed by the excited state while the photons associated with the 0–0 transition will be reabsorbed by the ground state. The ASE spectrum will therefore have the same spectral shape as the PL spectrum.
This reabsorption of the emitted photons by the ground and excited states remains as the origin for a high power of 14.5 mJ cm−2 is needed to start to see the ASE phenomenon. As indicated previously, this ASE threshold is rather large compared to other examples reported in the literature, with values that go from 30 μJ cm−2 to 5.3 mJ cm−2.13,27–29
To further unravel the dynamics of these compounds, global analysis (GA) was performed using the TA data (Fig. 4(d)–(f)). Interestingly, this analysis showed the presence of a new species in both, YZ–1 and YZ–2 that was obscured due to the large lifetime and intensity of the singlet states. This new species is formed in the nanosecond timescale (Fig. S3, ESI†) and remains constant at the technique time resolution.
To identify these new bands present in YZ–1 and YZ–2, we performed μs-TAS (Fig. S4, ESI†). The μs-TAS on YZ–1 shows a double band (at 540 and 640 nm) positive spectrum that decays with negligible changes with a lifetime of 360 μs. YZ–2 μs-TAS data show a less defined spectrum that decays in 170 μs. In both cases these signals were quenched in the presence of oxygen and fully recovered upon being purged with nitrogen; all being indications of the formation of triplet species. The similarity of the spectra obtained by μs-TAS to the spectra of the species formed in the nanosecond timescale seen in GA of fs-TA (Fig. S5, ESI†) clearly proved the identity of these species as triplet states.
In addition, the GA also gives information about the relative population of the states present in our molecules paying attention to the intensity to the decays associated with each species (Fig. S3, ESI†). In our case it is quite difficult to obtain suitable values as stimulated emission, triplet formation and non-radiative recombination occur in a very similar time scale. Still, we can do an estimation of the triplet yield for YZ–1: 0.1. The YZ–2 triplet is shown in a much lower intensity and it is difficult to calculate a suitable value for its formation, but it is lower than for YZ–1.
Fs-TAS data was also obtained in the same configuration used for the ASE characterisation (dispersed in a 1 wt% PS solid matrix, Fig. S6, ESI†). The solid state results were similar to those observed in solution. However, in the films, the singlet species relaxed faster to the ground state in line with their solid-state form. Their shorter singlet lifetime, which completely decays in 100 ps, allows us to clearly discern the spectral shape of triplets in YZ–1 and YZ–2. On the other hand, in YZ–3 the disappearance of the singlet left no signal, further demonstrating the inability of YZ–3 to form triplets.
Discussion
Despite the large ΦF obtained for the three molecules studied in this work, only YZ–3 displayed ASE. Given that the ΦF value of YZ–3 (0.52) is lower than that of YZ–1 (0.70) and similar to that of YZ–2 (0.55), one would expect similar ASE properties. Hence, the ΦF difference cannot explain the lack of ASE for YZ–1 and YZ–2. In addition, the fluorescence lifetimes associated with the studied molecules can neither explain the lack of ASE properties for YZ–1 and YZ–2. Indeed, YZ–3, the only molecule displaying ASE, is the one with the slowest fluorescence lifetime among the three, which is clearly unexpected as usually a shorter lifetime is associated with a larger ASE efficiency.The formation of triplets in these compounds, however, serves as a feasible explanation for the lack of ASE on YZ–1 and YZ–2. For ASE to work, a proper imbalance (towards population inversion) from the ground state and the first singlet state, which owes the emission, is needed. Formation of triplets, with their inherent large decay to ground state lifetime, impedes the re-population of singlets. According to the seminal work of Garnier et al.,37 for ASE to succeed 4 different vibronic levels are needed: S0,0, S0,v, S1,0, S1,v′ (Fig. 5). For amplified spontaneous emission to be effective, the vibration relaxation processes S1,v′ → S1,0 and S0,v → S0,0, processes 2 and 4, respectively, in Fig. 5, have to be extremely efficient. The importance of the efficiency of these processes lies in the prevention of the excessive population of any intermediate state that can hinder the population of the energy levels that are responsible of the ASE phenomenon (S0,0 and S1,0). However, the presence of triplets and their inherent large relaxation to ground state lifetime hampers the population of the ground state, and, hence, the repopulation of the S1,0 state. The lack of the unbalanced population due to the presence of triplet states and their breakage of the 4-level system represent the main reason for YZ–1 and YZ–2 molecules not displaying ASE properties. Another important point to add to the matter is the very low intensity of the triplet absorbance for YZ–1 and YZ–2 (Fig. 4). This clearly suggests a low population of triplet states, and, hence, a very low triplet formation yield. This low triplet yield is expected for such planar molecules and, consequently, low spin–orbit coupling. Even in a regime of low triplet yield, it was enough to completely inhibit the ASE properties of YZ–1 and YZ–2.
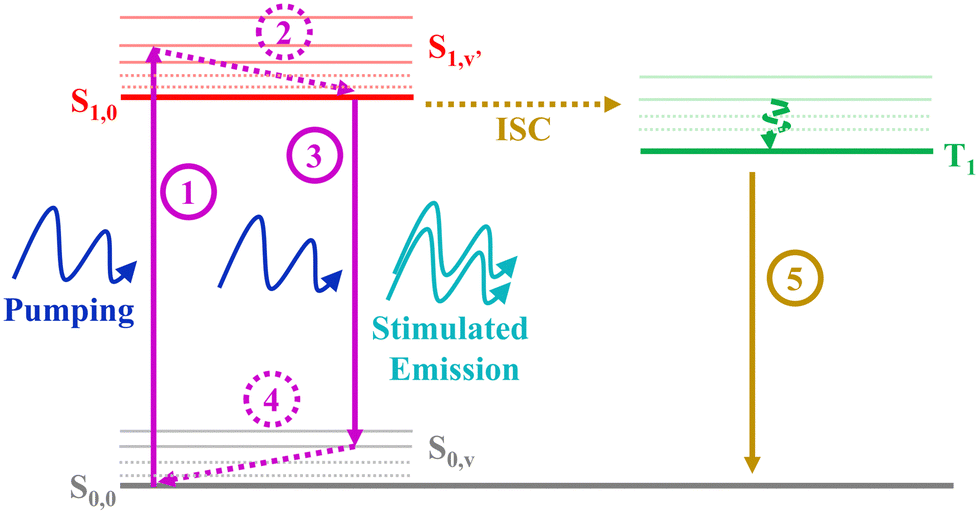 | ||
| Fig. 5 Scheme depicting the 4-level scheme for successful ASE and the effect of triplet level addition. | ||
Interestingly, the lowest ΦF value for the YZ–3 contrasts with the lack of triplet formation. We can write ΦF as 1 − Φnr − ΦT where Φnr and ΦT are the yields of non-radiative recombination and triplet formation, respectively. Therefore, with a lower ΦF, a larger ΦT can be expected. Hence, for YZ–3, to compensate the negligible ΦT, a much larger Φnr is expected. A similar case is expected for YZ–2, with a lower ΦF and lower intensity of the triplet states in comparison with YZ–1. The increase in the non-radiative Φnr from YZ–1 to YZ–3 (ca. 0.2, 0.4 and 0.5) is in line with the energy gap law.38 According to this law, when the closer two different states are in energetics, the rate of the internal conversion, i.e., non-radiative, between these states increases. Hence, the smaller the bandgap a molecule posseses, a larger yield of non-radiative relaxation is expected, which is in line with what was seen for YZ–n.
To further investigate the origin of this difference in triplet formation despite the little structural changes, the vertical transitions were calculated by TD-DFT at the B3LYP/6-31G level of theory (calculated energy levels are presented in Fig. 6 and molecular orbitals in Fig. S7, ESI†). The triplet formation in our compounds should go along the following path: the sample is excited to Sn to swiftly relax to the most stable singlet level, S1. Then, two different characteristics have to be accomplished in order to undergo intersystem crossing. The difference between singlet and triplet energy levels has to be minimal (lower than 0.2 eV). Also, a large difference in the orbital composition from the S1 to the Tn, this is, a large spin–orbit coupling (SOC), is needed.39–42 As seen by the calculation results (Fig. 6(b)), all three molecules show very little spin–orbit constant, indicating that the major weight of the ISC process has to rely on the density of available triplet states. YZ–1 has a triplet energy level almost isoenergetic to the S1, providing a favorable opportunity to ISC despite the very low SOC constant for this material. On the other hand, YZ–2 has two available states in the ±0.2 eV range, with one triplet energy level with the largest SOC constant among the three molecules. However, the YZ–3 has only one available triplet state at ±0.2 eV and its SOC is half of the one for the YZ–2.
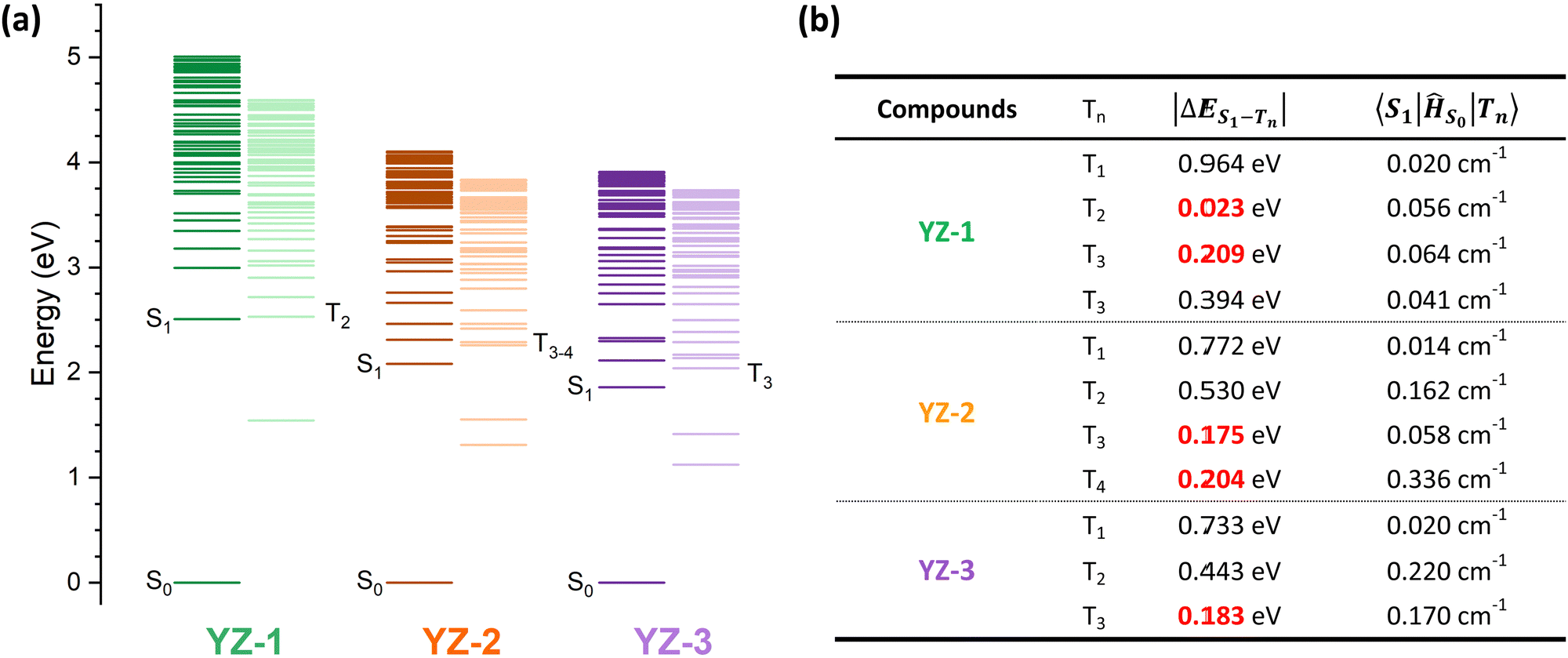 | ||
| Fig. 6 Results of the TD-DFT calculations at the B3LYP/6-31G** level of theory. (a) Calculated energy level of the singlet and triplet excited states for YZ–1, YZ–2 and YZ–3. (b) Table summarising the results of the TD-DFT calculations of the triplet energy levels including the difference in energy from the S1 and the spin–orbit coupling value for the transition from S1 to the corresponding triplet level. To help the eye, the triplet energy levels within ca. ±0.2 eV have been highlighted in red. Spin orbit coupling has been calculated using the ORCA software package. | ||
Noticeably, the calculated T1 energy level of YZ–3 is much closer to the S0. According to the energy gap law, as explained previously, a very fast relaxation from this T1 energy level to the ground state could be expected. Then, this could serve as an explanation of the lack of triplet species in TAS characterisation. The energy proximity of these states could increase the internal conversion rate from T1 to S0 by several orders of magnitude from the microsecond, time range where triplets usually decay, to the nanosecond time range where YZ–3 singlets decay. This similarity in timescales could obscure the presence of triplets in the TAS characterisation. However, this cannot be the case according to the TAS results of YZ–3 in the solid matrix (Fig. S6, ESI†). When YZ–n molecules are suspended in a solid matrix, the singlet states decay in a faster manner, clearly revealing the presence of triplet states for YZ–1 and YZ–2. However, in the case of YZ–3, the TA signal becomes zero in 1 ns, neglecting the hypothesis of a very short-lived triplet state that decays from a triplet energy state with small ΔES0–T1.
Conclusions
Three different perylene-based PAH molecules have been studied to characterise their ASE properties: YZ–1, YZ–2 and YZ–3. From these three compounds, only YZ–3 showed ASE at a wavelength of 648 nm with an energy threshold of 14.5 mJ cm−2. These differences could not be explained in terms of ΦF or fluorescence lifetime where YZ–1 showed the largest ΦF and shortest fluorescence lifetime. Transient absorption spectroscopy indicated that YZ–1 and YZ–2 were able to create triplets whereas YZ–3 could not. The origin of this behaviour was a lower density of triplet states for YZ–3 in comparison with the smaller molecules. Triplets, with their large lifetimes, represent a major loss mechanism because they neglect the 4-level diagram for ASE, where the relaxation from the non-emissive excited state to the ground state has to be largely efficient to lead to unbalanced population. This work clearly exemplifies how triplet large lifetime represents a major loss mechanism for ASE even at low yields. In addition, this work clearly shows how little changes in the molecular structure, as seen by the difference between YZ–2 and YZ–3, have massive importance in the density of excited states, and therefore, in the triplet formation and ASE properties. This work paves the way for the development of new molecules where triplet generation is completely inhibited to obtain largely efficient organic lasers.Author contributions
S. M. Q.: ground state spectroscopic characterisation and quantum calculations. J. M.-M. and P. B.: fabrication of films and optical and ASE characterisation. Y. Z.: molecule synthesis. M. D.-F.: PL and TAS characterisation. J. W.: synthesis supervision and manuscript revision. M. D.-G.: ASE device supervision and analysis; writing and editing. J. M.-B.: transient absorption characterisation and analysis, original draft, writing, editing and project supervision. J. C.: writing, editing and project supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. M.-B. wants to acknowledge the Spanish University Ministry and the European Union for his Maria Zambrano fellowship with NextGen-Eu funding. M. D.-F. acknowledges the Spanish Science and Innovation Ministry for his FPI fellowship. J. W. acknowledges financial support from A*STAR AME grant (A20E5c0089). The group at the University of Alicante is grateful for financial support from the “Ministerio de Ciencia e Innovación” (MCIN) of Spain and the European Regional Development Fund (grant No. PID2020-119124RB-I00) and from the Generalitat Valenciana through grant No. AICO/2021/093. Besides, this study is part of the Advanced Materials program supported by the Spanish MCIN with funding from European Union Next Generation EU and by Generalitat Valenciana (grant no. MFA/2022/045). The group at the UA thanks Dr J. M. Villalvilla and Dr J. A. Quintana for useful discussions. We also thank the Research Central Services (SCAI) of the University of Málaga for the access to the EVI, EEL and MENL to perform ground state and transient spectroscopic characterisation. J. C. is grateful for funding provided by MICIN/FEDER (PID2021-127127NB-I00 and PID2019-110305GB-I00) and Junta de Andalucía (PROYEXCEL-0328).References
- W. Lowrie, R. J. E. Westbrook, J. Guo, H. I. Gonev, J. Marin-Beloqui and T. M. Clarke, J. Chem. Phys., 2023, 158, 110901 CrossRef CAS PubMed
.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS
- Z. Ren, J. Yang, D. Qi, P. Sonar, L. Liu, Z. Lou, G. Shen and Z. Wei, Adv. Mater. Technol., 2021, 6, 2000889 CrossRef CAS
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed
- Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC
- Y. Zou, Y. Han, S. Wu, X. Hou, C. H. E. Chow and J. Wu, Angew. Chem., Int. Ed., 2021, 60, 17654–17663 CrossRef CAS PubMed
- C. Aumaitre and J. Morin, Chem. Rec., 2019, 19, 1142–1154 CrossRef CAS PubMed
- L. H. Nguyen and T. N. Truong, ACS Omega, 2018, 3, 8913–8922 CrossRef CAS PubMed
- J. P. Larkindale and D. J. Simkin, J. Chem. Phys., 1971, 55, 5668–5674 CrossRef CAS
- X.-L. Fan, X.-Q. Wang, J.-T. Wang and H.-D. Li, Phys. Lett. A, 2014, 378, 1379–1382 CrossRef CAS
- X. Feng, J. Wu, M. Ai, W. Pisula, L. Zhi, J. P. Rabe and K. Müllen, Angew. Chem., 2007, 119, 3093–3096 CrossRef
- J.-K. Bin and J.-I. Hong, Org. Electron., 2011, 12, 802–808 CrossRef CAS
- G. M. Paternò, Q. Chen, X. Wang, J. Liu, S. G. Motti, A. Petrozza, X. Feng, G. Lanzani, K. Müllen, A. Narita and F. Scotognella, Angew. Chem., Int. Ed., 2017, 56, 6753–6757 CrossRef PubMed
- Y. Zou, V. Bonal, S. Moles Quintero, P. G. Boj, J. M. Villalvilla, J. A. Quintana, G. Li, S. Wu, Q. Jiang, Y. Ni, J. Casado, M. A. Díaz-García and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 14927–14934 CrossRef CAS PubMed
- M. Misra, D. Andrienko, B. Baumeier, J.-L. Faulon and O. A. von Lilienfeld, J. Chem. Theory Comput., 2011, 7, 2549–2555 CrossRef CAS PubMed
- H. Narita, H. Choi, M. Ito, N. Ando, S. Ogi and S. Yamaguchi, Chem. Sci., 2022, 13, 1484–1491 RSC
- G. M. Paternò, L. Moretti, A. J. Barker, Q. Chen, K. Müllen, A. Narita, G. Cerullo, F. Scotognella and G. Lanzani, Adv. Funct. Mater., 2019, 29, 1805249 CrossRef
- V. Bonal, R. Muñoz-Mármol, F. Gordillo Gámez, M. Morales-Vidal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Nat. Commun., 2019, 10, 3327 CrossRef PubMed
- R. Muñoz-Mármol, F. Gordillo, V. Bonal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, A. M. Ross, G. M. Paternò, F. Scotognella, G. Lanzani, A. Derradji, J. C. Sancho-García, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Adv. Funct. Mater., 2021, 2105073 CrossRef
- J. M. Marin-Beloqui, K. J. Fallon, H. Bronstein and T. M. Clarke, J. Phys. Chem. Lett., 2019, 10, 3813–3819 CrossRef CAS PubMed
- P. Westacott, J. R. Tumbleston, S. Shoaee, S. Fearn, J. H. Bannock, J. B. Gilchrist, S. Heutz, J. deMello, M. Heeney, H. Ade, J. Durrant, D. S. McPhail and N. Stingelin, Energy Environ. Sci., 2013, 6, 2756 RSC
- M. Fujitsuka, D. W. Cho, T. Iwamoto, S. Yamago and T. Majima, Phys. Chem. Chem. Phys., 2012, 14, 14585 RSC
- B. Matarranz, G. Ghosh, R. Kandanelli, A. Sampedro, K. K. Kartha and G. Fernández, Chem. Commun., 2021, 57, 4890–4893 RSC
- M. Anni, A. Perulli and G. Monti, J. Appl. Phys., 2012, 111, 093109 CrossRef
- E. M. Calzado, M. G. Ramírez, P. G. Boj and M. A. D. García, Appl. Opt., 2012, 51, 3287 CrossRef CAS PubMed
- E. M. Calzado, J. M. Villalvilla, P. G. Boj, J. A. Quintana and M. A. Díaz-García, J. Appl. Phys., 2005, 97, 093103 CrossRef
- Y. Gu, V. Vega-Mayoral, S. Garcia-Orrit, D. Schollmeyer, A. Narita, J. Cabanillas-González, Z. Qiu and K. Müllen, Angew. Chem., Int. Ed., 2022, 61, e202201088 CrossRef CAS PubMed
- X. Xu, G. Serra, A. Villa, R. Muñoz-Mármol, S. Vasylevskyi, M. Gadea, A. Lucotti, Z. Lin, P. G. Boj, R. Kabe, M. Tommasini, M. Á. Díaz-García, F. Scotognella, G. M. Paternò and A. Narita, Chem. Sci., 2022, 13, 13040–13045 RSC
- G. M. Paternò, Q. Chen, R. Muñoz-Mármol, M. Guizzardi, V. Bonal, R. Kabe, A. J. Barker, P. G. Boj, S. Chatterjee, Y. Ie, J. M. Villalvilla, J. A. Quintana, F. Scotognella, K. Müllen, M. A. Díaz-García, A. Narita and G. Lanzani, Mater. Horiz., 2022, 9, 393–402 RSC
- Y. Zou, V. Bonal, S. Moles Quintero, P. G. Boj, J. M. Villalvilla, J. A. Quintana, G. Li, S. Wu, Q. Jiang, Y. Ni, J. Casado, M. A. Díaz-García and J. Wu, Angew. Chem., 2020, 132, 15037–15044 CrossRef
- R. Kabe, H. Nakanotani, T. Sakanoue, M. Yahiro and C. Adachi, Adv. Mater., 2009, 21, 4034–4038 CrossRef CAS
- A. Mahfoud, A. Sarangan, T. R. Nelson and E. A. Blubaugh, J. Lumin., 2006, 118, 123–130 CrossRef CAS
- J. Casado, V. Hernández, J. T. López Navarrete, M. Algarra, D. A. da Silva Filho, S. Yamaguchi, R. Rondão, J. S. Seixas de Melo, V. Navarro-Fuster, P. G. Boj and M. A. Díaz-García, Adv. Opt. Mater., 2013, 1, 588–599 CrossRef
- Y. Yang, Z. Jiang, Y. Liu, T. Guan, Q. Zhang, C. Qin, K. Jiang and Y. Liu, J. Phys. Chem. Lett., 2022, 13, 9381–9389 CrossRef CAS PubMed
- A. Portone, L. Ganzer, F. Branchi, R. Ramos, M. J. Caldas, D. Pisignano, E. Molinari, G. Cerullo, L. Persano, D. Prezzi and T. Virgili, Sci. Rep., 2019, 9, 7370 CrossRef PubMed
- X. Xu, R. Muñoz-Mármol, S. Vasylevskyi, A. Villa, G. Folpini, F. Scotognella, G. Maria Paternò and A. Narita, Angew. Chem., Int. Ed., 2023, 62, e202218350 CrossRef CAS PubMed
- G. Horowitz, F. Kouki, A. El Kassmi, P. Valat, V. Wintgens and F. Garnier, Adv. Mater., 1999, 11, 234–238 CrossRef CAS
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS
- S. Medina Rivero, M. J. Alonso-Navarro, C. Tonnelé, J. M. Marín-Beloqui, F. Suárez-Blas, T. M. Clarke, S. Kang, J. Oh, M. M. Ramos, D. Kim, D. Casanova, J. L. Segura and J. Casado, J. Am. Chem. Soc., 2023, 145, 27295–27306 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04740g |
| This journal is © The Royal Society of Chemistry 2024 |